Normal Hemostasis and Coagulation
After completion of this chapter, the reader will be able to:
1. List the systems that interact to provide hemostasis.
2. Describe the properties of the vascular intima in the initiation and regulation of hemostasis and fibrinolysis.
3. List the hemostatic functions of tissue factor–bearing cells and blood cells, especially platelets, in hemostasis.
4. Describe the relationships among platelet function, von Willebrand factor, and fibrinogen and their impact on hemostasis.
5. Describe the nature, origin, and function of each of the tissue and plasma factors necessary for normal coagulation.
6. Explain the role of vitamin K in the production and function of plasma clotting factors in the prothrombin group.
7. Diagram fibrinogen structure, fibrin formation, fibrin polymerization, and fibrin cross-linking.
8. Distinguish between serine proteases and cofactors in the coagulation pathway.
9. Describe the tissue factor pathway, amplification pathway, and contact activation in coagulation.
10. List the factors in order of reaction in the extrinsic, intrinsic, and common pathways.
11. Describe the in vivo coagulation process and the role of tissue factor–bearing cells and platelets.
12. Show how tissue factor pathway inhibitor, the protein C pathway, and the serine protease inhibitors function to regulate coagulation and prevent thrombosis.
13. Describe the fibrinolytic pathway, its regulators, and its products.
Overview of Hemostasis
Primary Hemostasis
Hemostasis involves the interaction of vasoconstriction, platelet aggregation, and coagulation enzymes to stop bleeding (Table 40-1). Primary hemostasis refers to the role of blood vessels and platelets in the initial formation of a platelet plug in response to a vascular injury. Primary hemostasis mechanisms are activated either by injuries to blood vessels or by the commonplace desquamation of dying or damaged endothelial cells. In primary hemostasis, the blood vessel contracts to seal the wound or reduce the blood flow (vasoconstriction). Procoagulant substances are exposed or released by the damaged endothelium. Platelets are activated, adhere to exposed collagen, secrete the contents of their granules, and aggregate with other platelets to form a platelet plug. Vasoconstriction and platelet plug formation are the initial, rapid, short-lived response to vessel damage, but by themselves they are inadequate to control major bleeding in the long term. Eventually the plug must be reinforced by fibrin. Defects in the primary hemostasis system such as thrombocytopenia, platelet disorders, or von Willebrand disease can cause debilitating, sometimes fatal, chronic hemorrhage.
TABLE 40-1
Primary and Secondary Hemostasis
| Primary Hemostasis | Secondary Hemostasis |
| Activated by desquamation and small injuries to blood vessels | Activated by large injuries to blood vessels and surrounding tissues |
| Involves vascular intima and platelets | Involves platelets and coagulation system |
| Rapid, short-lived response | Delayed, long-term response |
| Procoagulant substances exposed or released by damaged or activated endothelial cells | Tissue factor exposed on cell membranes |
Secondary Hemostasis
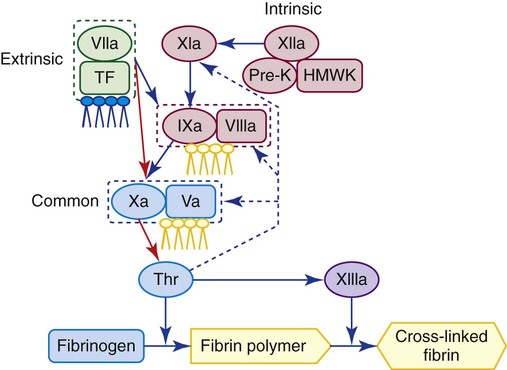
Secondary hemostasis is the activation of a series of plasma proteins in the coagulation system to form a fibrin clot. Blood plasma transports at least 16 glycoproteins, mostly trypsinlike enzymes called serine proteases. These proteins circulate as inactive zymogens that become activated during the process of coagulation and, in turn, form complexes that activate other zymogens to ultimately generate thrombin, an enzyme that converts fibrinogen to a localized fibrin clot. Some coagulation proteins are cofactors that join in forming a complex and function to stabilize and enhance enzymatic action. Still others are control proteins that regulate each step of the process and prevent excessive thrombin generation. Figure 40-1 shows a simplified sequence of activation of the coagulation proteins to form fibrin. Secondary hemostasis is triggered by tissue damage that exposes TF, a protein present in the membrane of subendothelial cells—fibroblasts and smooth muscle cells—but not on endothelial cells. Exposed TF forms a complex with activated factor VII, which activates factors IX and X. Factor IX combines with its cofactor, factor VIII, also to activate factor X. The complex formed by factor X and its cofactor, factor V, converts prothrombin to thrombin. Thrombin is the key enzyme in coagulation. It splits peptides from fibrinogen to produce fibrin, activates factor XIII to cross-link and stabilize the clot, activates platelets, and feeds back to activate factors V, VIII, and XI. Fibrin formation in secondary hemostasis is necessary to control bleeding, especially from large wounds incurred through trauma, surgery, or dental procedures. The coagulation system, similar to other humoral amplification mechanisms, is complex because it translates a diminutive physical or chemical stimulus into a profound lifesaving event.1 The absence of a single plasma procoagulant may doom the individual to lifelong anatomic hemorrhage, chronic inflammation, and transfusion dependence.
Fibrinolysis
The final event of hemostasis is fibrinolysis, the slow digestion and removal of the fibrin clot as healing occurs.2 Plasminogen is bound to fibrin during coagulation and is activated by tissue plasminogen activator (TPA) into the serine protease, plasmin. Plasmin slowly and systematically degrades the fibrin clot into fragments called fibrin degradation products, designated X, Y, D, E, and D-dimer.
Vascular Intima in Hemostasis
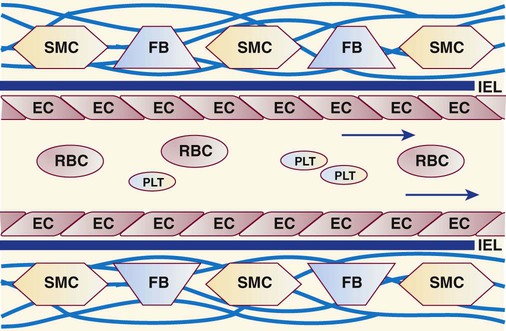
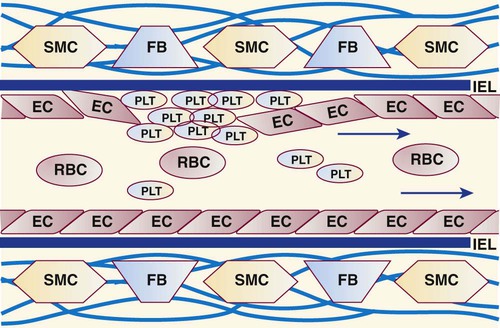
Intimal cells and their environment play a premier role in hemostasis.3 The innermost lining of blood vessels is a monolayer of metabolically active endothelial cells (Box 40-1, Figure 40-2).4 These form a smooth, unbroken surface that promotes the fluid passage of blood and prevents turbulence that otherwise may activate platelets and coagulation enzymes. An elastin-rich internal elastic lamina and its surrounding layer of connective tissues support the endothelial cells. In all blood vessels, fibroblasts occupy the connective tissue layer and produce collagen. Smooth muscle cells in arteries and arterioles, but not in the walls of veins, venules, or capillaries, contract during primary hemostasis.
