[level-membership-for-critical-care-medicine-category]
35 Nontraumatic Intracerebral and Subarachnoid Hemorrhage
 Intracerebral Hemorrhage
Intracerebral Hemorrhage
Spontaneous (nontraumatic) intracerebral hemorrhage (ICH) accounts for approximately 10% of all strokes in North America and about 20% to 30% in East Asia. It is associated with greater mortality and more severe neurologic deficits than any other stroke subtype.1–3 Nearly half of all patients die within the first 30 days; survivors often have significant residual disability.4
Pathophysiology
The pathophysiologic mechanisms of brain injury due to ICH are complex. The primary injury is one of local tissue destruction as rupture of a cerebral blood vessel introduces a sudden stream of blood into the brain parenchyma. In over one third of patients, continued bleeding or rebleeding results in hematoma enlargement and further mechanical injury within the first few hours after onset.5 The mass of the hematoma produces tissue shifts within the intracranial cavity.
Experimental models of ICH consistently suggest that ischemia is an important part of the pathophysiology of ICH.6,7 In clinical studies, peri-clot and ipsilateral hemispheric hypoperfusion have been demonstrated,8–10 but the hypoperfusion does not appear to represent ischemia.11,12 Positron emission tomography (PET) studies in humans performed 5 to 22 hours after symptom onset showed that perihematomal cerebral metabolism was reduced to a greater degree than cerebral blood flow (CBF), suggesting that the hypoperfusion reflects reduced metabolic demand of the damaged tissue surrounding the hematoma rather than ongoing ischemia.12 Magnetic resonance imaging (MRI) studies within 6 hours after symptom onset demonstrate hypoperfusion without restricted diffusion, findings that are inconsistent with ischemia.13
Cerebral edema has been demonstrated to occur within hours of experimental ICH, variably thought to result from the toxic effects of blood-derived enzymes, from increased osmotic pressure exerted by clot-derived serum proteins, or from ischemia.14–16 The presence, time course, and importance of edema formation in humans are debated, however. Signal changes on radiographic studies after ICH indicate increased water content in the area surrounding the clot, but the clinical and pathophysiologic significance of this is not known. Edema does not appear to contribute to early increases in mass effect17 and is not associated with worsened functional outcome or increased mortality.18,19
Causes and Risk Factors
The leading risk factor for ICH, occurring in over half of all cases, is chronic hypertension.20 Long-term adequate treatment of chronic hypertension significantly reduces this risk.21 Increasing age is another risk factor, with a doubling of the rate of hemorrhage with each decade of life until age 80, when the incidence plateaus at nearly 25 times that of the previous decade.22 In the United States, ICH is 2 to 3 times more common in African Americans and Hispanics than in Caucasians (incidence rates of 32, 35, and 10 to 15 per 100,000 population, respectively).23,24 The incidence in Asian countries is considerably greater (61 per 100,000 population).25
Low serum cholesterol has been implicated in a number of studies,26 and use of high-dose statins appears to increase the risk of ICH, particularly in those with prior history of ICH.27 The impact of smoking,28 alcohol abuse,29,30 and diabetes31,32 on the risk of ICH is disputed.
Hypertensive Hemorrhage
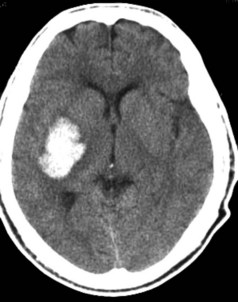
Hypertensive ICH predominantly occurs deep in the cerebral hemispheres, most often in the putamen33 (Figure 35-1). Other frequently involved sites include the thalamus, lobar white matter, cerebellum, and pons. The common link between these sites is that they are all supplied by small penetrating arteries,34 perpendicular branches directly off major arteries that are subject to high sheer stress and that have no collaterals. These features make them vulnerable to the effects of increased blood pressure. Chronic hypertension damages the tunica media, resulting in lipohyalinosis, fibrinoid necrosis, and microaneurysms (Charcot-Bouchard aneurysms). Although Charcot-Bouchard aneurysms have been demonstrated in the weakened vessel walls of patients with ICH, their pathogenetic role in vascular rupture is uncertain.35 The occurrence of ICH in an atypical location, in multiple locations, or in association with subarachnoid hemorrhage raises the suspicion of a non-hypertensive etiology, such as a cerebral vascular anomaly, blood dyscrasia, or trauma.
Intracranial Aneurysms and Vascular Malformations
Approximately half of intracranial AVMs in adults present with hemorrhage.36 In 60% of cases, the hemorrhage is parenchymal, involving virtually any location within the cerebrum, brainstem, or cerebellum.37 The majority of AVMs become symptomatic by age 40; thus hemorrhage due to AVM occurs in a younger population than that due to aneurysms or hypertension. Multiple calcified vascular channels may be seen within the hematoma on CT scan, suggesting the presence of an AVM. MRI and four-vessel cerebral angiography are useful adjuncts in the diagnosis of these lesions.
Other Causes
Cerebral amyloid angiopathy (CAA) is an important cause of predominantly lobar, often recurrent, ICH in the elderly. Histopathologic studies in CAA demonstrate the deposition of beta-amyloid protein in the media and adventitia of small meningeal and cortical vessels; deposition in the typical sites for hypertensive hemorrhage is rare but has been reported in the cerebellum.38 The prevalence of amyloid in cerebral vessels increases dramatically with age39,40 and may partially account for the exponential rise in the risk for ICH with increasing age. There is an overrepresentation of the apolipoprotein E ε2 and ε4 genotypes in CAA-related hemorrhage, and these alleles are associated with an earlier age of onset of first hemorrhage and a higher risk of early recurrence.41,42 Although neuropathologic examination remains the only means of definitively diagnosing CAA, the presence of multiple or recurrent lobar ICH (including asymptomatic microhemorrhages detected on gradient-echo MRI) in individuals 55 years or older without other known causes of hemorrhage strongly suggests the diagnosis.43 Recent PET studies with 11C-Pittsburgh compound B (PIB) suggest an occipital predominant increase in amyloid in such patients.44 Neuropathologic correlation remains to be demonstrated, but PIB-PET appears to be a promising tool for in vivo diagnosis of CAA.
Hematologic causes of ICH include the use of antithrombotic and thrombolytic agents as well as systemic disease (e.g., thrombocytopenia, leukemia, and hepatic and renal failure) and congenital or acquired factor deficiencies. The incidence of oral anticoagulant (OAC)-associated ICH has been increasing in parallel with the increased use of warfarin following pivotal trials in atrial fibrillation in the 1990s. A recent population-based study in the greater Cincinnati area identified a fivefold increased incidence of OAC-associated ICH between 1988 and 1999, such that this condition now accounts for 17% of all ICH cases.45 The incidence is anticipated to rise further in the coming years as the population ages. Although the risk of ICH is greater in the setting of very elevated international normalized ratio (INR), a significant number of hemorrhages occur when the INR is within the therapeutic range.46 Hematoma expansion may be more common in OAC-associated ICH and occur over a longer time frame because of persistent coagulopathy, contributing to the doubling of mortality rate compared to spontaneous ICH.47
The relationship between antiplatelet agent use and hematoma size, hematoma expansion, and outcome in ICH are active areas of investigation. Most studies suggest that antiplatelet use at ICH onset is not associated with larger hematoma size, hematoma growth, or poor clinical outcome48; however, an association has been demonstrated between reduced platelet activity and hematoma growth and poor outcome.49 Whether platelet transfusion is beneficial in this situation is unknown.
Hemorrhage from an underlying neoplasm is rare but occasionally occurs with malignant primary CNS tumors such as glioblastoma multiforme and lymphoma and with metastatic tumors such as melanoma, choriocarcinoma, renal cell carcinoma, and bronchogenic carcinoma.50 Benign tumors are almost never associated with ICH.
ICH may also occur in association with infection (e.g., infiltration of vessel wall by fungal organisms,51 necrotizing hemorrhagic encephalitis with herpes simplex,52 vasculitis,53 venous sinus occlusion,54 in a delayed fashion after head trauma,55 following reperfusion (e.g., after carotid endarterectomy or acute thrombolysis),56 and with the use of various drugs, particularly sympathomimetics (e.g., cocaine, amphetamines, pseudoephedrine, and phenylpropanolamine).57 Finally, some degree of hemorrhagic transformation of acute cerebral infarcts is common,58 though symptomatic ICH in this setting is rare in the absence of anticoagulation or thrombolytic therapy.
Clinical Features
The clinical presentation of ICH is often indistinguishable from that of ischemic stroke but more commonly includes altered level of consciousness, headache, and vomiting, reflecting the presence of increased intracranial pressure (ICP).33 Blood pressure is elevated in the majority of patients (see later discussion). Seizures occur in nearly one-third of patients at onset or within the first few days, particularly in those with lobar hemorrhages or underlying vascular or neoplastic lesions, and may be purely electrographic.59 Symptoms are maximal at onset or develop over minutes to hours. Neurologic deterioration within 48 hours after hospital admission has been reported to occur in 22% of patients with ICH.60 The cause for clinical worsening is not always evident, but it is predicted by clinical and biological markers of inflammation on admission and commonly associated with increased hematoma size and intraventricular bleeding.
Diagnostic Studies
Noncontrast computed tomography (CT) scanning has been the traditional gold standard for diagnosis of acute ICH. The typical CT appearance of an acute hematoma consists of a well-defined area of increased density surrounded by a rim of decreased density. Over time, the borders of both the high- and low-attenuation regions become increasingly indistinct such that the hematoma is isodense with adjacent brain parenchyma by 2 to 6 weeks.61 Peripheral contrast enhancement can often be seen at this time.62 By 2 to 6 months, there may be no CT evidence of previous hemorrhage or there may be an area of hypodensity or a slit-like scar.63
Recent studies have suggested that MRI has high sensitivity and specificity for the diagnosis of acute ICH.64–66 These studies have been criticized, however, for major methodological limitations including the basing of sensitivity estimates on only a small fraction of patients investigated, lack of CT comparator in many patients, and both incorporation and spectrum bias (highly selected patient sample) that may have overestimated diagnostic accuracy of MRI.67 In addition, MRI may not be feasible in a substantial number of patients with acute ICH because of impaired consciousness, hemodynamic compromise, or vomiting.68 The benefits of MRI over CT are its superior performance in the identification of associated vascular malformations, greater accuracy in determining the approximate age of a hematoma (because each hemoglobin oxidation state during evolution of the hematoma produces a predictable pattern of MR signal intensity),69 and its utility in demonstrating the iron-containing deposits of previous asymptomatic hemorrhages.70
Angiography is useful in evaluating the cause of ICH if an underlying aneurysm or vascular malformation is suspected, but the yield of such studies is extremely low when the patient has chronic hypertension and the hemorrhage is in one of the typical sites associated with hypertensive hemorrhage.71 Multidetector CT angiography is evolving as an alternative to conventional angiography. In a retrospective review of 623 patients, multidetector CT angiography identified a vascular etiology for ICH in 91 (15%), with a sensitivity of 96% and a specificity of 99%. The yield was higher in patients who were younger than 46 (47%), had lobar (20%) or infratentorial (16%) ICH locations, had lobar hemorrhages with intraventricular extension (25%), and had neither hypertension nor impaired coagulation (33%).72 In another study of 78 patients, CT angiography identified all but one of the 22 lesions seen on conventional angiography, with a sensitivity of 96% and a specificity of 100%.73
Multidetector CT angiography can also be used to identify the presence of active contrast extravasation into the hematoma, an indicator of active hemorrhage. Termed the “spot sign,” these foci of intralesional enhancement are seen in up to one third of patients with acute ICH74 and are associated with an increased risk of hematoma expansion, in-hospital mortality, and poor outcome in survivors.75
Treatment
Initial Stabilization
Acute ICH is a medical emergency requiring considerable attention to airway and respiratory management, hemodynamic status, and correction of any underlying coagulopathy. As many as half of all patients with ICH undergo mechanical ventilation.76 Blood pressure is often elevated at presentation, sometimes markedly so. Finally, given the frequency of hematoma enlargement over the first few hours, aggressive correction of coagulopathies might be helpful.
Airway and Respiratory Management
Initial airway management includes proper positioning, frequent suctioning, and placement of an oral or nasal airway. Frequent assessments for sonorous respiration, inability to manage oral secretions, or decreased oxygen saturation are necessary. If conservative measures are ineffective, intubation may be necessary. Intubation of patients with ICH requires adequate sedation and jaw relaxation as well as prevention of elevation of ICP. Several factors may conspire to raise ICP during intubation: hypoxia, hypercarbia, and direct tracheal stimulation causing systemic and intracranial hypertension. Intravenous (IV) lidocaine (1-1.5 mg/kg) has been recommended to block this response,77 although data supporting its use are lacking.78 Short-acting IV anesthetic agents (thiopental, 1-5 mg/kg; or etomidate, 0.1-0.5 mg/kg) also block this response79 and additionally suppress brain metabolic rate,80 theoretically improving tolerance of a transient fall in cerebral perfusion pressure (CPP) should it occur. Etomidate is generally preferred over thiopental, since it is less likely to lower blood pressure. Paralytic agents are usually unnecessary but, if needed, short-acting agents should be used.
Hemodynamics
Arterial blood pressure is elevated on admission in the majority of patients with ICH, even in the absence of a history of hypertension.33 Mean arterial pressure (MAP) is greater than 120 mm Hg in over two-thirds of patients and greater than 140 mm Hg in over one-third.81 Although this acute increase in blood pressure is often implicated as the cause of the hemorrhage, it may simply be a reflection of chronic hypertension, the brain’s attempt to maintain CPP in response to the sudden increase in ICP, pain and anxiety, and sympathetic activation. Even without pharmacologic intervention, blood pressure tends to decline to premorbid levels during the first 7 to 10 days after hemorrhage.82
There is substantial controversy over if and when to lower blood pressure after acute ICH and how aggressive any intervention should be.83 Proponents of rapid treatment of acute hypertension argue that high blood pressure may predispose to hematoma enlargement and may exacerbate vasogenic edema by increasing capillary hydrostatic pressure, especially in areas with a damaged blood-brain barrier. Yet, an association between hypertension and edema has never been demonstrated, and data on the effect of hypertension on hematoma enlargement have been inconsistent.84,85 Another potential reason to lower blood pressure is that hypertension during the acute phase of ICH has been shown to correlate with a poor prognosis in some studies.86,87 One compelling reason to consider lowering blood pressure in ICH patients with moderate to severe hypertension is the potential for end-organ damage. Such patients are at risk for systemic complications of elevated blood pressure, including myocardial ischemia, congestive heart failure, and acute renal failure.
The major argument against the treatment of elevated blood pressure is that lowering blood pressure might exacerbate ischemic damage in the tissue surrounding the hematoma by impairing blood flow.88 Chronic hypertension shifts the cerebral autoregulatory curve to the right such that a higher CPP is required to maintain adequate CBF.89,90 Lowering the blood pressure to “normal” levels in these patients might thus lead to inadequate CBF. Similarly, since CPP is equal to the difference between MAP and ICP, lowering blood pressure may reduce CPP below the autoregulatory limit in patients in whom ICP is elevated due to a large space-occupying clot or hydrocephalus.
Seeking to address the issue of whether lowering blood pressure produces cerebral ischemia in acute ICH, several studies of CBF autoregulation in patients with recent ICH and elevated blood pressure (MAP > 130-140 mm Hg) have been carried out.91–93 Taken together, these studies demonstrate that regional and global autoregulation are preserved after ICH down to a lower MAP limit that averages 110 mm Hg or about 80% of the admission MAP.
These observations set the stage for the INTERACT trial,94 a prospective trial of blood pressure management beginning within 6 hours of symptom onset in 404 patients with spontaneous ICH and elevated systolic blood pressure (150-220 mm Hg). Patients were randomized to an early intensive blood pressure–lowering strategy that targeted a reduction in systolic blood pressure to below 140 mm Hg within 1 hour, or control, with a target systolic pressure of less than 180 mm Hg. The primary efficacy endpoint was hematoma growth at 24 hours. After the first hour of treatment, mean blood pressure was 13.3 mm Hg lower (95% confidence interval [CI] 8.9-17.6 mm Hg; P <0.0001) in the intensive blood pressure–lowering group. Hematoma growth at 24 hours was 36.3% in the control group and 13.7% in the intensive treatment group, a 22.6% difference (95% CI 0.6%-44.5%; P=0.04). After adjusting for initial hematoma volume and time from onset to CT, median hematoma growth at 24 hours differed by 1.7 mL (95% CI 0.5-3.9, P=0.13). Intensive blood pressure–lowering treatment did not increase the risk of adverse events or improve 90-day clinical outcome. A much larger follow-on trial is currently underway.
