30 Critical Neuropathophysiology
 Elevated Intracranial Pressure
Elevated Intracranial Pressure
Physiology of Intracranial Pressure and Cerebral Blood Flow
The brain, spinal cord, cerebrospinal fluid (CSF), and blood are encased in the protecting but noncompliant skull and vertebral canal, constituting a nearly incompressible system (Figure 30-1). In a totally incompressible system, pressure would rise linearly with increased volume. However, there is capacitance in the system, thought to be provided by the intervertebral spaces. Once this capacitance is exhausted, the ICP increases dramatically with increased intracranial volume.
Based on the following relationship:
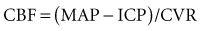
the concern is raised mathematically that increasing ICP is associated with decrements in CBF. However, the effect of increasing ICP on CBF is not straightforward, as mean arterial pressure (MAP) may increase with ICP elevations,1 and cerebral vascular resistance (CVR) adjusts with decreasing CPP (increasing cerebral vessel diameter) to maintain CBF until maximal vasodilatation occurs.2,3 This results in an increase in cerebral blood volume (CBV). This is thought to occur at a CPP less than 50 mm Hg, although considerable individual heterogeneity in this value exists with good reasons to believe that the lower limit of autoregulation (LLA) may be higher.4 Thus, increasing ICP initially is often associated with vasodilatation and/or increasing MAP to maintain CBF without a nutritive decrement.
Normal ICP is less than 10 mm Hg. ICP greater than 20 mm Hg is generally treated with ICP-reducing agents,5 but this is an epidemiologically derived action. Head trauma studies have indicated that patients with ICP over 20 mm Hg generally do poorly,5,6 although simply elevating ICP to above 20 mm Hg (in experimental animals) is not necessarily associated with decrements in CBF or permanent sequelae, provided the above-noted compensatory mechanisms occur,7,8 and venous ICP-related venous outflow obstruction with positive-feedback exacerbation of ICP does not occur.9
Nonetheless, increasing ICP due to mass lesions or obstruction of CSF outflow can exhaust compensatory mechanisms, with compromise of CBF. Initially, distal runoff of the cerebral circulation increases. As the process continues, the normally continuous (through systole and diastole) cerebral perfusion becomes discontinuous (systolic perfusion only) (Figure 30-2).10 Further compromise of CPP results in further oxygen extraction progressing to anaerobic metabolism, exacerbation of edema, and ultimately intracranial circulatory arrest.10 Thus, when ICP increases, early recognition is important to determine whether a deleterious sequence of events is starting.
Traditional notions of cerebral autoregulation, with CBF constant over a CPP range of approximately 50 to 150 mm Hg, has not gone without challenge.4 Drummond argues that this common notion derives from a figure in a review article by Lassen,11 which itself was an estimate based on data published by McCall in 195312 from pregnant volunteers undergoing blood pressure (BP) alteration with hydralazine and Veratrum viride. Despite the use of potentially cerebral vasoactive drugs, these observations remained unconfirmed in humans. Drummond suggests that most human data published since 1953 support an LLA of 70 mm Hg, with one investigator suggesting the onset of cerebral ischemia symptoms in normal humans to arise at a MAP of 55 mm Hg.13 Moreover, his closer review of published data suggests large interindividual variation in LLA. Drummond suggests that the only safe approach to an individual patient is to assume that no less than 75% of his/her resting MAP should be assumed to be the LLA. Symptoms of cerebral hypoperfusion tend to arise when MAP falls to about 50% of the resting value. These assertions of the need to individualize are increasingly being supported in the context of head injury with recent studies of the use of dynamic autoregulation assessment to determine optimal BP for a given patient.6,14–16
It is also of interest that LLA, based on CPP (MAP − ICP), may vary with ICP and with jugular venous pressure. McPherson et al., in a canine model, noted LLA was higher with elevated jugular venous pressure. This may, however, actually reflect the lack of knowledge regarding the proper definition for CPP, and that it may vary depending on the influence of the venous Starling resistor.8,17 Brady et al.,18 in an atraumatic immature piglet model of intracranial hypertension, found that LLA had a positive correlation with ICP. That is, LLA CPP was higher with higher levels of ICP. They suggest the possibility that compensating for an increase in ICP with an equivalent increase in arterial blood pressure (ABP) may not be sufficient to prevent a decrement in CBF and cerebral ischemia. Further studies in adults will be needed. Nonetheless, Brady et al.18 point out that Cremer et al.19 observed an LLA elevation in adult trauma patients with intracranial hypertension. The overall suggestion is that there is a need for individualized dynamic autoregulation assessment to determine each patient’s optimal CPP.6,14–16 Indeed, this may be only one component of a battery of multimodal monitoring, so-called integrative neuromonitoring, that is increasingly being advocated.20
Another approach to characterizing cerebral autoregulation has been espoused by Dewey et al.,21 Early et al.,22 and Burton et al.23 Using observations in pacemaker-dependent dogs and a beat-to-beat measure of brain blood flow, they observed that abrupt cessation of cardiac activity produced zero CBF well above the generally accepted LLA. Indeed, they reported that this critical closing pressure varied with the resting MAP, generally being about 40 to 50 mm Hg below MAP. They concluded that the normal cerebral circulation assumes a tonic state of contraction that varies with MAP and ICP, more tone at higher BP or lower ICP, less at lower BP or higher ICP, such that the true dynamic cerebral perfusion pressure is MAP − CCP, with CBF = (MAP − CCP)/CVR. Burton’s model can be use to describe CCP as: CCP = ICP + tension of arterial walls.21 The CCP is presumed to be altered by various drugs and disease states to thereby produce variations in CBF, despite otherwise unchanged traditional CPP (MAP − ICP). Thus the true definition and measurement of CPP may be a good deal more dynamic and complex than is understood at this time.
CCP was further studied more recently by Czosnyka et al.24 in humans with traumatic brain injury (TBI). If autoregulation is relatively intact, CCP-ICP remains high, but with injury sufficient to produce dysautoregulation, CCP-ICP decreases, indicating decreased tension in arterial walls.
Contributors to Intracranial Hypertension
Brain
The brain normally occupies about 80% of the contents of the skull, but its volume can be increased by edema. There are two types of edema, cytotoxic and vasogenic, referring to swelling produced by cellular or vascular processes, respectively.25 Any edema can increase ICP. It can be heterogeneously distributed such that pressure gradients occur, leading to a variety of herniation syndromes.
Cerebrospinal Fluid
CSF is generated in the choroid plexus and absorbed in the arachnoid villi. An equilibrium normally exists between production and absorption. Disruption of this equilibrium can lead to increased ICP with hydrocephalus, the condition wherein there is an excess of fluid in all or part of the CSF in the brain. Hydrocephalus is generally categorized as communicating or noncommunicating. In communicating hydrocephalus, the CSF circulation between the site of CSF production and absorption is intact. However, abnormally decreased absorption or increased production results in increased CSF accumulation. In noncommunicating hydrocephalus, the pathways are blocked such that CSF cannot circulate to the convexity of the brain to be absorbed. This results in accumulation of CSF in the ventricles, producing distension.26
Blood
Another mechanism of increased CBV occurs with obstruction of venous outflow. This results in brain engorgement and edema and CBV-mediated increased ICP, but without increased CBF27; this too is discussed in more detail later.
Venous Pathology
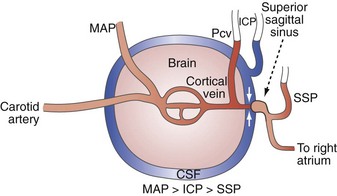
Venous pathology also plays a role in the genesis and propagation of intracranial hypertension. Blood coursing through the brain runs through arteries, capillaries, veins, sagittal and other dural sinuses, and then on to the internal jugular and other extracranial veins. In the context of a closed intracranial space, the relationship of these vessels to the tissue and CSF surrounding them becomes important. Notably, several investigators, in laboratory preparations, have observed a distinct drop off in intraluminal pressure in going from cerebral cortical veins to the sagittal sinus. This is most evident when ICP is elevated and indicates the presence of a vascular waterfall at a point just proximal to the sagittal sinus as the extraluminal high-pressure CSF acts to impede flow from cortical veins to the sagittal sinus.8,9,28,29 In fact, Nemoto9 and Nakagawa et al.28 have further observed that the cerebral venous pressure tends to be consistently higher than the ICP. The implications of these observations are that elevated ICP begets increased cerebral venous pressure. The increased cerebral venous pressure promotes and exacerbates brain edema, which may have been the initial cause of the intracranial hypertension. This then leads to a positive-feedback cycle wherein increased ICP increases cerebral venous pressure which then increases ICP.9,30 Thus any other factors that may promote brain edema or otherwise increase ICP in this tenuous situation (e.g., high extraventricular drain, systemic hypertension,31 hypoosmolarity) may initiate such a positive-feedback process.
Types of Intracranial Hypertension
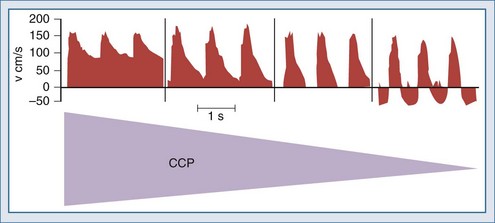
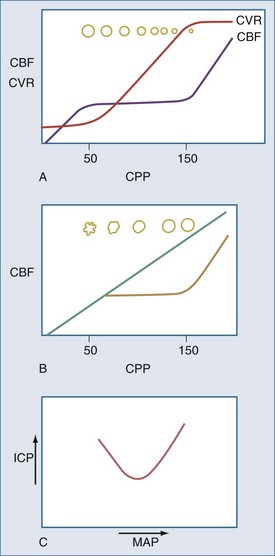
There are two types of intracranial hypertension, categorized according to CBF as hyperemic or oligemic (Figure 30-3).6 In the normal state, increases in CBF are not associated with increased ICP, because capacitive mechanisms compensate for the CBV-mediated increased intracranial volume. However, in the situation of disturbed intracranial compliance, small increases in intracranial volume produce significant increases in ICP.2,3
For many years it has been known that abrupt noxious stimuli briefly increase ICP in the setting of decreased intracranial compliance. Recent studies have revealed that such situations are associated with hyperemia, strongly suggesting that brief hyperemic intracranial hypertension is not a dangerous situation.32 However, it is reasonable to be concerned about such hyperemia for four reasons. First, elevated ICP due to hyperemia in one portion of the brain may increase ICP to compromise CBF in other areas of the brain in which CBF is marginal. Secondly, increased pressure in one area of the brain may produce gradients that might lead to a herniation syndrome. Thirdly, there is theoretical concern that inappropriate hyperemia predisposes the brain to worsened edema or hemorrhage as occurs with hyperperfusion syndromes. And fourthly, there is increasing evidence that increased ICP obstructs venous outflow to further exacerbate brain edema in a positive-feedback manner.9,30 Thus hyperemic intracranial hypertension has a theoretical potential to be deleterious, although this has yet to be demonstrated in a systematic fashion. For brief periods, as may occur during intubation or other limited exposure to noxious stimuli, it is suggested (but not proven) that it may not be problematic.33 An example of this conundrum is illustrated in Figure 30-4.
