7 Oligodendrogliomas
Frequency and Incidence
According to data collected by the Central Brain Tumor Registry of the United States (CBTRUS 2007-2008), OD, OA, and anaplastic oligodendroglioma (AO) accounted for 5.1%, 2.8%, and 2.8% respectively of neuroepithelial tumors and 2.0%, 1.0%, and 1.1% of all primary brain tumors. Therefore low-grade oligodendroglial tumors are three times more frequent than anaplastic tumors, and in total represent 4.1% of tumors compared with 8.5% for astrocytomas.1 Assuming a population of just over 58 million people in the United Kingdom and an incidence rate of between 15/100,0002 and 21/100,0003 for all primary brain tumors, there are approximately 428 new cases of OD/OA/AO per year.
Histology
The realization that oligodendrogliomas are more chemosensitive than astrocytomas has increased the enthusiasm amongst neuropathologists to search hard for evidence of oligodendroglial differentiation in biopsy and resection specimens.4 This in turn has ignited considerable debate about the minimum criteria for OD/OA.
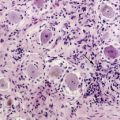
Oligodendrogliomas can present at any age and in any location in the brain, although they frequently occur in cortical locations in the frontotemporal lobes as partially calcified mass lesions. Macroscopically, they may be distinguished by the presence of a mucoid matrix that is softer than the firmer surrounding brain. They often infiltrate grey matter and change the appearance of the cerebral cortex. The most characteristic microscopic feature of OD are the sheets of uniformly rounded cells and perinuclear halos, giving the so-called fried egg appearance, due to a technical artifact whereby the cytoplasm swells up in paraffin sections after a delay in fixation (Figure 7-1). This is not always present, however, particularly if the tumor specimen is frozen prior to being embedded in paraffin, nor is this appearance pathognomic for OD, as it also occurs in ependymomas and central neurocytomas. The key pathological feature that distinguishes OD from astrocytomas is the fact that cell density and nuclear size and shape are much more uniform. Astrocytomas typically display considerable nuclear pleomorphism and, in particular, angulated nuclei with spindle-shaped cells. Another classic feature is the presence of a rich, thin-walled capillary network arranged in a way that resembles chicken wire. Other frequent features include calcification, either within the tumor or within the surrounding brain, and cortical invasion with perineuronal or perivascular cells, known as satellitosis.
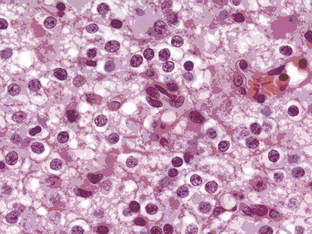
Grade II oligodendrogliomas have a low mitotic index (less than 5%), although there may be well-circumscribed foci of higher cellularity, an occasional mitosis, and cellular atypia.5 By definition, OD cannot have “significant mitotic activity,” but this is a vague term and the exact number of mitoses has not been defined, although in a recent large series, a cutoff value of 6 to 10 mitoses per high power field correlated with reduced survival.6
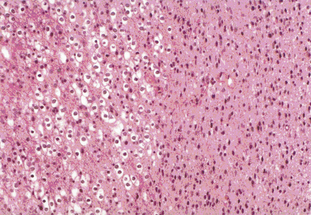
Tumors with a mixture of astrocytic and oligodendroglial components are called oligoastrocytomas (OA) or mixed gliomas, which, by definition, show divergent forms of glial differentiation. The two cell types may occur as regionally distinct and cytologically distinct populations of cells within the same tumor (Figure 7-2) or, more rarely, as intermixed populations containing cells with both astrocytic and oligodendroglial phenotypes. Many oligodendrogliomas contain GFAP-positive cells (i.e., cells of astrocytic lineage) and so the demonstration of astrocyte-like cells in an OD should not be regarded as necessarily diagnostic of an oligoastrocytoma, unless the proportion of the minority cell type exceeds some arbitrary figure ranging from 20% to 60%. Particular care should be taken when interpreting frozen specimens, as the process of freezing can change cells with an oligodendroglial morphology to cells with a more astrocytoma-like pattern with dark irregular nuclei. These tumors are regarded as morphologically ambiguous tumors and further characterization can only be carried out using molecular genetics. The variable criteria for the diagnosis of an OA has been succinctly reviewed by Burger.7
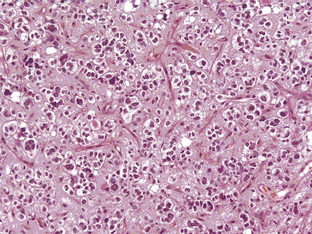
The grade III anaplastic OD (AO) still retains the characteristic nuclear uniformity of OD but is more cellular, and shows more nuclear atypia, frequent mitotic activity, and noticeable microvascular proliferation or hypertrophy (Figure 7-3). AO may present de novo or evolve from malignant transformation of a pure OD. Necrosis may be present and does not in itself indicate a grade IV glioma.8 The vessels are frequently thickened and hypertrophied but do not form the glomerular tufts seen in glioblastoma multiforme (GBM). Most AOs do not evolve into GBMs; however, there are occasional cases and, in these, it is not really possible to distinguish between a “highly malignant” AO and a GBM with some oligodendroglial features. In the most recent WHO classification (2007), anaplastic oligoastrocytoma (AOA) with necrosis was reclassified as GBM-O. Recent data from a large trial of AOs and AOAs suggest that these tumors have similar survival curves to that of GBM and confirm this reclassification. Whether this tumor is a separate clinicopathological and molecular genetic entity remains to be proven.
