Chapter 22 Lymph nodes, thymus and spleen
COMMON CLINICAL PROBLEMS FROM DISEASE OF THE LYMPH NODES, THYMUS AND SPLEEN
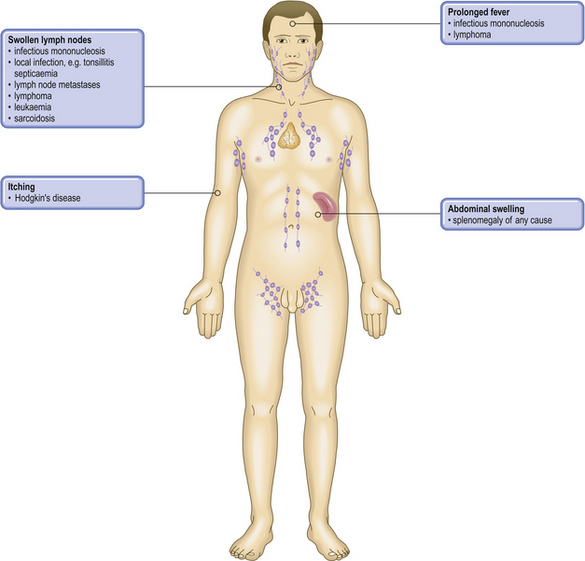
Pathological basis of signs and symptoms attributable to the lymphoreticular system
| Sign or symptom | Pathological basis |
|---|---|
| Enlarged lymph nodes |
Enlarged spleen
Susceptibility to infection
Weight loss/pyrexiaInterleukins produced by inflammatory or lymphomatous (i.e. type B symptoms) tissue acting on thermoregulatory centre in hypothalamusMuscle weakness (myasthenia gravis)Thymic hyperplasia or neoplasiaHowell–Jolly inclusions in red cellsPersistence of DNA fragments in red cells due to splenic atrophy
LYMPH NODES
NORMAL STRUCTURE AND FUNCTION
Lymph nodes are discrete encapsulated structures, usually ovoid and ranging in diameter from a few millimetres to several centimetres. They are situated along the course of lymphatic vessels and are more numerous where these vessels converge (e.g. roots of limbs, neck, pelvis and mediastinum).
Micro-architecture and functional anatomy
Lymph nodes are surrounded by a connective tissue capsule, with trabeculae that extend into the substance of the node and provide a framework for the contained cellular elements. Beneath the capsule is a slit-like space, the subcapsular sinus, into which the afferent lymphatics drain after penetrating the capsule. Lymph from the subcapsular sinus passes via the medullary cords to the hilum of the lymph node from which the efferent lymphatic drains.
Three distinct micro-anatomical regions can be recognised within normal lymph nodes (Ch. 9). These regions are:
The micro-anatomical regions of the lymph nodes are populated by a variety of specialised cells with different functional characteristics.
Germinal centres
The germinal centre is the principal site of B-cell activation in response to antigenic challenge. Antigen, bound to antibody, entering the lymph node via the afferent lymphatics is trapped upon the surface of specialised antigen-presenting cells called dendritic reticulum cells (DRCs) by their Fc receptors. DRCs are restricted to primary follicles and germinal centres and are binucleate cells with long cytoplasmic processes linked by desmosomes which form a network throughout the germinal centre. Antigen trapped on the surface of the DRC is presented to ‘virgin’ B-lymphocytes in the presence of T-helper cells (T-cell co-operation) and these B-cells subsequently undergo a series of morphological and functional changes (Table 22.1). After antigenic challenge, the initial step in B-cell transformation is the formation of the centroblast, which is a rapidly dividing cell that is responsible for expansion of the antigen-reactive B-cell clone; this then develops into a centrocyte (Fig. 22.1). During the germinal centre reaction the B-cell immunoglobulin genes undergo hypermutation to produce higher affinity immunoglobulin molecules; B-cells in which hypermutation does not achieve this undergo apoptosis. The number of B-cells that act as progenitors for the fully mature germinal centre is remarkably small and the mass of the germinal centre B-cell population is made up by the extensive proliferative activity of a small number of progenitor cells.
Table 22.1 Characteristics and nomenclature (Kiel scheme) of follicle centre cells and therefore B-cell lymphomas derived therefrom
| Cell features | Nomenclature |
|---|---|
| Small lymphocyte with round nucleus | Lymphocyte |
| Small or large cell with indented nucleus | Centrocyte |
| Large cell with round nucleus and usually multiple nucleoli | Centroblast |
| Large cell with round nucleus and large nucleolus | Immunoblast |
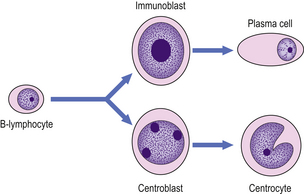
Fig. 22.1 Morphological changes of the germinal centre B-cell. Hypothetical follicle centre B-cell transformation. The virgin B-lymphocyte has two activation pathways. The first involves direct transformation to an immunoblast following exposure to an antigen, with subsequent differentiation to an immunoglobulin-secreting plasma cell; this pathway is independent of the germinal centre. The second pathway occurs within the germinal centre and involves the generation of a centroblast as the initial reaction to antigen exposure, with subsequent differentiation to a centrocyte.
The function of germinal centres is to generate immunoglobulin-secreting plasma cells in response to antigenic challenge. Within the lymph node, plasma cells are located principally within the medullary cords.
The fully formed germinal centre is seen histologically as a rounded, pale structure in the cortex of the lymph node, surrounded by a rim of small, round lymphocytes termed the mantle zone. Distinct zonation may be seen within the germinal centre: a pale zone faces towards the subcapsular sinus, is rich in centrocytes and T-cells, and contains the greatest density of DRCs; at the opposite pole of the germinal centre is a dark zone rich in rapidly dividing centroblasts mixed with tingible body macrophages which phagocytose the cellular debris generated by apoptosis of B-cells secondary to unsuccessful immunoglobulin gene hypermutation (Fig. 22.2). In florid B-cell reactions a population of post-germinal centre B-cells may accumulate adjacent to the mantle zone; these are termed marginal zone B-cells.
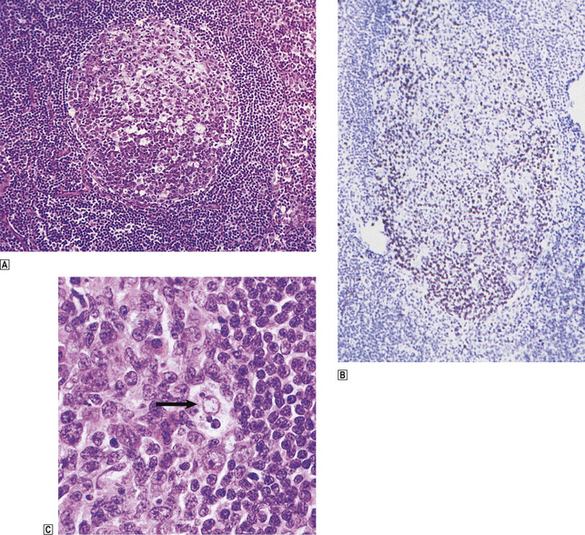
Fig. 22.2 Lymph node germinal centre.  Normal germinal centre showing distinct zoning, the lower half containing closely packed and rapidly dividing centroblasts.
Normal germinal centre showing distinct zoning, the lower half containing closely packed and rapidly dividing centroblasts.  A germinal centre stained with monoclonal antibody to proliferating cells (Ki-67). Numerous dividing cells (brown nuclei) are seen in the lower, centroblast-rich, region of the germinal centre.
A germinal centre stained with monoclonal antibody to proliferating cells (Ki-67). Numerous dividing cells (brown nuclei) are seen in the lower, centroblast-rich, region of the germinal centre.  High-power view of phagocytic or ‘tingible body’ macrophage (arrowed) engulfing apoptotic lymphoid cells. These are concentrated in the most proliferative part of the germinal centre.
High-power view of phagocytic or ‘tingible body’ macrophage (arrowed) engulfing apoptotic lymphoid cells. These are concentrated in the most proliferative part of the germinal centre.
Paracortex
The paracortex is the T-cell-dependent region of the lymph node and accordingly contains large numbers of T-lymphocytes with a predominance of the helper/inducer subset (CD4+). The cluster of differentiation (CD) 4 antigen is expressed by helper/inducer T-cells. As in the germinal centre, specialised antigen-presenting cells are present in the paracortex; these are called interdigitating reticulum cells (IDCs) and are different morphologically and functionally from the DRCs. IDCs possess abundant cytoplasm with complex membrane profiles which interdigitate with surrounding T-cells. Large amounts of class II human leukocyte antigen (HLA) substances are expressed on the surface of the IDC and this is important for interactions between immune cells, especially in antigen presentation to T-cells (particularly the helper T-cells).
Medulla
Lymph enters the marginal sinus of the node and drains to the hilum through sinuses that converge in the medullary region. The sinuses are lined by macrophages which phagocytose particulate material within the lymph. Between the sinuses in the medulla lie the medullary cords, which contain numerous plasma cells and are one of the main sites of antibody secretion within the lymph node.
LYMPH NODE ENLARGEMENT




Lymph node enlargement (lymphadenopathy) may be localised or widespread and is a common clinical problem that frequently requires a biopsy to establish a diagnosis. The causes of lymphadenopathy are varied and include:
NON-NEOPLASTIC LYMPHADENOPATHY
Lymph nodes respond to a wide variety of inflammatory stimuli by cellular proliferation which leads to node enlargement. The cell type that proliferates is dependent upon the antigenic stimulus, which may elicit:
Non-specific reactive hyperplasia
The pattern of cellular proliferation within a lymph node may give some clue to the aetiology of the lymphadenopathy (see below); however, in many instances these clues are absent and the features are termed non-specific reactive hyperplasia. On occasions the node enlargement may reach a considerable size and be difficult to distinguish clinically and macroscopically from neoplastic disorders. Microscopically, numerous enlarged germinal centres are seen; these may be present throughout the node and are not restricted to the outer cortex as in the normal state. The germinal centres are active, with a predominance of large blast cells, a high mitotic rate, and often contain numerous tingible body macrophages. There may be an expanded marginal zone which may form a thick rim around the germinal centre. The paracortex usually shows some degree of hyperplasia characterised by the presence of transformed, large lymphoid cells and vessels lined by large endothelial cells (high endothelial venules). The sinuses often show hyperplasia of the lining macrophages, termed sinus histiocytosis.
