[level-membership-for-pathology-category]
Chapter 5 Disorders of growth, differentiation and morphogenesis
Growth, differentiation and morphogenesis are the processes by which a single cell, the fertilised ovum, develops into a large complex multicellular organism, with co-ordinated organ systems containing a variety of cell types, each with individual specialised functions. Growth and differentiation continue throughout adult life, as many cells of the body undergo a constant cycle of death, replacement and growth in response to normal (physiological) or abnormal (pathological) stimuli.
There are many stages in human embryological development at which anomalies of growth and/or differentiation may occur, leading to major or minor abnormalities of form or function, or even death of the fetus. In post-natal and adult life, some alterations in growth or differentiation may be beneficial, as in the development of increased muscle mass in the limbs of workers engaged in heavy manual tasks. Other changes may be detrimental to health, as in cancer, where the outcome may be fatal.
This chapter explores the wide range of abnormalities of growth, differentiation and morphogenesis that may be encountered in clinical practice, relating them where possible to specific deviations from normal cellular functions or control mechanisms.
DEFINITIONS
Growth
Growth is the process of increase in size resulting from the synthesis of specific tissue components. The term may be applied to populations, individuals, organs, cells, or even subcellular organelles such as mitochondria.
Types of growth in a tissue (Fig. 5.1A) are:
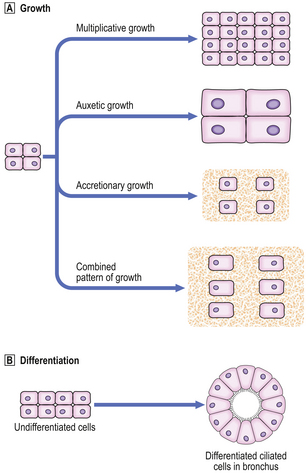
 Types of growth in a tissue.
Types of growth in a tissue.  Differentiation of undifferentiated cells into ciliated cells in bronchus.
Differentiation of undifferentiated cells into ciliated cells in bronchus.Differentiation
Differentiation is the process whereby a cell develops an overt specialised function or morphology that distinguishes it from its parent cell. There are many different cell types in the human body, but all somatic cells in an individual have identical genomes. Differentiation is the process by which genes are expressed selectively and gene products act to produce a cell with a specialised function (Fig. 5.1B). After fertilisation of the human ovum, and up to the eight-cell stage of development, all of the embryonic cells are apparently identical. Thereafter, cells undergo several stages of differentiation in their passage to fully differentiated cells, such as, for example, the ciliated epithelial cells lining the respiratory passages of the nose and trachea. Although the changes at each stage of differentiation may be minor, differentiation can be said to have occurred only if there has been overt change in cell morphology (e.g. development of a skin epithelial cell from an ectodermal cell), or an alteration in the specialised function of a cell (e.g. the synthesis of a hormone).
Morphogenesis




Morphogenesis is the highly complex process of development of structural shape and form of organs, limbs, facial features, etc. from primitive cell masses during embryogenesis. For morphogenesis to occur, primitive cell masses must undergo co-ordinated growth and differentiation, with movement of some cell groups relative to others, and focal programmed cell death (apoptosis) to remove unwanted features.
NORMAL AND ABNORMAL GROWTH IN SINGLE TISSUES
Within an individual organ or tissue, increased or decreased growth takes place in a range of physiological and pathological circumstances as part of the adaptive response of cells to changing requirements for growth. In both fetal and adult life, tissue growth depends upon the balance between the increase in cell numbers, due to cell proliferation, and the decrease in cell numbers, due to cell death. Non-proliferative cells are termed ‘quiescent’; such cells differentiate and adopt specific phenotypes capable of carrying out their specific function (Fig. 5.2).
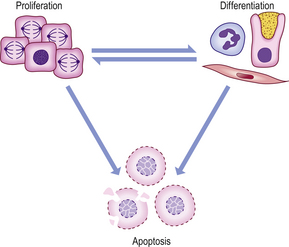
Fig. 5.2 Cell proliferation and death. Individual cells have three potential fates: proliferation, differentiation or apoptosis. After division, individual daughter cells may differentiate, and under some circumstances some differentiated cells may re-enter the cell cycle. The growth rate of a tissue is determined by the net balance between proliferation, differentiation and apoptosis.
In fetal life, growth is rapid and all cell types proliferate, but even in the fetus there is constant cell death, some of which is an essential (and genetically programmed) component of morphogenesis. In post-natal and adult life, however, the cells of many tissues lose their capacity for proliferation at the high rate of the fetus, and cellular replication rates are variably reduced. Some cells continue to divide rapidly and continuously, some divide only when stimulated by the need to replace cells lost by injury or disease, and others are unable to divide whatever the stimulus.
Regeneration





Regeneration enables cells or tissues destroyed by injury or disease to be replaced by functionally identical cells. These replaced ‘daughter’ cells are usually derived from a tissue reservoir of ‘parent’ stem cells (discussed below, p. 92). The presence of tissue stem cells, with their ability to proliferate, governs the regenerative potential of a specific cell type. Mammalian tissues fall into three classes according to their regenerative ability:
Labile cells proliferate continuously in post-natal life; they have a short life-span and a rapid ‘turnover’ time. Their high regenerative potential means that lost cells are rapidly replaced by division of stem cells. However, the high cell turnover renders these cells highly susceptible to the toxic effects of radiation or drugs (such as anti-cancer drugs) that interfere with cell division. Examples of labile cells include:
The high regenerative potential of the skin is exploited in the treatment of patients with skin loss due to severe burns. The surgeon removes a layer of skin which includes the dividing basal cells from an unburned donor site, and fixes it firmly to the burned graft site where the epithelium has been lost (Ch. 6). Dividing basal cells in the graft and the donor site ensure regeneration of squamous epithelium at both sites, enabling rapid healing in a large burned area where regeneration of new epithelium from the edge of the burn would otherwise be prolonged.
Stable cells (sometimes called ‘conditional renewal cells’) divide very infrequently under normal conditions, but their stem cells are stimulated to divide rapidly when such cells are lost. This group includes cells of the liver, endocrine glands, bone, fibrous tissue and the renal tubules.
Permanent cells normally divide only during fetal life, but their active stem cells do not persist long into post-natal life, and they cannot be replaced when lost. Cells in this category include neurones, retinal photoreceptors and neurones in the eye, cardiac muscle cells and skeletal muscle (although skeletal muscle cells do have a very limited capacity for regeneration).
The cell cycle
Successive phases of progression of a cell through its cycle of replication are defined with reference to DNA synthesis and cellular division. Unlike the synthesis of most cellular constituents, which occurs throughout the interphase period between cell divisions, DNA synthesis occurs only during a limited period of the interphase; this is the S phase of the cell cycle. A further distinct phase of the cycle is the cell-division stage or M phase (Fig. 5.3) comprising nuclear division (mitosis) and cytoplasmic division (cytokinesis). Following the M phase, the cell enters the first gap (G1) phase and, via the S phase, the second gap (G2) phase before entering the M phase again. Although initially regarded as periods of inactivity, it is now recognised that these ‘gap’ phases represent periods when critical processes occur, preparing the cells for DNA synthesis and mitosis.
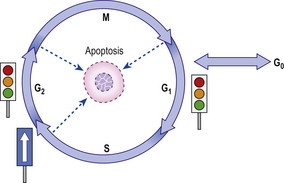
Fig. 5.3 The four stages of the cell cycle. G1 represents preparation for DNA synthesis (S phase), and G2 represents preparation for mitosis (M phase). After mitosis individual daughter cells may each re-enter the cycle at G1 if appropriate stimuli are present. Alternatively, they may permanently or temporarily enter G0 and differentiate. Progress around the cell cycle is one-way. ‘Checkpoints’ ensure one phase does not commence until the previous phase is completed. Failure of a phase to complete satisfactorily results in cell cycle arrest, or—if the problem is irretrievable—apoptosis.
After cell division (mitosis), individual daughter cells may re-enter G1 to undergo further division if appropriate stimuli are present. Alternatively, they may leave the cycle and become quiescent or ‘resting’ cells—a state often labelled as G0. Entry to G0 may be associated with a process of terminal differentiation, with loss of potential for further division and death at the end of the lifetime of the cell; this occurs in permanent cells, such as neurones. Other quiescent cells retain some ability to proliferate by re-entering G1 if appropriate stimuli are present.
Molecular events in the cell cycle
Cell division is a highly complex process and cells possess elaborate molecular machinery to ensure its successful completion. A number of internal ‘checkpoints’ exist to ensure that one phase is complete before the next commences (Fig. 5.3). This is vital to ensure, for example, that DNA replication has been performed accurately and that cells do not divide before DNA replication is complete. The various proteins and enzymes that carry out DNA replication, mitotic spindle formation, etc. are typically only present and active during the appropriate phases of the cycle. The timely production and activation of these proteins is regulated by the activity of a family of evolutionarily conserved proteins called cyclin dependent kinases (CDKs), which activate their target proteins by phosphorylation. The activity of CDKs is, in turn, regulated by a second family of proteins, the cyclins. Transitions from one phase of the cycle to the next are initiated by rises in the levels of specific cyclins. The transition from G0 to G1 at the initiation of the cell cycle, for example, is triggered by external signals such as growth factors leading to rises in the levels of cyclin D. Problems during cell division, such as faulty DNA replication, result in rises in the levels of a third family of proteins, the CDK inhibitors (CDKIs), which can prevent CDKs from triggering the next phase of cell division until the issue is resolved. In the face of major failures, cells will typically initiate apoptosis rather than permit the generation of improperly formed progeny. Damage to the genes that encode proteins involved in the regulation of cell-cycle progression is seen in many cancers (Ch. 11).
Duration of the cell cycle
In mammals, different cell types divide at very different rates, with observed cell cycle times (also called generation times) ranging from as little as 8 hours, in the case of gut epithelial cells, to 100 days or more, exemplified by hepatocytes in the normal adult liver. However, the duration of the individual phases of the cycle is remarkably constant and independent of the rate of cell division. The principal difference between rapidly dividing cells and those that divide slowly is the time spent temporarily in G0 between divisions; some cells remain in the G0 phase for days or even years between divisions, whilst others rapidly re-enter G1 after mitosis.
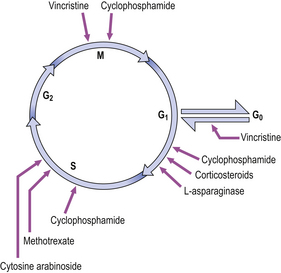
Therapeutic interruption of the cell cycle
Many of the drugs used in the treatment of cancer affect particular stages within the cell cycle (Fig. 5.4). These drugs inhibit the rapid division of cancer cells, although there is often inhibition of other rapidly dividing cells, such as the cells of the bone marrow and lymphoid tissues. Thus, anaemia, a bleeding tendency and suppression of immunity may be clinically important side-effects of cancer chemotherapy.
Cell death in growth and morphogenesis
It seems illogical to think of cell death as a component of normal growth and morphogenesis, although we recognise that the loss of a tadpole’s tail, which is mediated by the genetically programmed death of specific cells, is part of the metamorphosis of a frog. It is now clear that such cell death has an important role in human development and in the regulation of tissue size throughout life. Alterations in the rate at which cell death occurs are important in situations such as hormonal growth regulation, immunity and neoplasia.
Apoptosis
The term apoptosis is used to denote a physiological cellular process in which a defined and programmed sequence of intracellular events leads to the death of a cell without the release of products harmful to surrounding cells. It is a biochemically specific mode of cell death characterised by activation of non-lysosomal endogenous endonuclease which digests nuclear DNA into smaller DNA fragments. Morphologically, apoptosis is recognised as death of scattered single cells which form rounded, membrane-bound bodies; these are eventually phagocytosed (ingested) and broken down by adjacent unaffected cells.
The co-existence of apoptosis alongside mitosis within a cell population ensures a continuous renewal of cells, rendering a tissue more adaptable to environmental demands than one in which the cell population is static.
Apoptosis can be triggered by factors outside the cell or it can be an autonomous event (‘programmed cell death’). In embryological development, there are three categories of autonomous apoptosis:
Morphogenetic apoptosis is involved in alteration of tissue form. Examples include:
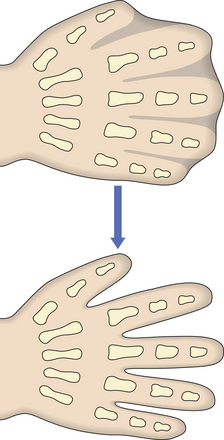
Fig. 5.5 Morphogenesis by apoptosis. Genetically programmed apoptosis (individual cell death) causing separation of the fingers during embryogenesis.
Failure of morphogenetic apoptosis in these four sites is a factor in the development of syndactyly (webbed fingers), cleft palate, spina bifida, and bladder diverticulum (pouch) or fistula (open connection) from the bladder to the umbilical skin, respectively.
Histogenic apoptosis occurs in the differentiation of tissues and organs, as seen, for example, in the hormonally controlled differentiation of the accessory reproductive structures from the Müllerian and Wolffian ducts. In the male, for instance, anti-Müllerian hormone produced by the Sertoli cells of the fetal testis causes regression of the Müllerian ducts (which in females form the fallopian tubes, uterus and upper vagina) by the process of apoptosis.
Phylogenetic apoptosis is involved in removing vestigial structures from the embryo; structures such as the pronephros, a remnant from a much lower evolutionary level, are removed by the process of apoptosis.