Anticoagulant Properties of Intact Vascular Intima
Normally, the intact vascular endothelium prevents bleeding and thrombosis by inhibiting platelet and coagulation activation and by promoting fibrinolysis. Intravascular thrombosis is prevented by several mechanisms (Box 40-2). First, endothelial cells are rhomboid and contiguous, providing a smooth inner surface that evens the blood flow and prevents harmful turbulence. Endothelial cells form a physical barrier between blood and the substances in the basement membrane that activate clotting, such as collagen, which promotes platelet activation and adhesion, and TF, which activates coagulation and fibrin formation. Endothelial cells synthesize and secrete a variety of substances that maintain normal blood flow. Prostacyclin, a platelet inhibitor, is synthesized through the eicosanoid pathway (see Chapter 13) and prevents unnecessary or undesirable platelet activation in intact vessels.5 Nitric oxide is synthesized in endothelial cells, vascular smooth muscle cells, neutrophils, and macrophages. Nitric oxide counteracts vasoconstriction and maintains healthy arterioles.6 Another important endothelial cell anticoagulant is tissue factor pathway inhibitor (TFPI), which controls the TF or extrinsic coagulation pathway. Finally, endothelial cells synthesize and secrete onto their cell surfaces two inhibitors of thrombin formation, thrombomodulin and heparan sulfate. Thrombomodulin is a membrane protein that activates the protein C pathway. The protein C pathway downregulates coagulation by digesting activated factors V and VIII, thereby inhibiting fibrin formation. Heparan sulfate is a glycosaminoglycan that retards coagulation by activating antithrombin, a coagulation regulatory protein.7 The pharmaceutical heparin, manufactured from porcine gut tissues, resembles heparan sulfate in its antithrombin activity. Heparin is used extensively as a therapeutic agent to prevent propagation of the thrombi that cause coronary thrombosis, strokes, deep vein thromboses, and pulmonary emboli.8
Procoagulant Properties of Damaged Vascular Intima
Although the intact endothelium has anticoagulant properties, when damaged, the vascular intima promotes coagulation. First, any harmful local stimulus, be it mechanical or chemical, induces vasoconstriction in arteries and arterioles (Table 40-2). Smooth muscle cells contract, the vascular lumen narrows or closes, and blood flow to the injured site is minimized. Although veins and capillaries do not have smooth muscle cells, bleeding into tissues creates some extravascular pressure on the blood vessel as well. Second, the subendothelial connective tissues of arteries and veins are rich in collagen, a flexible, elastic structural protein that binds and activates platelets. Some connective tissue degeneration occurs naturally in aging, which leads to an increased tendency toward bruising. Third, endothelial cells secrete VWF, a 600,000- to 20,000,000-D glycoprotein that is necessary for platelets to adhere to exposed subendothelial collagen in arterioles.9 Fourth, on activation, endothelial cells secrete and coat themselves with P-selectin, an adhesion molecule that promotes platelet and leukocyte binding.10 Endothelial cells also secrete immunoglobulin-like adhesion molecules called intercellular adhesion molecules (ICAMs) and platelet endothelial cell adhesion molecules (PECAMs) that promote leukocyte binding.11 Finally, subendothelial cells (i.e., smooth muscle cells and fibroblasts) support the constitutive surface protein TF.12 Exposed TF activates the coagulation system through factor VII. TF also appears on the surface of endothelial cells and on bloodborne monocytes during inflammation.13
TABLE 40-2
Procoagulant Properties of the Damaged Vascular Intima
| Structure | Procoagulant Property |
| Smooth muscle cells in arterioles and arteries | Induce vasoconstriction |
| Exposed subendothelial collagen | Binds VWF and platelets |
| Damaged or activated endothelial cells | Secrete VWF Secrete adhesion molecules: P-selectin, ICAMs, PECAMs |
| Exposed smooth muscle cells and fibroblasts | Tissue factor exposed on cell membranes |
| Endothelial cells in inflammation | Tissue factor is induced by inflammation |
Fibrinolytic Properties of Vascular Intima
Endothelial cells support fibrinolysis with the secretion of TPA. During thrombus formation, both TPA and plasminogen bind to polymerized fibrin. TPA activates fibrinolysis by converting plasminogen to plasmin, which slowly digests fibrin and restores blood flow. Endothelial cells, as well as other cells, may secrete plasminogen activator inhibitor 1 (PAI-1), a TPA control protein that inhibits fibrinolysis.14 Thrombin bound to thrombomodulin activates thrombin-activatable fibrinolysis inhibitor (TAFI), which increases the tendency for thrombus formation.
Although the significance of the vascular intima in hemostasis is well recognized, there are few valid laboratory methods to assess the integrity of endothelial cells, smooth muscle cells, fibroblasts, and their collagen matrix.15 The diagnosis of blood vessel disorders is often based on clinical symptoms, family history, and laboratory tests that rule out platelet or coagulation disorders.
Platelets
Platelets are produced from the cytoplasm of bone marrow megakaryocytes (see Chapter 13).16 Although platelets are only 2 to 3 µm in diameter on a fixed, stained peripheral blood film, they are complex, metabolically active cells that interact with their environment and initiate and control hemostasis.17
In an injury, platelets adhere, aggregate, and secrete the contents of their granules (Table 40-3).18,19 Adhesion is the property of binding to nonplatelet surfaces such as subendothelial collagen (Figures 40-3 and 40-4). VWF links platelets to collagen in areas of high shear stress such as arteries and arterioles, whereas platelets may bind directly to collagen in damaged veins and capillaries. VWF binds platelets through the glycoprotein (GP) Ib/IX/V receptor. The importance of platelet adhesion is underscored by bleeding disorders such as Bernard-Soulier syndrome in which the GP Ib/IX/V receptor is absent, and von Willebrand disease, in which VWF is missing or defective. Aggregation is the attachment of platelets to each other (Figure 40-5).20 When platelets are activated, a change in the GP IIb/IIIa receptor allows binding of fibrinogen, as well as VWF and fibronectin. Fibrinogen binds to GP IIb/IIIa receptors on adjacent platelets and cross-links them in the presence of Ca2+. Fibrinogen binding is essential for platelet aggregation, as evidenced by bleeding and compromised aggregation in patients with afibrinogenemia or in patients who lack GP IIb/IIIa (Glanzmann thrombasthenia). In in vitro platelet aggregation studies, the most commonly used agonists to induce aggregation are thrombin, arachidonic acid, adenosine diphosphate (ADP), collagen, and epinephrine, which bind to receptors on the platelet membrane. Platelets secrete the contents of their granules during adhesion and aggregation, although most secretion occurs late in the platelet activation process. Platelets secrete procoagulants, such as VWF, factor V, and fibrinogen, as well as control proteins, Ca2+, ADP, and other hemostatic molecules. See Table 40-4 for a summary of the contents of platelet α-granules and dense bodies.
TABLE 40-3
| Function | Characteristics |
| Adhesion: platelets roll and cling to nonplatelet surfaces | Reversible; seals endothelial gaps, some secretion of growth factors, in arterioles von Willebrand factor is necessary for adhesion |
| Aggregation: platelets adhere to each other | Irreversible; platelet plugs form, platelet contents are secreted, requires fibrinogen |
| Secretion: platelets discharge the contents of their granules | Irreversible; occurs during aggregation, platelet contents are secreted, essential to coagulation |
TABLE 40-4
| Platelet α-Granules | Platelet δ-Granules (Dense Bodies) |
| Large molecules β-Thromboglobulin Factor V Factor XI Protein S Fibrinogen Von Willebrand factor Platelet factor 4 (heparin inhibitor) Platelet-derived growth factor |
Small molecules Adenosine diphosphate (activates neighboring platelets) Adenosine triphosphate Calcium Serotonin (vasoconstrictor) |
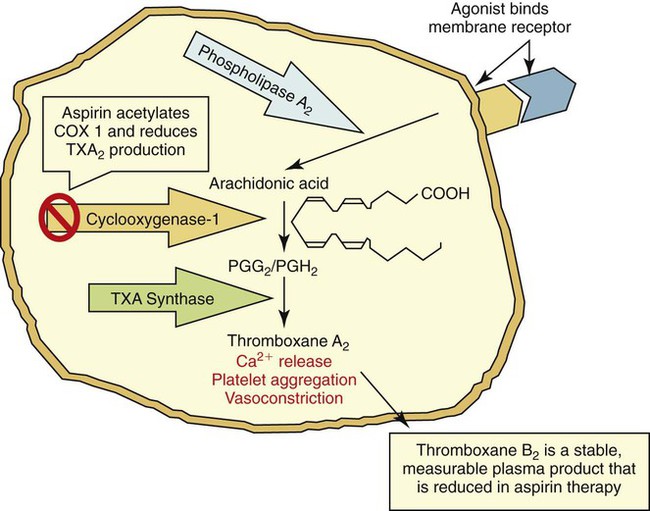
During activation, ADP and Ca2+ activate phospholipase A2, which converts membrane phospholipid to arachidonic acid. Cyclooxygenase converts arachidonic acid into prostaglandin endoperoxides. In the platelet, thromboxane synthetase converts prostaglandins into thromboxane A2, which causes Ca2+ to be released and promotes platelet aggregation and vasoconstriction (Figure 40-6). Aspirin permanently inactivates cyclooxygenase, blocking thromboxane A2 production and causing impairment of platelet function (aspirin effect).
See Chapter 13 for an in-depth description of platelet structure and function. Platelet disorders are considered in detail in Chapters 43 and 44.
