Chapter 23 Non–Laryngeal Mask Airway Supraglottic Airway Devices
I Introduction
Nothing is more fundamental to the practice of general anesthesia than the maintenance of a clear upper airway. The choice of device depends on several factors, including access to the airway, duration of surgery, and risk factors for aspiration. After placement, the cuffed ETT provides a secure airway and protects against aspiration, but placement and removal of an ETT require training and judgment. Although ETTs typically are used without incident, complications ranging from trivial to life-threatening can occur.1
Advanced airway management depends on many airway devices, several of which have been included in the American Society of Anesthesiologists (ASA) difficult airway algorithm.2 The Classic laryngeal mask airway (LMA Classic, LMA North America, San Diego, CA) was introduced into clinical practice in 1988. Since then and particularly in the past 10 years, there has been an explosion of supraglottic airway devices (SADs) designed to compete with the LMA Classic, especially single-use devices. The introduction of single-use devices has been driven by concern about the sterility of cleaned, reusable devices (e.g., elimination of proteinaceous material, risk of transmission of prion disease) and the inability to recycle the device enough to be cost-effective. More than 20 manufacturers produce single-use LMs. Other designs of SADs have been introduced, and they are the main focus of this chapter.
II Nomenclature
The term supraglottic airway device (SAD) is used to describe a group of airway devices designed to establish and maintain a clear airway during anesthesia. SADs have several roles, including maintenance of the airway during spontaneously breathing or controlled-ventilation anesthesia, airway rescue after failed intubation or out of the hospital, use during cardiopulmonary resuscitation, and use as a conduit to assist difficult tracheal intubation. Brimacombe recommended that the term extraglottic airway be used, because many of these devices have components that are infraglottic (i.e., hypopharynx and upper esophagus).3 This textbook describes all airway devices that have a ventilation orifice or orifices above the glottis as supraglottic and those that deliver anesthetic gases or oxygen below the vocal cords (e.g., transtracheal jet ventilation, cricothyrotomy) as infraglottic. Other terms and acronyms include supraglottic airway (SGA), extraglottic airway device (EAD), and periglottic airway device (PAD), but SAD is more widely accepted and is used in this chapter.
Brimacombe and Miller suggested there should be a classification system for this increasingly complex family of devices. Miller4 described three main sealing mechanisms: cuffed perilaryngeal sealers, cuffed pharyngeal sealers, and cuffless, anatomically preshaped sealers. Further subdivision can be made by considering whether the device is single use or reusable and whether protection from aspiration of gastric contents is offered. The practical value of this type of classification is uncertain. Chapters 22 and 27 review the LMA, its variants, and the Combitube.
Second-generation SADs have been designed with safety in mind, and they incorporate design features that aim to reduce the risk of aspiration.5 They include the ProSeal LMA (PLMA), i-gel, LMA Supreme, Laryngeal Tube Suction II (LTS-II), disposable version of the LTS (LTS-D), the Streamlined Liner of the Pharynx Airway (SLIPA), and the Baska mask. The efficacy of several of these designs has not been proven.
III Limitations of the Classic Laryngeal Mask Airway
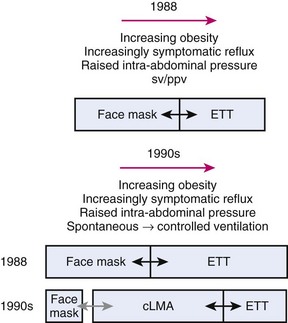
Prior to 1988, choices of airway devices essentially were limited to the face mask and endotracheal tube (ETT). The LMA Classic was designed by Archie Brain in the United Kingdom in the early 1980s, and it was introduced into anesthetic practice in 1988. Its introduction revolutionized airway management (Fig. 23-1). It was soon recognized to be a suitable device to use for many cases that previously were managed with a face mask or an ETT, because the LMA Classic had many advantages over both devices.6 It has been used in approximately 200 million episodes of anesthesia globally. More than 2500 studies on the device have been published. The LMA Classic is considered the benchmark against which other SADs are judged. A 2008-2009 UK census found that 56% of all episodes of general anesthesia were delivered with a SAD as the primary airway,7 and 90% of the devices were LMAs and LMs.
A Problems with Controlled Ventilation
The LMA Classic usually seals the pharynx with a pressure of 16 to 24 cm H2O, and this airway leak pressure is rarely above 30 cm H2O. This relatively low- pressure seal means that when positive pressure is applied to the LMA Classic, gas leakage is common. Studies have shown a 5% failure rate for achieving an expired tidal volume of 10 mL/kg,8 an audible leak in 48% of patients when ventilating to peak pressures of 17 to 19 cm H2O, and a detectable leak rate as high as 90%, with an 8% failure rate for adequate ventilation.9,10 Devitt and colleagues applied increasing peak airway pressures while ventilating through an LMA Classic. They found that as the airway pressure rose from 15 to 30 cm H2O, the incidence of audible leak rose from 25% to 95%, and the leak fraction ([inspired minute volume−expired minute volume]/inspired minute volume) rose from 13% to 27%.11 As the airway pressure increased, the incidence of airway leak into patients’ stomachs rose from 2% to 35%. These findings indicate that the LMA Classic has a relatively low-pressure airway (pharyngeal) seal and that as higher airway pressures are applied, there is a risk of loss of ventilating gases and gastric inflation. Loss of ventilating gases is associated with hypoventilation, loss of anesthetic agent, and environmental pollution, and gastric inflation that may increase the risk of regurgitation.
B Problems with Airway Protection
The LMA Classic is not regarded as providing protection against aspiration of regurgitated gastric contents and is contraindicated for patients who are not fasted or who may have a full stomach. The LMA Classic has a pharyngeal seal that is usually in the range of 16 to 24 cm H2O. Its tip obturates the upper esophagus, and it has an esophageal seal of 40 to 50 cm H2O, but it has no drain tube.12 Despite this design, cadaver work shows the LMA Classic protects the glottis from regurgitant esophageal fluid considerably more efficiently than the unprotected airway.13
Soon after the introduction of the LMA Classic, several small studies raised concerns about the ability of the LMA Classic to protect the airway from regurgitant matter and therefore from pulmonary aspiration. Early concerns were raised that the LMA Classic, sitting at the back of the throat, might stimulate a swallowing reflex, especially during light planes of anesthesia, leading to relaxation of the upper and lower esophageal sphincters and increasing the risk of regurgitation and aspiration. A physiologic study recorded a fall in the lower esophageal barrier pressure of 4 cm H2O during LMA Classic anesthesia, compared with a 2 cm H2O rise during face mask anesthesia.14 A study using swallowed methylene blue capsules demonstrated a 25% incidence of soiling of the inner portion of the LMA Classic on removal, and a small study reported a 2% incidence of aspiration, which occurred during spontaneous and controlled ventilation.15,16
As experience has accumulated, the evidence of a fundamental problem with aspiration has been reevaluated. Until 2004, 16 years after its introduction, there were no published reports of fatal aspiration during use of an LMA Classic. In 2004, Keller and colleagues published a series of three cases of serious morbidity, including one death from aspiration during LMA Classic anesthesia.17 Each case had risk factors for aspiration, and on reviewing all 20 published reports of aspiration during use of an LMA Classic, the investigators found identifiable risk factors in 19 of 20 cases. In the accompanying editorial, Asai listed more than 40 factors that increased the risk of aspiration.18 Several large studies have shown a low rate of aspiration; Verghese and Brimacombe reported a series of 11,910 uses (40% with controlled ventilation, 19% during intra-abdominal surgery, and 5% with a duration longer than 2 hours).19 Insertion success rate was 99.8%, the incidence of airway-related critical incidents was 0.16% during spontaneous ventilation and 0.14% during controlled ventilation, and there was one case of aspiration. Bernardini and Natalini reported three aspirations in a series of 35,630 LMA Classic uses for controlled ventilation (1 of 11,877).20 In an editorial, Sidaras and Hunter21 estimated an incidence of confirmed pulmonary aspiration during LMA Classic use of 1 in 11,000, and Brimacombe and Berry’s meta-analysis calculated a risk during elective surgery of 1 case in 4300 operations.6 This is similar to the rate of aspiration reported by Warner and colleagues in a study of 214,000 patients predating use of the LMA, in which aspiration occurred in 1 in 4000 elective operations.22
Although the risk of aspiration is relatively low in expert hands, this rate is achieved primarily by careful and appropriate case selection, expert insertion, and meticulous management of the airway after insertion. The Fourth National Audit Project of the Royal College of Anaesthetists and Difficult Airway Society (NAP4) in the United Kingdom studied major airway complications of 2.9 million episodes of general anesthesia and found that aspiration was the most common cause of airway-related deaths.1 One third of these complications occurred during maintenance with a LM or LMA in place, and for many of the patients, the risk of aspiration made this unwise.