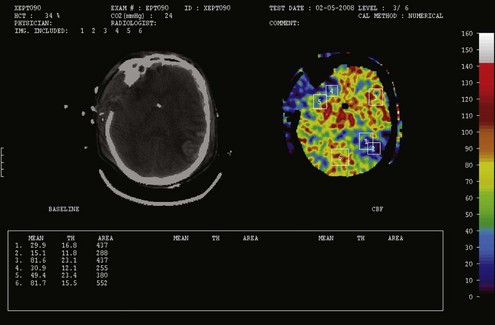
In contrast, oligemic intracranial hypertension is associated with compromised cerebral perfusion and is clearly deleterious.6,10 This is supported by the high mortality rate observed in head trauma patients in whom ICP rises due to brain edema with decrements in CBF.10,34 Transcranial Doppler echography and CBF studies on these patients have demonstrated that CBF is low and perfusion is discontinuous during the cardiac cycle (see Figure 30-2).10,35 Moreover, jugular venous bulb data indicate that oxygen extraction is markedly increased, suggesting loss of reserve with occurrence of anaerobic metabolism.35 In this setting, noxious stimuli can further increase the ICP, producing the situation of hyperemic added to oligemic intracranial hypertension. Presumably, in this setting the hyperemic rise in ICP acts to further reduce regional CBF in compromised areas with brain edema and may contribute to vasogenic edema.
Blood Pressure Effects on Intracranial Pressure: Plateau Waves
Lundberg, in a pioneering 1960 study,34 monitored ICP in hundreds of patients, identifying characteristic pressure waves. One category of these waves has been identified as plateau waves, which are known to be associated with increased CBV (Figure 30-5).2 Such waves occur when the ICP abruptly increases to systemic BP levels for about 15 to 30 minutes, occasionally accompanied by neurologic deterioration. Rosner3 synthesized the data and convincingly suggests that intracranial blood volume dysautoregulation is responsible for plateau waves. He induced mild head trauma in cats and subsequently intensively monitored the animals after the insult. With normal fluctuations in BP, while in the normal range, he observed that mild BP decrements to a mean of approximately 70 to 80 mm Hg preceded the development of plateau waves (Figure 30-6). Cerebral blood volume in normally autoregulating brain tissue increases with decreasing BP. However, the increase in CBV is nonlinear. There is an exponential increase in CBV as CPP decreases to levels of 80 mm Hg and below (Figure 30-7).3 A small decrease in BP, although in the normotensive range, produces exponential increases in CBV in a setting of abnormal intracranial compliance with the ICP at the elbow of the ICP-intracranial volume curve. Thus a small decrease in BP introduces an exponential CBV change upon an exponential ICP relationship such that ICP will increase abruptly and to a significant extent. Cremer et al.’s observations of ICP increases with deliberate ABP decreases in TBI patients provide further support that these concepts are relevant to clinical practice.19
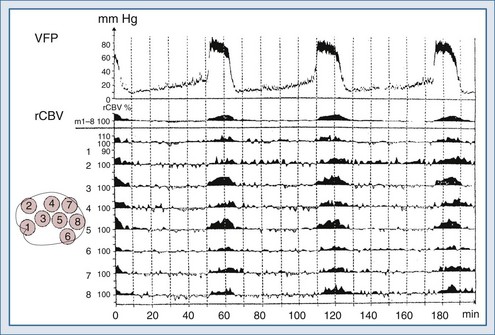
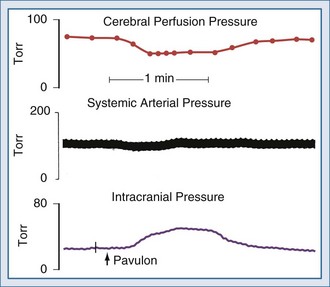
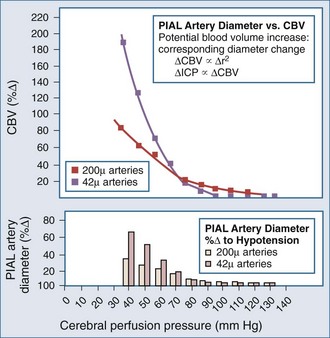
Figure 30-7 Cerebral vasodilatation occurs exponentially as cerebral perfusion pressure is reduced.
(From Rosner MJ, Becker DP. The etiology of plateau waves: a theoretical model and experimental observations. In: Ishii S, Nagai H, Brock M, editors. Intracranial Pressure V. New York: Springer-Verlag; 1983:301.)
Conversely, hypertension can also increase ICP, with animal models showing increased brain water with dopamine-induced increased blood pressure.31,36 Typically, within the normal autoregulatory range, changes in BP have no effect on ICP. However, with brain injury and associated vasoparalysis, BP increases mechanically to produce cerebral vasodilatation, increasing ICP (Figure 30-8).6,15,16,37–40
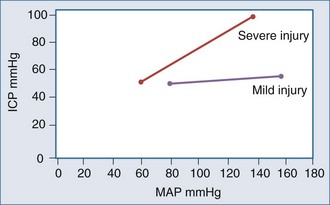
It appears that both increasing and decreasing BP can increase ICP, suggesting the presence of a CPP optimum for ICP—based on Rosner’s observations,3,6,41,42 probably about 80 to 100 mm Hg, although this has not been definitively determined experimentally (Figures 30-9 and 30-10). An alternate view that lower BP should be employed has been argued as the so-called Lund approach by Grände et al.,30 with much of its rationale based on the earlier discussion of the role of venous obstruction in intracranial hypertension. Indeed, recent studies are increasingly supporting the notion that the CPP optimum is highly variable and should be individually determined with emerging technologies.6,15,16
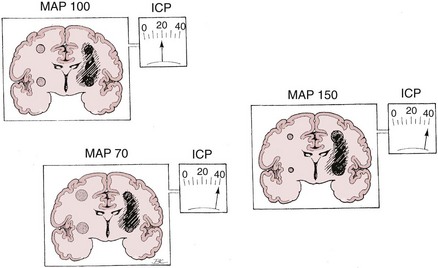
Blood Pressure, Brain Injury, and Intracranial Hypertension—Beyond Plateau Waves
Recent advances in transcranial Doppler (TCD) ultrasonography have allowed insights into dynamic, nearly instantaneous assessment of cerebral autoregulation in critically ill patients. Such technology has permitted the aforementioned observations of CCP in head-injured humans24 and the report of apparent diminution in arterial wall tension in patients with cerebral dysautoregulation. Moreover, Czosnyka et al.14 observed in TBI patients a U-shaped curvilinear relationship in flow velocity versus ABP, with worse autoregulation with ABP lower than 75 mm Hg and ABP higher than 125 mm Hg. They also noted increasing ABP to also increase ICP, further indicating a marker of dysautoregulation, the so-called PRx6,15,16 (see following paragraph). Dynamic time domain analysis of cerebrovascular autoregulation using near-infrared spectroscopy (NIRS) or TCD is a current topic of investigation with promising reports of potential efficacious and valid bedside use.15,39,43–45
The ICP pressure-reactivity index (PRx) is a more recently described quantitation of the earlier description of abnormal dynamic correlation of ICP changes with ABP changes and is another means to dynamically evaluate autoregulation,6,15,16,40 with reports indicating that PRx correlates well with other autoregulation indices.6,16,38,46 Steiner et al.15 reported on the use of PRx monitoring in TBI patients to determine the optimal CPP. Patients with better autoregulation as defined by PRx had better outcomes. Moreover, patients with dysautoregulation related to higher ABP with corresponding ICP elevation also had worse outcomes, suggesting that autoregulation monitoring to ensure adherence to an individual’s optimal CPP may be an outcome-altering intensive care unit (ICU) measure. Zweifel et al.6 report congruent observations. Notably, PRx, as with TCD-based autoregulation studies, also appears to undergo a U-shaped curvilinear relationship with variations in CPP, with it being abnormally high (i.e., ICP varies with ABP) at low (ischemic) and high (hyperemic) CPP in TBI patients. Further complementing this are observations of abnormally high oxygen extraction fraction (OEF) and low OEF at these respective ABP extremes. This is underscored by several reports of a significant ischemic burden in TBI patients,47–50 suggesting a delicate balance between hypotension-associated hypoperfusion and hypertension-associated edema exacerbation, both of which will worsen regional ischemia. Taken altogether, these autoregulation studies introduce the hypothesis that there is an individualized ABP optimum in TBI patients6 that should be a therapeutic goal.
Positive End-Expiratory Pressure and Intracranial Hypertension
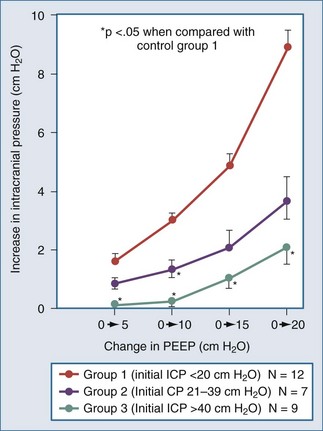
Positive end-expiratory pressure (PEEP) can increase ICP in two ways. The first is through impedance of venous return, increasing cerebral venous pressure and ICP. The second is through decreased BP and reflex increase of CBV, increasing ICP (Figure 30-11). The latter is likely the most common mechanism. Huseby’s data51 suggest that cerebral venous effects only occur with very high PEEP, a notion theoretically supported by the earlier discussion on the role of the veins in autoregulation of CBF and genesis of intracranial hypertension.
