Chapter 82 Non-antiarrhythmic Therapies for Cardiac Arrhythmias
Background
Heart disease in any form may predispose the patient to the development of ventricular arrhythmias, some of which are lethal and may lead to sudden cardiac death (SCD). As a major public health burden, SCD has an incidence of nearly a half-million cases every year in the United States.1 Considerable effort has been invested in devising therapies specifically aimed at reducing the incidence of ventricular arrhythmias, particularly in the setting of ischemic heart disease and heart failure, where these fatal arrhythmias are exceedingly common. To date, however, the preponderance of evidence shows that the strategy of using antiarrhythmic drug therapy to directly suppress arrhythmias fails to improve morbidity or mortality outcomes. In ischemic heart disease, the Cardiac Arrhythmia Suppression Trial (CAST), the Canadian Amiodarone Myocardial Infarction Arrhythmia Trial (CAMIAT), and the European Myocardial Infarct Amiodarone Trial (EMIAT) found that antiarrhythmic therapy either increased mortality or failed to reduce it.2–4 A similar observation was noted in the Survival Trial of Antiarrhythmic Therapy in Congestive Heart Failure (CHF-STAT) and the Antiarrhythmic Trial with Dronedarone in Moderate to Severe Congestive Heart Failure Evaluating Morbidity Decrease (ANDROMEDA) in the setting of heart failure.5,6 The use of an implantable cardioverter-defibrillator (ICD), a form of nonpharmaceutical antiarrhythmic therapy, has been shown to be beneficial in the long term but has failed to improve survival in the early postmyocardial infarction period when the incidence of SCD is highest.7
Growing evidence, however, suggests that certain therapies that are not used primarily for antiarrhythmic intent can actually be effective in preventing the development of ventricular arrhythmias, reducing their recurrence, and decreasing SCD incidence. Among these so-called non-antiarrhythmic therapies are β-adrenergic blocking agents, calcium channel blockers (CCBs), statins, fish oil, and renin-angiotensin-aldosterone system (RAAS) antagonists, and others (Figure 82-1). These agents are thought to target the underlying disease processes and reverse the basic pathophysiological substrates that promote arrhythmogenesis; this is expected to result in long-term cardiac electrophysiological stabilization.
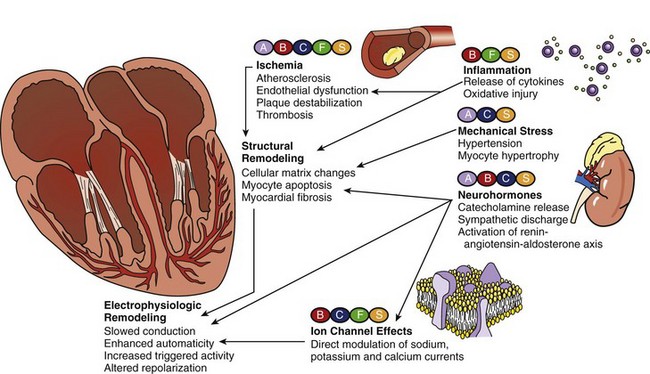
Sudden Cardiac Death and Ventricular Arrhythmias
SCD is the unexpected natural death from a cardiac cause a short time after onset of symptoms, defined epidemiologically as any cardiac death occurring out of the hospital or taking place in the emergency room. It accounts for 63% of all cardiovascular deaths, and 84% of SCDs are caused by ventricular arrhythmias, often starting as ventricular tachycardia, rapidly progressing to ventricular fibrillation.8,9 Although not all instances of SCD are caused by arrhythmias, it is used by most investigations on cardiovascular outcomes as a surrogate marker for lethal ventricular arrhythmias. Crude and imprecise as it may be, SCD incidence still serves as an important indicator that helps measure the clinical effects of various antiarrhythmic therapies.
β-Blockers
The role of β-adrenergic blocking agents in the prevention of ventricular arrhythmia and SCD is well established, especially in the setting of ischemic heart disease and heart failure. Early observational studies showed that the β-blockers timolol, metoprolol, and propranolol reduced the incidence of SCD by 30% to 45% after myocardial infarction. In a large randomized study, the Clopidogrel and Metoprolol in Myocardial Infarction Trial: The Second Chinese Cardiovascular Study (COMMIT-CCS2) found a 17% relative risk reduction in the incidence of ventricular fibrillation with the early use of metoprolol in patients with myocardial infarction.10 The Carvedilol Post-Infarct Survival Control in Left Ventricular Dysfunction (CAPRICORN) trial involving patients with myocardial infarction and heart failure also revealed that carvedilol was associated with a 77% reduction in the risk of malignant ventricular arrhythmias as well as a 23% reduction in overall mortality. In patients with heart failure, β-blockers were found to have a more pronounced and more consistent effect in reducing SCD than any other therapy, including angiotensin-converting enzyme (ACE) inhibitors. The Cardiac Insufficiency Bisoprolol Study II (CIBIS II) demonstrated that bisoprolol reduced SCD by 44% and total mortality by 34% in patients with severe symptomatic systolic heart failure. Similarly, β-blockers reduced SCD in heart failure by 43% in the Metoprolol CR/XL Randomized Intervention Trial in Chronic Heart Failure (MERIT-HF), in which long-acting metoprolol was used, and by 36% in the Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) trial, in which carvedilol was used.11 Some agents may have greater antiarrhythmic properties than others, as seen in the Carvedilol Or Metoprolol European Trial (COMET), where carvedilol reduced SCD incidence more than did metoprolol in patients with myocardial infarction.12
In ischemic heart disease, β-blockers reduce myocardial oxygen demand, optimize diastolic filling, and improve coronary blood flow as a result of their negative chronotropic and inotropic effects. Areas of ischemic myocardium are particularly prone to arrhythmogenesis, and the suppression of ventricular arrhythmias by β-blockers may be partly attributed to their anti-ischemic properties. Myocardial infarction induces a state of heightened catecholamine discharge and autonomic dysfunction, both of which lower the threshold for ventricular fibrillation. Antagonism of this high cardiac sympathetic tone may also explain the protective effect of β-blockers against SCD. Antiproliferative and antiatherogenic mechanisms have also been proposed for some agents. Studies on animal models of myocardial ischemia further suggest that certain β-blockers may have sodium channel–blocking actions that could stabilize the myocyte membrane and suppress ventricular arrhythmias, similar to class I antiarrhythmic agents.13 β-Blockers may even induce electrophysiological changes in action potential by blocking the L-type calcium current, the transient outward potassium current, and the rectifier potassium current, resembling the class III antiarrhythmic actions of amiodarone. Experimental models also postulate that specific β-blockers such as carvedilol could exert significant antioxidant activity that protects the myocardium from reperfusion arrhythmias through adenosine-mediated processes.14
Statins
In patients with atherosclerotic heart disease enrolled in the Antiarrhythmics versus Implantable Defibrillators (AVID) trial, lipid-lowering therapy was associated with a 40% relative risk reduction in ventricular tachyarrhythmias and cardiac mortality.15 This significant benefit was expressed early in follow-up and is unlikely to be from the effects on cholesterol levels. In the Multicenter Automatic Defibrillator Implantation Trial (MADIT)-II involving patients with coronary artery disease and heart failure, statin use was associated with risk reductions of 28% for ventricular arrhythmias and 35% for SCD, with no significant effect on non-SCD.16 In the setting of nonischemic cardiomyopathy, an analysis of the Defibrillators in Non-ischemic Cardiomyopathy Treatment Evaluation (DEFINITE) trial showed that statin therapy was associated with an 86% reduction in arrhythmic sudden death, 22% reduction in ICD shocks from ventricular arrhythmias, and 78% reduction in mortality.17 A meta-analysis of randomized controlled trials on pravastatin, simvastatin, atorvastatin, and fluvastatin showed that statin treatment decreased the risk of SCD by 19%.18 The findings of these observational studies were further supported by small randomized trials in which patients with advanced heart failure treated with statins had significantly lower rates of SCD and all-cause mortality but not of pump failure.19