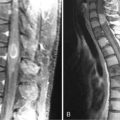
Oligodendrogliomas have a greater propensity for leptomeningeal spread than astrocytic tumors. This occurs in about 1% to 2% of cases, with systemic metastases also being occasionally reported. This may be a consequence of the longer survival of patients as compared to other glioma subtypes and of heterozygous deletions of CDKN2A/p 16 genes.9
Molecular genetics
Oligodendrogliomas are associated with specific genetic abnormalities, namely loss of the short arm of chromosome 1 (1 p) and the long arm of chromosome 19 (19 q). This was first demonstrated 10 years ago in a genomic wide analysis,10 and subsequently linked in retrospective studies to a durable response to a combination of procarbazine, CCNU, and Vincristine (PCV) as well as longer survival.11,12 In contrast, response rates for patients with isolated 1 p loss or no loss was less than 25%, although the presence of 1 p loss may also identify other treatment-sensitive malignant gliomas, including rare GBMs (see Table 7-1).13 The chromosomal locations involved in the favourable therapeutic response have been further narrowed down to lesions at 1p36 and 19q13.3.14 Of the two markers, 1 p has the greater specificity since 19 q losses have also been reported in high-grade astrocytomas and OA.15
TABLE 7-1 1 p and 19 q Losses versus Glioma Subtypes and Primary/Recurrent Status Adapted from Smith et al. 200013
The frequency of these losses varies between different grades of glioma and different histological subtypes (see Figure 7-1). As can be seen, the 1 p/19 q loss is not specific to OD as it also occurs in OA and a minority of astrocytomas; however, it appears to be a statistically significant predictor of prolonged survival in patients with pure OD, irrespective of tumor grade, but not in patients with OA or astrocytomas.16 The presence of a typical perinuclear halo in more than 50% of tumor cells and a “chicken wire” vascular pattern is seen in more than 90% of tumors showing LOH on 1 p or 19 q.17
More recently, mutations of two enzymes involved in the Krebs cycle, isocitrate dehydrogenase (IDH) 1 and 2, have been described in 84% of OD, 94% of AO, and 100% of OA and AOA, making this an even more common molecular signature than 1p19q.18 These mutations are rarely seen in primary GBM.
Clinical Features and Natural History
Most ODs are slow-growing tumors; they usually present in adults between the ages of 30 and 50 with partial or generalized seizures. The incidence of seizures is lower in AO than in OD/OA.19 Occasionally, they occur in older patients. In these cases, they more commonly present with symptoms of an expanding, infiltrating mass lesion (raised intracranial pressure with or without focal neurological deficits).20 They are more likely to present as an intracerebral hemorrhage than other low-grade gliomas, probably because of their thin-walled capillary network.
ODs and other low-grade gliomas account for up to 15% of patients with medically intractable seizures.21 Seizure control may vary and, particularly in tumors occurring in the motor strip, be resistant to anticonvulsant polypharmacy.22 In this situation, patients may be considered for “epilepsy surgery” on symptomatic grounds rather than on pure oncological grounds. The relationship between the molecular genetics and their mode of presentation is not clear, but the clinical phenotype is presumably related in some way to the speed of growth and the duration of time that the tumor has been present in the brain. This is supported by the observation that there is an inverse relationship between tumor grade and frequency of seizures—as a general rule, the more benign the tumor, the more likely it is to be associated with intractable epilepsy.
The prognosis of OD is significantly better than for astrocytomas, with a median survival of 10 years or more. As with astrocytomas, there is a wide range of survival times, presumably due to underlying genetic factors. Like other low-grade tumors, favourable prognostic factors for OD include age less than 40 years, presentation with seizures, normal neurological examination and, in some reports, extent of resection.23,24 In a retrospective review of 106 patients with OD (n=77) and OA (n=29 ) from the Memorial Sloan Kettering Cancer Center in New York, the overall median time to progression was 5.0 years (0.5 –14.2) and the overall survival, 16.7 years.25 Neither of these two markers of outcome were significantly affected by treatment.
Imaging
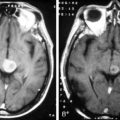
Oligodendrogliomas and oligoastrocytomas are space-occupying lesions that have a predilection for frontal lobes and often infiltrate up to the cortical surface. There are no specific features that distinguish OD/OA from astrocytomas, although the presence of calcification on CT scans and, specifically, a gyriform pattern (due to calcification along the cortical ribbon) is highly suggestive. MR spectroscopy may, in time, be able to distinguish OD/OA from astrocytomas; a study of 15 patients with OD and AO showed that the level of glutamine plus glutamate was significantly higher than in low-grade astrocytomas.26 Similarly, a study of diffusion-weighted imaging (DWI) showed that ODs had significantly lower group apparent diffusion coefficient (ADC) values than astrocytomas (A) and that up to 83% of the subjects could be correctly classified into the OD and A groups by reference to an ADC histogram.27
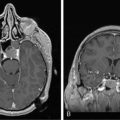
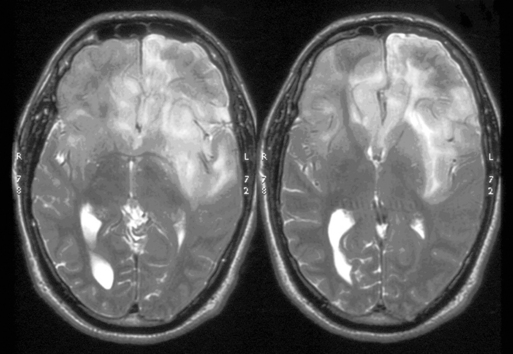
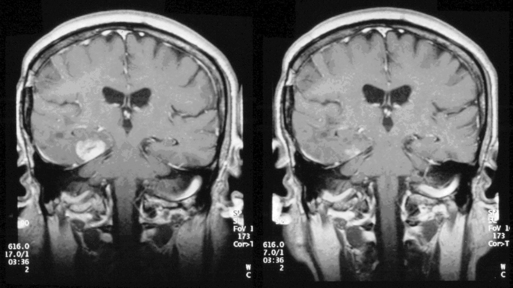
These tumors can be diffusely infiltrating (Figure 7-4) or well-demarcated (Figure 7-5), and appear on MR imaging as high-signal on T2W, PD, and FLAIR sequences and as intermediate-signal or low-signal on T1W sequences. Of these, FLAIR offers superior delineation of the tumor margins and is also better at showing different tumor components, at defining the borders of a postoperative cavity, and at demonstrating local spread to white matter tracts.28
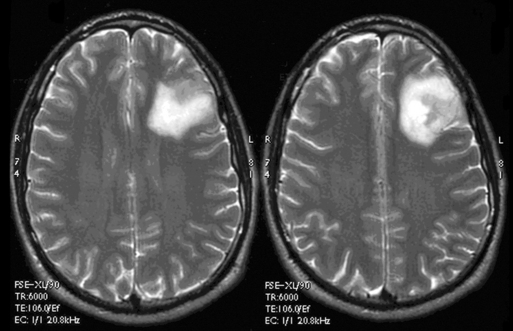
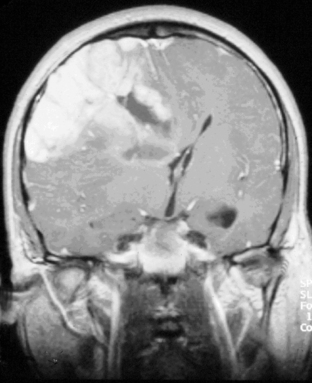
