[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Nonmalignant Leukocyte Disorders
After the completion of this chapter, the reader will be able to:
1. Describe the basic genetic defect and the morphologic consequences in Pelger-Huët anomaly.
2. Indicate with what cell type Pelger-Huët cells might be confused and discuss what is meant by “pseudo-Pelger Huët.”
3. Compare and contrast two genetic causes of neutrophilic hypersegmentation and indicate what other disorder must be ruled out.
4. Describe the basic genetic defect and the morphologic consequences in Alder-Reilly anomaly, Chédiak-Higashi syndrome, and May-Hegglin anomaly.
5. Indicate how inclusions in Alder-Reilly anomaly and May-Hegglin anomaly might be confused with reactive morphology.
6. Discuss the defect in and functional consequences of chronic granulomatous disease.
7. Discuss the basic defects in and functional consequences of leukocyte adhesion disorders.
8. Discuss at least three other miscellaneous granulocyte disorders.
9. Describe the characteristic macrophage morphology associated with the mucopolysaccharidoses, Gaucher disease, and Niemann-Pick disease.
10. Describe the basic defect in genetic disorders leading to decreased T-lymphocyte production, decreased B-lymphocyte production, and the combined decrease of T, B, and natural killer lymphocytes.
11. Define what is meant by neutrophilia, neutropenia, lymphocytosis, lymphocytopenia, monocytosis, monocytopenia, eosinophilia, eosinopenia, and basophilia; give some examples of conditions in which each occurs.
12. Describe the nuclear and cytoplasmic alterations in granulocytes, monocytes, and lymphocytes that indicate a reaction to infection, inflammation, or stress.
Case Studies
Case 1
1. What is the most likely cause of this child’s recurring infections?
2. Biochemically speaking, what is the specific nature of the problem?
3. Do all microorganisms produce equally severe disease in individuals affected with this disorder?
4. How is this disorder transmitted genetically in the majority of cases?
Case 2
1. Based on the differential results, what cells should the medical laboratory scientist look for on the patient’s blood film?
2. Where on the blood film should the examination be made and why?
3. What is the significance if the suspected cells are found?
4. What preparation other than a blood film might be helpful in this situation?
Genetic Leukocyte Abnormalities
Nuclear Abnormalities
Pelger-Huët Anomaly
Pelger-Huët anomaly (PHA) is an autosomal dominant disorder resulting in decreased nuclear segmentation that is most apparent in neutrophils. It is the most common genetic disorder of leukocytes and is caused by a mutation of the lamin B receptor. The lamin B receptor is an integral protein in the inner nuclear membrane,1 and the associated gene is located on chromosome band 1q41-43.2 PHA is one of a large number of so-called laminopathies ranging from a type of muscular dystrophy to premature aging.3
Heterozygous PHA is characterized by granulocytes whose nucleus is segmented only once, taking the form of “two very distinct segments connected by a very thin filament.”4 In addition, what appear to be band forms may be present; however, the chromatin shows a highly clumped pattern and stains densely. The heterozygous mutation does not affect the function of neutrophils.5
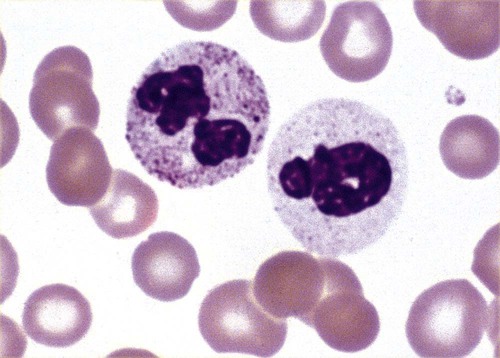
Homozygous PHA is relatively rare and is characterized by granulocytes with round or oval nuclei (no segmentation) as well as the band-form nuclei described earlier. The chromatin pattern is also very dense and clumped. The homozygous form may be accompanied by impaired cognitive development, skeletal abnormalities, and heart defects.5 Leukocyte differential counts performed on blood samples from patients with either heterozygous or homozygous PHA may mistakenly identify the abnormal cells as immature granulocytes if the highly clumped, dense chromatin pattern is not noticed (Figure 28-1).
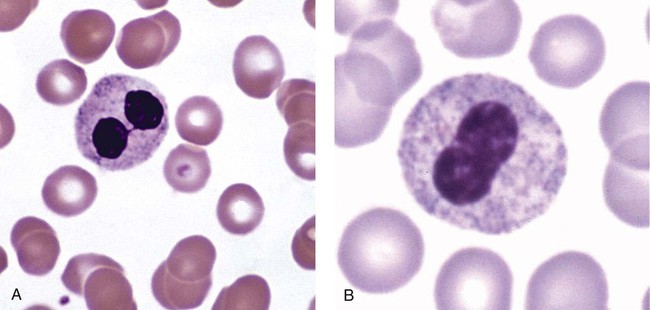
Medical laboratory scientists must distinguish inherited PHA from an acquired form of nuclear hyposegmentation known as pseudo–Pelger-Huët anomaly (pseudo-PHA) that may be seen in a variety of malignant myeloproliferative neoplasms.6 Two distinctions between inherited PHA and pseudo-PHA are the following: (1) the percentage of affected neutrophils is much higher in the inherited form (usually over 80%), whereas in the acquired form fewer than 50% are usually affected; and (2) pseudo-PHA alterations are usually accompanied by other morphologic indications of malignancy, such as the presence of blast forms.
Hereditary Neutrophil Hypersegmentation
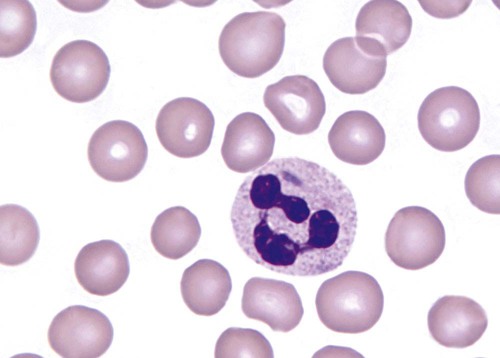
Hereditary neutrophil hypersegmentation designates a group of at least two disorders. The first is a benign autosomal dominant condition characterized by hypersegmented neutrophils (Figure 28-2) with no clinical signs or symptoms. The major concern is to distinguish this condition from the neutrophilic hypersegmentation found in megaloblastic anemia. In the hereditary form, a large majority of neutrophils are hypersegmented and there is no macrocytic anemia. In megaloblastic anemia, fewer than 50% of neutrophils are usually hypersegmented.
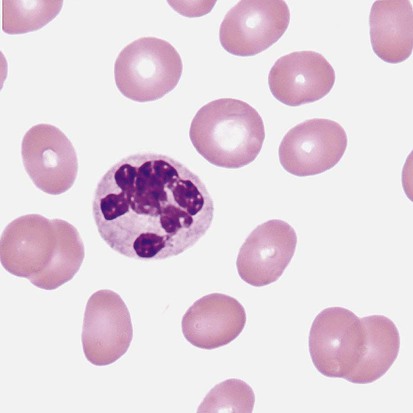
The second disorder is myelokathexis, an inherited form of neutropenia characterized by hypersegmented neutrophils and bone marrow hyperplasia of myeloid cells. There is increased programmed cell death (apoptosis) of granulocyte precursors.7 In this case the neutrophil nuclei, in addition to being hypersegmented, are pyknotic (to be described later), and the intrasegmental filaments are longer than normal.
Cytoplasmic Abnormalities
Alder-Reilly Anomaly
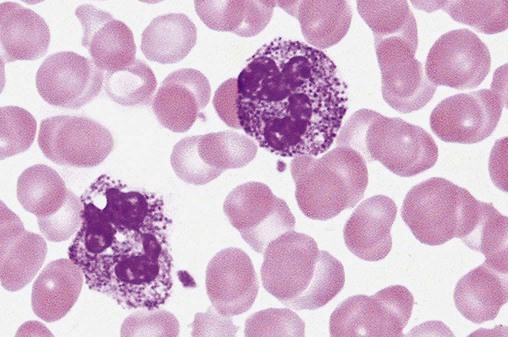
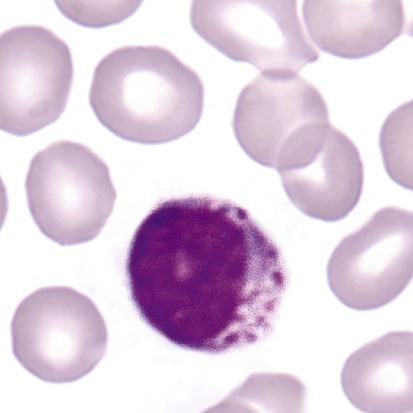
Alder-Reilly anomaly is transmitted as a recessive trait and is characterized by granulocytes with large, darkly staining metachromatic granules that may resemble toxic granulation (described later) (Figure 28-3). The granules are composed of mucopolysaccharides and are sometimes referred to as Alder-Reilly bodies or Reilly bodies. In more severe forms, the granules may also be found in monocytes and lymphocytes. The basic defect is the incomplete degradation of mucopolysaccharides (protein-carbohydrate complexes), and there may be a structural abnormality in the myeloperoxidase gene.8 These granules are also discussed later in the section on storage diseases.
