Non-astrocytic gliomas
OLIGODENDROGLIOMAS
Oligodendrogliomas usually present in adulthood. They show a broad range of behaviors; some neoplasms with bland cytologic features grow extremely slowly and are associated with a long history of epilepsy, yet anaplastic oligodendrogliomas can have a poor prognosis (Table 36.1). Attempts to categorize this heterogeneity in a variety of grading systems have been successful in separating groups of patients with distinct outcomes, but consensus supports the view that two broad categories are sufficient. These correspond to ‘oligodendroglioma’ and ‘anaplastic oligodendroglioma’, grade 2 and grade 3, respectively in the WHO classification.
Table 36.1
Prognostic indicators in oligodendrogliomas
Age (young age – favorable)
Postoperative Karnofsky score (high score – favorable)
Pathologic distinction between WHO grade 2 and WHO grade 3 (anaplastic) neoplasms (WHO grade 2 – favorable)
Ki-67 labeling index (<5% – favorable)
Chromosomes 1p/19q co-deletion (presence – favorable)
MGMT methylation (presence – favorable)
IDH1 mutation (presence – favorable in anaplastic oligodendroglioma)
Proneural gene expression profile (presence – favorable)
Anaplastic neoplasms with homozygous CDKN2A deletion (absence – favorable)
 OLIGODENDROGLIOMAS
OLIGODENDROGLIOMAS Constitute 7–10% of primary intracranial neoplasms.
Constitute 7–10% of primary intracranial neoplasms.
 Occur with equal incidence in males and females.
Occur with equal incidence in males and females.

 Occur most often in the frontal lobe.
Occur most often in the frontal lobe.
 Occur most frequently in patients aged 30–50 years.
Occur most frequently in patients aged 30–50 years.
 Most often present with epilepsy.
Most often present with epilepsy.
 Are probably more chemosensitive and radiosensitive than most other gliomas.
Are probably more chemosensitive and radiosensitive than most other gliomas.

MACROSCOPIC APPEARANCES
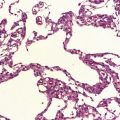
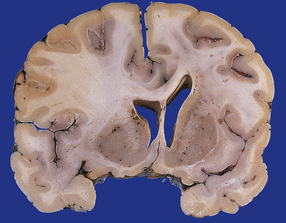
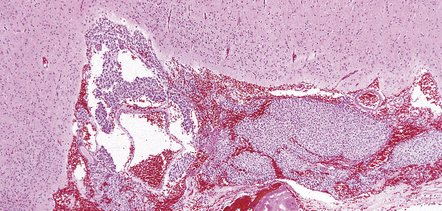
Oligodendrogliomas have a variety of macroscopic appearances. Small diffuse examples may be difficult to locate in resection specimens. Distortion of the normal cortical gray and white matter interface and discoloration of tissue provide clues to their location (Fig. 36.1). Other neoplasms are more obvious, appearing as moderately well-circumscribed, gray masses. The propensity of superficial oligodendrogliomas to invade the subarachnoid space may produce a neoplasm that sits partially on the cortical surface of the cerebrum.
MICROSCOPIC APPEARANCES
Oligodendrogliomas are composed of uniform cells (Figs 36.2–36.5). These readily infiltrate gray matter, but tend to produce a more abrupt edge in white matter (Fig. 36.2). A lobular architecture is sometimes seen and microcysts can be prominent, particularly in myxoid tumors. Small foci of dystrophic calcification are usually found, often at the edge of the neoplasm or in infiltrated gray matter. A delicate vasculature consisting of branching capillaries runs through typical oligodendrogliomas.