The presence of contrast enhancement in a glioma is often regarded as a sign of malignancy (Figure 7-6) and, in the case of OD, reduces the median survival from 11 years in nonenhancing tumors to 3 years in enhancing ones. For this reason, Daumas-Duport has suggested a grading classification for OD that includes the presence of contrast enhancement as a variable and which was found to be highly predictive of survival.29 Contrast enhancement reflects breakdown of the blood-brain barrier and correlates with angiogenesis. In a study examining the prognostic significance of both tumor enhancement and angiogenesis in OD, 10-year survival was 83% in patients with nonenhancing tumors, compared to only 14% in enhancing tumors; 79% of the tumors showing contrast enhancement had a high tumor angiogenesis index indicating a close relationship between contrast enhancement and endothelial surface area.30 Furthermore, a significant proportion of low-grade gliomas, and in particular OD, may display high relative cerebral blood volume (rCBV) foci on perfusion imaging not reflective of high-grade histopathology.31
Functional imaging, particularly using FDG-PET, has been adopted in some centers for the management of OD, in particular for determining the degree of malignancy and guiding the surgeon to the most malignant area, as well as for discrimination of recurrent tumor from radiation necrosis.32 However, whether it provides additional information over and above that of conventional MRI sequences is debatable.
The results of imaging are vital to determining the subsequent management of a patient with a radiologically suspected low-grade glioma. However, MRI scans may be falsely reassuring. Several studies have demonstrated that contrast enhancement in gliomas is only weakly predictive of tumor grade; in a study of 314 unselected patients with both malignant and low-grade gliomas, 58 lacked contrast enhancement and, of these, approximately one third were malignant, particularly in older patients.33 For this reason, biopsy is recommended for older patients presenting with seizures and a nonenhancing lesion, as they are more likely to be harbouring a high-grade glioma. For younger patients without neurological deficit whose tumor has not changed on a second scan 3 months after the first study, it is reasonable to adopt a watch and wait policy, although the precise scanning interval will vary according to local resources.
Imaging is therefore vital for monitoring the natural history of low-grade gliomas, particularly as these tumors are often not treated until there is clinical or radiological evidence of progression.34 Although it is well recognized that low-grade gliomas eventually undergo malignant transformation, very little is known about their radiological history and growth rates during their “stable” phase. In a study of 27 patients with untreated OD and OA, analysis of mean tumor diameters over time showed that these tumors were continuously growing with an average slope of 4.1 mm/year.35 We have extended this study to all types of grade II gliomas and have shown that even in nontransformers, an average growth rate of 13% per annum was observed, compared to a growth rate of transformers up until the penultimate scan (i.e., before transformation took place) of 26% per annum. This growth rate increased to 56% per annum in the final 6-month time period that included transformation, implying that low-grade gliomas that grow more rapidly in their “stable” phase are more likely to undergo malignant transformation.36
Management
SURGERY
Although surgery offers the possibility of cure in selected patients with low-grade gliomas (e.g., pilocytic astrocytomas), it is almost impossible to resect a diffusely infiltrating grade II tumor in its entirety without unacceptable neurological deficit. Although there are a large number of case series reporting retrospectively that patients with radical resections of low-grade gliomas live longer than those with partial resections or biopsies, any survival advantage may be due to patient selection and underlying tumor biology rather than the nature of the surgical procedure. More favourable results from surgery may be due to selection of young patients with good performance status who have small tumors in noneloquent regions. Even in these patients, however, recurrences at the resection margin occur some years after surgery despite an apparent gross total resection. Other arguments advanced for choosing resection over biopsy include reducing the radiotherapy treatment volume and improving the accuracy of histological diagnosis. However, whether these have any clear benefits overall is unknown.37
Advances in neurosurgery, including preoperative fMRI localisation of speech and motor areas, the ability to fuse this information with image-guidance data, and perioperative cortical localization using the technique of awake craniotomy, all serve to maximize the possibility of a safe resection by avoiding critical cortical structures. However, even in the most experienced hands, postoperative FLAIR imaging usually shows a small rim of residual tissue that was not seen by the surgeon, simply because tumor-infiltrated brain looks identical to normal brain. The prognostic importance of this high signal rim is not entirely clear. A recent surgical study retrospectively evaluated the survival of 170 patients with low-grade gliomas (WHO grade II astrocytomas, oligodendrogliomas, and oligoastrocytomas) and showed that gross total resection, defined as complete resection of the preoperative FLAIR signal abnormality, was associated with longer time to progression and malignant transformation than near total resections (less than 3 mm signal rim around resection cavity) and subtotal resections (residual nodular FLAIR abnormality). Overall survival at 5 and 10 years was improved independently of other prognostic factors in the GTR group over the NTR/STR groups, suggesting a survival advantage from resecting all visible tumor tissue.38 The advent of interventional MRI which permits the surgeon to assess the extent of resection perioperatively using on-table MR imaging in real time may improve the chances of a macroscopically complete resection but still will not address the problem of satellite tumor cells in radiologically normal-appearing peritumoral tissue. Furthermore, staying within the radiological margins of the tumor does not guarantee the absence of a postoperative deficit.