Chédiak-Higashi Syndrome
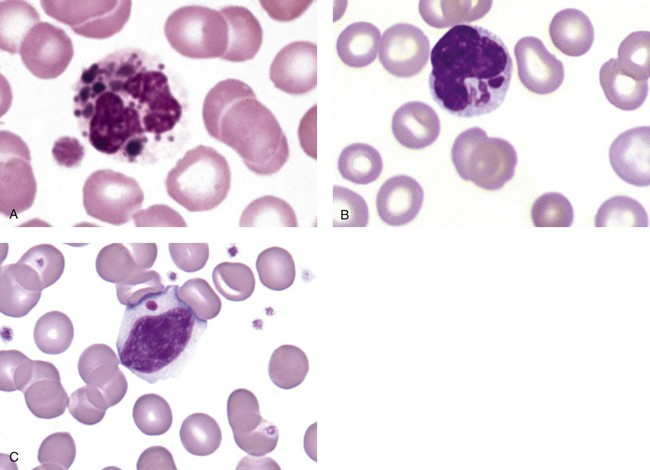
Chédiak-Higashi syndrome (CHS) is a rare and fatal autosomal recessive disease characterized by cells with enlarged lysosomal vesicles. All cells with lysosomal organelles are affected, including the melanosomes of melanocytes in the skin, the dense granules of platelets, and leukocyte granules. Patients usually have a tendency to bleed, frequent bacterial infections, symptoms of immunodeficiency, variable albinism, and progressive neurologic dysfunction.9 CHS is associated with a mutation in the LYST gene that encodes for a type of vesicle trafficking regulatory protein.10 Patients who do not die of bacterial infection develop a lymphohistiocytic infiltration into the major organs of the body, resulting in organ failure.9
Hematologic findings in CHS include giant lysosomal granules in granulocytes, monocytes, and lymphocytes (Figure 28-4). The platelet abnormality usually results in a prolonged bleeding time, and granule release by thrombin is impaired.11
May-Hegglin Anomaly
May-Hegglin anomaly is one of a group of at least four overlapping autosomal dominant platelet disorders: May-Hegglin anomaly (MHA), Sebastian syndrome (SBS), Fechtner syndrome (FS), and Epstein syndrome (EPS). All are caused by mutations of the nonmuscle myosin heavy-chain type IIA gene referred to as MYH9.12 MHA, SBS, and FS are characterized by large basophilic inclusions in leukocytes, thrombocytopenia, and giant platelets (Figure 28-5). FS and EPS are also associated with nephritis and deafness. Patients with MHA and SBS generally do not have renal disease or deafness; however, there have been a few reports of renal disease in individuals with MHA.13
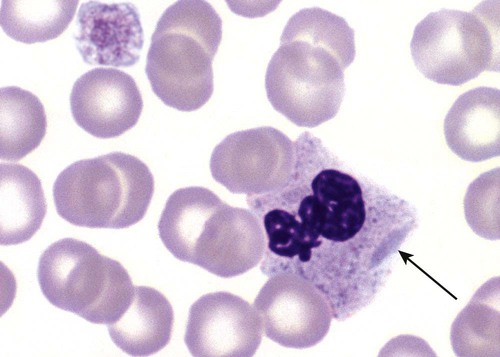
The leukocyte inclusions in MHA have been described as “Döhle body–like” (Döhle bodies are discussed later in this chapter); however, there are striking ultrastructural differences. Döhle bodies are made up of lamellar rows of rough endoplasmic reticulum, whereas the MHA inclusions consist of randomly placed rods in an amorphous background. In addition, Döhle bodies are found only in neutrophils, whereas MHA inclusions are seen in neutrophils, eosinophils, basophils, and monocytes.14 MHA inclusions may stain a very pale color with Wright stain and may be missed in monocytes whose cytoplasm is also blue-grey.
Functional Abnormalities
Chronic Granulomatous Disease
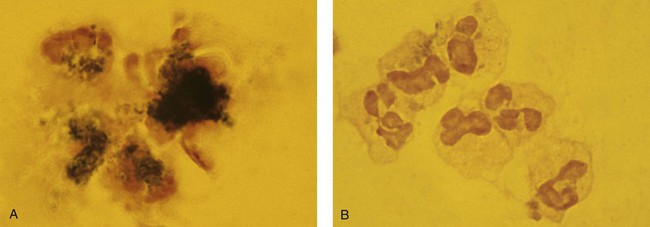
Chronic granulomatous disease (CGD) is a disorder caused by the inability of phagocytes to produce superoxide and reactive oxygen species. The basic defect is one or more mutations in any of four genes responsible for proteins that make up a complex known as NADPH oxidase (NADPH is the reduced form of nicotinamide adenine dinucleotide phosphate). The majority of cases (approximately 60% to 65%) are X-linked recessive and 35% to 40% are autosomal recessive, depending on which gene has been mutated. The X-linked forms tend to carry a higher mortality rate.15
In the nitroblue tetrazolium reduction test, normal neutrophils, when stimulated, reduce the yellow water-soluble nitroblue tetrazolium to a dark blue insoluble formazan. CGD neutrophils cannot perform this reduction (Figure 28-6). Flow cytometry uses a fluorescent probe, such as dihydrorhodamine-123, to measure intracellular production of reactive oxygen species.15
Leukocyte Adhesion Disorders
Leukocyte adhesion disorders (LADs) result in the inability of neutrophils and monocytes to adhere to endothelial cells and to transmigrate from the blood to the tissues. The consequence of this inability is increased and potentially lethal bacterial infections. The basic defect is a mutation in the genes responsible for the formation of cell adhesion molecules, including integrins and selectins (see Chapter 12). LADs have been subdivided into three subcategories. LAD-I is caused by a mutation in the gene(s) responsible for β2 integrin subunits (CD11/CD18), resulting in either decreased or defective β2 integrins, which are necessary for adhesion to endothelial cells, recognition of bacteria, and outside-in signaling.16 In addition to experiencing recurrent infections, patients with LAD-I frequently have neutrophilia.
LAD-II is considerably rarer than LAD-I and presents in a similar manner (recurrent infection, neutrophilia, and impaired pus formation); however, the leukocytes have normal β2 integrins. In addition, patients with LAD-II may have mental retardation, growth defects, and/or deficiency in H blood group antigen.17 In this case, the basic defect is faulty ligands for selectin adhesive molecules.
Patients with LAD-III present with Glanzmann thrombasthenia–like bleeding problems (see Chapter 44) as well as LAD-I symptoms despite normal integrin expression. A recent report indicates that the basic defect is a mutation in KINDLIN3, a gene responsible for a protein that binds to the β2 integrin subunit.18
Miscellaneous Granulocyte Disorders
Miscellaneous granulocyte disorders include several relatively rare diseases.