Statins have pleiotrophic effects that go beyond lipid lowering, including anti-inflammatory, anti-proliferative, anti-oxidative, and anti-thrombotic properties, which may as well explain their apparent antiarrhythmic effects. Myocardial ischemia is a potent trigger for lethal ventricular arrhythmias, and both atorvastatin and pravastatin have been shown to significantly reduce myocardial ischemia in the Study Assessing Goals in the Elderly (SAGE) and the Regression Growth Evaluation Statin Study (REGRESS).20,21 Proposed mechanisms include inhibition of luminal narrowing, restoration of endothelial function, plaque stabilization, anti-inflammatory effects, as well as modification of hemostatic processes that ultimately lead to improvement in myocardial perfusion and amelioration of ischemia-driven reperfusion injury. Ischemic heart disease is associated with cardiac electrophysiological remodeling leading to the formation of anatomic barriers, regions of slow conduction, and prolonged repolarization. In animal models of myocardial infarction and heart failure, successful inhibition of myocardial remodeling has been demonstrated with simvastatin and fluvastatin.22,23
Aside from their anti-ischemic properties, statins may also exert antiarrhythmic effects through various structural, neurohormonal, and electrophysiological mechanisms. The bilayer phospholipid structure of the membranes, as well as its functions, can be altered by changes in the dietary distribution of fatty acids.24 At least in theory, it may be possible for statins to exert direct antiarrhythmic effects by changing the lipid environment of the myocyte cell or organelle membranes, which results in altered channel protein function and ionic currents. Cardiomyocyte hypertrophy usually follows myocardial infarction and contributes to ventricular remodeling that, in turn, predisposes to ventricular arrhythmias. Pravastatin has been shown to decrease such hypertrophy in animal studies, probably by decreasing the secretion of endothelin-1. Dysfunctional intramyocardial calcium handling results in ventricular action potential abnormalities. By inhibiting intracellular calcium mobilization and transmembrane currents, statin therapy could stabilize ventricular repolarization and decrease the risk of arrhythmia.25 Overexposure of the myocardium to cathecholamines, in addition to worsening ischemia, also directly stimulates arrhythmogenesis. In experimental models, lipid-lowering therapy has been shown to normalize autonomic function and decrease sympathetic tone, as well as increase the cardiac response to parasympathetic stimulation.26,27 Diminished heart rate variability, an electrophysiological marker of endothelial dysfunction, is a known predictor of susceptibility to arrhythmias and SCD. Rosuvastatin and simvastatin have been shown in animal models to improve heart rate variability, likely through the modulation of nitric oxide synthase function.28 Increased QT variability, another marker of vulnerability to lethal arrhythmias, has been shown to improve with atorvastatin and fluvastatin therapy.29
Despite its well-established beneficial effect on cardiovascular mortality and evidence of reduction of SCD, the antiarrhythmic role of statins is still under debate because of conflicting reports. In the setting of myocardial ischemia, high-dose atorvastatin did not reduce the rates of resuscitated cardiac arrest in the Incremental Decrease in End Points Through Aggressive Lipid Lowering (IDEAL) and Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL) trials.30,31 In patients with stable coronary artery disease, statin therapy similarly did not reduce the incidence of resuscitated cardiac arrest with the use of atorvastatin in the Treat to New Targets (TNT) trial and simvastatin in the Scandinavian Simvastatin Survival Study (4S).32,33 In fact, the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT) showed a trend toward a higher incidence of malignant arrhythmias with atorvastatin in high-risk hypertensive patients.34 The small number of patients who presented with life-threatening arrhythmias in these negative studies may explain disparities in outcomes, but large randomized trials that address the issue directly are still lacking. For now, statin use for prevention of ventricular arrhythmias seems only to benefit patients who had acute myocardial infarction, those who underwent revascularization procedures, and those who received ICDs.
Fish Oil
Epidemiologic data, including that of the U.S. Physicians Health Study, indicate that ingestion of very-long-chain n-3 polyunsaturated fatty acids (n-3 PUFA), found in fish or fish oil, is associated with reduced cardiovascular mortality.35 This was further reinforced by clinical investigations, including the Diet and Reinfarction Trial, which showed that consumption of fatty fish was associated with a 30% reduction in all-cause mortality.36 This was largely thought to result from the stabilization of the atherosclerotic plaque by n-3 PUFA through its lipid-lowering, anti-thrombotic, and anti-inflammatory properties. The GISSI-Prevenzione trial found a 45% reduction in SCD in patients with previous malignant ventricular arrhythmias who consumed 1 g of n-3 PUFA daily.37 In this trial, the beneficial effect of n-3 PUFA was thought to be unrelated to its effects on the lipid profile because the reduction in SCD appeared very early, and no effect was observed on the rate of myocardial infarction. This finding was reaffirmed in the Cardiovascular Health Study, which noted that frequent fish consumption was associated with a 58% reduction in arrhythmic death without changing the risk of myocardial infarction.38 This led to the theory that n-3 PUFA acids may have antiarrhythmic and antifibrillatory properties that are independent of their antiatherosclerotic and anti-thrombotic effects.
Such hypothesis is also supported by animal and experimental evidence. In an animal model of ischemia-induced SCD, infusion of n-3 PUFA emulsion prior to coronary artery ligation prevented ventricular fibrillation by 75%.39 Long-term feeding studies in primate models showed that n-3 PUFA supplementation and successful incorporation of the fatty acids into the myocardial membrane significantly reduced myocardial vulnerability to arrhythmic stimuli induced by ischemia.40 Dietary n-3 PUFA also reduced drug-induced repolarization abnormalities and abolished ventricular arrhythmias in a rabbit model.41 These cardioprotective effects have been attributed specifically to eicosapentaenoic and docosahexaenoic acids, which make up the majority of n-3 PUFA in fish-derived fats.
The consistent reduction of cardiac mortality and SCD with n-3 PUFA in observational human studies and experimental investigations has brought to light a major role of fish oil in cardiovascular therapy. However, randomized controlled trials have so far failed to demonstrate a clinically relevant antiarrhythmic effect. The Fatty Acid Antiarrhythmia Trial randomized patients at high risk for fatal ventricular arrhythmias to supplementation with either fish oil or placebo and found a trend toward reduced risk of arrhythmic events with n-3 PUFA, but this did not reach statistical significance.42 A multicenter, double-blind study in the United States that randomized patients with recent malignant ventricular arrhythmias to fish oil or placebo showed that n-3 PUFA supplementation did not prevent episodes of ventricular tachycardia or fibrillation.43 In fact, a trend toward increased ventricular arrhythmia occurrence was seen, which suggests a proarrhythmic effect of fish oil. The Study on Omega-3 Fatty Acids and Ventricular Arrhythmia (SOFA), a similar multicenter, randomized, placebo-controlled study conducted in Europe, also found no evidence of a strong protective effect of n-3 PUFA against ventricular arrhythmia.44 It did, however, establish that fish oil treatment does not cause harmful proarrhythmic effects. Meta-analysis of randomized trials involving nearly 33,000 patients found that fish oil supplementation was associated with a significant (20%) reduction in cardiac death, but it had no effect on arrhythmias or overall mortality.45 Proposed explanations for the inconsistent data in clinical trials include differences in study protocols, lack of standardized dosing, and inconsistencies in duration of supplementation, among a host of other confounders. Since most of the benefits were seen in the setting of early ischemic heart disease, it is conceivable that the antiarrhythmic utility of n-3 PUFA may be confined only to prevention of ventricular arrhythmia in patients with recent myocardial infarction, when the amount of scar tissue is still limited.