Regulation of apoptosis
Apoptosis may be triggered by external signals, such as detachment from the extracellular matrix, the withdrawal of growth factors, or specific signals from other cells. This mode of activation of apoptosis is called the extrinsic pathway. By contrast, the intrinsic pathway is activated by intracellular signals, such as DNA damage or failure to conduct cell division correctly. Although apoptosis can be induced by diverse signals in a variety of cell types, a few genes appear to regulate a final common pathway. The most important of these are the members of the bcl-2 family (bcl-2 was originally identified at the t(14;18) chromosomal breakpoint in follicular B-cell lymphoma, and it can inhibit many factors that induce apoptosis). The bax protein (also in the bcl-2 family) forms bax–bax dimers which enhance apoptotic stimuli. The ratio of bcl-2 to bax determines the cell’s susceptibility to apoptotic stimuli, and constitutes a ‘molecular switch’ which determines whether a cell will survive, leading to tissue expansion, or undergo apoptosis.
The study of factors regulating apoptosis is of considerable importance in finding therapeutic agents to enhance cell death in malignant neoplasms.
Increased growth: hypertrophy and hyperplasia




The response of an individual cell to increased functional demand is to increase tissue or organ size (Fig. 5.6) by:
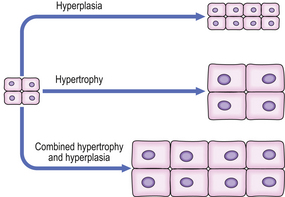
Fig. 5.6 Hyperplasia and hypertrophy. In hypertrophy, cell size is increased. In hyperplasia, cell number is increased. Hypertrophy and hyperplasia may co-exist.
The stimuli for hypertrophy and hyperplasia are very similar, and in many cases identical; indeed, hypertrophy and hyperplasia commonly co-exist. In permanent cells hypertrophy is the only adaptive option available under stimulatory conditions. In some circumstances, however, permanent cells may increase their DNA content (ploidy) in hypertrophy, although the cells arrest in the G2 phase of the cell cycle without undergoing mitosis; such a circumstance is present in severely hypertrophied hearts, where a large proportion of cells may be polyploid.
An important component of hyperplasia, which is often overlooked, is a decrease in cell loss by apoptosis; the mechanisms of control of this decreased apoptosis are unclear, although they are related to the factors causing increased cell production (Fig. 5.7).
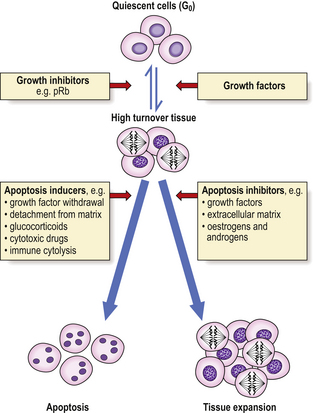
Fig. 5.7 Control of tissue growth by induction or inhibition of apoptosis. Quiescent (mitotically inactive) cells in G0 are recruited into a high turnover (mitotically active) state by growth factors. Their subsequent fate depends on the presence or absence of apoptosis inducers or inhibitors. The inducers and inhibitors are mediated by the bax and bcl-2 proteins respectively, among others.
Physiological hypertrophy and hyperplasia
Examples of physiologically increased growth of tissues include:
In addition to such physiologically increased tissue growth, hypertrophy and hyperplasia are also seen in tissues in a wide range of pathological conditions.
Pathological hypertrophy and hyperplasia
Many pathological conditions are characterised by hypertrophy or hyperplasia of cells. In some instances, this is the principal feature of the condition from which the disease is named. The more common examples are summarised in Table 5.1. For more detail, consult the relevant chapters.
Table 5.1 Examples of non-regenerative hypertrophy and hyperplasia
| Organ/tissue | Condition | Comment |
|---|---|---|
| Myocardium | Right ventricular hypertrophy | Response to pulmonary valve stenosis, pulmonary hypertension or ventricular septal defect (Ch. 13) |
| Left ventricular hypertrophy | Response to aortic valve stenosis or systemic hypertension (Ch. 13) | |
| Arterial smooth muscle | Hypertrophy of arterial walls | Occurs in hypertension (Ch. 13) |
| Capillary vessels | Proliferative retinopathy | Complication of diabetes mellitus (Ch. 26) |
| Bone marrow | Erythrocyte precursor hyperplasia | Response to increased erythropoietin production (e.g. due to hypoxia) (Ch. 23) |
| Cytotoxic T-lymphocytes | Hyperplastic expansion of T-cell populations | Involved in cell-mediated immune responses (Ch. 9) |
| Breast | Juvenile hypertrophy (females) | Exaggerated pubertal enlargement (Ch. 18) |
| Due to high oestrogen levels (e.g. in cirrhosis, iatrogenic, endocrine tumours) (Ch. 18) | ||
| Gynaecomastia (males) | ||
| Prostate | Epithelial and connective tissue hyperplasia | Relative excess of oestrogens stimulates oestrogen-sensitive central zone (Ch. 20) |
| Thyroid | Follicular epithelial hyperplasia | Most commonly due to a thyroid-stimulating antibody (Graves’ disease) (Ch. 17) |
| Adrenal cortex | Cortical hyperplasia | Response to increased ACTH production (e.g. from a pituitary tumour or, inappropriately, from a lung carcinoma) (Ch. 17) |
| Myointimal cells | Myointimal cell hyperplasia in atheromatous plaques | Myointimal cells in plaques proliferate in response to platelet-derived growth factor (Ch. 13) |
Apparently autonomous hyperplasias
In some apparently hyperplastic conditions, cells appear autonomous, and continue to proliferate rapidly despite the lack of a demonstrable stimulus or control mechanism. The question then arises as to whether these should be considered to be hyperplasias at all, or whether they are autonomous and hence neoplastic. If the cells can be demonstrated to be monoclonal (derived as a single clone from one cell), then this suggests that the lesion may indeed be neoplastic, but clonality is often difficult to establish.
Hyperplasia in tissue repair
The proliferation of vascular (capillary) endothelial cells and myofibroblasts in scar tissue, and the regeneration of specialised cells within a tissue, are the important components of the response to tissue damage.
Angiogenesis is the process whereby new blood vessels grow into damaged, ischaemic or necrotic tissues in order to supply oxygen and nutrients for cells involved in regeneration and repair (the term ‘vasculogenesis’ should be reserved specifically for the blood vessel proliferation that occurs in the developing embryo and fetus). In response to local tissue damage, vascular endothelial cells within pre-existing capillaries are activated by angiogenic growth factors such as vascular endothelial growth factor (VEGF), released by hypoxic cells or macrophages. These activated endothelial cells then migrate towards the angiogenic stimulus to form a ‘sprout’. Cell migration is facilitated by the secretion of enzymes including the matrix metalloproteinases, which selectively degrade extracellular matrix proteins. Adjacent sprouts connect to form vascular loops, which canalise and establish a blood flow. Later, mesenchymal cells—including pericytes and smooth muscle cells—are recruited to stabilise the vascular architecture, and the extracellular matrix is remodelled.
Two other initiating mechanisms exist in addition to the above ‘sprouting’ form of angiogenesis: existing vascular channels may be bisected by an extracellular matrix ‘pillar’ (intussusception), with the two channels subsequently being extended towards the angiogenic stimulus. The final mechanism involves circulating primordial stem cells which are recruited at sites of hypoxia and differentiate into activated vascular endothelial cells. Note that a similar process of angiogenesis occurs in response to tumour cells, as an essential component of the development of the blood supply of enlarging neoplasms. Such angiogenesis is a potential therapeutic target in the treatment of malignant neoplasms, although theoretically such drugs might impair angiogenesis and therefore delay healing of wounds.
Myofibroblasts often follow new blood vessels into damaged tissues, where they proliferate and produce matrix proteins such as fibronectin and collagen to strengthen the scar. Myofibroblasts eventually contract and differentiate into fibroblasts. The resulting contraction of the scar may cause important complications, such as:
Thus vascular endothelial cell and myofibroblast hyperplasia are important components of repair and regeneration at various sites in the body, as described below.
Skin
The healing of a skin wound is a complex process involving the removal of necrotic debris from the wound and repair of the defect by hyperplasia of capillaries, myofibroblasts and epithelial cells. Figure 5.8 illustrates some of the key events, most of which are mediated by growth factors.
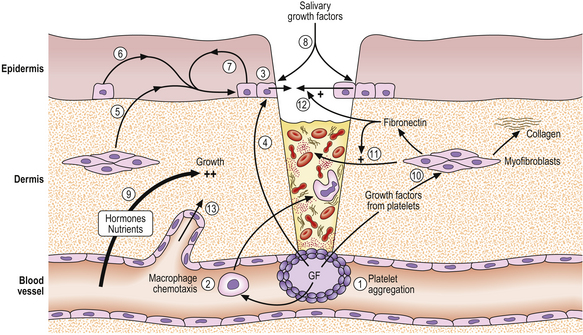
Fig. 5.8 Factors mediating wound healing. A wound is shown penetrating the skin and entering a blood vessel. (1) Blood coagulation and platelet degranulation, releasing growth factors (GF). (2) These are chemotactic for macrophages, which migrate into the wound to phagocytose bacteria and necrotic debris (3). Epidermal basal epithelial cells are activated by released growth factors from the platelets (4) and dermal myofibroblasts (5); from epidermal cells by paracrine (6) and autocrine (7) mechanisms; and from saliva (8) (if the wound is licked). Nutrients and oxygen (9) and circulating hormones and growth factors diffusing from blood vessels all contribute to epidermal growth. Growth factors from the platelets stimulate cell division in myofibroblasts (10), which produce collagen and fibronectin. Fibronectin stimulates migration of dermal myofibroblasts (11) and epidermal epithelial cells (12) into and over the wound. Angiogenic growth factors (not shown) stimulate the proliferation and migration of new blood vessels into the area of the wound (13).
When tissue injury occurs there is haemorrhage into the defect from damaged blood vessels; this is controlled by normal haemostatic mechanisms, during which platelets aggregate and thrombus forms to plug the defect in the vessel wall. Because of interactions between the coagulation and complement systems, inflammatory cells are attracted to the site of injury by chemotactic complement fractions. In addition, platelets release two potent growth factors, platelet-derived growth factor (PDGF) and transforming growth factor-beta (TGF-beta), which are powerfully chemotactic for inflammatory cells, including macrophages; these migrate into the wound to remove necrotic tissue and fibrin.
In the epidermis, PDGF acts synergistically with epidermal growth factor (EGF), derived from epidermal cells, and the somatomedins, insulin-like growth factor 1 (IGF-1) and insulin-like growth factor 2 (IGF-2), to promote proliferation of basal epithelial cells. EGF is also present in high concentrations in saliva and may reach wounds when they are licked. In the dermis, myofibroblasts proliferate in response to PDGF (and TGF-beta); collagen and fibronectin secretion is stimulated by TGF-beta, and fibronectin then aids migration of epithelial and dermal cells. Capillary budding and proliferation are stimulated by angiogenic factors such as VEGF. The capillaries ease the access of inflammatory cells and fibroblasts, particularly into large areas of necrotic tissue.
Hormones (e.g. insulin and thyroid hormones) and nutrients (e.g. glucose and amino acids) are also required. Lack of nutrients or vitamins, the presence of inhibitory factors such as corticosteroids or infection, or a locally poor circulation with low tissue oxygen concentrations, may all materially delay wound healing; these factors are very important in clinical practice.
Liver
In severe chronic hepatitis (Ch. 16) extensive hepatocyte loss is followed by scarring, as is the case in the skin or other damaged tissues. Like epidermal cells in the skin, hepatocytes have massive regenerative potential and surviving hepatocytes may proliferate to form nodules. Hyperplasia of hepatocytes and fibroblasts is presumably mediated by a combination of hormones and growth factors, although the mechanisms are far from clear. Regenerative nodules of hepatocytes and scar tissue are the components of cirrhosis of the liver.
Heart
Myocardial cells are permanent cells (i.e. they remain permanently in G0 and cannot enter G1), and so cannot divide in a regenerative response to tissue injury. In myocardial infarction, a segment of muscle dies and, if the patient survives, it is replaced by scar tissue. As the remainder of the myocardium must work harder for a given cardiac output, it undergoes compensatory hypertrophy (without cell division) (Fig. 5.9). Occasionally, there may be right ventricular hypertrophy as a result of left ventricular failure and consequent pulmonary hypertension.
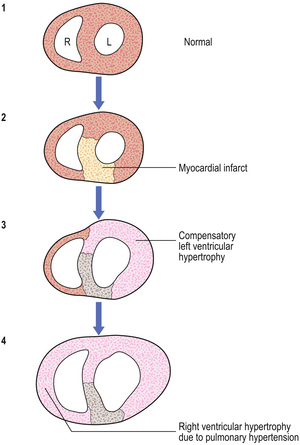
Fig. 5.9 Cardiac hypertrophy. A horizontal slice through the myocardium of the left (L) and right (R) ventricles. (1) Normal. (2) Area of anteroseptal left ventricular infarct. (3) Compensatory hypertrophy of the surviving left ventricle. (4) Right ventricular hypertrophy secondary to left ventricular failure and pulmonary hypertension.
Decreased growth: atrophy





Atrophy is the decrease in size of an organ or cell by reduction in cell size and/or reduction in cell numbers, often by a mechanism involving apoptosis. Tissues or cells affected by atrophy are said to be atrophic or atrophied. Atrophy is an important adaptive response to a decreased requirement of the body for the function of a particular cell or organ. It is important to appreciate that for atrophy to occur there must be not only a cessation of growth but also an active reduction in cell size and/or a decrease in cell numbers, mediated by apoptosis.
Atrophy occurs in both physiological and pathological conditions.
Physiological atrophy and involution
Physiological atrophy occurs at times from very early embryological life, as part of the process of morphogenesis. The process of atrophy (mediated by apoptosis of cells) contributes to the physiological involution of organs such as the thymus gland in early adult life, and late old age is accompanied by atrophy of various tissues (Table 5.2).
