Obesity
The Problem and Its Management
The role of nutrition in support of a vigorous, healthy life and the adverse consequences of excess food intake on body weight have been recognized and documented for thousands of years.1,2 Despite this long history, an unequivocal definition of obesity as a disease continues to generate controversy among scientists, clinicians, the general public, and policy makers. Given the substantial increase in the prevalence of excess body weight over the past 20 years, the relevance of this debate is of particular importance.3 An evaluation of this topic was recently commissioned by the Council of the Obesity Society.4 The writing group concluded that although one cannot scientifically prove that obesity is or is not a disease, the evidence supports the definition of obesity as a disease in that such a definition is likely to have more positive than negative consequences on both treatment and research into this complex condition.4
Epidemiology of Overweight and Obesity
Body adiposity is classified by the body mass index (BMI), which is calculated by dividing weight (in kilograms) by height (in meters squared), or by dividing weight (in pounds) by height (in inches squared) and multiplying the result by 704. A general guideline for classifying weight status by BMI has been put forth by the National Institutes of Health5 and World Health Organization6 (Table 2-1). Using this guideline, men and women are considered overweight if the BMI is between 25 and 29.9 kg/m2 and obese if the BMI ≥ 30 kg/m2. It is important to recognize that these categories represent somewhat artificial cutoffs within the continuous relationship between BMI and mortality derived from a primarily Caucasian U.S. population. Large epidemiologic studies have established that death rates are increased in overweight and obese subjects of differing ethnic backgrounds, but the categories of overweight and obese associated with increased mortality can differ from that in Caucasians.4,7 Thus it has been suggested that appropriate cutoffs for the overweight category begin at BMI ≥ 23 kg/m2 in Asians and BMI ≥ 24 kg/m2 in Hispanics.7
Table 2-1
Classification of Overweight and Obesity by BMI, Waist Circumference, and Associated Disease Risk
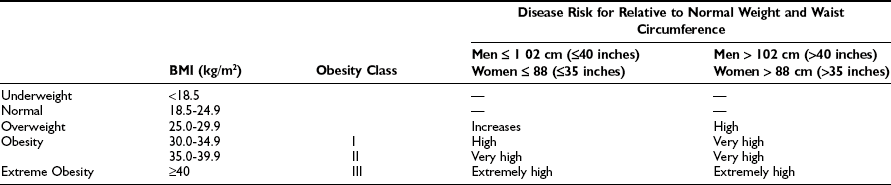
Data from www.nhlbi.nih.gov/guidelines/obesity/e_txtbk/intro/intro.htm.
Using the definitions in Table 2-1, the prevalence of obesity in U.S. adults doubled between 1980 and 2002.8 Findings from the most recent National Health and Nutrition Examination Survey (NHANES 2003–2004) indicate that 66.3% of the total U.S. population (men and women) was overweight, and 32.2% of the population was obese.3 The prevalence of overweight and obesity were greater for non-Hispanic blacks (76.1% and 45.0%) and Mexican Americans (75.8% and 36.8%). As illustrated in Table 2-2, minority women (non-Hispanic black and Mexican) were markedly more overweight and obese than Caucasian women or minority men.
Table 2-2
Prevalence of Overweight, Obesity, and Extreme Obesity in the U.S. Population for 2003-2004
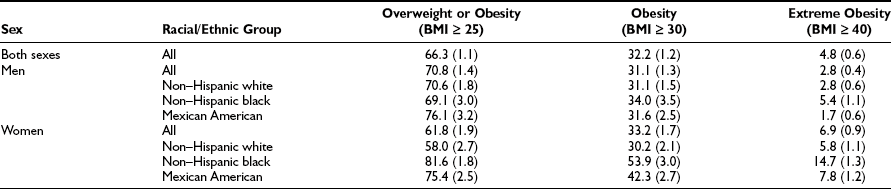
Data from Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal KM: Prevalence of overweight and obesity in the United States, 1999–2004. JAMA 295:1549–1555, 2006.
Excess body weight is also prevalent in children and adolescents (Fig. 2-1). Among children 2 to 19 years, 17.1% were overweight (defined as a BMI for age at or above the sex-specific 95th percentile) in NHANES 2003–2004. This was a significant increase compared to data from 1999–2000 and 2001–2002. Using a cutoff of BMI for age at or above the sex-specific 85th percentile, 34.8% of children and adolescents were at risk for overweight or were overweight. The prevalence of overweight in children and adolescents suggests that the need for medical management of obesity and associated comorbidities will not decline in the future.
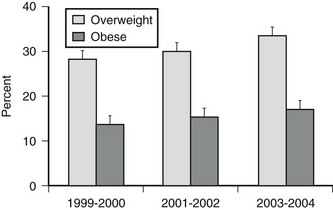
FIGURE 2-1 Prevalence of overweight (BMI for age at 85th percentile or higher) and obesity (BMI for age at 95th percentile or higher) in children and adolescents ages 2 to 19 years. (Data presented as mean ± SEM from Ogden CL, Carroll MD, Curtin LR, et al: Prevalence of overweight and obesity in the United States, 1999–2004. JAMA 295:1549–1555, 2006.)
Medical expenses for overweight and obesity accounted for 9.1% of total U.S. medical expenditures in 1998 and may have reached as high as $78.5 billion ($92.6 billion in 2002 dollars). Approximately half of these costs were paid by Medicaid and Medicare.9 It has also been estimated that annual medical expenditures incurred by obese adults are 37.4% higher ($732) than for normal weight adults.10 Additional economic costs of obesity include lost productivity and wages due to illness and absence from work.10
Diseases Associated With Obesity
Overweight and obesity are associated with the development of a number of comorbidities, many of which contribute to the higher rates of mortality observed in this population. A number of factors modify the relationship between obesity and disease.11 For example, the relative risk of mortality from obesity is significantly increased in elderly men and women (≥65 years) compared to younger age groups. However, in contrast to younger subjects, overweight does not confer increased risk of mortality in elderly individuals.12 Gender and ethnicity further modify the relationship of adiposity to disease.
The distribution of body fat into intraabdominal (visceral) depots versus that in peripheral subcutaneous depots is often a better predictor of disease in obesity than is total adiposity measured by BMI. Increased visceral adipose tissue is a significant determinant of risk for type 2 diabetes mellitus13 and cardiovascular disease.14
Cardiovascular and Cerebrovascular Disease
Overweight, obesity, and abdominal fat deposition increase the risk of both cardiovascular and cerebrovascular diseases in the United States11 and worldwide.15 Obesity results in significantly greater rates of cardiovascular mortality.11 The reasons for the increased risk for cardiovascular and cerebrovascular diseases include elevations of blood pressure, low-density lipoprotein cholesterol, triglycerides, small dense low-density lipoprotein cholesterol, total cholesterol, fibrinogen, plasminogen activator inhibitor-1, and insulin, together with decreases in high-density lipoprotein cholesterol. The cluster of three abnormalities is diagnostic of the metabolic syndrome.16 The metabolic syndrome is defined by the Third Report of the National Cholesterol Education Program Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) as having three or more of the following criteria: waist circumference greater than 102 cm in men and 88 cm in women; serum triglycerides of at least 150 mg/dL (1.69 mmol/L in men) and 50 mg/dL (1.29 mmol/L) in women; blood pressure of at least 130/85 mm Hg; or serum glucose level of at least 100 mg/dL (6.1 mmol/L). The age-adjusted prevalence of the metabolic syndrome is 25% in the U.S. population.17 Not only is this disorder highly prevalent, it also increases cardiovascular and all-cause mortality as demonstrated in an 11-year follow-up study.18
Hypertension
The INTERSALT study involving more than 10,000 men and women reported that a 10-kg increase in weight was associated with a 3 mm Hg rise in systolic blood pressure and a 2.3 mm Hg rise in diastolic blood pressure.19 This degree of blood pressure elevation has been associated with a 12% increase in coronary heart disease (CHD) and a 24% increase in stroke. The precise mechanism by which changes in weight alter blood pressure has not been established. Increased sympathetic activity due to elevated serum leptin appears to be an important component of obesity-related hypertension in rodent models.20
Dyslipidemia
Increases in BMI are associated with increases in total cholesterol, triglycerides, total low-density lipoprotein (LDL), and small dense LDL and with decreases in high-density lipoproteins.21 The risk of CHD is primarily due to increases in LDL. An increase in BMI of 10 units, starting from a level between 20 and 30 kg/m2, will raise LDL cholesterol levels between 10 and 20 mg/dL. Changes of this magnitude can be expected to increase the risk of CHD by 10% over a 5- to 10-year period. The risk may be particularly great for individuals with more prominent upper body obesity, in whom triglyceride, small dense LDL, and apolipoprotein B levels are high.