In contrast, CT-guided or MRI-guided stereotactic biopsy carries a very low risk of serious morbidity and mortality (1% to 2%), with a 93% chance of obtaining a definite histological diagnosis.39
The relationship between extent of resection (EOR) and survival is conflicting and seems to favor patients with oligodendrogliomas more than patients with astrocytomas.40 Unfortunately, there is little data that specifically analyzes the relationship between EOR and stratifies outcome for other end points such as time to progression and malignant transformation. Furthermore, assessment of EOR in retrospective series varies from the perioperative surgical estimate to calculation of volumes from early postoperative scans. In view of these considerations, aggressive surgical resection for patients with OD should not be undertaken by neurosurgeons without careful consideration of the long natural history and heightened chemosensitivity of these tumors and the role of other therapeutic modalities. The question of resective surgery in recurrent disease is even more controversial and should be limited to situations where there is a reasonable potential for improving the function and quality of life of the patient.
RADIOTHERAPY
The use of radiotherapy as adjuvant treatment following surgery is well-established for patients with AO. In contrast, the role of radiotherapy in the treatment of patients with low-grade OD is more controversial, particularly as there are concerns over long-term radiotherapy-induced toxicity, including dementia and radiation vasculopathy. It has been suggested that patients with OD who present with intracranial hypertension and/or progressive neurological deficit benefit more from radiotherapy than those who have tumors causing seizures without neurological deficit.41
The radiation dose for low-grade glioma is usually 4500 to 5000 cGy, preferably with three-dimensional conformal ports.42 The incidence of neurotoxicity may have been overestimated, as the older literature studied patients who had been given whole brain RT rather than focal RT, which is regarded as best practice nowadays. Cognitive deficits in patients with low-grade gliomas (LGG) may occur as a result of the tumor itself or as a result of the underlying seizure disorder and anticonvulsant medication, as well as the delayed effects of RT. There have been conflicting reports of late neurocognitive sequelae following RT. A recent neuropsychological cross-sectional study comparing 104 patients with LGGs who had had radiotherapy anywhere from 1 year to 22 years previously with 91 patients who had not received radiotherapy, 100 low-grade hematologic patients, and 195 healthy controls showed that overall the glioma patients had significantly lower scores on objective tests and much lower self-reported cognitive functioning than patients in both control groups. Furthermore, 34% of patients in the glioma group had moderate to severe cognitive disability compared with 22% of the hematologic controls. However, there were only slight differences in cognitive functioning between irradiated and nonirradiated glioma patients and, although a greater percentage of irradiated patients had cognitive deficits (39% vs. 29%), this difference was not statistically significant.43 This suggests that the underlying tumor, epilepsy, and antiepileptic drugs are more important factors in causing cognitive decline than the radiotherapy itself.
Most studies addressing the efficacy of RT in OD have been retrospective and, as with low-grade astrocytomas, have reached conflicting conclusions.44–47 It has now been established that the dose of radiotherapy used for LGGs does not influence survival. In 1985, the EORTC Radiotherapy Cooperative Group launched a randomized phase III study (EORTC 22844) comparing high-dose (59.4 Gy in 6.5 weeks) with low-dose (45 Gy in 5 weeks) radiotherapy in patients with histologically verified LGGs and found no difference in survival between the two doses.48 This finding has been confirmed in a more recent American study comparing survival and toxicity in patients treated with low-dose (50.4 Gy/28 fractions) and high-dose (64.8 Gy/36 fractions) radiotherapy.49 Both 2-year and 5-year survival were better in the low-dose group, albeit not significantly, while radiation necrosis, although rare, occurred in both groups, with one fatality in each arm. The actuarial incidence at 2 years was slightly higher (5% vs. 2.5%) in the high-dose arm. A quality-of-life questionnaire revealed lower levels of functioning and higher symptom burden in patients given higher doses of RT. These group differences were statistically significant for fatigue/malaise and insomnia immediately after radiotherapy and in leisure time and emotional functioning at 7 to 15 months after randomization. These findings suggest that for conventional radiotherapy for low-grade cerebral glioma, a schedule of 45 Gy in 5 weeks not only saves valuable resources, but also spares patients a prolonged treatment at no loss of clinical efficacy.50
The timing of radiotherapy as either primary or adjuvant treatment (i.e., given after surgery) has now been addressed in a recently published large European study, EORTC 22845.51 Over 300 patients with pathologically diagnosed grade II gliomas were randomized to either radiotherapy at a dose of 54 Gy in 6 weeks immediately after surgery or no treatment until tumor progression. Progression-free survival was 5.3 years in the early irradiated group compared to 3.4 years in the control group (p<0.0001) but there was no difference in overall survival (7.4 years vs. 7.2 years). Therefore, on present evidence adjuvant radiotherapy following incomplete tumor excision is not routinely recommended in patients without evidence of progressive disease. The doses and techniques usually employed are the same as for astrocytomas.