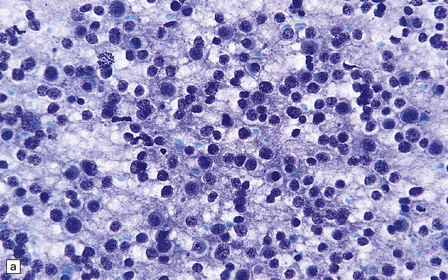
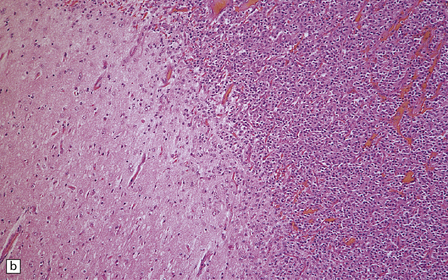
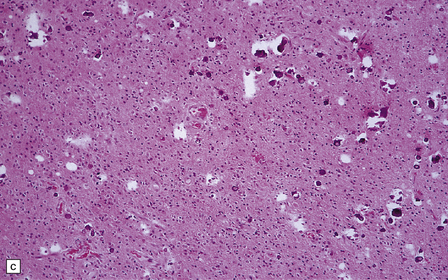
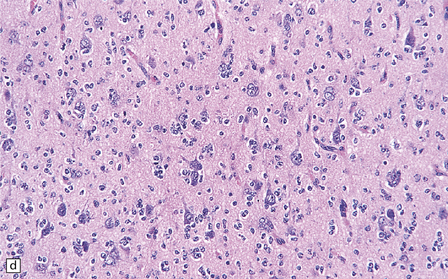
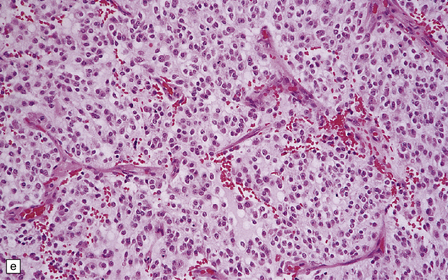
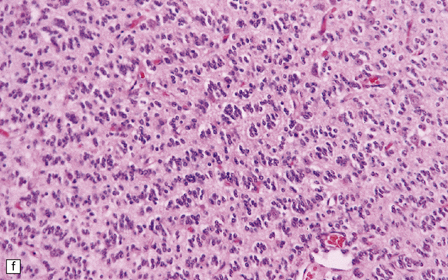
36.2 Oligodendroglioma.
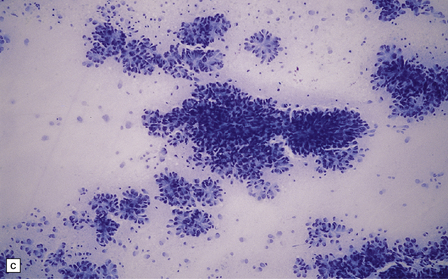
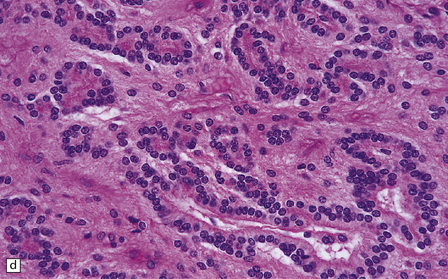
(a) Perioperative smear preparations generally reveal little cytologic pleomorphism. The round nuclei and ill-defined cytoplasm are typical. (b) A relatively circumscribed border to what is generally an infiltrating neoplasm may be observed in white matter. (c) Dystrophic calcification is a common feature. (d) The cerebral cortex is diffusely infiltrated by neoplastic cells, which encircle neurons (satellitosis). (e) A network of delicate bifurcating capillaries is typical, and sometimes divides the cells into lobules. (f) The oligodendroglioma is one of several neuroepithelial neoplasms that may show rhythmic nuclear palisading.
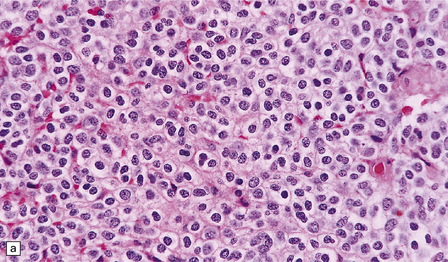
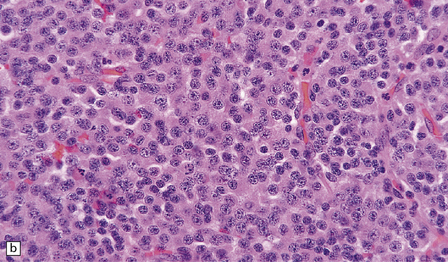
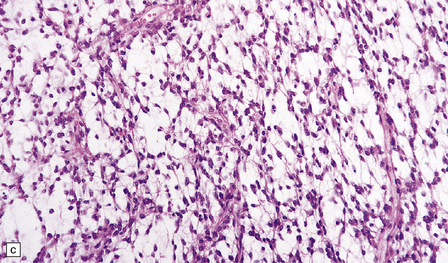
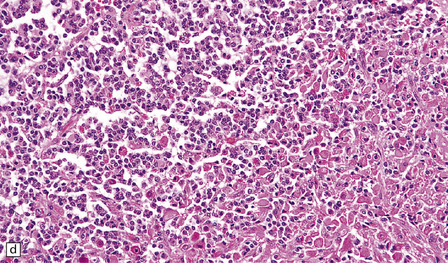
36.3 Oligodendroglioma.
Cytological features include round cells with (a) or without (b) artefactual clearing of the cytoplasm, and (c) loosely arranged elongated cells with uniform round nuclei. (d) Foci of small ‘minigemistocytic’ cells with eosinophilic cytoplasm and an eccentric, round nucleus are seen in some oligodendrogliomas. The nuclear form is similar to that in the round cells, and these foci do not indicate a diagnosis of mixed glioma.
 GENETIC ASPECTS OF GLIOMAS WITH OLIGODENDROGLIAL FEATURES
GENETIC ASPECTS OF GLIOMAS WITH OLIGODENDROGLIAL FEATURES
 A white matter oligodendrocyte progenitor cell has been implicated as the cell of origin for oligodendroglioma.
A white matter oligodendrocyte progenitor cell has been implicated as the cell of origin for oligodendroglioma.
 Isocitrate dehydrogenase 1 (IDH1) mutation is a common and early genetic event in oligodendroglial tumors (Fig. 36.6). This finding is shared with other low- or intermediate-grade astrocytic and oligoastrocytic neoplasms as well as secondary glioblastomas.
Isocitrate dehydrogenase 1 (IDH1) mutation is a common and early genetic event in oligodendroglial tumors (Fig. 36.6). This finding is shared with other low- or intermediate-grade astrocytic and oligoastrocytic neoplasms as well as secondary glioblastomas.
 Allelic loss on chromosome 1p and chromosome 19q is present in as many as 80% of both oligodendrogliomas and anaplastic oligodendrogliomas and is considered an early neoplastic event (Fig. 36.7). Combined 1p/19q loss is not specific for oligodendrogliomas, occurring in oligoastrocytomas. In contrast to IDH1 mutation, 1p/19q loss is rare in astrocytomas.
Allelic loss on chromosome 1p and chromosome 19q is present in as many as 80% of both oligodendrogliomas and anaplastic oligodendrogliomas and is considered an early neoplastic event (Fig. 36.7). Combined 1p/19q loss is not specific for oligodendrogliomas, occurring in oligoastrocytomas. In contrast to IDH1 mutation, 1p/19q loss is rare in astrocytomas.







 Homozygous deletion of CDKN2A (p16) on 9p occurs in up to 40% of anaplastic oligodendrogliomas.
Homozygous deletion of CDKN2A (p16) on 9p occurs in up to 40% of anaplastic oligodendrogliomas.


 Astrocytic and oligodendroglial components of an oligoastrocytoma show the same genetic profile.