Hyperimmunoglobulinemia E, also known as Job syndrome, is characterized by elevated levels of immunoglobulin E, skeletal abnormalities, and recurrent severe bacterial infections.19 Neutrophils in these patients have poor directional motility. Evidence points to dysregulation in cytokine production by T cells, resulting in imbalances between interferon-γ and transforming growth factor β in relation to interleukin-4 (IL-4).20
Lazy leukocyte syndrome is extremely rare and is characterized by neutropenia and poor neutrophil response to chemotactic agents.21 Abnormalities in neutrophil cytoplasmic actin and myosin microfilaments are responsible for the dsyfunction.22
Myeloperoxidase (MPO) deficiency is probably the most common neutrophil abnormality; however, symptoms are relatively mild in most patients. The discovery of MPO deficiency is credited to the automated hematology analyzers that use MPO to identify cells. The MPO gene is located on chromosome 17, and up to 10 mutations have been described.23
Diabetes mellitus can be either inherited or acquired. It has been associated with poor neutrophil function, especially when glucose levels are very high.24 The most consistent abnormality is abnormal oxidative burst activity.25
Monocyte/Macrophage Lysosomal Storage Diseases
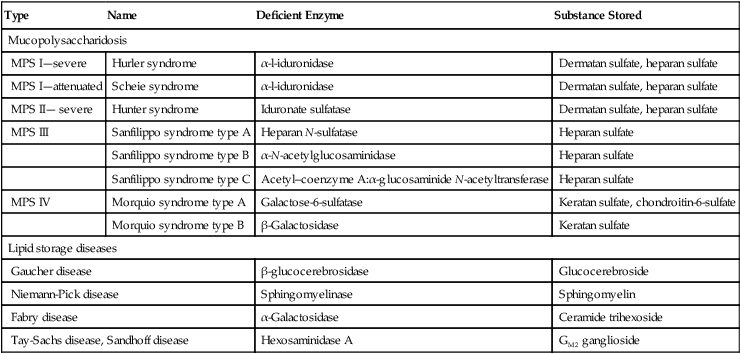
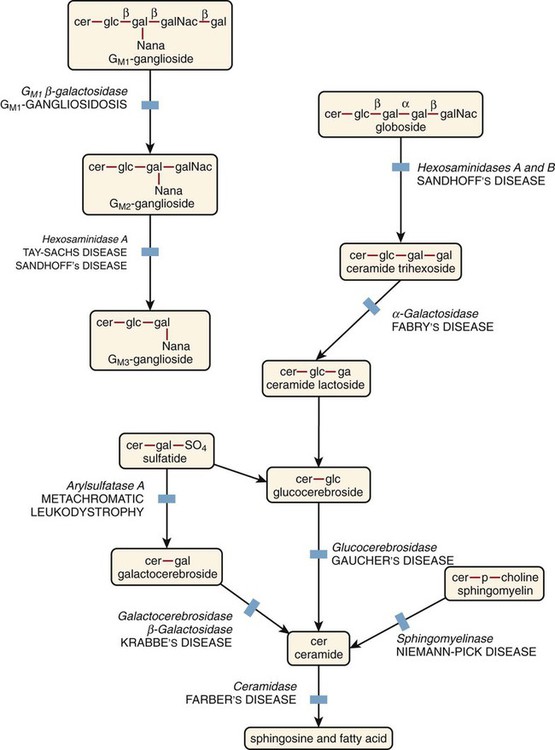
Monocyte/macrophage lysosomal storage diseases can be subdivided into mucopolysaccharide (or glycosaminoglycan [GAG]) storage diseases and lipid storage diseases (Table 28-1). As a group, they represent inherited enzyme deficiencies or defects that result in flawed degradation of phagocytized material and buildup of the partially digested material within the phagocyte. All cells containing lysosomes can be affected, including T lymphocytes.26
TABLE 28-1
Variants of Monocyte/Macrophage Lysosomal Storage Disorders
| Type | Name | Deficient Enzyme | Substance Stored |
| Mucopolysaccharidosis | |||
| MPS I—severe | Hurler syndrome | α-l-iduronidase | Dermatan sulfate, heparan sulfate |
| MPS I—attenuated | Scheie syndrome | α-l-iduronidase | Dermatan sulfate, heparan sulfate |
| MPS II— severe | Hunter syndrome | Iduronate sulfatase | Dermatan sulfate, heparan sulfate |
| MPS III | Sanfilippo syndrome type A | Heparan N-sulfatase | Heparan sulfate |
| Sanfilippo syndrome type B | α-N-acetylglucosaminidase | Heparan sulfate | |
| Sanfilippo syndrome type C | Acetyl–coenzyme A:α-glucosaminide N-acetyltransferase | Heparan sulfate | |
| MPS IV | Morquio syndrome type A | Galactose-6-sulfatase | Keratan sulfate, chondroitin-6-sulfate |
| Morquio syndrome type B | β-Galactosidase | Keratan sulfate | |
| Lipid storage diseases | |||
| Gaucher disease | β-glucocerebrosidase | Glucocerebroside | |
| Niemann-Pick disease | Sphingomyelinase | Sphingomyelin | |
| Fabry disease | α-Galactosidase | Ceramide trihexoside | |
| Tay-Sachs disease, Sandhoff disease | Hexosaminidase A | GM2 ganglioside | |
The mucopolysaccharidoses (MPSs) are a family of inherited disorders of GAG degradation. Each MPS is caused by deficient activity of an enzyme necessary for the degradation of dermatan sulfate, heparan sulfate, keratan sulfate, and/or chondroitin sulfate. The partially degraded GAG builds up in the lysosomes and eventually results in physical abnormality and sometimes mental retardation. The MPSs have been subdivided according to which enzyme is defective, which GAG is being stored, and whether the symptoms are severe or attenuated (see Table 28-1).27
The peripheral blood of a patient with MPS may appear relatively normal; however, metachromatic Reilly bodies may be seen in neutrophils, monocytes, and lymphocytes (Figure 28-7). Bone marrow may reveal macrophages with large amounts of metachromatic material. Diagnosis relies on assays for the specific enzymes involved. Treatment has consisted of enzyme replacement therapy or hematopoietic stem cell transplantation.27
Lipid storage diseases include a variety of disorders in which lipid catabolism is defective (Figure 28-8). Two of these disorders are characterized by macrophages with distinctive morphology and are discussed here.
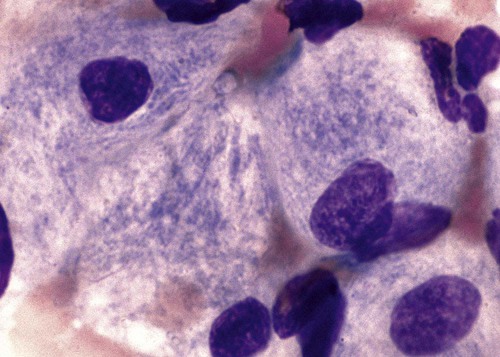
Gaucher disease is the most common of the lysosomal lipid storage diseases. It is an autosomal recessive disorder caused by a defect in the catabolic enzyme β-glucocerebrosidase. This results in accumulation of glucocerebroside in macrophages throughout the body, including osteoclasts in bone and microglia in the brain. Gaucher disease has been subdivided into three types based on clinical signs and symptoms (Table 28-2).28 Hematologic features may include anemia and thrombocytopenia reflecting the hypersplenism that is common in these patients. Bone marrow contains distinctive macrophages called Gaucher cells, occurring individually or in clusters, that have abundant fibrillar blue-grey cytoplasm with a striated or wrinkled appearance (sometimes described as onion skin–like) (Figure 28-9).
TABLE 28-2
Clinical Subtypes of Gaucher Disease
| Clinical Features | Type I: Nonneuropathic | Type II: Acute Neuropathic | Type III: Subacute Neuropathic |
| Clinical onset | Childhood/adulthood | Infancy | Childhood |
| Hepatosplenomegaly | + | + | + |
| Skeletal abnormality | + | – | + |
| Neurodegeneration | – | +++ | ++ |
| Death | Variable | By 2 years | 2nd-4th decade |
| Ethnic predilection | Ashkenazi Jews | Panethnic | Swedes |
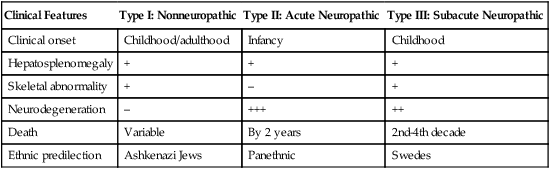
Note: Absence and severity of features are indicated by – to +++.
Treatment of Gaucher disease includes the use of recombinant glucocerebrosidase, which is quite expensive.29 Another approach is the use of pharmacologic “chaperones.”30
Pseudo-Gaucher cells that look remarkably similar to Gaucher cells under the light microscope can be seen in a variety of conditions in which cell turnover is increased. For example, they have been reported in thalassemia,31 chronic myelogenous leukemia,32 and acute lymphoblastic leukemia.33 Pseudo-Gaucher cells reflect an overwhelming of the glucocerebrosidase enzyme by the excessive cell turnover rather than a true decrease in the enzyme. Electron microscopy can distinguish between the two cells, because pseudo-Gaucher cells do not contain the tubular inclusions described in Gaucher cells.