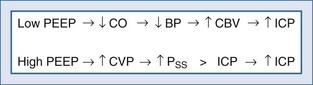
Shapiro and colleagues52 demonstrated increases in ICP in head-injured humans during intracranial hypertension with application of PEEP (Figure 30-12). Examination of their data suggests that the most profound decreases in CPP occurred in patients with PEEP-induced decrements in MAP. This is consistent with the view put forth by Rosner3 that decreases in BP increase CBV and ICP. Aidinis and colleagues,53 in studies on cats, confirmed these observations in a more controlled setting. In addition, they assessed the role of pulmonary compliance, finding that decreased pulmonary compliance induced by oleic acid injections results in less effect of PEEP to increase ICP. In situations in which PEEP is likely to be needed, with decrements in pulmonary compliance, such observations indicate that any adverse effects on ICP are less likely to be manifest. This may be related to observations that hemodynamic effects of PEEP are less apparent with noncompliant lungs,53,54 such that hypotensive-mediated increases in CBV do not occur.
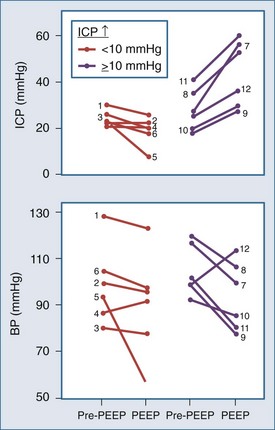
The intuitive notion that PEEP increases cerebral venous pressure to increase ICP is not as straightforward as it initially may seem. For PEEP to increase cerebral venous pressure to levels that will increase ICP, the cerebral venous pressure must at least equal the ICP, which affects the Starling resistor just proximal to the sagittal sinus.28 Thus the higher the ICP, the higher PEEP must be to have such a direct hydraulic effect on ICP. This concept was nicely proved by Huseby and colleagues51 in dog studies in which PEEP was increased progressively, with different starting levels of ICP (Figure 30-13). It is important to note that they prevented PEEP-induced decrements in BP, thus avoiding any reflex increases in CBV. They suggested a hydraulic model to better conceptualize this (Figure 30-14). For example, if all of a 10 cm H2O PEEP application were transmitted to the cerebral vasculature—which is unlikely given the decreased pulmonary compliance associated with the need for such PEEP—ICP will only be affected if it is less than 10 cm H2O (7.7 mm Hg), increasing to a level no higher than the applied PEEP. This presupposes no PEEP-induced arterial pressure decrement.
Antihypertensive Therapy Effects on Intracranial Pressure
Intracranial pressure can also be influenced by antihypertensive drugs. In general, vasodilator drugs such as nitroprusside,55,56 nitroglycerin,57 and nifedipine58 can be expected to increase ICP. Conversely, nonvasodilator antihypertensive drugs, generally sympatholytic drugs such as trimethaphan or beta-adrenergic blocking drugs such as esmolol or labetalol,59 can be expected to have little or no effect on ICP. These observations suggest that the rise in ICP due to vasodilators is caused by increased CBF with an attendant increase in CBV. Also, decreases in BP, as an indirect consequence, may produce vasodilation in autoregulation brain areas, with increased CBV and ICP as discussed earlier for plateau wave physiology. The increase in ICP by these direct and indirect mechanisms thus does not threaten ischemia directly, although herniation and hyperperfusion syndromes and the aforementioned issues with venous outflow obstruction may occur and might be problematic. There has been a report of neurologic deterioration with nitroprusside use despite no change in BP.56 Another consideration in the use of vasodilators is the propensity to reflexively increase endogenous plasma catecholamine concentrations.60 Such increases in plasma catecholamines may be deleterious to the marginally perfused injured brain.61–63
 Hyperperfusion Syndromes
Hyperperfusion Syndromes
In a variety of clinical situations, CBF may be inappropriately increased for a given BP. In the extreme case of such situations, vasoparalysis is present, and CBF becomes more or less a linear function of BP as described in recent dynamic autoregulation studies in TBI.6,14,15,38 Such hyperperfusion syndromes may occur early in cases of severe hepatic encephalopathy,64,65 2 to 3 days after severe head injury,35 after resection of large arteriovenous malformations (AVMs),66–68 after carotid endarterectomy of severely stenotic lesions with poor collaterals,69,70 after cerebral arterial thrombolysis,71 and possibly during administration of cerebral vasodilators at high systemic BP.
Fulminant hepatic failure produces widespread physiologic changes, including altered cerebral physiology.64,65 Aggarwal and coworkers64,65 systematically examined cerebral hemodynamics and metabolism in severe hepatic encephalopathy and during recovery after hepatic transplantation. They have identified phases that are traversed in the course of going from normal cerebral physiology to brain death. Patients initially demonstrate elevated CBF at normotension. This is usually followed by hyperemic (high CBF and/or CBV) intracranial hypertension, then edema with oligemic intracranial hypertension, and finally intracranial circulatory arrest and brain death. The data clearly suggest that the hyperemia may be deleterious, possibly contributing to the development of subsequent cerebral edema. This is supported by observations that the cerebral edema seems to be prevented through the use of barbiturates and hyperventilation during the hyperemic phase.
Several investigators, in the course of examining cerebrovascular physiology after head trauma, have observed that patients with severe head injury initially have normal or low CBF. This is followed a few days later by increased CBF, which is associated with intracranial hypertension.34 This may contribute to subsequent oligemic intracranial hypertension. This concept has been challenged by Marmarou et al. in a clinical study which did not reveal delayed hyperemia in most patients.72
The concept of normal perfusion pressure breakthrough indicates hyperperfusion at normal BP, such as after resection of a large AVM, when the remaining blood vessels lack the ability to constrict normally and regulate blood flow, resulting in abnormally high regional CBF. The pathogenesis is thought to be related to chronic arterial hypotension proximal to the AVM. The larger the AVM, the lower the intracranial BP to which the patient is acclimated (i.e., the cerebral vasculature locally down-regulates the CBF-MAP autoregulatory relationship). Removing the AVM abruptly exposes the cerebral arterial vessels and arterioles to pressure never before experienced.67 Thus despite the BP being within normal limits, the pressure-naïve vasculature is unable to autoregulate, and the physiology of malignant hypertension may ensue to cause cerebral edema and/or hemorrhage. This is an attractive hypothesis that makes physiologic sense. However, Young and coworkers68 report that autoregulation of the vascular bed after AVM resection is generally intact, indicating that vasoparalysis due to chronic hypotension may not be the most important contributor to normal perfusion pressure breakthrough.
One cause of neurologic deterioration after carotid endarterectomy is cerebral edema and/or hemorrhage. This is rather unusual, but the presence postoperatively of a unilateral throbbing headache suggests that it may be present. Blood flow studies reveal such patients to have cerebral hyperemia associated with removal of a large proximal obstruction. While normotension is usually well tolerated, hypertension probably increases the risk of hemorrhage, especially if there was a preoperative cerebral infarction. Similar to the AVM situation described earlier, vasculature that has acclimated to low proximal pressure now is presented with arterial pressure that is much higher, although within the epidemiologic norm.69
After thrombolysis of a cerebral artery, one important source of morbidity is edema or hemorrhage of the reperfused territory. With reperfusion of the ischemic tissue, hyperemia and dysautoregulation occurs for a period of time.71 If sustained, this suggests that irreversible endothelial damage has occurred, and the patient is at risk for secondary edema or hemorrhage, particularly if the depth of ischemia is sufficient to produce early changes on a computed tomography scan.73
Vasodilators such as nitroprusside are frequently used in patients with severe arterial hypertension. When CBF is measured, it is noted that nitroprusside has minimal CBF effect with induced hypotension.74 However, data are not available on its CBF-CBV effects with treatment of hypertension. Such vasodilators are known to cause an increase in ICP,56,75 suggesting an element of cerebral hyperemia. This is supported by reports of cerebral dysautoregulation induced by nitroprusside.76 This ICP elevation and hyperemia74,77 appear to decrease as BP is lowered. This notion is supported by observations during neurosurgery with cerebral swelling present when nitroprusside is administered.78 With its use for induced hypotension during neurosurgery, the brain is noted to be flaccid with no hyperemia evident. Thus cerebral vasodilators can produce a cerebral dysautoregulation/hyperperfusion syndrome, the extent of which is likely dependent on BP. Their use has not yet been reported to be associated with exacerbation of cerebral edema/hemorrhage.
All of the above syndromes describe a clinical course in humans consisting of inappropriate hyperemia for a given BP, followed by cerebral edema or hemorrhage. This suggests that the failure to autoregulate at normal pressure results in exposure of arterioles and capillaries to unacceptably high pressure. This then results in disruption of the blood-brain barrier, with consequent transudation of fluid or frank bleeding. The recent PRx data in humans with TBI and hyperemia with higher BPs in dysautoregulating brain6 further supports these concepts.
 Hyperthermia
Hyperthermia
Temperature management can be critical in neurointensive care. In animal models, hyperthermia has been shown to have deleterious effects on outcome after cerebral ischemia,79 head trauma,79 and seizure.80 Nonrandomized human studies in stroke, TBI, and spinal cord injury strongly suggest a negative effect of hyperthermia on outcome,81–86 with protective effects when induced normothermia is employed in TBI.84 Conversely, mild hypothermia has been shown to have potential for neuroprotection.87–94 The extent of hypothermia required to produce protection is modest (32°C to 36°C). The extent of protection is not adequately explained by reduction in cerebral metabolic rate,95 suggesting that hypothermia has additional beneficial effects such as decreased free radical production or reduction in neurotransmitter neurotoxicity.96
Preliminary reports from a single-center trial of head trauma patients indicated that moderate hypothermia confers cerebral protection when applied within 6 hours of insult and maintained for 24 to 48 hours.97 This observation was not confirmed in a subsequent multi-institutional trial, although head-injured patients who presented with hypothermia had a better outcome.98 In addition, two recent single-center reports of hypothermia after cardiac arrest provide strong support for the notion that mild hypothermia is protective after cerebral ischemia.99,100 Based on these reports, the American Heart Association has adopted hypothermia as a recommended therapy after resuscitation from cardiopulmonary arrest.101 Reports on its use in TBI are conflicting, but nonetheless the Brain Trauma Foundation suggests selective and cautious application of prophylactic moderate hypothermia to 32°C to 35°C for 48 hours may be useful.102
Further complicating the role of hypothermia, however, are the recent results of the IHAST trial103 showing no protection from mild hypothermia (used for all patients regardless of whether focal ischemia arose) during cerebral aneurysm surgery.