Tissue Factor–Bearing Cells, Erythrocytes, Monocytes, and Lymphocytes
TF is an intrinsic transmembrane receptor found on extravascular cells such as fibroblasts, smooth muscle cells, and pericytes but, under normal conditions, not on vessel endothelial cells.21 Vessel injury exposes blood to the subendothelial TF-bearing cells and leads to activation of coagulation through factor VIIa. TF is also expressed in high levels in the brain, lung, placenta, heart, kidney, and testis.
Erythrocytes, monocytes, and lymphocytes also participate in hemostasis. Erythrocytes add bulk and structural integrity to the fibrin clot; there is a tendency to bleed in anemia. In inflammatory conditions, monocytes and lymphocytes, as well as endothelial cells, provide surface-borne TF that triggers coagulation. Leukocytes also have a series of membrane integrins and selectins that bind adhesion molecules and help stimulate the production of inflammatory materials that promote the wound healing process.22
Coagulation System
Nomenclature of Procoagulants
Plasma transports at least 16 procoagulants, also called coagulation factors or clotting factors. Nearly all are glycoproteins synthesized in the liver, although a few are made by monocytes, endothelial cells, and megakaryocytes (Table 40-5, Figure 40-7). Eight are enzymes that circulate in an inactive form called zymogens. Others are cofactors that bind and stabilize their respective enzymes. During clotting, the procoagulants become activated and produce a localized thrombus. Additional plasma glycoproteins are controls that regulate the coagulation process.
TABLE 40-5
Plasma Procoagulants: Function, Molecular Weight, Plasma Half-Life, and Plasma Concentration
| Factor | Customary Name | Function | Molecular Weight (daltons) | Half-Life (hours) | Mean Plasma Concentration |
| I* | Fibrinogen | Thrombin substrate, polymerizes to form fibrin | 340,000 | 100-150 | 200-400 mg/dL |
| II* | Prothrombin | Serine protease | 71,600 | 60 | 10 mg/dL |
| III* | Tissue factor | Cofactor | 44,000 | Insoluble | None |
| IV* | Ionic calcium | Mineral | 40 | NA | 8-10 mg/dL |
| V | Labile factor | Cofactor | 330,000 | 24 | 1 mg/dL |
| VII | Stable factor | Serine protease | 50,000 | 6 | 0.05 mg/dL |
| VIII | Antihemophilic factor | Cofactor | 260,000 | 12 | 0.01 mg/dL |
| VWF | von Willebrand factor | Factor VIII carrier and platelet adhesion | 600,000-20,000,000 | 24 | 1 mg/dL |
| IX | Christmas factor | Serine protease | 57,000 | 24 | 0.3 mg/dL |
| X | Stuart-Prower factor | Serine protease | 58,800 | 48-52 | 1 mg/dL |
| XI | Plasma thromboplastin antecedent (PTA) | Serine protease | 143,000 | 48-84 | 0.5 mg/dL |
| XII | Hageman factor | Serine protease | 84,000 | 48-70 | 3 mg/dL |
| Prekallikrein | Fletcher factor, pre-K | Serine protease | 85,000 | 35 | 35-50 mcg/mL |
| High-molecular-weight kininogen | Fitzgerald factor, HMWK | Cofactor | 120,000 | 156 | 5 mg/dL |
| XIII | Fibrin-stabilizing factor (FSF) | Transglutaminase, transamidase | 320,000 | 150 | 2 mg/dL |
| Platelet factor 3 | Phospholipids, phosphatidylserine, PF3 | Assembly molecule | — | Released by platelets | — |
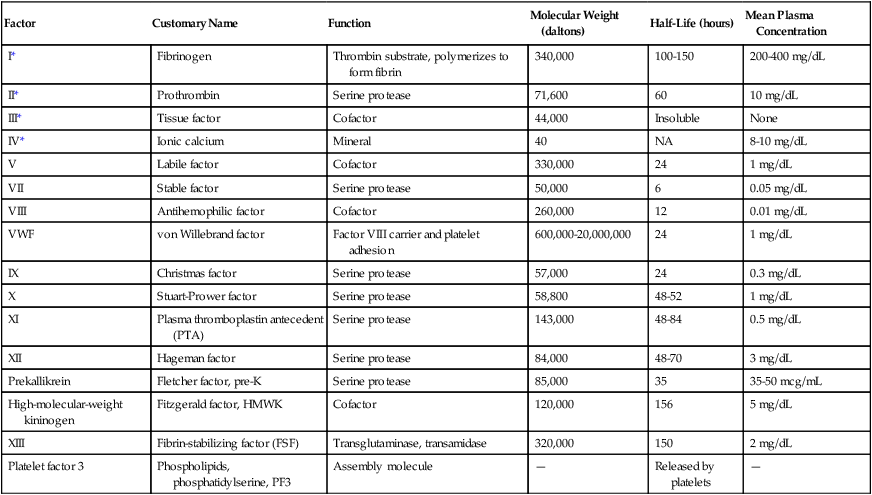
*These factors are customarily identified by name rather than Roman numeral.
From Greenberg DL, Davie EW: The blood coagulation factors: their complementary DNAs, genes, and expression. In Colman RW, Marder VJ, Clowes, AM, et al, editors: Hemostasis and thrombosis: basic principles and clinical practice, ed 5, Philadelphia, 2006, Lippincott Williams & Wilkins, pp 21-58.
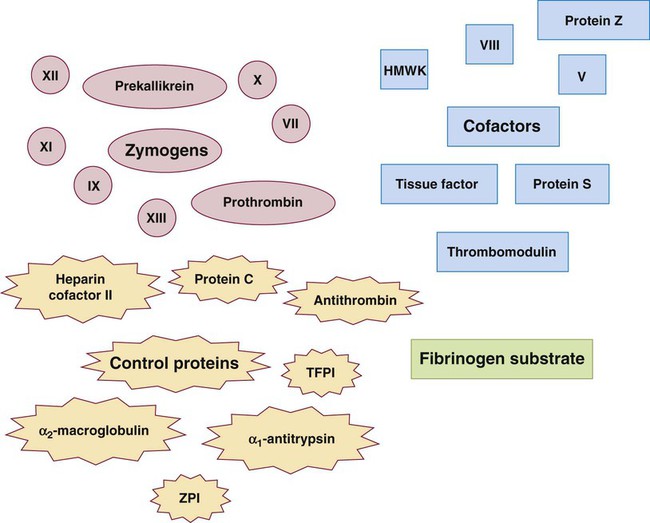
In 1958 the International Committee for the Standardization of the Nomenclature of the Blood Clotting Factors officially named the plasma procoagulants using Roman numerals in the order of their initial description or discovery.23 When a procoagulant becomes activated, a lower-case a appears behind the numeral; for instance, activated factor VII is VIIa. Both zymogens and cofactors become activated in the coagulation process.
We customarily call factor I fibrinogen and factor II prothrombin, although occasionally they are identified by their numerals. The numeral III was given to tissue thromboplastin, a crude mixture of TF and phospholipid. Now that the precise structure of TF has been described, the numeral designation is seldom used. The numeral IV identified the plasma cation calcium (Ca2+); however, calcium is referred to by its name or chemical symbol, not by its numeral. The numeral VI was assigned to a procoagulant that later was determined to be activated factor V; VI was withdrawn from the naming system and never reassigned. Factor VIII, antihemophilic factor, is a cofactor that circulates linked to a large carrier protein, VWF. Prekallikrein, also called Fletcher factor, and high-molecular-weight kininogen (HMWK), also called Fitzgerald factor, have never received Roman numerals because they belong to the kallikrein and kinin systems, and their primary functions lie within these systems. Platelet phospholipids, particularly phosphatidylserine, are required for the coagulation process but were given no Roman numeral; instead they were called collectively platelet factor 3. The molecular weights, plasma concentrations, and plasma half-lives of the procoagulants are given in Table 40-5. These essential pieces of clinical information assist in the interpretation of laboratory tests, monitoring of anticoagulant therapy, and design of effective replacement therapies in deficiency-related hemorrhagic diseases. For example, factor VIII has a short half-life of 12 hours, so replacement therapy for hemophilic individuals who are deficient in factor VIII is administered every 12 hours. For most factors, the level of hemostatic effectiveness is 25% to 30%. This is the minimum level that must be maintained to prevent bleeding in factor-deficient patients. Therapy for a hemophilic patient is designed to maintain the factor level above 30%. Depending on the patient’s condition, a higher level may be desirable, such as in a patient undergoing surgery. The half-life is also important in monitoring anticoagulant therapy, especially warfarin (Coumadin), because factor VII is reduced rapidly (6 hours), whereas reduction of prothrombin takes 4 to 5 days. Therefore, the full effect of warfarin is not realized until approximately 5 days after therapy has begun.