Table 5.2 Tissues involved in physiological atrophy and involution
| Embryo and fetus | Early adult |
| Branchial clefts | Thymus |
| Notochord | |
| Thyroglossal duct | Late adult and old age |
| Müllerian duct (males) | Uterus, endometrium (females) |
| Wolffian duct (females) | Testes (males) |
| Bone (particularly females) | |
| Neonate | Gums |
| Umbilical vessels | Mandible (particularly edentulous) |
| Ductus arteriosus | Cerebrum |
| Fetal layer adrenal cortex | Lymphoid tissue |
Pathological atrophy
There are several categories of pathological condition in which atrophy may occur.
Decreased function
As a result of decreased function as, for example, in a limb immobilised as a consequence of a fracture, there may be marked muscle atrophy (due to decrease in muscle fibre size). Extensive physiotherapy may be required to restore the muscle to its former bulk, or to prevent the atrophy.
In extreme cases of ‘disuse’ atrophy of a limb, bone atrophy may lead to osteoporosis and bone weakening; this is also a feature of conditions of prolonged weightlessness, such as occurs in astronauts.
Loss of innervation
Loss of innervation of muscle causes muscle atrophy, as is seen in nerve transection or in poliomyelitis, where there is loss of anterior horn cells of the spinal cord. In paraplegics, loss of innervation to whole limbs may also precipitate ‘disuse’ atrophy of bone, which becomes osteoporotic.
Loss of blood supply
This may cause atrophy as a result of tissue hypoxia, which may also be a result of a sluggish circulation. Epidermal atrophy is seen, for example, in the skin of the lower legs in patients with circulatory stagnation related to varicose veins or with atheromatous narrowing of arteries.
‘Pressure’ atrophy
This occurs when tissues are compressed, by either exogenous agents (atrophy of skin and soft tissues overlying the sacrum in bedridden patients producing ‘bed sores’) or endogenous factors (atrophy of a blood vessel wall compressed by a tumour). In both of these circumstances a major factor is actually local tissue hypoxia.
Lack of nutrition
Lack of nutrition may cause atrophy of adipose tissue, the gut and pancreas and, in extreme circumstances, muscle. An extreme form of systemic atrophy similar to that seen in severe starvation is termed ‘cachexia’; this may be seen in patients in the late stages of severe illnesses such as cancer. In some wasting conditions, such as cancer, cytokines such as tumour necrosis factor (TNF) are postulated to influence the development of cachexia.
Loss of endocrine stimulation
Atrophy of the ‘target’ organ of a hormone may occur if endocrine stimulation is inadequate. For example, the adrenal gland atrophies as a consequence of decreased ACTH secretion by the anterior pituitary; this may be caused by destruction of the anterior pituitary (by a tumour or infarction), or as a result of the therapeutic use of high concentrations of corticosteroids (in, for example, the treatment of cancer), with consequent ‘feedback’ reduction of circulating ACTH levels.
Hormone-induced atrophy
This form of atrophy may be seen in the skin, as a result of the growth-inhibiting actions of corticosteroids. When corticosteroids are applied topically in high concentrations to the skin, they may cause dermal and epidermal atrophy which may be disfiguring. All steroids, when applied topically, may also be absorbed through the skin to produce systemic side-effects, e.g. adrenal atrophy when corticosteroids are used.
Decreased growth: hypoplasia



Although the terms ‘hypoplasia’ and ‘atrophy’ are often used interchangeably, the former is better reserved to denote the failure in attainment of the normal size or shape of an organ as a consequence of a developmental failure. Hypoplasia is, therefore, a failure in morphogenesis, although it is closely related to atrophy in terms of its pathogenesis. An example of hypoplasia is the failure in development of the legs in adult patients with severe spina bifida and neurological deficit in the lower limbs.
Metaplasia





Metaplasia is the transformation of one type of differentiated cell into another fully differentiated cell type. It occurs in the context of alterations in the cellular environment, particularly if associated with chronic cellular injury and repair. Metaplasia may be due to the inappropriate activation or repression of groups of genes involved in the maintenance of cellular differentiation, or by mutations in such genes. The metaplastic ‘daughter’ cells replace the original cells, giving rise to a tissue type that may in some circumstances be better able to withstand the adverse environmental changes.
Examples of metaplasia in epithelial tissues include a change to squamous epithelium (squamous metaplasia) in a variety of tissues, including:
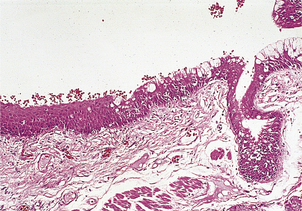
Fig. 5.10 Squamous metaplasia in the bronchus of a smoker. On the right is the mature ciliated pseudostratified columnar epithelium. On the left, the epithelium has undergone metaplasia to a thicker, mature, stratified squamous epithelium.
Another example is the replacement of normal squamous epithelium of the oesophagus by columnar glandular epithelium (glandular metaplasia), sometimes showing overt intestinal differentiation. This condition, known as Barrett’s oesophagus, is caused by the chronic reflux of gastro-duodenal contents, including acid and bile, into the oesophagus.
Examples of metaplasia in mesenchymal tissues are bone formation (osseous metaplasia):
Metaplasia is frequently associated with the subsequent development of malignancy within the metaplastic tissue. This is presumably because the environmental changes that initially caused the metaplasia may also induce dysplasia, which, if it is persistent, may progress to tumour formation.
Metaplasia is sometimes said to occur in tumours as, for example, in squamous or glandular ‘metaplasia’, which may occur in transitional carcinomas of the bladder. These examples of disordered cellular differentiation certainly do occur in tumours, but the term ‘metaplasia’ is best reserved for changes in non-neoplastic tissues.
Dysplasia




Dysplasia is a premalignant condition characterised by increased cell proliferation, the presence of cellular atypia, and decreased differentiation. It may be caused by longstanding irritation and injury of a tissue with chronic inflammation, or by exposure to carcinogenic substances. Whilst early mild forms of dysplasia may be reversible if the initial stimulus is removed, severe dysplasia is a reflection of underlying DNA damage and such lesions may progress to frank malignancy without appropriate treatment.
In affected tissues, dysplasia may be recognised by:
Dysplasia may occur in tissue that has coincident metaplasia (e.g. dysplasia developing in metaplastic squamous epithelium from the bronchus of smokers). Dysplasia may also develop without co-existing metaplasia, for example in squamous epithelium of the uterine cervix, glandular epithelium of the stomach, or the liver.
Dysplasia may be present for many years before a malignant neoplasm develops. This is the rationale for screening programmes, such as the cervical cancer screening programme, in which early dysplastic changes can be identified and appropriate treatment given before the condition progresses to malignancy.
The term ‘dysplasia’ is sometimes used misleadingly to denote the failure of differentiation of an organ which may retain primitive embryological structures. To avoid confusion, it is better to substitute the terms ‘maldifferentiation’ or ‘dysgenesis’ for this condition.
Polyps
The term ‘polyp’ is used to describe the macroscopic (‘naked eye’) appearance of a smooth mass of tissue that projects outwards from the surface of an organ. Polyps are described as ‘sessile’ when they are flat and ‘pedunculated’ when they have a stalk.
The term ‘polyposis’ is used to describe a condition or syndrome where there are multiple polyps in an organ (e.g. polyposis coli, affecting the colon) or an organ system (e.g. hamartomatous polyposis of the gastrointestinal tract in Peutz–Jeghers syndrome).
The term ‘polyp’, when used alone and without further qualification, is purely descriptive of the shape of a lesion and does not signify any specific underlying pathological process (such as hyperplasia, metaplasia, dysplasia or neoplasia). A polyp results from focal tissue expansion at a site at (or near) the organ surface, when the enlarging mass takes the line of least mechanical resistance as it expands outwards, rather than into the underlying tissue. The pathological process that causes both the focal tissue expansion and polyp formation may be either non-neoplastic (e.g. inflammation, hyperplasia, metaplasia, dysplasia) or neoplastic (e.g. neoplasms of epithelial, mesenchymal, lymphoid or other cellular origin). Non-neoplastic polyps and most neoplastic polyps are common and benign, but a small proportion of malignant neoplasms can have a polypoid appearance (e.g. lymphomatous polyposis of the gastrointestinal tract; polypoid adenocarcinoma of the large bowel).
Neoplasia




The word ‘neoplasia’ literally means ‘new growth’, and the lesion so produced is termed a neoplasm. A neoplasm is an abnormal tissue mass, the excessive growth of which is unco-ordinated with that of normal tissues, and which persists after the removal of the neoplasm-inducing stimulus. The term tumour is often used to denote a neoplasm. Numerous factors have been implicated in the development of human tumours, and these are discussed in detail in Chapter 11.
Cell proliferation is essential in development and in adult life. Carefully regulated cell proliferation permits the growth, maintenance and repair of tissues and organs, and allows the body to respond flexibly to various environmental stimuli. Neoplasia results from acquired damage to critical genes that encode the proteins responsible for regulating the proliferation of individual cells, and manifests as excessive and purposeless growth. There are multiple steps in the development of neoplasms, and many of these involve subversion of the normally controlled mechanisms of growth and cellular differentiation, e.g. hormones, growth factors and growth factor-simulating proteins such as some of the oncoproteins. The growth of neoplasms continues in an autonomous manner, in the absence of normal physiological stimuli and without normal negative feedback mechanisms to arrest the cellular proliferation. Finally, it is worth noting that in continually renewing tissues, such as skin or gut epithelium, the only cells that are likely to persist for sufficient time to acquire the number of genetic changes needed for development of such neoplasms are the stem cells (p. 92); cancers in these tissues can therefore be viewed as disorders of the relevant stem cells.
SYSTEMIC GROWTH DISORDERS
The most rapid normal growth occurs during fetal life, when the embryo undergoes the equivalent of some 42 cell divisions in progressing from a fertilised ovum to term (at 40 weeks), with only five more cell divisions needed to achieve adult size. In the first 2 months of embryological life, differing rates of growth, death and migration of cells are responsible for morphogenesis (development of form) within the developing fetus. Maximal growth velocity, however, does not occur until about 20 weeks (for body length) and 34 weeks (for body weight) of gestation; growth velocity then slows until term.
Despite apparently precise genetic programming of growth, marked variations in birth size can occur due to either normal physiological processes or disease. The most obvious manifestation of growth is an increase in height, which is largely a function of longitudinal skeletal growth. In infancy and childhood there may be variations in height between individuals of the same sex, but the most important normal variations are those between the heights of the two sexes. In childhood, girls are typically shorter than boys, but they become temporarily taller than boys as a result of their pre-pubertal growth spurt (9 cm/year), which starts at around 10.5 years of age. The boys’ growth spurt starts at about 12.5 years, but the higher growth velocity (10.3 cm/year) results in their overtaking the girls in height at about 14 years, and accounts for the final height advantage in boys.
Within an individual, different parts of the body do not follow the same pattern as longitudinal growth. Children have relatively larger heads compared with the trunk and legs than do adults. The reproductive organs grow little before puberty, but then increase rapidly in size. Lymphoid organs grow maximally before puberty, and their growth velocity decreases before skeletal growth velocity.
Growth is controlled by and susceptible to variations in:
Endocrinological growth control and its disorders
Cells may be stimulated into growth by the action of hormones and growth factors (Fig. 5.11). Hormones are synthesised and stored in specific tissues (glands) and released into the blood to exert their effect on distant target cells; this is the endocrine mechanism. Individual growth factors are synthesised in many different cell types throughout the body. They often act on the cell in which they are synthesised (an autocrine mechanism), or on nearby cells in the same tissue (a paracrine mechanism), but they may have additional endocrine actions on distant cells via the blood stream.
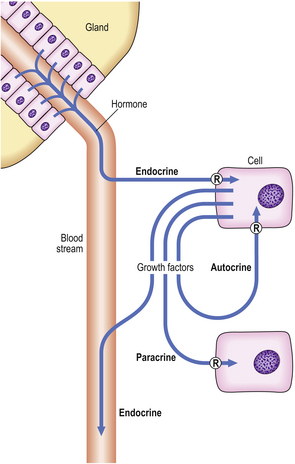
Fig. 5.11 Endocrine, paracrine and autocrine mechanisms of hormones and growth factors. In each case, the hormone and growth factors have their effects through specific receptors (R).
Individual hormones and growth factors require highly specific cellular receptors to mediate their actions on target cells. Steroid hormone receptors are intracellular, but receptors for peptide hormones and growth factors are located on the cell membrane. A high concentration of a receptor renders a cell highly susceptible to the actions of a hormone or growth factor; conversely, the absence of a receptor leads to hormone or growth factor insensitivity.
Post-natal growth
In post-natal life, growth is controlled by an endocrine pathway that regulates total body size (Fig. 5.12). Growth hormone (GH) is central to the endocrine control of post-natal growth.
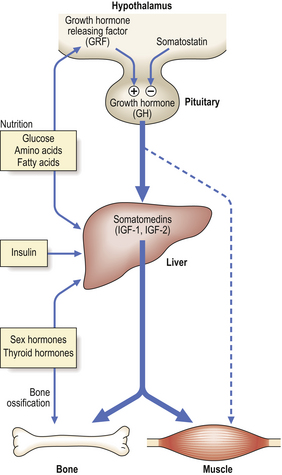
Fig. 5.12 Post-natal growth. The hormonal control of post-natal growth is mediated by hypothalamic and pituitary hormones, and liver somatomedins—the insulin-like growth factors 1 and 2 (IGF-1 and IGF-2).
The release of GH from the pituitary is regulated by the opposing actions of hypothalamic growth hormone releasing factor (GRF) and the inhibitory hormone somatostatin. Growth hormone acts via intermediary hormones—the somatomedins, IGF-1 and IGF-2; these are predominantly (but not exclusively) synthesised in the liver. Somatomedins may also be released from the liver under the influence of insulin, sex-steroid hormones, thyroid hormone and nutritional factors. Growth hormone may have a minor direct anabolic effect on non-skeletal tissues, but here too IGF-1 and IGF-2 are quantitatively more important.