Astrocytic and oligodendroglial components of an oligoastrocytoma show the same genetic profile.
 Allelic loss on 17p/TP53 mutation and 1p/19q loss are inversely associated in oligoastrocytomas.
Allelic loss on 17p/TP53 mutation and 1p/19q loss are inversely associated in oligoastrocytomas.
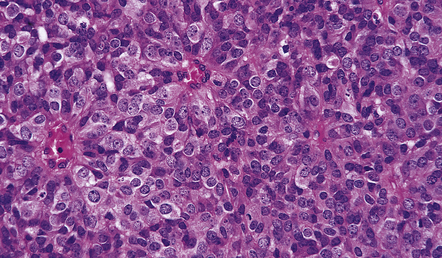
Cytoplasm is rather sparse and not easily identifiable in some cells. Artefactual clearing of the cytoplasm is common in paraffin-embedded material, particularly when fixation is delayed, and gives a ‘fried-egg’ appearance to the cells (Fig. 36.3a). Nuclei are round and usually contain faintly speckled chromatin. A single small nucleolus may be present. Cells occasionally adopt a slightly elongated form, but nuclei retain their characteristic uniformity. Some cells, termed ‘minigemistocytes’, have eccentrically placed nuclei and eosinophilic cytoplasm that is strongly immunoreactive for GFAP (Fig. 36.3d). This overlap with an astrocytic morphophenotype is not on its own indicative of mixed glioma status, but should prompt a search for anaplastic features. Exceptionally, ‘signet-ring’ cells may be evident. Invasion of the subarachnoid space is common (Fig. 36.4). Immunohistochemistry with antibodies to GFAP generally reveals labeling of only a small proportion of cells (Fig. 36.5). Immunoreactivity for myelin-specific proteins is rare. Mitoses are sparse, except in neoplasms with other anaplastic characteristics such as cytologic pleomorphism, microvascular proliferation, and areas of necrosis (Fig. 36.8).
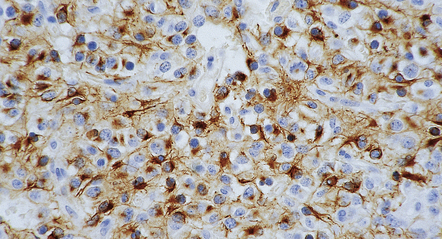
36.6 Anaplastic oligodendroglioma.
Widespread immunoreactivity for the isocitrate dehydrogenase 1 (IDH1) mutant protein (R132H) is present in infiltrating tumor cells.
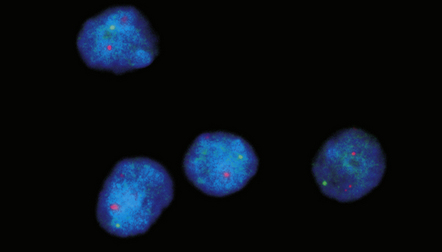
36.7 Anaplastic oligodendroglioma.
This fluorescence in situ hybridization preparation involving four neoplastic nuclei shows loss of chromosome 1p (2 red signals per nucleus from centromeric probes and 1 green signal per nucleus from subtelomeric probes).
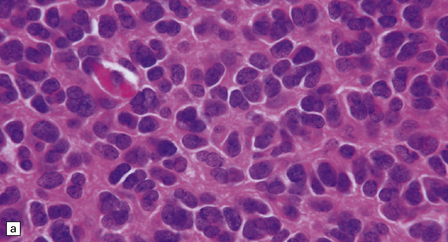
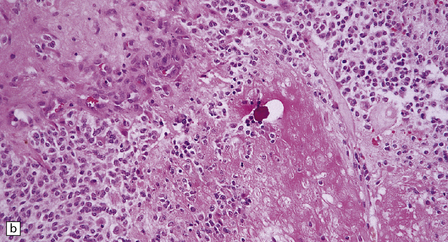
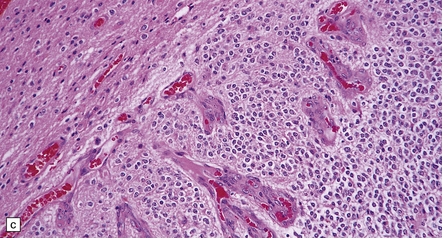
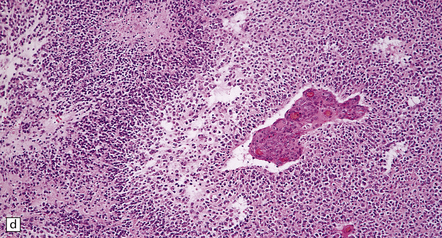
36.8 Anaplastic oligodendroglioma.
Increased cytologic pleomorphism (a) and (b), capillary endothelial proliferation (c) and (d), and micronecrosis (b) and (d) are evident in anaplastic tumors which can have an aggressive biologic behavior.
 DIFFERENTIAL DIAGNOSIS OF OLIGODENDROGLIOMA
DIFFERENTIAL DIAGNOSIS OF OLIGODENDROGLIOMA
 Macrophages in infarcts or in foci of demyelination can appear as scattered or grouped cells with small round nuclei and clear cytoplasm, but unlike neoplastic oligodendrocytes macrophages have a slightly granular cytoplasm, which contains lipid and PAS-positive material, and are immunoreactive for CD68.
Macrophages in infarcts or in foci of demyelination can appear as scattered or grouped cells with small round nuclei and clear cytoplasm, but unlike neoplastic oligodendrocytes macrophages have a slightly granular cytoplasm, which contains lipid and PAS-positive material, and are immunoreactive for CD68.