 Gas Exchange
Gas Exchange
Cerebrovascular reserve is compromised in many intracranial pathologic processes. Normally, the brain compensates for decrements in supply of oxygen and substrates by vasodilating to maintain or increase flow.104 Animal experiments indicate that it is possible to produce a condition in which cerebrovascular reserve is compromised with increased tendency to cerebral infarction. For example, occlusion of one carotid artery or inducing moderate hypoxemia does not produce symptoms as cerebral vasodilatation occurs to compensate. Indeed, some investigators contend that arterial hypoxemia occurring with normal cerebral vascular compensatory mechanisms does not cause brain damage. Of course, one contributing factor to this view is that hypoxic myocardial dysfunction produces circulatory collapse and death such that isolated posthypoxic (without ischemia) neuronal injury cannot occur. However, if hypoxemia is added to carotid occlusion, or vice versa, a stroke can occur because compensatory mechanisms, already fully utilized, cannot accommodate the further decrease in oxygen supply.105,106 Examples of variants of this situation abound clinically.107 Such examples of attenuated cerebrovascular reserve include cerebral edema, hypoxemia, carotid artery stenosis, peri-infarct penumbra, and anemia. Menon et al.,50 using perilesional OEF data, report impaired reserve around contusions in TBI patients, raising the notion of heterogeneous distribution of cerebrovascular reserve after TBI in humans. This further supports their observations in other reports of an increased ischemic burden with TBI.48,49 In each of these situations, although not easy to quantify, it is clear that added situations of compromised oxygen supply to the brain will risk neuronal injury.
Changes in PaCO2 have a profound impact on CBF. Normally, CBF varies linearly with PaCO2 between 20 and 60 mm Hg.107 PaCO2-mediated changes in CBF occur with corresponding changes in CBV. In situations of abnormal intracranial compliance in which small changes in intracranial volume have large ICP effects, decreasing PaCO2 reduces ICP, and increasing PaCO2 raises ICP.
The primary concern with raised ICP is that it may be associated with cerebral oligemia, so these effects of PaCO2 on ICP are paradoxical. That is, decreasing PaCO2 reduces ICP but at the expense of CBF (Figure 30-15).108 Minhas and colleagues109 report that mild hyperventilation in brain-injured patients produces dangerous perilesional CBF decrements. However, Gupta and colleagues,110 using tissue measures of brain-injured humans, reported sequential increases in PtiO2 with decreasing PaCO2, with an optimum at 26 to 30 mm Hg. Nonetheless, data from head-trauma studies indicate that routine use of hyperventilation can worsen outcome.111 Conversely, allowing hypercapnia to occur, although leading to increased ICP, is associated with increased CBF. These observations pertain to normally autoregulating tissue. The CBF effects in injured brain tissue can be unpredictable. For example, allowing PaCO2 to increase CBF in autoregulating brain areas by increasing ICP may compromise flow or produce venous outflow obstruction in other injured, already fully vasodilated regions.
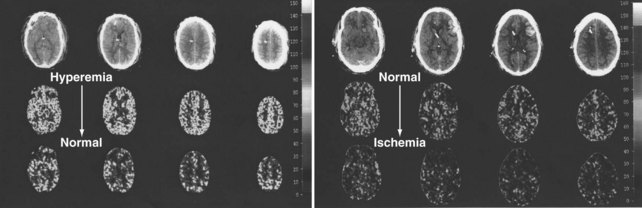
Related to these concerns is the growing practice of permissive hypercapnia in some types of respiratory failure, performed to reduce the risk of ventilator-mediated lung injury. Reports are somewhat conflicting regarding its safety in the brain-injured patient. In a non-trauma porcine model, van Huls and colleagues112 found that hypercapnia to 90 mm Hg increased tissue PO2 while increasing ICP from 20 to 30 mm Hg. Zhou et al.113 in a rodent ischemia model demonstrated neuroprotection with modest hypercapnia. These reports and those of others suggest no direct neurotoxic harmful effects, but theoretically it seems hyperemia-mediated increased ICP still might introduce a risk of hyperemia-mediated herniation or inducement of a positive-feedback cycle through venous outflow obstruction and worsened edema9 as discussed earlier. A recent report by Tasker and Peters,114 however, suggests that the negative hyperemic effects associated with hypercapnia resolve over a day or so such that the pulmonary benefits of the hypercapnia can be gained as the adverse neurologic effects subside. This does raise the possibility of an unacceptable respiratory alkalosis on cessation of the permissive hypercapnia. Moreover, in neonates, hypercapnia increases CBF115 that may lead to cerebral edema, increased ICP, and intraventricular hemorrhage.116–119 Concerns are also raised by a pediatric case report of nonaneurysmal subarachnoid hemorrhage associated with and seemingly caused by permissive hypercapnia.120
The possibility of a neuroprotective effect of respiratory acidosis has also been reported.113,121 Brain homogenates develop far fewer free radicals and less lipid peroxidation when pH is lowered by carbon dioxide than when it is lowered by hydrochloric acid,122 and greater inhibition of tissue lactate production occurs when lowered pH is due to carbon dioxide than when it is due to hydrochloric acid.123 Vanucci et al.124 report a protective effect of modest hypercapnia in an in vivo model of neuronal hypoxia. In trauma patients with multiple organ dysfunction, Gentilello and colleagues125 found permissive hypercapnia to increase ICP but adjusted the level of hypercapnia if ICP rises occurred. Similar problematic ICP increases were also observed in two head-injured patients by Levy et al.,126 which they managed using tracheal gas insufflation, which may be a compromise solution in this conundrum of conflicting physiology and no outcome data. In summary, the data are not conclusive regarding the safety of permissive hypercapnia in the presence of brain injury. It seems that the optimal approach would be to cautiously apply it and adjust according to the ICP response. If unacceptable ICP elevations arise, the options would include abandoning permissive hypercapnia, treating the ICP to allow normalization of the CBF response to the CO2 elevation over a few days, and possibly adding tracheal insufflation to the ventilator strategy to control PaCO2.
 Hyperglycemia
Hyperglycemia
Hyperglycemia has been associated with exacerbation of brain damage with both head trauma and cerebral ischemia,127–129 but it is not a straightforward issue. Clearly, neuronal damage after global cerebral ischemia is exacerbated by hyperglycemia.130 Some studies have suggested that a blood glucose level over 120 mg% is deleterious in stroke patients.127 However, subsequent studies with subhuman primates subjected to global ischemia have suggested a threshold of around 180 mg%.112 Clearly, a blood glucose concentration greater than 400 mg% causes striking worsening of neurologic outcome with global ischemia.128,131 The issue is further clouded by observations from brain microdialysis in human TBI patients of increased lactate/pyruvate ratio with aggressive control of blood glucose.132
With focal cerebral ischemia, the situation is less clear. There have been animal and human studies showing that brain damage is worsened, not affected, or lessened with hyperglycemia.133–137 One report by Prado and colleagues137 in rats suggested that the discriminating factor regarding worsened brain damage with hyperglycemia is whether there is collateral flow. Areas of the brain with minimal or absent collateral vessels were not affected or were improved with hyperglycemia. Brain areas with a continued trickle of flow sustained worse damage. Presumably, the continued substrate supply in anaerobic/oligemic (not ischemic) areas allowed greater accumulation of organic acids in the cells, leading to worsening brain damage.133,138 Unfortunately, these observations are difficult to apply clinically to individual patients with focal ischemia.
Hyperglycemia has not been shown to have either deleterious or protective effects in two animal models of status epilepticus.139,140 The model used in Swan’s report140 produced limbic system damage, whereas Kofke and colleagues139 used a model producing substantia nigra damage. Seizure-induced nigral damage in rats is associated with hypermetabolic lactic acidosis141 that was not exacerbated by hyperglycemia. The fact that nigral damage was not exacerbated with hyperglycemia suggests that metabolic acidosis may not be the sole factor in the development of brain damage after seizure.
 Sepsis
Sepsis
In animal models, sepsis is known to decrease CBF while inducing neuroinflammation142 with altered cerebral metabolic rate (up or down), mitochondrial disfunction,143–145 metabolomic and proteomic disturbances,146 and disruption of the blood-brain barrier (BBB).147–149 These alterations and others undoubtedly underlie the clinical syndrome of septic encephalopathy with associated cognitive impairment.150–152 In addition, it can decrease BP in a manner that may not be well tolerated by the brain with abnormal cerebrovascular reserve. Sepsis-induced decreases in BP can turn an area of cerebral oligemia into an area of ischemic cerebral infarction.
 Sodium
Sodium
Hypernatremia
Hypernatremia can occur in neurologic ICU patients because of nonketotic diabetic coma, dehydration from lack of fluid intake or diuretic use, hypertonic fluid administration, diabetes insipidus, or panhypopituitarism.153 It can be associated with thirst, irritability, seizures, intracranial hemorrhage, or coma, although the rate of increase in sodium concentration is thought to be an important factor in the clinical presentation. For example, a sodium level of 170 mEq/L can be associated with little neurologic symptomatology if the rise occurs over a prolonged period. Indeed, hypertonic saline is occasionally used as a primary therapy for raised ICP,154–157 in which case the elevation in sodium should be considered desirable, with desirable ICP, vasoregulatory, and neurochemical effects. Moreover, treating it could precipitate a rebound increase in ICP.