In the same way as with chemotherapy, loss of chromosome 1 p may also predict response to radiotherapy. Patients with 1 p LOH had significantly longer median progression-free survival after RT compared with those whose tumors had intact 1 p (55 vs. 6 months, p<0.01) implying a common mechanism of sensitivity to ionizing radiation and alkylating chemotherapy.52
CHEMOTHERAPY
Chemotherapy has traditionally been used as adjuvant treatment for high-grade gliomas, although the data in low-grade gliomas is sparse. A small study carried out in the 1980s by the Southwest Oncology Group was prematurely closed because of slow accrual after recruiting only 60 patients.53 Patients were randomized to receive radiotherapy only following an incomplete resection or radiotherapy plus lomustine (CCNU). There was a nonsignificant trend to improved median survival in the chemotherapy arm (7.4 years vs. 4.5 years) hinting at a possible benefit, but no further prospective studies have been done in this group of patients.
CHEMOTHERAPY FOR ANAPLASTIC OLIGODENDROGLIOMA
Interest in chemotherapy as an option for treating LGGs remained low until 1994 when a National Cancer Institute of Canada study reported a 75% response rate in patients with AO treated with the familiar and previously relatively unsuccessful PCV regimen (procarbazine, lomustine (CCNU), and vincristine) (Figure 7-7).5 Subsequently, temozolomide (TMZ) has also been found to have activity, with high response rates and durable responses.54,55 These responses correlate with loss of 1 p and 19 q LOH, and the presence of this unique genetic signature correlates with increased survival. The combination of 1 p and 19 q LOH is associated with a 100% response rate to PCV with median progression-free survival rates in excess of 31 months, and overall survival in excess of 123 months. The presence of 1 p loss only without 19 q conferred a high level of chemosensitivity but shorter progression-free survival and overall survival, whereas patients with an intact 1 p had lower responses and shorter survival times.56 AO may have other genetic abnormalities such as p53 or PTEN mutations, 10 q LOH, or amplification of the EGFR gene,57 but none of these appear to confer the same degree of responsiveness as 1 p and 19 q LOH. The relationship between 1 p loss and chemosensitivity has also been shown to occur in OA, albeit to a lesser extent.58,59
Because of the increasing interest in chemotherapy for AO, two large prospective trials investigating the role of PCV (procarbazine, CCNU, vincristine) chemotherapy were carried out comparing neoadjuvant and adjuvant chemotherapy against radiotherapy alone. Despite the different timing of chemotherapy with regard to radiotherapy, the results were broadly similar and somewhat surprising. In the neoadjuvant study of 289 patients with pure and mixed AO, there was no benefit in terms of overall survival between the two groups (median survival 4.9 years after PCV+RT vs. 4.7 years after RT alone). There was a slight prolongation of progression-free survival in the combined treatment group (2.6 years for PCV+RT vs. 1.7 years for RT alone), but this was at the expense of considerable acute toxicity in the PCV group (65% patients had Grade 3 or 4 toxicity and one patient died). Irrespective of treatment, patients with 1 p and 19 q loss lived longer than other patients (greater than 7 years vs. 2.8 years).60 Similarly, in the adjuvant study of 368 patients followed up for a median of 5 years, median survival was 40.3 months in the RT/PCV group vs. 30.6 months in the RT group alone, which was not significant. As in the neoadjuvant study, progression-free survival was longer in the RT/PCV group than the RT group (23 months vs. 13 months; p=0.018).61
Both studies identified a significant survival advantage for patients whose tumors had combined 1 p/19 q deletions, irrespective of treatment received. In fact, combined loss of 1 p/19 q is now regarded as the most important prognostic factor for anaplastic oligodendroglial tumors, with median survivals of 6 to 7 years in the presence of 1 p/19 q loss compared with 2 to 3 years in the absence of these codeletions. Similarly, in WHO grade II OD and OA, median survival was 10 to 15 years for 1 p/19 q codeleted tumors, compared to 5 to 8 years for patients without the deletion.62
CHEMOTHERAPY FOR LOW-GRADE OD/OA
Following some small observational studies that suggested that chemotherapy using a combination of PCV was beneficial in ordinary low-grade oligodendrogliomas63,64 three phase II studies of TMZ have recently been published.
The first study treated 46 patients with “progressive low-grade glioma” and reported an objective response rate of 61% (complete response [CR] 24% and partial response [PR] 37%), with an additional 35% having stable disease (SD).65 For the 20 patients with oligodendrogliomas, the response rate was 60% (CR 25%, PR 35%). Serious toxicity occurred in six patients. This study included heavily pretreated patients: 52% of the patients had had either subtotal or gross total resections, 15% had had prior radiotherapy, and 22% prior chemotherapy. Furthermore, 70% of patients had enhancing lesions on MRI scanning, suggesting a higher grade tumor.
In contrast, a single center study that only recruited untreated patients with histologically verified grade II gliomas (17 astrocytomas, 11 oligodendrogliomas and 2 mixed gliomas) showed much less impressive results. Of 29 evaluable patients, 10% had a partial response, 48% a minimal response (MR), 38% stable disease, and 4% progressive disease. The figures for the patients with OD were PR 20%, MR 30%, SD 50%, and PD 0%. The hematological toxicity was 3.5%.66 A third study treated 60 patients with both OD and OA and reported clinical improvement in 51% of patients, particularly those with intractable seizures, and “objective radiological response” in 31% of patients (17% PR and 14% MR).67 Interestingly the mean time to maximum tumor response was 12 months (range 5 to 20 months) and, as expected, tumor response correlated with loss of chromosome 1 p.