Coronary Artery Disease
In the Determinants of Atherosclerosis in Youth (PDAY) study, fatty streaks and advanced lesions in coronary arteries postmortem from subjects 15 to 34 years old were positively associated with obesity and abdominal fat deposition.22 Subjects undergoing cardiac catheterization today are more frequently obese, younger with more comorbidities, and present with more single-vessel disease.23 However, obesity is not associated with increased mortality or postoperative cerebrovascular events after cardiac surgery.24
Congestive Heart Failure
Both overweight and obesity have been shown to be independent risk factors for the development of congestive heart failure.25,26 Furthermore, because hypertension and diabetes are also associated with congestive heart failure, the overall risk when these dependent factors are taken into account is proportionally increased.27
Stroke
The risk of stroke is nearly twofold higher in women with a BMI greater than 32 kg/m2 than in women with a BMI less than 21 kg/m2.28 In men, each unit increase of BMI over 23 kg/m2 was associated with a significant 6% increase in the adjusted relative risks of total, ischemic, and hemorrhagic stroke.29 Adjustment for hypertension, diabetes, and hypercholesterolemia slightly attenuated the risk for stroke in this study.
Diabetes Mellitus
An association between increased weight and the development of type 2 diabetes mellitus is well established.30 In fact, the risk for diabetes increases at BMI levels below that established for the diagnosis of overweight. In the Nurses’ Health Study, BMI values above 22 kg/m2 were associated with an increased risk of diabetes.31 It has been estimated that the relative risk for diabetes increases by 25% for each unit of BMI above 22 kg/m. It has also been estimated that more than a quarter of all newly diagnosed cases of diabetes in the United States are due to weight gain of more than 5 kg.32 See Chapter 15 for additional information on the etiology of diabetes and obesity.
Nonalcoholic Fatty Liver Disease
Nonalcoholic fatty liver disease (NAFLD) describes a range of liver abnormalities that include hepatomegaly, abnormal liver biochemistry, steatosis, steatohepatitis, fibrosis, and cirrhosis.33 Estimates of the prevalence of NAFLD in obese subjects range from 30% to 100%, and there is a strong association with abdominal obesity and metabolic syndrome. The disease is equally represented in men and women and is also known to affect children. The progression from simple steatosis (for the most part considered benign) to steatohepatitis (necroinflammatory change and hepatocellular injury) has been suggested to follow a “two-hit” model.34 The first hit results from any interference in metabolism of free fatty acids, which then accumulate in the liver. The second hit may be due to oxidative stress resulting from metabolism of excess fatty acids or could result from proinflammatory cytokines. Thus the insulin resistance and subclinical inflammation present in obesity support the development of NAFLD. Weight loss and improved insulin sensitivity appear to reduce hepatic steatosis,35,36 although additional study is needed to fully understand the effects of various therapeutic interventions.
Cancer
There is a strong association between adiposity and increased risk of mortality from cancer.37 In reviewing the evidence in 2007, the World Cancer Research Fund (WCRF) concluded that body fatness is associated with increased risk of esophageal adenocarcinoma and with cancers of the pancreas, colorectum, postmenopausal breast, endometrium, and kidney.38 In a very recent meta-analysis,39 it was determined that in men, a 5 kg/m2 increase in BMI was strongly associated with esophageal adenocarcinoma and with thyroid, colon, and renal cancers. In women, strong associations were found between a 5 kg/m2 increase in BMI and endometrial, gallbladder, esophageal adenocarcinoma, and renal cancers. Weaker positive associations were also found between increased BMI and rectal cancer and malignant melanoma in men; postmenopausal breast, pancreatic, thyroid, and colon cancers in women; and leukemia, multiple myeloma, and non-Hodgkin lymphoma in both sexes. Possible mechanisms linking cancer with excess body weight could be increased insulin/IGF (which may alter the balance between cell proliferation and apoptosis), altered adipokines, localized inflammation, and oxidative stress.40
Gyenecologic Abnormalities
Polycystic ovarian syndrome, a disorder that includes hirsutism, obesity, ovulatory and menstrual dysfunction, and insulin resistance, is among the most common causes of infertility in women who are overweight.41 Even modest increases in weight in young women can adversely affect reproductive function.42
Obesity during pregnancy is also associated with excessive morbidity. Pregnant women with obesity have nearly a 10-fold excess risk of hypertension and a significant increase in the risk of gestational diabetes. Furthermore, the risk of congenital malformations, primarily neural tube defects, is increased in the pregnancy of obese women.43 Finally, increased weight before pregnancy has been shown to result in an increased risk of adverse fetal outcomes.44
Obstructive Sleep Apnea
Episodes of apnea and hypopnea during sleep occur due to partial or complete airway obstruction in obese subjects. A conservative estimate of the prevalence of obstructive sleep apnea in obesity is 30%.45 Diagnosis and treatment of sleep apnea in obese patients is particularly important because of the sequelae of hypoxia, hypertension, myocardial infarction, and cardiac arrhythmias associated with this condition. Obstructive sleep apnea is also independently associated with alterations in glucose metabolism which put obese subjects at greater risk for development of type 2 diabetes.46
Gallstones
For women with a BMI greater than 40 kg/m2, the risk of gallstones is nearly seven times higher than for women with a BMI less than 24 kg/m2.47 The risk of gallstones is increased with rapid weight loss such as with very-low-calorie diets or weight loss surgery.48
Osteoarthritis
Obesity is likely the single most important risk factor for development of severe osteoarthritis of the knee due to the additional load on this joint.49 A study in twins estimated that for every 1-kg rise in body weight, the risk of osteoarthritis increases by approximately 10%.50 Obesity is also a cofactor in development of osteoarthritis in non-weight-bearing joints, with hyperinsulinemia and proinflammatory cytokines suggested to contribute to disease mechanisms.49
Regulation of Energy Balance
Energy Expenditure
Total daily energy expenditure (TEE) can be divided into four major components, including resting metabolic rate (RMR), the thermic effect of food, physical energy expenditure, and non-exercise activity thermogenesis (see Chapter 1 for details). Fat-free mass (primarily skeletal muscle) is the most important contributor to TEE, so the greater an individual’s muscle mass, the greater their TEE.51 Fat-free mass is also the main determinant of RMR, which accounts for 60% to 70% of the total daily energy expenditure. After adjusting for fat-free mass, age and gender are also important predictors of TEE and RMR. The significant contribution of muscle mass to TEE underlies the recommended use of exercise in weight loss programs.
Energy expenditure has been rigorously examined in an attempt to detect defects that could contribute to the development of obesity. In adult Pima Indians, a population prone to the development of obesity, low relative RMR was associated with greater risk of becoming obese, compared to subjects with the highest RMR.51 However, low RMR as a significant cause of obesity has not been born out in other populations, and in most studies, obese subjects actually have a higher RMR than the lean subjects, owing to their greater skeletal muscle mass.52 TEE is also increased in obese subjects secondary to the greater energy expenditure required to move a larger body mass, although during non-weight-bearing exercise, obese individuals expend the same amount of energy as lean subjects doing equivalent work.53 Despite these physiologic observations, epidemiologic studies suggest that obese subjects are more likely to engage in sedentary behaviors than lean individuals.53,54 Thus defects in energy expenditure other than reduced physical activity are likely to contribute little to the development of obesity in humans.