Again, it is important to appreciate that each of the above hormone actions is mediated via individual specific cellular receptor proteins, the concentration of which principally determines the sensitivity of the target cell to the hormone.
Reduced growth
Reduced GH production due to hypopituitarism (of whatever pathological cause) in childhood leads to dwarfism, which can be corrected by regular GH injections given before puberty arrests skeletal growth by epiphyseal fusion. Dwarfism due to GH deficiency is characterised by normal body proportions, in contrast to the effects of reduced thyroid hormones (see below).
Reduced GH receptors are a feature of the rare Laron dwarfism. Although circulating GH concentrations are high, the liver is insensitive to GH, and circulating somatomedins are greatly reduced. Treatment with GH will not increase growth rate.
Reduced thyroid hormone secretion causes a reduction in hepatic IGF-1 secretion. In the resultant dwarfism, the head is of normal size but the limbs are stunted because bone ossification is reduced. Treatment with GH has no effect, but thyroxine is corrective if given before puberty.
Block between GH and IGF-1 release may occur in malnutrition or emotionally based growth retardation, although the metabolite-mobilising actions of GH are maintained. IGF-1 levels correlate positively with the protein content of the diet and nitrogen balance.
Inhibition of growth by corticosteroids may occur with endogenous corticosteroids, e.g. Cushing’s disease, or with exogenous corticosteroids used, for example, in the treatment of asthma or leukaemia.
Increased growth
Increased GH secretion from a normal pituitary or a pituitary tumour results in increased IGF-1 and increased growth. Before puberty this causes gigantism; after puberty, longitudinal skeletal growth cannot occur (due to maturation and ossification with resulting epiphyseal fusion), but the hands, feet and head increase in size to produce acromegaly.
Increased sex-steroid hormone secretion in childhood may lead to precocious puberty, with an initial increase in height resulting from a premature rise in pubertal IGF-1 levels. However, epiphyseal fusion in long bones is accelerated, and the final height may be below normal.
Embryo and fetal growth
The control of growth later in fetal life is very different from that in the post-natal child. The fetus is a self-contained unit with respect to growth, as maternal peptide and thyroid hormones do not cross the placenta in physiologically significant concentrations and, although sex-steroid hormones and other steroids do cross the placenta, they are generally metabolised into inactive forms.
The fetus produces its own GH, but this is not used to promote growth as the GH receptor is greatly reduced in the fetus (particularly in the liver). Indeed, fetal growth does not require any pituitary hormonal influence, and anencephalic human fetuses often attain normal weights for gestational age.
The most important growth-regulating hormone in the fetus is, without doubt, insulin (Fig. 5.13). As in the adult, blood insulin concentrations in the fetus are controlled by glucose concentrations, and both insulin and glucose are required for normal metabolic functions of the fetus and the placenta. In addition, however, insulin directly stimulates the production of growth factors (in particular the somatomedin IGF-2) in cells, and these act on the IGF-2-synthesising cells and adjacent cells, by autocrine and paracrine mechanisms respectively, to stimulate growth. Additional, but relatively less important, effects on growth factor synthesis are stimulated by human placental lactogen (HPL)—a hormone that has marked structural similarity to GH, and is synthesised by the placenta—and fetal thyroid hormones. Although fetal growth is not GH-mediated, somatomedins (particularly IGF-2 in the fetus) are important, although they are not yet under GH control. Other growth factors, such as EGF, PDGF, TGF-beta and nerve growth factor (NGF), are probably also involved.
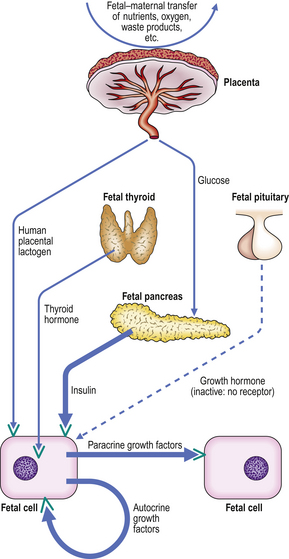
Birth weight
Many factors may affect fetal growth rate and ultimate size (Table 5.3). Although insulin levels are crucial, the most common causes of low birth weight are premature delivery (before 40 weeks’ gestation) and genetically controlled small size (a small baby born to small parents). It is therefore important to relate the birth weight first to gestational age and then to allow for parental size.
Table 5.3 Factors affecting fetal growth rate and size
| Factors | Examples |
|---|---|
| Decreased growth | |
| Genetic |
EndocrineReduced fetal insulin or insulin receptor levels (rare)NutritionMaternal intake <1500 kcal/dayIntra-uterine environment
Increased growth EndocrineFetal hyperinsulinaemia (e.g. in maternal diabetes mellitus)
Reduced growth
Reduced fetal insulin production may lead to a low birth weight, e.g. 1.2–2 kg (normal mean 3 kg). The causes are uncommon, and include:
Reduced fetal insulin receptors, with resultant insensitivity to circulating insulin, cause a similar growth reduction (the rare Leprechaun syndrome).
Increased growth
Increased growth occurs due to increased circulating insulin levels in the fetus (hyperinsulinaemia).
Diabetic mothers may have heavy infants, although birth lengths are not usually increased. Increased glucose diffuses passively into the fetus from the mother, stimulating fetal insulin secretion. The increased weight is due mainly to excess fat.
Infants with hyperinsulinaemia associated with the rare condition nesidioblastosis (an uncontrolled proliferation of pancreatic endocrine cells) or Beckwith–Weidemann syndrome (see below) are more obviously overgrown, e.g. birth weight 4.5–5.5 kg (normal mean 3 kg).
Genetic factors in growth control
The most important genetic factors that regulate the height of an individual are:
As such a large number of hormones, growth factors, receptors, enzymes and other proteins play a co-ordinated role in normal growth, it is not surprising that a wide range of chromosomal abnormalities can interfere with normal growth. Some of these conditions are inherited; others are the result of sporadic gene mutation or chromosomal aberration. Some conditions are incompatible with life; others are compatible with a normal life modified only by reduced stature.
Primary genetically mediated growth abnormalities are classified into two broad groups, according to whether growth of the limbs and/or trunk with respect to the head is proportionate or disproportionate.
Proportionate alterations of skeletal growth
Autosomal chromosomes
Proportionate alterations of skeletal growth may result from abnormalities of autosomal chromosomes including:
Disproportionate alterations of skeletal growth
Disproportionate shortness of stature at birth is often the result of the genetically mediated osteochondrodysplasias (specific disorders of growth of bone and/or cartilage). These can be classified into two groups, depending upon whether the disproportionate shortness of limbs is, or is not, accompanied by a significantly shortened spine. Examples of these conditions include the following.
Achondroplasia
Achondroplasia, the most common of the osteochondrodysplasias, is an autosomal dominant condition (although there are many sporadic cases). The genetically mediated defect is considered to be a primary disturbance of endochondral ossification which occurs in early life and is well established by birth (although severely affected fetuses may die towards the end of pregnancy). Patieuts with achondroplasia, if they survive the neonatal period, usually reach adult life but with reduced stature. They have severe shortening of the limbs (hypomelia or micromelia), with long bones as little as half the normal length. Epiphyses are greatly enlarged, and the shafts of long bones widen to surround the enlarged epiphyses at the ends of long bones. Accompanying changes in the base of the skull may cause narrowing of the foramen magnum, with spinal cord compression. The spine itself is not shortened.
Rare osteochondrodysplasias
Many (but not all) of these rare conditions have an autosomal recessive mode of inheritance. They include severe conditions such as achondrogenesis, which is incompatible with life, and conditions such as pseudoachondroplasia, where the spine is shortened in addition to the limbs.
Nutritional factors in growth control
Maternal nutrition, surprisingly, has a very small influence on human fetal size, and even severe food deprivation results in little more than a 200–300 g reduction in birth weight. In the Dutch winter famine of 1944, birth weight declined only when maternal nutritional input was less than 1500 kcal per day, and then by no more than 500 g.
The fetus is, however, highly susceptible to placental disorders such as infection or to partial detachment, both of which reduce the fetal intake of nutrients and impair gas exchange; the result, if not fatal, may be a severe reduction in birth weight. Small local defects, such as small haemangiomas, do not affect placental function, and hence do not affect growth.
In post-natal life, growth may be severely affected by low or poorly balanced nutritional intake. Starvation, in the form of kwashiorkor (protein deprivation) or marasmus (protein and total calorie deprivation; see Ch. 7), severely disturbs growth endocrinology, producing a negative nitrogen balance as part of a catabolic state.
Environmental factors in growth control
Fetal growth can be affected by several physiological and pathological environmental factors:
Post-natal environmental growth effects can be seen in children suffering from emotional deprivation or physical abuse, who may have a reduced rate of growth. Catch-up growth occurs following hospitalisation, without medication.
Effect of intercurrent disease on growth
The commonest growth alterations in childhood are those that are caused indirectly by a wide range of diseases, that decrease growth for the duration of the illness and are followed by a period of ‘catch-up’ growth. These diseases include common bacterial and viral illnesses experienced by many children. Some are relatively short-lived and the decreased growth may not be noticed. However, with increasing severity and chronicity of the illness, it is more likely that the growth disturbance will be noticeable and clinically significant. Thus, severe growth disturbance may be present in chronic cardiovascular or respiratory disease, renal disease, hepatic cirrhosis, chronic gastrointestinal disease (e.g. coeliac disease, Crohn’s disease), and chronic infections such as AIDS, malaria or tuberculosis.
The effect of intercurrent diseases may compound growth disturbances due to the nutritional or environmental factors discussed above, and/or primary endocrinological or genetic growth disorders.
DIFFERENTIATION AND MORPHOGENESIS IN HUMAN DEVELOPMENT
Differentiation is the process whereby a cell develops an overt specialised function that was not present in the parent cell. It is an important component of morphogenesis; this is the means by which limbs or organs are formed from primitive groups of cells. Thus, abnormalities of differentiation often lead to abnormal morphogenesis and fetal abnormality. It must be remembered, however, that growth also plays an important role in morphogenesis; cells that vary in their differentiation may have very different growth characteristics. Variations in differentiation may also affect the ability of some cells to migrate with respect to others. Thus, normal embryological development requires highly co-ordinated processes of differentiation, growth and cell migration which together comprise morphogenesis.
Control of normal differentiation



A fertilised ovum may develop into a male or female, a human or a blue whale; the outcome depends on the structure of the genome. There are many similarities between the corresponding cell types in different species. Individual cell types are distinct only because, in addition to the many universal proteins required by all cell types for ‘housekeeping’ functions such as cellular metabolism, each cell produces a characteristic set of specialised proteins which define that particular cell type.
Most differentiated cells contain the same genome as in the fertilised ovum. This has been demonstrated elegantly by injecting the nucleus of a differentiated tadpole gut epithelial cell into an unfertilised frog ovum, the nucleus of which was destroyed using ultraviolet light; the result was a normal frog with the normal variety of differentiated cell types (Fig. 5.14). More recently a variety of mammalian species—most notably a sheep—have been cloned from a single ovarian cell.
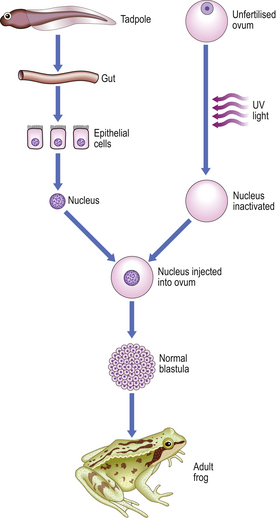
Fig. 5.14 Potential of the genome of somatic cells. Differentiated cells from the gut of a tadpole have the complete genome and potential for control of production of the whole frog. (After J B Gurdon.)
There are very few exceptions to the rule that differentiated cells contain an identical genome to that of the fertilised ovum. In humans, for example, they include B- and T-lymphocytes which have antigen receptor genes rearranged to endow them with a large repertoire of possible receptors (Ch. 9).
Transcriptional control
As most differentiated cells have an identical genome, differences between them cannot be due to amplification or deletion of genes. The cells of the body differ not in the range of genes present in each cell, but in how those genes are expressed, i.e. transcribed and translated into proteins. Genes are selectively switched on or off to control the synthesis of gene products.
The synthesis of a gene product can in theory be controlled at several levels:
In practice, the modulation of transcription is the main mechanism by which gene expression is regulated in many of the important ‘decision’ stages of differentiation in embryogenesis.
For a cell to differentiate in a particular way, multiple genes must be switched on whilst many others must be switched off. There is now ample evidence that the regulation of transcription of entire groups of genes is mediated by the gene products of a small number of ‘control’ genes (or transcription factors), which may themselves be regulated by the product of a single ‘master’ gene (Fig. 5.15).
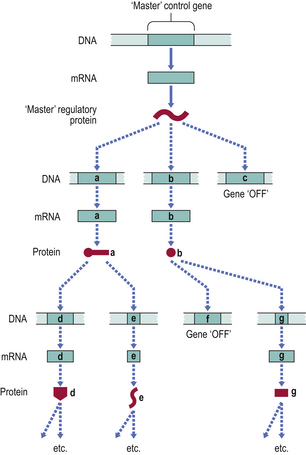
Positional control in early embryogenesis
Some insight into possible control mechanisms in human differentiation and morphogenesis has been gained from observations of the fruit fly, Drosophila. Disturbances of single ‘master’ genes in Drosophila have been shown to result in major malformations, such as the development of legs on the head in place of antennae, mediated by the response of many controlled genes to the alteration in ‘master’ gene product. Such a homeotic mutation (the transformation of one body part into another part that is usually found on a different body segment) highlights the importance of another factor in the control of differentiation and morphogenesis, namely the three-dimensional spatial co-ordinates (position) of a cell within an embryo at a given time.