The chemosensitivity of OD extends to gliomatosis cerebri, where there is diffuse infiltration of the brain by glioma cells, rendering surgery inappropriate and increasing the potential toxicity of large-field RT. In a recent retrospective study of 63 consecutive patients with gliomatosis cerebri (GC), oligodendroglial GC had a better prognosis in terms of progression-free and overall survival compared to astrocytic and oligoastrocytic GC, regardless of the chemotherapy regimen used.68
MANAGEMENT OF RECURRENT DISEASE
As indicated above, the response rates of patients with AO to PCV chemotherapy vary from 60% to 70%69,70 to 30% to 40%71 in patients with recurrent disease. In an EORTC phase II study of TMZ in 38 patients with recurrent OD and OA, the response rate was 53% and the median time to progression for responding patients 13.2 months.72 This is significantly better for other recurrent high-grade gliomas and implies that about 50% of tumors remain chemosensitive even at recurrence. In exceptional circumstances, tumors may be re-irradiated; response is best seen in patients with OD who have a good performance status.73
Prognosis
There are a large number of prognostic factors for patients with oligodendroglial tumors, the most important of which are age, performance status, histological grade, and 1 p/19 q status. The effect of age is the most striking, with patients less than 40 years consistently experiencing a longer survival; in one study, patients younger than 20 years at presentation had a median survival of 17.5 years compared to 13 months in patients over 60.74 Other clinical variables include frontal location, absence of neurological deficit at diagnosis, and presentation with seizures. Molecular prognostic factors, excluding 1 p/19 q status, include lower proliferative activity, loss of 10 q, and EGFR amplification.
The survival times from recently published series are significantly longer than from reports from over a decade ago. The 5-year and 10-year survival figures for OD are 73% and 49%, respectively, and for AO, 63% and 33%, respectively.75 Other studies have confirmed these figures.
1. Central Brain Tumor Registry of the United States. Primary brain tumors in the United States 1995–1999. URL: http://www.cbtrus.org/2007-2008/tables/report/2007
2. C.E. Counsell, D.A. Collie, R. Grant. Incidence of intracranial tumours in the Lothian region of Scotland 1989–90. J Neurol Neurosurg Psychiatry. 1996;61:143-150.
3. L.H. Pobereskin, J.B. Chadduck. Incidence of brain tumours in two English counties: a population based study. J Neurol Neurosurg Psychiatry. 2000;69:464-471.
4. P.C. Burger. What is an oligodendroglioma? Brain Pathol. 2002;12:257-259.
5. D.R. MacDonald. Low-grade gliomas, mixed gliomas and oligodendrogliomas. Semin Oncol. 1994;21:236-248.
6. C. Giannini, B.W. Scheithauer, A.L. Weaver, et al. Oligodendrogliomas: reproducibility and prognostic value of histologic diagnosis and grading. J Neuropathol Exp Neurol. 2001;60:248-262.
7. O.C. Burger. What is an oligodendroglioma? Brain Pathol. 2002;12:257-259.
8. P.C. Burger, C.E. Rawlings, E.B. Cox, et al. Clinicopathologic correlations in the oligodendroglioma. Cancer. 1986;59:1345-1352.
9. M.T. Giordana, C. Ghimenti, E. Leonardo, et al. Molecular genetic study of a metastatic oligodendroglioma. J Neurooncol. 2004;66:265-271.
10. J. Reifenberger, G. Reifenberger, L. Liu, et al. Molecular genetic analysis of oligodendroglial tumors show preferential allelic deletions on 19 q and 1 p. Am J Pathol. 1994;145:1175-1190.
11. G. Cairncross, D. Macdonald, S. Ludwin, et al. Chemotherapy for anaplastic oligodendroglioma: National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 1994;12:2013-2021.
12. M.J. van den Bent, L.HJ. Looijenga, K. Langenberg, et al. Chromosomal anomalies in oligodendroglial tumours are correlated with clinical features. Cancer. 2003;97:1276-1284.
13. Y. Ino, M.C. Zlatescu, H. Sakai, et al. Long patient survival and therapeutic responses in histologically disparate high grade gliomas with chromosome 1 p loss. J Neurosurg. 2000;92:983-990.
14. M.J. van den Bent. New perspectives for the diagnosis and treatment of oligodendroglioma. Expert Rev Anticancer Ther. 2001;1:348-356.
15. J.S. Smith, B. Alderete, y Minn, et al. Localisation of common deletion regions on 1 p and 19 q in human gliomas and their association with histological subtype. Oncogene. 1999;18:4144-4152.
16. J.S. Smith, A. Perry, T.J. Borell, et al. Alterations of chromosome arms 1 p and 19 q as predictors of survival in oligodendrogliomas, astrocytomas and mixed oligoastrocytomas. J Clin Oncol. 2000;18:636-645.
17. T. Watanabe, M. Nakamura, J.M. Kros, et al. Phenotype versus genotype correlation in oligodendrogliomas and low-grade diffuse astrocytomas. Acta Neuropathol. 2002;103:267-275.
18. H. Yan, W. Parsons, G. Jin, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765-773.
19. I.R. Whittle, A. Beaumont. Seizures in patients with supratentorial oligodendroglial tumours. Clinicopathological features and management considerations. Acta Neurochir (Wien). 1995;135:19-24.
20. Tucha O, Smely C, Preier M, et al. Cognitive deficits before treatment among patients with brain tumors. Neurosurgery 47:324–33.
21. D.D. Spencer, S.S. Spencer, R.H. Mattson, P.D. Williamson. Intracerebral masses in patients with intractable partial epilepsy. Neurology. 1984;34:432-436.