Niemann-Pick disease is a group of disorders that are subclassified into two categories, those with a primary deficiency in acid sphingomyelinase (ASM) and those with defective NPC1 and NPC2 genes that regulate intracellular processing and transport of low-density lipoprotein–derived cholesterol.34
ASM deficiency occurs in a broad clinical spectrum ranging from an infantile neurologic form that results in death by 3 years of age to a nonneurologic form compatible with survival into adulthood, with numerous intermediate forms in between.35 ASM is a lysosomal enzyme whose substrate is sphingomyelin, which accumulates in the lysosome if ASM is defective. Over 100 mutations have been reported in the gene for ASM. The bone marrow of individuals with Niemann-Pick disease contains numerous macrophages with a foamy cytoplasm packed with lipid-filled lysosomes that appear as vacuoles after staining (Figure 28-10). Although there is no satisfactory treatment at this time, phase 1 studies of enzyme replacement are being carried out, and gene therapy is being evaluated in mice.35
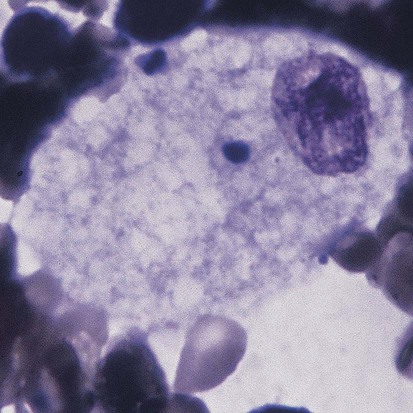
Discovery of the NPC1 gene36 in 1997 and a second (NPC2) gene37 in 2000 began a new wave of research and study on Niemann-Pick disease. The functions of the encoded proteins are unknown. When NPC1 protein is defective, there is an accumulation of sphingosine, glycosphingolipids, sphingomyelin, and cholesterol. Macrophages as described earlier can be found in most organs. One hypothesis is that the accumulation of sphingosine causes a decrease in lysosomal calcium, which, in turn, leads to defective transport and accumulation of cholesterol.38
B- and T-Lymphocyte Abnormalities
T-lymphocyte abnormality is best represented by a condition known as DiGeorge syndrome. This syndrome is characterized by the absence or underdevelopment of the thymus and hence markedly decreased numbers of T lymphocytes. It is associated with a microdeletion in chromosome band 22q11.2, which is the most common chromosome deletion and occurs in approximately 1 in 3500 births. Individuals with this deletion have a broad range of abnormalities, including defective parathyroid glands, cardiac abnormalities, abnormal facial development, and neurologic disorders. Fewer than 1% of patients with this deletion are athymic, a condition sometimes referred to as complete DiGeorge syndrome.39 Many of these patients are treated with thymus transplantation, with an approximately 75% survival rate.40
B-lymphocyte abnormality is represented by sex-linked agammaglobulinemia (XLA), which is caused most frequently by a mutation in the gene encoding Bruton tyrosine kinase (Btk). Such mutations result in a block in B-lymphocyte development.41 A large number of different mutations in the Btk gene have been reported; however, other mutations in the pre–B cell receptor complex proteins have also been reported in patients with presumed XLA, such as mutations in the µ heavy chain.42
Combined B-lymphocyte/T-lymphocyte abnormalities include severe combined immunodeficiency (SCID) and Wiskott-Aldrich syndrome. SCID can be divided into two types: adenosine deaminase deficiency and X-linked SCID. Both result in depletions of T, B, and natural killer (NK) lymphocytes. Adenosine deaminase deficiency results in excess amounts of its natural substrates (adenosine and 2′-deoxyadenosine), which cause lymphocyte depletion through a variety of mechanisms.43
X-linked SCID is the more common of the two and is caused by a mutation in the gene encoding the IL-2 receptor γ chain, which is shared by several interleukins. The mutation results in T lymphopenia, B cells that are dysfunctional, and a lack of NK cells.44
Wiskott-Aldrich syndrome is also X-linked and is caused by a mutation in a gene that encodes a protein called WASp. The mutation results in low levels or absence of the protein, and affected individuals have immunodeficiency, eczema, and thrombocytopenia. Absence of WASp affects migration, adhesion, and activation of a variety of leukocytes, including T cells, B cells, and NK cells.45
Reactive or Stress-Related Leukocyte Abnormalities
Quantitative Abnormalities
Neutrophils
An absolute increase in neutrophils above 8.7 × 109/L is referred to as neutrophilia and can be caused by a shift in neutrophils from the marginated pool to the circulating pool, such as is seen after strenuous exercise, during labor, in tachycardia, and with epinephrine or cortisone therapy. It can also be caused by pathologic conditions that result in a shift from the bone marrow storage pool to the peripheral blood and increased production of neutrophils. The latter is almost always accompanied by a shift to the left (presence of immature neutrophils). Table 28-3 lists the major types of disorders that result in neutrophilia.
TABLE 28-3
Causes of Neutrophilia and Neutropenia
| Neutrophilia | Neutropenia |
| Physiologic (e.g., strenuous exercise) Acute infections Inflammation (e.g., burns, after surgery, myocardial infarction) Intoxication (e.g., uremia, diabetic acidosis, poisoning, insect envenomation) Acute hemorrhage or hemolysis Response to fast-growing tumors Malignant myeloproliferative disorders Certain medications (e.g., steroids) |
Overwhelming infection Hemodialysis Physical agents/drugs (e.g., chemotherapy, radiation, chloramphenicol, any agent causing aplasia) Decreased or ineffective production (e.g., megaloblastic anemia) Splenic sequestration Autoimmune disorders Debilitated states (e.g., alcoholism) Hereditary disorders (e.g., cyclic neutropenia) |
The term leukemoid reaction refers to a leukocyte count above 50 × 109/L with neutrophilia and a marked left shift. Leukemoid reactions can be confused with chronic myelogenous leukemia. There are several distinguishing features between the two, and these are listed in Box 28-1.
An absolute decrease in neutrophils below 2.0 × 109/L is referred to as neutropenia. Neutropenia can be genetic or acquired. Genetic neutropenias (often referred to as congenital neutropenias) are a relatively rare group of disorders associated with decreased marrow production of neutrophils, often due to a mutation in ELA2, the gene coding for neutrophil elastase, and less frequently due to mutations in hematopoietic regulatory genes.46,47
Acquired neutropenia can result from decreased production caused by marrow aplasia, acute leukemia, or a megaloblastic process. The presence of autoimmune antibodies against neutrophils leads to neutropenia. Neutropenia can also be due to overwhelming infection and inflammation, which are often seen in immunocompromised patients and in the elderly. Table 28-3 lists causes of neutropenia.
Eosinophils
Eosinophilia is defined as an absolute eosinophil count above 0.4 × 109/L.
Increased eosinophils are seen in patients with parasite infestations, especially those due to tissue-invading parasites. Eosinophils are also increased in allergic disorders such as asthma48 and dermatitis.49
Certain tumors cause an increase in eosinophils both in the blood and around the tumor. Eosinophils are able to penetrate solid tumors, which allows tumoricidal cytokines to bring about tumor necrosis.50 Certain autoimmune disorders such as lupus erythematosus have also been associated with eosinophilia.51
If an individual is found to have eosinophilia without any discernible cause such as those listed above, and if the eosinophilia lasts more than 6 consecutive months, the diagnosis of hypereosinophilic syndrome, or HES, is used. HES is rare and is associated with damage to organs such as the skin, heart, and lungs as well as central and peripheral nervous systems. Consequently, once a diagnosis of HES is made, corticosteroids and monoclonal antibodies against IL-5 are administered to lower the eosinophil count.52 Table 28-4 lists the major nonmalignant causes of eosinophilia and eosinopenia.
TABLE 28-4
Causes of Eosinophilia and Eosinopenia
| Eosinophilia | Eosinopenia |
| Allergic reactions (e.g., asthma, hay fever, drug sensitivity) Parasitic infestations (especially with tissue-invading parasites) Hypersensitivity reactions (e.g., Loeffler syndrome) Skin diseases (e.g., dermatitis herpetiformis) Tropical eosinophilia Malignant myeloproliferative disorders Certain infections (e.g., scarlet fever) |
Infections or inflammation in which neutrophilia occurs Thymoma and hypogammaglobulinemia Rare inherited formsAutoimmune disorders with antieosinophil antibodies Unknown causes |
Eosinopenia is defined as an absolute eosinophil count of less than 0.09 × 109/L. The most common cause of decreased eosinophils is infection or inflammation that is accompanied by neutrophilia. Eosinophils move into the tissues under these circumstances, and marrow release of eosinophils may be inhibited. More rare causes of eosinopenia include autoimmune disorders that result in the formation of antieosinophil antibodies, hypogammaglobulinemia in the presence of thymoma, certain drug allergies, and urticaria.53
Basophils
Basophilia is defined as an absolute basophil count greater than 0.15 × 109/L. The most common cause of basophilia is the presence of a malignant myeloproliferative neoplasm such as chronic myelogenous leukemia, which is covered in Chapter 34. Nonmalignant causes of basophilia are relatively rare and include hypothyroidism, ulcerative colitis, some bee stings, and some types of nephrosis. Because the normal number of basophils is so low, basopenia is difficult to establish. Basopenia has been reported, however, in chronic urticaria.54
Monocytes
Monocytosis is defined as an absolute monocyte count greater than 1.1 × 109/L. Because monocytes are not stored in the bone marrow, relative monocytosis is frequently the first sign of recovery from acute overwhelming infection or severe neutropenia (agranulocytosis). Monocytosis is also seen during certain bacterial infections, especially tuberculosis, subacute bacterial endocarditis, syphilis, and brucellosis. In addition to monocytosis, subacute bacterial endocarditis is often characterized by the presence of circulating macrophages that can be seen in the edges of blood films due to their large size. Circulating macrophages can be found weeks before the blood culture is positive for an infectious agent.55 Some laboratories make nucleated cell concentrate films (buffy coat preparations) to increase the chance of finding the macrophages.