Under highly controlled experimental conditions, it has been demonstrated that changes in body weight result in compensatory changes in energy expenditure which attempt to restore the body to the original starting weight. For example, a 10% reduction in body weight in either lean or obese subjects is associated with a significant reduction in both resting and non-resting metabolic rate beyond that which would be predicted to result from the decrease in fat mass and fat-free mass.55 This greater-than-expected reduction in TEE, termed adaptive thermogenesis, likely contributes to a degree to the failure of weight loss programs. The environmental pollutant organochlorine and oxygen desaturation due to obstructive sleep apnea have recently been proposed to increase adaptive thermogenesis.56
Spontaneous physical activity or non-exercise-activity thermogenesis (NEAT—all activity other than volitional exercise) defends against body weight gain in certain individuals.57,58 Individuals who are resistant to weight gain have a greater activation of NEAT than do individuals who gain weight easily when an experimental increase in caloric intake is imposed. It has also been shown that lean individuals stand and walk 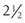 hours per day more than obese subjects.59 Imposing a weight gain in lean subjects does not alter the time standing or walking. In contrast, weight loss does not decrease the sitting time of obese subjects. These observations have prompted the theoretical model that individuals are genetically determined to be either NEAT activators who remain lean or NEAT conservers who become obese in the current environment.60 It has thus been suggested that increasing NEAT through both individual approaches (get up from the chair) and environmental re-engineering (remove the chair) are needed to reduce obesity.60
hours per day more than obese subjects.59 Imposing a weight gain in lean subjects does not alter the time standing or walking. In contrast, weight loss does not decrease the sitting time of obese subjects. These observations have prompted the theoretical model that individuals are genetically determined to be either NEAT activators who remain lean or NEAT conservers who become obese in the current environment.60 It has thus been suggested that increasing NEAT through both individual approaches (get up from the chair) and environmental re-engineering (remove the chair) are needed to reduce obesity.60
Energy Intake
In humans, the decision to eat is the result of the complex integration of environmental cues and higher cognitive function with internal physiologic signals regarding nutrient status and energy stores.61 The gastrointestinal tract, liver, pancreas, and adipose tissue provide information concerning the presence of nutrients and amount of energy stored in the body via humoral and neural signals to the CNS (Fig. 2-2). The most important targets within the CNS for the internal physiologic signals are thought to be the hypothalamus and brainstem (nucleus of solitary tract, area postrema, parabrachial nucleus). In addition, circulating free fatty acids and glucose also regulate hypothalamic function.62,63
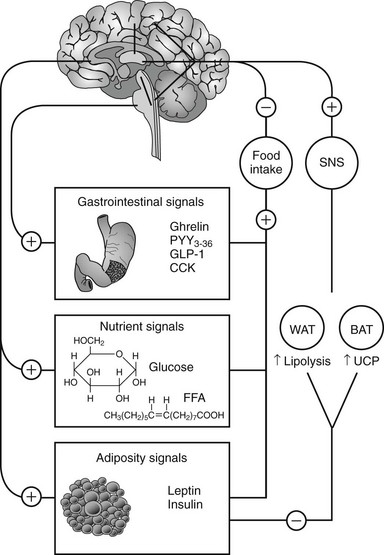
FIGURE 2-2 Integration between feeding-related signals to the brain and food intake and energy expenditure. Food intake initiates a series of signals that reach the hypothalamus (nutrient and adiposity signals) or the brainstem (gastrointestinal signals). The hypothalamus integrates these signals with other sensory, cognitive, and environmental information from the cerebral cortex. This integrated information, when sent back to the periphery, results in a decrease in food intake and activation of the sympathetic nervous system (SNS). The SNS stimulates lipolysis in white adipose tissue (WAT) and thermogenesis in brown adipose tissue of rodents (BAT) via activation of uncoupling protein-1 (UCP). CCK, Cholecystokinin; FFA, free fatty acids; GLP-1, glucagon-like peptide-1; PYY3-36, peptide YY3-36.
Adiposity Signals
Leptin and insulin convey information to the hypothalamus about the amount of energy stored within the body and satisfy the criteria for a negative-feedback system.64 These hormones circulate in the blood at concentrations proportional to body fat content. Leptin and insulin promote weight loss when administered to the CNS, and blocking the central neural activity of these hormones increases food intake and weight gain.
Gastrointestinal Signals
Cholecystokinin (CCK) was the first gut peptide demonstrated to regulate meal size in a dose-dependent manner.65 Whereas CCK reduced meal intake by nearly one half in rodents, a selective antagonist completely reversed this suppression. By itself, the CCK antagonist was able to increase food intake by approximately one third.66
Peptide YY (PYY) is a 36-amino-acid peptide structurally related to neuropeptide Y and pancreatic polypeptide. The major form of PYY released by L cells in the gut is the N-terminal truncated form PYY3-36. Peripheral administration of PYY3-36 at physiologic doses has been shown to reduce food intake in both rodents and humans.67
Glucagon-like peptide-1 (GLP-1) is a neuropeptide hormone produced by posttranslational processing of the preproglucagon gene by L cells in the gut. This incretin is of significant current interest for its ability to stimulate insulin release; a number of pharmaceutical compounds that prolong the lifetime of this hormone in the circulation are currently in use.68 The GLP-1 analog exendin-4 reduced body weight in clinical trials testing the compound’s ability to enhance insulin secretion.69
Oxyntomodulin is also a posttranslational product of the preproglucagon gene released into the circulation postprandially. Oxyntomodulin inhibits gastric acid secretion and reduces food intake in rodents and humans.70
Ghrelin, a 28-amino-acid peptide made in the stomach, is a ligand for the growth hormone–secretagogue receptor present in the hypothalamus and brainstem.71 In contrast to all other known gut peptides, ghrelin stimulates food intake in rodents72 and humans.73 Plasma levels of ghrelin increase during fasting and fall after eating, which has been interpreted to suggest that ghrelin is a hunger hormone.72 Plasma ghrelin concentrations are increased with diet-induced weight loss,74 and postprandial fall in circulating levels is attenuated or absent75 in obese subjects, suggesting that ghrelin may be involved in the pathophysiology of obesity. The active form of ghrelin contains a fatty acyl side chain attached to serine 3.71 The enzyme responsible for acylating ghrelin has recently been identified, providing a new therapeutic target to regulate the actions of this orexigenic hormone.76,77
Central Integration of Energy Homeostasis
As illustrated in Fig. 2-2, the signals of nutrient status and energy stores are communicated to the hypothalamus and brainstem via humoral and neural pathways. Within the arcuate nucleus of the hypothalamus, neurons co-expressing neuropeptide Y (NPY) and agouti-related protein (AgRP) stimulate food intake. Other neurons co-express α-melanocyte-stimulating hormone (α-MSH) and cocaine- and amphetamine-regulated transcript (CART) and activate melanocortin receptors to decrease food intake (see Chapter 1 for details). Leptin and insulin activate α-MSH/CART signaling and inhibit NPY/AgRP neurons to reduce food intake.62,78 Cross-talk exists between leptin/insulin-responsive arcuate neurons and brainstem neurons that receive input from the gastrointestinal satiety hormones.62 The importance of this interaction underlies current efforts to develop dual pharmacotherapy for weight loss.79
The reward value of food intake is mediated by dopamine and µ-opioid signaling in forebrain structures. Neurons from the ventral tegmental area and nucleus accumbens project to the hypothalamus, where reward is integrated with the signals of nutrient status.62
Efferent neural signals arising within the hypothalamus communicate with peripheral tissues to regulate food intake and energy expenditure. Activation of sympathetic neural activity increases lipolysis in white adipose tissue and energy expenditure in brown adipose tissue of rodents. Uncoupling protein (UCP-1) in brown adipose tissue functions to produce proton leak across the mitochondrial membrane, dissociating substrate oxidation from ATP synthesis.80 As humans have little brown fat, this mechanism for activation of energy expenditure may appear to be limited. However, a family of novel uncoupling proteins is expressed in human tissue including muscle. These proteins do not appear to mediate inducible mitochondrial proton leak in a manner similar to that of UCP-1, although UCP-3 has been suggested to facilitate fatty acid oxidation.81 In addition, recent findings using fluorodeoxyglucose positron emission tomography challenge the notion that adult humans lack brown fat.82 Distinct symmetrical areas of brown adipose tissue have been detected in the upper body and appear to be of sufficient quantity to influence energy expenditure, thus justifying investigation into the role of this tissue in regulating body weight.