22. A. Pace, L. Bove, P. Innocenti, et al. Epilepsy and gliomas: incidence and treatment in 119 patients. J Exp Clin Cancer Res. 1998;17:479-482.
23. E.G. Shaw, B.W. Scheithauer, J.R. O’Fallon, et al. Oligodendrogliomas: the Mayo clinic experience. J Neurosurg. 1992;76:428-434.
24. P. Celli, I. Nofrone, L. Palma, et al. Cerebral oligodendroglioma: prognostic factors and life history. Neurosurgery. 1994;35:1018-1034.
25. J.D. Olson, E. Riedel, L.M. DeAngelis. Long-term outcome of low-grade oligodendroglioma and mixed glioma. Neurology. 2000;54:1442-1448.
26. M. Rijpkema, J. Schuuring, Y. van der Meulen, et al. Characterization of oligodendrogliomas using short echo time 1 H MR spectroscopic imaging. NMR Biomed. 2003;16:12-18.
27. D.J. Tozer, H.R. Jäger, N. Danchaivijitr, et al. Apparent diffusion coefficient histograms may predict low-grade glioma subtype. NMR Biomed. 2007;20:49-57.
28. M. Bynevelt, J. Britton, H. Seymour, et al. FLAIR imaging in the follow-up of low-grade gliomas: time to dispense with the dual echo? Neuroradiology. 2001;43:129-133.
29. C. Daumas-Duport, M.L. Tucker, H. Kolles, et al. Oligodendrogliomas. Part II: A new grading system based on morphological and imaging criteria. J Neurooncol. 1997;34:61-78.
30. J. Vaquero, M. Zurita, C. Morales, et al. Prognostic significance of tumor-enhancement and angiogenesis in oligodendroglioma. Acta Neurol Scand. 2002;106:19-23.
31. M.H. Lev, Y. Ozsunar, J.W. Henson, et al. Glial tumor grading and outcome prediction using dynamic spin-echo MR susceptibility mapping compared with conventional contrast-enhanced MR: confounding effect of elevated rcbv of oligodendrogliomas. AJNR Am J Neuroradiol. 2004;25:214-221.
32. H.H. Engelhard, A. Stelea, A. Mundt. Oligodendroglioma and anaplastic oligodendroglioma: clinical features, treatment and prognosis. Surg Nurol. 2003;60:443-456.
33. J.N. Scott, P.M.A. Brasher, R.J. Sevick, et al. How often are nonenhancing supratentorial gliomas malignant? A population study. Neurology. 2002;59:947-949.
34. L.D. Recht, R. Lew, T.W. Smith. Suspected low-grade glioma: is deferring treatment safe? Ann Neurol. 1992;31:431-436.
35. E. Mandonnet, J-Y Delattre, M-L Tanguy, et al. Continuous growth of mean tumor diameter in a subset of grade II gliomas. Ann Neurol. 2003;53:524-528.
36. J.H. Rees, H. Watt, H.R. Jäger, et al. Volumes and growth rates of untreated adult low-grade gliomas indicate risk of malignant transformation. Eur J Radiol. 2008. Jul 14 Epub
37. I.R. Whittle. The dilemma of low-grade glioma. J Neurol Neurosurg Psychiatry. 2004;75(Suppl. 2):ii31-ii36.
38. M.J. McGirt, K.L. Chaichana, F.J. Attenello, et al. Extent of surgical resection is independently associated with survival in patients with hemispheric infiltrating low-grade gliomas. Neurosurgery. 2008;63:700-707.
39. D. Kondziolka, L.D. Lunsford. The role of stereotactic biopsy in the management of gliomas. J Neuro-oncol. 1999;42:205-213.
40. M.S. Berger, R.C. Rostomily. Low-grade gliomas: functional mapping resection strategies, extent of resection and outcome. J Neuro-oncol. 1997;34:85-101.
41. P. Celli, I. Nafrone, B.S. Lucio Palma, et al. Cerebral oligodendroglioma: prognostic factors and life history. Neurosurg. 1994;35:1018-1035.
42. E.J. Dropcho. Low-grade gliomas in adults. Curr Treat Options Neurol. 2004;6:265-271.
43. M. Klein, J.J. Heimans, N.K. Aaronson, et al. Effect of radiotherapy and other treatment-related factors on mid-term to long-term cognitive sequelae in low-grade glioms. A comparative study. Lancet. 2002;360:1361-1368.
44. T.S. Nijjar, W.J. Simpson, T. Gadalla, et al. Oligodendroglioma. The Princess Margaret Hospital experience (1958–84). Cancer. 1993;71:4002-4006.
45. K.E. Wallner, M. Gonzales, G.E. Sheline. Treatment of oligodendrogliomas with or without postoperative irradiation. J Neurosurg. 1988;68:684-688.
46. D.E. Bullard, C.E. Rawlings, B. Phillips, et al. Oligodendroglioma. An analysis of the value of radiation therapy. Cancer. 1987;60:2179-2188.
47. D.E. Gannett, W.M. Wisbeck, D.L. Silbergeld, M.S. Berger. The role of postoperative irradiation in the treatment of oligodendroglioma. Int J Radiat Oncol Biol Phys. 1994;30:567-573.
48. A.BMF. Karim, B. Maat, R. Hatlevoll, et al. A randomized trial on dose-response in radiation therapy of low-grade cerebral glioma: European Organization for Research and Treatment of Cancer (EORTC) study 22844. Int J Radiat Oncol Biol Phys. 1996;36:549-556.
49. E. Shaw, R. Arusell, B. Scheithauer, et al. Prospective randomized trial of low- versus high-dose radiation therapy in adults with supratentorial low-grade glioma: initial report of a North Central Cancer Treatment Group/Radiation Therapy Oncology Group/Eastern Cooperative Oncology Group study. J Clin Oncol. 2002;20:2223-2224.