Classification of Procoagulants
Physiologic Function of Procoagulants
The plasma procoagulants may be serine proteases or cofactors.24 Serine proteases are proteolytic enzymes of the trypsin family and include the procoagulants thrombin (factor IIa); factors VIIa, IXa, Xa, XIa, and XIIa; and prekallikrein (pre-K).25 Each member has a reactive seryl amino acid residue in its active site and acts on its substrate by hydrolyzing peptide bonds, digesting the primary backbone and producing small polypeptide fragments. Serine proteases are synthesized as inactive zymogens consisting of a single peptide chain (Table 40-6). Activation occurs when the zymogen is cleaved at one or more specific sites by the action of another protease during the coagulation process. Activation is a localized cell-surface process, limited to the site of injury and controlled by regulatory mechanisms. If zymogen activation is uncontrolled and generalized, the condition is called disseminated intravascular coagulation (DIC), a serious, often life-threatening condition (see Chapter 42).
TABLE 40-6
Plasma Procoagulant Serine Proteases
| Inactive Zymogen | Active Protease | Cofactor | Substrate |
| Prothrombin (II) | Thrombin (IIa) | — | Fibrinogen, V, VIII, XI, XIII |
| VII | VIIa | Tissue factor | IX, X |
| IX | IXa | VIIIa | X |
| X | Xa | Va | Prothrombin |
| XI | XIa | — | IX |
| XII | XIIa | High-molecular-weight kininogen | XI |
| Prekallikrein | Kallikrein | High-molecular-weight kininogen | XI |
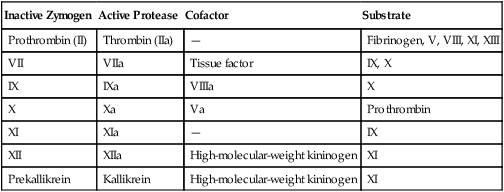
The coagulation cofactors are TF, factor V, factor VIII, and HMWK. Each cofactor binds its particular serine protease. When bound, serine proteases gain stability and increased reactivity (Table 40-7).26
TABLE 40-7
| Inactive Form | Active Form | Binds |
| Tissue factor | Exposed tissue factor | VIIa |
| V | Va | Xa |
| VIII | VIIIa | IXa |
| High-molecular-weight kininogen | Kinin | XIIa, Prekallikrein |
The remaining components of the coagulation pathway are fibrinogen, factor XIII, phospholipids, calcium, and VWF (Box 40-3). Fibrinogen is the ultimate substrate of the coagulation pathway. When hydrolyzed by thrombin, fibrinogen polymerizes to form the primary structural protein of the fibrin clot.27 Factor XIIIa is a transglutaminase that catalyzes the transfer of amino acids among the γ chains of fibrin polymers. This reaction cross-links fibrin polymers to provide physical strength to the fibrin clot. Factor XIIIa reacts with other plasma and cellular structural proteins and is essential to wound healing and tissue integrity. Factor XIII is a heterodimer whose α subunit is produced mostly from megakaryocytes and monocytes. The β subunit is produced in the liver.28
Coagulation occurs on the surface of platelet or endothelial cell membrane phospholipids and not in fluid phase. Any fluid phase reaction is inhibited by the coagulation control proteins. Serine proteases bind to negatively charged phospholipid surfaces, mostly phosphatidylserine, through positively charged calcium ions, so calcium is required for the assembly of coagulation complexes on phospholipid membranes. VWF is a large glycoprotein that participates in platelet adhesion and transports the procoagulant factor VIII. VWF is synthesized in megakaryocytes and endothelial cells.29
Biochemical Nature of Several Procoagulants
Vitamin K–Dependent Prothrombin Group
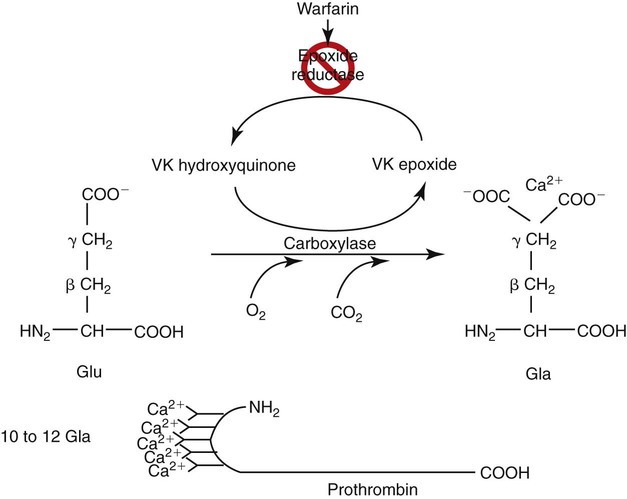
Prothrombin, factors VII, IX, and X, and the regulatory proteins protein C, protein S, and protein Z are vitamin K dependent (Table 40-8). These are named the prothrombin group because of their structural resemblance to prothrombin: all seven proteins have 10 to 12 glutamic acid units near their amino termini. Vitamin K is a quinone found in green leafy vegetables, fish, and liver, and is produced by the intestinal organisms Bacteroides fragilis and Escherichia coli. Vitamin K catalyzes an essential posttranslational modification of the prothrombin group proteins: γ-carboxylation of amino-terminal glutamic acids (Figure 40-8). Glutamic acid is modified to γ-carboxyglutamic acid when a second carboxyl group is added to the γ carbon. With two ionized carboxyl groups, the γ-carboxyglutamic acids gain a net negative charge, which enables them to bind ionic calcium (Ca2+). The bound calcium permits the vitamin K–dependent proteins to bind negatively charged phospholipids, such as phosphatidylserine. Phospholipid binding is essential to coagulation reactions.
TABLE 40-8
Vitamin K–Dependent Coagulation Factors
| Procoagulants | Regulatory Proteins |
| Prothrombin (II) | Protein C |
| VII | Protein S |
| IX | Protein Z |
| X |
Von Willebrand Factor: Factor VIII Complex
VWF is a multimeric glycoprotein composed of multiple subunits of 240,000 D each.30 The subunits are produced by endothelial cells and megakaryocytes, where they combine to form molecules that range from 600,000 to 20,000,000 D. VWF molecules are stored in α-granules in platelets and in storage sites called Weibel-Palade bodies in endothelial cells. The molecules are released from storage into the plasma, where they circulate at a concentration of 7 to 10 mcg/mL.
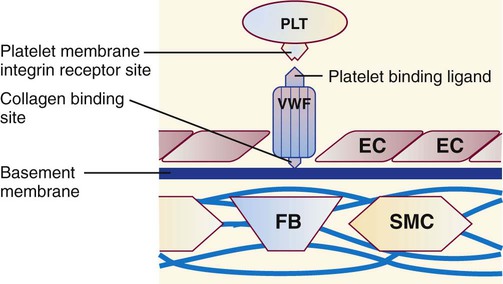
VWF has receptor sites for both platelets and collagen (Figure 40-9) and helps bind platelets to exposed subendothelial collagen during platelet adhesion, especially in arteries and arterioles where the flow of blood is faster. The primary platelet surface receptor for VWF is GP Ib/IX/V.31 Arginine–glycine–aspartic acid (RGD) sequences in VWF also bind a second platelet integrin, GP IIb/IIIa, during platelet aggregation.
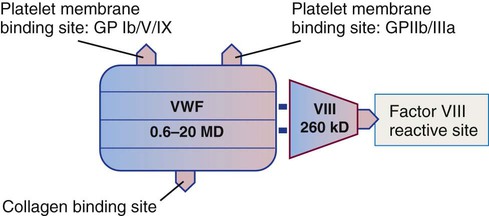
A third site binds to collagen, and a fourth site binds the plasma procoagulant cofactor, factor VIII. Factor VIII is produced by hepatocytes and other tissues. It is one of two plasma procoagulants whose production is sex linked, the other being factor IX. Factor VIII has a molecular mass of 260,000 D and circulates bound to VWF. Free factor VIII is unstable except during coagulation and cannot be detected in plasma. Factor VIII is labile and deteriorates over hours in stored blood despite being bound to VWF. Males with hemophilia A have diminished factor VIII activity but normal VWF levels.32 Because factor VIII depends on VWF for stability, individuals with von Willebrand disease who have diminished VWF also have diminished factor VIII activity levels. Typically, factor VIII levels decrease to hemorrhagic levels (less than 30%) only in severe von Willebrand disease.