Therapy for Obesity
The assessment of overweight and obese patients should include: (1) the history of weight gain and maximum body weight; (2) consideration of medications that may contribute to weight gain, such as corticosteroids, thiazolidinediones, and antipsychotic agents; (3) previous approaches to weight reduction; (4) patterns of food intake, including binge eating; and (5) physical activity levels. Measurement of waist circumference and evaluation of blood pressure, glycemia, cholesterolemia, and liver and cardiovascular function should be performed. A waist circumference greater than 102 cm in men and 88 cm in women is an independent predictor for obesity-associated risk factors.5 Treatment of coexisting conditions should follow standard guidelines. The patient’s readiness for weight reduction should be addressed, although the absence of readiness should not preclude communication between provider and patient about the importance of weight reduction.83
The general goals of the weight loss program are to reduce and maintain a lower body weight or prevent further weight gain in subjects who cannot lose weight. The three treatment options for weight loss are lifestyle modification (diet, exercise, behavioral modification), pharmacotherapy, and bariatric surgery. Table 2-3 presents a summary of the treatment guidelines put forth by the National Heart, Lung, and Blood Institute,5 which are generally consistent with the recommendations of a number of medical associations.83
Table 2-3
Weight-Loss Treatment Guidelines
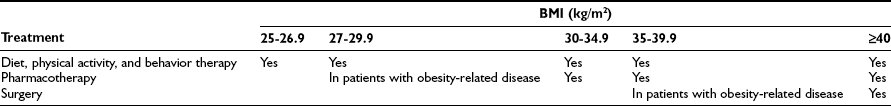
Data from Wadden TA, Butryn ML, Wilson C: Lifestyle modification for the management of obesity. Gastroenterology 132:2226-2238, 2007; and Eckel RH: Clinical practice. Nonsurgical management of obesity in adults. N Engl J Med 358:1941-1950, 2008.
Lifestyle Modification
A combination of reduced-calorie diet and increased physical activity is the initial recommendation to achieve weight loss and is considered the cornerstone of treatment for all overweight and obese individuals.84
Diet
For individuals with BMI < 34.9 kg/m2, a reduction in daily energy intake of 500 kcal/day will result in loss of approximately 1 pound per week and a 10% reduction of initial weight by 6 months. Patients with more severe obesity (BMI ≥ 35 kg/m2) should reduce intake by 500 to 1000 kcal/day to achieve weight loss of 1 to 2 pounds per week and 10% of initial body weight by 6 months.5 Although often viewed as failure by the obese patient, a 10% weight loss is associated with significant improvement in obesity-related comorbid conditions.85
A number of dietary options are available. The choice of a diet may be left in part to patient preference, as well as to cost considerations. One of the keys to successful weight control is to identify a reduced-calorie eating plan that is acceptable to the patient, making long-term adherence more likely.84
Very-Low-Calorie Diets (VLCD) provide 400 to 800 kcal/day in a liquid form containing large amounts of dietary protein. They are safe when provided with medical supervision but are associated with an increased risk of symptomatic gallstones. Although very-low-calorie diets result in significant weight loss, they are associated with substantial weight regain, such that long-term efficacy is not different from low-calorie diets.86
Low-Calorie Diets (LCD) provide 900 to 1500 kcal/day through combined use of liquid diet, nutrition bars, and conventional food. These diets have largely replaced VLCD because of similar efficacy with lower medical monitoring costs.84 The use of meal replacements (nutrition bars and liquid formula) provide patients with a fixed amount of food that requires little preparation and allows avoidance of problem foods. These factors appear to improve adherence to the diet.87 Portion-controlled servings of conventional foods are also more effective in inducing weight loss than self-selected table foods.84
Low-carbohydrate, high-fat diets facilitate dietary adherence and weight loss by simplifying food choices. In addition, the high protein content may increase satiation. A meta-analysis of six randomized controlled trials of low-carbohydrate versus low-fat diets showed that low-carbohydrate diets induced significantly greater weight loss in the first 6 months of treatment, but there was no difference in weight loss between the two diets at 1 year. Low-carbohydrate diets were associated with greater improvements in triglycerides and high-density lipoprotein cholesterol levels; low-fat diets were more effective in reducing low-density lipoprotein cholesterol and total cholesterol.88 A more recent comparison found a greater weight loss at 1 year with a low-carbohydrate diet.89 This study differed from those in the meta-analysis in that study size was larger and dietary adherence was better. Overall, these findings suggest that low-carbohydrate, high-fat diets are a safe and reasonable dietary option when used for up to 1 year. Further study of the safety of such diets over longer periods is recommended.84
Exercise
Increased physical activity is central to any weight loss program. Given findings suggesting that total caloric intake has not risen dramatically in the United States over the past 2 decades, many experts believe that the rise in prevalence of obesity is directly attributable to a decrease in the amount of physical activity.60 Because of the high volume and intensity of physical activity required to produce an energy deficit that results in an acceptable rate of weight loss, exercise in the absence of caloric restriction is relatively ineffective in inducing weight loss.90 However, exercise is essential for long-term weight management.91 Increased physical activity provides cardiovascular benefits, prevents loss of muscle mass during weight loss, may promote better dietary adherence through improved mood,84 and may directly reduce visceral fat mass.92 Physical activity can be increased through programmed exercise activities such as walking, running, and swimming. Recent guidelines from the U.S. Department of Health & Human Services recommend 30 minutes of moderate-intensity physical activity on most days of the week to reduce risk of chronic disease. Sixty minutes of moderate- to vigorous-intensity physical activity on most days is recommended to prevent weight gain. To maintain weight loss, 60 to 90 minutes of moderate- to vigorous-intensity physical activity daily is the suggested.93 Obese individuals can also increase energy expenditure through lifestyle modifications such as parking further away from store entrances and taking stairs instead of elevators.94
Behavioral Modification
Eating is often undertaken for personal, psychologic, or social reasons, and these behaviors are powerful forces that impede long-term caloric restriction and weight loss. Efforts at weight loss independent of changes in behavior are unlikely to succeed. Some strategies for behavioral modification include stimulus control (avoiding cues that prompt eating), self monitoring through daily records of food consumption and activity, positive thinking, and social support. Group therapy appears more effective than individual treatment for weight loss. Group sessions provide empathy, social support, and competition. Participation in weight maintenance sessions following weight loss prevents weight regain. Long-term patient-provider contact via telephone, mail, or email is also useful.84
Pharmacotherapy
Drug treatment for overweight and obese patients has a history of at least 100 years. Despite this, there are currently a limited number of pharmaceuticals available. Pharmacotherapy for weight loss was originally considered to be short term, but obesity is a chronic condition, and a number of studies have shown that patients regain weight when therapy is discontinued.5,6 In the mid-1990s, the U.S. Food and Drug Administration (FDA) recommended that weight loss drugs be studied for a 2-year period for approval.95 Thus pharmacotherapy for weight loss is now considered long term, and as in the pharmacologic treatment of all diseases, no risk-free, highly efficacious therapies are available. A balance must be achieved between risk and efficacy in a highly heterogeneous group of individuals with a wide range of risk factors for significant disease.85
Early weight loss therapy was based on the short-term use of amphetamines or structurally similar compounds. Currently, phentermine, benzphetamine, phendimetrazine, and diethylpropion (schedule III and IV drugs) are approved for short-term use of 12 weeks. These drugs act primarily to inhibit reuptake of norepinephrine and dopamine at nerve endings to enhance satiation (level of fullness during consumption of a meal) and satiety (level of hunger after consumption of a meal).85 Two additional anorexiants that patients currently seeking weight loss therapy may have taken in the past are fenfluramine and dexfenfluramine. These compounds were removed from the market in 1997 owing to increased incidence of valvular heart disease.96
Two drugs currently approved for long-term treatment of overweight patients, orlistat and sibutramine, are discussed in detail in the next sections. It is important to note that treatment outcomes are significantly better when pharmacotherapy is administered as part of a weight loss program that includes diet, exercise, and behavioral modification.5,6,84
Orlistat
A synthetic derivative of lipstatin (from Streptomyces toxytricini), orlistat inhibits gastric and pancreatic lipases, enzymes critical for the digestion and absorption of fat from the gastrointestinal tract. Inhibition of lipases causes a reduction in the absorption of fat and an increase in the excretion of triglycerides in feces. In a 2-year randomized, crossover study of 688 obese individuals (BMI ~ 36 kg/m2, weight ~ 100 kg), 120 mg of orlistat three times daily caused a 10.2% decrease from initial body weight after the first year versus a 6.1% decrease in the placebo group.97 Several meta-analyses have shown favorable effects of orlistat on weight loss and weight maintenance. In addition, pooled data show significant overall effects in reducing cholesterol, triglycerides, blood pressure, and hemoglobin A1c.85
Sibutramine
A highly selective inhibitor of serotonin and norepinephrine reuptake at nerve endings, sibutramine also inhibits dopamine reuptake to a lesser degree. In several published studies, it has been shown to cause weight loss through the mechanism of appetite suppression. In a dose-ranging, 12-week trial in obese patients (BMI ~ 32 kg/m2, weight ~ 85 kg), sibutramine caused a maximum weight loss of approximately 5 kg (~6% loss from initial weight) versus a weight loss of approximately 1 kg (~1% loss from initial weight) in the placebo group.98 In a 1-year placebo-controlled trial, analysis of patients who completed the study showed that 65% of patients taking 15 mg of sibutramine lost 5% of their initial body weight, whereas only 29% of placebo-treated patients lost as much.99 In the 2-year STORM study (Sibutramine Trial of Obesity Reduction and Maintenance), patients taking sibutramine maintained 80% of their initial weight loss compared to placebo controls who maintained only a 20% weight loss.100 A meta-analysis of sibutramine use in diabetic patients found a mean weight loss of 5.53 ± 2.2 kg compared to 0.90 ± 0.17 kg in placebo-treated patients.101 Sibutramine also improved glucose, hemoglobin A1c, triglycerides, and HDL cholesterol.