Prevention of Hemorrhage Extension
Because hemorrhage extension occurs within the first few hours after symptom onset in about one-third of patients (Figure 35-2), it seems appropriate that any coagulopathy should be corrected as rapidly as possible. Patients taking warfarin should receive IV vitamin K and enough fresh frozen plasma (FFP) to normalize the coagulation profile. Prothrombin complex concentrate may be a useful alternative. Care must be taken not to precipitate congestive heart failure, however, and diuretics may be required. Additionally there is risk of transfusion-related acute lung injury with the administration of fresh frozen plasma, which can complicate the process considerably. Correcting coagulopathy associated with thrombolytic-induced ICH is discussed later.
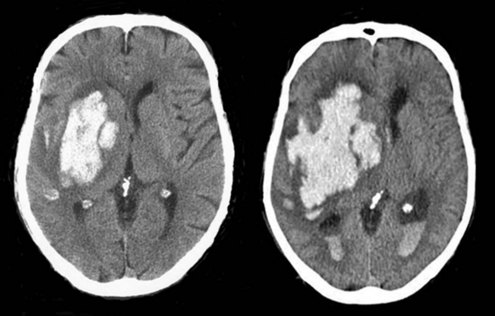
Even in those patients without coagulopathy, promoting early hemostasis might limit ongoing bleeding and decrease hematoma volume. Factor VIIa is a coagulation factor that interacts with tissue factor exposed in the wall of a damaged blood vessel to drive a burst of thrombin that initiates platelet aggregation and accelerates formation of a stable fibrin clot. A phase IIb placebo-controlled dose-ranging proof-of-concept study found that treatment with recombinant factor VIIa (rFVIIa) given as a single IV bolus within 4 hours of ICH onset decreased hematoma growth and improved clinical outcome despite a small increase in thromboembolic events. A much larger phase III trial comparing placebo to 20 and 80 µg/kg of rFVIIa followed. This study confirmed the ability of rFVIIa to reduce hematoma growth; however, at 90 days there was no difference in clinical outcome.95 A post hoc exploratory analysis suggested that a subgroup of younger patients who present earlier and have no significant intraventricular hemorrhage might benefit from rFVIIa, but this has not been tested.96
Intraventricular Hemorrhage and Hydrocephalus
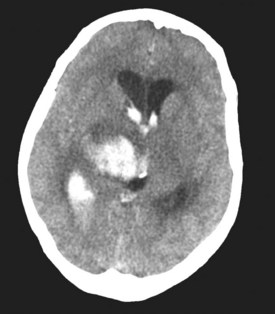
In approximately 40% of patients with ICH, blood extends into the ventricular system (intraventricular hemorrhage [IVH]).97 Mortality in these patients is high.98,99 IVH may contribute to poor outcome by blocking cerebrospinal fluid (CSF) pathways, with resultant hydrocephalus and increased ICP. In addition, intraventricular blood and/or its breakdown products may exert direct chemical irritative effects on periventricular structures. Hydrocephalus may develop after ICH either in association with IVH or because of direct mass effect on a ventricle (e.g., on the third ventricle with a thalamic hemorrhage) (Figure 35-3). External ventricular drainage (ventriculostomy) is frequently used to treat hydrocephalus and IVH, but its efficacy has never been established. Ventriculostomy in the setting of IVH is difficult to manage because the catheter frequently becomes obstructed with thrombus, interrupting drainage and raising ICP. Flushing the system helps remove thrombus from the catheter but increases the risk of ventriculitis. Recently, investigators have attempted to facilitate removal of blood from the ventricles via direct intraventricular administration of thrombolytic agents. Preliminary studies have been promising,100 and a multicenter randomized trial is currently underway.
Intracranial Hypertension
The incidence, impact, and appropriate management of intracranial hypertension in ICH are not well understood. Factors likely to contribute to elevated ICP in this population include large hematoma size, minimal degree of underlying cerebral atrophy, hydrocephalus, and edema, but the true incidence of intracranial hypertension is unclear, since routine ICP monitoring is not performed. Because the hematoma is localized and the increase in volume it produces can be compensated for to some degree by reduction in the size of the ventricles and subarachnoid space, a global increase in ICP may not be seen unless the hemorrhage is very large or is associated with marked hydrocephalus. However, mass effect from the hematoma and local tissue shifts can compress the brainstem or result in herniation in the absence of a global increase in ICP.101,102 Thus the utility of ICP monitoring has never been established.
Elevated ICP is often treated with osmotic agents (mannitol, hypertonic saline) and, if the ventricles are enlarged, CSF drainage. A recent case series suggested that rapid reversal of clinical transtentorial herniation (decreased level of consciousness and dilated pupil) with hyperventilation and osmotic agents improved long-term outcome.103 There are only a few small clinical trials of osmotic agents in ICH, which do not provide sufficient data to support their routine use.104 The best available data on corticosteroids in ICH indicate that they do not provide any benefit and increase the rate of complications.105
Surgical Evacuation
The rationale for surgical evacuation of a hematoma is that reducing mass effect and removing neurotoxic clot constituents should minimize injury to adjacent brain tissue and hence improve outcome. Unfortunately, several randomized controlled trials of surgery for supratentorial ICH dating back to 1961 have all failed to show a benefit of the intervention.106–109 A meta-analysis of three of these trials reported that patients undergoing surgical evacuation via open craniotomy had a higher rate of death or dependency at 6 months compared to those managed medically (83% versus 70%).110 Criticisms of these trials are that the surgical techniques used were outdated, patient selection was inadequate, and surgery was delayed too long.
Because open craniotomy is complicated by tissue damage sustained during the approach to the hematoma, a variety of new techniques for clot removal have been proposed, including an Archimedes screw, ultrasonic aspirator, modified endoscope, modified nucleotome, double track aspirator, intraoperative CT monitoring, and instillation of thrombolytics. However, the recurrence of bleeding due to the loss of tamponade effect on adjacent tissue that occurs in 10% of patients treated with open craniotomy remains an issue with the newer techniques. In addition, because the newer techniques involve limited surgical exposure, concern exists that rebleeding will be more difficult to control than with open craniotomy. One study comparing endoscopic aspiration to medical management found a better outcome in the surgical group (74% death or disability compared to 90%), but the benefit was limited to patients with lobar hematomas.111 Three studies addressed the feasibility of early craniotomy for ICH. In one,112 34 patients were treated within 12 hours of ICH. Mortality was 18% in the surgical group and 23% in the medical group. In another study,113 20 patients were randomized, with a median time to surgery of 8.5 hours from onset. Good outcome (Glasgow Outcome Scale score > 3) was 56% with surgery and 36% in the medically treated group (P = NS). The third, a study of ultra-early surgery (<4 hours), found a disturbingly high rate of postoperative rebleeding.114
A lack of benefit of surgery in ICH was also shown in a recently completed multicenter trial in which 1033 patients were randomized within 72 hours of ICH onset to surgical hematoma evacuation (open craniotomy or stereotactic aspiration, at surgeon’s discretion) or initial conservative management. Favorable outcome occurred in 26.1% in the surgery group and 23.8% in the initial conservative treatment group, a nonsignificant difference (odds ratio 0.89; 95% CI 0.66-1.19). There was also no difference in mortality (surgery 62.6% versus conservative treatment 63.7%). Subgroup analysis suggested a possible benefit of surgery in patients with superficial hematomas (less than 1 cm from cortical surface).115 A trial comparing surgical and medical management of superficial hematomas is currently underway.
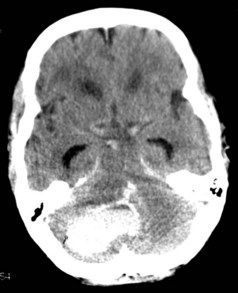
Cerebellar hemorrhages were excluded from the randomized trials of surgery, but nonrandomized case series report good outcomes for surgically treated patients with cerebellar hemorrhages that are large or associated with brainstem compression or obstruction of the fourth ventricle. Recommended criteria for when to evacuate a cerebellar hematoma have thus included diminished level of consciousness, large size of the hematoma (>3 cm3), midline location, compression of basal cisterns and/or brainstem, and presence of hydrocephalus (Figure 35-4).116–118 Patient selection is important as many patients with smaller hemorrhages do well with medical management.119
Management of Thrombolytic-Induced Ich
Associated with considerable morbidity and mortality, symptomatic ICH is a feared complication of thrombolytic therapy. Symptomatic ICH occurs after thrombolytic treatment of acute ischemic stroke in approximately 6% of patients.120 It is substantially less common after thrombolytic treatment of extracerebral thrombosis (myocardial infarction, pulmonary embolism, deep venous thrombosis, and arterial and graft occlusion)121 but results in a similarly poor outcome.122 Factors that increase the risk of symptomatic ICH include intraarterial versus IV route of administration, early ischemic changes on pretreatment CT, greater symptom severity, and elevated serum glucose or history of diabetes mellitus.123,124
In the setting of thrombolytic therapy, any new neurologic deficit, especially with a decline in consciousness, should be assumed to be due to hemorrhage. Management of a suspected ICH should begin with stopping the thrombolytic infusion, reassessing the patient’s airway, and obtaining an emergent CT scan. Blood studies (prothrombin time, partial thromboplastin time, thrombin, and fibrinogen levels) should be performed to assess fibrinolytic state. Preparations for giving FFP, cryoprecipitate, and platelets should be initiated at the first suspicion of hemorrhage so that they will be ready if needed; however, no reliable data are available to guide the choice of blood product. The NINDS rt-PA study125 stipulated 6 to 8 units of cryoprecipitate or FFP and 6 to 8 units of platelets, but only rarely was this amount of blood product given to an individual patient during the study. Although neurosurgical consultation was frequently obtained in patients with symptomatic ICH in the NINDS trial, only one patient in the study underwent surgery, and that patient died.
Management of Seizures
Although seizures may theoretically exacerbate ICH, they have not been demonstrated to alter outcome. Prophylactic anticonvulsants may reduce the risk of early seizures in patients with lobar ICH but do not affect the risk of developing epilepsy.126 Thus reasonable approaches include either a brief period of prophylaxis or treating only if seizures occur. As for any hospitalized patient, the treatment of clinical seizures typically begins with an IV benzodiazepine such as lorazepam followed by an IV agent such as fosphenytoin.
Supportive Care
Patients with ICH are prone to the same medical complications seen in patients with ischemic stroke, including fever, deep venous thrombosis (DVT), pulmonary embolism, and pneumonia.127,128 Given the association between fever and worsened outcome in experimental models of brain injury, it is reasonable for antipyretic medications to be administered in febrile patients with ICH. The use of pneumatic sequential compression devices and elastic stockings has been shown to significantly decrease the incidence of DVT in patients with acute ICH relative to elastic stockings alone.129 Subcutaneous heparin at a dose of 5000 U three times daily when initiated on day 2 after hemorrhage has been shown to significantly reduce the frequency of DVT relative to treatment begun on day 4 or 10, with no concomitant increase in hematoma expansion.130 In another study, subcutaneous enoxaparin (40 mg daily) initiated at 48 hours after ICH was also safe. A benefit of enoxaparin over compression stockings could not been detected because of the low incidence of DVT in both treatment groups.131
Prognostic Factors and Causes Of Mortality
Mortality following ICH is high (25%-50%), with over half of the deaths occurring in the first 48 hours. Although patients who have small hemorrhages and mild deficits may recover completely, the majority of ICH survivors have significant residual disability.4,132,133 A variety of clinical, laboratory, and radiographic predictors of poor outcome have been identified, the most consistent being impaired level of consciousness and large hematoma size on admission. Other predictive clinical features variably include increasing age, elevated admission blood pressure on admission, rapid decline in blood pressure over the first 24 hours, history of diabetes, antecedent OAC use, male gender, and in-hospital neurologic deterioration. Laboratory parameters identified in at least one study include hyperglycemia, elevated troponin level, elevated plasma S100B level, elevated plasma D-dimer level, elevated INR, low serum cholesterol and triglyceride levels, and apolipoprotein E ε2 or ε4 allele. Radiographic features include infratentorial hematoma location, intraventricular spread of blood, midline shift, hydrocephalus, hematoma growth, and presence of the spot sign on CT angiography. A number of prognostic models of varying complexity have been developed to allow risk stratification upon presentation with ICH. One easy to use model is the ICH score,134 which is based on point assignments for Glasgow Coma Scale score, ICH volume, presence of intraventricular hemorrhage, infratentorial location, and patient age and has been validated to accurately predict 30-day mortality. It has been demonstrated, however, that withdrawal of support in patients felt likely to have a poor outcome biases predictive models in ICH and negates the predictive value of all other variables.135 Thus, the most frequent cause of death after ICH is withdrawal of care, followed by early (within 48 hours) transtentorial herniation with progression to brain death. Medical complications of immobility (pulmonary embolism, pneumonia, sepsis) account for most of the other deaths.133 For survivors of ICH, the risk of recurrent stroke is approximately 4% per year. Recurrent ICH occurs about twice as often as ischemic stroke, especially in those with previous lobar hemorrhage.136
 Subarachnoid Hemorrhage
Subarachnoid Hemorrhage
Although it is the least common form of stroke, subarachnoid hemorrhage (SAH) has great impact on its sufferers. One-quarter of patients die before reaching medical attention,137 and because of the consequences of secondary insults—rebleeding, hydrocephalus, and delayed ischemia due to vasospasm—more than half of those that reach medical attention either die or are left with neurologic deficits.
Pathophysiology
In SAH, the primary site of bleeding is within the subarachnoid space, but may also involve hemorrhage into the brain parenchyma, ventricular system, or subdural space. Rupture of an intracranial saccular aneurysm (Figure 35-5) is by far the most common cause of spontaneous SAH. Saccular or berry aneurysms are small, rounded protrusions of the arterial wall occurring predominantly at bifurcations of the large arteries of the circle of Willis at the base of the brain. The most common sites of ruptured aneurysms are the distal internal carotid artery and its posterior communicating artery junction (41%); anterior communicating artery/anterior cerebral artery (34%); middle cerebral artery (20%); and vertebrobasilar arteries (4%).138 About 20% of patients have multiple aneurysms.
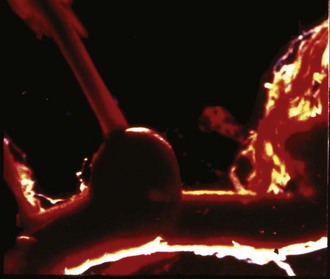
Figure 35-5 Autopsy specimen of intracranial aneurysm filled with pressurized blood to simulate subarachnoid hemorrhage.
The pathogenesis of aneurysms remains controversial, especially in regard to the relative important of developmental versus acquired factors. Proponents of the congenital theory suggest that aneurysms arise at sites of faulty fusion between muscular segments within the arterial wall. Supporters of the acquired-degenerative theory focus on the role of vascular damage caused by hemodynamic stress.139 The third possibility is that aneurysms develop at sites harboring congenital defects with superimposed degenerative changes.
Causes and Risk Factors
Genetic conditions that predispose to aneurysm formation include polycystic kidney disease, connective tissue disorders, and coarctation of the aorta. Recently it has become clear that in some patient populations, genetic factors play a role in aneurysm formation, without other associated conditions.140 There is a familial form as well.141 Other types of aneurysms that less commonly cause SAH include atherosclerotic, mycotic, and traumatic aneurysms.
The risk of SAH increases with age, peaking at 55 to 60 years. There is a slight male predominance in younger age groups and a slight female predominance among older patients.142 Potentially reversible risk factors for SAH include cigarette smoking, oral contraceptive use, alcohol abuse, and hypertension.143 Prospective cohort studies have reported a relative risk of SAH as high as 5.7 for female and 4.7 for male smokers,144 but no increased risk in former smokers. Oral contraceptive use, in addition to being an independent risk factor for SAH, dramatically increases the risk among smokers.145 A dose-response relationship exists between alcohol consumption and incidence of SAH.
Clinical Features
Presentation
The most common initial symptom of SAH, occurring in over 90% of patients, is a sudden severe headache. Less severe warning (“sentinel”) headaches146 may precede the presenting event in as many as half and are thought to represent minor leaks. In about half of patients, loss of consciousness accompanies the headache.147 The mechanisms thought to be responsible for the loss of consciousness are the sudden surge in ICP at the moment of hemorrhage or cardiac arrhythmias. Vomiting can be a prominent symptom. Seizure activity may be reported,148 but it is unclear whether this represents true epileptic seizures or reflex posturing related to the sudden rise in ICP. Focal deficits at the onset of hemorrhage occur in less than 10% of cases. After a few hours, a stiff neck can develop, reflecting the sterile meningeal inflammation induced by the presence of blood in the subarachnoid space.