Non-specific reactive hyperplasia may occur in lymph nodes draining sites of infection and, in some cases, pathogenic organisms may cause inflammatory changes within the substance of the node, termed lymphadenitis, which may progress to abscess formation.
Specific disorders
Some types of non-neoplastic lymphadenopathy exhibit histological features that allow the pathologist to make an exact diagnosis. These may be grouped into the following categories:
Granulomatous lymphadenitis
Granulomatous lymphadenitis can occur in a variety of clinical settings such as mycobacterial infection (Ch. 14), sarcoidosis (Ch. 14) and Crohn’s disease (Ch. 15). These are described elsewhere and will not be detailed here.
Infection with Toxoplasma gondii, a protozoal organism, in the immunocompetent host produces a flu-like illness of short duration and localised lymphadenopathy, usually occipital or high cervical, which persists for some weeks. The affected lymph node is enlarged and shows germinal centre hyperplasia with formation of ill-defined granulomas adjacent to them. In addition, there is florid marginal zone B-cell hyperplasia characterised by a proliferation of medium-sized, monomorphic B-cells. This histological triad of follicular hyperplasia with adjacent granulomas and marginal zone B-cell hyperplasia suggests a diagnosis of toxoplasmic lymphadenitis which should be confirmed serologically (Fig. 22.3).
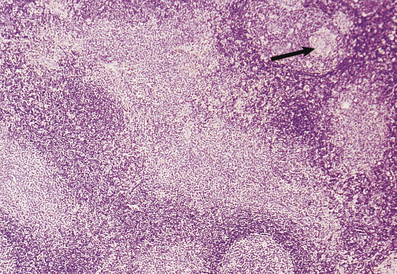
Fig. 22.3 Toxoplasmic lymphadenitis. There is follicular hyperplasia with epithelioid granulomas (arrowed) adjacent to germinal centres. A perifollicular proliferation of uniform B-cells, termed marginal zone B-cells, is also present.
Lymph nodes draining tumours occasionally show a granulomatous reaction in the absence of metastatic involvement, possibly a reaction to tumour antigens. It is particularly common in Hodgkin’s lymphoma. Lymph nodes may develop a granulomatous response to foreign particulate material; this most often occurs as a response to silicone compounds used in plastic surgery and joint replacement.
Necrotising lymphadenitis
A variety of diseases caused by infectious agents may lead to necrosis within lymph nodes. Examples are lymphogranuloma venereum and cat scratch disease. Lymphogranuloma venereum is a sexually transmitted chlamydial disease and most commonly affects the groin nodes. Cat scratch disease follows a bite or scratch from an infected cat. Days to weeks later, tender lymphadenopathy develops in the cervical or axillary regions; the groin is less commonly affected. Two organisms have been shown to be responsible for cat scratch disease: both are extracellular, pleomorphic coccobacilli. The commoner is Bartonella henselae, which causes up to 75% of cases; less common is Afipia felis. In immunosuppressed patients, particularly those with AIDS, infection with B. henselae may cause an unusual vascular proliferation termed bacillary angiomatosis. This may affect lymph nodes or extranodal sites. Lymphogranuloma venereum and cat scratch disease show histological similarities, with formation of stellate abscesses within the lymph node, surrounded by palisaded histiocytes (Fig. 22.4).
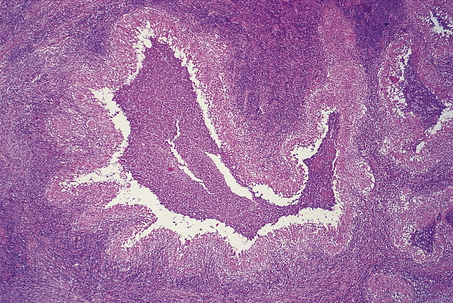
Fig. 22.4 Cat scratch disease. Lymph node showing central abscess formation surrounded by palisaded histiocytes.
A rare form of necrotising lymphadenitis is Kikuchi’s disease, in which tender cervical or occipital lymphadenopathy develops, most commonly in young adult women. The aetiology is unknown. Systemic lupus erythematosus may cause a necrotising lymphadenitis which is histologically very similar to Kikuchi’s disease.
Sinus histiocytosis
Sinus histiocytosis with massive lymphadenopathy (SHML or Rosai–Dorfman syndrome) is a rare condition of unknown aetiology which is more common in black populations than in others. It presents typically with bulky cervical lymphadenopathy in the first and second decades of life and may persist for several years. SHML may, however, affect any age and any organ. Histologically, the lymph node sinuses are grossly distended by an infiltrate of large histiocytic cells whose morphology is quite distinctive; admixed with these cells are lymphocytes and plasma cells, which are often seen in the cytoplasm of the histiocytes. Molecular analysis of SHML has shown it to be a polyclonal disorder. The disease often follows a benign course and may regress spontaneously. Some patients with extensive nodal and extranodal disease have pursued an aggressive course and fatalities due to SHML have occurred.
Langerhans’ cell histiocytosis (histiocytosis X) may affect lymph nodes and characteristically involves the sinuses initially, where clusters of typical, pale Langerhans’ cells with folded nuclei may be seen among giant cells and eosinophils. Langerhans’ cell histiocytosis is a clonal neoplastic disease which may present as a variety of clinical syndromes with uni- or multi-focal disease.
Paracortical hyperplasia
Paracortical hyperplasia is a prominent feature in many cases of lymphadenopathy. Two entities deserve special mention: dermatopathic lymphadenopathy and infectious mononucleosis.
Patients with exfoliative chronic skin conditions such as severe eczema or psoriasis, and patients with cutaneous T-cell lymphoma, quite commonly develop enlarged lymph nodes in the groin and axilla. This condition is dermatopathic lymphadenopathy. The enlarged lymph nodes may have a yellow or brown cut surface and, microscopically, the paracortex is expanded by pale histiocytes with the cytological features of interdigitating reticulum cells and Langerhans’ cells; lipid droplets and melanin pigment may also be apparent.
Infectious mononucleosis is due to Epstein–Barr virus. This causes widespread lymphadenopathy and is characterised, certainly in the later stages, by paracortical hyperplasia with numerous, large, transformed T-cells. The histological picture may be mistaken for high-grade non-Hodgkin’s lymphoma or Hodgkin’s lymphoma by the unwary pathologist.
Human immunodeficiency virus infection
The human immunodeficiency virus (HIV) specifically binds to the cluster of differentiation (CD) 4 antigen, which is expressed by helper/inducer T-cells and by cells of the mononuclear phagocytic system, and to a member of the chemokine receptor family CXCR4. The destruction of cells bearing the CD4 antigen causes a severe immune dysregulation, which ultimately leads to a profound immunodeficiency state called the acquired immune deficiency syndrome (AIDS).
Lymphadenopathy is extremely common in HIV infection and may be observed in association with systemic symptoms in the AIDS-related complex (Ch. 9) and in the persistent generalised lymphadenopathy (PGL) syndrome (defined as persistent, extra-inguinal lymphadenopathy, in two or more non-contiguous sites, of greater than 3 months’ duration and of no known aetiology other than HIV infection).
Morphology
Lymph node biopsies from patients infected with HIV show a spectrum of appearances that, although not absolutely specific, are virtually diagnostic in the appropriate clinical setting. Initially the follicles are hyperplastic and often markedly irregular in shape. Ultrastructurally, a proliferation of dendritic reticulum cells is observed, with complex branching of their processes. In between the DRC processes, retroviral particles can be identified. In some follicles there is focal destruction of the follicular dendritic cell (FDC) meshwork. This is associated with an infiltrate of small CD8+ T-lymphocytes and focal haemorrhage into the germinal centres (‘follicular lysis’). The paracortical reaction is usually disproportionately less than the degree of follicular activation and only scattered immunoblasts and transformed lymphocytes are observed. There is also a reversal of the normal CD4:CD8 ratio of T-cells, often with a preponderance of CD8+ cells. The sinuses may be filled with marginal zone B-cells.
In the later stages of HIV infection, involutional changes are apparent. There is loss of germinal centre B-cells and depletion of paracortical T-cells; sinus histiocytosis may be prominent. These involutional changes are a poor prognostic sign and portend the development of AIDS.
Complications
Lymphadenopathy in HIV infection may not be due solely to immune dysregulation and aberrant lymphocyte proliferation; a variety of neoplastic and infective conditions may also affect the lymph node. Lymphadenopathic Kaposi’s sarcoma and high-grade B-cell non-Hodgkin’s lymphoma (often with Burkitt-like morphology) are common. A wide variety of infectious agents may cause lymph node enlargement, of which atypical mycobacterial infection is frequently encountered (Fig. 22.5). Pneumocystis jiroveci may also be encountered in the lymph nodes of severely immunosuppressed patients.
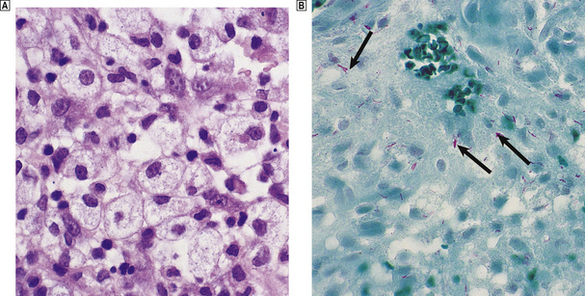
 Aggregates of large histiocytes with foamy cytoplasm.
Aggregates of large histiocytes with foamy cytoplasm.  A similar area stained for acid-fast bacilli (Ziehl–Neelsen stain), demonstrating numerous mycobacteria (stained red; arrowed).
A similar area stained for acid-fast bacilli (Ziehl–Neelsen stain), demonstrating numerous mycobacteria (stained red; arrowed).NEOPLASTIC LYMPHADENOPATHY
Neoplastic lymph node enlargement may occur in:
Hodgkin’s lymphoma
The first detailed account of the pathology of Hodgkin’s lymphoma was given by Thomas Hodgkin in 1832 in which he described the morbid anatomical appearances of lymph nodes. Earlier descriptions of a similar disease process exist but these lack sufficient detail to categorise them as a distinctive clinicopathological entity. The eponymous term Hodgkin’s disease was generously applied by Wilks in 1865 after he had discovered Hodgkin’s original paper. Hodgkin’s observations were based purely on the macroscopic appearances of the disease at postmortem and it was some years before microscopic studies were undertaken. Over the ensuing decade a gradual awareness of the histopathological features of Hodgkin’s lymphoma emerged with descriptions of the typical giant cell by Sternberg and Reed.
