[level-membership-for-hematology-oncology-and-palliative-medicine-category]
16 Neurotoxicity of Chemotherapy
Types of Neurological Damage
COGNITIVE DYSFUNCTION
Cancer patients have frequently recognized decreased cognitive function (“chemo-brain”) during chemotherapy, which, in the past, was attributed by their physicians to stress or depression. Patients report problems with memory retrieval, learning, and concentration, which may persist after treatment has finished or never fully resolve.1 The incidence of acute problems during treatment ranges from 15% to 70%, with 50% of patients in one study identifying persistent problems a year after treatment.2 Cross-sectional studies also suggest persistent cognitive dysfunction in 20% to 30% of patients 2 to 10 years posttreatment.3,4
Mechanisms for this functional decline are not fully understood, but recent work by Han et al.5 on 5-fluorouracil neurotoxicity in mice suggest extensive myelin damage and persistent suppression of both oligodendrocyte and progenitor cell proliferation in the subventricular zone, hippocampus, and corpus callosum.
ACUTE ENCEPHALOPATHY
Acute encephalopathy is a common problem in oncology patients; it has a wide range of precipitating factors including metabolic derangements, hypoxia, brain metastases, meningeal carcinomatosis, infection, paraneoplastic phenomena, and drugs.6 Presenting symptoms typically include lethargy, confusion, somnolence, seizures, or coma. The diagnosis of drug-induced encephalopathy is typically one of exclusion. A careful drug history should be taken, including recent administration of narcotic analgesia and antiemetic cover, used frequently for cancer patients undergoing chemotherapy and/or for symptom control. CNS infections should also be excluded, particularly in immunocompromised and neutropenic patients. Investigations should include urea/electrolytes, liver function, serum glucose, calcium magnesium, viral serology, and CSF examination. If focal neurological deficit is present, CT or MRI imaging may be helpful and, in any case, should be undertaken prior to a lumbar puncture. EEG is particularly helpful if seizures occur, and will typically show generalized slowing with delta wave activity.7 Encephalopathy due to cytotoxic drug exposure is generally self-limiting and recovers spontaneously. The only specific therapy is the use of methylene blue in ifosfamide-induced encephalopathy, which should be considered in any patients undergoing ifosfamide chemotherapy. Risk factors include extremes of age, dose/ schedule, previous cranial radiotherapy, and renal or hepatic dysfunction.8,9
LEUKOENCEPHALOPATHY
Leukoencephalopathy may follow on from acute encephalopathy, but may also be the first indication of neurotoxicity several months to years after administration of cytotoxic drugs. Patients present with cognitive deficits, which may progress to dementia, coma, and death.10–14 MRI imaging shows widespread changes throughout the white matter,15–18 and histologically, there is axonal swelling, demyelination, and neuronal death.19 Those most at risk are patients treated with methotrexate or cytarabine, particularly if given intrathecally or if cranial radiotherapy preceded cytotoxic administration.9 Elderly patients with primary central nervous system lymphoma undergoing treatment with high-dose methotrexate and whole-brain radiotherapy (WBRT) are at particularly high risk of developing this complication.20 There is no specific therapy that can halt the progressive decline, and overall the prognosis is poor. Elderly patients with primary CNS lymphoma should be informed about these risks; it should also be considered by the treating oncologists whether these patients can be treated with reduced-dose WBRT or avoid WBRT altogether after-high dose methotrexate.
CEREBELLAR DYSFUNCTION
Cerebellar signs in an oncology patient are usually due to direct spread of cancer, particularly if it is asymmetric. In rarer situations, this can be due to paraneoplastic syndromes, which tend to have a subacute onset and may be associated with the presence of antineuronal antibodies.6 Care should also be taken to distinguish cerebellar ataxia from sensory ataxia due to a severe sensory neuropathy. Cytarabine and 5-fluorouracil are the cytotoxics most likely to cause cerebellar dysfunction including truncal ataxia, gait disturbance, and ataxia.8,14,21,22 Acutely, the MRI tends to be normal, but subsequent scans may show chronic atrophy due to irreversible Purkinje cell loss.15–18
SPINAL CORD TOXICITY
Spinal cord toxicity can occur following intrathecal administration of certain cytotoxics in acute leukemias, lymphomas, and brain tumors. Intrathecal chemotherapy is administered either as part of a lumbar puncture procedure or into the ventricles, via an Ommaya reservoir.6 The drugs most commonly used are cytarabine, methotrexate, and hydrocortisone; they may be given singly, sequentially, or together as “triple therapy.” Symptoms usually arise after multiple cycles of therapy and include both spinal cord and nerve root signs. Loss of neurological function may progress upwards.23 Histologically, there are focal areas of necrosis, particularly at the periphery of the spinal cord, associated with axonal swelling and demyelination. Myelin basic protein levels in the CSF may be elevated.24 Typically, only half of those affected will show any sign of recovery.