Renin-Angiotensin-Aldosterone System Antagonists
The benefit of ACE inhibitors in reducing cardiovascular and overall mortality in the setting of heart failure and ischemic heart disease is firmly established. A favorable effect on SCD was also noted, suggesting possible antiarrhythmic properties of ACE inhibitors. In patients with chronic heart failure, the Vasodilator Heart Failure Trial II (V-HEFT-I) demonstrated that enalapril reduced ventricular arrhythmias by 27% and SCD by 52%.46 Post hoc analyses of the Survival and Ventricular Enlargement (SAVE) trial and the Studies of Left Ventricular Dysfunction (SOLVD) trials also showed a trend toward reduced arrhythmic events and SCD, albeit not statistically significant.47,48 In patients with acute myocardial infarction, however, the Acute Infarction Ramipril Efficacy (AIRE) trial found that ramipril not only reduced total mortality but also decreased the incidence of SCD by 30%.49 Even in patients without heart failure, the Heart Outcomes Prevention Evaluation (HOPE) trial demonstrated that ramipril was able to cut arrhythmic death by 21%.50 Analyses of the Chinese Cardiac Study (CCS-1), the Cooperative New Scandinavian Enalapril Survival Study (CONSENSUS)-II, the Fourth International Study of Infarct Survival (ISIS-4), and the Survival of Myocardial Infarction Long-Term Evaluation (SMILE) trial also showed a trend toward reduced arrhythmic events and SCD, but none achieved statistical significance.51–54
Depressed left ventricle (LV) systolic function and ventricular dilatation are the most important predictors for developing ventricular arrhythmias. ACE inhibitors are thought to reduce SCD primarily because of their beneficial effect in reversing LV remodeling and improving systolic function. Electrophysiological remodeling also accompanies these myocardial fibrosis and structural changes, leading to slow conduction, early after-depolarization (EAD), and other proarrhythmic repolarization abnormalities. ACE inhibitors have been shown to reduce the degree of ventricular dispersion of repolarization by maintaining myocyte sodium and pH homeostasis, acting analogously as a class I antiarrhythmic agent.55 ACE inhibitors are also hypothesized to exert antiarrhythmic effects through antiatherosclerotic, anti-thrombotic, and neurohormonal mechanisms. Stabilization and regression of atherosclerotic plaque has been shown to be associated with ACE inhibitor therapy.56 Angiotensin-II is a vasoactive peptide that can activate inflammatory cytokines, promote ultrastructural myocardial fibrosis, modulate myocyte ion channels, induce release of endogenous cathecholamines, decrease gap junction conduction, and increase transmural dispersion of refractoriness in the ventricles.57 These effects substantially increase the propensity for arrhythmogenesis and SCD but can be effectively attenuated with ACE inhibitor or angiotensin receptor blocker (ARB) therapy.
Although observational and small prospective randomized studies point toward a modest reduction in ventricular arrhythmias, strong evidence to confirm the long-term antiarrhythmic benefit of ACE inhibitors is currently lacking.58 It was postulated that ACE inhibitors only have a modest effect on ventricular arrhythmias and SCD because of incomplete blockade of angiotensin II. In the Evaluation of Losartan in the Elderly (ELITE) study comparing the ARB losartan and the ACE inhibitor captopril in patients with heart failure, a 46% risk reduction in total mortality was seen with losartan compared with captopril, primarily because of a 64% risk reduction in SCD.59 This difference in SCD and mortality outcomes, however, was no longer seen in the subsequent ELITE-II trial. In the Optimal Trial in Myocardial Infarction with Angiotensin II Antagonist Losartan (OPTIMAAL) of patients with acute myocardial infarction, losartan was associated with a 19% risk reduction in SCD compared with captopril, although there were fewer cardiovascular deaths with captopril.60 ARBs were thought to exert a more complete inhibition of angiotensin II, especially at the tissue level, and this may explain its apparent advantage over ACE inhibitors. The Candesartan in Heart Failure Assessment of Reduction in Mortality and Morbidity (CHARM) trial showed that the ARB candesartan, when added to standard heart failure therapy that includes ACE inhibitors (with or without β-blockers and aldosterone-antagonists), improves mortality and morbidity outcomes, perhaps because of a more comprehensive inhibition of the RAAS.61 Like ACE inhibitors, regression of left ventricle hypertrophy (LVH) is seen with ARB therapy, which can explain a large part of its perceived antiarrhythmic effects. The Losartan Intervention For Endpoint Reduction in Hypertension (LIFE) trial found that regression of LVH with losartan was associated with a 30% reduction in SCD, independent of blood pressure reduction and other cardiac risk factors.62
Aldosterone can induce arrhythmogenesis through mechanisms similar to that of angiotensin-II, including detrimental effects on LV structural and electrophysiological remodeling, myocardial fibrosis, endothelial dysfunction, platelet activation, and sympathetic tone. In patients with chronic heart failure, the aldosterone antagonist spironolactone was associated with a significant reduction in SCD by 29% and total mortality by 30% in the Randomized Aldactone Evaluation Study (RALES).63 In patients with myocardial infarction, the Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS) found a 21% reduction in SCD and 15% reduction in overall mortality with the selective aldosterone blocker eplerenone.64 Eplerenone reduced SCD by 37% within 30 days when given early (7 days average) after myocardial infarction.65 Concerns have been expressed, however, regarding the safety of aldosterone antagonist therapy because of significant increase in hyperkalemia-related morbidity after publication of the RALES study.66
Nondihydropyridine Calcium Channel Blockers
Analyses of landmark studies in hypertension indicated a favorable effect of nondihydropyridine CCBs on SCD, suggesting possible significant antiarrhythmic properties. In the Systolic Hypertension in Europe Trial, successful antihypertensive therapy using nitrendipine, with the possible addition of enalapril or hydrochlorothiazide, significantly reduced SCD by 26% and total mortality by 24%.67 The Avoiding Cardiovascular Events in Combination Therapy in Patients Living with Systolic Hypertension (ACCOMPLISH) trial of hypertensive patients found that the addition of amlodipine to ACE inhibitor therapy increased the rate of resuscitation after sudden cardiac arrest by 75% and reduced cardiac mortality by 20% when compared with combination ACE inhibitor-thiazide therapy.68 In high-risk hypertensive patients with diabetes and coronary artery disease, long-acting nifedipine was found to be as effective as ACE inhibitors in reducing cardiovascular events, with a trend toward better SCD outcomes.69 In the setting of chronic heart failure, the Prospective Randomized Amlodipine Survival Evaluation (PRAISE) trial showed that the addition of amlodipine to standard heart failure therapy was associated with a 21% reduction in sudden cardiac death, which was much higher than the reduction in other modes of cardiac death, pointing to a possible antiarrhythmic mechanism for this particular CCB.70
Nondihydropyridine CCBs are known to have antianginal and anti-ischemic effects, and this might partially explain some of their antiarrhythmic properties. In animal models of myocardial ischemia, nondihydropyridine CCBs were shown to reduce ventricular arrhythmias from reperfusion injury, possibly related to their attenuation of cardiac sympathetic nerve activity.71 Subgroup analysis of the PRAISE trial, however, found a 38% reduction in SCD with amlodipine in patients with heart failure caused by nonischemic etiologies, and it appears to have no effect on cause-specific mortality in ischemic cardiomyopathy. The apparent antiarrhythmic properties of nondihydropyridine CCBs may just be a reflection of their beneficial effects on regression of LV hypertrophy and remodeling, which are independent predictors of SCD.72 Hypertension, in itself, is associated with development of complex ventricular arrhythmias; thus control of hypertension with effective agents, including CCBs, is expected to have a favorable impact on SCD.73
Other Therapies
Ranozaline, a piperazine, is the first among a novel class of agents that exert antianginal effects by enhancing myocardial relaxation and reducing diastolic dysfunction. In addition to its anti-ischemic effects, ranozaline therapy was also shown by the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction (MERLIN-TIMI)-36 trial to be associated with a significant 36% reduction in ventricular arrhythmias in patients with acute coronary syndrome.74 Its anti-ischemic properties could be attributed to the inhibition of the late phase of the inward sodium current, which is responsible for cytosolic calcium accumulation that ultimately leads to impaired myocardial mechanical relaxation. The increased concentration of intracellular calcium also produces electrical instability and repolarization abnormalities that promote ventricular arrhythmogenesis, a process which ranozaline can potentially modulate. In experimental models, ranozaline was shown to induce ion channel alterations similar to that of amiodarone, leading to suppression of EADs and reduction in spontaneous ventricular arrhythmias.75 In spite of its favorable effects on ventricular arrhythmias, however, ranozaline has not yet been shown to reduce mortality or major adverse cardiovascular events, and data on SCD are lacking.