Many attempts were made to classify Hodgkin’s disease into clinically meaningful groups, the most successful of which was proposed by Lukes and Butler in 1966. This classification was felt to be too complicated for clinical use and was simplified at the Rye Conference. The essential concept of the Lukes and Butler classification was that nodular sclerosis was a distinctive entity and separate from other types of Hodgkin’s disease and that lymphocyte predominant, mixed cellularity and lymphocyte depleted represented a spectrum of disease of varying clinical aggressiveness which was reflected in the histopathological picture.
More recent studies have demonstrated that lymphocyte predominant nodular Hodgkin’s lymphoma is distinctive and that nodular sclerosis and mixed cellularity are closely related. This view is reflected in the World Health Organization classification, which clearly separates nodular lymphocyte predominant from other forms of Hodgkin’s lymphoma, which are collectively termed ‘classical Hodgkin’s lymphoma’:
The malignant cell of Hodgkin’s lymphoma forms only a small percentage of the cellular population within affected lymph nodes, the bulk of the tissue being made up of reactive lymphocytes, macrophages, plasma cells and eosinophils attracted into the cellular milieu by a variety of cytokines secreted by the Hodgkin’s and Reed–Sternberg (H-RS) cells. The relative paucity of the H-RS cell population has hampered efforts to define its origin. Many cell lineages have been postulated for the H-RS cell, including macrophages, follicular dendritic cells, interdigitating reticulum cells and, most recently, lymphocytes. Elegant microdissection studies have isolated single H-RS cells which have then been subjected to molecular biological analysis; this has shown that, in the vast majority of cases studied, the H-RS cells have a clonal immunoglobulin gene rearrangement, indicating that they are derived from B-lymphocytes. In addition, there is evidence of somatic hypermutation of the immunoglobulin genes which indicates that the cells are of germinal centre origin. The majority of cases of classical Hodgkin’s lymphoma have defects in the critical transcription factors required for immunoglobulin production, either OCT 2 or BOB 1 or both. Approximately 25% of patients with classical Hodgkin’s lymphoma acquire crippling mutations in their immunoglobulin genes. Both the lack of transcription factors and crippling mutations in the immunoglobulin genes prevent the production of a functional immunoglobulin molecule.
Nodular lymphocyte predominant Hodgkin’s lymphoma differs from classical Hodgkin’s lymphoma in having intact immunoglobulin transcription factors and an absence of crippling mutations. This form of Hodgkin’s lymphoma also shows the presence of ongoing immunoglobulin gene hypermutation in an identical form to that seen in the follicle centre cell non-Hodgkin’s lymphoma and again emphasises the difference between lymphocyte predominant and classical Hodgkin’s lymphoma.
The clear demonstration of a lymphoid cell of origin for classical Hodgkin’s lymphoma as well as the nodular lymphocyte predominant type has prompted replacement of the term Hodgkin’s disease with Hodgkin’s lymphoma.
Classical Hodgkin’s lymphoma
Clinical features
Hodgkin’s lymphoma shows a peak incidence in the third and fourth decades and is relatively rare in childhood and old age.
The commonest clinical presentation is one of lymphadenopathy, most often in the upper half of the body, with involvement of cervical and/or axillary lymph nodes. The enlarged nodes are typically rubbery, discrete and mobile, and may achieve a considerable size. Radiological evidence of mediastinal involvement is present in over half of patients and on occasion may be massive, causing respiratory embarrassment. A third of patients with Hodgkin’s lymphoma have systemic symptoms (weight loss greater than 10%, unexplained pyrexia of 39°C or more, and drenching night sweats) and in a small proportion the clinical picture will be dominated by these symptoms. Classical Hodgkin’s lymphoma appears to arise in lymph nodes or the thymus, and spreads, certainly in its early stages, via the lymphatics in a contiguous and predictable fashion. Involvement of the liver and bone marrow is rarely seen in the absence of splenic involvement and thus the spleen appears to be the key to haematogenous dissemination.
Stage is an important determinant in the treatment and prognosis of patients with Hodgkin’s lymphoma; the staging system currently used is that proposed at the Ann Arbor workshop in 1971 and modified at the Cotswolds meeting in 1989.
The absence or presence of the systemic symptoms described above is indicated by the suffix A or B respectively; E indicates involvement of a single extranodal site contiguous with or proximal to a known nodal site; X denotes the presence of bulky disease, > one-third widening of the mediastinum at T5–6 or >10cm maximum dimension of a nodal mass. The survival of patients declines with advancing stage, bulky disease (X) and the presence of systemic (B) symptoms.
Classical Hodgkin’s lymphoma is also associated with a variety of haematological and biochemical abnormalities such as anaemia, lymphocytopenia, a raised erythrocyte sedimentation rate (ESR) and a low serum albumin. These abnormalities are also indicators of a reduced survival.
Morphology
Classical Hodgkin’s lymphoma is principally a disease of lymph nodes. A lymph node biopsy is usually done to establish the diagnosis.
Macroscopically, affected lymph nodes are enlarged, with a smooth surface. Classical Hodgkin’s lymphoma, unlike the non-Hodgkin’s lymphomas, rarely breaches the lymph node capsule, a fact that accounts for the discrete nature of the lymphadenopathy upon palpation. The cut surface is usually homogeneously white (Fig. 22.6), although in some histological subtypes a nodular or fibrotic appearance may be present.
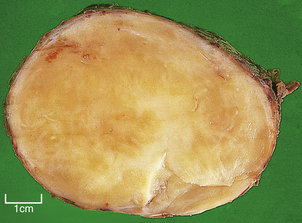
Fig. 22.6 Mixed cellularity Hodgkin’s lymphoma. Macroscopic appearances of an axillary lymph node from a 32-year-old man with mixed cellularity Hodgkin’s lymphoma. There is a homogeneous white cut surface. The capsule is not involved and there is no infiltration of surrounding fat.
Microscopically, affected lymph nodes show a partial or complete effacement of their normal architecture by a mixed infiltrate containing lymphocytes, histiocytes, plasma cells and eosinophils as well as the malignant cells of classical Hodgkin’s lymphoma; these are large cells and take the form of mononuclear Hodgkin’s cells or of Reed–Sternberg cells, which have a large, pale, multilobed nucleus and a prominent eosinophilic nucleolus about the size of a red blood cell (Fig. 22.7).
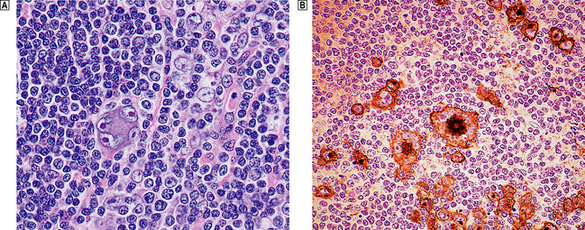
Fig. 22.7 Hodgkin’s lymphoma.  A classical Reed–Sternberg cell shows multiple nuclei with prominent nucleoli.
A classical Reed–Sternberg cell shows multiple nuclei with prominent nucleoli.  Reed–Sternberg cells identified by immunohistochemical staining for CD30 (Ki-1 antigen).
Reed–Sternberg cells identified by immunohistochemical staining for CD30 (Ki-1 antigen).
Nodular sclerosis
The term nodular sclerosis describes many of the histological features of this, the commonest subtype of classical Hodgkin’s lymphoma (75% of cases). The normal lymph node architecture is replaced by cellular nodules which are separated by bands of collagen (Fig. 22.8). Within the cellular nodules is a mixed infiltrate similar to other types of Hodgkin’s lymphoma but containing a distinctive Hodgkin’s cell variant termed the lacunar cell. The lacunar cell is so named because it appears to sit in a space or ‘lacuna’, caused by the disappearance of its lipid-rich cytoplasm during the process of creating a paraffin block of the tissue. It possesses the large nucleus and prominent eosinophilic nucleolus seen in other Hodgkin’s cells.
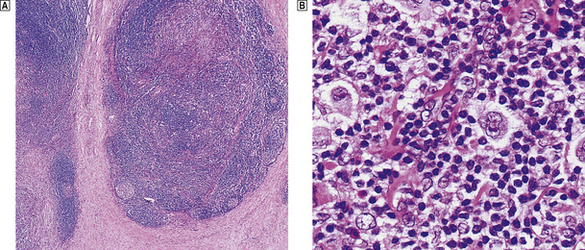
Fig. 22.8 Nodular sclerosing Hodgkin’s lymphoma.  The cellular nodules are surrounded by thick collagen bands.
The cellular nodules are surrounded by thick collagen bands.  Lacunar cells (Hodgkin’s cell surrounded by a clear space) are characteristic of nodular sclerosing Hodgkin’s lymphoma.
Lacunar cells (Hodgkin’s cell surrounded by a clear space) are characteristic of nodular sclerosing Hodgkin’s lymphoma.
Nodular sclerosis is a distinctive form and does not transform into any of the other subtypes of classical Hodgkin’s lymphoma. However, the cytological composition of the cellular nodules may vary from one in which the predominant cell is the small lymphocyte, with only scanty lacunar and Reed–Sternberg cells, to a histological picture that is dominated by Hodgkin’s cells with depletion of lymphocytes. This latter form has been correlated with an aggressive natural history and is termed grade 2 nodular sclerosis; all other histological subtypes are classified as grade 1 nodular sclerosis.
Nodular sclerosis displays distinct clinical differences from other subtypes of Hodgkin’s lymphoma, having an almost equal sex ratio (most forms of Hodgkin’s lymphoma show a marked male predominance), a striking propensity for mediastinal involvement at presentation (50% of patients) and an association with the bizarre syndrome of alcohol intolerance (5% of patients). There is now strong evidence from gene expression profiling studies that nodular sclerosing classical Hodgkin’s lymphoma shares many features in common with a subtype of large B-cell lymphoma presenting in the mediastinum, termed mediastinal large B-cell lymphoma. The similarities are further highlighted by the occurrence of composite tumours containing nodular sclerosing Hodgkin’s lymphoma and mediastinal large B-cell lymphoma within the same lymph node and by the presence of grey zone cases which share morphological and phenotypic similarities of both entities. There are also marked similarities in the patient population affected and the clinical distribution of disease.
Mixed cellularity
Mixed cellularity is the second commonest subtype of classical Hodgkin’s lymphoma (18% of cases). As the name suggests, the histological picture is an admixture of lymphocytes, histiocytes, plasma cells, eosinophils, Hodgkin’s cells and Reed–Sternberg cells; the latter are relatively abundant compared to lymphocyte-rich classical Hodgkin’s lymphoma.