Certain protozoal and rickettsial infections such as malaria and Rocky Mountain spotted fever may be associated with monocytosis. Noninfectious diseases associated with monocytosis include collagen vascular diseases such as lupus erythematosus, and malignancies of the ovary, stomach, and breast are frequently associated with monocytosis.56
Monocytopenia, defined as an absolute monocyte count of less than 0.4 × 109/L, can be seen following steroid therapy57 as well as during hemodialysis.58 Viral infections, especially those due to the Epstein-Barr virus (EBV), can cause monocytopenia (Table 28-5).59
TABLE 28-5
Causes of Monocytosis and Monocytopenia
| Monocytosis | Monocytopenia |
| Bacterial infections (e.g., tuberculosis, subacute bacterial endocarditis, syphilis)Recovery from acute infectionProtozoal and rickettsial infections (e.g., malaria, typhus)Certain carcinomasGranulomatous diseasesCollagen vascular diseases (e.g., lupus erythematosus)Malignancies of monocyte cell line | Overwhelming infections in immunocompromised patientsHemodialysisEpstein-Barr virus infectionSteroid therapy |
Lymphocytes
The definition of lymphocytosis varies with the age of the individual. Children older than 2 weeks and younger than 8 to 10 years normally have higher lymphocyte counts than adults. Lymphocytosis in young children is defined as an absolute lymphocyte count greater than 10.0 × 109/L, whereas in adults, it is defined as a count greater than 4.8 × 109/L. As can be seen from the tables inside the front cover of this book, newborn infants have lymphocyte counts very similar to those of adults. Lymphocytoses can be subdivided into those with and those without reactive morphologic alterations, as are described later. See Table 28-6 for a listing of disorders that result in lymphocytosis. Infectious lymphocytosis is a childhood disease characterized by large numbers of normal-looking small lymphocytes. If lymphoid malignancies are accompanied by morphologic alterations, the alterations are not reactive but dyspoietic and are often clonal in nature (i.e., all cells look alike).
TABLE 28-6
Causes of Lymphocytosis and Lymphocytopenia
| Lymphocytosis | Lymphocytopenia |
| Without Morphologic Alterations Bordetella pertussis infection Acute infectious lymphocytosis With Morphologic Alterations Infectious mononucleosis Infectious hepatitis Cytomegalovirus infection Viral influenza Lymphoid malignancies |
Steroid therapy Strenuous exercise Morphine administration Human immunodeficiency virus infection Genetic abnormalities |
Qualitative (Morphologic) Abnormalities
Granulocytes
Nuclear abnormalities caused by reaction to infection, inflammation, or stress include nuclear hypersegmentation and pyknosis. Hypersegmentation is often seen in chronic infections and may reflect borderline folate deficiency caused by long-term hyperproliferation of granulocytes in response to the infection or inflammation. Toxic hypersegmentation is differentiated from that seen in megaloblastic anemia because it is not accompanied by the presence of macrocytic red cells (Figure 28-11). Eosinophils may also become hypersegmented (three or more nuclear segments) when reacting to a toxic situation.
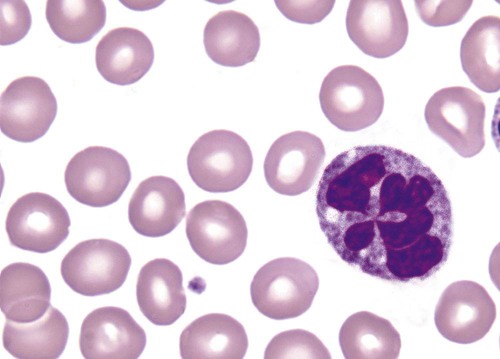
Pyknotic nuclei in neutrophils generally indicate imminent cell death. In a pyknotic nucleus, nuclear water has been lost and the chromatin becomes very dense and dark; however, filaments can still be seen between segements.60 Pyknotic nuclei should not be confused with necrotic nuclei found in dead cells. Necrotic nuclei are rounded fragments of nucleus with no filaments and no chromatin pattern (Figure 28-12).
Döhle bodies are cytoplasmic inclusions consisting of ribosomal ribonucleic acid (RNA) arranged in parallel rows.61 Döhle bodies are found in band and segmented neutrophils (Figure 28-13). With Wright staining, they appear as pale blue round or elongated bodies between 1 and 5 µm in diameter. They are usually located in close apposition to cellular membranes. Any time delay in preparing the blood film from the ethylenediaminetetraacetic acid (EDTA) tube can affect their appearance so that they may look more gray than blue and in some cases may not be visible. Döhle bodies are relatively nonspecific. Their presence has been associated with a wide range of conditions, including bacterial infections, sepsis,61 and normal pregnancy.62
Toxic granulation reflects an increase in mucosubstance within primary granules.63 There is a positive correlation between levels of C-reactive protein and the percentage of neutrophils with toxic granulation64; therefore, toxic granulation is considered an important marker of inflammation.65 Toxic granulation may be induced by increases in granulocyte colony-stimulating factor.66 Toxic granules are larger than specific granules and stain blue-black with Romanowsky stains. It is important to distinguish between true toxic granulation, poor staining, and the metachromatic granules found in inherited MPS (see earlier in chapter). Unlike the dark granules seen in the other two situations, true toxic granules tend to cluster within the neutrophil cytoplasm, and not all neutrophils are equally affected (Figure 28-14).
Vacuolation in granulocytes generally reflects phagocytosis, either of self (autophagocytosis) or of extracellular material. Autophagocytosis can be caused by drugs such as sulfonamides and chloroquine,59 by prolonged storage in EDTA, by autoantibodies,67 by acute alcoholism,68 and by exposure to high doses of radiation.69 Autophagocytic vacuoles tend to be small (approximately 2 µm) and distributed throughout the cytoplasm. Phagocytic vacuoles are often seen in septic processes caused by either bacteria or fungi. Phagocytic vacuoles are large (up to 6 µm) and are frequently outlined by visible toxic granules (Figure 28-15). It is important to distinguish between true vacuolation and that caused by excessive storage in EDTA. If the blood film was made more than 2 hours after venipuncture, artifactual vacuolation should be suspected.
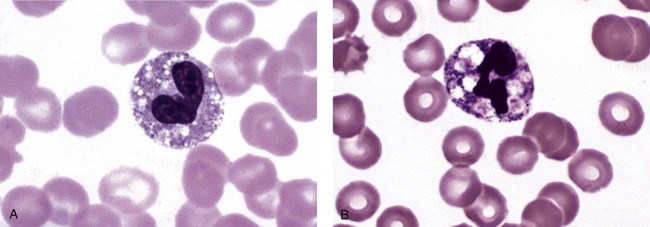
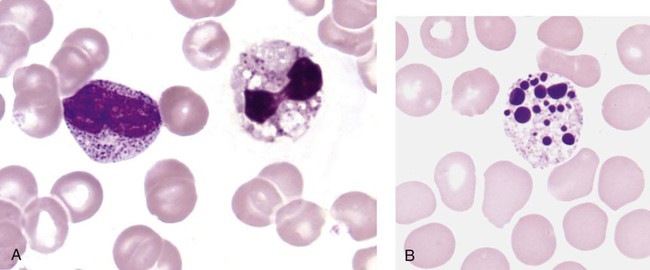
When phagocytic vacuoles are seen, a careful examination should be made for bacteria or yeast within the vacuoles. Cases of ehrlichiosis and anaplasmosis have been increasing in the United States over the past decade.70–72 Ehrlichia and Anaplasma are small obligate intracellular bacteria transmitted by ticks to humans and other vertebrate hosts. These organisms grow as a cluster (morula) in neutrophils (A. phagocytophilum and E. ewingii) (Figure 28-16, A) and in monocytes (E. chaffeensis) (Figure 28-16, B). Leukopenia, thrombocytopenia, and elevated liver enzymes are common laboratory findings, and anemia may develop in about half the cases of human monocytic ehrlichiosis after 2 weeks.70,73 Intracellular aggregates in neutrophils or monocytes may occasionally be detected in the first week of infection on a Wright-Giemsa–stained peripheral blood film or a buffy coat preparation, but immunofluorescent antibody titers or polymerase chain reaction testing is needed for confirmation.70 Early diagnosis is essential, because antibiotic treatment with doxycycline is very effective and can prevent serious complications.
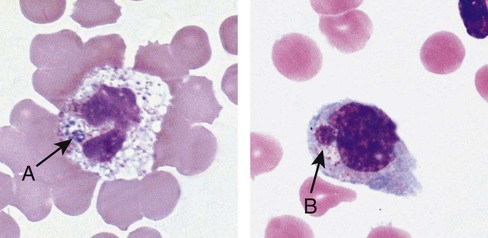
Degranulation is a common finding in activated neutrophils and eosinophils (Figure 28-17). Both primary and secondary granules are emptied into phagosomes, and secondary granules are also secreted into the extracellular space.74 Degranulation is often accompanied by disruption of cellular membranes during the process of making the blood film.

Cytoplasmic swelling may be caused by actual osmotic swelling of the cytoplasm or by increased adhesion to the glass slide by stimulated neutrophils. Regardless of cause, the result is a variation in neutrophil size, or neutrophil anisocytosis (Figure 28-18).
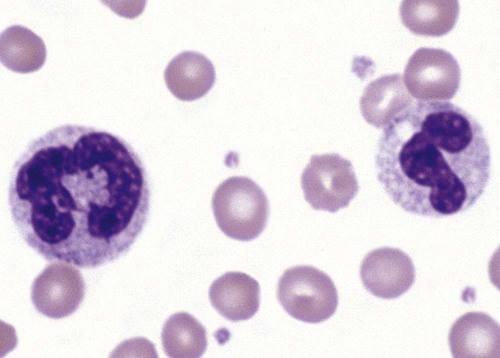
Monocytes
Occasional immature monocytes may be seen in the peripheral blood in response to infection or inflammation, but this is not as common as neutrophilic left shifts (Figure 28-19).