C Problems with Accessing the Airway for Intubation
The LMA Classic sits over the vocal cords in more than 90% of cases and may be used as a conduit for intubation, but several factors limit the ease of this application.23 The internal lumen of the device is relatively narrow, limiting the size of ETT that can be passed. Size 4 and 5 LMA Classic devices accommodate most manufacturers’ cuffed ETTs with internal diameters (IDs) of 6.0 and 6.5 mm, respectively. A tube of adequate length must be used to exit the LMA Classic and reach the midtrachea; an ETT of approximately 29 cm can be placed through a size 5 LMA Classic. Across the distal end of the airway tube are two flexible bars forming a grill that prevents the tongue from impeding insertion and the epiglottis from causing obstruction after placement; these bars may act as an impediment to intubation through an LMA Classic. The angle at which an ETT exits the mask of the LMA Classic means that blind insertion frequently leads to esophageal intubation. Brimacombe reported blind intubation with an ETT through the LMA Classic to have a first-time success rate of 52% and overall success rate of 59%.24 Use of a bougie is less successful (32% of first attempts and 45% overall), and even fiberoptically guided techniques have a failure rate of 18%. After intubation has been achieved, removal of the LMA Classic without displacement of the ETT is cumbersome. Overall, direct intubation through the LMA Classic is a far from ideal technique.
The technique is dramatically improved if an Aintree intubation catheter (AIC, Cook Critical Care, Bloomington, IN) is used.25 The hollow AIC (ID of 4.6 mm, external diameter [ED] of 7.0 mm, length of 46 cm) is placed over a fiberscope, the scope and AIC are negotiated through the LMA Classic into the midtrachea, and the fiberscope then is removed, followed by removal of the LMA Classic. Care must be taken to ensure the AIC is not advanced too far, especially if gases are passed through it, as this risks barotraumas. This technique can be performed with or without the use of a Bodai adapter (Sontek Medical, Lexington, MA). The Bodai adapter allows oxygen and gas administration through the attached breathing circuit during exchange of the LMA to an ETT. The AIC remains in place, and a suitably sized lubricated ETT is then advanced over the catheter. Although the AIC technique does not appear in current airway guidelines, its use is simple, has a high success rate, and is widely reported.26,27
D Reusable Design
The LMA Classic is reusable and designed to be used up to 40 times. An in vitro study suggested that the LMA Classic and ProSeal LMA may be reused up to an average of 130 and 80 times, respectively, before showing signs of failing the preuse tests recommended by the manufacturer, and in vivo work supports use up to 60 times.28,29
After use, the LMA Classic is cleaned (decontaminated) before sterilization by autoclave (up to 137° C for 3 minutes with the cuff fully deflated), and it is stored in sterile packaging thereafter. A 2001 bench-top study demonstrated that routine decontamination and sterilization failed to remove all proteinaceous material from airway devices and from the LMA Classic in particular.30 At the same time, there was increasing public awareness about variant Creutzfeldt-Jakob disease (vCJD), especially in the United Kingdom. Concerns grew that residual prions, the infective, misfolded proteins responsible for vCJD, might remain and be passed from patient to patient. Several national bodies recommended using single-use devices “wherever possible,”31 even though the estimated risk of such cross-contamination was 1 to 10 cases in 100,000 patients.32 Brimacombe and coworkers described the rush toward single-use LMs as “driven by fears of the unknown and scientific misinformation.”33 Since then, the risk of vCJD has fallen dramatically, and the risk of transmission is likely to be vanishingly small.34 This risk must be balanced against other risks introduced by alternative equipment.35 Blunt and Burchett found that even a small deterioration in safety as a result of using a single-use device of poorer quality in place of a reusable device increased the overall risk to patients and went against the recommendation of the Spongiform Encephalopathy Advisory Committee (SEAC).32
E Absence of a Bite Block
The LMA Classic lacks a bite block and is prone to obstruction by biting in the agitated patient during emergence. Use of a bite block (e.g., rolled gauze placed between the molar teeth) is recommended until the LMA Classic is removed, and failure to adhere to this recommendation can lead to airway obstruction, hypoxia, and postobstructive pulmonary edema.36 The LMA Flexible also has no bite block, but the intubating LMA (ILMA), ProSeal, and Supreme LMA do.
IV Efficacy, Safety, and Evaluation of Supraglottic Airway Devices
A small survey of SAD manufacturers in 2003 examined several devices introduced around that time35 and found that the number of patients in whom the device had been used before marketing was less than 150 all in cases but one. In most cases, no trials were published in peer-reviewed journals before launching the product. One device launched in 2001 and remained without published data 18 months later. Only two of seven devices were compared with the LMA Classic in randomized, controlled trials before marketing, and the largest of them enrolled only 60 patients. This situation has not changed, and many later devices have been introduced with little or no trial evidence of their efficacy.
What regulations govern the introduction of new medical devices, particularly airway devices? In the European Union, the use of medical devices is controlled by three European Directives as part of European law.36 Some directives are specifically applicable to airway devices, and adherence is overseen by a regulatory body in each member country. The statutory body has responsibility for ensuring that medical devices do not threaten patients’ health and safety. Statutory requirements are largely harmonized throughout Europe, and compliance with one country’s requirements allows distribution and marketing of a device throughout the European Union. Although many countries have mechanisms that are designed to critically examine the efficacy of new technologies (e.g., National Institute of Clinical Excellence [NICE] in the United Kingdom), these bodies often have specific conditions (e.g., for NICE, new technology for new procedures) such that new (airway) equipment designed to do an old job tends to fall outside their areas of inspection and regulation.
After a device is marketed, clinical trials are not required to demonstrate efficacy or quality of performance. Manufacturers are legally bound to report serious or potentially serious adverse incidents.37 The statutory body requires reporting of incidences in which “malfunction of or deterioration in the characteristics and performance of a device” leads to “actual or potential patient harm.”37 There is also a mechanism for voluntary reporting of incidents by users. Whether these mechanisms lead to reliable reporting of such incidents and whether these schemes identify devices that are poorly designed or underperform is not clear. Formal assessment of performance may come from postmarketing cohort or comparative studies. However, these studies are uncommon, and they usually are published at some interval after a device has been marketed.
A Desirable Features of Supraglottic Airway Devices
Many assume that reusable devices may be replaced by cheaper, single-use devices, and some think that single-use devices are intrinsically preferable. However, many single-use devices differ from the reusable devices they seek to replace in design and in the materials used. Some modifications appear to be minor, but the implications for performance have generally not been evaluated. The work on single-use laryngoscopes and intubation bougies provides evidence that changes in product material may alter performance considerably.38,39 Data on the current versions of the single-use LM and comparisons between these and the LMA Classic remain largely unavailable.
B Efficacy Versus Safety
Safety encompasses avoidance of complications occurring at all stages of anesthesia and afterward. Prevention of aspiration requires a good-quality seal within the laryngopharynx and esophagus (i.e., esophageal seal) to prevent gas leaking into the esophagus and stomach and to prevent regurgitant matter passing from the esophagus into the airway. A functioning drain tube enables regurgitant matter to bypass the larynx and be vented outside, protecting the airway and giving an early indication of regurgitation to the anesthesiologist. Studies have shown that the extent of esophageal seal varies considerably among SADs. Those with a drain tube can effectively vent regurgitant fluid if the drain is not occluded.12,40,41
C Structured Approaches to Evaluation of New Devices
New airway devices should undergo mandatory assessment of manufacturing quality and clinical performance before marketing. The characteristics of the ideal SAD outlined earlier provide a checklist against which function can be assessed. Several methods have been recommended.35,42,43 Cook described a three-stage evaluation process35:
Stage 1. Bench evaluation using manikins or models designed to test function and basic safety
Stage 2. A rigorous cohort study to determine whether the device is effective and to further exclude major concerns about safety
Stage 3. A randomized, controlled trial against the current gold standard for the procedure for which the new device is expected to be used (e.g., LMA Classic, PLMA, ILMA)
In stage 1, the bench models include airway manikins and others, such as those specifically designed to test aspiration risk.44 This stage is limited by lack of fidelity of available manikins.45 With the increasing use of SADs during resuscitation, during out-of-hospital rescue, and by non-anesthesiologists, there is an urgent need to develop realistic manikins for testing and training. Data acquired from such studies require intelligent interpretation and knowledge of the relative performance of different manikins.46–48 Results of manikins studies are considerably limited, and at best, they may be used to evaluate basic information on device performance and durability and to identify major conceptual or design problems. Appropriate bench testing may lead to further development of a device before starting clinical studies.