The pressor effects of sibutramine have been known since early trials.98,99 A meta-analysis of sibutramine use found that the drug produced significant weight loss and a significant increase in blood pressure.102 Subgroup analysis suggested a greater effect on systolic and diastolic blood pressure in subjects weighing ≥ 92 kg. A second meta-analysis found an effect of the drug only on diastolic blood pressure.103 The Sibutramine in Cardiovascular Outcomes Trial (SCOUT) was initiated in 2005 with the aim of studying patients for 5 years to test whether drug-induced weight loss reduces the risk for fatal and nonfatal cardiovascular disease. The trial enrolled and treated 10,742 subjects in the 6-week lead-in period.104 Of subjects enrolled, 97% had cardiovascular disease, 88% hypertension, and 84% type 2 diabetes. During the lead-in, there was a significant reduction in body weight and blood pressure (both systolic and diastolic), and heart rate increased 1.5 bpm. Two consecutive increases in blood pressure or pulse rate of >10 mm Hg/bpm were observed in 4.7% and 3.5% of subjects, respectively. The investigators thus conclude that treatment with sibutramine for 6 weeks appears efficacious, tolerable, and safe in a high-risk population for whom sibutramine is usually contraindicated. However until the SCOUT trial is complete and definitive recommendations made, blood pressure and heart rate should be closely monitored in patients taking sibutramine.
Bariatric Surgery
To date, surgery remains the most effective means of inducing significant weight loss in the extremely obese subject. Several studies have demonstrated that bariatric surgery–induced weight loss is effective in reducing obesity-related comorbidities. A benefit on overall and cause-specific mortality has also been demonstrated.105–107 In-house mortality rates are less than 1% but are significantly affected by the experience of the surgeon and hospital staff.108 The National Institute of Diabetes and Digestive Diseases has established the Longitudinal Assessment of Bariatric Surgery (LABS) consortium to facilitate refinement of risk prediction for both patients and surgical programs.109
The National Institutes of Health established guidelines for the surgical treatment of obesity in 1991.110 Eligible patients with a BMI ≥ 40 kg/m2 should be well informed and motivated. In addition, they should have demonstrated inability to lose weight with conventional therapy and have acceptable operative risks. Adults with a BMI ≥ 35 kg/m2 with a serious comorbidity such as type 2 diabetes, hypertension, cardiomyopathy, sleep apnea, or severe joint disease may also be surgical candidates. Bariatric surgery should be performed in conjunction with a comprehensive follow-up plan consisting of nutritional, behavioral, and medical monitoring.111
Bariatric surgical procedures can be divided by mechanism of action into those that cause malabsorption (Roux-en-Y gastric bypass and biliopancreatic diversion) and those that produce gastric restriction (vertical banded gastroplasty and laparoscopic adjustable gastric banding).108,112
Laparoscopic Adjustable Gastric Banding
Weight loss 10 years after surgery is greater with gastric bypass procedures than with gastric banding.108,113 This result is likely due to the combination of malabsorption and restriction of intake achieved with these procedures. Several estimates suggest that 10% to 40% of patients do not achieve successful long-term weight loss after bariatric surgery.108 This could be related to surgical procedure or lack of lifestyle modification (continued ingestion of high-calorie soft foods and liquids). Perioperative complications can also include persistent vomiting, dumping syndrome, and vitamin malabsorption.113 Hyperinsulinemic hypoglycemia with nesidioblastosis has also been observed,114 and there are concerns that bariatric surgery may predispose individuals to metabolic bone disease.115 All of these findings reinforce the need for a comprehensive postsurgical plan for nutritional, behavioral, and medical monitoring.
Bariatric surgery results in marked improvement in type 2 diabetes, with rapid improvements in glucose and insulin that occur within days to weeks following surgery.105,116 The majority of patients on insulin therapy prior to surgery discontinue insulin use by 6 weeks postsurgery.116 The rapid resolution of type 2 diabetes following bariatric procedures, independent of weight loss, suggests that surgery could be a viable therapeutic option for this disease.117 A case report has demonstrated resolution of type 2 diabetes following duodenal-jejunal bypass in two overweight subjects (BMI 29 and 30 kg/m2). Additional studies will be necessary to assess the long-term safety and efficacy of this procedure.118
Future Therapy
Cannabinoid type 1 (CB1) receptor antagonists represent a new class of potential antiobesity compounds.119 CB1 receptors are located throughout the CNS and on peripheral tissues such as adipocytes, hepatocytes, and muscle and endothelial cells. Endocannabinoids stimulate CB1 receptors in the CNS to increase appetite, resulting in weight gain. Rimonabant, an oral CB1 receptor antagonist, caused significant weight loss in four large clinical trials, but adverse events were higher with drug than in placebo. Rimonabant is approved for use in a number of countries but not the United States. Taranabant, a second compound in this class still in clinical trials, has been shown to produce significant weight loss through both inhibition of food intake and increased energy expenditure.120,121
A number of drugs approved by the FDA for other indications also cause weight loss. The antidepressants fluoxetine, sertraline, and bupropion and the antiepileptic topiramate all cause weight loss to varying degrees.85 Fluoxetine and sertraline are selective serotonin reuptake inhibitors, and bupropion inhibits reuptake of norepinephrine and dopamine. The modulation of GABA receptors by topiramate is hypothesized to be the mechanism through which this compound reduces food intake.
Metformin, pramlintide, and exenatide are all approved for use in treating type 2 diabetes (see Chapter 22 for details). In the Diabetes Prevention Trial (DPT), the metformin-treated group lost 2.5% of body weight.122 The effect of metformin on weight loss (~5%) is not large enough to meet FDA requirements for a weight loss drug, but use of metformin should help with weight loss in subjects with diabetes or prediabetes.85 Pramlintide reduced body weight in type 2 diabetics by 2.6 kg with 1 year of treatment.123 Exenatide treatment resulted in weight loss of 1.4 kg and 4.8 kg versus placebo and insulin, respectively, in a recent meta-analysis.124 Ongoing studies continue to evaluate the weight loss potential of these drugs.