In Drosophila, a group of genes, which individually cause a range of homeotic mutations, have been found to share a 60-amino-acid sequence domain which is common to genes controlling normal larval segmentation. This sequence, named the homeobox, has also been demonstrated in vertebrates, including humans (Ch. 3). Homeobox-containing genes (also known as homeobox genes) are transcriptional regulators influencing morphogenesis. Parts of human anatomy appear to be constructed on a segmental basis, for example rows of somites, teeth and limb segments, and here it is probable that homeobox genes have an important morphogenetic role.
Cell determination
The homebox genes, and other genes that regulate embryogenesis, act on the embryo at a very early stage, before structures such as limbs have begun to form. Nonetheless, by observing the effects of selective marking or obliteration of cells, a ‘fate map’ of the future development of cells in even primitive embryos can be constructed. Thus, some of the cells of somites become specialised at a very early stage as precursors of muscle cells, and migrate to their positions in primitive limbs. These muscle-cell precursors resemble many other cells of the limb rudiment, and it is only after several days that they differentiate and manufacture specialised muscle proteins. Thus, long before they differentiate, the developmental path of these cells is planned; such a cell which has made a developmental choice before differentiating is said to be determined. A determined cell must:
Determination therefore differs from differentiation, in which there must be demonstrable tissue specialisation.
Some cells which are determined, but not differentiated, may remain so for adult life; good examples are the stem cells, such as bone marrow haemopoietic cells or basal cells of the skin, which proliferate continuously and produce cells committed to a particular form of differentiation. Hypoplastic and aplastic anaemia, which result in anaemia, neutropenia and thrombocytopenia, are thought to be due to a failure or suppression of bone marrow haemopoietic stem cells (Ch. 23).
Cell position and inductive phenomena
As the fields of cells over which spatial chemical signals act are generally small, large-scale changes to the whole individual are the result of factors operating very early in embryonic development, whilst more specific minor features of differentiation within small areas of an organ or limb are specified later and depend on the position of the cell within the structure. Simple changes may occur in response to a diffusible substance (such as vitamin A in the developing limb bud), and serve to control local cell growth and/or differentiation according to the distance from the source. Additional differentiation changes may, however, occur as a result of more complex cellular interactions.
Many organs eventually contain multiple distinct populations of cells that originate separately but later interact. The pattern of differentiation in one cell type may be controlled by another, a phenomenon known as induction. Examples of induction include:
Inductive phenomena also occur in cell migrations, sometimes along pathways that are very long, controlled by generally uncertain mechanisms (although it is known, for example, that migrating cells from the neural crest migrate along pathways that are defined by the host connective tissue). Inductive phenomena control the differentiation of the migrating cell when it arrives at its destination—neural crest cells differentiate into a range of cell types, including sympathetic and parasympathetic ganglion cells, and some cells of the neuroendocrine (APUD) system.
Maintenance and modulation of an attained differentiated state
Once a differentiated state has been attained by a cell, it must be maintained. This is achieved by a combination of factors:
Even in the adult, minor changes to the differentiated state may occur if the local environment changes. These alterations to the differentiated state are rarely great, and most can be termed modulations, i.e. reversible interconversions between closely related cell phenotypes. An example of a modulation is the alteration in synthesis of certain liver enzymes in response to circulating corticosteroids. More substantial changes in cell phenotype represent metaplasia (see p. 82).
In the neonatal stage of development, cell maturation may involve modulations of the differentiated state. Examples are:
Normal differentiation and morphogenesis: summary
Differentiation
During development of an embryo, determination and differentiation occur in a cell by transcriptional modifications to the expression of the genome, without an increase or decrease in number of genes present. The factors involved are summarised in Figure 5.16. Expression of individual genes within the genome is modified during development by:
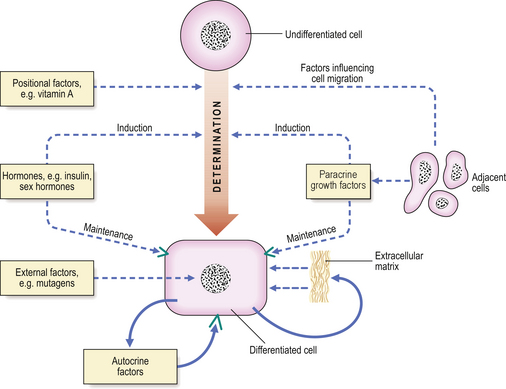
Fig. 5.16 Differentiation. Factors affecting determination, differentiation, maintenance and modulation of the differentiated state of a cell during embryogenesis include positional factors, hormones, paracrine growth factors and external factors such as teratogens. With the exception of positional factors, all of these are important in influencing the differentiated state of cells in post-natal and adult life.
External factors may cause alterations to the differentiated state of the cell, either during development or at any stage of adult life.
Morphogenesis
The main features of morphogenesis are summarised in Figure 5.17.
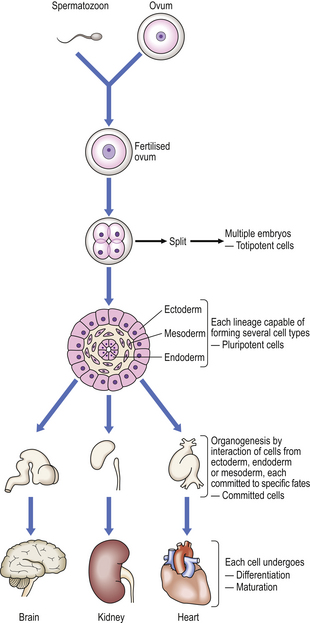
Stem cells and transdifferentiation
Stem cells are ‘parent’ cells that retain replicative potential, and whose progeny may differentiate into many different types of ‘daughter’ cell, although different stem cell types have varying potential for this:
The presence or absence of tissue stem cells within a single tissue is related to the ability of the cells of that tissue to regenerate after physiological or pathological cell loss or destruction. Thus haemopoietic stem cells in bone marrow replace the different blood cell types after haemorrhage (blood cells are ‘labile’ cells), while brain neurones (‘permanent’ cells) cannot be replaced, because there are no functioning neuronal stem cells in the adult brain.
When organs (such as the kidneys) or cells (such as brain neurones) fail because of ageing or disease, a patient may die or suffer increasing disability. Organ transplantation may be possible, although there are insufficient organ donors, and the transplanted organ may be rejected. In 1998, human embryonic stem (ES) cells were successfully extracted from blastocysts and aborted fetuses and grown in vitro. This raises the possibility that these ES cells could be induced to differentiate into organs or cells for transplantation. While some biotechnology companies can produce cells for simple bone or joint repairs from mesenchymal stem cells, creation of more complicated tissues or organs (such as the kidney) will be much more difficult. Because of the ethical issues associated with the use of embryonic stem cells, more recent research has focused on the possibility of inducing stem cells from one organ system, such as haemopoietic stem cells (bone marrow cells differentiating into red and white blood cells and platelets) to develop into cells of other organ systems (e.g. kidney, liver or brain) by a process of ‘transdifferentiation’ (Fig. 5.18). Through such ‘adult stem cell plasticity’, it is possible that in the future an adult patient’s own bone marrow stem cells could be induced artificially to transdifferentiate, and replace cells or organs (such as the kidney) that have been damaged by disease. This would also avoid the risk of immunological rejection of transplanted organs.
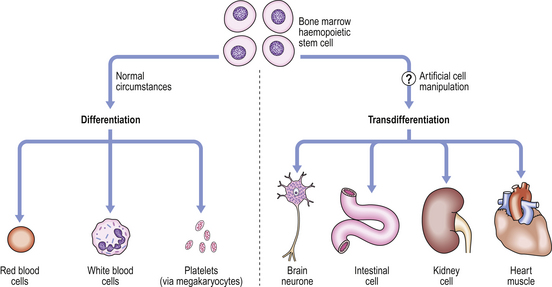
Figure 5.18 Stem cell differentiation and transdifferentiation. Under normal conditions, adult bone marrow haemopoietic stem cells differentiate into red blood cells (erythrocytes), white blood cells (neutrophils) and megakaryocytes to produce platelets. However, artificial manipulation of these cells in vitro may induce them to transdifferentiate into cells of other organs. This ability to make substantial changes of cell type is termed ‘plasticity’ of the stem cells. (Modified from Bonnet D 2002 Haemopoietic stem cells. Journal of Pathology 197: 430–440, Figure 2.)
Congenital disorders of differentiation and morphogenesis
A congenital disorder is defined as one present at birth. The term thus embraces chromosomal disorders, hereditary and spontaneous genetic diseases, non-genetically determined failures of differentiation and morphogenesis, and other conditions that have detrimental effects on the growth, development and well-being of the fetus.
The processes involved in human conception and development are so complex that it is perhaps remarkable that any normal fetuses are produced; the fact that they are produced is a result of the tight controls of growth and morphogenesis which are involved at all stages of development. The usual outcome of human conception is abortion; 70–80% of all human conceptions are lost, largely as a consequence of chromosomal abnormalities (Fig. 5.19). The majority of these abortions occur spontaneously in the first 6–8 weeks of pregnancy, and in most cases the menstrual cycle might appear normal, or the slight delay in menstruation causes little concern. Chromosomal abnormalities are present in 3–5% of live-born infants, and a further 2% have serious malformations that are not associated with chromosomal aberrations. The most common conditions in these two categories are illustrated in Table 5.4.
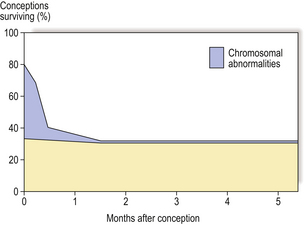
Fig. 5.19 Fate of human conceptions. Between 70% and 80% of human conceptions are lost by spontaneous abortion in the first 6–8 weeks of pregnancy, most as a consequence of chromosomal abnormality. Chromosomal abnormalities are present in 3–5% of live-born infants.
Table 5.4 Incidence of some congenital abnormalities
| Chromosomal abnormality | Incidence per 1000 live births |
|---|---|
| Down’s syndrome (47, +21) | 1.4 |
| Klinefelter’s syndrome (47XXY) | 1.3 |
| Double Y male (47XYY) | <1 |
| Multiple X female (47XXX) | <1 |
| Major malformations | Incidence per 1000 stillbirths + live births |
| Congenital heart defects | 6 |
| Pyloric stenosis | 3 |
| Spina bifida | 2.5 |
| Anencephaly | 2 |
| Cleft lip (± cleft palate) | 1 |
| Congenital dislocation of the hip | 1 |
Chromosomal abnormalities affecting whole chromosomes
Autosomal chromosomes
The three most common autosomal chromosome defects involve the presence of additional whole chromosomes (trisomy). The incidence of trisomies increases with maternal age, and to a lesser extent paternal age. Most trisomies are incompatible with life and result in early abortion. As the genome of every cell in the body has an increased number of genes, gene product expression is greatly altered and multiple abnormalities result during morphogenesis. Those trisomies that are compatible with life have more serious manifestations the larger the chromosome involved, presumably since a greater number of individual genes are involved.
Trisomy 21 (Down’s syndrome) affects approximately 1 in 1000 births; it is associated with mental retardation, a flattened facial profile and prominent epicanthic folds. The hands are short, with a transverse palmar crease. There are also abnormalities of the ears, trunk, pelvis and phalanges.
Trisomy 18 (Edwards’ syndrome) affects 1 in 5000 births. It is associated with ear and jaw, cardiac, renal, intestinal and skeletal abnormalities.
Trisomy 13 (Patau’s syndrome) affects 1 in 6000 births, with microcephaly and microphthalmia, hare lip and cleft palate, polydactyly, abnormal ears, ‘rocker-bottom’ feet, and cardiac and visceral defects. As with Edwards’ syndrome, most affected infants die in the first year of life.
Sex chromosomes
Chromosomal disorders affecting the sex chromosomes (X and Y) are relatively common, and usually induce abnormalities of sexual development and fertility. In general, variations in X chromosome numbers cause greater mental retardation.
Klinefelter’s syndrome (47XXY) affects 1 in 850 male births. There is testicular atrophy and absent spermatogenesis, eunuchoid bodily habitus, gynaecomastia, female distribution of body hair and mental retardation. Variants of Klinefelter’s syndrome (48XXXY, 49XXXXY, 48XXYY) are rare, and affected individuals have cryptorchidism and hypospadias, in addition to more severe mental retardation and radio-ulnar synostosis.
Double Y males (47XYY) form 1 in 1000 male births; they are phenotypically normal, although most are over 6 feet (1.8 m) tall. The condition is said to be associated with aggressive or criminal behaviour, although this may be a manifestation of lower intelligence.
Turner’s syndrome (gonadal dysgenesis; 45X) occurs in 1 in 3000 female births. About one-half are mosaics (45X/46XX) and some have 46 chromosomes and two X chromosomes, one of which is defective. Turner’s syndrome females may have short stature, primary amenorrhoea and infertility, webbing of the neck, broad chest and widely spaced nipples, cubitus valgus, low posterior hairline and coarctation of the aorta.
Multiple X females (47XXX, 48XXXX) comprise 1 in 1200 female births. They may be mentally retarded, and have menstrual disturbances, although many are normal and fertile.
True hermaphrodites (most 46XX, some 46XX/47XXY mosaics) have both testicular and ovarian tissue, with varying genital tract abnormalities.
Parts of chromosomes
The loss (or addition) of even a small part of a chromosome may have severe effects, especially if ‘controlling’ or ‘master’ genes are involved, as these affect many other genes. An example of a congenital disease in this group is cri-du-chat syndrome (46XX, 5p− or 46XY, 5p−). This rare condition (1 in 50000 births) is associated with deletion of the short arm of chromosome 5 (5p−), and was so named because infants have a characteristic cry like the miaow of a cat. There is microcephaly and severe mental retardation; the face is round, there is gross hypertelorism (increased distance between the eyes) and epicanthic folds.