In stage 2, a cohort study may be used for the first assessment of clinical performance in patients. This approach enables full clinical evaluation of the new device under routine clinical conditions. Functions that can be tested include ease of insertion, pharyngeal seal, airway resistance, stability of the device in different head and neck positions, ease of passage of a gastric tube, positioning of the airway over the larynx, and suitability for fiberscopic or catheter exchange techniques. Learning curves can be examined. A cohort study also enables assessment of function during spontaneous and controlled ventilation and determination of airway trauma or pharyngolaryngeal morbidity. The cohort must be large enough to enable identification of common problems, but unless it is very large, it cannot detect uncommon or rare problems. For instance, for an event that does not occur in a cohort study of n cases, the 95% confidence interval (CI) for frequency of that event is approximately 1 in 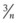 .49 For example, if no nerve injuries occur in a cohort study of 100 cases, the upper limit of the 95% CI for risk of nerve injury is 1 in 33. A cohort of at least 100 patients is a reasonable compromise between being large enough to identify important uncommon events and remaining a practical size.
.49 For example, if no nerve injuries occur in a cohort study of 100 cases, the upper limit of the 95% CI for risk of nerve injury is 1 in 33. A cohort of at least 100 patients is a reasonable compromise between being large enough to identify important uncommon events and remaining a practical size.
Stage 3 employs a randomized, controlled trial. After successful completion of bench and cohort evaluations, the need for further modifications of the device should be considered. Significant modifications necessitate repetition of the early evaluations. On successful completion of the early evaluations, the new device should be compared with its best existing competitor. In many cases, this is the LMA Classic. The randomized, controlled trial must be of adequate size to identify clinically important differences in function. Studies may be designed to test the hypothesis that the devices perform differently (i.e., superiority-inferiority trials) or that the test device does not perform significantly less well than the benchmark device (i.e., noninferiority trials).50 Power calculations can be based on data acquired from phase 2, but trials of at least 100 patients provide more comprehensive and clinically useful comparisons. Economic evaluation of cost-effectiveness of the new device may take place at this stage. Data from the three phases of evaluation can be used to determine what role the new airway device has in the market.
A second structured approach to evaluating and choosing new devices was made by Wilkes and colleagues.43 In this proposal, a central body of experts would coordinate research to evaluate new devices, review available evidence and provide national recommendations on devices reaching standards of acceptability. Although potentially of value for a large population (e.g., a country), the barrier it may create to free trade and the likelihood of legal challenges are problems.
The UK Difficult Airway Society (DAS) has proposed a guideline whereby purchasers could adopt a minimum level of evidence before making a pragmatic decision about the purchase or use of an airway device.42 This minimum level of evidence (i.e., level 3b: a case- or historical-controlled cohort study) would form the basis of a professional standard to guide those with responsibility for selecting airway devices.51 Devices without this minimum level of evidence would not be purchased. The investigators argue that widespread adoption of this professional standard would lead to situations in which it was in the interests of manufacturers and purchasers to acquire such evidence and the DAS would support both parties in setting up research with this aim. The strength of this approach lies in purchasers driving the need to raise the evidence bar and manufacturers being encouraged to perform clinical trials at an early stage in device development. This approach is not anticompetitive because it creates no barriers to manufacturers bringing a device to market, but it does raise the level of expectation of the community of purchasers about what they wish to purchase.
V First-Generation Supraglottic Airway Devices
A Generic Laryngeal Masks
For cost reasons, most single-use LMs are made with a PVC cuff, which increases the rigidity of the device. PVC is cheaper than silicone and reduces the cost of these devices for the manufacturer and purchaser. PVC is less permeable to nitrous oxide than silicone, and during nitrous oxide administration, the increase in LM cuff pressure in the early phase of anesthesia is considerably less than when using a silicone device.52,53
PVC has two disadvantages. First, it is more rigid than silicone. Brain examined PVC as a material for LMA construction in the mid-1980s but rejected it because of its rigidity.54 The increased rigidity of PVC LMs may cause problems, such as increased trauma to the airway during use and an increase in pharyngolaryngeal morbidity postoperatively. The rigidity, especially in masks with thicker cuffs, may lead to folds and wrinkles in the partially inflated cuff, which may affect the airway seal or create channels that enable regurgitant fluid to reach the larynx, altering efficacy and safety. The impact of these effects is not known for many devices because of the lack of published evidence. Some manufacturers use a softer form of siliconized PVC, which may obviate the problems described.
The second concern is the toxicity of phthalates, such as di-2-ethylhexyl phthalate (DEHP) or dioctyl phthalate (DOP), which are used in the manufacturing process to render the PVC softer. These chemicals are not linked to the plastic matrix and leach out slowly during use. They are considered potentially carcinogenic, mutagenic, and reprotoxic,55 and there is particular concern about phthalates in products that may be placed in the mouth (e.g., children’s toys, airway devices). The issue has been considered important enough by some for bills banning PVC in some products to be brought before the legislatures of some U.S. states and the European Parliament. Phthalates are banned from use in cosmetics in Europe, and in May 2005, the European Parliament voted to make permanent a temporary ban on six phthalates (including DEHP) in PVC used in children’s toys and called for investigation into health care equipment.55 It is easy to overstate the importance of phthalates in the safety of PVC LMs, but the unquantified low risk they pose bears comparison with the unquantified low risk of transmission of prion disease when using a reusable device. Alternative plasticizers, such as no-DOP formulas and di-isononyl-cyclohexane dicarboxylate (Hexamoll DINCH) are available for use in PVC applications, although at considerably increased cost. In the past few years, an increasing number of low-cost silicone LMs designed for single use and reuse have been introduced.
The greatest concern about the profusion of LMs on the market is the lack of rigorous evaluation. Typically, there is no robust evidence to inform whether the single-use or reusable LMs perform similarly to equivalent LMAs or to inform which LM performs best. The limited evidence does show that all LMs and LMAs are not equivalent. Of the approximately 30 devices available, only 2 have substantial publications that compare their efficacy with that of LMAs: the AuraOnce LM (Ambu Inc., Glen Burnie, MD) and the Portex Soft Seal LM (SSLM, Smiths Medical ASD, Keene, NH). For the other devices, there appears to be a void of published evidence. A publication from the National Health Service (NHS) Centre for Evidence-Based Purchasing illustrates the difficulty in determining the relative merits and demerits of these competitors to the LMA Classic. The document listed more than 25 alternative standard LMs and reported a total of 18 comparative trials for the devices. Some of these studies were of poor quality. This publication record contrasts with the more than 2500 publications on the LMA Classic.56
B Features of Laryngeal Masks
C Single-Use Laryngeal Masks
1 Ambu AuraOnce Laryngeal Mask
The Ambu AuraOnce is the most investigated model. It is a sterile, single-use product made of PVC with a preformed curve designed to replicate human anatomy (Fig. 23-2). This angled curve is designed to ensure that the patient’s head remains in a natural position when the mask is in use and to facilitate insertion without exerting force on the upper jaw. A reinforced tip reduces the risk of the device folding back during insertion. Reinforcing internal ribs built into the curve of the airway tube provide a degree of flexibility and enable the airway tube to conform to individual anatomic variations and adapt to a wide range of head positions without loss of function.
The Ambu Aura40 is the reusable, silicone version of the Ambu AuraOnce.40 Its built-in curve is designed o replicate the natural human anatomy and offers the same features as the Ambu AuraOnce. It can be steam autoclaved up to 40 times.
The Ambu Aura-i (Fig. 23-3) is a modification of the Ambu AuraOnce that is designed to facilitate intubation in a fashion similar to that of the ILMA. It has a shorter, wider, and more rigid airway tube than the Ambu AuraOnce, and proximally, a rigid plastic sleeve acts as a handle for insertion and as a bite block. It is designed for use during routine airway maintenance and as a device for airway rescue and fiberoptic intubation. Sizes 3, 4, and 5 accommodate ETTs up to sizes 6.5-, 7.5-, and 8-mm ID, respectively. Ambu has released a single-use videoscope that may be used with the Ambu Aura-i.
a Application
• The size of the Ambu AuraOnce must be appropriate for the patient. Use the manufacturer’s guidelines combined with clinical judgment to select the correct size. Always have a spare Ambu AuraOnce ready for use.
• Resistance or swallowing may indicate inadequate anesthesia or inappropriate technique, or both. Inexperienced users should choose a deeper level of anesthesia.
• The Ambu AuraOnce is inserted in a fashion similar to that described for the traditional LMAs in Chapter 22. Excess force must be avoided at all times. When the mask is fully inserted, resistance is felt. If the cuff fails to flatten or curls over as it is advanced, it is necessary to withdraw the mask and reinsert it. In case of tonsillar obstruction, a diagonal shift of the mask is often successful.
c Medical Literature
Approximately 25 publications about the Ambu AuraOnce are listed on PubMed.