Combination pharmacotherapy is also being evaluated. The discovery of leptin held great promise for a potential target at which to direct therapy. However, the observation that leptin levels reach very high levels in obese patients led to the conclusion that most patients with obesity are resistant to the effects of leptin on reducing appetite and increasing energy expenditure.125 Although leptin effectively reduces body weight in rodents, high pharmacologic doses only elicit marginal weight loss in obese humans.126,127 However, a 24-week proof-of-concept clinical study has demonstrated that the combination of leptin and pramlintide produced significantly greater weight loss (12.7 ± 0.9%) compared to leptin (8.2 ± 1.3%) or pramlintide alone (8.4 ± 0.9%).79 Combinations of bupropion with naltrexone and topiramate with phentermine are also being tested.85
Other potential pharmaceuticals take advantage of the meal-terminating properties of the gut hormones. PYY and oxyntomodulin continue to undergo study for feasibility as weight loss therapy.70 The enzyme responsible for acylating ghrelin to create the active form of the hormone has recently been identified, providing a potential new therapeutic target to reduce the actions of this orexigenic hormone.76,77 Regulators of food intake in the CNS under investigation include selective serotoninergic agents targeting the 5-HT2c receptor, a serotonin receptor subtype which appears to specifically inhibit food intake. Melanin-concentrating hormone receptor agonists inhibit food intake in rodents.85
Genome-wide association studies have finally identified the first gene contributing to common forms of human obesity, FTO (fat mass and obesity associated gene).128 The function of FTO is not fully understood, but it has been implicated in the central regulation of food intake and in regulation of lipolysis in adipose tissue. This gene or its downstream effectors are exciting new targets for pharmacotherapy.
Finally, it is important to point out that all therapies discussed previously, both approved and in development, target reduced physical activity and increased food intake as the major contributors to the obesity epidemic. Interestingly, a recent meta-analysis found supportive but not conclusive evidence for additional explanations for the increased prevalence of obesity.129 Additional potential causes of obesity include sleep deprivation, endocrine disruptors, reduced variability in ambient temperature, and decreased smoking, among others. These factors may interact with or contribute to decreased physical activity and increased food intake. Future research will be needed to determine if an intervention targeting these potential causes of obesity is warranted.
References
1. Bray, GA. Historical framework for the development of ideas about obesity. In: Bray GA, Bouchard C, eds. Handbook of Obesity. ed 2. New York: Marcel Dekker Inc; 2004:1–31.
2. Haslam, D. Obesity: a medical history. Obes Rev. 2007;8(Suppl 1)):31–36.
3. Ogden, CL, Carroll, MD, Curtin, LR, et al. Prevalence of overweight and obesity in the United States, 1999–2004. JAMA. 2006;295:1549–1555.
4. TOS Obesity as a Disease Writing GroupAllison, DB, Chair. Obesity as a Disease: A White Paper on Evidence and Arguments Commissioned by the Council of The Obesity Society. Obesity (Silver Spring). 2008;16:1161–1177.
5. National Institutes of HealthNational Heart, Lung and Blood Institute, Clinical Guidelines on the Identification, Evaluation, and Treatment of Overweight and Obesity in Adults: The Evidence Report. Obes Res, 6;Suppl 2), 1998:51S–209S. www.nhlbi.nih.gov/guidelines/obesity/e_txtbk/intro/intro.htm.
6. World Health Organization. Obesity: Preventing and Managing the Global Epidemic. WHO Technical Report Series No. 894. Geneva: WHO; 2000.
7. James, WP. The epidemiology of obesity: the size of the problem. J Intern Med. 2008;263:336–352.
8. Hedley, AA, Ogden, CL, Johnson, CL, et al. Prevalence of overweight and obesity among US children, adolescents, and adults, 1999–2002. JAMA. 2004;291:2847–2850.
9. Economic consequences of overweight and obesity. Centers for Disease Control and Prevention www.cdc.gov/nccdphp/dnpa/obesity/.
10. Finkelstein, EA, Ruhm, CJ, Kosa, KM. Economic causes and consequences of obesity. Annu Rev Public Health. 2005;26:239–257.
11. Flegal, KM, Graubard, BI, Williamson, DF, et al. Cause-specific excess deaths associated with underweight, overweight, and obesity. JAMA. 2007;298:2028–2037.
12. Janssen, I, Mark, AE. Elevated body mass index and mortality risk in the elderly. Obes Rev. 2007;8:41–59.
13. Bray, GA, Jablonski, KA, Fujimoto, WY, et al. Relation of central adiposity and body mass index to the development of diabetes in the Diabetes Prevention Program. Am J Clin Nutr. 2008;87:1212–1218.
14. Fox, CS, Massaro, JM, Hoffmann, U, et al. Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation. 2007;116:39–48.
15. Yusuf, S, Hawken, S, Ounpuu, S, et al. Obesity and the risk of myocardial infarction in 27,000 participants from 52 countries: a case-control study. Lancet. 2005;366:1640–1649.
16. Ford, ES, Giles, WH, Dietz, WH. Prevalence of the metabolic syndrome among US adults. JAMA. 2002;287:356–359.
17. Ford, ES, Giles, WH, Mokdad, AH. Increasing prevalence of the metabolic syndrome among U.S. adults. Diabetes Care. 2004;27:2444–2449.
18. Lakka, HM, Laaksonen, DE, Lakka, Ta, et al. The metabolic syndrome and total and cardiovascular disease mortality in middle-aged men. JAMA. 2002;288:2709–2716.
19. Dyer, AR, Elliott, P. The INTERSALT study: Relations of body mass index to blood pressure. INTERSALT Co-operative Research Group. J Hum Hypertens. 1989;3:299–308.
20. Rahmouni, K, Correia, ML, Haynes, WG, et al. Obesity-associated hypertension: new insights into mechanisms. Hypertension. 2005;45:9–14.
21. Després, JP. Cardiovascular disease under the influence of excess visceral fat. Crit Pathw Cardiol. 2007;6:51–59.
22. McGill, HC, Jr., McMahan, CA, Gidding, SS. Preventing heart disease in the 21st century: implications of the Pathobiological Determinants of Atherosclerosis in Youth (PDAY) study. Circulation. 2008;117:1216–1227.
23. Poirier, P, Giles, TD, Bray, GA, et al. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss. Arterioscler Thromb Vasc Biol. 2006;26:968–976.
24. Rockx, MA, Fox, SA, Stitt, LW, et al. Is obesity a predictor of mortality, morbidity and readmission after cardiac surgery? Can J Surg. 2004;47:34–38.
25. Hubert, HB, Feinleib, M, McNamara, PM, et al. Obesity as an independent risk factor for cardiovascular disease: a 26-year follow-up of participants in the Framingham Heart Study. Circulation. 1983;67:968–977.
26. Alpert, MA, Fraley, MA, Birchem, JA, et al. Management of obesity cardiomyopathy. Expert Rev Cardiovasc Ther. 2005;3:225–230.
27. He, J, Ogden, LG, Bazzano, LA, et al. Risk factors for congestive heart failure in US men and women: NHANES I epidemiologic follow-up study. Arch Intern Med. 2001;161:996–1002.
28. Rexrode, KM, Hennekens, CH, Willett, WC, et al. A prospective study of body mass index, weight change, and risk of stroke in women. JAMA. 1997;277:1539–1545.
29. Kurth, T, Gaziano, JM, Berger, K, et al. Body mass index and the risk of stroke in men. Arch Intern Med. 2002;162:2557–2562.
30. Harris, MI, Flegal, KM, Cowie, CC, et al. Prevalence of diabetes, impaired fasting glucose, and impaired glucose tolerance in U.S. adults. The Third National Health and Nutrition Examination Survey, 1988–1994. Diabetes Care. 1998;21:518–524.
31. Colditz, GA, Willett, WC, Stampfer, MJ, et al. Weight as a risk factor for clinical diabetes in women. Am J Epidemiol. 1990;132:501–513.
32. Ford, ES, Williamson, DF, Liu, S. Weight change and diabetes incidence: findings from a national cohort of US adults. Am J Epidemiol. 1997;146:214–222.
33. Yan, E, Durazo, F, Tong, M, et al. Nonalcoholic fatty liver disease: pathogenesis, identification, progression, and management. Nutr Rev. 2007;65:376–384.
34. Day, CP, James, OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845.
35. Shaffer, EA. Bariatric surgery: a promising solution for nonalcoholic steatohepatitis in the very obese. J Clin Gastroenterol. 2006;40:S44–50.
36. Larson-Meyer, DE, Newcomer, BR, Heilbronn, LK, et al. Effect of 6-Month Calorie Restriction and Exercise on Serum and Liver Lipids and Markers of Liver Function. Obesity (Silver Spring). 2008;16:1355–1362.