Practice Guidelines
The current guidelines on management of ventricular arrhythmias and prevention of SCD, published jointly by the American College of Cardiology (ACC), American Heart Association (AHA), and European Society of Cardiology (ESC), consider β-blockers to be the mainstay of antiarrhythmic therapy because of their safety and proven efficacy.76 The current guidelines also recognize the antiarrhythmic properties of statins and fish oil, and their potential role in SCD prevention. However, no definite recommendations were made on their utility because of conflicting clinical data on their effectiveness.
Non-antiarrhythmic Therapy of Atrial Fibrillation
The concept of AF cardioversion and maintaining patients on sinus rhythm is appealing, given the theoretical benefit on improved hemodynamics. However, multiple landmark trials in AF (including the AFFIRM, PIAF, RACE, STAF, and HOT-CAFÉ studies) repeatedly failed to demonstrate any long-term advantage of a rhythm-control strategy over the rate-control approach.77 Even in the setting of heart failure, where antiarrhythmic therapy was thought to have the greatest impact, the Atrial Fibrillation and Congestive Heart Failure (AF-CHF) trial found no clear evidence to recommend a routine rhythm-control approach.78 In fact, some of these studies even point toward worse morbidity outcomes with the use of pharmacologic antiarrhythmic agents.
β-Blockers in Atrial Fibrillation
The utility of β-adrenergic blocking agents for rate control in AF is well established. However, their role in preventing the development of AF and in maintaining sinus rhythm is less well defined. Such a role has been most documented in the setting of cardiac surgery as prophylaxis against postoperative AF. Metoprolol was associated with a 20% reduction in postoperative AF after elective cardiac surgery, while carvedilol was associated with a 15% lower incidence of AF after coronary artery bypass grafting.79,80 The excessive adrenergic stimulation wrought by cardiac surgery is thought to promote postoperative atrial arrhythmogenesis, a process β-blockers could effectively attenuate. Pericardial inflammation and oxidative injury also are known triggers for postoperative AF, and these can be modulated by the antioxidant actions of certain β-blockers, carvedilol in particular.81 In patients with heart failure, in addition to their known benefits on mortality and morbidity rates, the use of β-blockers was associated with 27% risk reduction in AF incidence.82
Small randomized controlled trials found that patients with AF taking metoprolol had significantly lower relapse rates compared with those on placebo after successful cardioversion and repeated cardioversion to sinus rhythm. However, nearly half of the subjects on this β-blocker still had AF relapse after 6 months of follow-up.83,84 Some evidence suggests that β-blockers only have significant antiatrial fibrillatory effects in those with hypertension.85 Even in hypertensive patients, however, the LIFE trial demonstrated that atenolol was inferior to losartan in preventing new-onset AF and stroke.86
Renin-Angiotensin-Aldosterone System Antagonists in Atrial Fibrillation
Retrospective analyses of large randomized trials point to a possible desirable effect of ACE inhibitors on AF pathogenesis. Trandolapril was associated with a 47% reduction in the incidence of new-onset AF in patients with systolic dysfunction after myocardial infarction who were enrolled in the Trandolapril Cardiac Evaluation (TRACE) study.87 Similarly, in the SOLVD trial, enalapril was found to be the most powerful predictor for AF risk reduction in patients with systolic dysfunction, accounting for a 78% decrease in AF incidence compared with placebo.88
A similar observation was noted in the post hoc analyses of ARB trials. Losartan was associated with a 33% risk reduction in new-onset AF compared with atenolol in hypertensive patients enrolled in the LIFE study, which translated to lower adverse cardiac events and stroke rates. Hypertensive patients in the Valsartan Antihypertensive Long-Term Use Evaluation (VALUE) trial had a 32% reduction in the incidence of persistent AF when treated with amlodipine. Patients with heart failure in the Val-HEFT study who were treated with valsartan had a 37% reduction in AF incidence.89 A more modest benefit was seen in the CHARM trial, where candesartan was associated with an 18% reduction in AF incidence compared with placebo in patients with symptomatic heart failure.90
In small prospective trials, the addition of ACE inhibitors or ARBs to amiodarone resulted in lower AF recurrence rates after cardioversion compared with amiodarone treatment alone.91,92 Meta-analyses of RAAS antagonist trials suggest that both ACE inhibitors and ARBs appear effective in preventing AF but may only be limited to patients with LV dysfunction or to those undergoing cardioversion.93
More recent randomized trials, however, provide contradictory data on the antifibrillatory effect of RAAS antagonists. Analysis of the HOPE trial showed that the ACE inhibitor ramipril did not reduce the incidence of AF compared with placebo.94 Similarly, the Telmisartan Randomised Assessment Study in ACE-intolerant Subjects with Cardiovascular Disease (TRANSCEND) found no effect of the ARB telmisartan on the incidence of new-onset AF compared with placebo in patients with high-risk cardiovascular diseases.95 The Ongoing Telmisartan Alone and in Combination with Ramipril Global Endpoint Trial (ONTARGET) demonstrated a weak trend toward a lower incidence of new-onset AF with telmisartan compared with ramipril, as well as minimal reduction in new-onset AF with combination telmisartan-ramipril therapy compared with either treatment alone, but these differences were statistically insignificant.96
Lipid-Lowering Agents in Atrial Fibrillation
Evidence exists to support the claim that inflammation, marked by increased cross-reactive protein (CRP) levels, is involved in the arrhythmogenesis and perpetuation of AF.97 Statins have been shown to reduce CRP levels independent of lipid-lowering, and it is plausible that statins exert clinically significant antifibrillatory properties because of their anti-inflammatory and antioxidant effects.98 Atorvastatin, in particular, has been shown to decrease the recurrence of AF after cardioversion, in part because of reduction in CRP levels.99 The Atorvastatin for Reduction of Myocardial Dysrhythmia after Cardiac Surgery 3 (ARMYDA-3) trial also found that elevated postoperative CRP levels were associated with a twofold increase in the risk of developing AF after cardiac surgery and that atorvastatin therapy started 1 week prior to surgery was associated with a 61% reduction in postoperative AF.100 Analysis of the ADVANCENT Registry of patients with LV systolic dysfunction demonstrated that statin therapy was associated with a 31% reduction in the prevalence of AF, which is larger than that seen in β-blockers and ACE inhibitors.101 This effect on AF was independent of lipid profile, suggesting possible anti-inflammatory and antioxidant mechanisms. Furthermore, experimental data suggest a significant interaction between RAAS and cholesterol homeostasis. Oxidized low-density lipoprotein was shown to upregulate the expression of ACE and angiotensin II. This neurohormonal “cross-talk,” which appeared to be attenuated with simvastatin treatment, was thought to be implicated in AF pathogenesis.102
In addition to their beneficial effects on lipid profile, fish oils also exert anti-inflammatory properties, as evidenced by the reduction in CRP and cytokine levels.103 Because of this anti-inflammatory effect, n3-PUFA may have clinical utility in AF therapy, similar to that seen with statins. A small randomized trial found that n3-PUFA treatment was associated with a significant 54% reduction in the incidence of postoperative AF in patients who underwent coronary artery bypass grafting (CABG).104 Observational data also suggest that dietary consumption of broiled or baked fish was associated with lower incidence of AF in an almost linear fashion.105 This finding, however, was refuted by larger epidemiologic studies that could not show a beneficial effect of fish consumption on atrial arrhythmias.106,107
Other Non-antiarrhythmic Therapies in Atrial Fibrillation
Because inflammation occupies a central role in the pathogenesis of AF, direct suppression of inflammation using corticosteroids has been explored. Prednisone has been shown to prevent experimentally induced AF and atrial flutter in animal models.108,109 A small prospective study demonstrated that prophylactic administration of steroids was associated with reduced incidence of postoperative AF in patients undergoing CABG, but this was associated with higher complication rates.110 In another small series, low-dose methylprednisolone therapy was also shown to successfully cut the incidence of recurrent AF through reduction of CRP levels.111 However, analysis of the Rotterdam study found that high-dose corticosteroid therapy was actually a strong risk factor for the development of AF.112 Safety issues associated with steroid therapy currently limits its use in AF management.