Mixed cellularity is a rather aggressive form of classical Hodgkin’s lymphoma when compared to grade 1 nodular sclerosis: it presents at an advanced stage relatively frequently (stages III and IV in > 50% of patients) and with systemic (B) symptoms (35% of patients) and a poor overall survival.
Lymphocyte-rich classical Hodgkin’s lymphoma
Lymphocyte-rich classical Hodgkin’s lymphoma (LRCHL) is a relatively recently described histological variant of classical Hodgkin’s lymphoma characterised by the presence of morphologically and phenotypically characteristic H-RS cells in a lymphocyte-rich cellular background. There may be a nodular or diffuse growth pattern. When nodular it is composed predominantly of mantle zone B-lymphocytes with scattered and sometimes scanty classical H-RS cells within the nodules. When diffuse there is a T-cell predominance. There is a close morphological overlap between LRCHL and nodular lymphocyte predominant Hodgkin’s lymphoma—the term is designed to alert the pathologist that everything that is lymphocyte rich is not lymphocyte predominant and that the clinical behaviour of LRCHL differs greatly from nodular lymphocyte predominant.
Lymphocyte depleted
Two histological patterns of lymphocyte depleted Hodgkin’s lymphoma may be recognised:
These two patterns of lymphocyte depletion may co-exist in the same lymph node and are closely allied conditions. Extensive necrosis is common in both subtypes.
Lymphocyte depletion is rare (< 1% of all cases of classical Hodgkin’s lymphoma) and carries the worst prognosis. Patients often present acutely with systemic (B) symptoms and usually at an advanced stage, with a high frequency of involvement of liver (60% of patients) and bone marrow (40% of patients). Lymphocyte depleted classical Hodgkin’s lymphoma appears to be becoming extremely rare in the developed world and cases are seldom seen outside the setting of immunodeficiency states.
Nodular lymphocyte predominant Hodgkin’s lymphoma
Clinical features
Nodular lymphocyte predominant Hodgkin’s lymphoma (N-LP) differs markedly in clinical presentation from classical Hodgkin’s lymphoma. There is a marked male predominance, over 80% of patients being men. The peak age incidence is a decade older for N-LP than for patients with classical Hodgkin’s lymphoma; most patients are in their thirties and forties. The majority of patients (nearly 90%) present with localised, asymptomatic disease (stages I and IIA) and the lymphadenopathy occurs in unusual locations such as the suprahyoid neck and inguinal regions, sites rarely involved by classical Hodgkin’s lymphoma. Mediastinal disease is rare. Late relapse is common in N-LP and may occur even after 20 years; the vast majority of relapses with classical Hodgkin’s lymphoma occur within 3 years. Relapses of N-LP occur at the site of original disease or in other typical N-LP locations. Patients with N-LP are at increased risk of developing secondary high-grade non-Hodgkin’s lymphoma (4% at 25 years), which is usually of B-cell lineage and may be clonally related to the original N-LP. The overall survival of patients with N-LP is excellent and superior to that in classical Hodgkin’s lymphoma.
Histologically, lymphocyte predominant Hodgkin’s lymphoma is characterised by a paucity of typical Hodgkin’s and Reed–Sternberg cells and abundant lymphocytes, sometimes admixed with bland histiocytes. There is a nodular growth pattern but occasionally diffuse areas may be present. Collagen band formation is not a feature. Nodular lymphocyte predominant Hodgkin’s lymphoma contains a distinctive Reed–Sternberg cell variant which is called the ‘popcorn’ cell because of its excessively lobulated nucleus. Phenotypically and genotypically the popcorn cell has been shown to be derived from a neoplastic germinal centre B-cell.
Survival in Hodgkin’s lymphoma
The overall survival figures for Hodgkin’s lymphoma vary between different treatment centres, but results from a British multi-centre trial show that approximately 75% of patients were still alive 5 years after diagnosis (Fig. 22.9). As relapse tends to occur early in the course of the disease (usually within 3 years), the majority of these patients are probably cured. It is now clear that treatment-related causes of death are becoming increasingly important for patients with Hodgkin’s lymphoma. Second epithelial malignancies and cardiovascular disease now rival the mortality from Hodgkin’s lymphoma itself.
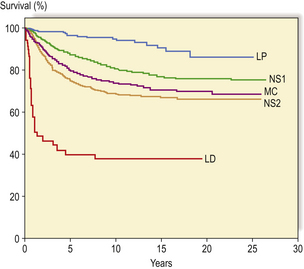
Fig. 22.9 Actuarial survival of patients with Hodgkin’s lymphoma. The data are subdivided according to a modified Rye histological classification with subdivision of nodular sclerosis (NS) into grades 1 and 2. (LP, lymphocyte predominant; MC, mixed cellularity; LD, lymphocyte depleted) (Data from the British National Lymphoma Investigation.)
Non-Hodgkin’s lymphomas




The non-Hodgkin’s lymphomas (NHLs) are malignant tumours of the immune system other than Hodgkin’s lymphoma. The vast majority are derived from lymphoid cells; solid tumours of the mononuclear phagocytic system (termed histiocytic lymphoma) are extremely rare.
NHLs form a wide spectrum of disease both clinically and biologically, ranging from slowly progressive neoplasms to rapidly growing destructive tumours. This diversity of clinical behaviour is reflected in the wide range of histological appearances exhibited by NHLs. Classification of NHL is based on the cell lineages in the normal immune system in which precursor lymphoid cells are processed in the thymus and bursa equivalent tissue and into lymphoid cells which are located in peripheral sites such as lymph nodes, spleen and mucosa-associated lymphoid tissue; these are termed peripheral T- and B-cells respectively (Table 22.2).
Table 22.2 World Health Organization classification of neoplastic diseases of the haematopoietic and lymphoid tissues (summary of main entries)
Precursor lymphoid neoplasms
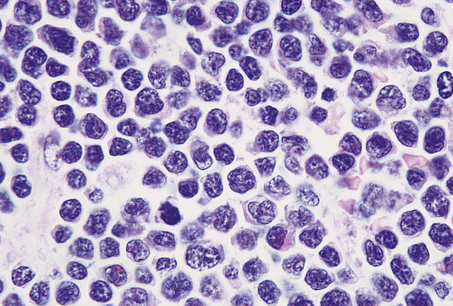
These are high-grade NHLs composed of diffuse sheets of medium-sized lymphoid cells possessing a high nucleo-cytoplasmic ratio. They may be of T-cell or B-cell lineage and express the nuclear antigen TdT (terminal deoxynucleotidyl transferase, an enzyme involved in amplification of antigen receptor diversity). These tumours form the spectrum of disease termed lymphoblastic lymphoma and lymphoblastic leukaemia, and these appear to be biologically the same disease process.
Lymphoblastic lymphomas are commonest in childhood but also occur in adults. Bone marrow infiltration is frequent and a leukaemic phase is often seen; this presents difficulties in separating acute lymphoblastic leukaemia (ALL) from lymphoblastic lymphoma. By convention, an arbitrary subdivision is made on the percentage of lymphoblasts present in the bone marrow (< 25% is lymphoma; >25% is ALL).
Precursor T-cell lymphomas/leukaemias tend to involve the mediastinum of adolescent boys; histologically, the lymphoblasts may have a distinct convoluted nuclear morphology (Fig. 22.10). Precursor B-cell lymphoma/leukaemia may present in leukaemic phase or have solid tumour deposits involving nodal and extranodal sites.
Peripheral non-Hodgkin’s lymphoma
Unlike the precursor or lymphoblastic lymphoma/leukaemia, the peripheral NHLs form a heterogeneous group of neoplasms with a wide range of histological appearances. They represent biologically distinctive tumours derived from B-cells, T-cells and natural killer (NK) cells and may arise in nodal or extranodal sites. As more is understood about the biology of the peripheral NHLs, so the complexity of this group of neoplasms increases; this is reflected in the proposed classification schemes (Table 22.2). The common subtypes of nodal, peripheral NHLs are:
B-cell chronic lymphocytic leukaemia/small lymphocytic lymphoma
B-cell chronic lymphocytic leukaemia/small lymphocytic lymphomas are tumours of immature small round lymphocytes. They are of B-cell lineage. The borderline between lymphocytic lymphoma and chronic lymphocytic leukaemia is blurred; there is close morphological and immunophenotypic homology between the neoplastic cells of the two conditions. Most now regard B-cell small lymphocytic lymphoma as a tissue manifestation of B-cell chronic lymphocytic leukaemia (CLL).
Affected lymph nodes are enlarged and usually smooth surfaced, with a homogeneous white cut surface. The normal nodal architecture is replaced by a monotonous infiltrate of small lymphocytes with scattered larger cells (called prolymphocytes) which may form aggregates termed pseudofollicles.
The disease is almost invariably disseminated, with a high frequency of splenic, liver and bone marrow infiltration.
B-CLL is exclusively a disease of adults. It is now clear that there is clinical and biological heterogeneity within B-CLL. In many it runs an indolent course with patients dying from unrelated causes; these cases are associated with immunoglobulin gene hypermutation. In some patients the disease is much more aggressive and these are commonly associated with unmutated immunoglobulin genes.
As the disease progresses it may cause death by extensive bone marrow infiltration and bone marrow failure. In 5–10% of patients the disease will transform into a high-grade pleomorphic large cell lymphoma (Richter’s syndrome) which is refractory to treatment and has a poor prognosis. Patients with B-CLL are frequently hypogammaglobulinaemic and are particularly susceptible to bacterial infections.
Follicular lymphoma
Follicular lymphomas are tumours derived from (or differentiated towards) germinal centre B-cells. These lymphomas contain an admixture of centroblasts and centrocytes and may have a purely follicular growth pattern or may have a mixed pattern with follicular and diffuse areas. In addition to the neoplastic B-cells, non-neoplastic T-lymphocytes and dendritic reticulum cells are also present within the neoplastic follicle, having been recruited by the neoplastic B-cell clone.
Important in the pathogenesis of follicular lymphoma is a specific chromosomal translocation involving the immunoglobulin heavy chain promoter region on chromosome 14 and the anti-apoptotic gene bcl-2 on chromosome 18 (t(14;18)(q32;q21)). This translocation causes the constitutive overexpression of bcl-2 protein, rendering follicular lymphoma cells relatively resistant to apoptosis.
Lymph nodes replaced by follicular lymphoma are usually enlarged and smooth surfaced. Cut surfaces are homogeneous grey/white with occasional areas of haemorrhage and necrosis. A faintly nodular pattern may be seen with a magnifying glass. Microscopically, the normal lymph node architecture is replaced by closely packed neoplastic follicles, often extending through the lymph node capsule into perinodal fat (Fig. 22.11); diffuse areas may also be present. The neoplastic follicles contain a mixture of centroblasts and centrocytes; the relative proportion of these cells forms the basis of grading these lymphomas. Tumours with relatively large numbers of centroblasts have a worse prognosis.