Single gene alterations
All of the inherited disorders of single genes are transmitted by autosomal dominant, autosomal recessive or X-linked modes of inheritance (Ch. 3). There are more than 2700 known Mendelian disorders; 80–85% of these are familial, the remainder are the result of new mutations. Sometimes the expression of the altered gene product has important effects on growth and morphogenesis, although in other cases a specific single abnormality in a particular biochemical pathway results.
Single gene disorders fall into three categories, discussed below.
Enzyme defects
An altered gene may result in decreased enzyme synthesis, or the synthesis of a defective enzyme (Ch. 7). A failure to synthesise the end products of a reaction catalysed by an enzyme may block normal cellular function. This occurs, for example, in albinism, caused by absent melanin production due to tyrosinase deficiency. Another effect may be the accumulation of the enzyme substrate, for example:
Defects in receptors or cellular transport
The lack of a specific cellular receptor causes insensitivity of a cell to substances such as hormones. In one form of male pseudohermaphroditism, for example, insensitivity of tissues to androgens, caused by lack of androgen receptor, prevents the development of male characteristics during fetal development.
Cellular transport deficiencies may lead to disorders such as cystic fibrosis (Ch. 7), a condition in which there is a defective cell membrane transport system across exocrine secretory cells.
Non-enzyme protein defects
Failure of production of important proteins, or production of abnormalities in proteins, has widespread effects. Thus, sickle cell anaemia is caused by the production of abnormal haemoglobin, and Marfan’s syndrome and Ehlers–Danlos syndrome are the result of defective collagen production.
Anomalies of fetal development
Abnormalities can occur at almost any stage of embryonic or fetal development; the mechanisms by which the anomaly occurs are sometimes unknown. Genetic factors may play a role in some conditions, but in many cases no simple genetic defect is identifiable. Anomalies of normal development caused by extrinsic physical forces (such as uterine constraint or amniotic bands) are termed deformations or disruptions. Intrinsic failures of morphogenesis, differentiation or growth are termed malformations.
The term syndrome refers to a collection of specific anomalies typically seen together but without an obvious single initiating localised defect. The term ‘sequence’ similarly refers to a condition with a constellation of typical individual features, but in which these features are secondary to an identified single localised primary anomaly which then leads to secondary effects elsewhere in the developing fetus. In the Potter sequence, for example, various primary causes of a decreased volume of amniotic fluid (oligohydramnios) all lead to fetal compression, with resultant deformations of the hands, feet, hips and facies. The sequential causal relationship between oligohydramnios, fetal compression and the observed resultant deformations distinguishes this condition as a syndrome rather than a sequence.
Embryo division abnormalities
Monozygotic twins result from the separation of groups of cells in the early embryo, well before the formation of the primitive streak. On occasion, there is a defect of embryo division, resulting in:
Teratogen exposure
Physical, chemical or infective agents can interfere with growth and differentiation, resulting in fetal abnormalities; such agents are known as teratogens. The extent and severity of fetal abnormality depend on the nature of the teratogen and the developmental stage of the embryo when exposed to the teratogen. Thus, if exposure occurs at the stage of early organogenesis (4–5 weeks of gestation), the effects on developing organs or limbs are severe.
Clinical examples of teratogenesis include the severe and extensive malformations associated with use of the drug thalidomide (absent/rudimentary limbs, defects of the heart, kidney, gastrointestinal tract, etc.), and the effects of rubella (German measles) on the fetus (cataracts, microcephaly, heart defects, etc.). Some other teratogens are listed in Table 5.5.
Table 5.5 Teratogens and their effects
| Teratogen | Teratogenic effect |
|---|---|
| Irradiation | Microcephaly |
| Drugs | |
| Thalidomide | Amelia/phocomelia (absent/rudimentary limbs), heart, kidney, gastrointestinal and facial abnormalities |
| Folic acid antagonists, e.g. 4-amino PGA | Anencephaly, hydrocephalus, cleft lip/palate, skull defects |
| Anticonvulsants | Cleft lip/palate, heart defects, minor skeletal defects |
| Warfarin | Nasal/facial abnormalities |
| Testosterone and synthetic progestagens | Virilisation of female fetus, atypical genitalia |
| Alcohol | Microcephaly, abnormal facies, oblique palpebral fissures, growth disturbance |
| Infections | |
| Rubella | Cataracts, microphthalmia, microcephaly, heart defects |
| Cytomegalovirus | Microcephaly |
| Herpes simplex | Microcephaly, microphthalmia |
| Toxoplasmosis | Microcephaly |
Failure of cell and organ migration
Failure of migration of cells may occur during embryogenesis.
Kartagener’s syndrome
In this rare condition there is a defect in ciliary motility, due to absent or abnormal dynein arms, the structures on the outer doublets of cilia that are responsible for ciliary movement. This affects cell motility during embryogenesis, which often results in situs inversus (congenital lateral inversion of the position of body organs resulting in, for example, left-sided liver and right-sided spleen). Complications in later life include bronchiectasis, and infertility due to sperm immobility.
Hirschsprung’s disease is a condition leading to marked dilatation of the colon and failure of colonic motility in the neonatal period, due to absence of Meissner’s and Auerbach’s nerve plexuses. It results from a selective failure of craniocaudal migration of neuroblasts in weeks 5–12 of gestation. It is, interestingly, 10 times more frequent in children with trisomy 21 (Down’s syndrome), and is often associated with other congenital anomalies.
Undescended testis (cryptorchidism) is the result of failure of the testis to migrate to its normal position in the scrotum. Although this may be associated with severe forms of Klinefelter’s syndrome (e.g. 48XXXY), it is often an isolated anomaly in an otherwise normal male. There is an increased risk of neoplasia in undescended testes.
Anomalies of organogenesis





Agenesis (aplasia)
The failure of development of an organ or structure is known as agenesis (aplasia). Obviously, agenesis of some structures (such as the heart) is incompatible with life, but agenesis of many individual organs is recorded. These include:
Atresia
Atresia is the failure of development of a lumen in a normally tubular epithelial structure. Examples include:
Hypoplasia
A failure in development of the normal size of an organ is termed hypoplasia. It may affect only part of an organ, e.g. segmental hypoplasia of the kidney. A relatively common example of hypoplasia affects the osseous nuclei of the acetabulum causing congenital dislocation of the hip, due to a flattened roof to the acetabulum.
Maldifferentiation (dysgenesis, dysplasia)
Maldifferentiation, as its name implies, is the failure of normal differentiation of an organ, which often retains primitive embryological structures. This disorder is often termed ‘dysplasia’, although this is a potential cause of confusion, as the more common usage of the term dysplasia implies the presence of a pre-neoplastic state (p. 83).
The best examples of maldifferentiation are seen in the kidney (‘renal dysplasia’) as a result of anomalous metanephric differentiation. Here, primitive tubular structures may be admixed with cellular mesenchyme and, occasionally, smooth muscle.
Ectopia, heterotopia and choristomas
Ectopic and heterotopic tissues are usually small areas of mature tissue from one organ (e.g. the gastric mucosa) that are present within another tissue (e.g. Meckel’s diverticulum) as a result of a developmental anomaly. Another clinically important example is endometriosis, in which endometrial tissue is found around the peritoneum in some women, causing abdominal pain at the time of menstruation.
A choristoma is a related form of heterotopia, where one or more mature differentiated tissues aggregate as a tumour-like mass at an inappropriate site. A good example of this is a complex choristoma of the conjunctiva (eye), which has varying proportions of cartilage, adipose tissue, smooth muscle, and lacrimal gland ascini. A conjunctival choristoma consisting of lacrimal gland elements alone could also be considered to be an ectopic (heterotopic) lacrimal gland.
Complex disorders of growth and morphogenesis
Three examples of complex multifactorial defects of growth and morphogenesis will be discussed: neural tube defects, disorders of sexual differentiation, and cleft palate and related disorders.
Neural tube defects
The development of the brain, spinal cord and spine from the primitive neural tube is highly complex and, not surprisingly, so too are the developmental disorders of the system (Fig. 5.20).
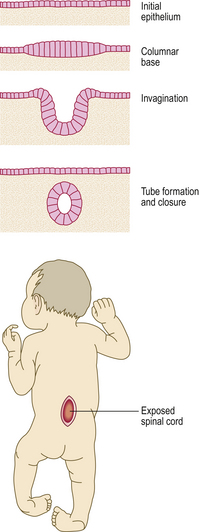
Fig. 5.20 Spina bifida. Top: Cross-section through the developing embryo. During the 4th week of development the neural tube is formed by invagination of the dorsal ectoderm. Failure of the neural tube to invaginate fully or of the overlying ectoderm to close afterwards results in neural tube defects such as spina bifida, in which the spinal cord is exposed (bottom). Deformity and hypoplasia of the legs results from the associated neurological deficit.
Neural tube malformations are relatively common in the UK and are found in about 1.3% of aborted fetuses, and 0.1% of live births. There are regional differences in incidence, and social differences, the condition being more common in social class V than in classes I or II. The pathogenesis of these conditions—anencephaly, hydrocephalus and spina bifida—is uncertain and probably multifactorial (Ch. 26), although dietary deficiency of folate (vitamin B9) during the early stages of embryogenesis is one established factor.
Disorders of sexual differentiation
Disorders of sexual differentiation are undoubtedly complex, and involve a range of individual chromosomal, enzyme and hormone receptor defects. The defects may be obvious and severe at birth, or they may be subtle, presenting with infertility in adult life.
Chromosomal abnormalities causing ambiguous or abnormal sexual differentiation have already been discussed (p. 94).
Female pseudohermaphroditism, in which the genetic sex is always female (XX), may be due to exposure of the developing fetus to the masculinising effects of excess testosterone or progestagens, causing abnormal differentiation of the external genitalia. The causes include:
Male pseudohermaphroditism, in which the genetic sex is male (XY), may be the result of several rare defects:
These defects result in the presence of a testis that is small and atrophic, and a female phenotype.
Cleft palate and related disorders
Cleft palate, and the related cleft (or hare) lip, are relatively common (about 1 per 1000 births). Approximately 20% of children with these disorders have associated major malformations. The important stages of development of the lips, palate, nose and jaws occur in the first 9 weeks of embryonic life. From about 5 weeks of gestational age the maxillary processes grow anteriorly and medially, and fuse with the developing fronto-nasal process at two points just below the nostrils, forming the upper lip. Meanwhile, the palate develops from the palatal processes of the maxillary processes, which grow medially to fuse with the nasal septum in the midline at about 9 weeks.
Failure of these complicated processes may occur at any stage, producing small clefts or severe facial deficits (Fig. 5.21). A cleft lip is commonly unilateral but may be bilateral; it may involve the lip alone, or extend into the nostril or involve the bone of the maxilla and the teeth. The mildest palatal clefting may involve the uvula or soft palate alone, but can lead to absence of the roof of the mouth. Cleft lip and palate occur singly or in combination, and severe combined malformations of the lips, maxilla and palate can be very difficult to manage surgically.
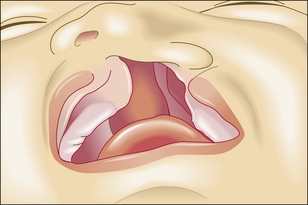
Fig. 5.21 Cleft palate. Diagram demonstrating a large defect involving the upper lip, the upper jaw and the palate.
Recently, lip and palate malformations have been extensively studied as a model of normal and abnormal states of morphogenesis in a complicated developmental system. It appears from the relatively high incidence of these malformations that the control of palatal morphogenesis is particularly sensitive to both genetic and environmental disturbances:
Recent experimental evidence has suggested that several cellular factors are involved in the fusion of the fronto-nasal and maxillary processes. The differentiation of epithelial cells of the palatal processes is of paramount importance in fusion of the processes. It is thought that the most important mechanism is mediated by mesenchymal cells of the palatal processes; these induce differentiation of the epithelial cells, to form either ciliated nasal epithelial cells or squamous buccal epithelial cells, or to undergo programmed cell death by apoptosis to allow fusion of underlying mesothelial cells. Positional information of a genetic and chemical (paracrine) nature is important in this differentiation, and is mediated via mesenchymal cells (and possibly epithelial cells). In addition, the events may be modified by the actions of EGF and other growth factors through autocrine or paracrine mechanisms, and by the endocrine actions of glucocorticoids and their intercellular receptors.
As yet, the precise way in which all of these factors interact in normal palatal development or cleft palate is unclear. In the mouse, it is known that physiological concentrations of glucocorticoids, their receptors and EGF are required for normal development, but that altered concentrations may precipitate cleft palate.
Commonly confused conditions and entities relating to growth, differentiation and morphogenesis
| Commonly confused | Distinction and explanation |
|---|---|
| Hyperplasia and hypertrophy | Both result in organ enlargement, in hyperplasia by cellular proliferation and in hypertrophy by cellular enlargement. Generally, the innate replicative capacity (i.e. permanent, stable or labile) of the organ’s cells determines whether it responds by hyperplasia or hypertrophy. |
| Agenesis, aplasia, atresia, achalasia and atrophy | Agenesis and aplasia imply a failure of formation of an organ or cell lineage; if the structure has a lumen, then atresia is often used (e.g. biliary atresia). Achalasia is failure of a sphincter to relax, causing proximal dilatation; it is not a disorder of growth. Atrophy is shrinkage of an organ; when it occurs naturally during maturation and ageing, the process is often called ‘involution’. |
| Dysplasia and dystrophy | Dysplasia means disordered growth or differentiation and, in epithelia, is often considered to be a precursor of neoplasia. Dystrophy means either abnormal development (e.g. muscular dystrophy) or, less commonly, a degenerative change (e.g. dystrophic calcification). |
| Congenital and genetic | Congenital refers to a condition present at birth, and often is used specifically to denote a condition with a visible morphological manifestation. Genetic conditions are those caused by abnormalities in genes or chromosomes, although the influence of these abnormalities may be modulated by environmental factors. |
Alberts B, Johnson A, Lewis J, Raff M, Roberts M, Walter O. Molecular biology of the cell, 4th edn. New York: Academic Press; 2002.