Fibrinogen Structure and Fibrin Formation
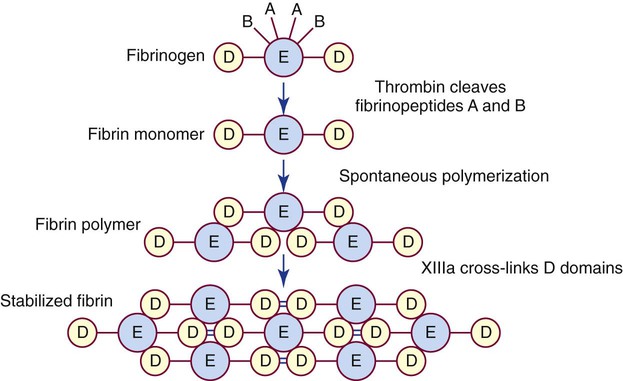
Fibrinogen is the primary substrate of thrombin. It is a 340,000-D glycoprotein synthesized in the liver. The normal plasma concentration of fibrinogen ranges from 200 to 400 mg/dL, the highest concentration of all the plasma procoagulants. Platelet α-granules absorb, transport, and release abundant fibrinogen.33
The fibrinogen molecule is a mirror-image dimer, with each half consisting of three nonidentical polypeptides, designated α, β, and γ, united by disulfide bonds (Figure 40-10). The six amino termini assemble to form a bulky central region called the E domain. The carboxy termini assemble at the two ends of the molecule to form two D domains.34
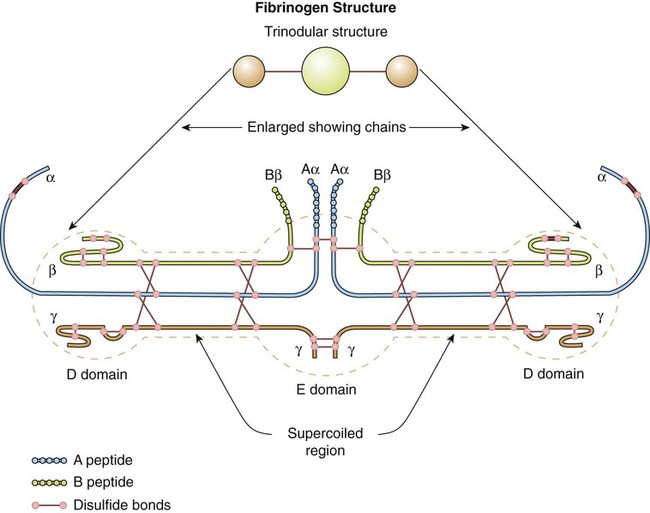
Thrombin cleaves fibrinopeptides A and B from the protruding amino termini of each of the two α and β chains, reducing the overall molecular weight by 10,000 D. The cleaved fibrinogen is called fibrin monomer. The exposed fibrin monomer α and β chain ends (E domain) have an immediate affinity for portions of the D domain of neighboring monomers, spontaneously polymerizing to form fibrin polymer (Figure 40-11).
Coagulation Pathways
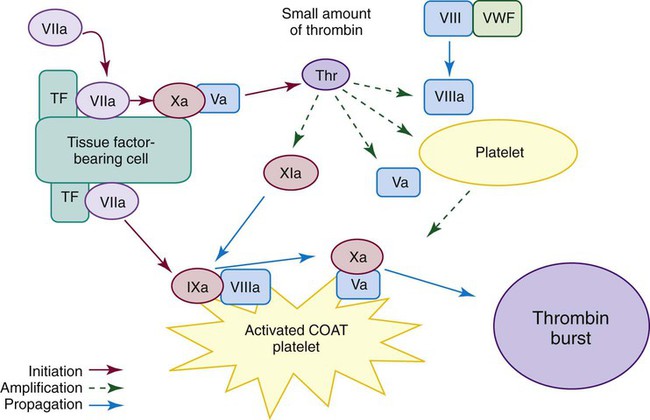
Tissue Factor Pathway
TF, a membrane receptor for factor VIIa, is present on vascular cells that are not normally in contact with blood, such as fibroblasts. Coagulation is triggered by the exposure of TF to plasma proteins on injury.35 In inflammatory conditions, leukocytes and other cells can also express TF and initiate coagulation. Factor VIIa binds to TF in the presence of phospholipid and calcium and triggers the activation of both factor IX and factor X (Figures 40-1 and 40-12). Factor IXa binds factor VIIIa on platelet phospholipid surfaces; this complex also activates factor X.
Factor Xa, activated by both TF : VIIa and IXa : VIIIa, binds factor Va on phospholipid surfaces. This Xa : Va complex activates prothrombin in a multistep hydrolytic process that releases a peptide fragment from prothrombin called prothrombin 1+2. Activated prothrombin is termed thrombin. Thrombin cleaves fibrinopeptides A and B from plasma fibrinogen; this produces fibrin monomer, which then polymerizes. Thrombin also activates factor XIII, which cross-links and stabilizes fibrin (see Figure 40-11).
The tissue factor pathway is characterized by three multimolecular complex formations. The first complex is composed of TF, factor VIIa, phospholipid, and Ca2+ on the TF-bearing cell; this complex activates IX and X. The second is composed of factor IXa, factor VIIIa, phospholipid, and Ca2+ on platelets, sometimes called tenase; this complex also activates factor X. The third complex is composed of factor Xa, factor Va, phospholipid, and Ca2+ and is often called prothrombinase; this complex converts prothrombin to thrombin (Table 40-9).
TABLE 40-9
| Components | Function | |
| First complex | VIIa, tissue factor, phospholipid, and Ca2+ | Cleaves IX and X |
| Second complex | IXa, VIIIa, phospholipid, and Ca2+ | Cleaves X, called tenase |
| Third complex | Xa, Va, phospholipid, and Ca2+ | Cleaves prothrombin, called prothrombinase |
Thrombin
The primary function of thrombin is to cleave fibrinopeptides A and B from the α and β chains of the fibrinogen molecule, triggering fibrin polymerization (see Figure 40-11). In addition, thrombin amplifies the coagulation mechanism by activating cofactors V and VIII and factor XI. Thrombin also activates factor XIII, the fibrin-stabilizing factor. Factor XIIIa forms covalent bonds between the D domains of the fibrin polymer to cross-link and stabilize the fibrin clot. Thrombin also initiates aggregation of platelets. Thrombin bound to thrombomodulin activates the protein C pathway to suppress coagulation, and it activates TAFI to suppress fibrinolysis. Thrombin therefore plays a role in coagulation (fibrin), in platelet activation, in coagulation control (protein C), and in fibrinolysis (TAFI). Because of its multiple autocatalytic functions, thrombin is considered the chief protease of the coagulation pathway.
Extrinsic, Intrinsic, and Common Pathways
Before 1992, two alternative coagulation pathways were described, both of which activated factor X in a common pathway leading to thrombin generation. Most coagulation experts identified the activation of factor XII as the primary step in coagulation because this factor could be found in blood, whereas TF could not. Consequently, the reaction system that begins with factor XII and culminates in fibrin polymerization has been called the intrinsic pathway. The coagulation factors of the intrinsic pathway, in order of reaction, are XII, pre-K, HMWK, XI, IX, VIII, X, V, prothrombin, and fibrinogen (see Figure 40-1). The laboratory test that screens for these factors is the activated partial thromboplastin time (APTT or PTT). We now know that the contact factors XII, pre-K, and HMWK do not play a significant role in in vivo coagulation, although their deficiencies cause abnormal results in in vitro laboratory tests of the intrinsic pathway, such as the PTT.