Diabetes insipidus can occur when disease processes affect the pituitary gland or its vascular supply. It should be suspected when urine output is inappropriately increased. Typically, urine output can increase abruptly to greater than 1 L per hour and be associated with severe hypernatremia and hypovolemic hypotension. Diagnosis of diabetes insipidus is based on continued output of dilute urine in the context of hypertonic serum. The specific gravity of urine will be close to 1.001, with osmolarity less than 200 mOsm/L despite serum osmolarity that may be greater than 320 mOsm/L.158
Hyponatremia
Hyponatremia can occur because of the syndrome of inappropriate secretion of antidiuretic hormone, so-called cerebral salt wasting, or excessive free water administration. Syndrome of inappropriate secretion of antidiuretic hormone is generally associated with hypervolemia and cerebral salt wasting with hypovolemia. Both syndromes can be associated with elevated urinary sodium concentrations, making differentiation between the two difficult in routine clinical practice.159 Rapidly increasing the sodium concentration can produce permanent neurologic damage due to central pontine myelinolysis.160 When the sodium level achieved with such overcorrection is extreme (i.e., 168 to 195 mmol/L), extrapontine myelinolysis has also been reported.161
 Catecholamines
Catecholamines
Subarachnoid hemorrhage is an entity particularly notable for catecholamine effects, some of which are described elsewhere in this book. However, catecholamine effects also occur with increased ICP, stroke, head trauma, or any situation of compromised midbrain-hindbrain oxygen delivery. Notably, intraarterial catecholamine infusions into human cerebral arteries without evident BBB disruption has little effect on CBF and CMR.162 However, subhuman primate studies in which BBB was disrupted indicate that such disruption followed by norepinephrine infusion produces significant increases in CBF and CMR,163 indicating apparent neuroactivation by intravenous catecholamines.
Johnston et al.164 evaluated a small group of patients for neurochemical effects of dopamine versus norepinephrine in brain-injured humans. Norepinephrine, but not dopamine, was associated with decreased arterial-venous oxygen differences and increased brain tissue oxygen but without differences in microdialysis indicators of anaerobiasis. In another report, dopamine increased ICP without having an impact on blood flow velocity or SjvO2 compared to norepinephrine in humans with TBI.165 Nonetheless, catecholamine-based vasopressor therapy is extremely common, although there remain significant knowledge gaps regarding the effects of these drugs, which basically are intravenous (IV) neurotransmitters, by and large, on the brain. A review specifically of their interaction in SAH illustrates many of these issues.
The dramatic increase in serum catecholamine levels after SAH peak at the same time as the peak incidence of post-SAH vasospasm, with symptom development corresponding to serum catecholamine levels.166–170 This leads to the notion that hypothalamic injury with excess catecholamine release may be an important factor in the genesis of post-SAH spasm and stroke.167 Several lines of evidence further support this hypothesis:
 Summary
SummaryKey Points
Grände PO, Asgeirsson B, Nordström CH. Volume-targeted therapy of increased intracranial pressure: the Lund concept unifies surgical and non-surgical treatments. Acta Anaesth Scand. 2002;46(8):929-941.
Huseby JS, Luce JM, Cary JM, et al. Effects of positive end-expiratory pressure on intracranial pressure in dogs with intracranial hypertension. J Neurosurg. 1981;55(5):704-705.
Levine S. Anoxic-ischemic encephalopathy in rats. Am J Pathol. 1960;36:1-17.
Lundberg N. Continuous recording and control of ventricular fluid pressure in neurosurgical practice. Acta Psychiatr Scand Suppl. 1960;36(149):1-193.
Nakagawa Y, Tsuru M, Yada K. Site and mechanism for compression of the venous system during experimental intracranial hypertension. J Neurosurg. 1974;41(4):427-434.
Neil-Dwyer G, Walter P, Cruickshank JM. Beta-blockade benefits patients following a subarachnoid hemorrhage. Eur J Clin Pharmacol. 1985;28(Suppl):25-29.
Nemoto EM. Dynamics of cerebral venous and intracranial pressures. Acta Neurochir Suppl. 2006;96:435-437.
Rosner MJ, Becker DP. Origin and evolution of plateau waves. Experimental observations and a theoretical model. J Neurosurg. 1984;60(2):312-324.
Steiner LA, Czosnyka M, Piechnik SK, et al. Continuous monitoring of cerebrovascular pressure reactivity allows determination of optimal cerebral perfusion pressure in patients with traumatic brain injury. Crit Care Med. 2002;30(4):733-738.
Werner C, Hoffman WE, Thomas C, et al. Ganglionic blockade improves neurologic outcome from incomplete ischemia in rats: partial reversal by exogenous catecholamines. Anesthesiology. 1990;73(5):923-929.
Zweifel C, Lavinio A, Steiner LA, et al. Continuous monitoring of cerebrovascular pressure reactivity in patients with head injury. Neurosurg Focus. 2008;25(4):E2.
1 Cushing H. Concerning a definite regulatory mechanism of the vaso-motor centre which controls blood pressure during cerebral compression. Johns Hopkins Hospital Bulletin. 1901;No 126:290-292.
2 Risberg J, Lundberg N, Ingvar D. Regional cerebral blood volume during acute rises in the intracranial pressure(plateau waves). J Neurosurg. 1969;31:303.
3 Rosner M, Becker D. Origin and evolution of plateau waves. Experimental observations and a theoretical model. J Neurosurg. 1984;50:312-324.
4 Drummond JC. The lower limit of autoregulation: time to revise our thinking? Anesthesiology. 1997 Jun;86(6):1431-1433.
5 Miller J, Becker D, Ward J, Sullivan H, Asams W, Rosner M. Significance of intracranial hypertension in severe head injury. J Neurosurg. 1977;47:503.
6 Zweifel C, Lavinio A, Steiner LA, et al. Continuous monitoring of cerebrovascular pressure reactivity in patients with head injury. Neurosurg Focus. 25(4), 2008.
7 Giulioni M, Ursino M, Alvisi C. Correlations among intracranial pulsatility, intracranial hemodynamics, and transcranial Doppler wave form: Literature review and hypothesis for future studies. Neurosurgery. 1988;22:807.
8 McPherson RW, Koehler RC, Traystman RJ. Effect of jugular venous pressure on cerebral autoregulation in dogs. American J Physiol. 1988;255(6 Pt 2):1516-1524.
9 Nemoto EM. Dynamics of cerebral venous and intracranial pressures. Acta Neurochir Suppl. 2006;96:435-437.
10 Hassler W, Steinmetz H, Gawlowski J. Transcranial Doppler ultrasonography in raised intracranial pressure and in intracranial circulatory arrest. J Neurosurg. 1988;68:745.
11 Lassen NA. Cerebral blood flow and oxygen consumption in man. Physiol Rev. 1959;39(2):183-238.
12 McCall ML. Cerebral circulation and metabolism in toxemia of pregnancy; observations on the effects of veratrum viride and Apresoline (1-hydrazinophthalazine). Am J Obstet Gynecol. 1953;66(5):1015-1030.
13 Moyer J, Morris G, Smith C. Cerebral hemodynamics during controlled hypotension induced by the continuous infusion of ganglionic blocking agents (hexamethonium, pendiomide and arfonad). J Clin Invest. 1954;33:1081-1088.
14 Czosnyka M, Smielewski P, Piechnik S, Steiner LA, Pickard JD. Cerebral autoregulation following head injury. J Neurosurg. 2001;95(5):756-763.
15 Steiner LA, Czosnyka M, Piechnik SK, et al. Continuous monitoring of cerebrovascular pressure reactivity allows determination of optimal cerebral perfusion pressure in patients with traumatic brain injury. Crit Care Med. 2002;30(4):733-738.
16 Czosnyka M, Brady K, Reinhard M, Smielewski P, Steiner LA. Monitoring of cerebrovascular autoregulation: facts, myths, and missing links. Neurocrit Care. 2009;10(3):373-386.
17 Piechnik SK, Czosnyka M, Richards HK, et al. Cerebral venous blood outflow: a theoretical model based on laboratory simulation. Neurosurgery. 2001 Nov;49(5):1214-1222. discussion 22-3
18 Brady KM, Lee JK, Kibler KK, et al. The lower limit of cerebral blood flow autoregulation is increased with elevated intracranial pressure. Anesth Analg. 2009;108(4):1278-1283.
19 Cremer OL, van Dijk GW, Amelink GJ, de Smet AMGA, Moons KGM, Kalkman CJ. Cerebral hemodynamic responses to blood pressure manipulation in severely head-injured patients in the presence or absence of intracranial hypertension. Anesth Analg. 2004;99(4):1211-1217.
20 De Georgia MA, Deogaonkar A. Multimodal monitoring in the neurological intensive care unit. Neurologist. 2005;11(1):45-54.
21 Dewey R, Pieper H, Hunt W. Experimental cerebral hemodynamics. Vasomotor tone, critical closing pressure, and vascular bed resistance. J Neurosurg. 1974;41:597.
22 Early C, Dewey R, Peiper H, Hunt W. Dynamic pressure-flow relationships in the monkey. J Neurosurg. 1974;41:590.
23 Burton AC, Burton AC. On the physical equilibrium of small blood vessels. Am J Physiol. 1951 Feb;164(2):319-329.
24 Czosnyka M, Smielewski P, Piechnik S, et al. Critical closing pressure in cerebrovascular circulation. J Neurol Neurosurg Psychiatry. 1999 May;66(5):606-611.
25 Klatzo I. Evolution of brain edema concepts. Acta Neurochir. 1994;60:3-6.
26 von Haken M, Aschoff A. Acute obstructive hydrocephalus. In: Hacke W, Hanley D, Einhaupl K, Bleck T, Diringer M, Ropper A, editors. Neuro Critical Care. New York: Springer-Verlag; 1994:869.
27 Bederson J, Wiestler O, Brustle O, Roth P, Frick R, Yasargil M. Intracranial venous hypertension and the effects of venous outflow obstruction in a rat model of arteriovenous fistula. Neurosurgery. 1991;29:341-350.
28 Nakagawa Y, Tsuru M, Yada K. Site and mechanism for compression of the venous system during experimental intracranial hypertension. J Neurosurg. 1974;41(4):427-434.
29 Piechnik SK, Czosnyka M, Richards HK, Whitfield PC, Pickard JD. Cerebral venous blood outflow: a theoretical model based on laboratory simulation. Neurosurgery. 2001;49(5):1214-1222.