Alison M .R., Poulsom R., Wright N .A., editors. Stem cells. Journal of Pathology. 2002;197:417-565
Alison M. R, Lovell M., Direckze N., Poulsom R. 2005 Adult stem cells and transdifferentiation. In: Recent advances in histopathology. vol 21. London: RSM Press.
Berry C. L. 1996. Paediatric pathology. 3rd edn. London: Springer.
Drury P .L., Howlett T .A.. Endocrinology. In In: Kumar P., Clark M., editors: Clinical medicine, 5th edn, Edinburgh: WB Saunders, 2002.
Eckfeldt C. E, Mendenhall E .M., Verfaillie C. M. The molecular repertoire of the ‘almighty’ stem cell. Nature Reviews. Molecular Cell Biology. 2005;6:726-737.
Gilbert S .F., Singer S. R. Developmental biology, 8th edn. Sunderland, MA: Sinauer, 2001.
Hall P. A. 1992 Differentiation, stem cells and tumour histogenesis. In: Anthony P .P., MacSween R. N. M, editors. Recent advances in histopathology, vol 15. Churchill Livingstone Edinburgh, pp 1-15
Iles R.K., Kumar P.J.. Cell and molecular biology and genetic disorders. In In: Kumar P., Clark M., editors: Clinical medicine, 5th edn, Edinburgh: WB Saunders, 2002.
Lawrence P .A.. The making of a fly. The genetics of animal design. Oxford: Blackwell Scientific Publications; 1992.
Lodish H., Berk A., Kaiser C.A., Kriege M.. Molecular cell biology, 6th edn. Basingstoke: WH Freeman, 2008.
Michalides R .J. A .M.. Cell cycle regulators: mechanisms and their role in aetiology, prognosis, and treatment of cancer. Journal of Clinical Pathology. 1999;52:555-568.
Sadler T.W.. Langman’s medical embryology, 10th edn. Baltimore: Lippincott Williams & Wilkins, 2006.
Thompson C B. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456-1462.
Tosk D., Slack J.M W. How cells change their phenotype. Nature Reviews. Molecular Cell Biology. 2002;3:187-194.
Wilmut I., Schnieke A E, McWhir J., Kind A J, Campbell K H S. Viable offspring derived from fetal and adult mammalian cells. Nature. 1997;385:810-813.
Wyllie A H. Apoptosis: an overview. British Medical Bulletin. 1997;53:451-465.
Browder L 1998 Dynamic development: http://www.ucalgary.ca/UofC/eduweb/virtualembryo/dev_biol.html
Gilbert S F 1997: http://zygote.swarthmore.edu/index.html
Hill M A (2007) Embryology; University of New South Wales: http://embryology.med.unsw.edu.au/
Online Mendelian Inheritance in Man (OMIM Home Page): http://www.ncbi.nlm.nih.gov/omim
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
Chapter 5 Disorders of growth, differentiation and morphogenesis
Growth, differentiation and morphogenesis are the processes by which a single cell, the fertilised ovum, develops into a large complex multicellular organism, with co-ordinated organ systems containing a variety of cell types, each with individual specialised functions. Growth and differentiation continue throughout adult life, as many cells of the body undergo a constant cycle of death, replacement and growth in response to normal (physiological) or abnormal (pathological) stimuli.
There are many stages in human embryological development at which anomalies of growth and/or differentiation may occur, leading to major or minor abnormalities of form or function, or even death of the fetus. In post-natal and adult life, some alterations in growth or differentiation may be beneficial, as in the development of increased muscle mass in the limbs of workers engaged in heavy manual tasks. Other changes may be detrimental to health, as in cancer, where the outcome may be fatal.
This chapter explores the wide range of abnormalities of growth, differentiation and morphogenesis that may be encountered in clinical practice, relating them where possible to specific deviations from normal cellular functions or control mechanisms.
DEFINITIONS
Growth
Growth is the process of increase in size resulting from the synthesis of specific tissue components. The term may be applied to populations, individuals, organs, cells, or even subcellular organelles such as mitochondria.
Types of growth in a tissue (Fig. 5.1A) are:
Differentiation
Differentiation is the process whereby a cell develops an overt specialised function or morphology that distinguishes it from its parent cell. There are many different cell types in the human body, but all somatic cells in an individual have identical genomes. Differentiation is the process by which genes are expressed selectively and gene products act to produce a cell with a specialised function (Fig. 5.1B). After fertilisation of the human ovum, and up to the eight-cell stage of development, all of the embryonic cells are apparently identical. Thereafter, cells undergo several stages of differentiation in their passage to fully differentiated cells, such as, for example, the ciliated epithelial cells lining the respiratory passages of the nose and trachea. Although the changes at each stage of differentiation may be minor, differentiation can be said to have occurred only if there has been overt change in cell morphology (e.g. development of a skin epithelial cell from an ectodermal cell), or an alteration in the specialised function of a cell (e.g. the synthesis of a hormone).
Morphogenesis
Morphogenesis is the highly complex process of development of structural shape and form of organs, limbs, facial features, etc. from primitive cell masses during embryogenesis. For morphogenesis to occur, primitive cell masses must undergo co-ordinated growth and differentiation, with movement of some cell groups relative to others, and focal programmed cell death (apoptosis) to remove unwanted features.
NORMAL AND ABNORMAL GROWTH IN SINGLE TISSUES
Within an individual organ or tissue, increased or decreased growth takes place in a range of physiological and pathological circumstances as part of the adaptive response of cells to changing requirements for growth. In both fetal and adult life, tissue growth depends upon the balance between the increase in cell numbers, due to cell proliferation, and the decrease in cell numbers, due to cell death. Non-proliferative cells are termed ‘quiescent’; such cells differentiate and adopt specific phenotypes capable of carrying out their specific function (Fig. 5.2).
Fig. 5.2 Cell proliferation and death. Individual cells have three potential fates: proliferation, differentiation or apoptosis. After division, individual daughter cells may differentiate, and under some circumstances some differentiated cells may re-enter the cell cycle. The growth rate of a tissue is determined by the net balance between proliferation, differentiation and apoptosis.
In fetal life, growth is rapid and all cell types proliferate, but even in the fetus there is constant cell death, some of which is an essential (and genetically programmed) component of morphogenesis. In post-natal and adult life, however, the cells of many tissues lose their capacity for proliferation at the high rate of the fetus, and cellular replication rates are variably reduced. Some cells continue to divide rapidly and continuously, some divide only when stimulated by the need to replace cells lost by injury or disease, and others are unable to divide whatever the stimulus.
Regeneration
Regeneration enables cells or tissues destroyed by injury or disease to be replaced by functionally identical cells. These replaced ‘daughter’ cells are usually derived from a tissue reservoir of ‘parent’ stem cells (discussed below, p. 92). The presence of tissue stem cells, with their ability to proliferate, governs the regenerative potential of a specific cell type. Mammalian tissues fall into three classes according to their regenerative ability:
Labile cells proliferate continuously in post-natal life; they have a short life-span and a rapid ‘turnover’ time. Their high regenerative potential means that lost cells are rapidly replaced by division of stem cells. However, the high cell turnover renders these cells highly susceptible to the toxic effects of radiation or drugs (such as anti-cancer drugs) that interfere with cell division. Examples of labile cells include:
The high regenerative potential of the skin is exploited in the treatment of patients with skin loss due to severe burns. The surgeon removes a layer of skin which includes the dividing basal cells from an unburned donor site, and fixes it firmly to the burned graft site where the epithelium has been lost (Ch. 6). Dividing basal cells in the graft and the donor site ensure regeneration of squamous epithelium at both sites, enabling rapid healing in a large burned area where regeneration of new epithelium from the edge of the burn would otherwise be prolonged.
Stable cells (sometimes called ‘conditional renewal cells’) divide very infrequently under normal conditions, but their stem cells are stimulated to divide rapidly when such cells are lost. This group includes cells of the liver, endocrine glands, bone, fibrous tissue and the renal tubules.
Permanent cells normally divide only during fetal life, but their active stem cells do not persist long into post-natal life, and they cannot be replaced when lost. Cells in this category include neurones, retinal photoreceptors and neurones in the eye, cardiac muscle cells and skeletal muscle (although skeletal muscle cells do have a very limited capacity for regeneration).
The cell cycle
Successive phases of progression of a cell through its cycle of replication are defined with reference to DNA synthesis and cellular division. Unlike the synthesis of most cellular constituents, which occurs throughout the interphase period between cell divisions, DNA synthesis occurs only during a limited period of the interphase; this is the S phase of the cell cycle. A further distinct phase of the cycle is the cell-division stage or M phase (Fig. 5.3) comprising nuclear division (mitosis) and cytoplasmic division (cytokinesis). Following the M phase, the cell enters the first gap (G1) phase and, via the S phase, the second gap (G2) phase before entering the M phase again. Although initially regarded as periods of inactivity, it is now recognised that these ‘gap’ phases represent periods when critical processes occur, preparing the cells for DNA synthesis and mitosis.
Fig. 5.3 The four stages of the cell cycle. G1 represents preparation for DNA synthesis (S phase), and G2 represents preparation for mitosis (M phase). After mitosis individual daughter cells may each re-enter the cycle at G1 if appropriate stimuli are present. Alternatively, they may permanently or temporarily enter G0 and differentiate. Progress around the cell cycle is one-way. ‘Checkpoints’ ensure one phase does not commence until the previous phase is completed. Failure of a phase to complete satisfactorily results in cell cycle arrest, or—if the problem is irretrievable—apoptosis.
After cell division (mitosis), individual daughter cells may re-enter G1 to undergo further division if appropriate stimuli are present. Alternatively, they may leave the cycle and become quiescent or ‘resting’ cells—a state often labelled as G0. Entry to G0 may be associated with a process of terminal differentiation, with loss of potential for further division and death at the end of the lifetime of the cell; this occurs in permanent cells, such as neurones. Other quiescent cells retain some ability to proliferate by re-entering G1 if appropriate stimuli are present.
Molecular events in the cell cycle
Cell division is a highly complex process and cells possess elaborate molecular machinery to ensure its successful completion. A number of internal ‘checkpoints’ exist to ensure that one phase is complete before the next commences (Fig. 5.3). This is vital to ensure, for example, that DNA replication has been performed accurately and that cells do not divide before DNA replication is complete. The various proteins and enzymes that carry out DNA replication, mitotic spindle formation, etc. are typically only present and active during the appropriate phases of the cycle. The timely production and activation of these proteins is regulated by the activity of a family of evolutionarily conserved proteins called cyclin dependent kinases (CDKs), which activate their target proteins by phosphorylation. The activity of CDKs is, in turn, regulated by a second family of proteins, the cyclins. Transitions from one phase of the cycle to the next are initiated by rises in the levels of specific cyclins. The transition from G0 to G1 at the initiation of the cell cycle, for example, is triggered by external signals such as growth factors leading to rises in the levels of cyclin D. Problems during cell division, such as faulty DNA replication, result in rises in the levels of a third family of proteins, the CDK inhibitors (CDKIs), which can prevent CDKs from triggering the next phase of cell division until the issue is resolved. In the face of major failures, cells will typically initiate apoptosis rather than permit the generation of improperly formed progeny. Damage to the genes that encode proteins involved in the regulation of cell-cycle progression is seen in many cancers (Ch. 11).
Duration of the cell cycle
In mammals, different cell types divide at very different rates, with observed cell cycle times (also called generation times) ranging from as little as 8 hours, in the case of gut epithelial cells, to 100 days or more, exemplified by hepatocytes in the normal adult liver. However, the duration of the individual phases of the cycle is remarkably constant and independent of the rate of cell division. The principal difference between rapidly dividing cells and those that divide slowly is the time spent temporarily in G0 between divisions; some cells remain in the G0 phase for days or even years between divisions, whilst others rapidly re-enter G1 after mitosis.
Therapeutic interruption of the cell cycle
Many of the drugs used in the treatment of cancer affect particular stages within the cell cycle (Fig. 5.4). These drugs inhibit the rapid division of cancer cells, although there is often inhibition of other rapidly dividing cells, such as the cells of the bone marrow and lymphoid tissues. Thus, anaemia, a bleeding tendency and suppression of immunity may be clinically important side-effects of cancer chemotherapy.
Cell death in growth and morphogenesis
It seems illogical to think of cell death as a component of normal growth and morphogenesis, although we recognise that the loss of a tadpole’s tail, which is mediated by the genetically programmed death of specific cells, is part of the metamorphosis of a frog. It is now clear that such cell death has an important role in human development and in the regulation of tissue size throughout life. Alterations in the rate at which cell death occurs are important in situations such as hormonal growth regulation, immunity and neoplasia.
Apoptosis
The term apoptosis is used to denote a physiological cellular process in which a defined and programmed sequence of intracellular events leads to the death of a cell without the release of products harmful to surrounding cells. It is a biochemically specific mode of cell death characterised by activation of non-lysosomal endogenous endonuclease which digests nuclear DNA into smaller DNA fragments. Morphologically, apoptosis is recognised as death of scattered single cells which form rounded, membrane-bound bodies; these are eventually phagocytosed (ingested) and broken down by adjacent unaffected cells.
The co-existence of apoptosis alongside mitosis within a cell population ensures a continuous renewal of cells, rendering a tissue more adaptable to environmental demands than one in which the cell population is static.
Apoptosis can be triggered by factors outside the cell or it can be an autonomous event (‘programmed cell death’). In embryological development, there are three categories of autonomous apoptosis:
Morphogenetic apoptosis is involved in alteration of tissue form. Examples include:
Fig. 5.5 Morphogenesis by apoptosis. Genetically programmed apoptosis (individual cell death) causing separation of the fingers during embryogenesis.