37. Calle, EE, Rodriguez, C, Walker-Thurmond, K, et al. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–1638.
38. World Cancer Research Fund. Food, Nutrition, Physical Activity, and the Prevention of Cancer: A Global Perspective, ed 2. Washington, DC: American Institute for Cancer Research; 2007.
39. Renehan, AG, Tyson, M, Egger, M, et al. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371:569–578.
40. Calle, EE, Kaaks, R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4:579–591.
41. Chang, RJ. The reproductive phenotype in polycystic ovary syndrome. Nat Clin Pract Endocrinol Metab. 2007;3:688–695.
42. Metwally, M, Li, TC, Ledger, WL. The impact of obesity on female reproductive function. Obes Rev. 2007;8:515–523.
43. Prentice, A, Goldberg, G. Maternal obesity increases congenital malformations. Nutr Rev. 1996;54:146–150.
44. Cnattingius, S, Bergstrom, R, Lipworth, L, et al. Prepregnancy weight and the risk of adverse pregnancy outcomes. N Engl J Med. 1998;338:147–152.
45. Pillar, G, Shehadeh, N. Abdominal fat and sleep apnea: the chicken or the egg? Diabetes Care. 2008;31(Suppl 2)):S303–309.
46. Tasali, E, Mokhlesi, B, Van Cauter, E. Obstructive sleep apnea and type 2 diabetes: interacting epidemics. Chest. 2008;133:496–506.
47. Stampfer, MJ, Maclure, KM, Colditz, GA, et al. Risk of symptomatic gallstones in women with severe obesity. Am J Clin Nutr. 1992;55:652–658.
48. Sugerman, HJ, Brewer, WH, Shiffman, ML, et al. A multicenter, placebo-controlled, randomized, double-blind, prospective trial of prophylactic ursodiol for the prevention of gallstone formation following gastric-bypass-induced rapid weight loss. Am J Surg. 1995;169:91–96.
49. Bliddal, H, Christensen, R. The management of osteoarthritis in the obese patient: practical considerations and guidelines for therapy. Obes Rev. 2006;7:323–331.
50. Cicuttini, FM, Baker, JR, Spector, TD. The association of obesity with osteoarthritis of the hand and knee in women: a twin study. J Rheumatol. 1996;23:1221–1226.
51. Ravussin, E, Bogardus, C. Energy balance and weight regulation: genetics versus environment. Br J Nutr. 2000;83(Suppl 1)):S17–S20.
52. Prentice, AM. Obesity and its potential mechanistic basis. Br Med Bull. 2001;60:51–67.
53. Prentice, A. Are defects in energy expenditure involved in the causation of obesity? Obesity Rev. 2007;8(Suppl 1):89–91.
54. Fox, KR, Hillsdon, M. Physical activity and obesity. Obesity Rev. 2007;8(Suppl 1):115–121.
55. Leibel, RL, Rosenbaum, M, Hirsch, J. Changes in energy expenditure resulting from altered body weight. N EngJ, 332. Med, 1995.
56. Major, GC, Doucet, E, Trayhurn, P, et al. Clinical significance of adaptive thermogenesis. Int J Obes (Lond). 2007;31:204–212.
57. Zurlo, R, Ferraro, R, Fontvieille, AM, et al. Spontaneous physical actvity and obesity: cross-sectional and longitudinal studies in Pima Indians. Am J Physiol. 1992;263:E296–E300.
58. Levine, JA, Eberhardt, NL, Jensen, MD. Role of nonexercise activity thermogenesis in resistance to fat gain in humans. Science. 1999;283:212–214.
59. Levine, JA, Lanningham-Foster, LM, McCrady, SK, et al. Interindividual variation in posture allocation: possible role in human obesity. Science. 2005;307:584–586.
60. Levine, JA. Nonexercise activity thermogenesis: liberating the life-force. J Intern Med. 2007;262:273–287.
61. Blundell, JE, Stubbs, J. Diet composition and the control of food intake in humans. In: Bray GA, Bouchard C, eds. Handbook of Obesity. ed 2. New York: Marcel Dekker Inc; 2004:427–460.
62. Morton, GJ, Cummings, DE, Baskin, DG, et al. Central nervous system control of food intake and body weight. Nature. 2006;443:289–295.
63. Cota, D, Proulx, K, Seeley, RJ. The role of CNS fuel sensing in energy and glucose regulation. Gastroenterology. 2007;132:2158–2168.
64. Schwartz, MW, Woods, SC, Porte, D, Jr., et al. Central nervous system control of food intake. Nature. 2000;404:661–671.
65. Gibbs, J, Young, RC, Smith, GP. Cholecystokinin decreases food intake in rats. J Comp Physiol Psychol. 1973;84:488–495.
66. Reidelberger, RD, O’Rourke, MF. Potent cholecystokinin antagonist L 364718 stimulates food intake in rats. Am J Physiol. 1989;257:R1512–R1518.
67. Batterham, RL, Cohen, MA, Ellis, SM, et al. Inhibition of food intake in obese subjects by peptide YY3–36. N Engl J Med. 2003;349:941–948.
68. Drucker, DJ, Nauck, MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidylpeptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696–1705.
69. DeFronzo, RA, Ratner, RE, Han, J, et al. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care. 2005;28:1092–1100.
70. Murphy, KG, Dhillo, WS, Bloom, SR. Gut peptides in the regulation of food intake and energy homeostasis. Endocr Rev. 2006;27:719–727.
71. Kojima, M, Hosoda, H, Date, Y, et al. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–660.
72. Tschop, M, Smiley, DL, Heiman, ML. Ghrelin induces adiposity in rodents. Nature. 2000;407:908–913.
73. Druce, MR, Neary, NM, Small, CJ, et al. Subcutaneous administration of ghrelin stimulates energy intake in healthy lean human volunteers. Int J Obes (Lond). 2006;30:293–296.
74. Cummings, DE, Weigle, DS, Frayo, RS, et al. Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N Engl J Med. 2002;346:1623–1630.
75. le Roux, CW, Patterson, M, Vincent, RP, et al. Postprandial plasma ghrelin is suppressed proportional to meal calorie content in normal-weight but not obese subjects. J Clin Endocrinol Metab. 2005;90:1068–1071.
76. Gutierrez, JA, Solenberg, PJ, Perkins, DR, et al. Ghrelin octanoylation mediated by an orphan lipid transferase. Proc Natl Acad Sci U S A. 2008;105:6320–6325.
77. Yang, J, Brown, MS, Liang, G, et al. Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell. 2008;132:387–396.
78. Hill, JW, Elmquist, JK, Elias, CF. Hypothalamic pathways linking energy balance and reproduction. Am J Physiol Endocrinol Metab. 2008;294:E827–E832.
79. Roth, JD, Roland, BL, Cole, RL, et al. Leptin responsiveness restored by amylin agonism in diet-induced obesity: evidence from nonclinical and clinical studies. Proc Natl Acad Sci U S A. 2008;105:7257–7262.
80. Dulloo, AG, Seydoux, J, Jacquet, J. Adaptive thermogenesis and uncoupling proteins: a reappraisal of their roles in fat metabolism and energy balance. Physiol Behav. 2004;83:587–602.
81. Harper, ME, Green, K, Brand, MD. The efficiency of cellular energy transduction and its implications for obesity. Annu Rev Nutr. 2008;28:13–33.
82. Nedergaard, J, Bengtsson, T, Cannon, B. Unexpected evidence for active brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab. 2007;293:E444–E452.
83. Eckel, RH. Clinical practice. Nonsurgical management of obesity in adults. N Engl J Med. 2008;358:1941–1950.
84. Wadden, TA, Butryn, ML, Wilson, C. Lifestyle modification for the management of obesity. Gastroenterology. 2007;132:2226–2238.
85. Bray, GA, Greenway, FL. Pharmacological treatment of the overweight patient. Pharmacol Rev. 2007;59:151–184.
86. Tsai, AG, Wadden, TA. Systematic review: an evaluation of major commercial weight loss programs in the United States. Ann Intern Med. 2005;142:56–66.
87. Gilden Tsai, A, Wadden, TA. The evolution of very-low-calorie diets: an update and meta-analysis. Obesity (Silver Spring). 2006;14:1283–1293.