30 Grande P, Asgeirsson B, Nordstrom C. Volume-targeted therapy of increased intracranial pressure: the Lund concept unifies surgical and non-surgical treatments. Acta Anaesth Scand. 2002;46(8):929-941.
31 Kongstad L, Grande PO. Arterial hypertension increases intracranial pressure in cat after opening of the blood-brain barrier. J Trauma. 2001;51(3):490-496.
32 Kofke W, Dong M, Bloom M, Policare R, Janosky J, Sekhar L. Transcranial Doppler ultrasonography with induction of anesthesia for neurosurgery. J Neurosurg Anaesthesiol. 1994;6:89.
33 Michenfelder J. The 27th Rovenstine Lecture: Neuroanesthesia and the achievement of professional respect. Anesthesiology. 1989;70(4):695.
34 Lundberg N. Continuous recording and control of ventricular fluid pressure in neurosurgical practice. Acta Psychiatr Scand Suppl. 1960;36(149):1-193.
35 Jaggi JL, Obrist WD, Gennarelli TA, Langfitt TW. Relationship of early cerebral blood flow and metabolism to outcome in acute head injury. J Neurosurg. 1990 Feb;72(2):176-182.
36 Beaumont A, Hayasaki K, Marmarou A, Barzo P, Fatouros P, Corwin F. Contrasting effects of dopamine therapy in experimental brain injury. J Neurotrauma. 2001;18(12):1359-1372.
37 Matakas F, Von Waechter R, Knupling R, Potolicchio SJJr. Increase in cerebral perfusion pressure by arterial hypertension in brain swelling. A mathematical model of the volume-pressure relationship. J Neurosurg. 1975 Mar;42(3):282-289.
38 Czosnyka M, Smielewski P, Kirkpatrick P, Laing RJ, Menon D, Pickard JD. Continuous assessment of the cerebral vasomotor reactivity in head injury. Neurosurgery. 1997;41(1):11-17.
39 Lang EW, Mehdorn HM, Dorsch NWC, Czosnyka M. Continuous monitoring of cerebrovascular autoregulation: a validation study. J Neurol Neurosurg Psychiatry. 2002;72(5):583-586.
40 Czosnyka M, Pickard JD. Monitoring and interpretation of intracranial pressure. J Neurol Neurosurg Psychiatry. 2004;75(6):813-821.
41 Rosner M, Becker D. The etiology of plateau waves: A theoretical model and experimental observations. In: Ishii S, Nagai H, Brock M, editors. Intracranial Pressure. New York: Springer-Verlag; 1983:301-306.
42 Rosner MJ, Daughton S. Cerebral perfusion pressure management in head injury. J Trauma. 1990 Aug;30(8):933-940. discussion 40-1
43 Brady KM, Lee JK, Kibler KK, et al. Continuous time-domain analysis of cerebrovascular autoregulation using near-infrared spectroscopy. Stroke. 2007;38(10):2818-2825.
44 Joshi B, Brady K, Lee J, et al. Impaired autoregulation of cerebral blood flow during rewarming from hypothermic cardiopulmonary bypass and its potential association with stroke. Anesth Analg. 2010;110(2):321-328.
45 Lang EW, Lagopoulos J, Griffith J, et al. Noninvasive cerebrovascular autoregulation assessment in traumatic brain injury: validation and utility. J Neurotrauma. 2003;20(1):69-75.
46 Lang EW, Lagopoulos J, Griffith J, et al. Cerebral vasomotor reactivity testing in head injury: the link between pressure and flow. J Neurol Neurosurg Psychiatry. 2003;74(8):1053-1059.
47 Coles J, Minhas P, Fryer T, et al. Effect of hyperventilation on cerebral blood flow in traumatic head injury: clinical relevance and monitoring correlates. Crit Care Clin. 2002;30(9):1950-1959.
48 Coles JP, Fryer TD, Smielewski P, et al. Incidence and mechanisms of cerebral ischemia in early clinical head injury. J Cereb Blood Flow Metab. 2004 Feb;24(2):202-211.
49 Coles JP, Fryer TD, Smielewski P, et al. Defining ischemic burden after traumatic brain injury using 15O PET imaging of cerebral physiology. J Cereb Blood Flow Metab. 2004 Feb;24(2):191-201.
50 Menon DK, Coles JP, Gupta AK, et al. Diffusion limited oxygen delivery following head injury. Crit Care Med. 2004;32(6):1384-1390.
51 Huseby J, Luce J, Cary J, Pavlin E, Butler J. Effects of positive end-expiratory pressure on intracranial pressure in dogs with intracranial hypertension. J Neurosurg. 1981;55(5):704.
52 Shapiro H, Marshall L. Intracranial pressure responses to PEEP in head-injured patients. J Trauma. 1978;18:254.
53 Aidinis S, Lafferty J, Shapiro H. Intracranial responses to PEEP. Anesthesiology. 1976;45(3):275-286.
54 Harken A, Brennan M, Smith B, et al. The hemodynamic response to positive end-expiratory ventilation in hypovolemic patients. Surgery. 1974;76:786-793.
55 Griswold W, Roznik V, Mendoza S. Nitroprusside induced intracranial hypertension. JAMA. 1981;246:2679.
56 Marsh M, Shapiro H, Smith R, et al. Changes in neurologic status and intracranial pressure associated with sodium nitroprusside administration. Anesthesiology. 1979;51:336.
57 Dohi S, Matsumoto M, Takahashi K. The effects of nitroglycerin on cerebrospinal fluid pressure in awake and anesthetized humans. Anesthesiology. 1981;54:511.
58 Hayashi M, et al. Treatment of systemic hypertension and intracranial hypertension and intracranial hypertension in cases of brain hemorrhage. Stroke. 1988;19:314.
59 Van Aken H, Puchstein C, Schweppe M-L, et al. Effect of labetalol on intracranial pressure in dogs with and without intracranial hypertension. Acta Anaesth Scand. 1982;26:615.
60 Stanek B, Zimpfer M, Fitzal S, Raberger G. Plasma catecholamines, plasma renin activity and haemodynamics during sodium nitroprusside-induced hypotension and additional beta-blockage with bunitrolol. Eur J Clin Pharmacol. 1981;19:317.
61 Werner C, Hoffman W, Thomas C, Miletich D, Albrecht R. Ganglionic blockade improves neurologic outcome from incomplete ischemia in rats: partial reversal by exogenous catecholamines. Anesthesiology. 1990;73:923.
62 Neil-Dwyer G, Walter P, Cruickshank J. Beta-blockade benefits patients following a subarachnoid hemorrhage. Eur J Clin Pharmacol. 1985;28:25.
63 Hoffman W, Kochs E, Werner C, Thomas C, Albrecht R. Dexmedetomidine improves neurologic outcome from incomplete ischemia in the rat. Reversal by the alpha 2-adrenergic antagonist atipamezole. Anesthesiology. 1991;75(2):328.
64 Aggarwal S, Kramer D, Yonas H, et al. Cerebral hemodynamic and metabolic changes in fulminant hepatic failure: a retrospective study. Hepatology. 1994;19:80.
65 Aggarwal S, Obrist W, Yonas H, et al. Cerebral hemodynamic and metabolic profiles in fulminant hepatic failure: relationship to outcome. Liver Transplant. 2005 Nov;11(11):1353-1360.
66 Batjer H, Devous MS, Meyer Y, Purdy P, Samson D. Cerebrovascular hemodynamics in arteriovenous malformation complicated by normal perfusion pressure breakthrough. Neurosurgery. 1988;22:503.
67 Young W, Solomon R, Prohovnik I, Ornstein E, Weinstein J, Stein B. 133Xe blood flow monitoring during arteriovenous malformation resection: a case of intraoperative hyperperfusion with subsequent brain swelling. Neurosurgery. 1988;22:765.
68 Young W, Kader A, Prohovnik I, et al. Pressure autoregulation is intact after arteriovenous malformation resection. Neurosurgery. 1993;32:491.
69 Reigel M, Hollier L, Sundt TJ, Piepgras D, Sharbrough F, Cherry K. Cerebral hyperperfusion syndrome: a cause of neurologic dysfunction after carotid endarterectomy. J Vasc Surg. 1987;5:628.
70 Schroeder T, Sillesen H, Srensen O, Engell H. Cerebral hyperperfusion following carotid endarterectomy. J Neurosurg. 1987;66:824.
71 Reinhard M, Wihler C, Roth M, et al. Cerebral autoregulation dynamics in acute ischemic stroke after rtPA thrombolysis. Cerebrovasc Dis. 2008;26(2):147-155.
72 Marmarou A, Fatouros PP, Barzó P, et al. Contribution of edema and cerebral blood flood volume to traumatic brain swelling in head-injured patients. J Neurosurg. 2000 Aug;93(2):183-193.
73 del Zoppo G. Thrombolysis in acute stroke. Neurologia. 1995;10(2):37-47.
74 Henriks C, Harmsen A, Christensen P, Sorensen M, Lester J, Paulson O. Controlled hypotension with sodium nitroprusside: effects on cerebral blood flow and Cerebral venous blood gases in patients operated for cerebral aneurysms. Acta Anaesth Scand. 1983;27:62-67.
75 Cottrell J, Patel K, Ransahoff J, et al. Intracranial pressure changes induced by sodium nitroprusside in patients with intracranial mass lesions. J Neurosurg. 1978;48:329.
76 Weiss M, Spence J, Apuzzo M, Heiden J, McComb J, Kurze T. Influence of nitroprusside on cerebral pressure autoregulation. Neurosurgery. 1979;4:56-59.
77 Candia G, Heros R, Lavyne M, Zervas N, Nelson C. Effect of intravenous sodium nitroprusside on cerebral blood flow and intracranial pressure. Neurosurgery. 1978;3:50-53.
78 Theard M, Cheng M, Crowder C, Tempelhoff R. Control of blood pressure during intracranial procedures: Comparison between nicardipine and nitroprusside. J Neurosurg Anesth. 1997;9:388.
79 Dietrich W. The importance of brain temperature in cerebral injury. J Neurotrauma. 1992;9(Suppl 2):S475-S485.
80 Blennow G, Brierley J, Meldrum B, Siesjo B. Epileptic brain damage: the role of systemic factors that modify cerebral energy metabolism. Brain. 1978;101:687.