The AF burden may be associated with increased atrial oxidative stress and peroxynitrite formation, processes that can be attenuated by vitamin C, which possesses antioxidant and anti-inflammatory properties.113 Some data suggest that vitamin C supplementation might reduce postoperative AF in patients undergoing CABG and also reduce the recurrence of AF after cardioversion.114,115 More reliable clinical evidence, however, is lacking at this time.
Non-antiarrhythmic Practice Guidelines in Atrial Fibrillation
The current guidelines on AF management, published jointly by the ACC, the AHA, and the ESC, describe β-blockers not only as rate-control agents (their primary role in AF) but also as possible first-line treatment in maintaining sinus rhythm in patients with myocardial infarction, heart failure, and hypertension.116 Although β-blockers may reduce subacute recurrences of AF after cardioversion, no further recommendations were made because they are unlikely to enhance the success of cardioversion or to suppress immediate or late recurrences of AF. The current guidelines also recognize the potential role of ACE inhibitors, ARBs, and statins in the long-term maintenance of sinus rhythm in high-risk patients; however, further clarification in randomized clinical trials is required before they can be routinely recommended.
Key References
Deedwania P, Stone PH, Bairey Merz CN, et al. Effects of intensive versus moderate lipid-lowering therapy on myocardial ischemia in older patients with coronary heart disease: Results of the Study Assessing Goals in the Elderly (SAGE). Circulation. 2007;115(6):700-707.
Ducharme A, Swedberg K, Pfeffer MA, et al. Prevention of atrial fibrillation in patients with symptomatic chronic heart failure by candesartan in the Candesartan in Heart failure: Assessment of Reduction in Mortality and morbidity (CHARM) program. Am Heart J. 2006;151(5):985-991.
Fletcher RD, Cintron GB, Johnson G, et al. Enalapril decreases prevalence of ventricular tachycardia in patients with chronic congestive heart failure. The V-HeFT II VA Cooperative Studies Group. Circulation. 1993;87(6 Suppl):VI49-V155.
Fuster V, Rydén LE, Cannom DS, et al. ACC/AHA/ESC 2006 Guidelines for the Management of Patients with Atrial Fibrillation: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Revise the 2001 Guidelines for the Management of Patients With Atrial Fibrillation): Developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation. 2006;114(7):e257-e354.
HOPE Investigators. Impact of ramipril on the incidence of atrial fibrillation: Results of the Heart Outcomes Prevention Evaluation study. Am Heart J. 2007;154(3):448-453.
Maggioni AP, Latini R, Carson PE, et al. Val-HeFT Investigators: Valsartan reduces the incidence of atrial fibrillation in patients with heart failure: Results from the Valsartan Heart Failure Trial (Val-HeFT). Am Heart J. 2005;149(3):548-557.
Patti G, Chello M, Candura D, et al. Randomized trial of atorvastatin for reduction of postoperative atrial fibrillation in patients undergoing cardiac surgery: Results of the ARMYDA-3 (Atorvastatin for Reduction of MYocardial Dysrhythmia After cardiac surgery) study. Circulation. 2006;114(14):1455-1461.
Pedersen OD, Bagger H, Kober L, Torp-Pedersen C. Trandolapril reduces the incidence of atrial fibrillation after acute myocardial infarction in patients with left ventricular dysfunction. Circulation. 1999;100(4):376-380.
Roy D, Talajic M, Nattel S, et al. Rhythm control versus rate control for atrial fibrillation and heart failure. N Engl J Med. 2008;358(25):2667-2677.
Scirica BM, Morrow DA, Hod H, et al. Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non ST-segment elevation acute coronary syndrome: Results from the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) randomized controlled trial. Circulation. 2007;116(15):1647-1652.
Singh SN, Fletcher RD, Fisher SG, et al. Amiodarone in patients with congestive heart failure and asymptomatic ventricular arrhythmia. Survival Trial of Antiarrhythmic Therapy in Congestive Heart Failure. N Engl J Med. 1995;333(2):77-82. 13
Vermes E, Tardif JC, Bourassa MG, et al. Enalapril decreases the incidence of atrial fibrillation in patients with left ventricular dysfunction: Insight from the Studies Of Left Ventricular Dysfunction (SOLVD) trials. Circulation. 2003;107(23):2926-2931.
Wachtell K, Lehto M, Gerdts E, et al. Angiotensin II receptor blockade reduces new-onset atrial fibrillation and subsequent stroke compared to atenolol: The Losartan Intervention For End Point Reduction in Hypertension (LIFE) study. J Am Coll Cardiol. 2005;45(5):712-719.
Zheng ZJ, Croft JB, Giles WH, et al. Sudden cardiac death in the United States, 1989 to 1998. Circulation. 2001;104(18):2158-2163.
Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: A report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death): Developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation. 2006;114(10):e385-e484.
1 Chugh SS, Jui J, Gunson K, et al. Current burden of sudden cardiac death: Multiple source surveillance versus retrospective death certificate-based review in a large U.S. community. J Am Coll Cardiol. 2004;44(6):1268-1275.
2 The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: Effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989;321(6):406-412.
3 Canadian Amiodarone Myocardial Infarction Arrhythmia Trial Investigators. Randomised trial of outcome after myocardial infarction in patients with frequent or repetitive ventricular premature depolarisations: CAMIAT. Lancet. 1997;349(9053):675-682.
4 European Myocardial Infarct Amiodarone Trial Investigators. Randomised trial of effect of amiodarone on mortality in patients with left-ventricular dysfunction after recent myocardial infarction: EMIAT. Lancet. 1997;349(9053):667-674.
5 Singh SN, Fletcher RD, Fisher SG, et al. Amiodarone in patients with congestive heart failure and asymptomatic ventricular arrhythmia. Survival Trial of Antiarrhythmic Therapy in Congestive Heart Failure. N Engl J Med. 1995;333(2):77-82.
6 Køber L, Torp-Pedersen C, McMurray JJ, et al. Dronedarone Study Group: Increased mortality after dronedarone therapy for severe heart failure. N Engl J Med. 2008;358(25):2678-2687.
7 Hohnloser SH, Kuck KH, Dorian P, et al. Prophylactic use of an implantable cardioverter-defibrillator after acute myocardial infarction, DINAMIT Investigators. N Engl J Med. 2004;351(24):2481-2488.
8 Zheng ZJ, Croft JB, Giles WH, et al. Sudden cardiac death in the United States, 1989 to 1998. Circulation. 2001;104(18):2158-2163.
9 Bayés de Luna A, Coumel P, Leclercq JF. Ambulatory sudden cardiac death: Mechanisms of production of fatal arrhythmia on the basis of data from 157 cases. Am Heart J. 1989;117(1):151-159.
10 . Clopidogrel and metoprolol in myocardial infarction trial: The Second Chinese Cardiovascular Study (COMMIT/CCS-2). Available at http://www.commit-ccs2.org
11 Domanski MJ, Krause-Steinrauf H, Massie BM, et al. A comparative analysis of the results from 4 trials of beta-blocker therapy for heart failure: BEST, CIBIS-II, MERIT-HF, and COPERNICUS. J Card Fail. 2003;9(5):354-363.