Cohort Studies
A multicenter study of the clinical performance of the Ambu AuraOnce was conducted by Hagberg and colleagues to evaluate ease of insertion, insertion success, airway seal, and ventilation.57 Device placement was successful in all 118 nonparalyzed, anesthetized patients on the first or second attempt (92.4% and 7.6%, respectively). Adequate ventilation was achieved in all patients, and the vocal cords could be visualized by fiberoptic endoscopy in 91.5% of patients. Complications and patients’ complaints were minor and were quickly resolved. The investigators reported that the curvature of the tube facilitated insertion and that the large, soft cuff allowed a higher oropharyngeal leak pressure than is commonly found in other LMs. The reinforced tip of the Ambu AuraOnce may avoid folding, which can lead to air leakage, a common problem with other LMs.
Genzwuerker and coworkers studied ventilation using the Ambu AuraOnce in different head positions in 30 patients.58 Five different head positions were used: head on a standard pillow, head rotated 90 degrees to the left side, head rotated 90 degrees to the right side, head with chin lift on a standard pillow, and head flat on a table without a pillow. No changes in the performance of the device were observed in any position. The Ambu AuraOnce may be a useful SAD for cases in which head movement may be necessary during surgery.
Randomized, Controlled Studies of Efficacy
Shariffuddin and Wang compared the Ambu AuraOnce with the LMA Classic during controlled ventilation in 40 patients in a randomized, crossover design.59 The mean ± standard deviation oropharyngeal leak pressure was higher for the Ambu AuraOnce (19 ± 7.5 cm H2O) than for the LMA Classic (15 ± 5.2 cm H2O, P = 0.004). The Ambu AuraOnce also required fewer insertion attempts (P = 0.02) but more manipulations to achieve a patent airway (P = 0.045). Values for time to insert the device and intraoperative performance were similar.
Sudhir and associates performed a randomized, crossover study comparing the Ambu AuraOnce and LMA Classic in 50 patients.60 Success rates for the first attempt success were similar (92% for the Ambu AuraOnce and 84% for the LMA Classic, P = 0.22). The volumes of air required to inflate the cuff to produce an effective airway seal were similar, but the cuff pressure was lower for the Ambu AuraOnce (median, 18 cm H2O) compared with LMA Classic (27 cm H2O, P = 0.007). Complications were similar in both groups.
Ng and coworkers compared the Ambu AuraOnce with the LMA Classic and reported the Ambu AuraOnce to be easier to insert, but they found no difference in time to insertion,61 successful insertion on the first attempt, oropharyngeal leak pressure, hemodynamic response to insertion, or complications of placement.
Francksen and colleagues compared the performance of the Ambu AuraOnce and the LMA Unique in 80 patients undergoing minor routine gynecologic surgery.62 They demonstrated that the time to insertion and failure rate were comparable and that oxygenation and ventilation variables were adequate with either device. Median airway leak pressures were slightly higher with the Ambu AuraOnce than with the LMA Unique (18 versus 16 cm H2O, P < 0 .013). No gastric inflation was observed with either device in this small study.
Francksen and coworkers also studied 120 patients scheduled for routine minor obstetric surgery who were randomly allocated to size 4 Ambu AuraOnce, LMA Unique, or SSLM.63 The Ambu AuraOnce was fastest to insert by a few seconds. All three had high first-attempt insertion success rates (all >95%). Rates of subjectively rated excellent insertion were 75% for the LMA Unique, 70% for the Ambu AuraOnce, and 65% for the SSLM. All devices were judged acceptable.
Lopez and colleagues compared four single-use LMs (i.e., Ambu AuraOnce, Solus LM, LMA Unique, and SSLM) in 200 patients with ASA physical status class I, II, or III who were undergoing elective ambulatory surgery and for whom airway management was performed by inexperienced residents.64 The Ambu AuraOnce (78%) and LMA Unique (80%) performed best in terms of ease of insertion, with the Solus requiring most reinsertions. Optimal ventilation was best with the LMA Unique (94%). Airway leak pressure was 27.3 mm Hg for the SSLM, 23.7 mm Hg for the Ambu AuraOnce, 22.1 mm Hg for the LMA Unique, and 20.9 mm Hg for the Solus. Blood staining occurred most frequently with the SSLM (38%).
Other Studies
Gernoth and colleagues compared the performance of the Ambu AuraOnce with the LMA Classic in 60 patients whose cervical spines were immobilized with an extrication collar before elective ambulatory interventions.65 Insertion time, number of insertion attempts, and airway leak pressures were comparable (25.6 ± 5.3 cm H2O for the Ambu AuraOnce and 26.5 ± 6.5 cm H2O for the LMA Classic). The investigators concluded that both devices were suitable for rapid and reliable airway management in patients with cervical immobilization.
Using an in vitro test, Zaballos and coworkers examined the Ambu AuraOnce, LMA Classic, PLMA, LMA Unique, and i-gel using magnetic resonance imaging (MRI).66 The Ambu AuraOnce and i-gel did not lead to artifacts, whereas each of the LMAs did.
Maino and associates examined the effect of nitrous oxide exposure on intracuff pressure of PVC and silicone LMs and the LMA Classic in vitro.67 All cuffs were initially inflated with air to a pressure of 60 cm H2O. The findings were not surprising given the physicochemical properties of PVC and silicone. When exposed to 66% nitrous oxide, the cuff pressure rose considerably more in the silicone cuffs than in the PVC devices. Among the PVC devices, the lowest increases in cuff pressure after 60 minutes of exposure were found in the Solus and LMA Unique (13 and 15 cm H2O, respectively), and the increases in the cuffs of the Ambu AuraOnce and SSLM were notably higher (28 and 31 cm H2O, respectively).
2 Soft Seal Laryngeal Mask
The SSLM is a tubular oropharyngeal airway with a mask and an inflatable peripheral cuff attached to the distal end (Fig. 23-4). It is designed to produce an airtight seal around the laryngeal inlet to provide a secure airway suitable for spontaneous or controlled ventilation during general anesthesia.
a Application
Insertion technique for the SSLM is similar to that recommended for LMAs in terms of patient position, device checking, preparation, and lubrication. Although many insertion techniques are used, insertion with the cuff partially inflated is recommended for the SSLM. If a muscle relaxant is administered before device insertion, use of a triple airway maneuver (i.e., mouth opening, head extension, and jaw thrust) should decrease the incidence of epiglottic downfolding.68 The cuff should be inflated with air until a “just-seal” pressure is obtained. A maximum intracuff pressure of 60 cm H2O is recommended.
c Medical Literature
There are approximately 27 publications about the SSLM in the medical literature.
Randomized, Controlled Studies of Efficacy
Van Zundert and colleagues performed a randomized, controlled trial of the use of the size 4 SSLM and LMA Classic during spontaneously breathing anesthesia in 200 adult patients.53 Insertion time, success, and fiberoptic position were equivalent. Further analysis of the cuff pressure change during nitrous oxide anesthesia, with the cuff pressure initially established at 45 mm Hg, showed that it increased in the LMA Classic to a mean of 100 mm Hg and that the mean final pressure in the SSLM was 47 mm Hg (P < 0.001).69 There was a greater incidence of sore throat in the LMA Classic group at 2 hours postoperatively but not at 24 hours.
Similar to the low rates of trauma reported in the study by van Zundert’s team, Hagberg and colleagues demonstrated that partial cuff inflation (30 mL of air) enhanced ease of insertion and minimized mucosal trauma.70,71
Shafik and associates performed a crossover comparison of the SSLM and LMA Classic in 60 patients.72 The primary outcome measure was first-attempt insertion, for which success rates were equivalent (92% for the SSLM and 96% for the LMA Classic), as was ease of insertion.
Lopez and coworkers compared the SSLM with the LMA Classic in a randomized trial enrolling 60 patients.73 Most performance characteristics were equivalent between devices, with the exception of a difference in airway seal (23 ±4 cm H2O for the SSLM and 20 ±4 cm H2O for the LMA Classic) and the fact that three patients in the SSLM group required the airway changed to an ETT.
Hanning and colleagues studied the SSLM and LMA Classic using a crossover design in a study of 35 healthy patients during paralyzed ventilation anesthesia.74 The oropharyngeal leak pressure was higher with the SSLM than the LMA Classic (21 versus 16 cm H2O).
In a crossover study, Brimacombe and colleagues compared the SSLM and LMA Unique in 90 healthy, paralyzed, anesthetized patients undergoing routine peripheral surgery.75 The LMA Unique was superior to the SSLM in terms of ease of insertion, fiberoptic position, and mucosal trauma but similar in terms of oropharyngeal leak pressure and ease of ventilation.
In another randomized, crossover study, Paech and associates compared the SSLM and the LMA Unique in 168 anesthetized, spontaneously breathing patients.52 The investigators reported that although both devices performed equivalently for first-time placement, the SSLM was subjectively rated more difficult to insert and more likely to cause mucosal trauma. However, the fiberoptic view of the larynx was better through the SSLM, and it more frequently provided a ventilation seal at 20 cm H2O. In contrast to the LMA Unique, its cuff pressure did not increase during nitrous oxide anesthesia. In this study, there was a larger proportion of females, a smaller mask size was used for male and female patients, and the SSLM was inserted with a partially inflated cuff.