Cell-Based (In vivo) Coagulation
A spectacular combination of cellular and biochemical events function in harmony to keep blood liquid within the veins and arteries, prevent blood loss from injuries by the formation of thrombi, and reestablish blood flow during the healing process.37 As noted earlier, the series of cascading proteolytic reactions traditionally known as the extrinsic and intrinsic coagulation pathways do not fully describe how coagulation occurs in vivo. These pathways are not distinct independent alternative mechanisms for generating thrombin, but are actually very interdependent. For example, a deficiency of factor VII in the extrinsic pathway can cause significant bleeding, even when the intrinsic pathway is normal. Similarly, deficiencies of some intrinsic pathway proteins, notably factors VIII and IX, may cause severe bleeding problems, regardless of the presence of a normal extrinsic pathway.38
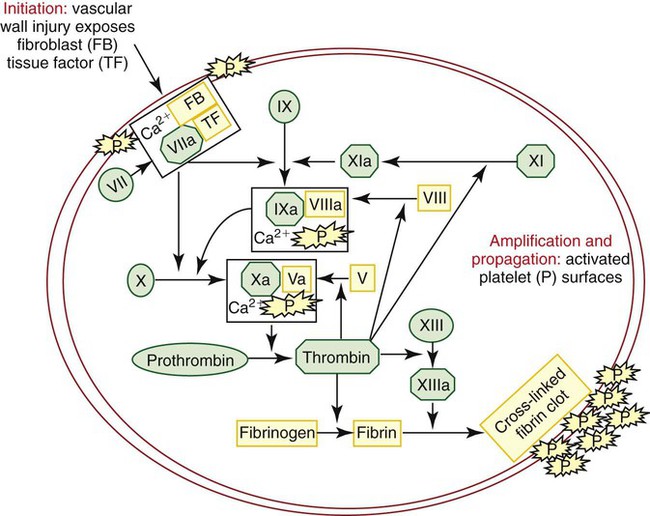
Current thinking about the physiologic process of coagulation recognizes the contribution of specific cells in regulating hemostasis. The cell-based model of hemostasis (Figure 40-13) describes the interaction of two cell types: platelets, within the blood circulation, and extravascular cells that bear TF.39 The cell-based model of coagulation is often described as occurring in three separate but overlapping phases: initiation, amplification, and propagation.
Initiation
Continuous low-level activation of the TF pathway occurs on TF-bearing cells outside the circulation, even in the absence of injury. Fibroblasts and other subendothelial cells carry TF, a transmembrane protein that is a cofactor to factor VII. Smaller plasma coagulation proteins, such as factors VII, IX, and X, are present in extravascular fluid as well as in the circulation. About 1% or less of VII is present normally in the activated form.20 VIIa binds to TF, and the TF : VIIa complex activates small amounts of both factor IX and factor X. Factor Xa complexes with Va to form prothrombinase, Xa : Va, and generates a limited amount of thrombin. Factor Va comes from the activation of factor V by Xa, or by platelets if there has been an injury, or by noncoagulation proteases.40 Xa : Va bound to the cell is protected from inactivation by control proteins. However, if Xa : Va dissociates from the cell, it is rapidly inactivated by the protease inhibitors TFPI, antithrombin, and protein Z–dependent protease inhibitor (ZPI). Factor IXa, on the other hand, can move to other cells through the extravascular fluid, because it is inhibited more slowly by antithrombin and is not inhibited by TFPI or ZPI. Because the larger components of hemostasis—platelets and VIII : VWF—are not present in the extravascular fluid, factor IX activation does not proceed further until there is an injury. The low level of thrombin generated by the cell-bound Xa : Va in the initiation phase does not produce a fibrin clot.
Amplification
When an injury and bleeding occur, platelets and VIII : VWF spill into the extravascular space. Coagulation is primed and ready to go. Platelets are activated at the site of injury by both the low-level thrombin generated in the initiation phase and by adherence to exposed collagen. They are sometimes referred to as COAT platelets, platelets partially activated by collagen and thrombin.40 These partially activated COAT platelets have a higher level of procoagulant activity than platelets exposed to collagen alone. Thrombin, in addition to activating platelets, activates factor V released from platelet α-granules, activates factor VIII and dissociates it from VWF, and activates factor XI.
Propagation
In the cell-based model, it may be helpful to think of the extrinsic or TF pathway as occurring on the TF-bearing cell and the intrinsic pathway (minus factor XII, HMWK, and pre-K) as occurring on the platelet surface. However, these are not totally separate pathways. TF : VIIa activates both factor IX and factor X. Factor XI is activated by the thrombin produced in the initiation phase, bypassing factor XII and the contact factors of the intrinsic pathway. Both pathways are necessary for normal coagulation, and deficiencies of VII, IX, VIII, X, V, or II can cause serious bleeding problems. Factor XI functions to supplement or boost factor IX activation, so deficiencies of factor XI usually produce less severe clinical manifestations than deficiencies of the other factors.36 The contact factors XII, HMWK, and pre-K are not involved in physiologic hemostasis, so deficiencies of these factors do not cause bleeding, although they do prolong the PTT.
Coagulation Regulatory Mechanisms
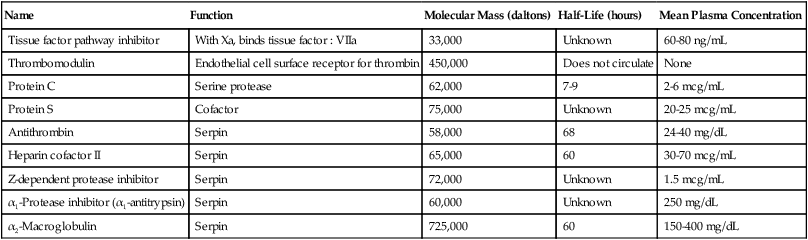
Serine proteases and cofactors in the coagulation system are regulated by inhibitors, cofactors, and feedback loops to maintain a complex and delicate balance between thrombosis and abnormal bleeding. The principal regulators are TFPI, antithrombin, and the protein C pathway. These function as natural anticoagulants, because they inhibit the action of procoagulants and prevent excessive clot formation or thrombosis. Acquired or inherited deficiencies of these proteins may be associated with increased incidence of venous thromboembolic disease. Figure 40-14 illustrates coagulation mechanism regulatory points. Characteristics of these and other coagulation regulatory proteins are summarized in Table 40-10.
TABLE 40-10
Coagulation Regulatory Proteins
| Name | Function | Molecular Mass (daltons) | Half-Life (hours) | Mean Plasma Concentration |
| Tissue factor pathway inhibitor | With Xa, binds tissue factor : VIIa | 33,000 | Unknown | 60-80 ng/mL |
| Thrombomodulin | Endothelial cell surface receptor for thrombin | 450,000 | Does not circulate | None |
| Protein C | Serine protease | 62,000 | 7-9 | 2-6 mcg/mL |
| Protein S | Cofactor | 75,000 | Unknown | 20-25 mcg/mL |
| Antithrombin | Serpin | 58,000 | 68 | 24-40 mg/dL |
| Heparin cofactor II | Serpin | 65,000 | 60 | 30-70 mcg/mL |
| Z-dependent protease inhibitor | Serpin | 72,000 | Unknown | 1.5 mcg/mL |
| α1-Protease inhibitor (α1-antitrypsin) | Serpin | 60,000 | Unknown | 250 mg/dL |
| α2-Macroglobulin | Serpin | 725,000 | 60 | 150-400 mg/dL |
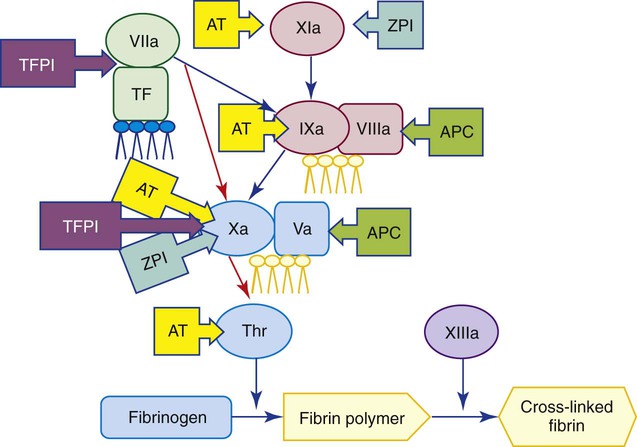
Tissue Factor Pathway Inhibitor
Factor VIIa and TF combine to activate factors IX and X in the tissue factor pathway. Factor Xa reacts with TF : VIIa and binds TFPI, an important coagulation regulatory protein (Figure 40-15). TFPI is synthesized by endothelial cells.41 About 80% is bound to the vessel wall and 20% circulates in the plasma. Most of the plasma TFPI is truncated and bound to low-density lipoproteins. The full-length free form, comprising only 2% of the total TFPI, is an effective inhibitor of the tissue factor pathway. TFPI inhibits coagulation in a two-step process by inactivating Xa and then binding to TF : VIIa to prevent it from activating additional Xa.42,43 TFPI provides feedback inhibition, because it is not functional until X is activated. Protein S, the cofactor of activated protein C (APC), is also a cofactor of TFPI and stimulates Xa inhibition by TFPI.44 Because of the inhibitory action of TFPI, the TF : VIIa : Xa reaction is short-lived. Coagulation pathway amplification occurs primarily through IXa,45 because Xa activation by IXa : VIIIa is negligibly affected by TFPI.