EPENDYMOMAS




 EPENDYMOMAS
EPENDYMOMAS












MACROSCOPIC APPEARANCES
Classic ependymomas grow as circumscribed soft gray masses that are generally related to the ventricular system. They are commonest in the posterior fossa, where they may fill the fourth ventricle and pass through its exit foramina (Fig. 36.9).
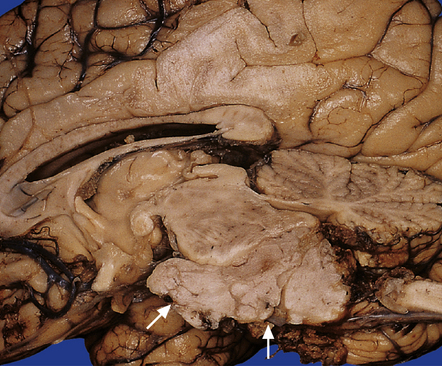
36.9 Ependymoma.
This fourth ventricular tumor has distorted the brain stem and formed an exophytic ventral mass (arrows).
Myxopapillary ependymomas generally appear as well-defined, lozenge-shaped neoplasms among the cauda equina, which they can sometimes envelop (Fig. 36.10). This behavior can hinder a complete surgical removal. Sectioning reveals tissue with a mucoid texture. Spontaneous hemorrhage into these neoplasms is quite common.
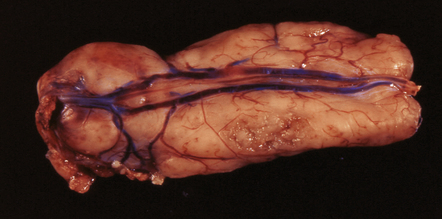
36.10 Myxopapillary ependymoma.
This well-circumscribed lozenge-shaped tumor with a smooth surface was related to the conus medullaris.
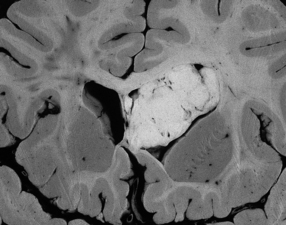
Subependymomas are firm lobulated intraventricular masses and are found predominantly in the fourth ventricle (Fig. 36.11).
MICROSCOPIC APPEARANCES
Ependymomas are generally composed of sheets of uniform cells with indistinct cytoplasmic borders and round or oval nuclei (Fig. 36.12). They are not diffuse gliomas; infiltration of adjacent tissues is minimal, except in some anaplastic examples. The nuclear:cytoplasmic ratio varies; cell density may be high, but infrequent nodules of densely packed cells may be scattered throughout paucicellular areas (Fig. 36.12c). Cells in these nodules may show more nuclear pleomorphism than surrounding cells, and an increased mitotic count. Mitoses should be infrequent and microvascular proliferation absent in classic ependymomas. Foci of micronecrosis without nuclear palisading may be found in classic ependymomas.
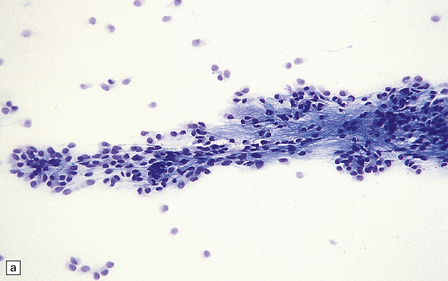
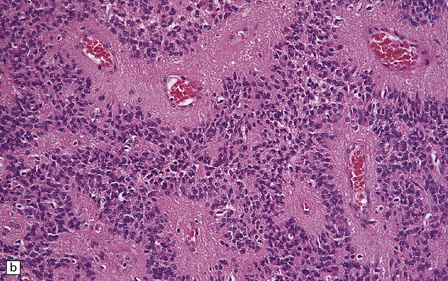
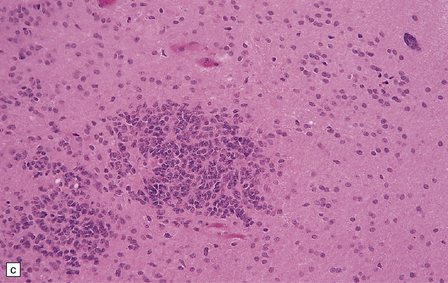
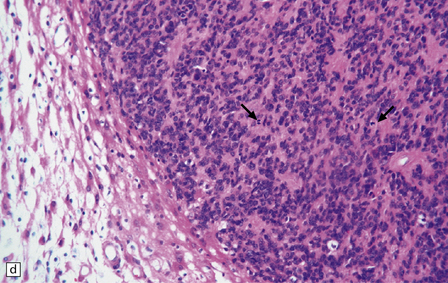
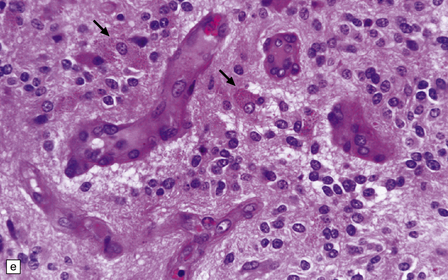
36.12 Classic ependymoma.
(a) Smear preparation showing cells with oval nuclei and fine cytoplasmic processes. (b) Sheets of small uniform cells with a moderately high nuclear:cytoplasmic ratio are interrupted by pseudorosettes around blood vessels. (c) The nuclear: cytoplasmic ratio may be low in some ependymomas, but nodules of densely packed cells are sometimes evident in the paucicellular areas, as illustrated here. (d) Ependymomas push against adjacent brain eliciting reactive changes, such as astrocytosis, and do not infiltrate in the same manner as diffuse astrocytic or oligodendroglial tumors. Here, the uniformity of round tumor cell nuclei is evident, along with several mitotic figures (arrows). (e) Tiny eosinophilic globules (arrows) may be found in a few cells.