12 Remme WJ, Cleland JG, Erhardt L, et al. Effect of carvedilol and metoprolol on the mode of death in patients with heart failure. Eur J Heart Fail. 2007;9(11):1128-1135.
13 Paletta MJ, Braham S, Beatch GN, et al. Mechanisms underlying the antiarrhythmic properties of β-adrenoreceptor blockade against ischemia-induced arrhythmias in acutely prepared rats. Br J Pharm. 1989;98:87-94.
14 El-Sherif N, Turitto G. Electrophysiologic effects of carvedilol: Is carvedilol an antiarrhythmic agent? PACE. 2005;28:985-990.
15 Mithell LB, Powell JL, Gillis AM, et al. Are lipid-lowering drugs also antiarrhythmic drugs? An analysis of the Antiarrhythmics versus Implantable Defibrillators (AVID) Trial. J Am Coll Cardiol. 2003;42:81-87.
16 Vyas AK, Guo H, Moss AJ, et al. Reduction in ventricular tachyarrhythmias with statins in the Multicenter Automatic Defibrillator Implantation Trial (MADIT)-II. J Am Coll Cardiol. 2006;47:769-773.
17 Goldberger JJ, Subacius H, Schaechter A, et al. Effects of statin therapy on arrhythmic events and survival in patients with nonischemic dilated cardiomyopathy. J Am Coll Cardiol. 2006;48:1228-1233.
18 Levantesi G, Scarano M, Marfisi RM, et al. Meta-analysis of effect of statin treatment on risk of sudden death. Am J Cardiol. 2007;100:1644-1650.
19 Vrtovec B, Okrajsek R, Golicnik A, et al. Atorvastatin therapy may reduce the incidence of sudden cardiac death in patients with advanced chronic heart failure. J Cardiac Fail. 2008;14:140-144.
20 Deedwania P, Stone PH, Bairey Merz CN, et al. Effects of intensive versus moderate lipid-lowering therapy on myocardial ischemia in older patients with coronary heart disease: Results of the Study Assessing Goals in the Elderly (SAGE). Circulation. 2007;115(6):700-707.
21 van Boven AJ, Jukema JW, Zwinderman AH, et al. Reduction of transient myocardial ischemia with pravastatin in addition to the conventional treatment in patients with angina pectoris. Circulation. 1996;94:1503-1505.
22 Patel R, Nagueh SF, Tsybouleva N, et al. Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation. 2001;104(3):317-324.
23 Hayashidani S, Tsutsui H, Shiomi T, et al. Fluvastatin, a 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitor, attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation. 2002;105(7):868-873.
24 Lamers JM, Hartog JM, Verdouw PD, et al. Dietary fatty acids and myocardial function. Basic Res Cardiol. 1987;82(Suppl 1):209-221.
25 Yokoyama K, Ishibashi T, Ohkawara H, et al. HMG-CoA reductase inhibitors suppress intracellular calcium mobilization and membrane current induced by lysophosphatidylcholine in endothelial cells. Circulation. 2002;105(8):962-967.
26 Gao L, Wang W, Li YL, et al. Simvastatin therapy normalizes sympathetic neural control in experimental heart failure: Roles of angiotensin II type 1 receptors and NAD(P)H oxidase. Circulation. 2005;112(12):1763-1770.
27 Welzig CM, Shin DG, Park HJ, et al. Lipid lowering by pravastatin increases parasympathetic modulation of heart rate: Galpha(i2), a possible molecular marker for parasympathetic responsiveness. Circulation. 2003;108(22):2743-2746.
28 Pelat M, Dessy C, Massion P, et al. Rosuvastatin decreases caveolin-1 and improves nitric oxide-dependent heart rate and blood pressure variability in apolipoprotein E−/− mice in vivo. Circulation. 2003;107(19):2480-2486.
29 Vrtovec B, Okrajsek R, Golicnik A, et al. Atorvastatin therapy increases heart rate variability, decreases QT variability, and shortens QTc interval duration in patients with advanced chronic heart failure. J Card Fail. 2005;11(9):684-690.
30 Pedersen TR, Faergeman O, Kastelein JJ, et al. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: The Incremental Decrease in End Points Through Aggressive Lipid Lowering (IDEAL) study. A randomized controlled trial. JAMA. 2005;294(19):2437-2445.
31 Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: The Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL) study. A randomized controlled trial. JAMA. 2001;285(13):1711-1718.
32 LaRosa JC, Grundy SM, Waters DD, et al. Treating to New Targets (TNT) Investigators: Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med. 2005;352(14):1425-1435.
33 Scandinavian Simvastatin Survival Study group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). Lancet. 1994;344(8934):1383-1389.
34 Sever PS, Dahlöf B, Poulter NR, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial—Lipid Lowering Arm (ASCOT-LLA): A multicentre randomised controlled trial. Lancet. 2003;361(9364):1149-1158.
35 Morris MC, Manson JE, Rosner B, et al. Fish consumption and cardiovascular disease in the physicians’ health study: A prospective study. Am J Epidemiol. 1995;142(2):166-175.
36 Burr ML, Fehily AM, Gilbert JF, et al. Effects of changes in fat, fish, and fibre intakes on death and myocardial reinfarction: Diet and reinfarction trial (DART). Lancet. 1989;2(8666):757-761.
37 Marchioli R, Barzi F, Bomba E, et al. Early protection against sudden death by n-3 polyunsaturated fatty acids after myocardial infarction: Time-course analysis of the results of the Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico (GISSI)-Prevenzione. Circulation. 2002;105(16):1897-1903.
38 Mozaffarian D, Lemaitre RN, Kuller LH, et al. Cardiac benefits of fish consumption may depend on the type of fish meal consumed: The Cardiovascular Health Study. Circulation. 2003;107(10):1372-1377.
39 Billman GE, Kang JX, Leaf A. Prevention of sudden cardiac death by dietary pure omega-3 polyunsaturated fatty acids in dogs. Circulation. 1999;99(18):2452-2457.
40 McLennan PL, Bridle TM, Abeywardena MY, et al. Comparative efficacy of n-3 and n-6 polyunsaturated fatty acids in modulating ventricular fibrillation threshold in marmoset monkeys. Am J Clin Nutr. 1993;58(5):666-669.
41 Dujardin KS, Dumotier B, David M, et al. Ultrafast sodium channel block by dietary fish oil prevents dofetilide-induced ventricular arrhythmias in rabbit hearts. Am J Physiol Heart Circ Physiol. 2008;295(4):H1414-H1421.
42 Leaf A, Albert CM, Josephson M, et al. Prevention of fatal arrhythmias in high-risk subjects by fish oil n-3 fatty acid intake. Circulation. 2005;112(18):2762-2768.
43 Raitt MH, Connor WE, Morris C, et al. Fish oil supplementation and risk of ventricular tachycardia and ventricular fibrillation in patients with implantable defibrillators: A randomized controlled trial. JAMA. 2005;293(23):2884-2891.
44 Brouwer IA, Zock PL, Camm AJ, et al. Effect of fish oil on ventricular tachyarrhythmia and death in patients with implantable cardioverter defibrillators: The Study on Omega-3 Fatty Acids and Ventricular Arrhythmia (SOFA) randomized trial. JAMA. 2006;295(22):2613-2619.
45 León H, Shibata MC, Sivakumaran S, et al. Effect of fish oil on arrhythmias and mortality: Systematic review. BMJ. 2008;337:a2931.
46 Fletcher RD, Cintron GB, Johnson G, et al. Enalapril decreases prevalence of ventricular tachycardia in patients with chronic congestive heart failure. The V-HeFT II VA Cooperative Studies Group. Circulation. 1993;87(6 Suppl):VI49-V155.
47 Pfeffer MA, Braunwald E, Moye LA, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 1992;327:669-677.
48 Pratt CM, Gardner M, Pepine C, et al. for the SOLVD Investigators: Lack of long-term ventricular arrhythmia reduction by enalapril in heart failure. Am J Cardiol. 1995;75:1244-1249.