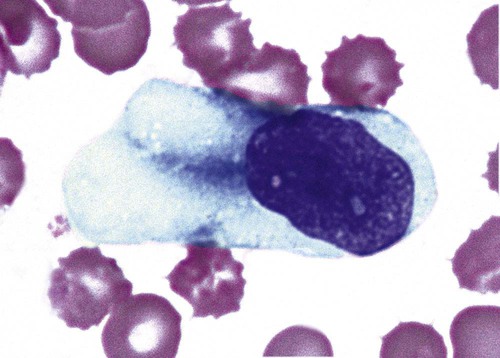
Nuclear alterations in activated monocytes are not common. Nuclear contortion is the only nuclear alteration that is described here. In these cases, the monocyte nucleus becomes quite thin and bandlike in areas and may appear to be segmenting (Figure 28-20).

Cytoplasmic alterations in toxic monocytes are those changes associated with transformation into macrophages. These include increases in cytoplasmic volume and spreading ability, increased numbers of cytoplasmic granules, and evidence of phagocytic activity (cytoplasmic vacuolation, intracellular debris, and highly irregular cytoplasmic borders) (Figure 28-21).
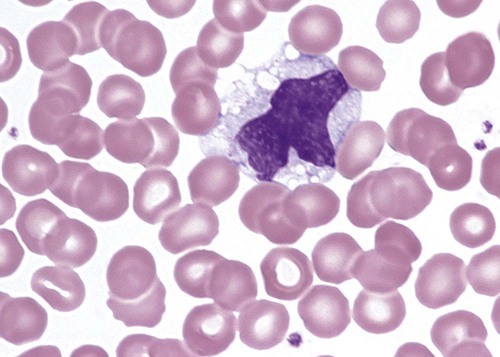
Lymphocytes
Nuclear alterations in reactive lymphocytes depend on the stage of blastogenesis and whether the cell is a T lymphocyte or a B lymphocyte. When a small resting lymphocyte is stimulated by the presence of antigen, it begins to enlarge, becoming a medium-sized lymphocyte and then a large lymphocyte. Meanwhile the chromatin pattern becomes less clumped and more homogeneous until nucleoli appear and the cell is recognized as a blast. A general rule of thumb when performing differential counts and encountering what looks like a blast form is that if all other findings are normal, the blast form is most likely a reactive lymphocyte undergoing blastogenesis and not anything more sinister (Figure 28-22).
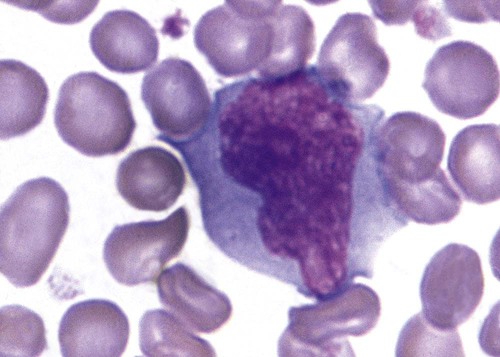
If the cell is a B lymphocyte, the blast form could be called an immunoblast. The blast divides to form a memory cell for that particular antigen and a proplasmacyte that then matures into a plasma cell or a plasmacytoid lymphocyte with its highly clumped nuclear chromatin. (See Figure 12-19.)
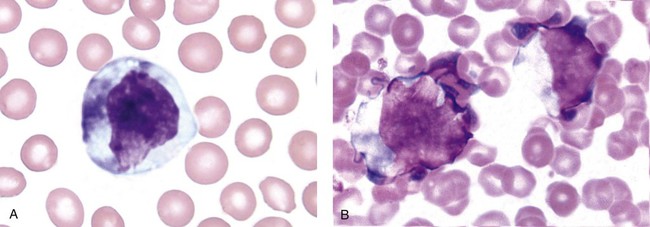
If the cell is a T lymphocyte, the blast divides to form a memory cell for the particular antigen and an effector T cell—a wide-bodied cell that is sometimes referred to as an infectious mononucleosis cell or viral lymphocyte because it is a cell characteristic of infectious mononucleosis. The nucleus of the effector T cell is somewhat fragile and has a tendency to adhere to the inner plasma membrane, and this effect becomes quite exaggerated when the sample is held in EDTA for a prolonged period (Figure 28-23).
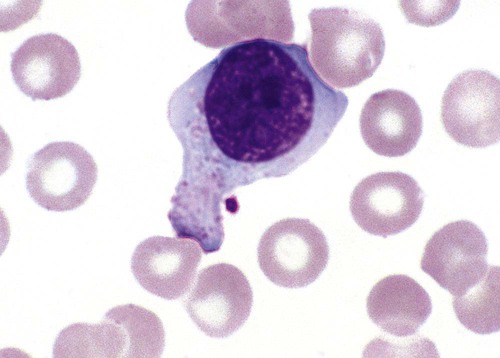
Cytoplasmic alterations in reactive lymphocytes are mainly increases in cytoplasmic RNA. If the cell is an effector B cell, which is a synthesizer of protein, there is considerable RNA in the cytoplasm and it therefore appears very blue with Romanowsky staining. If the cell is an effector T cell, the RNA appears to be localized in patches of blue along the periphery of the cell as well as radiating out from the nucleus. Because of this, the reactive T cell has been said to look like a flared skirt or a fried egg. Another cytoplasmic feature seen in cytotoxic T lymphocytes as well as NK lymphocytes is the presence of azure granules (Figure 28-24).
Infectious Mononucleosis
Although reactive lymphocytes may be seen in other disorders such as infections by cytomegalovirus, viral influenza, or hepatitis A and B, infectious mononucleosis is a more frequent cause. Infectious mononucleosis is associated with the EBV and has an incidence in the United States of 500 cases per 100,000 annually. It has its highest frequency in young adults, aged 15 to 24 years.75 The virus is found in body fluids, especially saliva, which gives rise to one of its nicknames, “the kissing disease.”
The incubation period of infectious mononucleosis is about 3 to 7 weeks. Presenting symptoms include the classic triad of pharyngitis (sore throat), fever, and lymphadenopathy.76 Fatigue, malaise, chills and fever, loss of appetite, and nausea are frequently cited. Splenomegaly is common but often only detected by ultrasound examination. Although there may be some elevation of liver function tests, hepatomegaly is rare. The majority of patients recover without complications within 4 weeks with only supportive care, but fatigue may continue for several months longer. Complications that may occur are generally mild and include hemolytic anemia, moderate thrombocytopenia, aplastic anemia, disseminated intravascular coagulation, thrombotic thrombocytopenic purpura, or hemolytic uremic syndrome. In rare cases, Guillain-Barré syndrome or other neurologic complications may occur.77 Hematologic findings resemble those seen in many viral disorders. There may be mild normocytic, normochromic anemia and a slight thrombocytopenia. The WBC count is usually elevated to a range of 11 to 20 × 109/L with approximately 15% of patients having leukocyte counts in excess of 20 × 109/L.78 The WBC differential shows a relative neutropenia with a relative and absolute lymphocytosis, possible monocytosis, and mild eosinophilia. There is a wide variation in lymphocytes, with up to 50% or higher exhibiting reactive features such as increased cytoplasm, retained basophilia, lobulated or oval nuclei with strands, and clumps of chromatin such as described earlier as effector T cells. The overall lymphocyte morphology in infectious mononucleosis is reflecting a strong T cell immune response to the infection of B cells by the EBV.
Testing for infectious mononucleosis includes rapid screening tests for the detection of heterophile antibodies, antibodies stimulated by the EBV that cross react with antigens found on sheep and horse red cells. However, not everyone with infectious mononucleosis will produce heterophile antibodies, especially children. Definitive testing for EBV infection can be performed for detection of IgM and IgG antibodies.77
Summary
• PHA is a genetic disorder of leukocytes, and the cells can be confused with immature neutrophils. An acquired form of hyposegmentation called pseudo-PHA can be seen in some malignant myeloproliferative neoplasms.
• Hereditary hypersegmentation is rare, but it can be confused with a megaloblastic process.
• Alder-Reilly anomaly is a manifestation of the mucopolysaccharidosis characterized by metachromatic granules in leukocytes.
• Other inherited leukocyte disorders include CHS, a lethal disorder characterized by giant lysosomes in all cells, and MHA, which may or may not have clinical symptoms and is characterized by leukocyte inclusions similar to Döhle bodies, as well as giant platelets.
• CGD of childhood is an inherited disorder of the NADPH oxidase system resulting in neutrophils that are incapable of killing many microorganisms.
• LADs are a group of disorders caused by mutations in the genes for adhesive molecules required for cells to migrate from the blood into the tissues.