Complications
Early Complications
Rebleeding
Rebleeding is heralded by a sudden worsening of headache, vomiting, blood pressure elevation, development of a new neurologic deficit, or arrhythmia. It occurs in up to one-third of patients and is often fatal. The risk of rebleeding is greatest during the first 24 hours, declining rapidly over the next 2 weeks.149 Rates of rebleeding are highest in women, those who are a poor clinical grade, those in poor medical condition, and those with elevated systolic blood pressure.
Hydrocephalus
Hydrocephalus occurs after SAH because of disturbances of CSF flow or reabsorption: subarachnoid blood may impair CSF reabsorption at the arachnoid granulations, and ventricular blood may obstruct its flow. Acute hydrocephalus can develops within hours of SAH,150 often in the absence of intraventricular blood. It usually manifests as a gradual decline in level of consciousness and can easily be treated by placement of an external ventricular drain. Delayed hydrocephalus may also develop gradually days to weeks later. Hydrocephalus must be distinguished from metabolic derangements, infection, and vasospasm. CT scan is essential in making the diagnosis. The natural history of untreated acute hydrocephalus is that about one third of patients progress, one-third spontaneously improve, and one third remain static.151
Delayed Complications
Vasospasm
The term vasospasm refers to complex changes in intracerebral vessels, with segmental or diffuse narrowing of the lumen due to arterial wall thickening, vasoconstriction, and impaired relaxation that reduce CBF. If the reduction in flow is severe enough, ischemia and infarction follow. The term delayed cerebral ischemia (DCI) describes the clinical situation where these and other factors conspire to produce ischemia. Other factors that may contribute to DCI include impaired autoregulation, hypovolemia, and microthrombosis.152 DCI is a leading cause of morbidity and mortality following SAH.
Arterial narrowing can be detected angiographically (Figure 35-6) in up to 70% of patients,153 of whom almost half become symptomatic. The pathogenesis of arterial vasospasm is complex and not fully understood, but sustained exposure of vessels to extraluminal blood constituents and catecholamines is thought to play a role. It involves structural changes in the vessel walls and in adrenergic nerve fibers. The onset of vasospasm is delayed, most commonly developing 5 to 10 days after initial hemorrhage, and may persist for up to 3 weeks. The strongest predictor of vasospasm is the amount of subarachnoid blood on the initial CT scan, with the greatest risk occurring in those having thick subarachnoid clots and intraventricular blood (graded using Fisher Scale and modified Fisher Scale; Table 35-1).154,155 Focal neurologic deficits resulting from vasospasm may appear abruptly or gradually and may fluctuate, exacerbated by hypovolemia or hypotension. If untreated, infarction may occur.
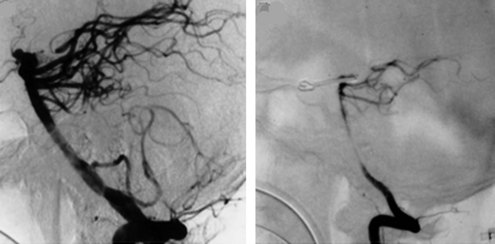
TABLE 35-1 Fisher Grade of Subarachnoid Hemorrhage on Initial Computed Tomography
CT, computed tomography; IVH, intraventricular hemorrhage; SAH, subarachnoid hemorrhage.
Medical Complications
Disturbances in sodium and water balance occur in approximately one-third of patients, and hyponatremia and volume depletion after SAH are correlated with an increased risk of symptomatic vasospasm and poor outcome.156 Although hyponatremia was previously attributed to inappropriate secretion of antidiuretic hormone (SIADH) and was therefore treated with fluid restriction, later evidence suggested that both sodium and water are lost. In fact, when administered normal “maintenance” volumes of fluid (2-3 L/day), as many as half of patients develop intravascular volume contraction.157
Cardiac abnormalities are common in the first 48 hours after SAH. Electrocardiographic (ECG) changes (Figure 35-7) including tall peaked T waves (“cerebral T waves”), diffuse T-wave inversion, ST-segment depression, and prolonged QT segments158 occur frequently and have been linked to elevated levels of circulating catecholamines. It appears that these changes usually do not represent myocardial ischemia, as the myocardial lesions reported are pathologically distinct from ischemia. Cardiac enzymes may be mildly elevated.159 Cardiac rhythm disturbances occur in about 30% to 40% of patients, especially on the day of hemorrhage or in the postoperative period. Arrhythmias are typically benign but can be life threatening in about 5%.160,161 In rare cases, “stunned myocardium” may occur, with impairment of myocardial contractility leading to a fall in cardiac output, hypotension, and pulmonary edema.162 This phenomenon can be dramatic but is transient, usually lasting 2 to 3 days, after which cardiac function returns to baseline.163 Management is the same as with other causes of cardiogenic shock.164 During hemodynamic treatment for vasospasm, pulmonary edema may occur in up to one-quarter of patients,165 though its incidence is lower with careful monitoring.166
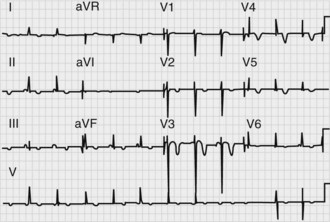
Figure 35-7 Electrocardiogram in a patient with acute subarachnoid hemorrhage, demonstrating diffuse T-wave inversions.
In a review of over 450 patients with SAH, Solenski et al.167 reported some degree of hepatic dysfunction in 24%. The majority had only mild abnormalities of hepatic enzymes without clinical accompaniment, but severe hepatic dysfunction occurred in 4%. Thrombocytopenia was found in 4% of patients, usually occurring in the setting of systemic sepsis. Renal dysfunction occurred in 7% of patients.
Fever, anemia, hyperglycemia, pneumonia, and hypertension occur frequently after SAH. Potential treatments include maintaining normothermia with antipyretics and possibly systemic cooling devices, administration of erythropoietin to prevent anemia, and preserving normoglycemia.168
Diagnostic Studies
CT is the imaging modality of choice in screening for SAH, having a sensitivity of better than 90%.169 Blood appears as high attenuation within the perimesencephalic and interpeduncular cisterns surrounding the brainstem (the basal cisterns), Sylvian fissure, and sulci (Figure 35-8).
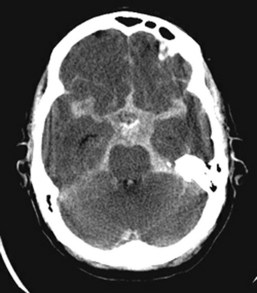
CT may fail to demonstrate SAH if the volume of blood is very small, if the hemorrhage occurred several days prior to the CT scan, or if the hematocrit is extremely low.170 Lumbar puncture for CSF analysis is indicated if CT is negative and clinical suspicion is high. Red blood cells in the CSF are indicative of SAH but can also be seen with traumatic puncture. The common technique of comparing cell counts in the first and last tubes collected is not reliable; however, the presence of yellow pigment (xanthochromia), resulting from red cell breakdown, can be helpful in distinguishing between the two.171 Xanthochromia develops 2 to 6 hours after hemorrhage and persists for 1 to 4 weeks. It can also be seen in the setting of high protein levels due to diabetes, renal failure or infection, in which case spectrophotometric analysis to identify hemoglobin breakdown products improves diagnostic accuracy.172
Once SAH has been diagnosed, cerebral angiography should be performed as soon as possible to identify the responsible vascular lesion, search for other lesions (multiple aneurysms are found in 20% to 30% of patients with aneurysmal SAH), and assist in operative management. Angiography does not identify a source of bleeding in 10% to 15% of patients with nontraumatic SAH. In some cases, this may be due to vasospasm or inadequate views to detect a subtle aneurysm, especially in the region of the anterior communicating artery or in the posterior circulation. Repeat angiography in about one week is recommended.173 There is a subset of patients in whom the blood on CT is localized to the perimesencephalic cisterns. In these cases, angiography is usually negative, and the bleeding is thought to be venous in origin; the prognosis is excellent, and repeat angiography is almost always negative.174
With its wide availability, ease of use, and safety profile, CT angiography is increasingly being used as the initial diagnostic tool in the investigation of SAH. Overall sensitivity is 90% or greater compared to conventional angiography but is notably lower for aneurysms smaller than 5 mm175; thus, a negative CT angiogram should be followed by conventional catheter angiography. A negative CT angiogram alone may be sufficient in the case of perimesencephalic SAH.174 Magnetic resonance angiography (MRA) has good sensitivity for detecting medium and large aneurysms, but sensitivity falls to less than 40% for small aneurysms. In addition, MRA is impractical for many acutely ill patients with SAH because of logistics, movement artifact, need for sedation, and difficulty in monitoring clinical status in the scanner.
Treatment
Initial Stabilization
The initial steps in the evaluation of a patient with suspected SAH should include assessment of airway, hemodynamic status, and the level of neurologic function. The Hunt and Hess Scale176 and the World Federation of Neurological Surgeons Scale177 provide standardized measures of the patient’s clinical condition (Tables 35-2 and 35-3).
TABLE 35-2 Hunt & Hess Clinical Classification of Subarachnoid Hemorrhage
TABLE 35-3 World Federation of Neurologic Surgeons Clinical Classification of Subarachnoid Hemorrhage
| Grade | Glasgow Coma Scale | Motor Deficits |
|---|---|---|
| I | 15 | Absent |
| II | 13-14 | Absent |
| III | 13-14 | Present |
| IV | 7-12 | Present or absent |
| V | 3-6 | Present or absent |
Routine Care and Monitoring
The routine monitoring of all patients with acute SAH should include serial neurologic examinations, continuous ECG monitoring, and frequent determinations of blood pressure, electrolytes, body weight, and fluid balance. The role of prophylactic anticonvulsants in patients who have not had a seizure is controversial. Initial use of anticonvulsants is generally recommended; however, the duration of administration should be limited to several days during the periprocedural period.178 Recent retrospective studies have suggested that routine use of anticonvulsants for a longer duration is associated with worse neurologic outcome.179 Dexamethasone is widely used to reduce meningeal irritation and intra- and postoperative edema, but there is no convincing evidence documenting its efficacy.
Fluid Management
A stable intravascular volume should be maintained by hydration with isotonic saline and daily monitoring of fluid balance, body weight, and hematocrit. Monitoring of fluid balance alone may not be adequate to prevent hypovolemia, and combining multiple clinical indicators of volume status are needed.180,181 In some patients with severe cerebral salt wasting, large volumes of fluid are required to prevent intravascular volume contraction.182 Hyponatremia can often be managed with restriction of free water by administering only isotonic IV fluids, minimizing oral liquids, and using concentrated enteral feedings. It is important to adjust the tonicity of the fluid, not the volume of fluids administered. Fludrocortisone is of marginal benefit in treating salt wasting183,184; however, one study suggested that hydrocortisone may be helpful.185 Persistent hyponatremia can be treated by using mildly hypertonic solutions (1.25%-2% saline) as the sole IV fluid. There may be a role for ADH antagonists such as conivaptan, but since they increase urine volume, extreme caution must be exercised to avoid hypovolemia.186
Hypertension
Magnesium Sulfate
Magnesium antagonizes calcium and thus could reduce vasospasm. Almost 40% of patients with SAH have low serum magnesium levels on presentation, leading to speculation that the administration of magnesium may improve outcome of SAH patients. Advantages of magnesium include ease of administration, low cost, and favorable safety profile.187 Several studies have suggested benefit,188,189 but controlled trials have been inconclusive.190,191 A large phase III randomized controlled international trial is currently underway.
Statins
Statins may be beneficial in SAH through their ability to induce nitric oxide synthetase, leading to dilation of cerebral vessels, or through their antiinflammatory effects. Some preliminary studies have suggested that they may reduce vasospasm and improve outcome,192,193 while others have not.194–196 A multicenter placebo-controlled double-blinded phase III trial is underway.
Management of Secondary Complications
Rebleeding
Multiple clinical trials have demonstrated that antifibrinolytic agents such as epsilon aminocaproic acid and tranexamic acid reduce the risk of rebleeding, but this benefit is offset by an increased incidence of vasospasm and hydrocephalus.197,198 With the advent of early surgery and now endovascular treatment of aneurysms, the use of these agents has declined dramatically. More recently, there has been interest in a shorter course of antifibrinolytic therapy while awaiting surgery or endovascular treatment. Tranexamic acid begun immediately upon SAH diagnosis and continued only until the aneurysm was secured (always within 72 hours) reduced the risk of rebleeding from 10.8% to 2.4% and did not increase risk of DCI.199
The definitive way to prevent rebleeding is to repair the aneurysm by surgical or endovascular means. Endovascular techniques involving electrolytically detachable platinum coils that thrombose the aneurysm are now routinely used to repair acutely ruptured aneurysms (Figure 35-9).
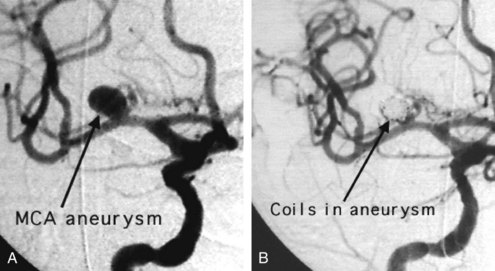
The International Subarachnoid Aneurysm Trial was a multinational, prospective, randomized trial that compared surgical clipping with endovascular coiling of acutely ruptured intracranial aneurysms. Participating centers were required to treat at least 60 SAH patients per year and offer both treatment modalities. Patients were eligible to be enrolled only if there was clinical equipoise regarding the best method to repair the aneurysm. Initial results favored endovascular coiling, with 23.7% dead or dependent at 1 year compared to 30.6% in the surgery group.200 Long-term follow-up indicated an increased risk of recurrent bleeding from a coiled aneurysm compared with a clipped aneurysm. At 5 years, the risk of death remained significantly lower in the coiled group than in the clipped group, but the proportion of survivors who were independent did not differ between the two groups.201
Vasospasm
Monitoring for vasospasm involves serial neurologic exams, serial transcranial Doppler (TCD) measurement of blood flow velocities,202,203 and catheter angiography. Neurologic signs may be vague, such as a global decline in responsiveness, or consist of focal deficits such as hemiparesis or language disturbance. Symptoms may wax and wane, being exacerbated by hypovolemia or hypotension. Vasospasm can be identified on TCD by an increase in linear blood flow velocity (LBFV): mild (>120 cm/sec), moderate (>160 cm/sec), or severe (>200 cm/sec) vasospasm.204 Alternatively, the rate of rise in the LBFV is used to define the onset of vasospasm. The sensitivity of TCD in detecting vasospasm is about 80% when compared to angiography, at least partly because TCD samples only a small segment of the vasculature.205 It has a very high negative predictive value, and the presence of normal velocities usually indicates the absence of vasospasm. Newer CT and MRI techniques including angiography and perfusion may have a role in assessing for delayed ischemia in the future.
Prevention
Routine measures taken to prevent or ameliorate the effects of vasospasm include mechanical removal of subarachnoid blood at the time of aneurysm surgery or by CSF drainage, administration of the centrally acting calcium channel antagonist nimodipine, and avoidance of intravascular volume contraction (see earlier) and hypotension. Nimodipine (60 mg orally every 4 hours) for 3 weeks after SAH reduces the impact of symptomatic vasospasm and improves outcome.206,207 It is not clear whether this beneficial effect is due to action on the cerebral vessels or to prevention of calcium influx into ischemic neurons. Any hypotension developing with nimodipine administration can usually be managed with fluids or adjusting the dosage schedule to 30 mg every 2 hours. In patients receiving hemodynamic augmentation for symptomatic vasospasm, nimodipine may have to be discontinued if it interferes with maintenance of blood pressure goals.
While there is general agreement that hypovolemia must be avoided, the use of prophylactic hypervolemia is more controversial.208–210 In a prospective controlled study, prophylactic volume expansion with albumin failed to reduce the incidence of clinical or TCD-defined vasospasm, did not improve CBF, and had no effect on outcome.211 Costs and complications may be higher with the use of prophylactic hypervolemia.
Prophylactic use of transluminal balloon angioplasty has recently been evaluated.212 Although it reduced the need for therapeutic angioplasty and reduced ischemic deficits, these benefits were offset by procedure-related vessel complications.
Treatment of Delayed Ischemic Deficits
The trigger for instituting more aggressive interventions varies widely. Some centers actively intervene in the setting of rising TCD velocities213 or angiographic vasospasm in asymptomatic patients,214 whereas others institute aggressive measures in the setting of clinical deterioration. Aggressive measures include both hemodynamic and endovascular manipulations.215–217 The goal is to improve CBF in ischemic regions. Since patients with SAH tend to become hypovolemic and lose pressure autoregulation,218 it has been inferred that hypervolemia, induced hypertension, and augmentation of cardiac output would accomplish that goal.