Cook and colleagues compared the SSLM with the LMA Unique in a randomized trial enrolling 100 patients.76 The study was stopped early because of a high rate of airway trauma in the SSLM group. The investigators reported that the SSLM required more attempts for successful insertion (P = 0.041), more manipulations (P < 0.0001), failed more often (P = 0.013), and caused more complications (P = 0.048) than the LMA Unique. In 14% of SSLM uses, insertion or ventilation failed, and its use had to be abandoned. Leak pressure was higher with the SSLM (26.5 versus 20.5 cm H2O, P = 0.005). Ventilation and fiberoptic view in those successfully inserted were not different for the two devices. The investigators also reported more complications during maintenance and sore throat postoperatively in recovery and at 24 hours for the SSLM. The SSLM was fully deflated before insertion in this study.
Francksen and coworkers studied 120 patients scheduled for routine minor obstetric surgery who were randomly allocated to the size 4 SSLM, Ambu AuraOnce, or LMA Unique.63 The SSLM was slightly slower to insert than the Ambu AuraOnce and as fast as the LMA Unique. All three had high success rates (all >95%) for first-attempt insertions. Rates of subjectively designated excellent insertions were 75% for the LMA Unique, 70% for the Ambu AuraOnce, and 65% for the SSLM. All devices were judged acceptable.
Van Zundert and colleagues compared the SSLM with the LMA Unique and the CobraPLA in a study of 320 patients breathing spontaneously.77 Insertion with the SSLM or LMA Unique was easier than with the CobraPLA (P < 0.02), but success rates and overall time to ventilation were similar. The SSLM and CobraPLA had higher airway seal than the LMA Unique (P < 0.001). Anatomic position was best with the CobraPLA, followed by the SSLM and LMA Unique. Blood staining occurred most frequently with the CobraPLA.
Hein and coworkers performed a crossover, randomized comparison of the SSLM and the SLIPA when inserted by trained medical students in 36 anesthetized patients.78 Success rates for first-attempt insertions were similar (67% for the SSLM and 83% for the SLIPA), as were overall success rates (89% for the SSLM and 94% for the SLIPA). Time to ventilation with the SLIPA was faster, and there was an overall preference (67%) among participants for the SLIPA.
Tan and associates performed a randomized comparison of the SSLM, LMA Unique, and LMA Classic inserted by novice medical officers for anesthesia.79 The novices had a total of five attempts with each mask. The SSLM was significantly slower to insert than the LMA Classic. The SSLM also had the lowest insertion success rate (80% for the LMA Classic, 77% for the LMA Unique, and 62% for the SSLM), although differences were not statistically significant. Although the SSLM achieved an airway seal of 4 to 5 cm H2O higher than the other devices, blood on the airway was most common with the SSLM (32%) compared with the LMA Unique (9%) and the LMA Classic (6%), and sore throats were reported most frequently after use of the SSLM (42%) or LMA Classic (41%) compared with the LMA Unique (14%).
Other Studies
Boonmak and colleagues studied the use of the SSLM (sizes 3 and 4) for guiding fiberoptic intubation with a 6.0- or 6.5-mm ETT in 60 patients with normal airways.80 The glottis was fully visible in 45% of patients after SSLM placement. Blind tracheal intubation succeeded in 5% of attempts, but with fiberoptic guidance, the success rate rose to 85%.
Danha and associates evaluated fiberoptically guided intubation through the SSLM and LMA Classic in 42 healthy patients with normal airways; a 6.0-mm, nasal, right-angle ETT was used for all evaluations.81 Total intubation times and overall success rates were not different for the two devices. The success rate for first-attempt intubation through the SSLM was 76%.
Kuvaki and coworkers compared two insertion techniques for the SSLM in 100 patients randomized to different insertion techniques.82 The SSLM was inserted in the two groups by a direct or a rotational technique, both without intraoral digital manipulation. The primary outcome measure was successful insertion at first attempt. The success rate for first-attempt insertion was higher with the direct technique (98%) than with the rotational technique (75%, P = 0.002), but insertion time was a few seconds faster with the latter method (P = 0.035). Final mask position over the larynx and airway morbidity were similar in both groups.
Keller and colleagues examined the pressure exerted in cadavers by the SSLM and LMA Unique on the larynx and pharyngeal wall during sequential inflation.83 The SSLM had a lower elastance and lower intracuff pressure for a given inflation volume. Mucosal pressure increased with cuff inflation at most locations, and values were not different for the two devices at any site or cuff volume.
D Intubating Laryngeal Airway
The ILA is manufactured of medical-grade silicon and is latex free (Fig. 23-5). Ridges are located in the airway tube and the mask. The ridges below the airway connector were designed to improve the tube seal. They also allow easy removal of the connector during intubation through the device. The airway tube of the ILA is curved in a manner designed to mimic the anatomic curve of the upper airway; the design aims to eliminate the need to bend the tube further during use, which can lead to kinking.

After intubation, the ILA is removed using the Cookgas ILA Removal Stylet (Fig. 23-6), which is specifically designed for this use. The stylet stabilizes the previously inserted ETT and allows controlled removal of the ILA without dislodging the ETT from the trachea. The removal stylet consists of an adapter connected to a rod. The adapter is tapered from bottom to top and has horizontal ridges and vertical grooves. The ridges engage and grip the ETT, and the grooves create an airway passage for spontaneously breathing patients during removal of the ILA. The taper enables the stylet to accommodate standard ETTs in many sizes (5.0 to 8.5 mm). The stylet is manufactured from polypropylene and is reusable up to 10 times after washing in detergent, but cannot be autoclaved.

1 Application
After checking the patency of the device and integrity of the cuff by completely deflating and reinflating the cuff with the maximum recommended volume of air (Table 23-1), the device should be fully deflated before insertion. Performing this while pressing the anterior portion of the mask onto a sterile flat surface ensures an appropriately formed cuff for insertion. The posterior portion of the device should be prepared by applying a sterile, water-based lubricant just before insertion.
| Mask Size | Patient Weight (kg) | Maximum Endotracheal Tube Internal Diameter (mm) |
|---|---|---|
| 2.5 | 20-50 | 6.5 |
| 3.5 | 50-70 | 7.5 |
| 4.5 | 70-100 | 8.5 |
Courtesy of Tyco Healthcare, Schaffhausen, Switzerland.
• Fiberoptic technique. A fiberoptic bronchoscope is passed through the ETT and into the trachea. When the carina is seen, the fiberscope can be left in place and the ETT advanced over it using the fiberscope as a guide (i.e., railroading). The ETT cuff can then be inflated, the scope removed, and the ETT connector replaced.
• Bougie technique. An intubation bougie (e.g., Eschmann tracheal tube introducer, Frova intubation catheter) is passed through the ETT and into the trachea. By directing the angulated distal end of the bougie anteriorly, the success rate is likely increased. By gently placing fingers over the cricoid cartilage, the stylet may be felt as it passes through the cricoid ring. When the bougie has been placed in the trachea, the ETT is passed over the bougie, through the laryngeal inlet, and into the trachea.
• Blind technique. The ETT is slowly advanced through the ILA. For spontaneously breathing patients, the anesthetic circuit can be attached to the ETT connector, and capnography may be used as a guide to successful tracheal placement. The Beck Airway Airflow Monitor (Great Plains Ballistics, Lubbock, TX) can also be used to facilitate blind tracheal intubation. If resistance to further advancement is encountered, the ILA should be repositioned.
2 Indications, Advantages, and Disadvantages
The ILA was designed for airway management as a SAD or as a conduit for blind, stylet-guided, or fiberoptically guided tracheal intubation. The ILA has a theoretical advantage over many other SADs because it is specifically designed to facilitate tracheal intubation. Its larger bowl and curved tube are designed to enhance entry into the mouth and passage along the oropharyngeal curve. The curve of its leading edge is designed to facilitate passage behind the epiglottis and arytenoid cartilages and into the upper esophageal inlet without the need for special deflation techniques or insertion devices. Each ILA accepts routinely used PVC ETTs (see Table 23-1).
3 Medical Literature
There is a limited but expanding literature on use of the ILA in adults.
a Cohort Studies
Klein and Jones performed the first cohort study in 28 patients scheduled for gynecologic surgery; 22 of them were also intubated through the ILA.84 The ILA was successfully placed (96.4%) on the first attempt in all but one patient. Leaks during manual ventilation were observed but were corrected with slight withdrawal of the device. The glottis was visible with the fiberscope in all patients, but a degree of epiglottic downfolding was observed in most cases. The investigators used the “Klein maneuver” to correct epiglottic downfolding: this involves jaw lift and withdrawal of the ILA, followed by reinsertion. Intubation through the ILA using fiberoptic guidance was 100% successful. The investigators reported that use of a flexible, reinforced ETT (Mallinckrodt) enhanced success during blind intubation.