• The mucopolysaccharidoses are a group of disorders each of which is associated with a specific defect in an enzyme necessary for the degradation of GAGs such as heparan sulfate, keratan sulfate, dermatan sulfate, and chondroitin sulfate. The result is the buildup of partially digested GAGs within macrophages and leukocytes.
• The lipid storage diseases are a group of disorders each of which is associated with a specific defect in an enzyme necessary for the degradation of lipids. The two lipid storage diseases with characteristic macrophage morphology are Gaucher disease and Niemann-Pick disease.
• Inherited lymphocyte disorders include DiGeorge syndrome, in which the lack or underdevelopment of the thymus results in decreased T cells; XLA, in which the lack of a kinase results in blocked B-cell development; and two types of SCID. Wiskott-Aldrich syndrome is a third inherited disorder affecting both T and B cells.
• Reactive changes in granulocytes include a left shift, hypersegmentation, Döhle bodies, toxic granulation, vacuoles, degranulation, and cytoplasmic swelling.
• Reactive changes in monocytes include occasional immature forms, contorted nuclei, and cytoplasmic evidence of transformation into macrophages.
• Reactive changes in lymphocytes include the presence of effector B cells (plasma cells and plasmacytoid lymphocytes), effector T cells (also known as infectious mononucleosis cells), and occasional blast forms reflecting blastogenesis.
Review Questions
1. Which of the following inherited leukocyte disorders is caused by a mutation in the lamin B receptor?
2. Which of the following inherited leukocyte disorders is one of a group of disorders with mutations in nonmuscle myosin heavy-chain IIA?
3. Which of the following inherited leukocyte disorders might be seen in Hurler syndrome?
4. Which of the following lysosomal storage diseases is characterized by macrophages with striated cytoplasm and storage of glucocerebroside?
5. The neutrophils in CGD are incapable of producing:
6. Individuals with X-linked SCID have a mutation that affects their ability to synthesize:
7. An absolute lymphocytosis with reactive lymphocytes suggests which of the following conditions?
8. What leukocyte cytoplasmic inclusion is composed of ribosomal RNA?
9. The expected complete blood count (CBC) results for women in active labor would include:
a. High total white blood cell (WBC) count with increased lymphocytes
b. High total WBC count with a slight shift to the left in neutrophils
10. Which of the following is true of the reactive lymphocytes whose cytoplasm has been described as looking like a flared skirt or fried egg?
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Nonmalignant Leukocyte Disorders
After the completion of this chapter, the reader will be able to:
1. Describe the basic genetic defect and the morphologic consequences in Pelger-Huët anomaly.
2. Indicate with what cell type Pelger-Huët cells might be confused and discuss what is meant by “pseudo-Pelger Huët.”
3. Compare and contrast two genetic causes of neutrophilic hypersegmentation and indicate what other disorder must be ruled out.
4. Describe the basic genetic defect and the morphologic consequences in Alder-Reilly anomaly, Chédiak-Higashi syndrome, and May-Hegglin anomaly.
5. Indicate how inclusions in Alder-Reilly anomaly and May-Hegglin anomaly might be confused with reactive morphology.
6. Discuss the defect in and functional consequences of chronic granulomatous disease.
7. Discuss the basic defects in and functional consequences of leukocyte adhesion disorders.
8. Discuss at least three other miscellaneous granulocyte disorders.
9. Describe the characteristic macrophage morphology associated with the mucopolysaccharidoses, Gaucher disease, and Niemann-Pick disease.
10. Describe the basic defect in genetic disorders leading to decreased T-lymphocyte production, decreased B-lymphocyte production, and the combined decrease of T, B, and natural killer lymphocytes.
11. Define what is meant by neutrophilia, neutropenia, lymphocytosis, lymphocytopenia, monocytosis, monocytopenia, eosinophilia, eosinopenia, and basophilia; give some examples of conditions in which each occurs.
12. Describe the nuclear and cytoplasmic alterations in granulocytes, monocytes, and lymphocytes that indicate a reaction to infection, inflammation, or stress.
Case Studies
Case 1
1. What is the most likely cause of this child’s recurring infections?
2. Biochemically speaking, what is the specific nature of the problem?
3. Do all microorganisms produce equally severe disease in individuals affected with this disorder?
4. How is this disorder transmitted genetically in the majority of cases?
Case 2
1. Based on the differential results, what cells should the medical laboratory scientist look for on the patient’s blood film?
2. Where on the blood film should the examination be made and why?
3. What is the significance if the suspected cells are found?
4. What preparation other than a blood film might be helpful in this situation?
Genetic Leukocyte Abnormalities
Nuclear Abnormalities
Pelger-Huët Anomaly
Pelger-Huët anomaly (PHA) is an autosomal dominant disorder resulting in decreased nuclear segmentation that is most apparent in neutrophils. It is the most common genetic disorder of leukocytes and is caused by a mutation of the lamin B receptor. The lamin B receptor is an integral protein in the inner nuclear membrane,1 and the associated gene is located on chromosome band 1q41-43.2 PHA is one of a large number of so-called laminopathies ranging from a type of muscular dystrophy to premature aging.3
Heterozygous PHA is characterized by granulocytes whose nucleus is segmented only once, taking the form of “two very distinct segments connected by a very thin filament.”4 In addition, what appear to be band forms may be present; however, the chromatin shows a highly clumped pattern and stains densely. The heterozygous mutation does not affect the function of neutrophils.5
Homozygous PHA is relatively rare and is characterized by granulocytes with round or oval nuclei (no segmentation) as well as the band-form nuclei described earlier. The chromatin pattern is also very dense and clumped. The homozygous form may be accompanied by impaired cognitive development, skeletal abnormalities, and heart defects.5 Leukocyte differential counts performed on blood samples from patients with either heterozygous or homozygous PHA may mistakenly identify the abnormal cells as immature granulocytes if the highly clumped, dense chromatin pattern is not noticed (Figure 28-1).
Medical laboratory scientists must distinguish inherited PHA from an acquired form of nuclear hyposegmentation known as pseudo–Pelger-Huët anomaly (pseudo-PHA) that may be seen in a variety of malignant myeloproliferative neoplasms.6 Two distinctions between inherited PHA and pseudo-PHA are the following: (1) the percentage of affected neutrophils is much higher in the inherited form (usually over 80%), whereas in the acquired form fewer than 50% are usually affected; and (2) pseudo-PHA alterations are usually accompanied by other morphologic indications of malignancy, such as the presence of blast forms.
Hereditary Neutrophil Hypersegmentation
Hereditary neutrophil hypersegmentation designates a group of at least two disorders. The first is a benign autosomal dominant condition characterized by hypersegmented neutrophils (Figure 28-2) with no clinical signs or symptoms. The major concern is to distinguish this condition from the neutrophilic hypersegmentation found in megaloblastic anemia. In the hereditary form, a large majority of neutrophils are hypersegmented and there is no macrocytic anemia. In megaloblastic anemia, fewer than 50% of neutrophils are usually hypersegmented.
The second disorder is myelokathexis, an inherited form of neutropenia characterized by hypersegmented neutrophils and bone marrow hyperplasia of myeloid cells. There is increased programmed cell death (apoptosis) of granulocyte precursors.7 In this case the neutrophil nuclei, in addition to being hypersegmented, are pyknotic (to be described later), and the intrasegmental filaments are longer than normal.
Cytoplasmic Abnormalities
Alder-Reilly Anomaly
Alder-Reilly anomaly is transmitted as a recessive trait and is characterized by granulocytes with large, darkly staining metachromatic granules that may resemble toxic granulation (described later) (Figure 28-3). The granules are composed of mucopolysaccharides and are sometimes referred to as Alder-Reilly bodies or Reilly bodies. In more severe forms, the granules may also be found in monocytes and lymphocytes. The basic defect is the incomplete degradation of mucopolysaccharides (protein-carbohydrate complexes), and there may be a structural abnormality in the myeloperoxidase gene.8 These granules are also discussed later in the section on storage diseases.
Chédiak-Higashi Syndrome
Chédiak-Higashi syndrome (CHS) is a rare and fatal autosomal recessive disease characterized by cells with enlarged lysosomal vesicles. All cells with lysosomal organelles are affected, including the melanosomes of melanocytes in the skin, the dense granules of platelets, and leukocyte granules. Patients usually have a tendency to bleed, frequent bacterial infections, symptoms of immunodeficiency, variable albinism, and progressive neurologic dysfunction.9 CHS is associated with a mutation in the LYST gene that encodes for a type of vesicle trafficking regulatory protein.10 Patients who do not die of bacterial infection develop a lymphohistiocytic infiltration into the major organs of the body, resulting in organ failure.9
Hematologic findings in CHS include giant lysosomal granules in granulocytes, monocytes, and lymphocytes (Figure 28-4). The platelet abnormality usually results in a prolonged bleeding time, and granule release by thrombin is impaired.11
May-Hegglin Anomaly
May-Hegglin anomaly is one of a group of at least four overlapping autosomal dominant platelet disorders: May-Hegglin anomaly (MHA), Sebastian syndrome (SBS), Fechtner syndrome (FS), and Epstein syndrome (EPS). All are caused by mutations of the nonmuscle myosin heavy-chain type IIA gene referred to as MYH9.12 MHA, SBS, and FS are characterized by large basophilic inclusions in leukocytes, thrombocytopenia, and giant platelets (Figure 28-5). FS and EPS are also associated with nephritis and deafness. Patients with MHA and SBS generally do not have renal disease or deafness; however, there have been a few reports of renal disease in individuals with MHA.13
The leukocyte inclusions in MHA have been described as “Döhle body–like” (Döhle bodies are discussed later in this chapter); however, there are striking ultrastructural differences. Döhle bodies are made up of lamellar rows of rough endoplasmic reticulum, whereas the MHA inclusions consist of randomly placed rods in an amorphous background. In addition, Döhle bodies are found only in neutrophils, whereas MHA inclusions are seen in neutrophils, eosinophils, basophils, and monocytes.14 MHA inclusions may stain a very pale color with Wright stain and may be missed in monocytes whose cytoplasm is also blue-grey.