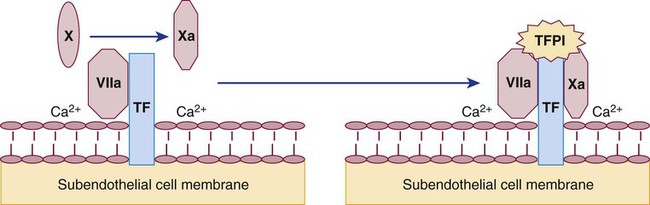
Protein C Regulatory System
During thrombosis, thrombin propagates the clot as it cleaves fibrinogen and activates factors V, VIII, XI, and XIII. In intact normal vessels, where coagulation would be inexpedient, thrombin binds the endothelial cell membrane protein thrombomodulin and triggers an essential coagulation regulatory system called the protein C system.46 The thrombin-thrombomodulin complex activates the zymogen protein C to a serine protease (Figure 40-16). APC binds its cofactor, free plasma protein S. The stabilized APC–protein S complex hydrolyzes and inactivates factors Va and VIIIa, slowing or blocking coagulation.
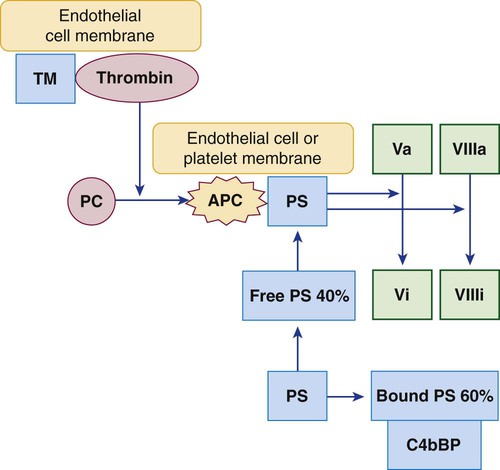
Protein S, the cofactor that binds and stabilizes APC, is synthesized in the liver and circulates in the plasma in two forms: free and covalently bound to the complement control protein C4b-binding protein (C4bBP). Bound protein S cannot participate in the protein C anticoagulant pathway; only free plasma protein S can serve as the APC cofactor. Protein S–C4bBP binding is of particular interest in inflammatory conditions, because C4bBP is an acute phase reactant. When the plasma C4bBP level increases, additional protein S is bound, and free protein S levels become proportionally decreased, which increases the risk of thrombosis. Chronic acquired or inherited protein C or protein S deficiency, or mutations of protein C, protein S, or factor V, compromise downregulation of Va and VIIIa and may be associated with recurrent venous thromboembolic disease, which underscores the importance of the protein C regulatory system (see Chapter 42).
Serine Protease Inhibitors (Serpins)
Antithrombin was the first of the coagulation regulatory proteins to be identified and the first to be assayed routinely in the clinical hemostasis laboratory.47 Other members of the serpin family are heparin cofactor II, ZPI, protein C inhibitor, α1-protease inhibitor (α1-antitrypsin), α2-macroglobulin, α2-antiplasmin, and PAI-1.48
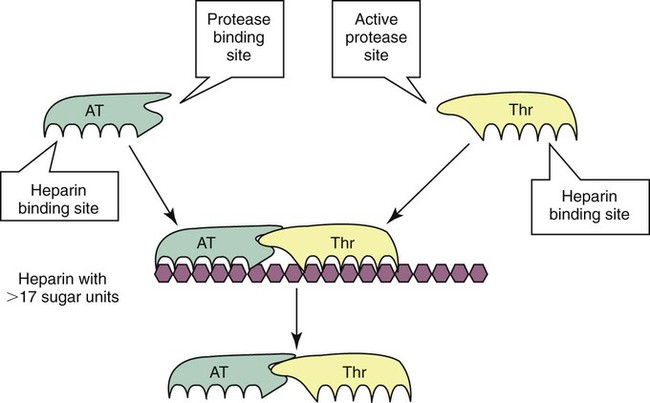
Antithrombin is a serine protease inhibitor (serpin) that binds and neutralizes all serine proteases except factor VIIa, including thrombin (factor IIa) and factors IXa, Xa, XIa, XIIa, kallikrein, and plasmin. Heparin cofactor II is a serpin that inactivates primarily thrombin. Antithrombin and heparin cofactor II both require heparin for effective anticoagulant activity. In vivo, heparin is available from endothelial associated mast cell granules or as endothelial cell heparan sulfate, a natural glycosaminoglycan that activates antithrombin, although not to the same intensity as unfractionated heparin. Therapeutically, unfractionated heparin, low-molecular-weight heparin, and heparin pentasaccharide are administered as anticoagulants. Unfractionated heparin is a heterogeneous glycosaminoglycan composed of 15 to 30 saccharide units. Unfractionated heparin increases the ability of antithrombin to neutralize thrombin and factors IXa, Xa, XIa, and XIIa by 1000-fold. Any heparin molecule of 17 or more saccharide units simultaneously binds antithrombin and thrombin. The antithrombin covalently binds and inactivates a thrombin molecule, forming an inactive thrombin-antithrombin complex, which is released from the heparin molecule. Laboratory measurement of thrombin-antithrombin is used as an indicator for thrombosis. Unfractionated heparin’s catalytic effect is to approximate thrombin to antithrombin (i.e., to bring the two molecules into proximity with each other) and to induce allostery, or change in steric conformation, of the antithrombin molecule (Figure 40-17). Heparin fractions of less than 17 saccharide units, such as therapeutic low-molecular-weight heparin, are able to bind antithrombin or thrombin separately but cannot bring them into the necessary spatial configuration to bind each other. Nevertheless, the allosteric changes induced in the antithrombin still make it capable of binding and inactivating factor Xa and other serine proteases. To summarize, with unfractionated heparin therapy, antithrombin preferentially inactivates thrombin, and with low-molecular-weight heparin therapy, antithrombin preferentially inactivates factor Xa.
ZPI, in the presence of its cofactor, protein Z, is a potent inhibitor of factor Xa.49–51 ZPI covalently binds protein Z and Xa in a complex with Ca2+ and phospholipid. Protein Z is a vitamin K–dependent plasma glycoprotein that is synthesized in the liver. Although protein Z has a structure similar to that of other vitamin K–dependent proteins VII, IX, X, and protein C, it lacks an activation site and, like protein S, is nonproteolytic. Protein Z increases the ability of ZPI to inhibit Xa 1000-fold. ZPI also inhibits factor XIa, in a separate reaction that does not require protein Z, phospholipid, and Ca2+. The inhibition of XIa is accelerated twofold by the presence of heparin.
Protein C inhibitor is a nonspecific, heparin-binding serpin that inhibits a variety of proteases, including APC, thrombin, thrombin-thrombomodulin complex, TPA, and urokinase.48 It is found not only in plasma but in many other body fluids and organs. Depending on its target, it can function as an anticoagulant (inhibits thrombin), as a procoagulant (inhibits thrombin-thrombomodulin and APC), or as a fibrinolytic inhibitor (inhibits TPA).
The serpins α1-protease inhibitor and α2-macroglobulin are able to inhibit serine proteases reversibly. See Table 40-11 and the section on fibrinolysis for further information on α2-antiplasmin and PAI-1.
TABLE 40-11
Proteins of the Fibrinolysis Pathway
| Name | Function | Molecular Mass (daltons) | Half-Life | Mean Plasma Concentration |
| Plasminogen | Plasma serine protease, plasmin digests fibrin/fibrinogen | 90,000 | 24-26 hr | 15-21 mg/dL |
| Tissue plasminogen activator | Serine protease secreted by activated endothelium, activates plasminogen | 68,000 | Unknown | 4-7 mcg/dL |
| Urokinase | Serine protease secreted by kidney, activates plasminogen | 54,000 | Unknown | — |
| Plasminogen activator inhibitor 1 | Secreted by endothelium, inhibits tissue plasminogen activator | 52,000 | 1 hr | 14-28 mg/dL |
| α2-Antiplasmin | Inhibits plasmin | 51,000 | Unknown | 7 mg/dL |
| Thrombin-activatable fibrinolysis inhibitor | Suppresses fibrinolysis | 55,000 | 8-10 min | 5 mcg/mL |

Fibrinolysis
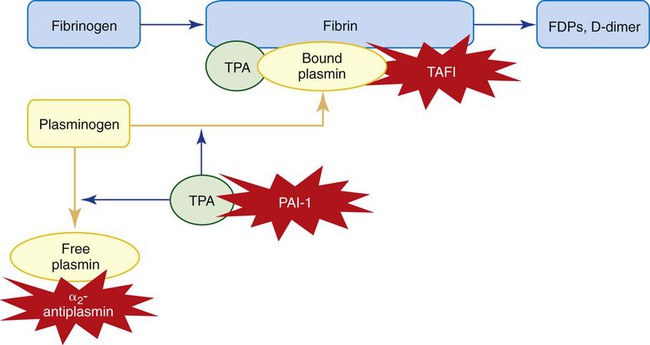
Fibrinolysis, the final stage of coagulation (Figure 40-18), begins a few hours after fibrin polymerization and cross-linking. Plasminogen, plasmin, TPA, and PAI-1, discussed later, are incorporated into the fibrin clot by binding to lysine through “kringle” loops. Fibrinolysis is the systematic, accelerating hydrolysis of fibrin by bound plasmin, which cleaves peptide bonds at arginine and lysine moieties in regions connecting fibrin’s D and E domains (Figure 40-19).52 TPA and urokinase activate fibrin-bound plasminogen several hours after thrombus formation, degrading fibrin and restoring normal blood flow during vascular repair. Again, there is a delicate balance between activators and inhibitors. Excessive fibrinolysis can lead to bleeding due to premature clot lysis before wound healing is established, whereas inadequate fibrinolysis can lead to clot extension and thrombosis.