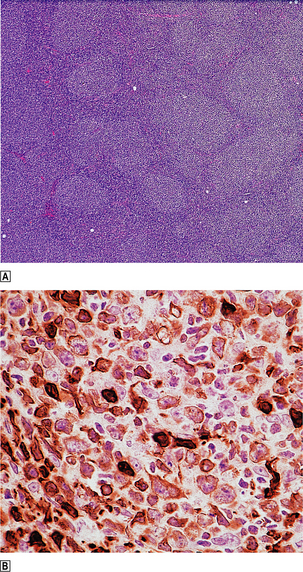
Fig. 22.11 Follicular lymphoma.  Numerous rounded lymphoid aggregates are seen throughout the lymph node.
Numerous rounded lymphoid aggregates are seen throughout the lymph node.  Immunohistochemical evidence of overexpression of bcl-2, an inhibitor of apoptosis.
Immunohistochemical evidence of overexpression of bcl-2, an inhibitor of apoptosis.
Follicular lymphoma is one of the commonest types of non-Hodgkin’s lymphoma and is a disease of late adult life with a peak age incidence in the sixth and seventh decades. It is exceptionally rare in children and young adults. Most patients with follicular lymphoma will present with painless, slowly progressive lymphadenopathy, usually with disseminated disease (stages III and IV); 40% of patients will have systemic symptoms.
Involvement of bone marrow, spleen and liver is common in follicular lymphoma. The earliest morphological manifestations of marrow infiltration are localised collections of irregular small lymphoid cells adjacent to bone trabeculae (termed paratrabecular infiltration). Extensive bone marrow replacement may occur with consequent marrow failure. A leukaemic phase of follicular lymphoma is seen in a significant percentage of patients, when neoplastic B-cells may be found in peripheral blood.
Splenic involvement may either be minimal or cause massive splenomegaly with hypersplenism. The neoplastic cells of follicular lymphoma maintain some degree of physiological homing function and selectively infiltrate the B-cell areas of the white pulp of the spleen, progressively replacing and expanding these structures such that the cut surface of the organ may be seen to contain numerous discrete white nodules separated by normal red pulp.
Follicular lymphoma is an indolent disease but it is generally regarded as incurable when disseminated, and the majority of patients die as a direct result of their lymphoma. The exact mode of death is sometimes difficult to determine but in at least 30% of patients the disease will transform into a diffuse large B-cell lymphoma. This lymphoma behaves aggressively and is usually refractory to treatment. In the remainder, following a clinical course of multiple remissions and relapses, the tumour becomes unresponsive to further chemotherapy, with a progressively increasing tumour burden leading to death.
Although it is difficult to determine prognosis for an individual patient with follicular lymphoma, factors that are associated with a reduced survival are rapid disease progression, the presence of systemic (B) symptoms and infiltration of vital organs. There is now good evidence that the non-neoplastic constituents of the follicles of follicular lymphoma are important determinants of prognosis. Follicles that contain a high percentage of reactive T-cells are associated with a favourable prognosis and those containing large numbers of macrophages are associated with a reduced survival. This important role of ‘bystander’ elements in determining the biological behaviour of tumours may be recognised in other lymphoma subtypes in the future.
Mantle cell lymphoma
Mantle cell lymphoma is the term currently used for a low-grade B-cell lymphoma that is derived from or differentiates towards the mantle zone of the germinal centre. The term encompasses entities such as centrocytic lymphoma and intermediately differentiated lymphocytic lymphoma in previous classification schemes.
Mantle cell lymphoma may have a nodular or diffuse architecture and is composed of a mixture of irregular small B-cells, follicular dendritic cells and T-cells; the B-cell component is monoclonal.
Mantle cell lymphoma has a distinctive chromosomal translocation—t(11;14)(q13;q32). This leads to the deregulation of the PRAD-1 gene with subsequent overexpression of the important cell cycle control protein cyclin D1.
Mantle cell lymphoma is not uncommon (6% of all NHLs) and has a peak age incidence in the seventh decade. It is generally regarded as a low-grade lymphoma but its natural history appears to be more aggressive than other low-grade NHLs and it has the ability to transform into a ‘blastoid’ form which has a very poor survival. The disease usually presents with lymphadenopathy but a distinctive pattern is seen in the gastrointestinal tract, where it is termed lymphomatous polyposis. Most patients have advanced stage and symptomatic disease. Leukaemic overspill is quite common.
Diffuse large B-cell lymphoma
Diffuse large B-cell lymphoma forms the commonest type of high-grade NHL; it may arise de novo or as a result of the transformation of a low-grade B-cell lymphoma (usually follicular lymphoma). As the name implies, this lymphoma is characterised by a diffuse outgrowth of large B-cells which may display centroblastic or immunoblastic cytology (Fig. 22.12).
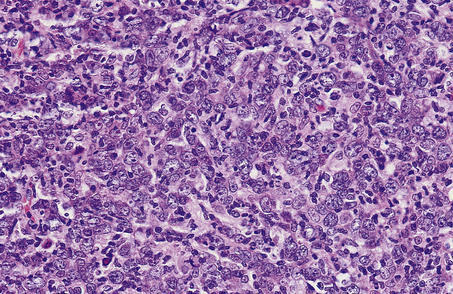
Fig. 22.12 Diffuse large cell lymphoma. The majority of the cells, growing in a diffuse pattern, are large B-cells (centroblasts) with multiple nucleoli often peripherally located.
Diffuse large B-cell lymphoma is usually a disease of adults but may occur in childhood. The majority of patients present with rapidly progressive nodal disease but extranodal involvement is common; involvement of the gastrointestinal tract and Waldeyer’s ring is most frequent.
Unlike the low-grade lymphomas, diffuse large B-cell lymphoma is frequently localised (stages I and II in 45% of patients) and curable even when advanced (5-year survival for patients with stage III and IV disease is approximately 35%). A variety of cytogenetic abnormalities are seen in diffuse large B-cell lymphoma; the commonest are the t(14;18) translocation resulting in deregulation of the bcl-2 gene and translocations involving the 3q27 breakpoint with overexpression and mutations of the bcl-6 gene. Gene expression profiling in diffuse large B-cell lymphoma has led to the identification of two potentially distinct biological forms of this disease, one termed germinal centre-like large B-cell lymphoma and one activated B-cell-like large B-cell lymphoma. The latter is associated with a relatively poor survival. Abnormalities of the bcl-6 gene have been associated with extranodal presentations and a more favourable clinical course.
Burkitt’s lymphoma
Burkitt’s lymphoma is a distinctive type of B-cell lymphoma, associated with a specific chromosomal translocation involving the c-myc gene at 8p24. The translocation involving c-myc usually occurs into the immunoglobulin heavy chain gene locus at 14q32 (t(8;14)(p24;q32)) but variant translocations involving the kappa and lambda light chain gene loci also occur. Three categories of Burkitt’s lymphoma are recognised: endemic, sporadic and immunodeficiency-associated. Endemic Burkitt’s lymphoma occurs in para-equatorial Africa and New Guinea and occurs much less commonly in other regions. The disease affects children and adolescents, is associated with Epstein–Barr virus infection and malaria (Ch. 11), and involves extranodal sites, particularly the jaw, gastrointestinal tract and gonads. The histological appearances are distinctive, with tightly packed medium-sized lymphoid cells interspersed with phagocytic macrophages which impart a ‘starry sky’ appearance to histological sections. There is a very high proliferation rate, with almost 100% of cells being in cycle. The histological and phenotypic features of sporadic and immunodeficiency-associated Burkitt’s lymphoma are identical. They have a propensity to involve the gastrointestinal tract and oropharynx.
Peripheral T-cell lymphoma
T-cell lymphomas are relatively uncommon in Europe and America, making up no more than 10% of NHL cases. In Japanand the Caribbean region they are much more common. This increased disease prevalence is due to the presence of an endemic retrovirus, the human T-cell leukaemia/lymphoma virus (HTLV-I), which appears to be a causative agent in some forms of T-cell malignancy.
The spectrum of T-cell NHL appears as broad as that of B-cell tumours and complicated classification schemes have been proposed. More recently it has been appreciated that the histological recognition of different subtypes of nodal peripheral T-cell lymphoma is difficult to perform reliably and that morphology is not a good indicator of clinical behaviour. For this reason the majority of these tumours have been categorised as peripheral T-cell lymphoma unspecified. Within this group of tumours there is wide variation in the size and shape of the neoplastic T-cells and they are frequently mixed with other non-neoplastic components of the paracortex, such as interdigitating reticulum cells and high endothelial venules; aggregates of epithelioid histiocytes and an eosinophil infiltrate may also be prominent.
Within the heterogeneous group of T-cell lymphomas there are several distinctive clinicopathological entities:
Mycosis fungoides (MF) and Sézary syndrome are closely allied cutaneous T-cell lymphomas. The neoplastic T-cells usually have a ‘helper’ cell phenotype (CD4) and form a band-like upper dermal infiltrate with a moderate degree of epidermal infiltration, often forming small aggregates of cells within the epidermis (termed Pautrier’s micro-abscesses), usually in association with epidermal Langerhans’ cells. The neoplastic T-cells are larger than normal lymphocytes and usually have a markedly irregular nuclear profile imparting a cerebriform appearance.
Cutaneous T-cell lymphoma is a disease of adult life with a tendency to involve older age groups.
Mycosis fungoides clinically progresses through three stages:
The density of the lymphoid infiltrate increases from the patch to the tumour stage and in some cases of tumour stage MF there may be transformation to a large-celled cytological pattern. Although MF is initially confined to the skin, lymph node and visceral organ involvement become clinically apparent later in the course of the disease and are particularly common in the tumour stage. This is a bad prognostic feature with a median survival of only 2.5 years compared to a 12-year median survival for patients with limited extent cutaneous disease.
The Sézary syndrome variant of CTCL is characterised by the presence of generalised erythroderma, lymphadenopathy and at least 10% of peripheral blood mononuclear cells having an atypical cerebriform morphology. The prognosis of patients with the Sézary syndrome is poor.