Bakker and colleagues performed a cohort evaluation of controlled ventilation and tracheal intubation in 59 healthy patients undergoing elective surgery.85 ILA insertion was successful in 100%, with a mean leak pressure of 19 ± 5 cm H2O. Blind tracheal intubation was attempted in 19 patients. The first-attempt success rate was 58%, and the overall success rate for intubation was 74%. Postoperatively, 10% of patients had dysphagia, and one patient was diagnosed with bilateral lingual nerve injury, although this complication fully resolved after 4 weeks. The investigators concluded that the ILA was an adequate SAD for insertion and ventilation but that the proposed advantage of ease of tracheal intubation would require further investigation.
Joffe and associates reported a 70-patient cohort study of single-use and reusable ILAs.86 The ILA insertion success rate was 100%, and in 57 patients, it was used as a primary airway only. The median airway leak pressures were 25 and 30 cm H2O for the single-use and reusable devices, respectively. Fiberoptically guided intubation was successful in 12 (92%) of 13 attempts. Postoperatively, 26% of patients complained of mild sore throat.
Yang and coworkers studied 60 patients who were anticipated to be difficult to intubate and in whom the ILA was used for intubation; in one half, it was guided by a fiberscope and in one half by a Shikani Optical Stylet (SOS).87 Insertion and ventilation through the ILA was successful in all patients. With the fiberscope, all but two were intubated on the first attempt, and they were successfully intubated on the second or third attempt. In the SOS group, 18 patients were intubated on the first attempt and 7 on the second attempt. The five failures in this group were all successfully intubated with the fiberscope. The fiberscope enabled faster intubation with a higher success rate. The hemodynamic changes recorded during intubation were minimal.
b Randomized, Controlled Studies of Efficacy
Using a noninferiority approach, Karim and colleagues compared the ILA and single-use ILMA during attempts at blind intubation in 154 healthy adults undergoing elective surgery.88 The primary outcome measure was successful intubation within two attempts. This was achieved with the ILA in 77% (60 of 78) and with the ILMA in 99% (75 of 76) of patients (95% CI for the difference, 12% to 32%; P < 0.0001). After two failed blind attempts, fiberoptic guidance was used. In the ILA group, this enabled successful intubation of another 14 patients, but it failed in 4 patients. The overall intubation success rate using the ILA with three attempts (one with a fiberscope) was 95%. The investigators concluded that the single-use ILMA appears to be superior to the ILA as a conduit to facilitate blind tracheal intubation. A notable feature of this study was that one half of the patients had a body mass index (BMI) of 30 to 40 kg/m2 including one fourth who had a BMI of more than 35 kg/m2. BMI did not appear to influence the success rate of the ILA.
Erlacher and associates compared the ILA, ILMA, and CobraPLUS (Engineered Medical Systems, Indianapolis, IN) for ventilation and blind intubation in 180 healthy adults.89 Ventilation was excellent with all devices, and minor repositioning was required in no more than 5% of patients in any group. Blind intubation was successful using the ILMA in 95% of cases, the ILA in 57%, and the CobraPLUS in 47%. Fiberoptic intubation was possible in all but one patient. Standard measures for predicting difficulty during routine intubation were not useful for predicting difficulty during blind intubation through these SADs.
c Other Studies
Wong reported the use of an ILA combined with a lighted stylet to achieve intubation in a patient with a potentially difficult airway.90 The patient had Hallermann-Streiff syndrome with oculomandibulofacial dystocia (i.e., birdlike appearance, mouth opening of 4 cm, receding chin, and Mallampati class III score). The lighted stylet (with the introducer removed) was placed inside the ETT, and transillumination was used to identify tracheal intubation.
E Laryngeal Tube
The VBM Laryngeal Tube (VBM Medizintechnik, Sulz, Germany) and King Laryngeal Tube (King Systems, Noblesville, IN) are SAD devices that were introduced to the European market in 1999 and to the United States in February 2003.91 Between 1999 and 2002, several modifications were made to the original version of the laryngeal tube (LT), including a softer tip, a change from cuff inflation by separate pilot tubes to a single pilot tube, and alterations to the ventilation orifices and proximal cuff. Among the several distinct LTs, some are first-generation and others second-generation SADs.
The LT consists of an airway tube with two inflatable balloons or cuffs designed to lie above and below the laryngeal inlet (i.e., pharyngeal and esophageal, respectively), and both cuffs are inflated through a single pilot tube and balloon (Fig. 23-7).91,92 When the device is inserted, it lies along the length of the tongue and extends distally into the proximal esophagus. Proximal and distal cuffs sit in the oropharynx and esophageal inlet, respectively. Inflation creates a seal, and ventilation occurs through orifices between the cuffs. Proximally, a 15-mm standard male adapter enables connection to the anesthetic circuit, and distally, several small airway orifices lie between the balloons (Fig. 23-8). Three black lines proximally indicate approximately correct depth of insertion.

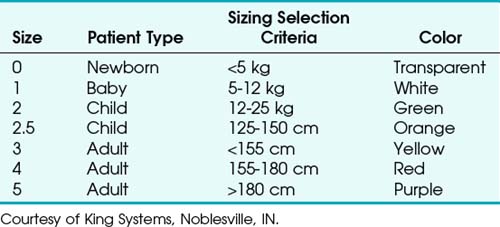
The LT is made of silicon and is designed to be reused up to 50 times. The airway tube is slim, curved, and relatively short, with an average diameter of 11.5 mm and a blind tip. Seven sizes are available for use in neonates up to large adults (Table 23-2).91,92 LT size selection should be based on the patient’s weight for sizes 0 to 2 and on height for sizes 3 to 5. The disposable version (LT-D) is sold in sizes 2 to 5. Adult and child sizes (but not infant and newborn sizes) are supplied with a reusable, silicone bite block.
2 Indications, Advantages, and Disadvantages
The LT is designed for use during spontaneous or controlled ventilation. Because the slim profile of the LT allows easy insertion through a narrow orifice, it can be considered for airway management in patients with restricted mouth opening. Insertion is relatively easy and provides a clear airway in most patients on the first attempt. Extensive training is unnecessary. Its insertion and performance characteristics have led to its being included as an airway device for use in the management of cardiac arrest in the 2010 guidelines of the International Liaison Committee on Resuscitation (ILCOR).93
Because of the design and length of the LT, inadvertent tracheal intubation is unlikely, although it has been reported with some versions of the device.94 The LT is associated with a low incidence of minor traumatic sequelae, such as sore throat, hoarseness, or blood on the device after use. The slim design, soft materials used in construction, and high-volume, low-pressure cuffs offer advantages. These high-volume, low-pressure cuffs provide a good seal and reduce the risk of ischemic damage when used appropriately. Another advantage is the ability to insert the device from a variety of positions relative to the patient, making it useful in emergency situations.
3 Medical Literature
a Cohort Studies and Studies of General Performance
Using earlier versions of the LT, Asai,95 Doerges,96 and their colleagues determined that after blind insertion, the device provided a patent airway in most patients at the first attempt. The LT can be inserted quickly without extensive training; it is considered a suitable airway management device with a high rate of successful insertion requiring a mouth opening as limited as 23 mm.97,98 Acceptance of the ease of LT placement is high among physicians, nurses, and paramedics, although correct positioning may require more adjustments in patients with an increased BMI.99–101 Insertion time is reported to be comparable to that for the LMA Classic.96
Several studies confirmed the efficacy of controlled ventilation with the LT and found that its airway seal was modestly better than that of the LMA Classic, with some patients achieving a seal above 30 cm H2O.96,102–105 Although numerous studies have shown the LT can be effective during mechanical ventilation, some studies, most notably that of Miller (inventor of the SLIPA), have found the LT to be unsatisfactory for spontaneous ventilation.106 The findings of Miller and colleagues were based on a frequent failure rate (7 of 17 patients) caused by loss of airway control during surgery.106 The investigators assessed use of a first-generation LT that did not feature a second, large ventilation aperture and two lateral ventilatory openings that are part of the newer version.