Hemodynamic Augmentation
Data supporting the use of hypervolemia are scant. As described earlier, prophylactic hypervolemia had no impact on CBF, vasospasm, or outcome.211 In one study of patients with symptomatic vasospasm, hypervolemia was reported to improve CBF, but a proper control group was not used.219 Other studies question whether hypervolemia adds further benefit beyond correction of hypovolemia and report that the impact of volume expansion on CBF is modest compared to induced hypertension.220
Hemodilution is perhaps the least understood component of triple-H therapy. The rationale is to augment CBF by reducing blood viscosity. The tradeoff is that oxygen-carrying capacity is reduced, reducing oxygen delivery. It has been suggested that a hematocrit of 30% provides the optimal balance between oxygen-carrying capacity and viscosity; however, one study found that despite a rise in CBF, oxygen delivery fell with hemodilution.221
Blood pressure augmentation may be the most effective hemodynamic intervention. Studies have demonstrated a consistent rise in CBF in response to blood pressure elevation with dopamine and phenylephrine, although the optimal target has not yet been identified.220 Under normal conditions, cardiac output does not influence CBF; however, with cerebral ischemia or impaired autoregulation, changes in cardiac output may alter CBF. Dobutamine or milrinone may be effective in improving cardiac output and CBF in some patients.
Endovascular Treatments
The endovascular approach to vasospasm involves treatment of constricted vessels with either balloon angioplasty or intraarterial infusion of vasodilating agents.180,222 Angioplasty on the proximal segments of vasospastic cerebral vessels yields impressive angiographic changes (Figure 35-10) that appear to be long lasting.223,224 Vasoconstriction in more distal vessels usually cannot be reached by angioplasty catheters and can be treated with intraarterial infusion of vasodilators.
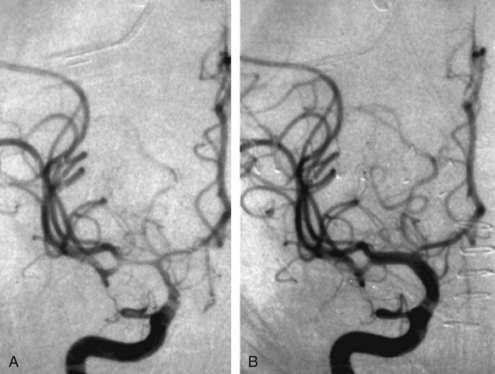
Intraarterial papaverine has an immediate and dramatic effect on blood vessels, but reversal of clinical deficits is inconsistent.225–227 The use of papaverine has largely been abandoned because of its short-lived effect and complications including increased ICP, apnea, worsening of vasospasm, neurologic deterioration and seizures.228 It has been replaced by nicardipine, verapamil, nimodipine, and milrinone.229–231
The timing of when to initiate endovascular therapy is debated. It is generally used if after a few hours, the response to hemodynamic augmentation is inadequate, but it may be the initial therapy in patients with poor cardiac function who are at high risk of complications of hemodynamic augmentation.232
Prognostic Factors and Causes Of Mortality
Untreated aneurysmal SAH carries a poor prognosis, with an estimated mortality rate of approximately 50%. Of those who make it to medical attention, mortality is 20% to 40%. Causes of death are about equally distributed among direct effects of the initial hemorrhage, rebleeding, vasospasm, and medical complications. Overall, less than one-third of patients achieve good neurologic recovery. Predictors of poor prognosis include loss of consciousness or poor neurologic condition (i.e., high Hunt & Hess grade) on admission, older age, hypertension, preexisting medical illness, ≥1 mm thickness of subarachnoid blood on CT (Fisher grade 3), seizures, cerebral edema, aneurysm location in the basilar artery, and symptomatic vasospasm.235–239 Scales quantifying degree of physiologic illness are also predictive of outcome in patients with SAH.240 Long-term survivors of the initial hemorrhage continue to suffer a 3% annual risk of re-hemorrhage.
Key Points
Acknowledgments
This work was supported by grants from the National Institutes of Health (NS35966 and NS044885).
Frontera JA, Claassen J, Schmidt JM, Wartenberg KE, Temes R, Connolly ESJr, et al. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified Fisher scale. Neurosurgery. 2006;59:21-27.
Macdonald RL, Pluta RM, Zhang JH. Cerebral vasospasm after subarachnoid hemorrhage: the emerging revolution. Nat Clin Pract Neurol. 2007;3:256-263.
Mayer SA, Brun NC, Begtrup K, Broderick J, Davis S, Diringer MN, et al. Efficacy and safety of recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med. 2008;358:2127-2137.
Suarez JI, Tarr RW, Selman WR. Aneurysmal subarachnoid hemorrhage. N Engl J Med. 2006;354:387-396.
This paper provides a comprehensive review of aneurysmal SAH.
Treggiari MM, Deem S. Which H is the most important in triple-H therapy for cerebral vasospasm? Curr Opin Crit Care. 2009;15:83-86.
Vergouwen MD, de Haan RJ, Vermeulen M, Roos YB. Effect of statin treatment on vasospasm, delayed cerebral ischemia, and functional outcome in patients with aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis update. Stroke. 2010;41:e47-e52.
Anderson CS, Huang Y, Wang JG, Arima H, Neal B, Peng B, et al. Intensive blood pressure reduction in acute cerebral haemorrhage trial (INTERACT): a randomised pilot trial. Lancet Neurol. 2008;7:391-399.
Prasad K Prasad K, Mendelow AD, Gregson B. Surgery for primary supratentorial intracerebral haemorrhage. Cochrane Database Syst Rev 2008;CD000200.
1 Foulkes MA, Wolf PA, Price TR, et al. The Stroke Data Bank: design, methods, and baseline characteristics. Stroke. 1988;19:547-554.
2 Suzuki K, Kutsuzawa T, Takita K, et al. Clinico-epidemiologic study of stroke in Akita, Japan. Stroke. 1987;18:402-406.
3 Kay R, Woo J, Kreel L, et al. Stroke subtypes among Chinese living in Hong Kong: the Shatin Stroke Registry. Neurology. 1992;42:985-987.
4 Feldmann E. Intracerebral hemorrhage. Current concepts of cerebrovascular disease and stroke. 1991;22:684-691.
5 Brott T, Broderick J, Kothari R, et al. Early hemorrhage growth in patients with intracerebral hemorrhage. Stroke. 1997;28:1-5.
6 Mendelow AD, Bullock R, Teasdale GM, et al. Intracranial haemorrhage induced at arterial pressure in the rat. Part 2: Short term changes in local cerebral blood flow measured by autoradiography. Neurol Res. 1984;6:189-193.
7 Nath FP, Kelly PT, Jenkins A, et al. Effects of experimental intracerebral hemorrhage on blood flow, capillary permeability, and histochemistry. J Neurosurg. 1987;66:555-562.
8 Sills C, Villar-Cordova C, Pasteur W, et al. Demonstration of hypoperfusion surrounding intracerebral hematoma in humans. J Stroke Cerebrovasc Dis. 1996;6:17-24.
9 Videen TO, Dunford-Shore JE, Diringer MN, et al. Correction for partial volume effects in regional blood flow measurements adjacent to hematomas in humans with intracerebral hemorrhage: implementation and validation. J Comput Assist Tomogr. 1999;23:248-256.
10 Pascual AM, Lopez-Mut JV, Benlloch V, et al. Perfusion-weighted magnetic resonance imaging in acute intracerebral hemorrhage at baseline and during the 1st and 2nd week: a longitudinal study. Cerebrovasc Dis. 2007;23:6-13.
11 Hirano T, Read SJ, Abbott DF, et al. No evidence of hypoxic tissue on 18F-fluoromisonidazole PET after intracerebral hemorrhage. Neurology. 1999;53:2179-2182.
12 Zazulia AR, Diringer MN, Videen TO, et al. Hypoperfusion without ischemia surrounding acute intracerebral hemorrhage. J Cereb Blood Flow Metab. 2001;21:804-810.
13 Schellinger PD, Fiebach JB, Hoffmann K, et al. Stroke MRI in intracerebral hemorrhage: is there a perihemorrhagic penumbra? Stroke. 2003;34:1674-1679.
14 Lee KR, Kawai N, Kim S, et al. Mechanisms of edema formation after intracerebral hemorrhage: effects of thrombin on cerebral blood flow, blood-brain barrier permeability, and cell survival in a rat model. J Neurosurg. 1997;86:272-278.
15 Wagner K, Xi G, Hua Y, de Courten-Myers G, Myers R, Brott T, et al. White matter edema in experimental lobar intracerebral hemorrhage. Stroke. 1995;26:178.
16 Yang GY, Betz AL, Chenevert TL, et al. Experimental intracerebral hemorrhage: relationship between brain edema, blood flow, and blood-brain barrier permeability in rats. J Neurosurg. 1994;81:93-102.
17 Zazulia AR, Diringer MN, Derdeyn CP, et al. Progression of mass effect after intracerebral hemorrhage. Stroke. 1999;30:1167-1173.
18 Gebel JMJr, Jauch EC, Brott TG, et al. Relative edema volume is a predictor of outcome in patients with hyperacute spontaneous intracerebral hemorrhage. Stroke. 2002;33:2636-2641.
19 Arima H, Wang JG, Huang Y, et al. Significance of perihematomal edema in acute intracerebral hemorrhage: the INTERACT trial. Neurology. 2009;73:1963-1968.
20 Brott T, Thalinger K, Hertzberg V. Hypertension as a risk factor for spontaneous intracerebral hemorrhage. Stroke. 1986;17:1078-1083.
21 Randomised trial of a perindopril-based blood-pressure-lowering regimen among 6,105 individuals with previous stroke or transient ischaemic attack. Lancet. 2001;358:1033-1041.
22 Ojemann RG, Heros RC. Spontaneous brain hemorrhage. Stroke. 1983;14:468-475.
23 Gross CR, Kase CS, Mohr JP, et al. Stroke in south Alabama: incidence and diagnostic features—a population based study. Stroke. 1984;15:249-255.
24 Bruno A, Carter S, Qualls C, et al. Incidence of spontaneous intracerebral hemorrhage among Hispanics and non-Hispanic whites in New Mexico. Neurology. 1996;47:405-408.
25 Tanaka H, Ueda Y, Date C, et al. Incidence of stroke in Shibata, Japan: 1976-1978. Stroke. 1981;12:460-466.
26 Segal AZ, Chiu RI, Eggleston-Sexton PM, et al. Low cholesterol as a risk factor for primary intracerebral hemorrhage: A case-control study. Neuroepidemiology. 1999;18:185-193.
27 Goldstein LB, Amarenco P, Szarek M, et al. Hemorrhagic stroke in the Stroke Prevention by Aggressive Reduction in Cholesterol Levels study. Neurology. 2008;70:2364-2370.
28 Kurth T, Kase CS, Berger K, et al. Smoking and the risk of hemorrhagic stroke in men. Stroke. 2003;34:1151-1155.
29 Juvela S, Hillbom M, Palomaki H. Risk factors for spontaneous intracerebral hemorrhage. Stroke. 1995;26:1558-1564.
30 Sturgeon JD, Folsom AR, Longstreth WTJr, et al. Risk factors for intracerebral hemorrhage in a pooled prospective study. Stroke. 2007;38:2718-2725.
31 Burchfiel CM, Curb JD, Rodriguez BL, et al. Glucose intolerance and 22-year stroke incidence. The Honolulu Heart Program. Stroke. 1994;25:951-957.
32 Rodriguez BL, D’Agostino R, Abbott RD, et al. Risk of hospitalized stroke in men enrolled in the Honolulu Heart Program and the Framingham Study: A comparison of incidence and risk factor effects. Stroke. 2002;33:230-236.
33 Mohr JP, Kistler JP, et al. Intracranial aneurysms. In: Mohr JP, editor. Stroke: pathophysiology, diagnosis and management. London: Churchill Livingstone; 1985:643-677.
34 Fisher CM. Pathological observations in hypertensive cerebral hemorrhage. J Neuropathol Exp Neurol. 1971;30:536-550.
35 Fisher CM. Cerebral miliary aneurysms in hypertension. Am J Pathol. 1972;66:313-330.
36 Brown RDJr, Wiebers DO, Torner JC, et al. Frequency of intracranial hemorrhage as a presenting symptom and subtype analysis: a population-based study of intracranial vascular malformations in Olmsted County, Minnesota. J Neurosurg. 1996;85:29-32.
37 Hook O, Johanson C. Intracranial arteriovenous aneurysms: a follow-up study with particular attention to their growth. Arch Neurol Psychiatry. 1958;80:39.
38 Itoh Y, Yamada M, Hayakawa M, et al. Cerebral amyloid angiopathy: a significant cause of cerebellar as well as lobar cerebral hemorrhage in the elderly. J Neurol Sci. 1993;116:135-141.
39 Masuda J, Tanaka K, Ueda K, et al. Autopsy study of incidence and distribution of cerebral amyloid angiopathy in Hisayama, Japan. Stroke. 1988;19:205-210.
40 Vinters HV, Gilbert JJ. Cerebral amyloid angiopathy: incidence and complications in the aging brain. II. The distribution of amyloid vascular changes. Stroke. 1983;14:924-928.
41 Greenberg SM, Vonsattel JP, Segal AZ, et al. Association of apolipoprotein E epsilon2 and vasculopathy in cerebral amyloid angiopathy. Neurology. 1998;50:961-965.
42 O’Donnell HC, Rosand J, Knudsen KA, et al. Apolipoprotein E genotype and the risk of recurrent lobar intracerebral hemorrhage. N Engl J Med. 2000;342:240-245.
43 Knudsen KA, Rosand J, Karluk D, et al. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology. 2001;56:537-539.
44 Ly JV, Donnan GA, Villemagne VL, et al. 11C-PIB binding is increased in patients with cerebral amyloid angiopathy-related hemorrhage. Neurology. 2010;74:487-493.
45 Flaherty ML, Kissela B, Woo D, et al. The increasing incidence of anticoagulant-associated intracerebral hemorrhage. Neurology. 2007;68:116-121.
46 Rosand J, Eckman MH, Knudsen KA, et al. The effect of warfarin and intensity of anticoagulation on outcome of intracerebral hemorrhage. Arch Intern Med. 2004;164:880-884.
47 Flibotte JJ, Hagan N, O’Donnell J, et al. Warfarin, hematoma expansion, and outcome of intracerebral hemorrhage. Neurology. 2004;63:1059-1064.
48 Sansing LH, Messe SR, Cucchiara BL, et al. Prior antiplatelet use does not affect hemorrhage growth or outcome after ICH. Neurology. 2009;72:1397-1402.
49 Naidech AM, Jovanovic B, Liebling S, et al. Reduced platelet activity is associated with early clot growth and worse 3-month outcome after intracerebral hemorrhage. Stroke. 2009;40:2398-2401.
50 Lieu AS, Hwang SL, Howng SL, et al. Brain tumors with hemorrhage. J Formos Med Assoc. 1999;98:365-367.
51 Walsh TJ, Hier DB, Caplan LR. Aspergillosis of the central nervous system: clinicopathological analysis of 17 patients. Ann Neurol. 1985;18:574-582.
52 Erdem G, Vanderford PA, Bart RD. Intracranial hemorrhage in herpes simplex encephalitis: an unusual presentation. Pediatr Neurol. 2002;27:221-223.
53 Younger DS, Hays AP, Brust JC, et al. Granulomatous angiitis of the brain. An inflammatory reaction of diverse etiology. Arch Neurol. 1988;45:514-518.
54 de Bruijn SF, de Haan RJ, Stam J. Clinical features and prognostic factors of cerebral venous sinus thrombosis in a prospective series of 59 patients. For The Cerebral Venous Sinus Thrombosis Study Group. J Neurol Neurosurg Psychiatry. 2001;70:105-108.
55 Alvarez-Sabin J, Turon A, Lozano-Sanchez M, et al. Delayed posttraumatic hemorrhage. “Spat-apoplexie”. Stroke. 1995;26:1531-1535.
56 Henderson RD, Phan TG, Piepgras DG, et al. Mechanisms of intracerebral hemorrhage after carotid endarterectomy. J Neurosurg. 2001;95:964-969.
57 Martin-Schild S, Albright KC, Hallevi H, et al. Intracerebral hemorrhage in cocaine users. Stroke. 2010;41:680-684.
58 Aviv RI, d’Esterre CD, Murphy BD, et al. Hemorrhagic transformation of ischemic stroke: prediction with CT perfusion. Radiology. 2009;250:867-877.
59 Claassen J, Jette N, Chum F, et al. Electrographic seizures and periodic discharges after intracerebral hemorrhage. Neurology. 2007;69:1356-1365.