Adult T-cell leukaemia/lymphoma (ATLL) is one of the few human malignancies for which there is strong evidence of a viral aetiology. The disease is endemic in the islands of southern Japan, the Caribbean basin, the tropical islands of the Pacific Ocean and in the Seychelles. Outside endemic areas the majority of affected patients have been black. The disease is associated with the retrovirus HTLV-I whose p40 tax protein leads to transcriptional activation of a range of genes in infected T-cells that confer a survival advantage. The disease presents with lymphadenopathy and a leukaemic blood picture, often associated with cutaneous infiltration and hypercalcaemia (secondary to the production of parathyroid hormone-related peptide). Low-grade and chronic forms of ATLL have a more indolent course. The affected lymph nodes are infiltrated by a pleomorphic T-cell proliferation, indistinguishable from other non-virus-associated T-cell lymphomas. The disease has a poor prognosis, with a median survival of less than 1year; most patients die from opportunistic infection due to severe immunosuppression.
Angioimmunoblastic T-cell lymphoma (AILD) is a rare but distinctive form of T-cell lymphoma. Although originally it was not thought to be neoplastic, the demonstration of T-cell receptor gene rearrangement in the majority of cases studied has indicated that this lesion should be regarded as a lymphoma. Patients usually present with widespread lymphadenopathy, systemic symptoms, skin rashes, polyclonal hypergammaglobulinaemia and evidence of immunosuppression. The prognosis for AILD is poor, although occasional spontaneous or steroid-induced remissions are described.
Anaplastic large cell lymphoma (ALCL) is a recently described high-grade NHL that is often of T-cell lineage but may fail to express lineage-associated markers; the latter are usually genotypically T-cell in origin. ALCL may arise de novo or may be a high-grade transformation of a low-grade NHL. Primary ALCL is most common in children and young adults, although any age may be affected. The disease may be nodal but there is frequently extranodal disease as well. Common sites of extranodal disease include the skin, gastrointestinal tract and Waldeyer’s ring. Histologically, ALCL shows a wide spectrum of appearances, and many different subtypes have been described, including common, small cell, lymphohistiocytic, giant cell and sarcomatoid. Despite this marked divergence in appearance, ALCL appears to be a unified biological entity and all cases contain a characteristic cell termed by some the ‘hallmark’ cell (Fig. 22.13). ALCL is associated with a particular chromosomal translocation, t(2;5)(p23;q35), which leads to the aberrant expression of a fusion protein containing a tyrosine kinase called anaplastic lymphoma kinase (ALK) and a ‘housekeeping’ protein, nucleophosphomin; this translocation occurs in 70–80% of ALK protein-expressing ALCLs. Variant translocations also occur in 20–30% of ALCLs, which fuse the ALK gene with other partners. Despite its very aggressive histological appearance, primary ALCL has a relatively good overall survival (approximately 80% of patients alive at 5 years). This favourable survival appears independent of traditional prognostic factors.
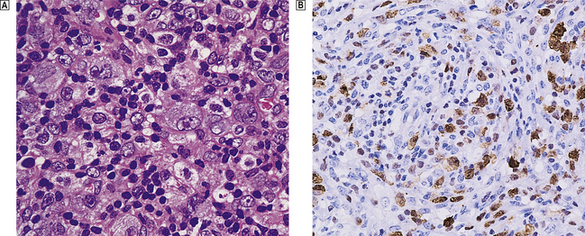
 Pleomorphic large cells infiltrate this lymph node.
Pleomorphic large cells infiltrate this lymph node.  Expression of anaplastic lymphoma kinase, a fusion protein associated with the characteristic chromosomal translocation.
Expression of anaplastic lymphoma kinase, a fusion protein associated with the characteristic chromosomal translocation.Extranodal lymphoma
Extranodal lymphoid tissue is widely distributed in the body, often located adjacent to mucosal surfaces (e.g. Peyer’s patches in the terminal ileum). The structure of this mucosa-associated lymphoid tissue (MALT) differs from that in peripheral lymph nodes and there are differences in function and patterns of lymphocyte recirculation. Many extranodal lymphomas show features in common with MALT and it is now clear that these are distinctive forms of NHL. They are currently termed extranodal marginal zone lymphoma.
Extranodal lymphomas make up at least a quarter of all NHLs and occur most commonly in the gastrointestinal tract (Ch. 15); other common sites include skin, salivary gland, thyroid and orbit. Most extranodal lymphomas occur in sites normally devoid of lymphoid tissue; the acquisition of lymphoid tissue as the result of either an autoimmune (such as Hashimoto’s thyroiditis) or infective (as seen in Helicobacter gastritis) process is the essential forerunner to the development of lymphoma in these locations.
Extranodal lymphomas of marginal zone type share common histological and cytogenetic features wherever they occur. Histologically, they are characterised by the presence of reactive germinal centres surrounded by a population of neoplastic B-cells which show cytological similarities to lymph node marginal zone B-cells. These marginal zone B-cells infiltrate epithelial structures to form lympho-epithelial lesions which are characteristic of extranodal marginal zone lymphomas (Fig. 22.14). These lymphomas often show impressive degrees of plasma cell differentiation, usually adjacent to the mucosal surface. Many extranodal lymphomas are low grade but some show features of high-grade lymphoma, usually of diffuse large B-cell type. These high-grade lymphomas may arise de novo or be associated with a low-grade extranodal marginal zone component.
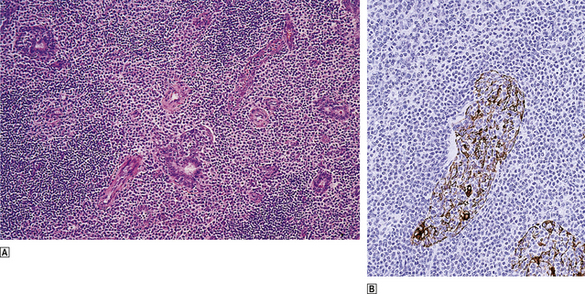
Fig. 22.14 Low-grade MALT lymphoma of the salivary gland.  Centrocyte-like cells infiltrate the glands and form lympho-epithelial lesions characteristic of low-grade MALT lymphoma.
Centrocyte-like cells infiltrate the glands and form lympho-epithelial lesions characteristic of low-grade MALT lymphoma.  Immunohistochemical demonstration of cytokeratin in the surviving glandular epithelium infiltrated by lymphoma cells.
Immunohistochemical demonstration of cytokeratin in the surviving glandular epithelium infiltrated by lymphoma cells.
MALT lymphomas lack the common cytogenetic abnormalities seen in nodal lymphomas (t(14;18) and t(11;14)) and are associated with novel karyotypic changes such as t(11;18) and t(1;14); both of these translocations are associated with overactivity of the NF kappa B transcription factor which appears important in the pathogenesis of this lymphoma subtype.
There are marked differences in the clinical behaviour of extranodal marginal zone lymphomas compared with that of nodal lymphomas. They appear to remain localised for long periods and have an indolent natural history, often with a very good prognosis. The pattern of relapse is also different, with a tendency to recur in extranodal locations.
The remarkable similarities in histology, cytogenetics and clinical course of extranodal lymphomas from different sites strongly suggest that they represent a common biological entity. The recognition and characterisation of the extranodal marginal zone lymphoma group is an important advance in lymphoma pathology.
Not all extranodal lymphomas correspond to the extranodal marginal zone pattern; some are identical to nodal lymphomas and some are distinctive clinicopathological entities that present in extranodal locations.
Lymphomatous polyposis is a manifestation of mantle cell lymphoma in the gastrointestinal tract. It is characterised by the presence of multiple mucosal polyps throughout the alimentary system; these may range in size from half a millimetre to many centimetres in diameter and tend to centre around the ileocaecal valve (Fig. 22.15). This is an aggressive lymphoma which frequently becomes leukaemic at some point in its natural history.
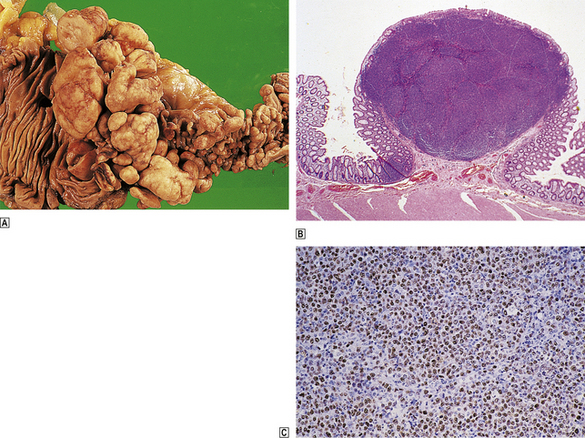
Fig. 22.15 Multiple lymphomatous polyposis of gut (mantle cell lymphoma).  The terminal ileum (right) and ascending colon are affected. These are characteristic macroscopic appearances with multiple mucosal polyps increasing in size towards the ileocaecal valve.
The terminal ileum (right) and ascending colon are affected. These are characteristic macroscopic appearances with multiple mucosal polyps increasing in size towards the ileocaecal valve.  Low-power histology showing nodular infiltration of the lamina propria by mantle cell lymphoma.
Low-power histology showing nodular infiltration of the lamina propria by mantle cell lymphoma.  Immunohistochemical detection of cyclin D1 overexpression, a characteristic feature of mantle cell lymphoma.
Immunohistochemical detection of cyclin D1 overexpression, a characteristic feature of mantle cell lymphoma.
Enteropathy-type T-cell lymphoma is a pleomorphic T-cell lymphoma that develops in some patients with coeliac disease. Enteropathy-type T-cell lymphoma has a predilection for jejunal involvement, where it often presents as multifocal lesions—ulcers, fissures and perforation. The tumour is probably derived from intramucosal T-cells. This is an aggressive lymphoma with a poor survival.
Classification of the non-Hodgkin’s lymphomas
The classification of malignant lymphoma has undergone dramatic changes in recent years, which have come to fruition with the publication of the new WHO classification of tumours of haematopoietic and lymphoid tissues. This classification is based on the previously published Revised European–American Lymphoma classification and classifies lymphomas according to lineage. Within each category, distinct diseases are defined according to a combination of morphology, phenotype, and genetic and clinical features with the aim of identifying lymphoma entities that can be recognised by pathologists and have clinical relevance (see Table 22.2).
THYMUS
NORMAL STRUCTURE AND FUNCTION
The thymus develops from the third and occasionally the fourth pharyngeal pouches. It is a pyramidal, bilobed, encapsulated organ situated in the anterior superior mediastinum. The relative weight of the thymus in comparison to body weight is greatest in the neonate (20–30 g). The absolute thymic weight peaks around puberty (40–50 g) and thereafter declines such that in the elderly adult the thymus is atrophic and composed largely of adipose tissue. The significance and the mechanisms of thymic atrophy are largely unknown.