81 Schmidt K, Chan C. Thermoregulation and fever in normal persons and in those with spinal cord injuries. [Review]. Mayo Clin Proc. 1992;67:469.
82 Azzimondi G, Bassein L, Nonino F, et al. Fever in acute stroke worsens prognosis: a prospective study. Stroke. 1995;26:2043-2050.
83 Greer DM, Funk SE, Reaven NL, et al. Impact of fever on outcome in patients with stroke and neurologic injury: a comprehensive meta-analysis. Stroke. 2008 Nov;39(11):3029-3035.
84 Schwarz S, Aschoff A, Schwab S. Incidence and prognostic significance of fever following cerebral hemorrhage. Neurology. 2000;54:354-361.
85 Puccio A, Fischer M, Jankowitz B, Yonas H, Darby J, Okonkwo D. Induced Normothermia Attenuates Intracranial Hypertension and Reduces Fever Burden after Severe Traumatic Brain Injury. Neurocrit Care. 2009.
86 Kammersgaard LP, Jorgensen HS, Rungby JA, et al. Admission body temperature predicts long-term mortality after acute stroke: the Copenhagen Stroke Study.[see comment]. Stroke. 2002 Jul;33(7):1759-1762.
87 Wang SQ, Lakatta EG, Cheng H, et al. Adaptive mechanisms of intracellular calcium homeostasis in mammalian hibernators. J Exp Biol. 2002 Oct;205(Pt 19):2957-2962.
88 Bavaria JE, Brinster DR, Gorman RC, Woo YJ, Gleason T, Pochettino A. Advances in the treatment of acute type A dissection: an integrated approach. Ann Thorac Surg. 2002 Nov;74(5):S1848-S1852. discussion S57-63
89 Storey KB. Cold ischemic organ preservation: lessons from natural systems. Journal of Investigative Medicine. 2004 Jul;52(5):315-322.
90 Maher J, Hachinski V. Hypothermia as a potential treatment for cerebral ischemia. [Review]. Cerebrovasc Brain Metab Rev. 1993;5:277.
91 Froehler MT, Geocadin RG. Hypothermia for neuroprotection after cardiac arrest: mechanisms, clinical trials and patient care. J Neurol Sci. 2007 Oct 15;261(1-2):118-126.
92 Gunn AJ, Thoresen M. Hypothermic neuroprotection. NeuroRx. 2006 Apr;3(2):154-169.
93 Sterz F, Safar P, Tisherman S, et al. Mild hypothermic cardiopulmonary resuscitation improves outcome after prolonged cardiac arrest in dogs. Crit Care Med. 1991;19:379-389.
94 Liu L, Yenari MA. Therapeutic hypothermia: neuroprotective mechanisms. Front Biosci. 2007;12:816-825.
95 Nemoto E, Klementavicius R, Melick J, Yonas H. Effect of mild hypothermia on active and basal cerebral oxygen metabolism and blood flow. Adv Exp Med Biol. 1994;361:469.
96 Busto R, Globus M, Dietrich W, Martinez E, Valdes I, Ginsberg M. Effect of mild hypothermia on ischemia-induced release of neurotransmitters and free fatty acids in rat brain. Stroke. 1989;20(7):904-910.
97 Marion D, Penrod L, Kelsey S, et al. Treatment of traumatic brain injury with moderate hypothermia. N Engl J Med. 1997;336(8):540-546.
98 Clifton G, Miller E, Choi S, et al. Hypothermia on admission in patients with severe brain injury. J Neurotrauma. 2002;19(3):293-301.
99 Bernard S, Gray T, Buist M, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346(8):557-563.
100 Group HaCAS. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346(8):549-556.
101 Nolan J, Morley P, Vanden Hoek T, et al. International Liaison Committee on Resuscitation. Therapeutic hypothermia after cardiac arrest: an advisory statement by the advanced life support task force of the International Liaison Committee on Resuscitation. Circulation. 2003;108(1):118-121.
102 Brain Trauma Foundation, American Association of Neurological Surgeons, Congress of Neurological Surgeons, Joint Section on Neurotrauma and Critical Care, AANS/CNSBratton SL, Chestnut RM, Ghajar J, et al. Guidelines for the management of severe traumatic brain injury. III. Prophylactic hypothermia. [Erratum appears in J Neurotrauma 2008 Mar;25(3):276-8]. J Neurotrauma. 2007;24(Suppl 1):S21-S25.
103 Todd M, Hindman B, Clarke W, Torner J. Intraoperative Hypothermia for Aneurysm Surgery Trial (IHAST) Investigators. Mild intraoperative hypothermia during surgery for intracranial aneurysm. N Engl J Med. 2005;352(2):135-145.
104 Kogure K, Scheinberg P, Reinmuth O, Fujishima M, Bustro R. Mechanisms of cerebral vasodilation in hypoxia. J Appl Physiol. 1970;29:223.
105 Levine S. Anoxic-ischemic encephalopathy in rats. Am J Pathol. 1960;36:1.
106 Hojer-Pedersen E. Effect of acetazolamide on CBF in subacute and chronic cerebrovascular disease. Stroke. 1987;18(5):887.
107 Tominaga S, Strandgaard S, Uemura K, Ito K, Kutsuzawa T. Cerebrovascular CO2 reactivity in normotensive and hypertensive man. Stroke. 1976;7(5):507.
108 Stringer W, Hasso A, Thompson J, Hinshaw D, Jordan K. Hyperventilation-induced cerebral ischemia in patients with acute brain lesions: demonstration by xenon-enhanced CT. Am J Neurorad. 1993;14(2):475-484.
109 Minhas P, Menon D, Herrod N, et al. Cerebral ischemia associated with hyperventilation: A PET study. J Neurosurg Anesth. 1997;9:380.
110 Gupta A, Gupta S, Swart M, Al-Rawi P, Hutchinson P, Kirkpatrick P. Comparison of brain tissue oxygen with jugular venous oxygen saturation during hyperventilation in head-injured patients. J Neurosurg Anesth. 9(399), 1997.
111 Muizelaar J, Marmarou A, Ward J, et al. Adverse effects of prolonged hyperventilation in patients with severe head injury: A randomized clinical trial. J Neurosurg. 1991;75:731-739.
112 van Hulst R, Hasan D, Lachmann B. Intracranial pressure, brain PCO2, PO2, and pH during hypo- and hyperventilation at constant mean airway pressure in pigs. Intensive Care Med. 2002;28(1):68-73.
113 Zhou Q, Cao B, Niu L, et al. Effects of permissive hypercapnia on transient global cerebral ischemia-reperfusion injury in rats. Anesthesiology. 2010;112(2):288-297.
114 Tasker R, Peters M. Combined lung injury, meningitis and cerebral edema: how permissive can hypercapnia be? Intensive Care Med. 1998;24(6):616-619.
115 Hansen N, Brubakk A, Bratlid D, Oh W, Stonestreet B. The effects of variations in PaCO2 on brain blood flow and cardiac output in the newborn piglet. Pediatr Res. 1984;18:1132-1136.
116 Skouteli H, Kuban K, Leviton A. Arterial blood gas derangements associated with death and intracranial hemorrhage in premature babies. J Perinatol. 1988;18:336-341.
117 Szymonowicz W, Yu V, Wilson F. Antecedents of periventricular haemorrhage in infants weighing 1250g or less at birth. Arch Dis Child. 1984;59:13-17.
118 Wallin L, Rosenfeld C, Laptook A, et al. Neonatal intracranial hemorrhage. 11. Risk factor analysis in an inborn population. Early Hum Dev. 1990;23:129-137.
119 Van De Bor M, Van Bel F, Lineman R, Ruys J. Perinatal factors and periventricular hemorrhage in preterm infants. Am J Dis Child. 1986;140:1125-1130.
120 Edmunds S, Harrison R. Subarachnoid hemorrhage in a child with status asthmaticus: significance of permissive hypercapnia. Pediatric Crit Care Med. 2003;4(1):100-103.
121 Laffey J, Kavanagh B. Carbon dioxide and the critically ill – too little of a good thing? Lancet. 1999;354:1283-1286.
122 Rehncrona S, Hauge H, Siesjö B. Enhancement of iron-catalyzed free radical formation by acidosis in brain homogenates: difference in effect by lactic acid and CO2. J Cereb Blood Flow Metab. 1989;9:65-70.
123 Abu Romeh S, Tannen R. Amelioration of hypoxia-induced lactic acidosis by superimposed hypercapnia or hypochloride acid infusion. Am J Physiol. 1986;250:702-709.
124 Vannucci R, Towfighi J, Heitjan D, Brucklacher R. Carbon dioxide protects the perinatal brain from hypoxic-ischemic damage: an experimental study in the immature rat. Pediatrics. 1995;95:868-874.
125 Gentilello L, Anardi D, Mock C, Arreola-Risa C, Maier R. Permissive hypercapnia in trauma patients. J Trauma. 1995;39(5):846-852.
126 Levy B, Bollaert P, Nace L, Larcan A. Intracranial hypertension and adult respiratory distress syndrome: usefulness of tracheal gas insufflation. J Trauma. 1995;39(4):799-801.
127 Pulsinelli W, Levy D, Sigsbee B, Scherer P, Plum F. Increased damage after ischemic stroke in patients with hyperglycemia with or without established diabetes mellitus. Am J Med. 1983;74:540.
128 Siemkowicz E. Hyperglycemia in the reperfusion period hampers recovery from cerebral ischemia. Acta Neurol Scand. 1981;64:207.
129 De Salles A, Muizelaar J, Young H. Hyperglycemia, cerebrospinal fluid lactic acidosis, and CBF in severely head-injured patients. Neurosurgery. 1987;21:45.
130 Lanier W, Stangland K, Scheithauer B, Milde J, Michenfelder J. The effects of dextrose infusion and head position on neurologic outcome after complete cerebral ischemia in primates: examination of a model. Anesthesiology. 1987;66(1):39.
131 Siemkowicz E, Gjedde A. Post-ischemic coma in rat: effect of different pre-ischemic blood glucose levels on cerebral metabolic recovery after ischemia. Acta Physiol Scand. 1980;110:225.