In another study by Hagberg and colleagues, the LT was demonstrated to provide a reliable airway during elective surgery with spontaneous ventilation.105 In this study, the mean depth of insertion was greater than expected for each size of the LT, and as a result, the company now manufactures this device 1 cm longer in the silicon version and 2 cm larger in the disposable version. The success rate for first-time placement was 86%, consistent with the rates of 85% to 95% found in previous studies.96,102–104
The dose of propofol required for LT insertion is approximately the same as for the LMA Classic.107 Nitrous oxide diffuses into the cuffs, and cuff pressures should be monitored during use. During 30 minutes of nitrous oxide anesthesia, Asai and coworkers demonstrated an increase of 15 cm H2O in the intracuff pressure.104,108
Because of the ease of insertion and a good airtight seal, the LT has been studied for airway management during cardiopulmonary resuscitation. There is considerable data on its use by medical staff and paramedics for primary airway management out of hospitals and for airway management during cardiopulmonary resuscitation.109–113 The LT is included in the 2010 ILCOR resuscitation guidelines.93 Dengler and associates reported some cases of massive pulmonary aspiration and one of gastric overinflation during use of the LT in the emergency setting.114 This followed ventilation with high peak airway pressures, and the investigators questioned whether the LTS-II or LTS-D would have been a more appropriate device.
Langlois and associates performed a cohort study of the LT-D in 50 anesthetized patients.115 Insertion was successful in 94%, and the median insertion time was 38 seconds. Insertion difficulty occurred in 25%. Mean oropharyngeal leak pressure increased from 26 cm H2O (range, 22 to 32.5 cm H2O) to 34 cm H2O (range, 29 to 40 cm H2O) at the end of surgery. No cases of gastric inflation, regurgitation, or hypoxia were reported. The incidence of moderate sore throat was 6% in the recovery room and 0% at 24 hours.
b Randomized, Controlled Studies of Efficacy
Amini and colleagues performed a study comparing the LT with the LT-D in 100 anesthetized, paralyzed patients.116 Both devices showed similar clinical performance in terms of insertion success (90% for the LT-D and 96% for the LT) and insertion time (28.4 and 23.6 seconds, respectively). There were no differences in airway leak pressure, fiberoptic position, or postoperative sore throat and dysphagia.
Yilidz and associates compared the LT with the LMA Classic in 132 patients.117 Oxygenation and ventilation were possible in all patients. Insertion success rates after the first, second, and third attempts were 84.8% (n = 56), 12.1% (n = 8), and 3% (n = 2) for the LT compared with 56.1% (n = 37), 25.8% (n = 17), and 18.2% (n = 12) for the LMA Classic (P = 0.001). Blood on the cuff was seen at removal in one patient with the LT and in 10 patients with the LMA Classic. Six patients in the LMA Classic group complained of hoarseness (P = 0.012). The success rates for LMA Classic insertion are markedly lower in this study than in many others, which may influence many of the results.
Noor and coworkers compared the LT and LMA Classic for ventilation during manual in-line stabilization in 40 healthy, anesthetized, and paralyzed patients.118 Three attempts were allowed. The success rates (100%), adequacy of ventilation, and hemodynamic changes were not different for the two devices. The LT has a higher first-attempt success rate (100% versus 85%) and was significantly faster to insert (25 versus 36 seconds, P = 0.001).
Kurola and colleagues studied insertion of the LT, ILMA, and CobraPLA by paramedical students in 96 anesthetized, paralyzed patients.119 The success rates for first-attempt insertion were 75% for the ILMA, 44% for the LT, and 22% for the CobraPLA. Overall success rates were 97% for the ILMA and 79% for the LT and the CobraPLA. In this small study, the numeric differences were not statistically significant.
c Other Studies
No studies have been published examining whether the lower cuff protects against aspiration. Khazin and associates studied hypopharyngeal pH changes as a marker of regurgitation during anesthesia with several SADs (e.g., LT, CobraPLA, LMA Classic, ILMA, and PLMA) and with ETTs in 180 patients.120 One to five patients in each study group of 30 had regurgitation episodes, but the rates were not statistically different between groups. The clinical relevance of the study, particularly its relevance to aspiration protection, is questionable. Bercker and coworkers designed a study to compare the seal of seven SADs in a cadaver model of elevated esophageal pressure. SADs included the LMA Classic, PLMA, ILMA, LT, LTS-II, Combitube, and EasyTube.40 All were inserted into unfixed human cadavers with an exposed esophagus that had been connected to a 130-cm water column. Slow and fast increases of esophageal pressure were performed, and the water pressure at which leakage appeared was documented. The Combitube, EasyTube, and ILMA withstood the water pressure up to more than 120 cm H2O. The PLMA, LT, and LTS-II blocked the esophagus despite 72 to 82 cm H2O of pressure. The LMA Classic leaked at 48 cm H2O, but only minor leakage was found in the trachea. Devices with an additional esophageal drain tube removed fluid sufficiently without pulmonary aspiration. The LTS-II and LTS-D are likely to provide better protection than the LT.
Ulrick-Pur and colleagues examined the mucosal pressures exerted by several SADs in fresh cadavers.121 Using maximum cuff volumes according to the manufacturers’ guidelines, the highest pharyngeal pressures were found with the ILMA. The LMA Classic, ILMA, and PLMA induced significantly higher pharyngeal pressures than the LT, EasyTube, or Combitube at maximum inflation. The maximum esophageal pressures were significantly higher using the EasyTube than with the Combitube. Tracheal mucosal pressures were significantly higher using the Combitube compared with the ETT and the EasyTube.
Asai and coworkers reported successful use of the LT in three patients in whom insertion of the LMA had failed, and they suggested the relative width of the devices was responsible for failure or success.122 In these cases, the pharyngeal space was narrowed by swollen tonsils, goiter, and redundant oropharyngeal tissue, and the investigators recommended that when LMA insertion is difficult or impossible because of a narrowed pharynx, insertion of the LT should be attempted before considering tracheal intubation. Reported side effects with the LT are few but do include tongue engorgement.123
F Cobra Perilaryngeal Airway
The Cobra perilaryngeal airway (CobraPLA, Engineered Medical Systems, Indianapolis, IN) (Fig. 23-9) was designed by David Alfery. It was based on a modification of the Guedel oral airway and was marketed in 1997. The initial idea was to modify the Guedel airway to accomplish mask ventilation in the most difficult airways encountered. It has since been adapted to create a SAD. The proximal portion of the airway was modified to enable attachment to an airway circuit, a circumferential cuff was added proximal to the distal breathing hole, and the distal end of the device was modified to form a cobra head shape. Later refinements included a distal flexible tip (tongue) and an internal ramp inside the cobra head to help guide an ETT toward the glottis. A modification of the CobraPLA, the CobraPLUS, incorporates an integrated temperature probe and a distal gas-sampling port.
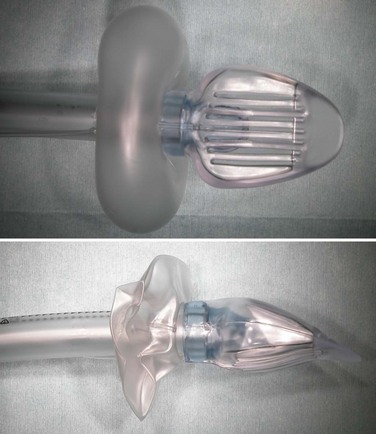
Figure 23-9 The Cobra perilaryngeal airway (CobraPLA) with cuff inflated (top) and deflated (bottom).
Agro and colleagues suggested a modified method of size selection: no. 3 for less than 60 kg, no. 4 for 60 to 80 kg, and no. 5 for more than 80 kg.100 In this study, relatively large CobraPLAs were used by skilled operators with techniques that included a combination of scissoring the mouth open and performing a jaw lift (i.e., Agro maneuver) in patients who had been given muscle relaxants. If using Agro’s range or a relatively large CobraPLA, the cuff inflation volume can be reduced from the maximum recommended by the manufacturer. Use of the larger sizes achieves a higher degree of airway seal.
The CobraPLUS is a modified version of the CobraPLA which in its adult version has an integrated core temperature–measuring device and in its pediatric version also has a distal gas-sampling post within the device head. This is of particular interest in newborns and infants, in whom very rapid respiratory rates and low tidal volumes result in inaccurate gas-sampling values if using more proximal sampling sites.124
1 Application
Some practical tips are worth considering. First, the patient should be at an adequate depth of anesthesia before insertion is attempted. Laryngospasm may occur during insertion if the patient is at too light a level of anesthesia. Second, if one CobraPLA is found to be unsuitable, an alternative size should be inserted before abandoning this technique. Third, if the CobraPLA is not inserted far enough, cuff inflation may cause tongue protrusion and a poor airway seal; the device should be advanced further or a smaller CobraPLA chosen. The cuff should not be visible at the base of the tongue when the mouth is opened. Fourth, if the CobraPLA is inserted too far, past the laryngeal inlet, ventilation is impossible. This may occur while learning the technique or if too small a CobraPLA is used. Pulling back the airway 1 to 2 cm usually resolves the situation. The CobraPLA is considered to have a steep learning curve, and the technique can be mastered in 5 to 10 insertions.100
A standard ETT may be used for intubation through a CobraPLA, ideally guided by a fiberscope. As the fiberscope and ETT exit the ventilation orifice, the soft bars of the grill separate easily without impediment to advancement. The internal ramp directs the fiberoptic bronchoscope anteriorly, and if the larynx is anterior to the ventilation orifice, this approach should enable prompt intubation. The large diameter of the CobraPLA permits the passage of an adequately sized ETT (Table 23-3). The CobraPLA can be left in place with its cuff deflated if desired.