Ependymomas label variably with antibodies to GFAP (Fig. 36.13). Focal immunoreactivity for epithelial membrane antigen is sometimes seen around canals. Ultrastructural examination reveals microvilli and cilia on the luminal surfaces, and zonulae adherentes.
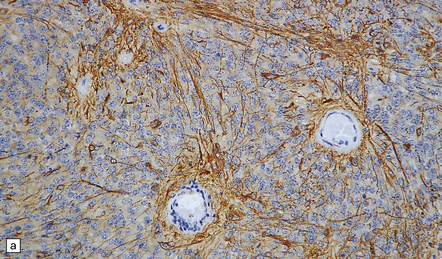
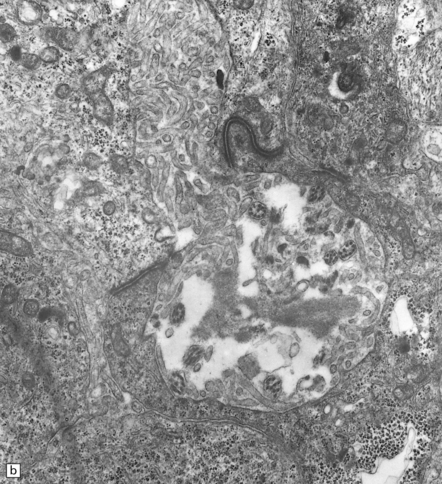
36.13 Ependymoma.
(a) Immunoreactivity for GFAP is most pronounced in fine cytoplasmic processes, particularly those that converge on blood vessels. (b) Ultrastructural examination reveals microvilli in lumina and intercellular junctions.
The pseudorosette is a perivascular anuclear zone of fibrillary processes that taper towards the vessel (Fig. 36.14). Pseudorosettes are a frequent, though not entirely specific, finding in ependymomas. Ependymal (true) rosettes with a central lumen and a halo of neoplastic cells are more specific, but found less consistently (Fig. 36.14). Rosettes and gland-like canals are a manifestation of epithelial differentiation in ependymomas.
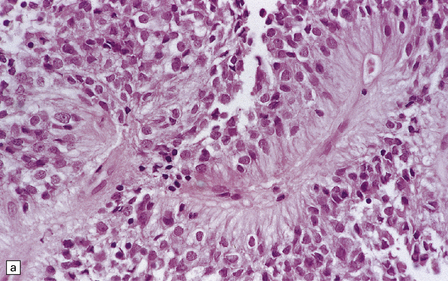
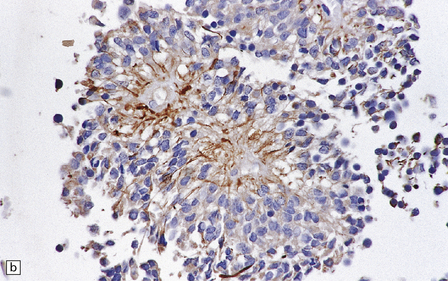
36.14 Ependymal rosettes.
(a) Pseudorosettes are characterized by fine cellular processes that taper towards the blood vessel. (b) The processes are immunoreactive for GFAP. (c) Smear preparations quite often contain perivascular pseudorosettes. True rosettes, as illustrated in this figure, are much less common. (d) Rosettes and canals are found in a minority of ependymomas.
Cellular ependymomas contain cells with a high nuclear: cytoplasmic ratio (Fig. 36.15). Few pseudorosettes are present, and cellular ependymomas do not contain paucicellular areas. Differentiating this variant from an anaplastic ependymoma is difficult, and depends on the identification of an increased mitotic count and cytologic pleomorphism in the latter. Microvascular proliferation and abundant necrosis are also features of most anaplastic ependymomas.
36.15 Cellular ependymoma.
A high nuclear:cytoplasmic ratio characterizes the cells of this neoplasm, and pseudorosettes are sparse. The mitotic count is low.
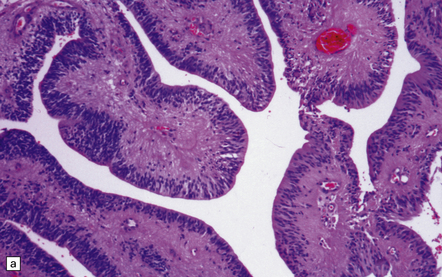
Rarely, intraventricular ependymomas exhibit a papillary pattern, and choroid plexus papilloma (CPP) then enters the differential diagnosis (Fig. 36.16). Papillary ependymomas do not have a subepithelial basement membrane over a fibrovascular core like the CPP, while the CPP does not contain pseudorosettes with tapering GFAP-positive processes.
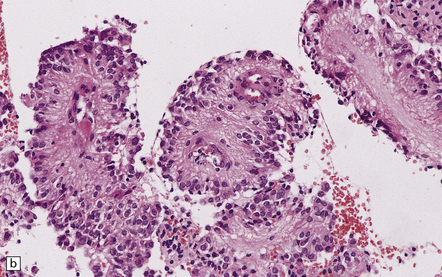
36.16 Papillary ependymoma.
(a) These broad papillae contain nuclei with a centrifugal distribution, and fibrillary processes characterize their vascular cores. (b) This neoplasm showed many papillary structures that, in cross section, consisted of pseudorosettes surrounded by uniform cells. Note the absence of a basement membrane beneath the circumferentially arranged cells. This pattern must not be confused with the artefactual pseudopapillae sometimes produced in sections of classic ependymoma.
Clear cell ependymoma may be confused with an oligodendroglioma, but the presence of pseudorosettes should declare its identity (Fig. 36.17). Some ependymomas are composed largely of elongated cells with an astrocytic morphology and have been referred to as tanycytic ependymomas (Fig. 36.18).