88. Nordmann, AJ, Nordmann, A, Briel, M, et al. Effects of low-carbohydrate vs low-fat diets on weight loss and cardiovascular risk factors: a meta-analysis of randomized controlled trials. Arch Intern Med. 2006;166:285–293.
89. Gardner, CD, Kiazand, A, Alhassan, S, et al. Comparison of the Atkins, Zone, Ornish, and LEARN diets for change in weight and related risk factors among overweight premenopausal women: the A TO Z Weight Loss Study: a randomized trial. JAMA. 2007;297:969–977.
90. Wadden, TA, Butryn, ML, Byrne, KJ. Efficacy of lifestyle modification for long-term weight control. Obes Res. 2004;12(Suppl):151S–162S.
91. Catenacci, VA, Wyatt, HR. The role of physical activity in producing and maintaining weight loss. Nat Clin Pract Endocrinol Metab. 2007;3:518–529.
92. Ohkawara, K, Tanaka, S, Miyachi, M, et al. A dose-response relation between aerobic exercise and visceral fat reduction: systematic review of clinical trials. Int J Obes (Lond). 2007;31:1786–1797.
93. U.S. Dept. of Health & Human Services. U.S. Dept. of Agriculture Dietary Guidelines for Americans. www.healthierus.gov/dietaryguidelines/, 2005.
94. Goldberg, JH, King, AC. Physical activity and weight management across the lifespan. Annu Rev Public Health. 2007;28:145–170.
95. Colman, E. Anorectics on trial: a half century of federal regulation of prescription appetite suppressants. Ann Intern Med. 2005;143:380–385.
96. Khan, MA, Herzog, CA, St Peter, JV, et al. The prevalence of cardiac valvular insufficiency assessed by transthoracic echocardiography in obese patients treated with appetite-suppressant drugs. N Engl J Med. 1998;339:713–718.
97. Sjostrom, L, Rissanen, A, Andersen, T, et al. European Multicentre Orlistat Study Group. Randomised placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. Lancet. 1998;352:167–172.
98. Hanotin, C, Thomas, F, Jones, SP, et al. Efficacy and tolerability of sibutramine in obese patients: a dose-ranging study. Int J Obes Relat Metab Disord. 1998;22:32–38.
99. Lean, ME. Sibutramine—a review of clinical efficacy. Int J Obes Relat Metab Disord. 1997;21(Suppl 1):30–39.
100. James, WP, Astrup, A, Finer, N, et al. Sibutramine Trial of Obesity Reduction and Maintenance (STORM) Study Group. Effect of sibutramine on weight maintenance after weight loss: a randomized trial. Lancet. 2000;356:2119–2125.
101. Vettor, R, Serra, R, Fabris, R, et al. Effect of sibutramine on weight management and metabolic control in type 2 diabetes: a meta-analysis of clinical studies. Diabetes Care. 2005;28:942–949.
102. Kim, SH, Lee, YM, Jee, SH, et al. Effect of sibutramine on weight loss and blood pressure: a meta-analysis of controlled trials. Obes Res. 2003;11:1116–1123.
103. Jordan, J, Scholze, J, Matiba, B, et al. Influence of sibutramine on blood pressure: evidence from placebo-controlled trials. Int J Obes (Lond). 2005;29:509–516.
104. Torp-Pedersen, C, Caterson, I, Coutinho, W, et al. Cardiovascular responses to weight management and sibutramine in high-risk subjects: an analysis from the SCOUT trial. Eur Heart J. 2007;28:2915–2923.
105. Buchwald, H, Avidor, Y, Braunwald, E, et al. Bariatric surgery: a systematic review and meta-analysis. JAMA. 2004;292:1724–1737.
106. Maggard, MA, Shugarman, LR, Suttorp, M, et al. Meta-analysis: surgical treatment of obesity. Ann Intern Med. 2005;142:547–559.
107. Sjöström, L, Narbro, K, Sjöström, CD, et al. Effects of bariatric surgery on mortality in Swedish obese subjects. N Engl J Med. 2007;357:741–752.
108. Elder, KA, Wolfe, BM. Bariatric surgery: a review of procedures and outcomes. Gastroenterology. 2007;132:2253–2271.
109. Belle, SH, Berk, PD, Courcoulas, AP, et al. Safety and efficacy of bariatric surgery: longitudinal assessment of bariatric surgery. Surg Obes Relat Dis. 2007;3:116–126.
110. National Institutes of Health. Gastrointestinal surgery for severe obesity: National Institutes of Health Consensus Development Conference Statement. Am J Clin Nutr. 1992;55(Suppl 2)):615S–619S.
111. McMahon, MM, Sarr, MG, Clark, MM, et al. Clinical management after bariatric surgery: value of a multidisciplinary approach. Mayo Clin Proc. 2006;81(10 Suppl):S34–S45.
112. DeMaria, EJ. Bariatric surgery for morbid obesity. N Engl J Med. 2007;356:2176–2183.
113. Bult, MJ, van Dalen, T, Muller, AF. Surgical treatment of obesity. Eur J Endocrinol. 2008;158:135–145.
114. Service, GJ, Thompson, GB, Service, FJ, et al. Hyperinsulinemic hypoglycemia with nesidioblastosis after gastric-bypass surgery. N Engl J Med. 2005;353:249–254.
115. Schweitzer, DH. Mineral metabolism and bone disease after bariatric surgery and ways to optimize bone health. Obes Surg. 2007;17:1510–1516.
116. Cummings, S, Apovian, CM, Khaodhiar, L. Obesity surgery: evidence for diabetes prevention/management. J Am Diet Assoc. 2008;108(4 Suppl 1):S40–S44.
117. Moo, TA, Rubino, F. Gastrointestinal surgery as treatment for type 2 diabetes. Curr Opin Endocrinol Diabetes Obes. 2008;15:153–158.
118. Cohen, RV, Schiavon, CA, Pinheiro, JS, et al. Duodenal-jejunal bypass for the treatment of type 2 diabetes in patients with body mass index of 22–34 kg/m2: a report of 2 cases. Surg Obes Relat Dis. 2007;3:195–197.
119. Isoldi, KK, Aronne, LJ. The challenge of treating obesity: the endocannabinoid system as a potential target. J Am Diet Assoc. 2008;108:823–831.
120. Addy, C, Li, S, Agrawal, N, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamic properties of taranabant, a novel selective cannabinoid-1 receptor inverse agonist, for the treatment of obesity: results from a double-blind, placebo-controlled, single oral dose study in healthy volunteers. J Clin Pharmacol. 2008;48:418–427.
121. Addy, C, Wright, H, Van Laere, K, et al. The acyclic CB1R inverse agonist taranabant mediates weight loss by increasing energy expenditure and decreasing caloric intake. Cell Metab. 2008;7:68–78.
122. Knowler, WC, Barrett-Connor, E, Fowler, SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403.
123. Maggs, D, Shen, L, Strobel, S, et al. Effect of pramlintide on A1C and body weight in insulin-treated African Americans and Hispanics with type 2 diabetes: a pooled post hoc analysis. Metabolism. 2003;52:1638–1642.
124. Amori, RE, Lau, J, Pittas, AG. Efficacy and safety of incretin therapy in type 2 diabetes: systematic review and meta-analysis. JAMA. 2007;298:194–206.
125. Considine, RV, Sinha, MK, Heiman, ML, et al. Serum immunoreactive leptin concentrations in normal weight and obese humans. New Engl J Med. 1996;334:292.
126. Heymsfield, SB, Greenberg, AS, Fujioka, K, et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled dose-escalation trial. JAMA. 1999;282:1568–1575.
127. Hukshorn, CJ, Saris, WHM, Westerterp-Plantenga, MS, et al. Weekly subcutaneous pegylated recombinant native human leptin (PEG-OB) administration in obese men. J Clin Endocrinol Metab. 2000;85:4003–4009.
128. Loos, RJ, Bouchard, C. FTO: the first gene contributing to common forms of human obesity. Obes Rev. 2008;9:246–250.
129. Keith, SW, Redden, DT, Katzmarzyk, PT, et al. Putative contributors to the secular increase in obesity: exploring the roads less traveled. Int J Obes (Lond). 2006;30:1585–1594.