60 Leira R, Davalos A, Silva Y, et al. Early neurologic deterioration in intracerebral hemorrhage: predictors and associated factors. Neurology. 2004;63:461-467.
61 Dolinskas CA, Bilaniuk LT, Zimmerman RA, et al. Computed tomography of intracerebral hematomas. I. Transmission CT observations on hematoma resolution. AJR Am J Roentgenol. 1977;129:681-688.
62 Takasugi S, Ueda S, Matsumoto K. Chronological changes in spontaneous intracerebral hematoma—an experimental and clinical study. Stroke. 1985;16:651-658.
63 Franke CL, van Swieten JC, van Gijn J. Residual lesions on computed tomography after intracerebral hemorrhage. Stroke. 1991;22:1530-1533.
64 Chalela JA, Kidwell CS, Nentwich LM, et al. Magnetic resonance imaging and computed tomography in emergency assessment of patients with suspected acute stroke: a prospective comparison. Lancet. 2007;369:293-298.
65 Oppenheim C, Touze E, Hernalsteen D, et al. Comparison of five MR sequences for the detection of acute intracranial hemorrhage. Cerebrovasc Dis. 2005;20:388-394.
66 Fiebach JB, Schellinger PD, Gass A, et al. Stroke magnetic resonance imaging is accurate in hyperacute intracerebral hemorrhage: a multicenter study on the validity of stroke imaging. Stroke. 2004;35:502-506.
67 Brazzelli M, Sandercock PA, Chappell FM, et al. Magnetic resonance imaging versus computed tomography for detection of acute vascular lesions in patients presenting with stroke symptoms. Cochrane Database Syst Rev 2009;4:CD007424.
68 Singer OC, Sitzer M, du Mesnil de RR, et al. Practical limitations of acute stroke MRI due to patient-related problems. Neurology. 2004;62:1848-1849.
69 Zimmerman RD, Heier LA, Snow RB, et al. Acute intracranial hemorrhage: intensity changes on sequential MR scans at 0.5 T. AJR Am J Roentgenol. 1988;150:651-661.
70 Tsushima Y, Aoki J, Endo K. Brain microhemorrhages detected on T2-weighted gradient-echo MR images. AJNR Am J Neuroradiol. 2003;24:88-96.
71 Zhu XL, Chan MS, Poon WS. Spontaneous intracranial hemorrhage: which patients need diagnostic cerebral angiography? A prospective study of 206 cases and review of the literature. Stroke. 1997;28:1406-1409.
72 Delgado Almandoz JE, Schaefer PW, Forero NP, et al. Diagnostic accuracy and yield of multidetector CT angiography in the evaluation of spontaneous intraparenchymal cerebral hemorrhage. AJNR Am J Neuroradiol. 2009;30:1213-1221.
73 Yoon DY, Chang SK, Choi CS, et al. Multidetector row CT angiography in spontaneous lobar intracerebral hemorrhage: a prospective comparison with conventional angiography. AJNR Am J Neuroradiol. 2009;30:962-967.
74 Delgado Almandoz JE, Yoo AJ, Stone MJ, et al. The spot sign score in primary intracerebral hemorrhage identifies patients at highest risk of in-hospital mortality and poor outcome among survivors. Stroke. 2010;41:54-60.
75 Almandoz JE, Yoo AJ, Stone MJ, et al. The spot sign score in primary intracerebral hemorrhage identifies patients at highest risk of in-hospital mortality and poor outcome among survivors. Stroke. 2010;41:54-60.
76 Gujjar AR, Deibert E, Manno EM, et al. Mechanical ventilation for ischemic stroke and intracerebral hemorrhage: indications, timing, and outcome. Neurology. 1998;51:447-451.
77 Brucia J, Rudy E. The effect of suction catheter insertion and tracheal stimulation in adults with severe brain injury. Heart Lung. 1996;25:295-303.
78 Robinson N, Clancy M. In patients with head injury undergoing rapid sequence intubation, does pretreatment with intravenous lignocaine/lidocaine lead to an improved neurological outcome? A review of the literature. Emerg Med J. 2001;18:453-457.
79 Modica PA, Tempelhoff R. Intracranial pressure during induction of anaesthesia and tracheal intubation with etomidate-induced EEG burst suppression. Can J Anaesth. 1992;39:236-241.
80 Hoffman WE, Charbel FT, Ausman JI. Cerebral blood flow and metabolic response to etomidate and ischemia. Neurol Res. 1997;19:41-44.
81 Carlberg B, Asplund K, Hagg E. The prognostic value of admission blood pressure in patients with acute stroke. Stroke. 1993;24:1372-1375.
82 Wallace JD, Levy LL. Blood pressure after stroke. JAMA. 1981;246:2177-2180.
83 Dandapani BK, Suzuki S, Kelley RE, et al. Relation between blood pressure and outcome in intracerebral hemorrhage. Stroke. 1995;26:21-24.
84 Fujii Y, Tanaka R, Takeuchi S, et al. Hematoma enlargement in spontaneous intracerebral hemorrhage. J Neurosurg. 1994;80:51-57.
85 Maruishi M, Shima T, Okada Y, et al. Involvement of fluctuating high blood pressure in the enlargement of spontaneous intracerebral hematoma. Neurol Med Chir (Tokyo). 2001;41:300-304.
86 Fogelholm R, Avikainen S, Murros K. Prognostic value and determinants of first-day mean arterial pressure in spontaneous supratentorial intracerebral hemorrhage. Stroke. 1997;28:1396-1400.
87 Terayama Y, Tanahashi N, Fukuuchi Y, et al. Prognostic value of admission blood pressure in patients with intracerebral hemorrhage. Keio Cooperative Stroke Study. Stroke. 1997;28:1185-1188.
88 Powers WJ. Acute hypertension after stroke: the scientific basis for treatment decisions. Neurology. 1993;43:461-467.
89 Strandgaard S, Olesen J, Skinhoj E, et al. Autoregulation of brain circulation in severe arterial hypertension. Br Med J. 1973;1:507-510.
90 Strandgaard S. Autoregulation of cerebral blood flow in hypertensive patients. The modifying influence of prolonged antihypertensive treatment on the tolerance to acute, drug-induced hypotension. Circulation. 1976;53:720-727.
91 Kaneko T, Sawada T, Niimi T, et al. Lower limit of blood pressure in treatment of acute hypertensive intracerebral hemorrhage. J Cereb Blood Flow Metab. 1983;3:S51-S52.
92 von Helden A, Schneider GH, Unterberg A, et al. Monitoring of jugular venous oxygen saturation in comatose patients with subarachnoid haemorrhage and intracerebral haematomas. Acta Neurochir Suppl (Wien). 1993;59:102-106.
93 Powers WJ, Zazulia AR, Videen TO, et al. Autoregulation of cerebral blood flow surrounding acute intracerebral hemorrhage. Neurology. 2001;57:18-24.
94 Anderson CS, Huang Y, Wang JG, et al. Intensive blood pressure reduction in acute cerebral haemorrhage trial (INTERACT): a randomised pilot trial. Lancet Neurol. 2008;7:391-399.
95 Mayer SA, Brun NC, Begtrup K, et al. Efficacy and safety of recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med. 2008;358:2127-2137.
96 Mayer SA, Davis SM, Skolnick BE, et al. Can a subset of intracerebral hemorrhage patients benefit from hemostatic therapy with recombinant activated factor VII? Stroke. 2009;40:833-840.
97 Tuhrim S, Horowitz DR, Sacher M, et al. Volume of ventricular blood is an important determinant of outcome in supratentorial intracerebral hemorrhage. Crit Care Med. 1999;27:617-621.
98 de Weerd AW. The prognosis of intraventricular hemorrhage. J Neurol. 1979;222:46-51.
99 Portenoy RK, Lipton RB, Berger AR, et al. Intracerebral haemorrhage: a model for the prediction of outcome. J Neurol Neurosurg Psychiatry. 1987;50:976-979.
100 Naff NJ, Carhuapoma JR, Williams MA, et al. Treatment of intraventricular hemorrhage with urokinase : effects on 30-Day survival. Stroke. 2000;31:841-847.
101 Andrews BT, Chiles BWIII, Olsen WL, et al. The effect of intracerebral hematoma location on the risk of brain-stem compression and on clinical outcome. J Neurosurg. 1988;69:518-522.
102 Janny P, Papo I, Chazal J, et al. Intracranial hypertension and prognosis of spontaneous intracerebral haematomas. A correlative study of 60 patients. Acta Neurochir (Wien). 1982;61:181-186.
103 Qureshi AI, Geocadin RG, Suarez JI, et al. Long-term outcome after medical reversal of transtentorial herniation in patients with supratentorial mass lesions. Crit Care Med. 2000;28:1556-1564.
104 Bereczki D, Fekete I, Prado GF, et al. Mannitol for acute stroke. Cochrane Database Syst Rev 2007;CD001153.
105 Poungvarin N, Bhoopat W, Viriyavejakul A, et al. Effects of dexamethasone in primary supratentorial intracerebral hemorrhage. N Engl J Med. 1987;316:1229-1233.
106 McKissock W, Richardson A, Taylor J. Primary intracerebral hemorrhage: a controlled trial of surgical and conservative treatment in 180 unselected cases. Lancet. 1961;2:221-226.
107 Batjer HH, Reisch JS, Allen BC, et al. Failure of surgery to improve outcome in hypertensive putamenal hemorrhage. A prospective randomized trial. Arch Neurol. 1990;47:1103-1106.
108 Juvela S, Heiskanen O, Poranen A, et al. The treatment of spontaneous intracerebral hemorrhage. A prospective randomized trial of surgical and conservative treatment. J Neurosurg. 1989;70:755-758.
109 Prasad K, Mendelow AD, Gregson B. Surgery for primary supratentorial intracerebral haemorrhage. Cochrane Database Syst Rev 2008;CD000200.
110 Hankey GJ, Hon C. Surgery for primary intracerebral hemorrhage: is it safe and effective? A systematic review of case series and randomized trials. Stroke. 1997;28:2126-2132.
111 Auer LM, Deinsberger W, Niederkorn K, et al. Endoscopic surgery versus medical treatment for spontaneous intracerebral hematoma: a randomized study. J Neurosurg. 1989;70:530-535.
112 Morgenstern LB, Frankowski RF, Shedden P, et al. Surgical treatment for intracerebral hemorrhage (STICH): a single-center, randomized clinical trial. Neurology. 1998;51:1359-1363.
113 Zuccarello M, Brott T, Derex L, et al. Early surgical treatment for supratentorial intracerebral hemorrhage: a randomized feasibility study. Stroke. 1999;30:1833-1839.
114 Morgenstern LB, Demchuk AM, Kim DH, et al. Rebleeding leads to poor outcome in ultra-early craniotomy for intracerebral hemorrhage. Neurology. 2001;56:1294-1299.
115 Mendelow AD, Gregson BA, Fernandes HM, et al. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the International Surgical Trial in Intracerebral Haemorrhage (STICH): a randomised trial. Lancet. 2005;365:387-397.
116 Kirollos RW, Tyagi AK, Ross SA, et al. Management of spontaneous cerebellar hematomas: a prospective treatment protocol. Neurosurgery. 2001;49:1378-1386.
117 Morioka J, Fujii M, Kato S, et al. Surgery for spontaneous intracerebral hemorrhage has greater remedial value than conservative therapy. Surg Neurol. 2006;65:67-72.
118 Qureshi AI, Tuhrim S, Broderick JP, et al. Spontaneous intracerebral hemorrhage. N Engl J Med. 2001;344:1450-1460.
119 Kobayashi S, Sato A, Kageyama Y, et al. Treatment of hypertensive cerebellar hemorrhage—surgical or conservative management? Neurosurgery. 1994;34:246-250.
120 Hacke W, Donnan G, Fieschi C, et al. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet. 2004;363:768-774.
121 Patel SC, Mody A. Cerebral hemorrhagic complications of thrombolytic therapy. Prog Cardiovasc Dis. 1999;42:217-233.
122 Mahaffey KW, Granger CB, Sloan MA, et al. Neurosurgical evacuation of intracranial hemorrhage after thrombolytic therapy for acute myocardial infarction: experience from the GUSTO-I trial. Global Utilization of Streptokinase and tissue-plasminogen activator (tPA) for Occluded Coronary Arteries. Am Heart J. 1999;138:493-499.
123 Kase CS, Furlan AJ, Wechsler LR, et al. Cerebral hemorrhage after intra-arterial thrombolysis for ischemic stroke: the PROACT II trial. Neurology. 2001;57:1603-1610.
124 Lansberg MG, Albers GW, Wijman CA. Symptomatic intracerebral hemorrhage following thrombolytic therapy for acute ischemic stroke: a review of the risk factors. Cerebrovasc Dis. 2007;24:1-10.
125 National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. N Engl J Med. 1995;333:1581-1587.
126 Passero S, Rocchi R, Rossi S, et al. Seizures after spontaneous supratentorial intracerebral hemorrhage. Epilepsia. 2002;43:1175-1180.
127 Zurasky JA, Aiyagari V, Zazulia AR, et al. Early mortality following spontaneous intracerebral hemorrhage. Neurology. 2005;64:725-727.
128 Bamford J, Dennis M, Sandercock P, et al. The frequency, causes and timing of death within 30 days of a first stroke: the Oxfordshire Community Stroke Project. J Neurol Neurosurg Psychiatry. 1990;53:824-829.
129 Lacut K, Bressollette L, Le Gal G, et al. Prevention of venous thrombosis in patients with acute intracerebral hemorrhage. Neurology. 2005;65:865-869.
130 Boeer A, Voth E, Henze T, et al. Early heparin therapy in patients with spontaneous intracerebral haemorrhage. J Neurol Neurosurg Psychiatry. 1991;54:466-467.
131 Orken DN, Kenangil G, Ozkurt H, et al. Prevention of deep venous thrombosis and pulmonary embolism in patients with acute intracerebral hemorrhage. Neurologist. 2009;15:329-331.
132 Daverat P, Castel JP, Dartigues JF, et al. Death and functional outcome after spontaneous intracerebral hemorrhage. A prospective study of 166 cases using multivariate analysis. Stroke. 1991;22:1-6.
133 Counsell C, Boonyakarnkul S, Dennis M, et al. Primary Intracerebral haemorrhage in the Oxfordshire Community Stroke Project; 2. Prognosis. Cerebrovasc Dis. 1995;5:26-34.
134 Hemphill JCIII, Bonovich DC, Besmertis L, et al. The ICH score: a simple, reliable grading scale for intracerebral hemorrhage. Stroke. 2001;32:891-897.
135 Becker KJ, Baxter AB, Cohen WA, et al. Withdrawal of support in intracerebral hemorrhage may lead to self- fulfilling prophecies. Neurology. 2001;56:766-772.
136 Bailey RD, Hart RG, Benavente O, et al. Recurrent brain hemorrhage is more frequent than ischemic stroke after intracranial hemorrhage. Neurology. 2001;56:773-777.
137 Weir B. Intracranial aneurysms and subarachnoid hemorrhage: an overview. In: Wilkins RW, Rengachary SS, editors. Neurosurgery. New York: McGraw-Hill; 1985:1308-1329.
138 Suarez JI, Tarr RW, Selman WR. Aneurysmal subarachnoid hemorrhage. N Engl J Med. 2006;354:387-396.
139 Sekhar LN, Heros RC. Origin, growth, and rupture of saccular aneurysms: a review. Neurosurgery. 1981;8:248-260.
140 Zacharia BE, Hickman ZL, Grobelny BT, et al. Epidemiology of aneurysmal subarachnoid hemorrhage. Neurosurg Clin N Am. 2010;21:221-233.
141 Brown RDJr, Huston J, Hornung R, et al. Screening for brain aneurysm in the Familial Intracranial Aneurysm study: frequency and predictors of lesion detection. J Neurosurg. 2008;108:1132-1138.
142 Weir B. Epidemiology. In: Weir B, editor. Aneurysms affecting the nervous system. Baltimore: Williams and Wilkins; 1987:19-53.
143 Lazaridis C, Naval N. Risk factors and medical management of vasospasm after subarachnoid hemorrhage. Neurosurg Clin N Am. 2010;21:353-364.
144 Morris KM, Shaw MD, Foy PM. Smoking and subarachnoid haemorrhage: a case control study. Br J Neurosurg. 1992;6:429-432.
145 Longstreth WT, Nelson LM, Koepsell TD, et al. Subarachnoid hemorrhage and hormonal factors in women. A population-based case-control study. Ann Intern Med. 1994;121:168-173.