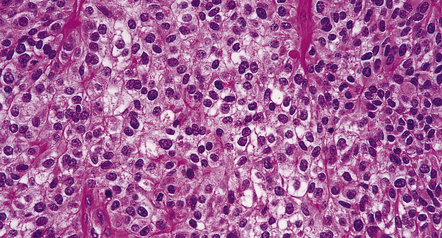
36.17 Clear cell ependymoma.
This may mimic an oligodendroglioma or a neurocytoma. Round cells with cytoplasmic clearing may be widespread in some ependymomas. Site and the presence of pseudorosettes help to distinguish this variant from oligodendroglioma, and ependymomas are not immunoreactive for synaptophysin, like the neurocytoma.
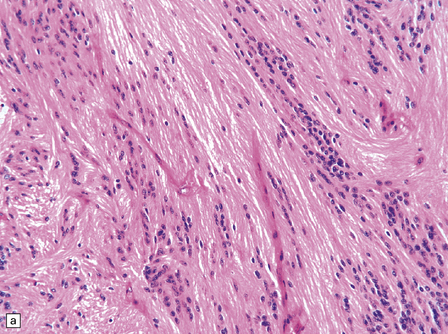
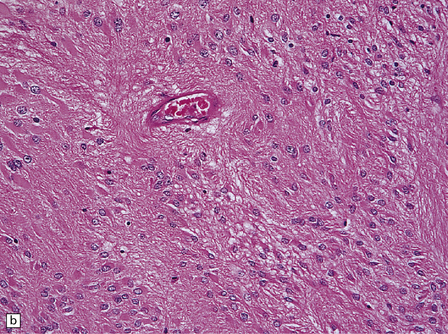
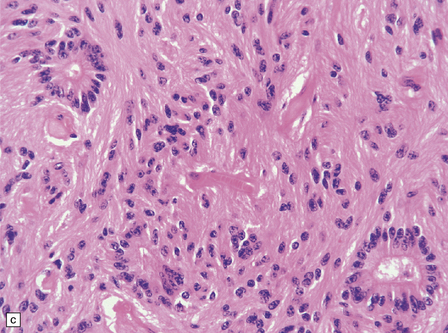
36.18 Tanycytic ependymoma.
(a) This variant is characterized by elongated cells with delicate cytoplasmic processes. It resembles a pilocytic or fibrillary astrocytoma, but its true identity is revealed by the presence of pseudorosettes (b) and occasional true rosettes (c).
Criteria for the diagnosis of anaplastic ependymoma are rather subjective. Necrosis and capillary endothelial proliferation may be widespread in anaplastic ependymomas (Fig. 36.19). The WHO classification emphasizes atypical cytologic features (i.e. nuclear pleomorphism), a high cell density, and plentiful mitoses in its diagnosis. It is important to note that no histologic feature of ependymomas correlates reliably with prognosis.
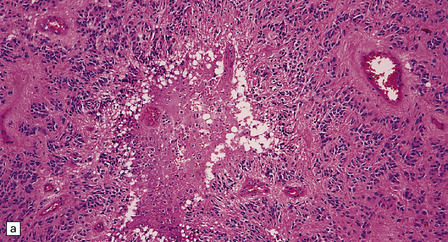
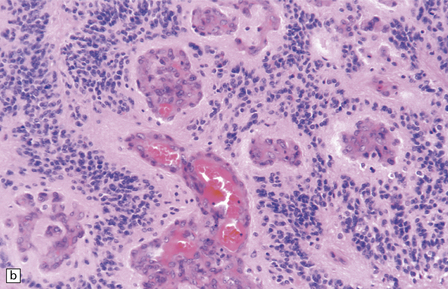
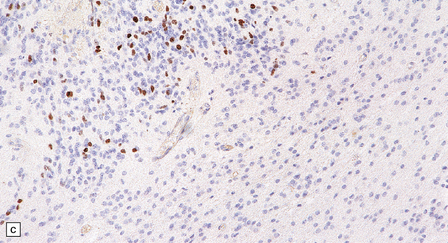
36.19 Anaplastic ependymoma.
(a) Multiple foci of necrosis and (b) capillary endothelial proliferation are features of anaplastic ependymomas. The WHO criteria for anaplastic ependymoma are met when these features accompany obvious cytologic abnormalities. (c) Immunohistochemistry with a Ki-67 antibody typically shows a high labeling index in areas with a high nuclear:cytoplasmic ratio. Ki-67 labeling index in ependymomas has shown a significant positive correlation with prognosis in several studies.
Myxopapillary ependymomas are characterized by abundant mucin, which envelops and separates clusters and fronds of tumor cells (Fig. 36.20). These tumors usually have monomorphic round nuclei, and may show either epithelioid or glial differentiation. Regions of glial differentiation contain cells with elongated fibrillary processes. Cells with an epithelioid morphophenotype usually form papillary structures with a vascular core. Evidence of previous hemorrhage may be found in the form of hemosiderin-laden macrophages. Intratumoral blood vessels tend to have thick hyalinized walls. Mitoses and foci of necrosis are rare.