132 Oddo M, Schmidt JM, Carrera E, et al. Impact of tight glycemic control on cerebral glucose metabolism after severe brain injury: a microdialysis study. Crit Care Med. 2008;36(12):3233-3238.
133 Sieber F, Traystman R. Special issues: glucose and the brain. [Review]. Crit Care Med. 1992;20:104.
134 Zasslow M, Pearl R, Shuer L, Steinberg G, Lieberson R, Larson CJ. Hyperglycemia decreases acute neuronal ischemic changes after middle cerebral artery occlusion in cats. Stroke. 1989;20:519.
135 de Courten-Myers G, Myers R, Schoolfield L. Hyperglycemia enlarges infarct size in cerebrovascular occlusion in cats. Stroke. 1988;19:623.
136 Nedergaard M. Mechanisms of brain damage in focal cerebral ischemia. [Review]. Acta Neurol Scand. 1988;77:81.
137 Prado R, Ginsberg M, Dietrich W, Watson B, Busto R. Hyperglycemia increases infarct size in collaterally perfused but not end-arterial vascular territories. J Cereb Blood Flow Metab. 1988;8:186.
138 Sapolsky R, Trafton J, Tombaugh G. Excitotoxic neuron death, acidotic endangerment, and the paradox of acidotic protection. Adv Neurol. 1996;71:237-244.
139 Kofke W, Ahdab-Barmada M, Rose M, Clyde C, Nemoto E. Substantia nigra damage after flurothyl-induced seizures in rats worsens after post seizure recovery: no exacerbation with hyperglycemia. Neurol Res. 1993;15:333.
140 Swan J, Meldrum B, Simon R. Hyperglycemia does not augment neuronal damage in experimental status epilepticus. Neurology. 1986;36(10):1351.
141 Ingvar M, Folbegrova J, Siesjo B. Metabolic alterations underlying the development of hypermetabolic necrosis in the substantia nigra in status epilepticus. J Cereb Blood Flow Metab. 1987;7(1):103.
142 Semmler A, Hermann S, Mormann F, et al. Sepsis causes neuroinflammation and concomitant decrease of cerebral metabolism. J Neuroinflam. 2008;5:38.
143 Comim CM, Rezin GT, Scaini G, Di-Pietro PB, Cardoso MR, Petronilho FC, et al. Mitochondrial respiratory chain and creatine kinase activities in rat brain after sepsis induced by cecal ligation and perforation. Mitochondrion. 2008 Sep;8(4):313-318.
144 d’Avila JC, Santiago AP, Amancio RT, et al. Sepsis induces brain mitochondrial dysfunction. Crit Care Med. 2008 Jun;36(6):1925-1932.
145 Hopkins RO. Sepsis, oxidative stress, and brain injury. Crit Care Med. 2007 Sep;35(9):2233-2234.
146 Hinkelbein J, Feldmann REJr, Peterka A, et al. Alterations in cerebral metabolomics and proteomic expression during sepsis. Curr Neurovasc Res. 2007 Nov;4(4):280-288.
147 Ekstrom-Jodal B, Haggendal E, Larsson L. CBF and oxygen uptake in endotoxic shock. An experimental study in dogs. Acta Anaesth Scand. 1982;26(3):163-170.
148 Hofer S, Bopp C, Hoerner C, et al. Injury of the blood brain barrier and up-regulation of icam-1 in polymicrobial sepsis. J Surg Res. 2008 May 15;146(2):276-281.
149 Sharshar T, Carlier R, Bernard F, et al. Brain lesions in septic shock: a magnetic resonance imaging study. Intensive Care Med. 2007 May;33(5):798-806.
150 Comim CM, Constantino LC, Barichello T, et al. Cognitive impairment in the septic brain. Curr Neurovasc Res. 2009 Aug;6(3):194-203.
151 Pytel P, Alexander JJ. Pathogenesis of septic encephalopathy. Curr Opin Neurol. 2009 Jun;22(3):283-287.
152 Eidelman LA, Putterman D, Putterman C, et al. The spectrum of septic encephalopathy. Definitions, etiologies, and mortalities. JAMA. 1996 Feb 14;275(6):470-473.
153 Oh M, Carroll H. Disorders of sodium metabolism: hypernatremia and hyponatremia.[Review]. Crit Care Med. 1992;20(1):94-103.
154 Doyle J, Davis D, Hoyt D. The Use of Hypertonic Saline in the Treatment of Traumatic Brain Injury. J Trauma. 2001;50:367-383.
155 Khanna S, Daniel D, Bradley P, et al. Use of hypertonic saline in the treatment of severe refractory posttraumatic intracranial hypertension in pediatric traumatic brain injury. Crit Care Med. 2000;28(4):1144-1151.
156 Suarez JI. Hypertonic saline for cerebral edema and elevated intracranial pressure. Cleve Clin J Med. 2004 Jan;71(Suppl 1):S9-13.
157 Suarez JI, Qureshi AI, Bhardwaj A, et al. Treatment of refractory intracranial hypertension with 23.4% saline. Crit Care Med. 1998 Jun;26(6):1118-1122.
158 Buonocore C, Robinson A. The diagnosis and management of diabetes insipidus during medical emergencies. Endocr Metab Clin North Am. 1993;22:411-423.
159 Maroon J, Nelson P. Hypovolemia in patients with subarachnoid hemorrhage: therapeutic implications. Neurosurgery. 1979;4:223.
160 Laureno R, Kapp B. Pontine and extrapontine myelinolysis following rapid correction of hyponatremia. Lancet. 1998;1:1439.
161 Brown W, Caruso J. Extrapontine myelinolysis with involvement of the hippocampus in three children with severe hypernatremia. J Child Neurology. 1999;14(7):428-433.
162 Olesen J. The effect of intracarotid epinephrine, norepinephrine, and angiotensin on the regional CBF in man. Neurology. 1972;22:978-987.
163 MacKenzie E, McCulloch J, Harper A. Influence of endogenous norepinephrine on CBF and metabolism. Am J Physiol. 1976;231:489.
164 Johnston AJ, Steiner LA, Chatfield DA, et al. Effect of cerebral perfusion pressure augmentation with dopamine and norepinephrine on global and focal brain oxygenation after traumatic brain injury. Intensive Care Med. 2004;30(5):791-797.
165 Ract C, Vigue B. Comparison of the cerebral effects of dopamine and norepinephrine in severely head-injured patients. Intensive Care Med. 2001;27(1):101-106.
166 Cruickshank J, Neil-Dwyer G, Stott A. Possible role of catecholamines, corticosteroids, and potassium in production of electro-cardiographic abnormalities associated with subarachnoid hemorrhage. Br Heart J. 1974;36:697.
167 Loach A, Benedict C. Plasma catecholamine concentration associated with cerebral vasospasm. J Neurol Sci. 1980;45:261.
168 Cruickshank J, Neil-Dwyer G, Brice J. Electrocardiographic changes and their prognostic significance in subarachnoid hemorrhage. J Neurol Neurosurg Psychiatry. 1974;37:755.
169 Neil-Dwyer G, Cruickshank J. The urinary catecholamine and plasma cortisol levels in patients with subarachnoid hemorrhage. J Neurol Sci. 1974;22:375.
170 Fraser R, Stein B, Barrett R, Pool J. Noradrenergic mediation of experimental cerebrovascular spasm. Stroke. 1970;1:356.
171 Peerless S, Kendall M. The innervation of the cerebral blood vessels. In: Smith R, Robertson J, editors. Subarachnoid Hemorrhage and Cerebrovascular Spasm. Springfield, Il: Charles C. Thomas; 1975:38-54.
172 Marion D, Segal R, Thompson M. Subarachnoid hemorrhage and the heart. Neurosurgery. 1986;18:101.
173 Cruickshank J, Neil-Dwyer G, Lane J. The effect of oral propranolol upon the ECG changes occurring in subarachnoid hemorrhage. Cardiovasc Res. 1975;9:236.
174 Kono T, Morita H, Kuroiwa T, Onaka H, Takatsuka H, Fujiwara A. Left ventricular wall motion abnormalities in patients with subarachnoid hemorrhage: neurogenic stunned myocardium. J Am Coll Cardiol. 1994;24:636.
175 Kolin A, Norris J. Myocardial damage from acute cerebral lesions. Stroke. 1984;15:990.
176 Svengaard N, Brismar J, Delgado T, Rosengren E. Subarachnoid hemorrhage in the rat: Effect on the development of vasospasm of selective lesions of the catecholamine systems in the lower brain stem. Stroke. 1985;16:602.
177 Cheshire WPJr, Saper CB. The insular cortex and cardiac response to stroke. Neurology. 2006 May 9;66(9):1296-1297.
178 Ay H, Koroshetz WJ, Benner T, et al. Neuroanatomic correlates of stroke-related myocardial injury. Neurology. 2006 May 9;66(9):1325-1329.
179 Laowattana S, Zeger SL, Lima JA, et al. Left insular stroke is associated with adverse cardiac outcome. Neurology. 2006 Feb 28;66(4):477-483. discussion 63
180 Darby J, Yonas H, Marks E, Durham S, Snyder R, Nemoto E. Acute CBF response to dopamine-induced hypertension after subarachnoid hemorrhage. J Neurosurg. 1994;80:857.
181 Svengaard N, Delgado T, Arbab M. Catecholaminergic and peptidergic systems underlying cerebral vasospasm: CBF and CMRgl changes following an experimental subarachnoid hemorrhage in the rat. Proceedings of the Charlottesville Conference Held April 29-May 1, 1987. In: Wilkins R, editor. Cerebral Vasospasm. New York: Raven Press; 1988:175.
182 Werner C, Hoffman W, Kochs E, Rabito S, Miletich D. Captopril improves neurologic outcome from incomplete cerebral ischemia in rats. Stroke. 1991;22:910.
183 Busto R, Harik S, Yoshida S, Scheinberg P, Ginsberg M. Cerebral norepinephrine depletion enhances recovery after brain ischemia. Ann Neurol. 1985;18:329.
184 Kofke W, Garman R, Garman R, Rose M. Opioid neurotoxicity: Role of neurotransmitter systems[Abstract]. J Neurosurg Anaesthesiol. 1995;7:321.
185 Stein S, Cracco R. Cortical injury without ischemia produced by topical monoamines. Stroke. 1982;13:74.