146 Gillingham FJ. The management of ruptured intracranial aneurysms. Scott Med J. 1967;12:377-383.
147 Fisher CM, Roberson GH, Ojemann RG. Cerebral vasospasm with ruptured saccular aneurysm—the clinical manifestations. Neurosurgery. 1977;1:245-248.
148 Huff JS, Perron AD. Onset seizures independently predict poor outcome after subarachnoid hemorrhage. Neurology. 2001;56:1423-1424.
149 Winn HR, Richardson AE, Jane JA. The long-term prognosis in untreated cerebral aneurysms: I. The incidence of late hemorrhage in cerebral aneurysm: a 10-year evaluation of 364 patients. Ann Neurol. 1977;1:358-370.
150 Hasan D, Vermeulen M, Wijdicks EF, et al. Management problems in acute hydrocephalus after subarachnoid hemorrhage. Stroke. 1989;20:747-753.
151 Vermeij FH, Hasan D, Vermeulen M, et al. Predictive factors for deterioration from hydrocephalus after subarachnoid hemorrhage. Neurology. 1994;44:1851-1855.
152 Macdonald RL, Pluta RM, Zhang JH. Cerebral vasospasm after subarachnoid hemorrhage: the emerging revolution. Nat Clin Pract Neurol. 2007;3:256-263.
153 Kassell NF, Sasaki T, Colohan AR, et al. Cerebral vasospasm following aneurysmal subarachnoid hemorrhage. Stroke. 1985;16:562-572.
154 Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery. 1980;6:1-9.
155 Frontera JA, Claassen J, Schmidt JM, et al. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified fisher scale. Neurosurgery. 2006;59:21-27.
156 Diringer MN. Neuroendocrine regulation of sodium and volume following subarachnoid hemorrhage. Clin Neuropharmacol. 1995;18:114-126.
157 Mori T, Katayama Y, Kawamata T, et al. Improved efficiency of hypervolemic therapy with inhibition of natriuresis by fludrocortisone in patients with aneurysmal subarachnoid hemorrhage. J Neurosurg. 1999;91:947-952.
158 Brouwers PJ, Wijdicks EF, Hasan D, et al. Serial electrocardiographic recording in aneurysmal subarachnoid hemorrhage. Stroke. 1989;20:1162-1167.
159 Deibert E, Barzilai B, Braverman AC, et al. Clinical significance of elevated troponin I levels in patients with nontraumatic subarachnoid hemorrhage. J Neurosurg. 2003;98:741-746.
160 Andreoli A, di Pasquale G, Pinelli G, et al. Subarachnoid hemorrhage: frequency and severity of cardiac arrhythmias. A survey of 70 cases studied in the acute phase. Stroke. 1987;18:558-564.
161 Estanol BV, Dergal EB, Cesarman E, San Martin OM, Loyo M, Ortego RP. Cardiac arrhythmias associated with subarachnoid hemorrhage: prospective study. Neurosurgery. 1969;5:675-680.
162 Banki NM, Kopelnik A, Dae MW, et al. Acute neurocardiogenic injury after subarachnoid hemorrhage. Circulation. 2005;112:3314-3319.
163 Lee VH, Connolly HM, Fulgham JR, et al. Tako-tsubo cardiomyopathy in aneurysmal subarachnoid hemorrhage: an underappreciated ventricular dysfunction. J Neurosurg. 2006;105:264-270.
164 Jain R, Deveikis J, Thompson BG. Management of patients with stunned myocardium associated with subarachnoid hemorrhage. AJNR Am J Neuroradiol. 2004;25:126-129.
165 Shimoda M, Oda S, Tsugane R, et al. Intracranial complications of hypervolemic therapy in patients with a delayed ischemic deficit attributed to vasospasm. J Neurosurg. 1993;78:423-429.
166 Miller JA, Dacey RGJr, Diringer MN. Safety of hypertensive hypervolemic therapy with phenylephrine in the treatment of delayed ischemic deficits after subarachnoid hemorrhage. Stroke. 1995;26:2260-2266.
167 Solenski NJ, Haley ECJr, Kassell NF, et al. Medical complications of aneurysmal subarachnoid hemorrhage: a report of the multicenter, cooperative aneurysm study. Participants of the Multicenter Cooperative Aneurysm Study. Crit Care Med. 1995;23:1007-1017.
168 Wartenberg KE, Mayer SA. Medical complications after subarachnoid hemorrhage: new strategies for prevention and management. Curr Opin Crit Care. 2006;12:78-84.
169 Boesiger BM, Shiber JR. Subarachnoid hemorrhage diagnosis by computed tomography and lumbar puncture: are fifth generation CT scanners better at identifying subarachnoid hemorrhage? J Emerg Med. 2005;29:23-27.
170 Kirkwood JR. Intracranial aneurysms and subarachnoid hemorrhage. In: Kirkwood JR, editor. Essentials of neuroimaging. New York: Churchill Livingstone; 1990:101-118.
171 Van Der Meulen JP. Cerebrospinal fluid xanthochromia: an objective index. Neurology. 1966;16:170-178.
172 Beetham R. CSF spectrophotometry for bilirubin—why and how? Scand J Clin Lab Invest. 2009;69:1-7.
173 Andaluz N, Zuccarello M. Yield of further diagnostic work-up of cryptogenic subarachnoid hemorrhage based on bleeding patterns on computed tomographic scans. Neurosurgery. 2008;62:1040-1046.
174 van Gijn J, Rinkel GJ. Subarachnoid haemorrhage: diagnosis, causes and management. Brain. 2001;124:249-278.
175 Marshall SA, Kathuria S, Nyquist P, et al. Noninvasive imaging techniques in the diagnosis and management of aneurysmal subarachnoid hemorrhage. Neurosurg Clin N Am. 2010;21:305-323.
176 Hunt WE, Hess RM. Surgical risk as related to time of intervention in the repair of intracranial aneurysms. J Neurosurg. 1968;28:14-20.
177 Report of World Federation of Neurological Surgeons Committee on a Universal Subarachnoid Hemorrhage Grading Scale. J Neurosurg. 1988;68:985-986.
178 Zubkov AY, Wijdicks EF. Antiepileptic drugs in aneurysmal subarachnoid hemorrhage. Rev Neurol Dis. 2008;5:178-181.
179 Rosengart AJ, Huo JD, Tolentino J, et al. Outcome in patients with subarachnoid hemorrhage treated with antiepileptic drugs. J Neurosurg. 2007;107:253-260.
180 Hoff R, Rinkel G, Verweij B, et al. Blood volume measurement to guide fluid therapy after aneurysmal subarachnoid hemorrhage: a prospective controlled study. Stroke. 2009;40:2575-2577.
181 Kramer AH. Statins in the management of aneurysmal subarachnoid hemorrhage—not (yet) a standard of care. Stroke. 2009;40:e80-e81.
182 Diringer MN, Wu KC, Verbalis JG, et al. Hypervolemic therapy prevents volume contraction but not hyponatremia following subarachnoid hemorrhage. Ann Neurol. 1992;31:543-550.
183 Hasan D, Lindsay KW, Wijdicks EF, et al. Effect of fludrocortisone acetate in patients with subarachnoid hemorrhage. Stroke. 1989;20:1156-1161.
184 Woo MH, Kale-Pradhan PB. Fludrocortisone in the treatment of subarachnoid hemorrhage-induced hyponatremia. Ann Pharmacother. 1997;31:637-639.
185 Katayama Y, Haraoka J, Hirabayashi H, et al. A randomized controlled trial of hydrocortisone against hyponatremia in patients with aneurysmal subarachnoid hemorrhage. Stroke. 2007;38:2373-2375.
186 Murphy T, Dhar R, Diringer M. Conivaptan bolus dosing for the correction of hyponatremia in the neurointensive care unit. Neurocrit Care. 2009;11:14-19.
187 Boet R, Chan MT, Poon WS, et al. Intravenous magnesium sulfate to improve outcome after aneurysmal subarachnoid hemorrhage: interim report from a pilot study. Acta Neurochir Suppl. 2005;95:263-264.
188 Wong GK, Chan MT, Boet R, et al. Intravenous magnesium sulfate after aneurysmal subarachnoid hemorrhage: a prospective randomized pilot study. J Neurosurg Anesthesiol. 2006;18:142-148.
189 Schmid-Elsaesser R, Kunz M, Zausinger S, et al. Intravenous magnesium versus nimodipine in the treatment of patients with aneurysmal subarachnoid hemorrhage: a randomized study. Neurosurgery. 2006;58:1054-1065.
190 van den Bergh WM, Algra A, van Kooten F, et al. Magnesium sulfate in aneurysmal subarachnoid hemorrhage: a randomized controlled trial. Stroke. 2005;36:1011-1015.
191 Prevedello DM, Cordeiro JG, de Morais AL, et al. Magnesium sulfate: role as possible attenuating factor in vasospasm morbidity. Surg Neurol. 2006;65(Suppl 1):S1.
192 Tseng MY, Czosnyka M, Richards H, et al. Effects of acute treatment with pravastatin on cerebral vasospasm, autoregulation, and delayed ischemic deficits after aneurysmal subarachnoid hemorrhage: a phase II randomized placebo-controlled trial. Stroke. 2005;36:1627-1632.
193 Lynch JR, Wang H, McGirt MJ, et al. Simvastatin reduces vasospasm after aneurysmal subarachnoid hemorrhage: results of a pilot randomized clinical trial. Stroke. 2005;36:2024-2026.
194 Kramer AH, Gurka MJ, Nathan B, et al. Statin use was not associated with less vasospasm or improved outcome after subarachnoid hemorrhage. Neurosurgery. 2008;62:422-427.
195 Chou SH, Smith EE, Badjatia N, et al. A randomized, double-blind, placebo-controlled pilot study of simvastatin in aneurysmal subarachnoid hemorrhage. Stroke. 2008;39:2891-2893.
196 Vergouwen MD, de Haan RJ, Vermeulen M, et al. Statin treatment and the occurrence of hemorrhagic stroke in patients with a history of cerebrovascular disease. Stroke. 2008;39:497-502.
197 Kassell NF, Torner JC, Adams HPJr. Antifibrinolytic therapy in the acute period following aneurysmal subarachnoid hemorrhage. Preliminary observations from the Cooperative Aneurysm Study. J Neurosurg. 1984;61:225-230.
198 Solomon RA, Fink ME. Current strategies for the management of aneurysmal subarachnoid hemorrhage. Arch Neurol. 1987;44:769-774.
199 Hillman J, Fridriksson S, Nilsson O, et al. Immediate administration of tranexamic acid and reduced incidence of early rebleeding after aneurysmal subarachnoid hemorrhage: a prospective randomized study. J Neurosurg. 2002;97:771-778.
200 Molyneux A, Kerr R, Stratton I, et al. International Subarachnoid Aneurysm Trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2143 patients with ruptured intracranial aneurysms: a randomised trial. Lancet. 2002;360:1267-1274.
201 Molyneux AJ, Kerr RS, Birks J, et al. Risk of recurrent subarachnoid haemorrhage, death, or dependence and standardised mortality ratios after clipping or coiling of an intracranial aneurysm in the International Subarachnoid Aneurysm Trial (ISAT): long-term follow-up. Lancet Neurol. 2009;8:427-433.
202 Vajkoczy P, Horn P, Thome C, et al. Regional cerebral blood flow monitoring in the diagnosis of delayed ischemia following aneurysmal subarachnoid hemorrhage. J Neurosurg. 2003;98:1227-1234.
203 Rigamonti A, Ackery A, Baker AJ. Transcranial Doppler monitoring in subarachnoid hemorrhage: a critical tool in critical care. Can J Anaesth. 2008;55:112-123.
204 Vora YY, Suarez-Almazor M, Steinke DE, et al. Role of transcranial Doppler monitoring in the diagnosis of cerebral vasospasm after subarachnoid hemorrhage. Neurosurgery. 1999;44:1237-1247.
205 Sloan MA, Haley ECJr, Kassell NF, et al. Sensitivity and specificity of transcranial Doppler ultrasonography in the diagnosis of vasospasm following subarachnoid hemorrhage. Neurology. 1989;39:1514-1518.
206 Petruk KC, West M, Mohr G, et al. Nimodipine treatment in poor-grade aneurysm patients. Results of a multicenter double-blind placebo-controlled trial. J Neurosurg. 1988;68:505-517.
207 Barker FG, Ogilvy CS. Efficacy of prophylactic nimodipine for delayed ischemic deficit after subarachnoid hemorrhage: a metaanalysis. J Neurosurg. 1996;84:405-414.
208 Muench E, Horn P, Bauhuf C, et al. Effects of hypervolemia and hypertension on regional cerebral blood flow, intracranial pressure, and brain tissue oxygenation after subarachnoid hemorrhage. Crit Care Med. 2007;35:1844-1851.
209 Raabe A, Beck J, Keller M, et al. Relative importance of hypertension compared with hypervolemia for increasing cerebral oxygenation in patients with cerebral vasospasm after subarachnoid hemorrhage. J Neurosurg. 2005;103:974-981.
210 Capampangan DJ, Wellik KE, Aguilar MI, et al. Does prophylactic postoperative hypervolemic therapy prevent cerebral vasospasm and improve clinical outcome after aneurysmal subarachnoid hemorrhage? Neurologist. 2008;14:395-398.
211 Lennihan L, Mayer SA, Fink ME, et al. Effect of hypervolemic therapy on cerebral blood flow after subarachnoid hemorrhage : a randomized controlled trial. Stroke. 2000;31:383-391.
212 Zwienenberg-Lee M, Hartman J, Rudisill N, et al. Effect of prophylactic transluminal balloon angioplasty on cerebral vasospasm and outcome in patients with Fisher grade III subarachnoid hemorrhage: results of a phase II multicenter, randomized, clinical trial. Stroke. 2008;39:1759-1765.
213 Manno EM, Gress DR, Schwamm LH, et al. Effects of induced hypertension on transcranial Doppler ultrasound velocities in patients after subarachnoid hemorrhage. Stroke. 1998;29:422-428.
214 Nolan CP, Macdonald RL. Can angiographic vasospasm be used as a surrogate marker in evaluating therapeutic interventions for cerebral vasospasm? Neurosurg Focus. 2006;21:E1.
215 Keyrouz SG, Diringer MN. Clinical review: Prevention and therapy of vasospasm in subarachnoid hemorrhage. Crit Care. 2007;11:220.
216 Koebbe CJ, Veznedaroglu E, Jabbour P, et al. Endovascular management of intracranial aneurysms: current experience and future advances. Neurosurgery. 2006;59:S93-102.
217 Kirmani JF, Alkawi A, Ahmed S, et al. Endovascular treatment of subarachnoid hemorrhage. Neurol Res. 2005;27(Suppl 1):S103-S107. S103-S107
218 Jaeger M, Schuhmann MU, Soehle M, et al. Continuous monitoring of cerebrovascular autoregulation after subarachnoid hemorrhage by brain tissue oxygen pressure reactivity and its relation to delayed cerebral infarction. Stroke. 2007;38:981-986.
219 Mori K, Arai H, Nakajima K, et al. Hemorheological and hemodynamic analysis of hypervolemic hemodilution therapy for cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Stroke. 1995;26:1620-1626.
220 Treggiari MM, Deem S. Which H is the most important in triple-H therapy for cerebral vasospasm? Curr Opin Crit Care. 2009;15:83-86.
221 Ekelund A, Reinstrup P, Ryding E, et al. Effects of iso- and hypervolemic hemodilution on regional cerebral blood flow and oxygen delivery for patients with vasospasm after aneurysmal subarachnoid hemorrhage. Acta Neurochir (Wien). 2002;144:703-712.
222 Eddleman CS, Hurley MC, Naidech AM, et al. Endovascular options in the treatment of delayed ischemic neurological deficits due to cerebral vasospasm. Neurosurg Focus. 2009;26:E6.
223 Eskridge JM, Newell DW, Pendleton GA. Transluminal angioplasty for treatment of vasospasm. Neurosurg Clin N Am. 1990;1:387-399.
224 Fujii Y, Takahashi A, Yoshimoto T. Effect of balloon angioplasty on high grade symptomatic vasospasm after subarachnoid hemorrhage. Neurosurg Rev. 1995;18:7-13.
225 Cross DTIII, Moran CJ, Angtuaco EE, et al. Intracranial pressure monitoring during intraarterial papaverine infusion for cerebral vasospasm. AJNR Am J Neuroradiol. 1998;19:1319-1323.
226 Milburn JM, Moran CJ, Cross DTIII, et al. Effect of intraarterial papaverine on cerebral circulation time. AJNR Am J Neuroradiol. 1997;18:1081-1085.
227 Sayama CM, Liu JK, Couldwell WT. Update on endovascular therapies for cerebral vasospasm induced by aneurysmal subarachnoid hemorrhage. Neurosurg Focus. 2006;21:E12.