49 Cleland JG, Erhardt L, Murray G, et al. Effect of ramipril on morbidity and mode of death among survivors of acute myocardial infarction with clinical evidence of heart failure. A report from the AIRE Study Investigators. Eur Heart J. 1997;18(1):41-51.
50 Teo KK, Mitchell LB, Pogue J, et al. HOPE Investigators: Effect of ramipril in reducing sudden deaths and nonfatal cardiac arrests in high-risk individuals without heart failure or left ventricular dysfunction. Circulation. 2004;110(11):1413-1417.
51 Chinese Cardiac Study Collaborative Group. Oral captopril versus placebo among 13,634 patients with suspected acute myocardial infarction: Interim report from the Chinese cardiac study. Lancet. 1995;345:686-687.
52 Swedberg K, Held P, Kjekshus J, et al. Effects of the early administration of enalapril on mortality in patients with acute myocardial infarction: Results of the cooperative new Scandinavian enalapril survival study II (CONSENSUS II). N Engl J Med. 1992;327:678-684.
53 ISIS-4 (Fourth International Study of Infarct Survival) Collaborative Group. ISIS-4: A randomised factorial trial assessing early oral captopril, oral mononitrate, and intravenous magnesium sulphate in 58,050 patients with suspected acute myocardial infarction. Lancet. 1995;345(8951):669-685.
54 Ambrosioni E, Borghi C, Magnani B, et al. for the Survival of Myocardial Infarction Long-Term Evaluation (SMILE) Study Investigators: The effect of the angiotensin-converting-enzyme inhibitor zofenopril on mortality and morbidity after anterior myocardial infarction. N Engl J Med. 1995;332(2):80-85.
55 Kassotis J, Mongwa M, Reddy CV. Effects of angiotensin-converting enzyme inhibitor therapy on QT dispersion post acute myocardial infarction. Pacing Clin Electrophysiol. 2003;26(4 Pt 1):843-848.
56 Bae JH, Rihal CS, Edwards BS, et al. Association of angiotensin-converting enzyme inhibitors and serum lipids with plaque regression in cardiac allograft vasculopathy. Transplantation. 2006;82(8):1108-1111.
57 Goette A, Lendeckel U. Electrophysiological effects of angiotensin II. Part I: Signal transduction and basic electrophysiological mechanisms. Europace. 2008;10:238-241.
58 Søgaard P, Gøtzsche C, Ravkilde J, et al. Ventricular arrhythmias in the acute and chronic phases after acute myocardial infarction: Effect on intervention with captopril. Circulation. 1994;90:101-107.
59 Pitt B, Segal R, Martinez FA, et al. Randomised trial of losartan versus captopril in patients over 65 with heart failure (Evaluation of Losartan in the Elderly Study, ELITE). Lancet. 1997;349(9054):747-752.
60 Dickstein K, Kjekshus J, OPTIMAAL Steering Committee of the OPTIMAAL Study Group. Effects of losartan and captopril on mortality and morbidity in high-risk patients after acute myocardial infarction: The OPTIMAAL randomised trial. Optimal Trial in Myocardial Infarction with Angiotensin II Antagonist Losartan. Lancet. 2002;360(9335):752-760.
61 Pfeiffer MA, Swedberg K, Granger CB, et al. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: The CHARM-Overall programme. Lancet. 2003;362:759-766.
62 Wachtell K, Okin PM, Olsen MH, et al. Regression of electrocardiographic left ventricular hypertrophy during antihypertensive therapy and reduction in sudden cardiac death: The LIFE Study. Circulation. 2007;116(7):700-705.
63 Pitt B, The Randomized Aldactone Evaluation Study Investigators. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med. 1999;341:709-717.
64 Pitt B, Remme W, Zannad F, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348(14):1309-1321.
65 Pitt B, the EPHESUS Investigators. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol. 2005;46(3):425-431.
66 Juurlink DN, Mamdani MM, Lee DS, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med. 2004;351:543-551.
67 Staessen JA, Fagard R, Thijs L, et al. Subgroup and per-protocol analysis of the randomized European Trial on Isolated Systolic Hypertension in the Elderly. Arch Intern Med. 1998;158(15):1681-1691.
68 Jamerson K, Weber MA, Bakris GL, et al. ACCOMPLISH Trial Investigators: Benazepril plus amlodipine or hydrochlorothiazide for hypertension in high-risk patients. N Engl J Med. 2008;359(23):2417-2428.
69 Yui Y, Sumiyoshi T, Kodama K, et al. Nifedipine retard was as effective as angiotensin converting enzyme inhibitors in preventing cardiac events in high-risk hypertensive patients with diabetes and coronary artery disease: The Japan Multicenter Investigation for Cardiovascular Diseases-B (JMIC-B) subgroup analysis. Hypertens Res. 2004;27(7):449-456.
70 O’Connor CM, Carson PE, Miller AB, et al. Effect of amlodipine on mode of death among patients with advanced heart failure in the PRAISE trial. Prospective Randomized Amlodipine Survival Evaluation. Am J Cardiol. 1998;82(7):881-887.
71 Nagai H, Minatoguchi S, Chen XH, et al. Cilnidipine, an N+L-type dihydropyridine Ca channel blocker, suppresses the occurrence of ischemia/reperfusion arrhythmia in a rabbit model of myocardial infarction. Hypertens Res. 2005;28(4):361-368.
72 Spirito P, Bellone P, Harris KM, et al. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med. 2000;342(24):1778-1785.
73 Verdecchia P, Borgioni C, Ciucci A, et al. Association between persistent pressure overload and ventricular arrhythmias in essential hypertension. Hypertension. 1996;28(2):284-289.
74 Scirica BM, Morrow DA, Hod H, et al. Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non ST-segment elevation acute coronary syndrome: Results from the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) randomized controlled trial. Circulation. 2007;116(15):1647-1652.
75 Antzelevitch C, Belardinelli L, Zygmunt AC, et al. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation. 2004;110(8):904-910.
76 Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 Guidelines for Management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: A report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop Guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death): Developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation. 2006;114(10):e385-e484.
77 Hagens VE, Van Gelder IC, Crijns HJ, RAte Control Versus Electrical Cardioversion Of Persistent Atrial Fibrillation (RACE) Study Group. The RACE study in perspective of randomized studies on management of persistent atrial fibrillation. Card Electrophysiol Rev. 2003;7(2):118-121.
78 Roy D, Talajic M, Nattel S, et al. Rhythm control versus rate control for atrial fibrillation and heart failure. N Engl J Med. 2008;358(25):2667-2677.
79 Connolly SJ, Cybulsky I, Lamy A, et al. Double-blind, placebo-controlled, randomized trial of prophylactic metoprolol for reduction of hospital length of stay after heart surgery: The beta-Blocker Length Of Stay (BLOS) study. Am Heart J. 2003;145(2):226-232.
80 Tsuboi J, Kawazoe K, Izumoto H, et al. Postoperative treatment with carvedilol, a beta-adrenergic blocker, prevents paroxysmal atrial fibrillation after coronary artery bypass grafting. Circ J. 2008;72(4):588-591.
81 Arumanayagam M, Chan S, Tong S, Sanderson JE. Antioxidant properties of carvedilol and metoprolol in heart failure: A double-blind randomized controlled trial. J Cardiovasc Pharmacol. 2001;37:48-54.
82 Nasr IA, Bouzamondo A, Hulot JS, et al. Prevention of atrial fibrillation onset by beta-blocker treatment in heart failure: A meta-analysis. Eur Heart J. 2007;28(4):457-462.
83 Kühlkamp V, Schirdewan A, Stangl K, et al. Use of metoprolol CR/XL to maintain sinus rhythm after conversion from persistent atrial fibrillation: A randomized, double-blind, placebo-controlled study. J Am Coll Cardiol. 2000;36(1):139-146.