Plasminogen and Plasmin
Plasminogen is a 92,000-D plasma zymogen produced by the liver (see Table 40-11).53,54 It is a single-chain protein possessing five glycosylated loops termed kringles. Kringles enable plasminogen to bind fibrin lysine residues during polymerization. This binding is essential to fibrinolysis. Plasminogen likewise binds plasma α2-antiplasmin, the major fibrinolysis control protein. Fibrin-bound plasminogen becomes converted into a two-chain active plasmin molecule when cleaved between arginine at position 561 and valine at position 562 by neighboring bound TPA or urokinase. Plasmin is a serine protease that systematically digests fibrin polymer by hydrolysis of arginine-related and lysine-related peptide bonds. As fibrin becomes digested, the exposed carboxy-terminal lysine residues bind additional plasmin, which incrementally accelerates clot digestion. Plasmin is capable of digesting fluid-phase fibrinogen and factors V and VIII; however, localization to fibrin through lysine binding prevents systemic activity. Plasma α2-antiplasmin rapidly binds and inactivates free plasmin. Several aminocarboxalic acids have an affinity for plasminogen’s kringles, which accounts for the antifibrinolytic properties of therapeutic tranexamic acid and ε-aminocaproic acid.
Plasminogen Activation and Control
Tissue Plasminogen Activator
Thrombin-Activatable Fibrinolysis Inhibitor
TAFI is a plasma procarboxypeptidase that becomes activated by the thrombin-thrombomodulin complex. This is the same complex that activates the protein C pathway; however, the two functions are independent. Activated TAFI inhibits fibrinolysis by cleaving exposed carboxy-terminal lysine residues from partially degraded fibrin, preventing the binding of TPA and plasminogen and blocking the formation of plasmin. In coagulation factor–deficient states, decreased thrombin production may reduce the activation of TAFI, resulting in increased fibrinolysis that contributes to bleeding. Conversely, in thrombotic disorders, increased thrombin generation may increase the activation of TAFI. The resulting decreased fibrinolysis may contribute further to thrombosis. TAFI also may play a role in regulating inflammation and wound healing.55
Fibrin Degradation Products and D-Dimer
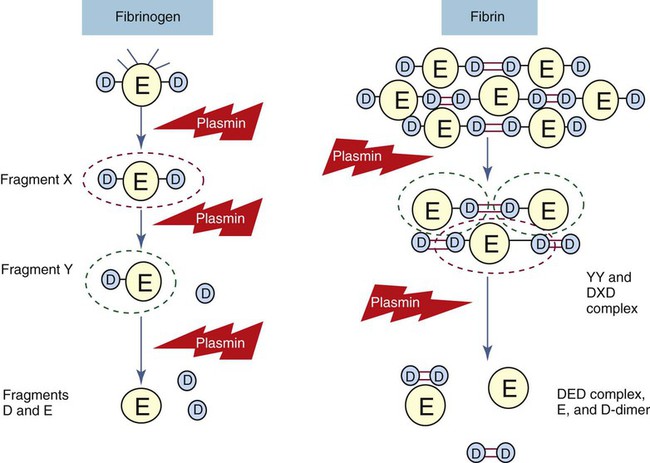
Fibrinolysis produces a series of identifiable fibrin fragments, X, Y, D, E, and D-D (see Figure 40-19).56 Several of these fragments inhibit hemostasis and contribute to hemorrhage by preventing platelet activation and by hindering fibrin polymerization. Fragment X is described as the central E domain with the two D domains (D-E-D), minus some peptides cleaved by plasmin. Fragment Y is the E domain after cleavage of one D domain (D-E). Eventually these fragments are further digested to individual D and E domains. The D-D fragment, called D-dimer, is two D domains from separate fibrin molecules cross-linked by the action of factor XIIIa. Fragments X, Y, D, and E are produced by digestion of either fibrin or fibrinogen by plasmin, but D-dimer is a product of digestion of fibrin. The various fragments may be detected by quantitative or semiquantitative immunoassay to reveal fibrinolytic activity. D-dimer is separately detectable by a quantitative monoclonal antibody immunoassay. The D-dimer immunoassay is used to identify chronic and acute DIC and to rule out venous thromboembolism in suspected cases of deep venous thrombosis or pulmonary embolism.
Summary
• The vascular intima, platelets, tissue factor–bearing cells, and coagulation and fibrinolytic proteins interact to maintain hemostasis.
• Intact vascular intima prevents coagulation through synthesis of prostacyclin, nitric oxide, TFPI, thrombomodulin, and heparan sulfate.
• Damaged intima promotes coagulation by vasoconstriction, exposure of TF and collagen, and secretion of VWF and other adhesion molecules.
• Platelets function in primary and secondary hemostasis through adhesion, aggregation, and secretion of granular contents.
• Platelets adhere to collagen through VWF and use fibrinogen to aggregate.
• Most coagulation factors are produced in the liver.
• The plasma factors of the prothrombin group (prothrombin; factors VII, IX, and X; protein C; protein S; and protein Z) require vitamin K in their production.
• Plasma coagulation factors include trypsinlike enzymes called serine proteases and cofactors that stabilize the proteases. Factor XIIIa is a transamidase.
• The extrinsic pathway of coagulation consists of the membrane receptor TF and coagulation factors VII, X, V, II, and I. The PT is a screening test for these factors.
• The intrinsic pathway factors are XII, pre-K, HMWK, XI, IX, VIII, X, V, II, and I. The PTT is a screening test for these factors.
• Activation of coagulation pathways produces thrombin, which converts fibrinogen to a fibrin clot. Thrombin also activates platelets and factors V, VIII, XI, and XIII, and binds to thrombomodulin to activate protein C and TAFI.
• Fibrinogen is cleaved by thrombin to form first fibrin monomer, then fibrin polymer, and finally, when acted on by factor XIIIa, cross-linked fibrin.
• In vivo, coagulation is initiated on TF-bearing cells. TF : VIIa activates factors IX and X, generating enough thrombin to activate platelets and factors V, VIII, and XI. Coagulation proceeds on activated platelet phospholipid membranes with the formation of IXa : VIIIa and Xa : Va complexes, which produces a burst of thrombin that cleaves fibrinogen to fibrin.
• The coagulation pathway is regulated by TFPI, APC, and the serpins, including antithrombin and ZPI. These control proteins prevent thrombosis and confine clotting to the site of injury.
• The fibrinolytic pathway digests the thrombus. Plasminogen is converted to plasmin by TPA. Plasmin degrades fibrin to fragments X, Y, D, and E, and D-dimer. Control proteins are PAI-1, α2-antiplasmin, and TAFI.
Review Questions
1. What intimal cell synthesizes and stores VWF?
2. What subendothelial structural protein triggers coagulation through activation of factor VII?
3. What coagulation plasma protein should be assayed when platelets fail to aggregate properly?
4. What role does vitamin K play for the prothrombin group factors?
a. Provides a surface on which the proteolytic reactions of the factors occur
b. Protects them from inappropriate activation by compounds such as thrombin
c. Accelerates the binding of the serine proteases and their cofactors
5. What is the source of fibrinopeptides A and B?
a. Plasmin proteolysis of fibrin
b. Thrombin proteolysis of fibrinogen
6. What serine protease forms a complex with factor VIIIa, and what is the substrate of this complex?
7. What protein secreted by endothelial cells activates fibrinolysis?
8. What two regulatory proteins form a complex that digests activated factors V and VIII?