228 Stiefel MF, Spiotta AM, Udoetuk JD, et al. Intra-arterial papaverine used to treat cerebral vasospasm reduces brain oxygen. Neurocrit Care. 2006;4:113-118.
229 Brisman JL, Eskridge JM, Newell DW. Neurointerventional treatment of vasospasm. Neurol Res. 2006;28:769-776.
230 Fraticelli AT, Cholley BP, Losser MR, et al. Milrinone for the treatment of cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Stroke. 2008;39:893-898.
231 Pierot L, Aggour M, Moret J. Vasospasm after aneurysmal subarachnoid hemorrhage: recent advances in endovascular management. Curr Opin Crit Care. 2010.
232 Frontera JA, Fernandez A, Schmidt JM, et al. Clinical response to hypertensive hypervolemic therapy and outcome after subarachnoid hemorrhage. Neurosurgery. 2010;66:35-41.
233 Vajkoczy P, Meyer B, Weidauer S, et al. Clazosentan (AXV-034343), a selective endothelin A receptor antagonist, in the prevention of cerebral vasospasm following severe aneurysmal subarachnoid hemorrhage: results of a randomized, double-blind, placebo-controlled, multicenter phase IIa study. J Neurosurg. 2005;103:9-17.
234 Macdonald RL, Kassell NF, Mayer S, et al. Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage (CONSCIOUS-1): randomized, double-blind, placebo-controlled phase 2 dose-finding trial. Stroke. 2008;39:3015-3021.
235 Claassen J, Carhuapoma JR, Kreiter KT, et al. Global cerebral edema after subarachnoid hemorrhage: frequency, predictors, and impact on outcome. Stroke. 2002;33:1225-1232.
236 Dennis LJ, Claassen J, Hirsch LJ, et al. Nonconvulsive status epilepticus after subarachnoid hemorrhage. Neurosurgery. 2002;51:1136-1143.
237 Lagares A, Gomez PA, Lobato RD, et al. Prognostic factors on hospital admission after spontaneous subarachnoid haemorrhage. Acta Neurochir (Wien). 2001;143:665-672.
238 Franz G, Brenneis C, Kampfl A, et al. Prognostic value of intraventricular blood in perimesencephalic nonaneurysmal subarachnoid hemorrhage. J Comput Assist Tomogr. 2001;25:742-746.
239 Qureshi AI, Sung GY, Razumovsky AY, et al. Early identification of patients at risk for symptomatic vasospasm after aneurysmal subarachnoid hemorrhage. Crit Care Med. 2000;28:984-990.
240 Schuiling WJ, de Weerd AW, Dennesen PJ, et al. The simplified acute physiology score to predict outcome in patients with subarachnoid hemorrhage. Neurosurgery. 2005;57:230-236.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
35 Nontraumatic Intracerebral and Subarachnoid Hemorrhage
Intracerebral Hemorrhage
Spontaneous (nontraumatic) intracerebral hemorrhage (ICH) accounts for approximately 10% of all strokes in North America and about 20% to 30% in East Asia. It is associated with greater mortality and more severe neurologic deficits than any other stroke subtype.1–3 Nearly half of all patients die within the first 30 days; survivors often have significant residual disability.4
Pathophysiology
The pathophysiologic mechanisms of brain injury due to ICH are complex. The primary injury is one of local tissue destruction as rupture of a cerebral blood vessel introduces a sudden stream of blood into the brain parenchyma. In over one third of patients, continued bleeding or rebleeding results in hematoma enlargement and further mechanical injury within the first few hours after onset.5 The mass of the hematoma produces tissue shifts within the intracranial cavity.
Experimental models of ICH consistently suggest that ischemia is an important part of the pathophysiology of ICH.6,7 In clinical studies, peri-clot and ipsilateral hemispheric hypoperfusion have been demonstrated,8–10 but the hypoperfusion does not appear to represent ischemia.11,12 Positron emission tomography (PET) studies in humans performed 5 to 22 hours after symptom onset showed that perihematomal cerebral metabolism was reduced to a greater degree than cerebral blood flow (CBF), suggesting that the hypoperfusion reflects reduced metabolic demand of the damaged tissue surrounding the hematoma rather than ongoing ischemia.12 Magnetic resonance imaging (MRI) studies within 6 hours after symptom onset demonstrate hypoperfusion without restricted diffusion, findings that are inconsistent with ischemia.13
Cerebral edema has been demonstrated to occur within hours of experimental ICH, variably thought to result from the toxic effects of blood-derived enzymes, from increased osmotic pressure exerted by clot-derived serum proteins, or from ischemia.14–16 The presence, time course, and importance of edema formation in humans are debated, however. Signal changes on radiographic studies after ICH indicate increased water content in the area surrounding the clot, but the clinical and pathophysiologic significance of this is not known. Edema does not appear to contribute to early increases in mass effect17 and is not associated with worsened functional outcome or increased mortality.18,19
Causes and Risk Factors
The leading risk factor for ICH, occurring in over half of all cases, is chronic hypertension.20 Long-term adequate treatment of chronic hypertension significantly reduces this risk.21 Increasing age is another risk factor, with a doubling of the rate of hemorrhage with each decade of life until age 80, when the incidence plateaus at nearly 25 times that of the previous decade.22 In the United States, ICH is 2 to 3 times more common in African Americans and Hispanics than in Caucasians (incidence rates of 32, 35, and 10 to 15 per 100,000 population, respectively).23,24 The incidence in Asian countries is considerably greater (61 per 100,000 population).25
Low serum cholesterol has been implicated in a number of studies,26 and use of high-dose statins appears to increase the risk of ICH, particularly in those with prior history of ICH.27 The impact of smoking,28 alcohol abuse,29,30 and diabetes31,32 on the risk of ICH is disputed.
Hypertensive Hemorrhage
Hypertensive ICH predominantly occurs deep in the cerebral hemispheres, most often in the putamen33 (Figure 35-1). Other frequently involved sites include the thalamus, lobar white matter, cerebellum, and pons. The common link between these sites is that they are all supplied by small penetrating arteries,34 perpendicular branches directly off major arteries that are subject to high sheer stress and that have no collaterals. These features make them vulnerable to the effects of increased blood pressure. Chronic hypertension damages the tunica media, resulting in lipohyalinosis, fibrinoid necrosis, and microaneurysms (Charcot-Bouchard aneurysms). Although Charcot-Bouchard aneurysms have been demonstrated in the weakened vessel walls of patients with ICH, their pathogenetic role in vascular rupture is uncertain.35 The occurrence of ICH in an atypical location, in multiple locations, or in association with subarachnoid hemorrhage raises the suspicion of a non-hypertensive etiology, such as a cerebral vascular anomaly, blood dyscrasia, or trauma.
Intracranial Aneurysms and Vascular Malformations
Approximately half of intracranial AVMs in adults present with hemorrhage.36 In 60% of cases, the hemorrhage is parenchymal, involving virtually any location within the cerebrum, brainstem, or cerebellum.37 The majority of AVMs become symptomatic by age 40; thus hemorrhage due to AVM occurs in a younger population than that due to aneurysms or hypertension. Multiple calcified vascular channels may be seen within the hematoma on CT scan, suggesting the presence of an AVM. MRI and four-vessel cerebral angiography are useful adjuncts in the diagnosis of these lesions.
Other Causes
Cerebral amyloid angiopathy (CAA) is an important cause of predominantly lobar, often recurrent, ICH in the elderly. Histopathologic studies in CAA demonstrate the deposition of beta-amyloid protein in the media and adventitia of small meningeal and cortical vessels; deposition in the typical sites for hypertensive hemorrhage is rare but has been reported in the cerebellum.38 The prevalence of amyloid in cerebral vessels increases dramatically with age39,40 and may partially account for the exponential rise in the risk for ICH with increasing age. There is an overrepresentation of the apolipoprotein E ε2 and ε4 genotypes in CAA-related hemorrhage, and these alleles are associated with an earlier age of onset of first hemorrhage and a higher risk of early recurrence.41,42 Although neuropathologic examination remains the only means of definitively diagnosing CAA, the presence of multiple or recurrent lobar ICH (including asymptomatic microhemorrhages detected on gradient-echo MRI) in individuals 55 years or older without other known causes of hemorrhage strongly suggests the diagnosis.43 Recent PET studies with 11C-Pittsburgh compound B (PIB) suggest an occipital predominant increase in amyloid in such patients.44 Neuropathologic correlation remains to be demonstrated, but PIB-PET appears to be a promising tool for in vivo diagnosis of CAA.
Hematologic causes of ICH include the use of antithrombotic and thrombolytic agents as well as systemic disease (e.g., thrombocytopenia, leukemia, and hepatic and renal failure) and congenital or acquired factor deficiencies. The incidence of oral anticoagulant (OAC)-associated ICH has been increasing in parallel with the increased use of warfarin following pivotal trials in atrial fibrillation in the 1990s. A recent population-based study in the greater Cincinnati area identified a fivefold increased incidence of OAC-associated ICH between 1988 and 1999, such that this condition now accounts for 17% of all ICH cases.45 The incidence is anticipated to rise further in the coming years as the population ages. Although the risk of ICH is greater in the setting of very elevated international normalized ratio (INR), a significant number of hemorrhages occur when the INR is within the therapeutic range.46 Hematoma expansion may be more common in OAC-associated ICH and occur over a longer time frame because of persistent coagulopathy, contributing to the doubling of mortality rate compared to spontaneous ICH.47
The relationship between antiplatelet agent use and hematoma size, hematoma expansion, and outcome in ICH are active areas of investigation. Most studies suggest that antiplatelet use at ICH onset is not associated with larger hematoma size, hematoma growth, or poor clinical outcome48; however, an association has been demonstrated between reduced platelet activity and hematoma growth and poor outcome.49 Whether platelet transfusion is beneficial in this situation is unknown.
Hemorrhage from an underlying neoplasm is rare but occasionally occurs with malignant primary CNS tumors such as glioblastoma multiforme and lymphoma and with metastatic tumors such as melanoma, choriocarcinoma, renal cell carcinoma, and bronchogenic carcinoma.50 Benign tumors are almost never associated with ICH.
ICH may also occur in association with infection (e.g., infiltration of vessel wall by fungal organisms,51 necrotizing hemorrhagic encephalitis with herpes simplex,52 vasculitis,53 venous sinus occlusion,54 in a delayed fashion after head trauma,55 following reperfusion (e.g., after carotid endarterectomy or acute thrombolysis),56 and with the use of various drugs, particularly sympathomimetics (e.g., cocaine, amphetamines, pseudoephedrine, and phenylpropanolamine).57 Finally, some degree of hemorrhagic transformation of acute cerebral infarcts is common,58 though symptomatic ICH in this setting is rare in the absence of anticoagulation or thrombolytic therapy.
Clinical Features
The clinical presentation of ICH is often indistinguishable from that of ischemic stroke but more commonly includes altered level of consciousness, headache, and vomiting, reflecting the presence of increased intracranial pressure (ICP).33 Blood pressure is elevated in the majority of patients (see later discussion). Seizures occur in nearly one-third of patients at onset or within the first few days, particularly in those with lobar hemorrhages or underlying vascular or neoplastic lesions, and may be purely electrographic.59 Symptoms are maximal at onset or develop over minutes to hours. Neurologic deterioration within 48 hours after hospital admission has been reported to occur in 22% of patients with ICH.60 The cause for clinical worsening is not always evident, but it is predicted by clinical and biological markers of inflammation on admission and commonly associated with increased hematoma size and intraventricular bleeding.
Diagnostic Studies
Noncontrast computed tomography (CT) scanning has been the traditional gold standard for diagnosis of acute ICH. The typical CT appearance of an acute hematoma consists of a well-defined area of increased density surrounded by a rim of decreased density. Over time, the borders of both the high- and low-attenuation regions become increasingly indistinct such that the hematoma is isodense with adjacent brain parenchyma by 2 to 6 weeks.61 Peripheral contrast enhancement can often be seen at this time.62 By 2 to 6 months, there may be no CT evidence of previous hemorrhage or there may be an area of hypodensity or a slit-like scar.63
Recent studies have suggested that MRI has high sensitivity and specificity for the diagnosis of acute ICH.64–66 These studies have been criticized, however, for major methodological limitations including the basing of sensitivity estimates on only a small fraction of patients investigated, lack of CT comparator in many patients, and both incorporation and spectrum bias (highly selected patient sample) that may have overestimated diagnostic accuracy of MRI.67 In addition, MRI may not be feasible in a substantial number of patients with acute ICH because of impaired consciousness, hemodynamic compromise, or vomiting.68 The benefits of MRI over CT are its superior performance in the identification of associated vascular malformations, greater accuracy in determining the approximate age of a hematoma (because each hemoglobin oxidation state during evolution of the hematoma produces a predictable pattern of MR signal intensity),69 and its utility in demonstrating the iron-containing deposits of previous asymptomatic hemorrhages.70
Angiography is useful in evaluating the cause of ICH if an underlying aneurysm or vascular malformation is suspected, but the yield of such studies is extremely low when the patient has chronic hypertension and the hemorrhage is in one of the typical sites associated with hypertensive hemorrhage.71 Multidetector CT angiography is evolving as an alternative to conventional angiography. In a retrospective review of 623 patients, multidetector CT angiography identified a vascular etiology for ICH in 91 (15%), with a sensitivity of 96% and a specificity of 99%. The yield was higher in patients who were younger than 46 (47%), had lobar (20%) or infratentorial (16%) ICH locations, had lobar hemorrhages with intraventricular extension (25%), and had neither hypertension nor impaired coagulation (33%).72 In another study of 78 patients, CT angiography identified all but one of the 22 lesions seen on conventional angiography, with a sensitivity of 96% and a specificity of 100%.73
Multidetector CT angiography can also be used to identify the presence of active contrast extravasation into the hematoma, an indicator of active hemorrhage. Termed the “spot sign,” these foci of intralesional enhancement are seen in up to one third of patients with acute ICH74 and are associated with an increased risk of hematoma expansion, in-hospital mortality, and poor outcome in survivors.75
Treatment
Initial Stabilization
Acute ICH is a medical emergency requiring considerable attention to airway and respiratory management, hemodynamic status, and correction of any underlying coagulopathy. As many as half of all patients with ICH undergo mechanical ventilation.76 Blood pressure is often elevated at presentation, sometimes markedly so. Finally, given the frequency of hematoma enlargement over the first few hours, aggressive correction of coagulopathies might be helpful.
Airway and Respiratory Management
Initial airway management includes proper positioning, frequent suctioning, and placement of an oral or nasal airway. Frequent assessments for sonorous respiration, inability to manage oral secretions, or decreased oxygen saturation are necessary. If conservative measures are ineffective, intubation may be necessary. Intubation of patients with ICH requires adequate sedation and jaw relaxation as well as prevention of elevation of ICP. Several factors may conspire to raise ICP during intubation: hypoxia, hypercarbia, and direct tracheal stimulation causing systemic and intracranial hypertension. Intravenous (IV) lidocaine (1-1.5 mg/kg) has been recommended to block this response,77 although data supporting its use are lacking.78 Short-acting IV anesthetic agents (thiopental, 1-5 mg/kg; or etomidate, 0.1-0.5 mg/kg) also block this response79 and additionally suppress brain metabolic rate,80 theoretically improving tolerance of a transient fall in cerebral perfusion pressure (CPP) should it occur. Etomidate is generally preferred over thiopental, since it is less likely to lower blood pressure. Paralytic agents are usually unnecessary but, if needed, short-acting agents should be used.
Hemodynamics
Arterial blood pressure is elevated on admission in the majority of patients with ICH, even in the absence of a history of hypertension.33 Mean arterial pressure (MAP) is greater than 120 mm Hg in over two-thirds of patients and greater than 140 mm Hg in over one-third.81 Although this acute increase in blood pressure is often implicated as the cause of the hemorrhage, it may simply be a reflection of chronic hypertension, the brain’s attempt to maintain CPP in response to the sudden increase in ICP, pain and anxiety, and sympathetic activation. Even without pharmacologic intervention, blood pressure tends to decline to premorbid levels during the first 7 to 10 days after hemorrhage.82
There is substantial controversy over if and when to lower blood pressure after acute ICH and how aggressive any intervention should be.83 Proponents of rapid treatment of acute hypertension argue that high blood pressure may predispose to hematoma enlargement and may exacerbate vasogenic edema by increasing capillary hydrostatic pressure, especially in areas with a damaged blood-brain barrier. Yet, an association between hypertension and edema has never been demonstrated, and data on the effect of hypertension on hematoma enlargement have been inconsistent.84,85 Another potential reason to lower blood pressure is that hypertension during the acute phase of ICH has been shown to correlate with a poor prognosis in some studies.86,87 One compelling reason to consider lowering blood pressure in ICH patients with moderate to severe hypertension is the potential for end-organ damage. Such patients are at risk for systemic complications of elevated blood pressure, including myocardial ischemia, congestive heart failure, and acute renal failure.