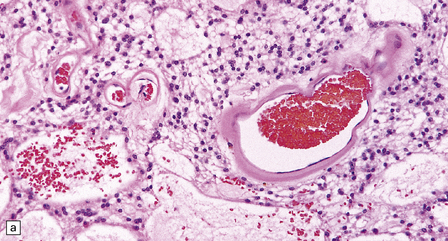
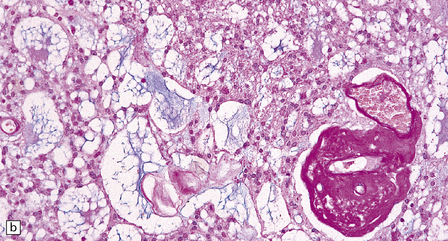
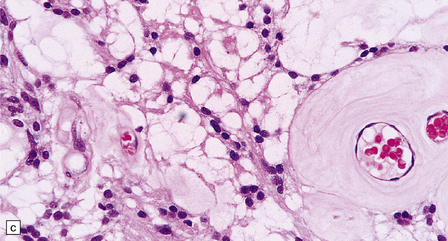
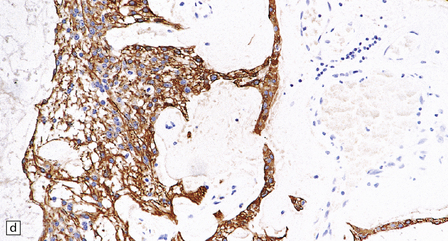
36.20 Myxopapillary ependymoma.
(a) and (b) Loosely arranged cells with delicate fibrillary cytoplasm surround pools of mucin in this myxopapillary ependymoma. (c) Blood vessels sometimes have markedly thickened walls. (d) The fibrillary cytoplasm is immunoreactive for GFAP.
Subependymomas are composed of cells with uniform, round or oval nuclei and generally scanty perinuclear cytoplasm. The cells are clustered in small, scattered groups and separated by broad bands of closely packed fibrillary processes. Occasional cells have more abundant, homogeneous, perinuclear cytoplasm. Microcysts are a common finding (Fig. 36.21). Mitoses are absent.
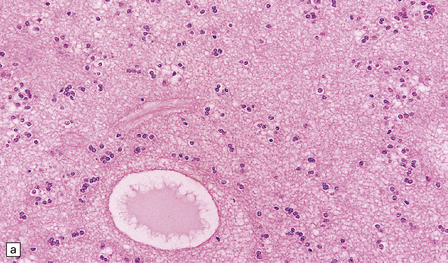
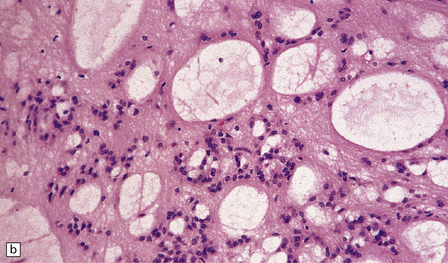
36.21 Subependymoma.
(a) Small groups of nuclei are surrounded by masses of fibrillary processes. (b) Clusters of microcysts in a subependymoma.
 GENETIC ASPECTS OF EPENDYMOMAS
GENETIC ASPECTS OF EPENDYMOMAS
 The genetic profiles of adult (mainly spinal cord) and childhood (mainly intracranial) ependymomas are distinct. This likely reflects distinct origins from site-specific progenitor or neural stem cells in spinal cord and embryonic cerebrum respectively. In particular, distinct populations of radial glia are thought to give rise to these tumors.
The genetic profiles of adult (mainly spinal cord) and childhood (mainly intracranial) ependymomas are distinct. This likely reflects distinct origins from site-specific progenitor or neural stem cells in spinal cord and embryonic cerebrum respectively. In particular, distinct populations of radial glia are thought to give rise to these tumors.





MIXED GLIOMAS
 DIFFERENTIAL DIAGNOSIS OF EPENDYMOMA
DIFFERENTIAL DIAGNOSIS OF EPENDYMOMA
 The presence of perivascular anuclear zones (pseudorosettes) with fine fibrillary GFAP-positive processes is the key to differentiating ependymomas from these neoplasms, which may be confused with ependymoma if biopsied tissue is limited. In these neoplasms, either pseudorosettes do not occur or fibrillary perivascular structures are immunoreactive for synaptophysin.
The presence of perivascular anuclear zones (pseudorosettes) with fine fibrillary GFAP-positive processes is the key to differentiating ependymomas from these neoplasms, which may be confused with ependymoma if biopsied tissue is limited. In these neoplasms, either pseudorosettes do not occur or fibrillary perivascular structures are immunoreactive for synaptophysin.







MACROSCOPIC AND MICROSCOPIC APPEARANCES
Oligoastrocytoma and anaplastic oligoastrocytoma
Classifications have invoked the diagnosis of oligoastrocytoma in two circumstances (Fig. 36.22): when a glioma consists of two regionally and cytologically distinct populations of cells, one with a clear astrocytic morphophenotype, and the other with features of an oligodendroglioma, and when a glioma consists of cells that exhibit cytologic features that appear to combine astrocytic and oligodendroglial phenotypes. The latter is uncommon. Oligoastrocytomas may contain a small number of mitoses, but if clinical progression is associated with increased cytologic pleomorphism, frequent mitoses, and microvascular proliferation, the term anaplastic oligoastrocytoma is appropriate (Table 36.2). When a tumor has features of an anaplastic oligoastrocytoma but also contains necrosis, the current WHO grading scheme considers such neoplasms as glioblastoma with an oligodendroglial component. The prognosis of these high-grade mixed gliomas with necrosis (glioblastoma with oligodendroglial component) is worse than that of anaplastic mixed gliomas without necrosis, but superior to that of the typical glioblastoma.