50. D. Curran, G.M. Kiebert, F. Aaronson, et al. Quality of life after radiation therapy of cerebral low-grade gliomas of the adult: results of a randomized phase III trial on dose response. Eur J Cancer. 1998;34:1902-1909.
51. M.J. van den Bent, D. Afra, O. de Witte, et al. Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: the EORTC 22845 randomised trial. Lancet. 2005;366:985-990.
52. G.S. Bauman, Y. Ino, K. Ueki, et al. Allelic loss of chromosome 1 p and radiotherapy plus chemotherapy in patients with oligodendrogliomas. Int J Radiat Oncol Biol Phys. 2000;48:825-830.
53. H.J. Eyre, J.R. Eltringham, J. Crowley, et al. A randomised trial of radiotherapy versus radiotherapy plus CCNU for incompletely resected low-grade gliomas: Southwest Oncology Group study. J Neurosurg. 1993;78:909-914.
54. O.L. Chinot, S. Honore, H. Dufour, et al. Safety and efficacy of temozolomide in patients with recurrent anaplastic oligodendroglioma after standard radiotherapy and chemotherapy. J Clin Oncol. 2001;19:2449-2455.
55. M.J. van den Bent, M.J.B. Taphoorn, A.A. Brandes, et al. Phase II study of first-line chemotherapy with temozolomide in recurrent oligodendroglial tumours. The European Organisation for Research and Treatment of Cancer Brain Tumour Group Study 26971. J Clin Oncol. 2003;21:2525-2528.
56. J.G. Cairncross, K. Ueki, M.C. Zlatescu, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90:1473-1479.
57. Y. Ino, R.A. Betensky, M.C. Zlatescu, et al. Molecular subtypes of anaplastic oligodendroglioma: implications for patient management at diagnosis. Clin Cancer Research. 2001;7:839-845.
58. L. Bissola, M. Eoli, B. Pollo, et al. Association of chromosome 1 losses and negative response in oligoastrocytomas. Ann Neurol. 2002;52:842-845.
59. J.S. Smith, A. Perry, T.J. Borell, et al. Alterations of chromosome arms 1 p and 19 q as predictors of survival in oligodendrogliomas, astrocytomas and mixed oligoastrocytomas. J Clin Oncol. 2000;18:636-645.
60. Intergroup Radiation Therapy Oncology Group Trial 9402G. Cairncross, B. Berkey, E. Shaw, et al. Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol. 2006;24:2707-2714.
61. M.J. van den Bent, A.F. Carpentier, A.A. Brandes, et al. Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer phase III trial. J Clin Oncol. 2006;24:2715-2722.
62. K.B. Fallon, C.A. Palmer, K.A. Roth, et al. Prognostic value of 1 p, 19 q, 9 p, 10 q and EGFR-FISH analyses in recurrent oligodendrogliomas. J Neuropathol Exp Neurol. 2004;63:314-322.
63. W.P. Mason, G.S. Krol, L.M. DeAngelis. Low-grade oligodendroglioma responds to chemotherapy. Neurology. 1996;46:203-207.
64. J. Streffer, M. Schabet, M. Bamberg, et al. A role for preirradiation PCV chemotherapy for oligodendroglial brain tumors. J Neurol. 2000;247:297-302.
65. J.A. Quinn, D.A. Reardon, A.H. Friedman, et al. Phase II trial of temozolomide in patients with progressive low-grade glioma. J Clin Oncol. 2003;21:646-651.
66. M. Brada, L. Viviers, C. Abson, et al. Phase II study of primary temozolomide chemotherapy in patients with WHO II gliomas. Annals of Oncology. 2003;14:1715-1721.
67. K. Hoang-Xuan, L. Capelle, M. Kujas, et al. Temozolomide as initial treatment for adults with low-grade oligodendrogliomas or oligoastrocytomas and correlation with chromosome 1 p deletions. J Clin Oncol. 2004;22:3133-3138.
68. M. Sanson, S. Cartaleat-Carel, N.A. Taillibert, et al. Initial chemotherapy in gliomatosis cerebri. Neurology. 2004;63:270-275.
69. N.A. Paleologos, D.R. Macdonald, N.A. Vick, J.G. Cairncross. Neoadjuvant procarbazine, CCNU and vincristine for anaplastic and aggressive oligodendroglioma. Neurology. 1999;53:1141-1143.
70. M. van den Bent, J. Kros, J. Heimans, et al. Response rate and prognostic factors of recurrent oligodendroglioma treated with procarbazine, CCNU and vincristine chemotherapy. Neurology. 1998;51:1140-1145.
71. R. Sofietti, R. Ruda, G. Bradac, D. Schiffer. PCV chemotherapy for recurrent oligodendrogliomas and oligoastrocytomas. Neurosurgery. 1998;43:1066-1073.
72. M.J. van den Bent, M.JB. Taphoorn, A.A. Brandes, et al. Phase II study of first-line chemo with temozolomide in recurrent oligodendroglial tumors: the European Organization for Research and Treatment of Cancer Brain Tumor Group Study 26971. J Clin Oncol. 2003;21:2525-2528.
73. T. Veninga, H.A. Langendijk, B.J. Slotman, et al. Reirradiation of primary brain tumours: survival, clinical response and prognostic factors. Radiother Oncol. 2001;59:127-137.
74. L. Westergaard, F. Gjerris, L. Klinken. Prognostic factors in oligodendrogliomas. Acta Neurochir (Wien). 1997;139:600-605.
75. K.H. Henderson, E.G. Shaw. Randomized trials of radiation therapy in adult low-grade gliomas. Semin Oncol. 2001;11:145-151.