The lobes of the thymus are divided into lobules by connective tissue septa which grow in from the fibrous capsule. Subdivision of the thymus into an outer, dark cortex and inner, pale medulla is apparent macroscopically.
The thymus is a central lymphoid organ and is responsible for the induction of cell-mediated immune function in developing lymphoid cells (Ch. 9). Most of this inductive activity appears to be located in the cortex, which contains densely packed medium-sized lymphoid cells, scattered epithelial cells and abundant interdigitating reticulum cells. The thymic epithelial cells are responsible for the secretion of thymic hormones, such as thymosin and thymopoietin, and critical cytokines which are necessary for T-cell lineage commitment and maturation. During the acquisition of immunocompetence the cortical lymphoid population is rapidly dividing, but many cells die in situ and relatively few migrate to the medulla and then to peripheral lymphoid organs. The thymic medulla is far less cellular than the cortex. In addition, the thymic medulla contains structures termed Hassall’s corpuscles, which are concentrically arranged squamous epithelial cells with central keratinisation; their function is unknown. A small and morphologically inconspicuous population of B-cells is present within the medulla and may be important in the pathogenesis of myasthenia gravis and mediastinal B-cell lymphoma.
DISORDERS OF THE THYMUS
Agenesis and hypoplasia
Agenesis and hypoplasia may occur because of a failure of either the epithelial or the lymphoid component of the thymus to develop properly.
In the DiGeorge and Nezelof syndromes there is defective development of the epithelial component of the thymus from the third pharyngeal pouch. The DiGeorge syndrome has, in addition, defective development of the fourth pharyngeal pouch, which results in an absence of the parathyroid glands. It is associated with deletion of the 22q11.2 chromosomal region. The thymus is either completely absent in these two syndromes or is represented by a fibrous streak.
Abnormalities of lymphoid colonisation of the thymus occur in the severe combined immune deficiency syndromes, ataxia–telangiectasia and reticular dysgenesis. In reticular dysgenesis the thymus is small, weighing little more than a few grams, and is composed of disordered aggregates of epithelial cells.
Acquired hypoplasia may be seen as a natural ageing phenomenon, as a response to stress and in the acquired immune deficiency syndrome following infection with HIV.
Hyperplasia
Thymic hyperplasia is strongly associated with autoimmune disease (in particular, myasthenia gravis). Hyperplasia is difficult to diagnose from the thymus size or weight alone, owing to extreme variation in these indices in the general population. The most reliable criterion is the formation of germinal centres within the thymus. The germinal centres are located principally in the medulla, from which they expand and may cause cortical atrophy. They are identical to the germinal centres in peripheral lymphoid sites.
Neoplasms
A wide variety of neoplasms occur within the thymus:
Thymic epithelial tumours
A variety of epithelial tumours arise from thymic epithelial cells. Those that appear histologically benign are termed thymomas. A percentage of thymomas, despite appearing histologically bland, may demonstrate an invasive growth pattern and are termed invasive thymomas.
Thymic carcinomas show the cytological features of malignancy in addition to usually having an invasive growth pattern.
Thymomas
Only those neoplasms in which the neoplastic cell is derived from thymic epithelium and appears histologically benign are termed thymomas. The vast majority of thymomas arise within the thymus, but thymomas from ectopic thymus tissue have been described in the soft tissues of the neck, hilum of the lung, other sites within the mediastinum and, rarely, the thyroid.
In addition to the neoplastic epithelial component, thymomas contain variable numbers of lymphoid cells; those that contain large numbers are termed ‘lymphocyte-rich’ and those with scanty numbers are termed ‘lymphocyte-poor’. The lymphocyte-rich thymomas to some extent recapitulate the structure of the cortical thymus in their microscopic appearances and the phenotype of the lymphoid cells present. Conversely, the lymphocyte-poor thymomas show similarities with the thymic medulla. In addition to variations in the degree of lymphocytic infiltration, there are also variations in the cytological appearances of the neoplastic epithelial cells, which may range from round to spindle-shaped.
Many thymomas are asymptomatic and are detected by chest X-ray performed for other reasons; some present with signs of local disease such as dyspnoea, cough and stridor, and the remainder present with autoimmune disease. The majority of thymomas (60–80%) are benign and complete surgical excision is curative. The remaining cases are termed invasive thymomas. There is a good correlation between survival and the extent of spread or presence of metastases in malignant thymomas.
Thymomas may be associated with a variety of disorders, including myasthenia gravis, pure red cell aplasia, neutropenia, thrombocytopenia, hypogammaglobulinaemia and systemic lupus erythematosus.
Other thymic neoplasms
Thymic involvement by Hodgkin’s lymphoma is relatively common and occurs particularly in the nodular sclerosing subtype.
Non-Hodgkin’s lymphomas originating in the thymus tend to be high-grade tumours of either T-lymphoblastic or large B-cell types. The T-lymphoblastic lymphomas of the thymus occur in young and adolescent boys, often possess a characteristic convoluted nuclear morphology, and usually develop a leukaemic phase. Their phenotype shows similarities to the cortical thymic lymphoid cell. The large B-cell lymphomas of the thymus tend to occur in women in the second and third decades of life. It has been postulated that they arise from thymic medullary B-cells. The biological similarities between mediastinal large B-cell lymphoma and nodular sclerosing classical Hodgkin’s lymphoma have been discussed previously. The prognosis of mediastinal large B-cell lymphoma, when accurately classified, appears to be more favourable than other variants of diffuse large B-cell lymphoma. Low-grade NHLs with histological features of extranodal marginal zone lymphoma of MALT type also occurin the thymus.
The same spectrum of germ cell neoplasia is apparent in the thymus as in the gonad and accounts for 10–15% of all primary mediastinal tumours. These neoplasms are thought to arise from primitive germ cells that have become misplaced during migration to the developing gonadal anlage. Mediastinal germ cell tumours are also rarely associated with haemopoietic neoplasms such as acute myeloid leukaemia, myelodysplasia, systemic mastocytosis and malignant histiocytosis. The mature teratomas are benign and complete surgical excision is curative. Teratomas with an immature component are usually of low stage and have a good prognosis. Seminomas are radio- and chemosensitive and have a relatively good prognosis. Non-seminomatous malignant germ cell tumours of the thymus have a significantly worse prognosis than their testicular counterparts.
A variety of benign and malignant mesenchymal tumours may originate in the thymus and these are identical to their counterparts outside the thymus; the commonest are lipomas. Thymolipomas are unique to the thymus and are circumscribed stromal tumours composed of an admixture of mature fat and thymic tissue. They are benign but may reach a substantial size, often weighing over half a kilogram. They may be associated with paraneoplastic syndromes such as myasthenia gravis, pure red cell aplasia and hypogammaglobulinaemia.
Thymic carcinoids and small cell carcinomas are rare neoplasms thought to arise from neuroendocrine cells scattered in the organ. About a third of thymic carcinoids are associated with Cushing’s syndrome. Both thymic carcinoids and small cell carcinomas are aggressive malignant neoplasms.
SPLEEN
NORMAL STRUCTURE AND FUNCTION
The spleen is an encapsulated organ normally weighing 100–150g in the adult; it is situated in the left upper quadrant of the abdomen, mostly concealed by the lower ribs. It first appears in the fifth gestational week and is mesodermally derived. The spleen has two functions: it is a lymphoid organ, and it has a great capacity for phagocytosing particulate material in the circulation and culling senescent red cells. These two functions of the spleen are architecturally distinct: the lymphoid function occurs in the white pulp and the phagocytic activity resides in the red pulp.
White pulp
The splenic artery is derived from the coeliac axis and enters the spleen at the hilum, then branches and follows the trabeculae of the fibrous capsule into the substance of the organ. Leaving the trabeculae as central arteries and arterioles, these branches become ensheathed in lymphoid cells. These aggregates of lymphoid cells are termed the white pulp and can be seen on the cut surface of the spleen as 1–2 mm diameter white nodules in the deep-red background of the red pulp. As in lymph nodes, the lymphocytes within the white pulp of the spleen show distinct micro-architectural segregation of different functional subsets. T-cells are found in the immediate vicinity of the central arterial vessel of the white pulp and are termed the peri-arteriolar lymphoid sheath. The B-cell follicle is eccentrically placed within the white pulp and may be composed predominantly of small lymphocytes, or may form a germinal centre when stimulated. At the junction of the red and white pulp, and surrounding the peri-arteriolar lymphoid sheath and the B-cell follicle, is a group of specialised B-cells termed marginal zone lymphocytes; their precise function is unknown. The white pulp is a major site of antibody production.
Red pulp
Most of the spleen is occupied by the red pulp, whose main function appears to be destruction of senescent red cells and phagocytosis of particulate material. The red pulp has a dual circulation, with a ‘closed’ sinusoidal pathway and an ‘open’ system through the splenic pulp cords.
The splenic arterial supply, having traversed the white pulp, flows through the penicillary arteries into the splenic sinuses or pulp cords. The sinuses are narrow channels with a discontinuous endothelial lining which allows adjacent macrophages access to the red cells as they traverse sinuses and drain into the trabecular veins. The open pulp-cord circulation places the red cells in prolonged intimate contact with serried ranks of macrophages before entering the splenic sinuses. Within the pulp cords, macrophages remove any intracytoplasmic inclusions (such as Howell–Jolly bodies—a process termed pitting) and excess surface membrane from the red blood cells. The environment within the pulp cords is hostile and the red cells must possess marked deformability and intact metabolic machinery to survive. Those cells that do not survive are phagocytosed and broken down.
DISORDERS OF THE SPLEEN
Congenital abnormalities
Congenital abnormalities in the form of accessory spleens or splenunculi are relatively common, occurring in about 10% of the population. They are rounded, encapsulated structures up to several centimetres in size and usually located near the spleen. Congenital asplenia and polysplenia are rare and often associated with other congenital malformations, particularly of the cardiovascular system.
Hypersplenism
The term ‘hypersplenism’ is applied to the association between a peripheral blood pancytopenia and splenic enlargement. It may be primary or secondary to a wide variety of pathological processes.
Primary hypersplenism is a poorly understood condition of unknown aetiology, characterised by marked and often massive splenomegaly and pancytopenia, in which leukopenia is particularly pronounced. Within the spleen there is marked lymphoid hyperplasia. The haematological response to splenectomy is excellent, although some patients may remain leukopenic.
Secondary splenomegaly (see below) may also be associated with hypersplenism.