84 Nergårdh AK, Rosenqvist M, Nordlander R, Frick M. Maintenance of sinus rhythm with metoprolol CR initiated before cardioversion and repeated cardioversion of atrial fibrillation: A randomized double-blind placebo-controlled study. Eur Heart J. 2007;28(11):1351-1357.
85 Van Noord T, Tieleman RG, Bosker HA, et al. Beta-blockers prevent subacute recurrences of persistent atrial fibrillation only in patients with hypertension. Europace. 2004;6(4):343-350.
86 Wachtell K, Lehto M, Gerdts E, et al. Angiotensin II receptor blockade reduces new-onset atrial fibrillation and subsequent stroke compared to atenolol: The Losartan Intervention For End Point Reduction in Hypertension (LIFE) study. J Am Coll Cardiol. 2005;45(5):712-719.
87 Pedersen OD, Bagger H, Kober L, Torp-Pedersen C. Trandolapril reduces the incidence of atrial fibrillation after acute myocardial infarction in patients with left ventricular dysfunction. Circulation. 1999;100(4):376-380.
88 Vermes E, Tardif JC, Bourassa MG, et al. Enalapril decreases the incidence of atrial fibrillation in patients with left ventricular dysfunction: Insight from the Studies Of Left Ventricular Dysfunction (SOLVD) trials. Circulation. 2003;107(23):2926-2931.
89 Maggioni AP, Latini R, Carson PE, et al. Val-HeFT Investigators: Valsartan reduces the incidence of atrial fibrillation in patients with heart failure: Results from the Valsartan Heart Failure Trial (Val-HeFT). Am Heart J. 2005;149(3):548-557.
90 Ducharme A, Swedberg K, Pfeffer MA, et al. Prevention of atrial fibrillation in patients with symptomatic chronic heart failure by candesartan in the Candesartan in Heart failure: Assessment of Reduction in Mortality and morbidity (CHARM) program. Am Heart J. 2006;151(5):985-991.
91 Ueng KC, Tsai TP, Yu WC, et al. Use of enalapril to facilitate sinus rhythm maintenance after external cardioversion of long-standing persistent atrial fibrillation. Results of a prospective and controlled study. Eur Heart J. 2003;24(23):2090-2098.
92 Madrid AH, Bueno MG, Rebollo JM, et al. Use of irbesartan to maintain sinus rhythm in patients with long-lasting persistent atrial fibrillation: A prospective and randomized study. Circulation. 2002;106(3):331-336.
93 Healey JS, Baranchuk A, Crystal E, et al. Prevention of atrial fibrillation with angiotensin-converting enzyme inhibitors and angiotensin receptor blockers: A meta-analysis. J Am Coll Cardiol. 2005;45:1832-1839.
94 HOPE Investigators. Impact of ramipril on the incidence of atrial fibrillation: results of the Heart Outcomes Prevention Evaluation study. Am Heart J. 2007;154(3):448-453.
95 Telmisartan Randomised AssessmeNt Study in ACE iNtolerant subjects with cardiovascular Disease (TRANSCEND) Investigators. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: A randomised controlled trial. Lancet. 2008;372(9644):1174-1183.
96 ONTARGET Investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358(15):1547-1559.
97 Chung MK, Martin DO, Sprecher D, et al. C-reactive protein elevation in patients with atrial arrhythmias: Inflammatory mechanisms and persistence of atrial fibrillation. Circulation. 2001;104(24):2886-2891.
98 Plenge JK, Hernandez TL, Weil KM, et al. Simvastatin lowers C-reactive protein within 14 days: An effect independent of low-density lipoprotein cholesterol reduction. Circulation. 2002;106(12):1447-1452.
99 Ozaydin M, Varol E, Aslan SM, et al. Effect of atorvastatin on the recurrence rates of atrial fibrillation after electrical cardioversion. Am J Cardiol. 2006;97(10):1490-1493.
100 Patti G, Chello M, Candura D, et al. Randomized trial of atorvastatin for reduction of postoperative atrial fibrillation in patients undergoing cardiac surgery: Results of the ARMYDA-3 (Atorvastatin for Reduction of MYocardial Dysrhythmia After cardiac surgery) study. Circulation. 2006;114(14):1455-1461.
101 Hanna IR, Heeke B, Bush H, et al. Lipid-lowering drug use is associated with reduced prevalence of atrial fibrillation in patients with left ventricular systolic dysfunction. Heart Rhythm. 2006;3(8):881-886.
102 Li D, Singh RM, Liu L, et al. Oxidized-LDL through LOX-1 increases the expression of angiotensin converting enzyme in human coronary artery endothelial cells. Cardiovasc Res. 2003;57(1):238-243.
103 Pischon T, Hankinson SE, Hotamisligil GS, et al. Habitual dietary intake of n-3 and n-6 fatty acids in relation to inflammatory markers among US men and women. Circulation. 2003;108(2):155-160.
104 Calò L, Bianconi L, Colivicchi F, et al. N-3 Fatty acids for the prevention of atrial fibrillation after coronary artery bypass surgery: A randomized, controlled trial. J Am Coll Cardiol. 2005;45(10):1723-1738.
105 Mozaffarian D, Psaty BM, Rimm EB, et al. Fish intake and risk of incident atrial fibrillation. Circulation. 2004;110(4):368-373.
106 Brouwer IA, Heeringa J, Geleijnse JM, et al. Intake of very long-chain n-3 fatty acids from fish and incidence of atrial fibrillation. The Rotterdam Study. Am Heart J. 2006;151(4):857-862.
107 Frost L, Vestergaard P. n-3 Fatty acids consumed from fish and risk of atrial fibrillation or flutter: The Danish Diet, Cancer, and Health Study. Am J Clin Nutr. 2005;81(1):50-54.
108 Shiroshita-Takeshita A, Brundel BJ, Lavoie J, Nattel S. Prednisone prevents atrial fibrillation promotion by atrial tachycardia remodeling in dogs. Cardiovasc Res. 2006;69(4):865-875.
109 Goldstein RN, Ryu K, Khrestian C, et al. Prednisone prevents inducible atrial flutter in the canine sterile pericarditis model. J Cardiovasc Electrophysiol. 2008;19(1):74-81.
110 Prasongsukarn K, Abel JG, Jamieson WR, et al. The effects of steroids on the occurrence of postoperative atrial fibrillation after coronary artery bypass grafting surgery: A prospective randomized trial. J Thorac Cardiovasc Surg. 2005;130(1):93-98.
111 Dernellis J, Panaretou M. Relationship between C-reactive protein concentrations during glucocorticoid therapy and recurrent atrial fibrillation. Eur Heart J. 2004;25(13):1100-1107.
112 van der Hooft CS, Heeringa J, Brusselle GG, et al. Corticosteroids and the risk of atrial fibrillation. Arch Intern Med. 2006;166(9):1016-1020.
113 Carnes CA, Chung MK, Nakayama T, et al. Ascorbate attenuates atrial pacing-induced peroxynitrite formation and electrical remodeling and decreases the incidence of postoperative atrial fibrillation. Circ Res. 2001;89(6):E32-E38.
114 Eslami M, Badkoubeh RS, Mousavi M, et al. Oral ascorbic acid in combination with beta-blockers is more effective than beta-blockers alone in the prevention of atrial fibrillation after coronary artery bypass grafting. Tex Heart Inst J. 2007;34(3):268-274.
115 Korantzopoulos P, Kolettis TM, Kountouris E, et al. Oral vitamin C administration reduces early recurrence rates after electrical cardioversion of persistent atrial fibrillation and attenuates associated inflammation. Int J Cardiol. 2005;102(2):321-326.
116 Fuster V, Rydén LE, Cannom DS, et al. ACC/AHA/ESC 2006 Guidelines for the Management of patients with atrial fibrillation: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Revise the 2001 Guidelines for the management of patients with atrial fibrillation): Developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation. 2006;114(7):e257-e354.