PERIPHERAL NEUROPATHY
Peripheral neuropathies are the most common neurological complications in patients receiving chemotherapy, especially with regimens containing taxanes (taxol, docataxel), platinum (cisplatin, carboplatin, oxaliplatin), and vinca alkaloids (vincristine). The neuropathy tends to be predominantly sensory in nature, with a glove and stocking distribution. Generally, symptoms are self-limiting, but in some patients the symptoms persist. The effect is cumulative,6 and patients frequently complain of acute subjective paresthesia of the extremities 2 to 3 days after chemotherapy. With subsequent treatment cycles, symptoms may progress to permanent paresthesia, with decreased sensation to pinprick, light touch, and vibration on formal testing.9,25–29 At this stage, chemotherapy should be stopped or the dose reduced, as continuing can lead to difficulty with activities of daily living. The neuropathy is largely reversible over several months but many patients may be left with some degree of paresthesia. In rare instances, neuropathy may be paraneoplastic in origin. Oxaliplatin is unusual in that it causes acute cold dysesthesias, as well as pharyngolaryngospasm, which usually starts shortly after administration of chemotherapy and then resolves.
Grading Toxicity
There is no universal grading system for the evaluation of patients with neurological toxicity although two neurotoxicity scoring systems are frequently used: NCI-CTC 3 (National Cancer Institute Common Toxicity Criteria version 3) and ECOG (Eastern Cooperative Oncology Group).30 The NCI-CTCAE scores a variety of symptoms from ataxia to motor neuropathy on a scale of 0 to 5, with 0 being normal, 1 mild self-limiting, 2 moderate, 3 severe undesirable, 4 life-threatening, and 5 death induced by adverse event. The full version can be downloaded from www.fda.gov. The ECOG criteria are organized in a similar manner with a scale from 0 to 4 and can be viewed at www.ecog.org. Generally patients with a toxicity score of 1 to 2 can continue with their treatment unmodified, while those with a score of 3 or 4 require dose modification or cessation of treatment.
Prevention
The patients most at risk are those receiving a high cumulative dose or intensive schedule, particularly if there is a preexisting condition such as diabetes mellitus, hereditary neuropathy, or multiple sclerosis.31 Previous radiotherapy may also increase the risk of developing neurotoxicity if patients are subsequently treated with cisplatin or methotrexate.32,33
Patients with HIV-related malignancies are also at increased risk of cytotoxic-induced neuropathy, since both HIV and the drugs used to treat it (highly active antiretroviral therapy, or HAART) can cause neurological damage independently.34 Distal sensory neuropathy is the commonest form of HIV-associated neuropathy and can be difficult to distinguish from that caused by specific nucleoside antiretrovirals. In addition, compounded neurological toxicities frequently occur because of reduced clearance of vincristine, vinblastine, and taxane; these complications can be reduced by changing these patients’ antiretroviral medications. More rarely, patients can develop inflammatory demyelinating polyneuropathy, mononeuritis multiplex, or neuronal damage due to opportunistic infections such as CMV and HZV.
Currently there are few therapies able to prevent neurological toxicity preemptively. Infusions of methylene blue are used prophylatically in patients receiving ifosfamide who have previously developed acute encephalopathy. A number of trials have investigated the benefits of agents such as calcium-magnesium infusions, carbamazepine, gabapentin, amifostine, and glutathione.7,35–52 However, the trials are small studies and no specific prophylaxis can be routinely recommended.
Treatment of Neurotoxicity
Apart from dose reduction or discontinuing the drugs implicated in the development of neurotoxicity, there is very little in the way of specific pharmacological management to reverse their side effects. Methylene blue is used for ifosfamide-induced encephalopathy and infusional calcium with magnesium may lessen the severity of established peripheral neuropathy due to oxaliplatin. However, considerable input from the multidisciplinary team is required in the holistic care of the patient,53 including analgesia for neuropathic pain, maximization of mobility (with occupational and physiotherapist involvement where there is loss of balance, sensory loss, or muscle weakness), and good nursing care to manage bowel or bladder dysfunction, protection of pressure areas in immobilized patients, and maintenance of a safe environment if patients are confused.