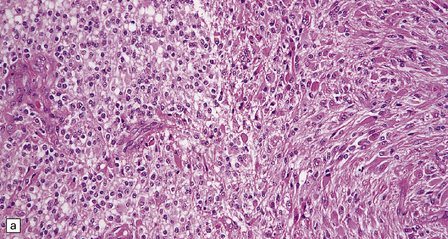
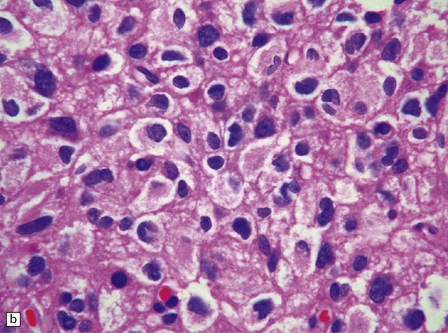
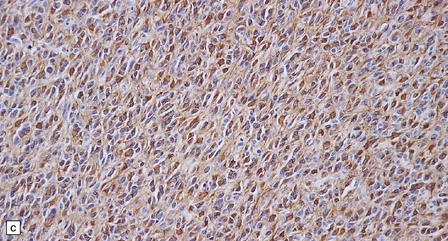
36.22 Mixed glioma/oligoastrocytoma.
(a) Oligodendroglial differentiation and distinct astrocytic differentiation are seen in one neoplasm. (b) Very occasionally a mixed glioma will demonstrate a morphophenotype that combines the features of astrocytic and oligodendroglial cells. (c) Immunoreactivity for GFAP is usually evident, but the ubiquitous pattern shown here should be distinguished from the circumscribed dense cytoplasmic reactivity of oligodendroglioma cells that show minigemistocytic differentiation.
REFERENCES
Bromberg, J.E., van den Bent, M.J. Oligodendrogliomas: molecular biology and treatment. Oncologist.. 2009;14:155–163.
Gilbert, M.R., Ruda, R., Soffietti, R. Ependymomas in adults. Curr Neurol Neurosci Rep.. 2010;10:240–247.
Jansen, M., Yip, S., Louis, D.N. Molecular pathology in adult gliomas: diagnostic, prognostic, and predictive markers. Lancet Neurol.. 2010;9:717–726.
Kilday, J.P., Rahman, R., Dyer, S., et al. Pediatric ependymoma: biological perspectives. Mol Cancer Res.. 2009;7:765–786.
Louis, D.N., Ohgaki, H., Wiestler, O.D., et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol (Berl).. 2007;114:97–109.
Aldape, K., Burger, P.C., Perry, A. Clinicopathologic aspects of 1p/19q loss and the diagnosis of oligodendroglioma. Arch Pathol Lab Med.. 2007;131:242–251.
Ducray, F., Idbaih, A., de Reyniès, A., et al. Anaplastic oligodendrogliomas with 1p19q codeletion have a proneural gene expression profile. Mol Cancer.. 2008;7:41.
Giannini, C., Burger, P.C., Berkey, B.A., et al. Anaplastic oligodendroglial tumors: refining the correlation among histopathology, 1p 19q deletion and clinical outcome in Intergroup Radiation Therapy Oncology Group Trial 9402. Brain Pathol.. 2008;18:360–369.
Griffin, C.A., Burger, P., Morsberger, L., et al. Identification of der(1;19)(q10;p10) in five oligodendrogliomas suggests mechanism of concurrent 1p and 19q loss. J Neuropathol Exp Neurol.. 2006;65:988–994.
Godfraind, C., Kaczmarska, J.M., Kocak, M., et al. Distinct disease-risk groups in pediatric supratentorial and posterior fossa ependymomas. Acta neuropathologica.. 2012;124:247–257.
Jenkins, R.B., Blair, H., Ballman, K.V., et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res.. 2006;66:9852–9861.
Persson, A.I., Petritsch, C., Swartling, F.J., et al. Non-stem cell origin for oligodendroglioma. Cancer Cell.. 2010;18:669–682.
Raghavan, R., Balani, J., Perry, A., et al. Pediatric oligodendrogliomas: a study of molecular alterations on 1p and 19q using fluorescence in situ hybridization. J Neuropathol Exp Neurol.. 2003;62:530–537.
van den Bent, M.J., Dubbink, H.J., Sanson, M., et al. MGMT promoter methylation is prognostic but not predictive for outcome to adjuvant PCV chemotherapy in anaplastic oligodendroglial tumors: a report from EORTC Brain Tumor Group Study 26951. J Clin Oncol.. 2009;27:5881–5886.
Watanabe, T., Nobusawa, S., Kleihues, P., et al. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol.. 2009;174:1149–1153.
Weller, M., Berger, H., Hartmann, C., et al. Combined 1p/19q loss in oligodendroglial tumors: predictive or prognostic biomarker? Clin Cancer Res.. 2007;13:6933–6937.
Hegi, M.E., Janzer, R.C., Lambiv, W.L., et al. Presence of an oligodendroglioma-like component in newly diagnosed glioblastoma identifies a pathogenetically heterogeneous subgroup and lacks prognostic value: central pathology review of the EORTC_26981/NCIC_CE.3 trial. Acta neuropathologica.. 2012;123:841–852.
Johnson, R.A., Wright, K.D., Poppleton, H., et al. Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nature.. 2010;466:632–636.
Korshunov, A., Witt, H., Hielscher, T., et al. Molecular staging of intracranial ependymoma in children and adults. J Clin Oncol.. 2010;28:3182–3190.
Mack, S.C., Taylor, M.D. The genetic and epigenetic basis of ependymoma. Childs Nerv Syst.. 2009;25:1195–1201.
Metellus, P., Guyotat, J., Chinot, O., et al. Adult intracranial WHO grade II ependymomas: long-term outcome and prognostic factor analysis in a series of 114 patients. Neuro-oncol.. 2010;12:976–984.
Zacharoulis, S., Moreno, L. Ependymoma: an update. J Child Neurol.. 2009;24:1431–1438.
Von Deimling, A., Reifenberger, G., Kros, J.M., et al. Oligoastrocytoma. In: Louis D.N., Ohgaki H., Wiestler O.D., et al, eds. WHO classification of tumours of the central nervous system. Lyon: IARC Press; 2007:63–65.
Von Deimling, A., Reifenberger, G., Kros, J.M., et al. Anaplastic oligoastrocytoma. In: Louis D.N., Ohgaki H., Wiestler O.D., et al, eds. WHO classification of tumours of the central nervous system. Lyon: IARC Press; 2007:66–67.