TABLE 23-3 Sizing for Passage of an Endotracheal Tube in a CobraPLA
| Cobra Size | ETT Size (mm) |
|---|---|
 |
3.0 |
| 1 | 4.5 |
 |
4.5 |
| 2 | 6.5 |
| 3 | 6.5 |
| 4 | 8.0 |
| 5 | 8.0 |
| 6 | 8.0 |
ETT, Endotracheal tube; PLA, perilaryngeal airway.
An alternative technique involves use of a lightwand to guide intubation, as has been described for other SADs.99,125,126 The technique is described in the “Laryngeal Tube” section. Blind intubation through the CobraPLA by advancing an ETT through it or by using a bougie guide may be successful, but it has a significant failure rate, and the preceding techniques are far preferable when the equipment is available.
2 Indications, Advantages, and Disadvantages
The relative ease of insertion is an advantage of the CobraPLA, which may make it suitable for use in airway emergencies even when it is used by personnel with little or no experience in the use of SADs. Some clinical evidence supports the efficacy of use by nonanesthesiologist physicians with minimal training in airway management.127 Pediatric sizes of the CobraPLUS can monitor core body temperature and distal carbon dioxide levels.
3 Medical Literature
Approximately 40 publications have described the CobraPLA or CobraPLUS. The CobraPLA was modified to aid insertion in 2006, as reported by the inventors, Alfery and Szmuk, in 2007.128 The manufacturers modified the CobraPLA and the CobraPLUS by adding a distal bend in the breathing tube. It is likely that only publications after 2007 used the updated device, and this should be considered in interpreting these studies.
a Cohort Studies
The first report of the use of the CobraPLA in a peer-reviewed journal was by Agro and coworkers in 2003; the study included 28 anesthetized and mechanically ventilated patients.129 After mannequin training, the CobraPLA was inserted in patients within 10 ± 3 seconds with a 100% success rate. Immediate ventilation was achieved in 57% of patients, and 43% required a positioning maneuver (e.g., pulling back).
In a later study, Agro and colleagues studied 110 patients and reported 100% successful insertion in a mean time of 6.8 ± 2 seconds.100 There were no adverse events or significant complications. Mean airway seal was 34 cm H2O using very low cuff inflation volumes by choosing relatively large CobraPLAs.
In contrast, Cook and Lowe reported an aborted study with the CobraPLA.130 During preparation for two studies, a total of 29 CobraPLAs were inserted, and two cases of significant aspiration were identified. The investigators raised concerns about the design of the CobraPLA and its safety in terms of aspiration protection. The manufacturers robustly rejected these assertions.131–133
Park and associates studied the effect of changing head and neck positions on SAD performance, including the CobraPLA, in 139 patients.134 Oropharyngeal leak pressure and cuff pressure were evaluated in four head and neck positions: neutral, 45 degrees of flexion, 45 degrees of extension, and 45 degrees of right rotation. Adverse events such as difficult ventilation or gastric insufflation were assessed. Airway leak pressures were well maintained with the CobraPLA, but “gastric insufflations occurred before the oropharyngeal leak in 37 of 45 patients.”134 The investigators concluded that caution is warranted when changing the position of the head and neck while using the Cobra-PLA because gastric insufflation may occur.
Nam and coworkers reported a case series of 50 uses of the CobraPLA in paralyzed patients undergoing elective surgery.135 The success rate for first-attempt insertion was 82%, with a mean insertion time of approximately 16 seconds. Airway leak pressure (22.5 cm H2O) was lower than other reports. The point of leakage was recorded as the neck (52%), abdomen (46%), or both sites (2%). The glottis was visible in 88% of patients through the CobraPLA. Postoperative blood staining was seen in 22%, mild dysphonia in 6%, mild dysphagia in 10%, and mild and moderate sore throat in 44% and 4%, respectively.
b Randomized, Controlled Studies of Efficacy
Akça and colleagues compared the CobraPLA with the LMA Unique in a randomized series of 81 patients, and the insertion times, airway adequacy, number of repositioning episodes, and minor complications were similar in both groups.136 However, the cuff leak pressure of the CobraPLA was significantly greater than that of the LMA Unique (23 versus 18 cm H2O). The investigators found lower airway leak pressures than Agro did, most likely because the Akça study used smaller CobraPLAs.
Gaitini and others, including the inventor, compared the CobraPLA with the LMA Unique during spontaneous ventilation in 80 anesthetized patients.137 They reported no statistically significant difference between the devices regarding ventilatory variables, number and type of airway interventions required for placement, fiberoptic view, or incidence of adverse events. Reported benefits of the CobraPLA were a significantly higher airway leak pressure (27 versus 21 cm H2O) and higher oxygen saturation (98% versus 97%) than for the LMA Unique. Conversely time for insertion was significantly shorter for the LMA Classic (24 versus 27 seconds), and insertion was slightly easier than with the CobraPLA.
In a crossover study between the CobraPLA and LMA Classic during controlled ventilation, Nam and associates reported a faster median insertion time for the CobraPLA (15 versus 25 seconds) and a higher median airway seal (23 versus 15 cm H2O).138 Other aspects of performance were not different.
Schebesta and coworkers compared the CobraPLA with the LMA Classic in 60 anesthetized patients.139 The study focused on airway sealing and gas leakage, and despite confirming a higher airway leak pressure with the CobraPLA (24 versus 20 cm H2O), use of the CobraPLA was associated with higher environmental nitrous oxide (but not sevoflurane) levels during anesthesia. The study also reported the LMA Classic was associated with easier positioning and a lower peak airway pressure.
Galvin and colleagues studied the CobraPLA and LMA Classic for gynecologic laparoscopy.140 Insertion characteristics, adverse events, and rates of throat morbidity were similar between devices. Peak airway pressures were higher with the LMA Classic than with the CobraPLA. Blood was observed on 40% of CobraPLAs after removal.
Turan and associates compared the CobraPLA with the LT and LMA Classic in 90 patients during short surgical procedures.141 There were similar results for insertion times, number and type of airway interventions, and hemodynamic variables. Success rates for first-attempt insertion were highest in the CobraPLA group (95%) and lowest in the LMA Classic group (57%). However, blood staining was seen in 50% of cases using the CobraPLA and only 17% of cases using the LMA Classic or LT. The rate of sore throat was also higher in the CobraPLA group than with either of the other SADs.
c Other Studies
Ben-Abraham and coworkers studied the ease of insertion of a CobraPLA in anaesthetized patients by residents in scrubs or in antichemical personal protective equipment (PPE).127 Wearing PPE caused only a modest slowing of insertion time (i.e., median time increased 14 seconds) for anesthesia residents and nonanesthesia residents after training. After insertion, 42% of airway devices leaked, preventing adequate positive-pressure ventilation, and 13 devices (26% of the total) required replacement.
The CobraPLA has been used for difficult airway management in adults:
• For securing an obstructed airway and facilitating fiberoptically guided intubation after thyroidectomy142
• For rescuing the airway during cardiopulmonary resuscitation143
• For maintaining the airway during urgent percutaneous, dilatational cricothyroidotomy while enabling visualization of the procedure through a fiberscope144
• For a series of five percutaneous tracheostomies in a similar manner145
• For airway rescue followed by fiberoptically guided tracheal intubation in a “difficult to intubate, difficult to ventilate” patient undergoing rapid-sequence induction of anesthesia146
Complications reported during use of the CobraPLA include aspiration, hypoglossal nerve injury,147 tube obstruction in a patient with a fixed flexed neck, and obstruction by incarceration of the epiglottis in the epiglottic bars.148,149
G Tulip Airway Device
Like the CobraPLA, the Tulip was named to reflect its overall shape and appearance. It is a simple airway tube with a distal airway orifice and surrounding distal cuff (Fig. 23-10). The device has a proximal, 15-mm standard connector. It is made of PVC and is designed for single use. The distal cuff is described as an inflatable polyhedral beveled cuff, which is designed to inflate below the soft palate, behind the tongue, above the epiglottis, and within the oropharynx.

3 Medical Literature
The literature for the Tulip is limited to a single abstract, which describes very effective and swift deployment in a manikin with ventilation and positioning over the manikin’s larynx as good as an i-gel and single-use LM.150 The relevance of these provisional findings to clinical use are unknown. Early clinical experience confirmed its ease of use as a SAD, and clinical trials are being undertaken in the United Kingdom (Amer Shaikh, personal communication, 2009).