Splenomegaly



The causes of secondary splenomegaly are numerous and include the following basic pathological processes:
Congestive splenomegaly
Conditions that lead to a persistent elevation of splenic venous blood pressure are capable of causing splenomegaly. The splenic venous pressure may be raised due to pre-hepatic, hepatic and post-hepatic causes.
Pre-hepatic causes include thrombosis of the extrahepatic portion of the portal vein or of the splenic vein.
Very marked splenomegaly occurs in longstanding portal hypertension associated with cirrhosis (Ch. 16).
Post-hepatic causes of congestive splenomegaly are associated with a raised pressure in the inferior vena cava, which is transmitted to the spleen via the portal system. These are usually associated with ascites and hepatomegaly. Decompensated right-sided heart failure and pulmonary or tricuspid valve disease are the usual post-hepatic causes of congestive splenomegaly.
The spleen in congestive splenomegaly is variably enlarged and may reach a massive size, weighing a kilogram or more. The capsule may be thickened and fibrotic, but it is in the red pulp that the major pathological alterations occur. The cut surface of the spleen has a beefy-red colour with an inconspicuous white pulp, often containing scattered, firm brown nodules; these are Gamna–Gandy nodules and represent areas of healed infarction, composed of fibrous and elastic tissue with abundant haemosiderin and dystrophic calcification. In the early stages of congestive splenomegaly, the sinusoids are distended with red cells. Later, fibrosis occurs around the sinusoids, which appear ectatic and empty. Foci of extramedullary haemopoiesis may be seen and it is postulated that these are secondary to local hypoxia within the spleen.
Infection
Systemic infection may cause moderate splenomegaly characterised by congestion and macrophage hyperplasia within the red pulp. The white pulp is usually prominent macroscopically and shows reactive changes microscopically, often with germinal centre formation.
Some viral diseases, in particular infectious mononucleosis, may produce more severe splenomegaly. In addition to the changes described above, the splenomegaly may also show immunoblastic infiltration of the red pulp. In infectious mononucleosis, the spleen is susceptible to rupture (Fig. 22.16).
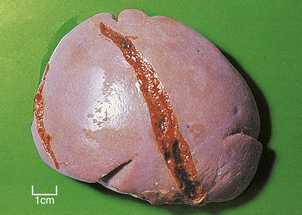
Fig. 22.16 Splenic rupture in infectious mononucleosis. This spleen was removed from an adolescent with infectious mononucleosis who sustained minor abdominal trauma.
Chronic malarial infection may lead to massive splenomegaly. The splenic capsule is thickened and fibrotic and the cut surface has a slate-grey coloration due to the abundant, iron-containing malarial pigment. Microscopically there is pronounced red pulp macrophage hyperplasia containing malarial parasites.
Immune disorders
A variety of immune disorders may lead to splenomegaly, in particular rheumatoid disease and systemic lupus erythematosus. Hypersplenism may ensue, and, in the case of rheumatoid disease in adults, is called Felty’s syndrome.
Neoplasms
Splenic infiltration is a common feature of a wide variety of haematological neoplasms including:
The pattern of splenic involvement is characteristic of each group of neoplasms.
Acute leukaemias preferentially infiltrate the red pulp, although minor degrees of white pulp involvement may be seen, particularly in acute lymphoblastic leukaemia. The splenic red pulp cords and sinuses are filled with numerous primitive haemopoietic blast cells.
Chronic myeloid leukaemia (CML) and the myeloproliferative syndromes may lead to massive degrees of splenomegaly. In CML, the red pulp is filled with myeloid precursors which are predominantly mature and, in the myeloproliferative disorders, the red pulp contains areas of extramedullary haemopoiesis. The red pulp expansion in these disorders gradually effaces the white pulp, such that the cut surface of the spleen has a homogeneous brick-red appearance. In contrast, chronic lymphocytic leukaemia infiltrates both the red and white pulp.
Classical Hodgkin’s lymphoma preferentially invades the white pulp and forms expansile nodules which encroach upon the red pulp. These may be single or multiple and range in size from a few millimetres to several centimetres in diameter (Fig. 22.17). The earliest site of involvement in the white pulp is the T-cell-dependent region of the peri-arteriolar lymphoid sheath, which progressively expands to obliterate the normal white pulp architecture.
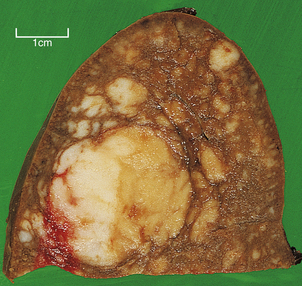
Fig. 22.17 Spleen infiltrated by Hodgkin’s lymphoma. Note the discrete white nodules of tumour ranging in size from a few millimetres to several centimetres.
Non-Hodgkin’s lymphomas also principally affect the white pulp, but their pattern of disease varies. The low-grade lymphomas form multiple small and medium-sized nodules that expand and replace the normal white pulp; the cut surface of the spleen is seen to be studded with numerous relatively even-sized nodules (Fig. 22.18). In contrast, high-grade lymphomas form small numbers of large destructive nodules. These differences in the pattern of splenic involvement by low- and high-grade NHLs are probably due to the physiological homing of the recirculating lymphoid cells of low-grade lymphomas to their natural milieu within the white pulp.
Fig. 22.18 Spleen infiltrated by low-grade non-Hodgkin’s lymphoma.  Macroscopic appearance. The cut surface is studded by small white nodules which stand out against the red pulp.
Macroscopic appearance. The cut surface is studded by small white nodules which stand out against the red pulp.  Low-power view showing numerous lymphoid aggregates with adjacent unaffected red pulp.
Low-power view showing numerous lymphoid aggregates with adjacent unaffected red pulp.
Although haemopoietic malignancies frequently involve the spleen, other tumours rarely metastasise to the spleen, and then only as part of a widely disseminated malignancy. Primary tumours of the spleen are rare and include hamartomas, benign and malignant vascular tumours and occasionally other mesenchymal neoplasms.
Storage disorders
Several storage disorders may cause splenomegaly: these include Niemann–Pick disease, Gaucher’s disease and the mucopolysaccharidoses (Ch. 7). Characteristically, there is marked red pulp expansion by macrophages whose cytoplasm is distended with the abnormal storage product (Fig. 22.19).
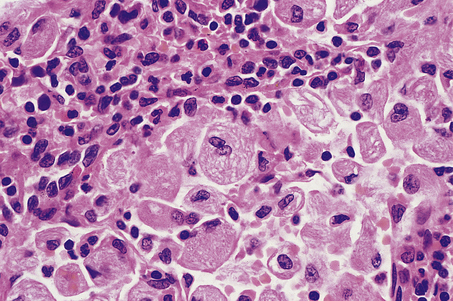
Splenic infarction
Splenic infarction follows occlusion of the splenic artery or its branches and is usually secondary to emboli that arise in the heart (Ch. 8). Occasionally, splenic infarction may be due to local thrombosis, as in sickle cell disease, myeloproliferative disorders and malignant infiltrates.
Splenic infarcts are macroscopically pale and wedge-shaped with the base adjacent to the splenic capsule. They may be single or multiple, and heal forming depressed scars.
Rupture of the spleen
Rupture of the spleen is usually caused by blunt abdominal trauma, particularly automobile accidents. Massive, life-threatening intraperitoneal haemorrhage may follow splenic rupture, necessitating emergency splenectomy.
Spontaneous rupture of the spleen may occur, particularly in infectious mononucleosis and in spleens enlarged by haemopoietic proliferations such as myelofibrosis.
Splenic atrophy
Splenic atrophy may occur in association with intestinal malabsorption states such as coeliac disease. In splenic atrophy, the spleen is small and weighs less than 50g. There is evidence of hyposplenism with numerous target cells and Howell–Jolly inclusion bodies in red cells.
Patients with sickle cell disease suffer multiple splenic infarcts and the spleen is greatly reduced in size and function.
Commonly confused conditions and entities relating to the lymph nodes, thymus and spleen
| Commonly confused | Distinction and explanation |
|---|---|
| Mantle cell and marginal zone lymphomas | Both are B-cell lymphomas and both can occur in the gastrointestinal tract. Mantle cell lymphomas have features of the cells in the mantle zone of germinal centres and a characteristic chromosomal translocation resulting in overexpression of cyclin D1. Marginal zone lymphomas, the most common type of extranodal lymphoma, contain cells resembling those in the marginal zone of lymph nodes, often with plasma cell differentiation. |
| Mycosis fungoides and Sézary syndrome | Both are cutaneous T-cell lymphomas (and have nothing to do with fungal infection). In mycosis fungoides the neoplastic T-cells are initially confined to focal skin lesions, whereas in Sézary syndrome there is more generalised involvement including atypical cells in the blood. |
| Thymoma and thymic lymphoma | Although thymomas often contain numerous lymphocytes, they are usually benign lesions derived from thymic epithelium. Thymic lymphomas may be of Hodgkin’s or non-Hodgkin’s type. |
| Hypersplenism and splenomegaly | Splenomegaly is enlargement of the spleen. If this is accompanied by blood pancytopenia, then the combination of splenic enlargement and functional disturbance is called hypersplenism. |
De Paepe P., De Wolf-Peeters C.. Diffuse large B-cell lymphoma: a heterogeneous group of non-Hodgkin lymphomas comprising several distinct clinicopathological entities. Leukemia. 2007;21:37-43.
Ekström-Smedby K.. Epidemiology and etiology of non-Hodgkin lymphoma. Acta Oncologica. 2006;45:258-271.
Higgins R.A., Blankenship J.E., Kinney M.C.. Application of immunohistochemistry in the diagnosis of non-Hodgkin and Hodgkin lymphoma. Archives of Pathology and Laboratory Medicine. 2008;132:441-461.
Kapatai G, Murray P 2007 Contribution of the Epstein–Barr virus to the molecular pathogenesis of Hodgkin lymphoma. Journal of Clinical Pathology 60: 1342–1349
Marcus R., Sweetenham J.W., Williams M.E.. Lymphoma: pathology, diagnosis and treatment. Cambridge: Cambridge University Press; 2007.
Martin P, Leonard J P 2007 Targeted therapies for non-Hodgkin lymphoma: rationally designed combinations. Clinical Lymphoma and Myeloma 7 (Suppl 5): S192–198
Tward J., Glenn M., Pulsipher M., Barnette P., Gaffney D.. Incidence, risk factors, and pathogenesis of second malignancies in patients with non-Hodgkin lymphoma. Leukemia and Lymphoma. 2007;48:1482-1495.