Failure of morphogenetic apoptosis in these four sites is a factor in the development of syndactyly (webbed fingers), cleft palate, spina bifida, and bladder diverticulum (pouch) or fistula (open connection) from the bladder to the umbilical skin, respectively.
Histogenic apoptosis occurs in the differentiation of tissues and organs, as seen, for example, in the hormonally controlled differentiation of the accessory reproductive structures from the Müllerian and Wolffian ducts. In the male, for instance, anti-Müllerian hormone produced by the Sertoli cells of the fetal testis causes regression of the Müllerian ducts (which in females form the fallopian tubes, uterus and upper vagina) by the process of apoptosis.
Phylogenetic apoptosis is involved in removing vestigial structures from the embryo; structures such as the pronephros, a remnant from a much lower evolutionary level, are removed by the process of apoptosis.
Regulation of apoptosis
Apoptosis may be triggered by external signals, such as detachment from the extracellular matrix, the withdrawal of growth factors, or specific signals from other cells. This mode of activation of apoptosis is called the extrinsic pathway. By contrast, the intrinsic pathway is activated by intracellular signals, such as DNA damage or failure to conduct cell division correctly. Although apoptosis can be induced by diverse signals in a variety of cell types, a few genes appear to regulate a final common pathway. The most important of these are the members of the bcl-2 family (bcl-2 was originally identified at the t(14;18) chromosomal breakpoint in follicular B-cell lymphoma, and it can inhibit many factors that induce apoptosis). The bax protein (also in the bcl-2 family) forms bax–bax dimers which enhance apoptotic stimuli. The ratio of bcl-2 to bax determines the cell’s susceptibility to apoptotic stimuli, and constitutes a ‘molecular switch’ which determines whether a cell will survive, leading to tissue expansion, or undergo apoptosis.
The study of factors regulating apoptosis is of considerable importance in finding therapeutic agents to enhance cell death in malignant neoplasms.
Increased growth: hypertrophy and hyperplasia
The response of an individual cell to increased functional demand is to increase tissue or organ size (Fig. 5.6) by:
Fig. 5.6 Hyperplasia and hypertrophy. In hypertrophy, cell size is increased. In hyperplasia, cell number is increased. Hypertrophy and hyperplasia may co-exist.
The stimuli for hypertrophy and hyperplasia are very similar, and in many cases identical; indeed, hypertrophy and hyperplasia commonly co-exist. In permanent cells hypertrophy is the only adaptive option available under stimulatory conditions. In some circumstances, however, permanent cells may increase their DNA content (ploidy) in hypertrophy, although the cells arrest in the G2 phase of the cell cycle without undergoing mitosis; such a circumstance is present in severely hypertrophied hearts, where a large proportion of cells may be polyploid.
An important component of hyperplasia, which is often overlooked, is a decrease in cell loss by apoptosis; the mechanisms of control of this decreased apoptosis are unclear, although they are related to the factors causing increased cell production (Fig. 5.7).
Fig. 5.7 Control of tissue growth by induction or inhibition of apoptosis. Quiescent (mitotically inactive) cells in G0 are recruited into a high turnover (mitotically active) state by growth factors. Their subsequent fate depends on the presence or absence of apoptosis inducers or inhibitors. The inducers and inhibitors are mediated by the bax and bcl-2 proteins respectively, among others.
Physiological hypertrophy and hyperplasia
Examples of physiologically increased growth of tissues include:
In addition to such physiologically increased tissue growth, hypertrophy and hyperplasia are also seen in tissues in a wide range of pathological conditions.
Pathological hypertrophy and hyperplasia
Many pathological conditions are characterised by hypertrophy or hyperplasia of cells. In some instances, this is the principal feature of the condition from which the disease is named. The more common examples are summarised in Table 5.1. For more detail, consult the relevant chapters.
Table 5.1 Examples of non-regenerative hypertrophy and hyperplasia
| Organ/tissue | Condition | Comment |
|---|---|---|
| Myocardium | Right ventricular hypertrophy | Response to pulmonary valve stenosis, pulmonary hypertension or ventricular septal defect (Ch. 13) |
| Left ventricular hypertrophy | Response to aortic valve stenosis or systemic hypertension (Ch. 13) | |
| Arterial smooth muscle | Hypertrophy of arterial walls | Occurs in hypertension (Ch. 13) |
| Capillary vessels | Proliferative retinopathy | Complication of diabetes mellitus (Ch. 26) |
| Bone marrow | Erythrocyte precursor hyperplasia | Response to increased erythropoietin production (e.g. due to hypoxia) (Ch. 23) |
| Cytotoxic T-lymphocytes | Hyperplastic expansion of T-cell populations | Involved in cell-mediated immune responses (Ch. 9) |
| Breast | Juvenile hypertrophy (females) | Exaggerated pubertal enlargement (Ch. 18) |
| Due to high oestrogen levels (e.g. in cirrhosis, iatrogenic, endocrine tumours) (Ch. 18) | ||
| Gynaecomastia (males) | ||
| Prostate | Epithelial and connective tissue hyperplasia | Relative excess of oestrogens stimulates oestrogen-sensitive central zone (Ch. 20) |
| Thyroid | Follicular epithelial hyperplasia | Most commonly due to a thyroid-stimulating antibody (Graves’ disease) (Ch. 17) |
| Adrenal cortex | Cortical hyperplasia | Response to increased ACTH production (e.g. from a pituitary tumour or, inappropriately, from a lung carcinoma) (Ch. 17) |
| Myointimal cells | Myointimal cell hyperplasia in atheromatous plaques | Myointimal cells in plaques proliferate in response to platelet-derived growth factor (Ch. 13) |
Apparently autonomous hyperplasias
In some apparently hyperplastic conditions, cells appear autonomous, and continue to proliferate rapidly despite the lack of a demonstrable stimulus or control mechanism. The question then arises as to whether these should be considered to be hyperplasias at all, or whether they are autonomous and hence neoplastic. If the cells can be demonstrated to be monoclonal (derived as a single clone from one cell), then this suggests that the lesion may indeed be neoplastic, but clonality is often difficult to establish.
Hyperplasia in tissue repair
The proliferation of vascular (capillary) endothelial cells and myofibroblasts in scar tissue, and the regeneration of specialised cells within a tissue, are the important components of the response to tissue damage.
Angiogenesis is the process whereby new blood vessels grow into damaged, ischaemic or necrotic tissues in order to supply oxygen and nutrients for cells involved in regeneration and repair (the term ‘vasculogenesis’ should be reserved specifically for the blood vessel proliferation that occurs in the developing embryo and fetus). In response to local tissue damage, vascular endothelial cells within pre-existing capillaries are activated by angiogenic growth factors such as vascular endothelial growth factor (VEGF), released by hypoxic cells or macrophages. These activated endothelial cells then migrate towards the angiogenic stimulus to form a ‘sprout’. Cell migration is facilitated by the secretion of enzymes including the matrix metalloproteinases, which selectively degrade extracellular matrix proteins. Adjacent sprouts connect to form vascular loops, which canalise and establish a blood flow. Later, mesenchymal cells—including pericytes and smooth muscle cells—are recruited to stabilise the vascular architecture, and the extracellular matrix is remodelled.
Two other initiating mechanisms exist in addition to the above ‘sprouting’ form of angiogenesis: existing vascular channels may be bisected by an extracellular matrix ‘pillar’ (intussusception), with the two channels subsequently being extended towards the angiogenic stimulus. The final mechanism involves circulating primordial stem cells which are recruited at sites of hypoxia and differentiate into activated vascular endothelial cells. Note that a similar process of angiogenesis occurs in response to tumour cells, as an essential component of the development of the blood supply of enlarging neoplasms. Such angiogenesis is a potential therapeutic target in the treatment of malignant neoplasms, although theoretically such drugs might impair angiogenesis and therefore delay healing of wounds.
Myofibroblasts often follow new blood vessels into damaged tissues, where they proliferate and produce matrix proteins such as fibronectin and collagen to strengthen the scar. Myofibroblasts eventually contract and differentiate into fibroblasts. The resulting contraction of the scar may cause important complications, such as:
Thus vascular endothelial cell and myofibroblast hyperplasia are important components of repair and regeneration at various sites in the body, as described below.
Skin
The healing of a skin wound is a complex process involving the removal of necrotic debris from the wound and repair of the defect by hyperplasia of capillaries, myofibroblasts and epithelial cells. Figure 5.8 illustrates some of the key events, most of which are mediated by growth factors.
Fig. 5.8 Factors mediating wound healing. A wound is shown penetrating the skin and entering a blood vessel. (1) Blood coagulation and platelet degranulation, releasing growth factors (GF). (2) These are chemotactic for macrophages, which migrate into the wound to phagocytose bacteria and necrotic debris (3). Epidermal basal epithelial cells are activated by released growth factors from the platelets (4) and dermal myofibroblasts (5); from epidermal cells by paracrine (6) and autocrine (7) mechanisms; and from saliva (8) (if the wound is licked). Nutrients and oxygen (9) and circulating hormones and growth factors diffusing from blood vessels all contribute to epidermal growth. Growth factors from the platelets stimulate cell division in myofibroblasts (10), which produce collagen and fibronectin. Fibronectin stimulates migration of dermal myofibroblasts (11) and epidermal epithelial cells (12) into and over the wound. Angiogenic growth factors (not shown) stimulate the proliferation and migration of new blood vessels into the area of the wound (13).
When tissue injury occurs there is haemorrhage into the defect from damaged blood vessels; this is controlled by normal haemostatic mechanisms, during which platelets aggregate and thrombus forms to plug the defect in the vessel wall. Because of interactions between the coagulation and complement systems, inflammatory cells are attracted to the site of injury by chemotactic complement fractions. In addition, platelets release two potent growth factors, platelet-derived growth factor (PDGF) and transforming growth factor-beta (TGF-beta), which are powerfully chemotactic for inflammatory cells, including macrophages; these migrate into the wound to remove necrotic tissue and fibrin.
In the epidermis, PDGF acts synergistically with epidermal growth factor (EGF), derived from epidermal cells, and the somatomedins, insulin-like growth factor 1 (IGF-1) and insulin-like growth factor 2 (IGF-2), to promote proliferation of basal epithelial cells. EGF is also present in high concentrations in saliva and may reach wounds when they are licked. In the dermis, myofibroblasts proliferate in response to PDGF (and TGF-beta); collagen and fibronectin secretion is stimulated by TGF-beta, and fibronectin then aids migration of epithelial and dermal cells. Capillary budding and proliferation are stimulated by angiogenic factors such as VEGF. The capillaries ease the access of inflammatory cells and fibroblasts, particularly into large areas of necrotic tissue.
Hormones (e.g. insulin and thyroid hormones) and nutrients (e.g. glucose and amino acids) are also required. Lack of nutrients or vitamins, the presence of inhibitory factors such as corticosteroids or infection, or a locally poor circulation with low tissue oxygen concentrations, may all materially delay wound healing; these factors are very important in clinical practice.
Liver
In severe chronic hepatitis (Ch. 16) extensive hepatocyte loss is followed by scarring, as is the case in the skin or other damaged tissues. Like epidermal cells in the skin, hepatocytes have massive regenerative potential and surviving hepatocytes may proliferate to form nodules. Hyperplasia of hepatocytes and fibroblasts is presumably mediated by a combination of hormones and growth factors, although the mechanisms are far from clear. Regenerative nodules of hepatocytes and scar tissue are the components of cirrhosis of the liver.
Heart
Myocardial cells are permanent cells (i.e. they remain permanently in G0 and cannot enter G1), and so cannot divide in a regenerative response to tissue injury. In myocardial infarction, a segment of muscle dies and, if the patient survives, it is replaced by scar tissue. As the remainder of the myocardium must work harder for a given cardiac output, it undergoes compensatory hypertrophy (without cell division) (Fig. 5.9). Occasionally, there may be right ventricular hypertrophy as a result of left ventricular failure and consequent pulmonary hypertension.
Fig. 5.9 Cardiac hypertrophy. A horizontal slice through the myocardium of the left (L) and right (R) ventricles. (1) Normal. (2) Area of anteroseptal left ventricular infarct. (3) Compensatory hypertrophy of the surviving left ventricle. (4) Right ventricular hypertrophy secondary to left ventricular failure and pulmonary hypertension.
Decreased growth: atrophy
Atrophy is the decrease in size of an organ or cell by reduction in cell size and/or reduction in cell numbers, often by a mechanism involving apoptosis. Tissues or cells affected by atrophy are said to be atrophic or atrophied. Atrophy is an important adaptive response to a decreased requirement of the body for the function of a particular cell or organ. It is important to appreciate that for atrophy to occur there must be not only a cessation of growth but also an active reduction in cell size and/or a decrease in cell numbers, mediated by apoptosis.
Atrophy occurs in both physiological and pathological conditions.
Physiological atrophy and involution
Physiological atrophy occurs at times from very early embryological life, as part of the process of morphogenesis. The process of atrophy (mediated by apoptosis of cells) contributes to the physiological involution of organs such as the thymus gland in early adult life, and late old age is accompanied by atrophy of various tissues (Table 5.2).
Table 5.2 Tissues involved in physiological atrophy and involution
| Embryo and fetus | Early adult |
| Branchial clefts | Thymus |
| Notochord | |
| Thyroglossal duct | Late adult and old age |
| Müllerian duct (males) | Uterus, endometrium (females) |
| Wolffian duct (females) | Testes (males) |
| Bone (particularly females) | |
| Neonate | Gums |
| Umbilical vessels | Mandible (particularly edentulous) |
| Ductus arteriosus | Cerebrum |
| Fetal layer adrenal cortex | Lymphoid tissue |
[/not-level-membership-for-pathology-category]