The major argument against the treatment of elevated blood pressure is that lowering blood pressure might exacerbate ischemic damage in the tissue surrounding the hematoma by impairing blood flow.88 Chronic hypertension shifts the cerebral autoregulatory curve to the right such that a higher CPP is required to maintain adequate CBF.89,90 Lowering the blood pressure to “normal” levels in these patients might thus lead to inadequate CBF. Similarly, since CPP is equal to the difference between MAP and ICP, lowering blood pressure may reduce CPP below the autoregulatory limit in patients in whom ICP is elevated due to a large space-occupying clot or hydrocephalus.
Seeking to address the issue of whether lowering blood pressure produces cerebral ischemia in acute ICH, several studies of CBF autoregulation in patients with recent ICH and elevated blood pressure (MAP > 130-140 mm Hg) have been carried out.91–93 Taken together, these studies demonstrate that regional and global autoregulation are preserved after ICH down to a lower MAP limit that averages 110 mm Hg or about 80% of the admission MAP.
These observations set the stage for the INTERACT trial,94 a prospective trial of blood pressure management beginning within 6 hours of symptom onset in 404 patients with spontaneous ICH and elevated systolic blood pressure (150-220 mm Hg). Patients were randomized to an early intensive blood pressure–lowering strategy that targeted a reduction in systolic blood pressure to below 140 mm Hg within 1 hour, or control, with a target systolic pressure of less than 180 mm Hg. The primary efficacy endpoint was hematoma growth at 24 hours. After the first hour of treatment, mean blood pressure was 13.3 mm Hg lower (95% confidence interval [CI] 8.9-17.6 mm Hg; P <0.0001) in the intensive blood pressure–lowering group. Hematoma growth at 24 hours was 36.3% in the control group and 13.7% in the intensive treatment group, a 22.6% difference (95% CI 0.6%-44.5%; P=0.04). After adjusting for initial hematoma volume and time from onset to CT, median hematoma growth at 24 hours differed by 1.7 mL (95% CI 0.5-3.9, P=0.13). Intensive blood pressure–lowering treatment did not increase the risk of adverse events or improve 90-day clinical outcome. A much larger follow-on trial is currently underway.
Prevention of Hemorrhage Extension
Because hemorrhage extension occurs within the first few hours after symptom onset in about one-third of patients (Figure 35-2), it seems appropriate that any coagulopathy should be corrected as rapidly as possible. Patients taking warfarin should receive IV vitamin K and enough fresh frozen plasma (FFP) to normalize the coagulation profile. Prothrombin complex concentrate may be a useful alternative. Care must be taken not to precipitate congestive heart failure, however, and diuretics may be required. Additionally there is risk of transfusion-related acute lung injury with the administration of fresh frozen plasma, which can complicate the process considerably. Correcting coagulopathy associated with thrombolytic-induced ICH is discussed later.
Even in those patients without coagulopathy, promoting early hemostasis might limit ongoing bleeding and decrease hematoma volume. Factor VIIa is a coagulation factor that interacts with tissue factor exposed in the wall of a damaged blood vessel to drive a burst of thrombin that initiates platelet aggregation and accelerates formation of a stable fibrin clot. A phase IIb placebo-controlled dose-ranging proof-of-concept study found that treatment with recombinant factor VIIa (rFVIIa) given as a single IV bolus within 4 hours of ICH onset decreased hematoma growth and improved clinical outcome despite a small increase in thromboembolic events. A much larger phase III trial comparing placebo to 20 and 80 µg/kg of rFVIIa followed. This study confirmed the ability of rFVIIa to reduce hematoma growth; however, at 90 days there was no difference in clinical outcome.95 A post hoc exploratory analysis suggested that a subgroup of younger patients who present earlier and have no significant intraventricular hemorrhage might benefit from rFVIIa, but this has not been tested.96
Intraventricular Hemorrhage and Hydrocephalus
In approximately 40% of patients with ICH, blood extends into the ventricular system (intraventricular hemorrhage [IVH]).97 Mortality in these patients is high.98,99 IVH may contribute to poor outcome by blocking cerebrospinal fluid (CSF) pathways, with resultant hydrocephalus and increased ICP. In addition, intraventricular blood and/or its breakdown products may exert direct chemical irritative effects on periventricular structures. Hydrocephalus may develop after ICH either in association with IVH or because of direct mass effect on a ventricle (e.g., on the third ventricle with a thalamic hemorrhage) (Figure 35-3). External ventricular drainage (ventriculostomy) is frequently used to treat hydrocephalus and IVH, but its efficacy has never been established. Ventriculostomy in the setting of IVH is difficult to manage because the catheter frequently becomes obstructed with thrombus, interrupting drainage and raising ICP. Flushing the system helps remove thrombus from the catheter but increases the risk of ventriculitis. Recently, investigators have attempted to facilitate removal of blood from the ventricles via direct intraventricular administration of thrombolytic agents. Preliminary studies have been promising,100 and a multicenter randomized trial is currently underway.
Intracranial Hypertension
The incidence, impact, and appropriate management of intracranial hypertension in ICH are not well understood. Factors likely to contribute to elevated ICP in this population include large hematoma size, minimal degree of underlying cerebral atrophy, hydrocephalus, and edema, but the true incidence of intracranial hypertension is unclear, since routine ICP monitoring is not performed. Because the hematoma is localized and the increase in volume it produces can be compensated for to some degree by reduction in the size of the ventricles and subarachnoid space, a global increase in ICP may not be seen unless the hemorrhage is very large or is associated with marked hydrocephalus. However, mass effect from the hematoma and local tissue shifts can compress the brainstem or result in herniation in the absence of a global increase in ICP.101,102 Thus the utility of ICP monitoring has never been established.
Elevated ICP is often treated with osmotic agents (mannitol, hypertonic saline) and, if the ventricles are enlarged, CSF drainage. A recent case series suggested that rapid reversal of clinical transtentorial herniation (decreased level of consciousness and dilated pupil) with hyperventilation and osmotic agents improved long-term outcome.103 There are only a few small clinical trials of osmotic agents in ICH, which do not provide sufficient data to support their routine use.104 The best available data on corticosteroids in ICH indicate that they do not provide any benefit and increase the rate of complications.105
Surgical Evacuation
The rationale for surgical evacuation of a hematoma is that reducing mass effect and removing neurotoxic clot constituents should minimize injury to adjacent brain tissue and hence improve outcome. Unfortunately, several randomized controlled trials of surgery for supratentorial ICH dating back to 1961 have all failed to show a benefit of the intervention.106–109 A meta-analysis of three of these trials reported that patients undergoing surgical evacuation via open craniotomy had a higher rate of death or dependency at 6 months compared to those managed medically (83% versus 70%).110 Criticisms of these trials are that the surgical techniques used were outdated, patient selection was inadequate, and surgery was delayed too long.
Because open craniotomy is complicated by tissue damage sustained during the approach to the hematoma, a variety of new techniques for clot removal have been proposed, including an Archimedes screw, ultrasonic aspirator, modified endoscope, modified nucleotome, double track aspirator, intraoperative CT monitoring, and instillation of thrombolytics. However, the recurrence of bleeding due to the loss of tamponade effect on adjacent tissue that occurs in 10% of patients treated with open craniotomy remains an issue with the newer techniques. In addition, because the newer techniques involve limited surgical exposure, concern exists that rebleeding will be more difficult to control than with open craniotomy. One study comparing endoscopic aspiration to medical management found a better outcome in the surgical group (74% death or disability compared to 90%), but the benefit was limited to patients with lobar hematomas.111 Three studies addressed the feasibility of early craniotomy for ICH. In one,112 34 patients were treated within 12 hours of ICH. Mortality was 18% in the surgical group and 23% in the medical group. In another study,113 20 patients were randomized, with a median time to surgery of 8.5 hours from onset. Good outcome (Glasgow Outcome Scale score > 3) was 56% with surgery and 36% in the medically treated group (P = NS). The third, a study of ultra-early surgery (<4 hours), found a disturbingly high rate of postoperative rebleeding.114
A lack of benefit of surgery in ICH was also shown in a recently completed multicenter trial in which 1033 patients were randomized within 72 hours of ICH onset to surgical hematoma evacuation (open craniotomy or stereotactic aspiration, at surgeon’s discretion) or initial conservative management. Favorable outcome occurred in 26.1% in the surgery group and 23.8% in the initial conservative treatment group, a nonsignificant difference (odds ratio 0.89; 95% CI 0.66-1.19). There was also no difference in mortality (surgery 62.6% versus conservative treatment 63.7%). Subgroup analysis suggested a possible benefit of surgery in patients with superficial hematomas (less than 1 cm from cortical surface).115 A trial comparing surgical and medical management of superficial hematomas is currently underway.
Cerebellar hemorrhages were excluded from the randomized trials of surgery, but nonrandomized case series report good outcomes for surgically treated patients with cerebellar hemorrhages that are large or associated with brainstem compression or obstruction of the fourth ventricle. Recommended criteria for when to evacuate a cerebellar hematoma have thus included diminished level of consciousness, large size of the hematoma (>3 cm3), midline location, compression of basal cisterns and/or brainstem, and presence of hydrocephalus (Figure 35-4).116–118 Patient selection is important as many patients with smaller hemorrhages do well with medical management.119
Management of Thrombolytic-Induced Ich
Associated with considerable morbidity and mortality, symptomatic ICH is a feared complication of thrombolytic therapy. Symptomatic ICH occurs after thrombolytic treatment of acute ischemic stroke in approximately 6% of patients.120 It is substantially less common after thrombolytic treatment of extracerebral thrombosis (myocardial infarction, pulmonary embolism, deep venous thrombosis, and arterial and graft occlusion)121 but results in a similarly poor outcome.122 Factors that increase the risk of symptomatic ICH include intraarterial versus IV route of administration, early ischemic changes on pretreatment CT, greater symptom severity, and elevated serum glucose or history of diabetes mellitus.123,124
In the setting of thrombolytic therapy, any new neurologic deficit, especially with a decline in consciousness, should be assumed to be due to hemorrhage. Management of a suspected ICH should begin with stopping the thrombolytic infusion, reassessing the patient’s airway, and obtaining an emergent CT scan. Blood studies (prothrombin time, partial thromboplastin time, thrombin, and fibrinogen levels) should be performed to assess fibrinolytic state. Preparations for giving FFP, cryoprecipitate, and platelets should be initiated at the first suspicion of hemorrhage so that they will be ready if needed; however, no reliable data are available to guide the choice of blood product. The NINDS rt-PA study125 stipulated 6 to 8 units of cryoprecipitate or FFP and 6 to 8 units of platelets, but only rarely was this amount of blood product given to an individual patient during the study. Although neurosurgical consultation was frequently obtained in patients with symptomatic ICH in the NINDS trial, only one patient in the study underwent surgery, and that patient died.
Management of Seizures
Although seizures may theoretically exacerbate ICH, they have not been demonstrated to alter outcome. Prophylactic anticonvulsants may reduce the risk of early seizures in patients with lobar ICH but do not affect the risk of developing epilepsy.126 Thus reasonable approaches include either a brief period of prophylaxis or treating only if seizures occur. As for any hospitalized patient, the treatment of clinical seizures typically begins with an IV benzodiazepine such as lorazepam followed by an IV agent such as fosphenytoin.
Supportive Care
Patients with ICH are prone to the same medical complications seen in patients with ischemic stroke, including fever, deep venous thrombosis (DVT), pulmonary embolism, and pneumonia.127,128 Given the association between fever and worsened outcome in experimental models of brain injury, it is reasonable for antipyretic medications to be administered in febrile patients with ICH. The use of pneumatic sequential compression devices and elastic stockings has been shown to significantly decrease the incidence of DVT in patients with acute ICH relative to elastic stockings alone.129 Subcutaneous heparin at a dose of 5000 U three times daily when initiated on day 2 after hemorrhage has been shown to significantly reduce the frequency of DVT relative to treatment begun on day 4 or 10, with no concomitant increase in hematoma expansion.130 In another study, subcutaneous enoxaparin (40 mg daily) initiated at 48 hours after ICH was also safe. A benefit of enoxaparin over compression stockings could not been detected because of the low incidence of DVT in both treatment groups.131
Prognostic Factors and Causes Of Mortality
Mortality following ICH is high (25%-50%), with over half of the deaths occurring in the first 48 hours. Although patients who have small hemorrhages and mild deficits may recover completely, the majority of ICH survivors have significant residual disability.4,132,133 A variety of clinical, laboratory, and radiographic predictors of poor outcome have been identified, the most consistent being impaired level of consciousness and large hematoma size on admission. Other predictive clinical features variably include increasing age, elevated admission blood pressure on admission, rapid decline in blood pressure over the first 24 hours, history of diabetes, antecedent OAC use, male gender, and in-hospital neurologic deterioration. Laboratory parameters identified in at least one study include hyperglycemia, elevated troponin level, elevated plasma S100B level, elevated plasma D-dimer level, elevated INR, low serum cholesterol and triglyceride levels, and apolipoprotein E ε2 or ε4 allele. Radiographic features include infratentorial hematoma location, intraventricular spread of blood, midline shift, hydrocephalus, hematoma growth, and presence of the spot sign on CT angiography. A number of prognostic models of varying complexity have been developed to allow risk stratification upon presentation with ICH. One easy to use model is the ICH score,134 which is based on point assignments for Glasgow Coma Scale score, ICH volume, presence of intraventricular hemorrhage, infratentorial location, and patient age and has been validated to accurately predict 30-day mortality. It has been demonstrated, however, that withdrawal of support in patients felt likely to have a poor outcome biases predictive models in ICH and negates the predictive value of all other variables.135 Thus, the most frequent cause of death after ICH is withdrawal of care, followed by early (within 48 hours) transtentorial herniation with progression to brain death. Medical complications of immobility (pulmonary embolism, pneumonia, sepsis) account for most of the other deaths.133 For survivors of ICH, the risk of recurrent stroke is approximately 4% per year. Recurrent ICH occurs about twice as often as ischemic stroke, especially in those with previous lobar hemorrhage.136
Subarachnoid Hemorrhage
Although it is the least common form of stroke, subarachnoid hemorrhage (SAH) has great impact on its sufferers. One-quarter of patients die before reaching medical attention,137 and because of the consequences of secondary insults—rebleeding, hydrocephalus, and delayed ischemia due to vasospasm—more than half of those that reach medical attention either die or are left with neurologic deficits.
Pathophysiology
In SAH, the primary site of bleeding is within the subarachnoid space, but may also involve hemorrhage into the brain parenchyma, ventricular system, or subdural space. Rupture of an intracranial saccular aneurysm (Figure 35-5) is by far the most common cause of spontaneous SAH. Saccular or berry aneurysms are small, rounded protrusions of the arterial wall occurring predominantly at bifurcations of the large arteries of the circle of Willis at the base of the brain. The most common sites of ruptured aneurysms are the distal internal carotid artery and its posterior communicating artery junction (41%); anterior communicating artery/anterior cerebral artery (34%); middle cerebral artery (20%); and vertebrobasilar arteries (4%).138 About 20% of patients have multiple aneurysms.
Figure 35-5 Autopsy specimen of intracranial aneurysm filled with pressurized blood to simulate subarachnoid hemorrhage.
The pathogenesis of aneurysms remains controversial, especially in regard to the relative important of developmental versus acquired factors. Proponents of the congenital theory suggest that aneurysms arise at sites of faulty fusion between muscular segments within the arterial wall. Supporters of the acquired-degenerative theory focus on the role of vascular damage caused by hemodynamic stress.139 The third possibility is that aneurysms develop at sites harboring congenital defects with superimposed degenerative changes.
Causes and Risk Factors
Genetic conditions that predispose to aneurysm formation include polycystic kidney disease, connective tissue disorders, and coarctation of the aorta. Recently it has become clear that in some patient populations, genetic factors play a role in aneurysm formation, without other associated conditions.140 There is a familial form as well.141 Other types of aneurysms that less commonly cause SAH include atherosclerotic, mycotic, and traumatic aneurysms.
The risk of SAH increases with age, peaking at 55 to 60 years. There is a slight male predominance in younger age groups and a slight female predominance among older patients.142 Potentially reversible risk factors for SAH include cigarette smoking, oral contraceptive use, alcohol abuse, and hypertension.143 Prospective cohort studies have reported a relative risk of SAH as high as 5.7 for female and 4.7 for male smokers,144
[/not-level-membership-for-critical-care-medicine-category]