Functional Abnormalities
Chronic Granulomatous Disease
Chronic granulomatous disease (CGD) is a disorder caused by the inability of phagocytes to produce superoxide and reactive oxygen species. The basic defect is one or more mutations in any of four genes responsible for proteins that make up a complex known as NADPH oxidase (NADPH is the reduced form of nicotinamide adenine dinucleotide phosphate). The majority of cases (approximately 60% to 65%) are X-linked recessive and 35% to 40% are autosomal recessive, depending on which gene has been mutated. The X-linked forms tend to carry a higher mortality rate.15
In the nitroblue tetrazolium reduction test, normal neutrophils, when stimulated, reduce the yellow water-soluble nitroblue tetrazolium to a dark blue insoluble formazan. CGD neutrophils cannot perform this reduction (Figure 28-6). Flow cytometry uses a fluorescent probe, such as dihydrorhodamine-123, to measure intracellular production of reactive oxygen species.15
Leukocyte Adhesion Disorders
Leukocyte adhesion disorders (LADs) result in the inability of neutrophils and monocytes to adhere to endothelial cells and to transmigrate from the blood to the tissues. The consequence of this inability is increased and potentially lethal bacterial infections. The basic defect is a mutation in the genes responsible for the formation of cell adhesion molecules, including integrins and selectins (see Chapter 12). LADs have been subdivided into three subcategories. LAD-I is caused by a mutation in the gene(s) responsible for β2 integrin subunits (CD11/CD18), resulting in either decreased or defective β2 integrins, which are necessary for adhesion to endothelial cells, recognition of bacteria, and outside-in signaling.16 In addition to experiencing recurrent infections, patients with LAD-I frequently have neutrophilia.
LAD-II is considerably rarer than LAD-I and presents in a similar manner (recurrent infection, neutrophilia, and impaired pus formation); however, the leukocytes have normal β2 integrins. In addition, patients with LAD-II may have mental retardation, growth defects, and/or deficiency in H blood group antigen.17 In this case, the basic defect is faulty ligands for selectin adhesive molecules.
Patients with LAD-III present with Glanzmann thrombasthenia–like bleeding problems (see Chapter 44) as well as LAD-I symptoms despite normal integrin expression. A recent report indicates that the basic defect is a mutation in KINDLIN3, a gene responsible for a protein that binds to the β2 integrin subunit.18
Miscellaneous Granulocyte Disorders
Miscellaneous granulocyte disorders include several relatively rare diseases.
Hyperimmunoglobulinemia E, also known as Job syndrome, is characterized by elevated levels of immunoglobulin E, skeletal abnormalities, and recurrent severe bacterial infections.19 Neutrophils in these patients have poor directional motility. Evidence points to dysregulation in cytokine production by T cells, resulting in imbalances between interferon-γ and transforming growth factor β in relation to interleukin-4 (IL-4).20
Lazy leukocyte syndrome is extremely rare and is characterized by neutropenia and poor neutrophil response to chemotactic agents.21 Abnormalities in neutrophil cytoplasmic actin and myosin microfilaments are responsible for the dsyfunction.22
Myeloperoxidase (MPO) deficiency is probably the most common neutrophil abnormality; however, symptoms are relatively mild in most patients. The discovery of MPO deficiency is credited to the automated hematology analyzers that use MPO to identify cells. The MPO gene is located on chromosome 17, and up to 10 mutations have been described.23
Diabetes mellitus can be either inherited or acquired. It has been associated with poor neutrophil function, especially when glucose levels are very high.24 The most consistent abnormality is abnormal oxidative burst activity.25
Monocyte/Macrophage Lysosomal Storage Diseases
Monocyte/macrophage lysosomal storage diseases can be subdivided into mucopolysaccharide (or glycosaminoglycan [GAG]) storage diseases and lipid storage diseases (Table 28-1). As a group, they represent inherited enzyme deficiencies or defects that result in flawed degradation of phagocytized material and buildup of the partially digested material within the phagocyte. All cells containing lysosomes can be affected, including T lymphocytes.26
TABLE 28-1
Variants of Monocyte/Macrophage Lysosomal Storage Disorders
| Type | Name | Deficient Enzyme | Substance Stored |
| Mucopolysaccharidosis | |||
| MPS I—severe | Hurler syndrome | α-l-iduronidase | Dermatan sulfate, heparan sulfate |
| MPS I—attenuated | Scheie syndrome | α-l-iduronidase | Dermatan sulfate, heparan sulfate |
| MPS II— severe | Hunter syndrome | Iduronate sulfatase | Dermatan sulfate, heparan sulfate |
| MPS III | Sanfilippo syndrome type A | Heparan N-sulfatase | Heparan sulfate |
| Sanfilippo syndrome type B | α-N-acetylglucosaminidase | Heparan sulfate | |
| Sanfilippo syndrome type C | Acetyl–coenzyme A:α-glucosaminide N-acetyltransferase | Heparan sulfate | |
| MPS IV | Morquio syndrome type A | Galactose-6-sulfatase | Keratan sulfate, chondroitin-6-sulfate |
| Morquio syndrome type B | β-Galactosidase | Keratan sulfate | |
| Lipid storage diseases | |||
| Gaucher disease | β-glucocerebrosidase | Glucocerebroside | |
| Niemann-Pick disease | Sphingomyelinase | Sphingomyelin | |
| Fabry disease | α-Galactosidase | Ceramide trihexoside | |
| Tay-Sachs disease, Sandhoff disease | Hexosaminidase A | GM2 ganglioside | |
The mucopolysaccharidoses (MPSs) are a family of inherited disorders of GAG degradation. Each MPS is caused by deficient activity of an enzyme necessary for the degradation of dermatan sulfate, heparan sulfate, keratan sulfate, and/or chondroitin sulfate. The partially degraded GAG builds up in the lysosomes and eventually results in physical abnormality and sometimes mental retardation. The MPSs have been subdivided according to which enzyme is defective, which GAG is being stored, and whether the symptoms are severe or attenuated (see Table 28-1).27
The peripheral blood of a patient with MPS may appear relatively normal; however, metachromatic Reilly bodies may be seen in neutrophils, monocytes, and lymphocytes (Figure 28-7). Bone marrow may reveal macrophages with large amounts of metachromatic material. Diagnosis relies on assays for the specific enzymes involved. Treatment has consisted of enzyme replacement therapy or hematopoietic stem cell transplantation.27
Lipid storage diseases include a variety of disorders in which lipid catabolism is defective (Figure 28-8). Two of these disorders are characterized by macrophages with distinctive morphology and are discussed here.
Gaucher disease is the most common of the lysosomal lipid storage diseases. It is an autosomal recessive disorder caused by a defect in the catabolic enzyme β-glucocerebrosidase. This results in accumulation of glucocerebroside in macrophages throughout the body, including osteoclasts in bone and microglia in the brain. Gaucher disease has been subdivided into three types based on clinical signs and symptoms (Table 28-2).28 Hematologic features may include anemia and thrombocytopenia reflecting the hypersplenism that is common in these patients. Bone marrow contains distinctive macrophages called Gaucher cells, occurring individually or in clusters, that have abundant fibrillar blue-grey cytoplasm with a striated or wrinkled appearance (sometimes described as onion skin–like) (Figure 28-9).
TABLE 28-2
Clinical Subtypes of Gaucher Disease
| Clinical Features | Type I: Nonneuropathic | Type II: Acute Neuropathic | Type III: Subacute Neuropathic |
| Clinical onset | Childhood/adulthood | Infancy | Childhood |
| Hepatosplenomegaly | + | + | + |
| Skeletal abnormality | + | – | + |
| Neurodegeneration | – | +++ | ++ |
| Death | Variable | By 2 years | 2nd-4th decade |
| Ethnic predilection | Ashkenazi Jews | Panethnic | Swedes |
Note: Absence and severity of features are indicated by – to +++.
Treatment of Gaucher disease includes the use of recombinant glucocerebrosidase, which is quite expensive.29
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