Cytotoxic Agents and Their Neurological Toxicities (See Table 16-1)
5-FLUOROURACIL
5-fluorouracil (5-FU), a pyrimidine analogue, is widely used in the treatment of gastrointestinal malignancies; it is administered either as a short intravenous bolus or as a prolonged continuous infusion. Neurotoxicity is rare, but may include acute cerebellar dysfunction in 3% to 7% of patients, causing gait ataxia, nystagmus, and scanning speech.54,55 Symptoms tend to resolve spontaneously within a few days of treatment cessation although administration of thiamine may be helpful.22 Patients may also develop acute confusion in the absence of cerebellar signs, which may recur on reexposure to 5-FU.56 The main risk factor for development of neurotoxicity is deficiency of the enzyme dihydropyrimidine dehydrogenase which metabolizes 5-FU.57–59

A subacute form of leukoencephalopathy occurs in approximately 2% of patients receiving 5-FU in combination with levamisole (an immunomodulatory agent).60–64 Presentation is with focal neurological abnormalities and cognitive impairment that may be mistaken for metastatic disease. MRI shows patchy abnormalities within the white matter that enhance with gadolinium. Histologically, these patches contain an intense inflammatory infiltration with demyelination but axonal sparing. It is thought that the levamisole disrupts the blood-brain barrier potentiating 5-FU’s access to the CNS. Although treatment with steroids has been advocated, this syndrome is frequently self-limiting and patients usually recover completely over the course of several weeks without specific therapy. In some patients, thymidine has also been used successfully.58 Rarely, a peripheral sensory neuropathy has also been reported.65 Because many patients received adjuvant 5-FU based treatments for a variety of cancers, including breast, gastrointestinal, and bowel cancers, patients should be monitored for possible long-term neurocognitive damage.5
ASPARAGINASE
Asparaginase (either as the L- or pegylated formulation) is a component of remission-induction therapy used to treat acute lymphoblastic leukemia (ALL). It may cause cerebrovascular accidents during the first few weeks of its administration.66 This is due to depletion of plasma proteins involved in coagulation, such as fibrinogen and antithrombin III. Thrombosis typically occurs within the dural sinuses and cerebral veins, leading to secondary hemorrhage or infarction.67 Patients present with a range of symptoms from mild headache to coma and death, which are caused by rapid increases in intracranial pressure. Once diagnosed, the asparaginase should be stopped and the patient anticoagulated unless hemorrhage is present.
Asparaginase acts to cleave asparagine, an essential amino acid required by rapidly proliferating cells (hence its antimitotic action) and also as a neurotransmitter. Therefore depletion of asparagine during treatment has also been associated with the development of neuropsychiatric symptoms such as depression and hallucinations.68
CYCLOPHOSPHAMIDE
Cyclophosphamide is a prodrug, which alkylates DNA after hepatic activation.69 It is used in a wide range of malignancies, including lymphoma, breast cancer, and testicular cancer. When given in high doses, it may lead to inappropriate secretion of ADH (SIADH), and hence a secondary metabolic encephalopathy may occur, with confusion, seizures, or coma.70–72 There have been a few case reports of cyclophosphamide also being associated with blurred vision, dizziness, and confusion in the absence of SIADH.73,74
CYTARABINE
Cytarabine is an analogue of adenosine, causing chain termination during DNA synthesis; it is one of the most effective cytotoxic drugs in the treatment of acute leukemia. It is used either at a conventional dose of 100 to 200 mg/m2/day or at high doses of 2 to 6 g/m2/day.69 Neurotoxicity is particularly common at the higher dose levels, affecting 16% to 50% of patients;8 it predominantly affects the central nervous system. The risk of neurotoxicity is increased by age, dose/ schedule (particularly cumulative dose), renal or hepatic impairment, and the concurrent use of neurotropic antiemetics such as phenothiazines.8,75,76
The mechanism of toxicity is not well-understood, but it appears that cytarabine directly causes neuronal death, possibly by the inhibition of cytidine-dependent neurotropic signal transduction,77 although it has also been shown to stimulate the production of reactive oxygen species that may also damage DNA directly by inducing strand breaks.78
Acute cerebellar dysfunction is the commonest central neurotoxicity, occurring in approximately 14% of patients; they typically present with dysarthria, nystagmus, gait ataxia, and confusion. Onset usually occurs during administration of a multi-day regimen, particularly above a cumulative dose of 36 g/m2 and generally resolves rapidly once cytarabine is withdrawn.8,9,14 However, toxicity may be permanent once more than 8% to 20% of Purkinje cells have been lost.8 Acute encephalopathy is also common, presenting with somnolence or seizures. MRI scanning reveals diffuse high-intensity lesions within the central matter on T2-weighting that may be reversible.18 The encephalopathy should rapidly resolve entirely on stopping cytarabine; however, damage may be permanent and progress to leukoencephalopathy in a minority of patients, usually those with preexisting organ dysfunction or neurological problems.8 Less commonly, optic neuropathy, anosmia, and an incompletely reversible myelopathy have been reported.14 As with methotrexate, the intrathecal administration of cytarabine may cause ascending myelitis.8,79–81 There have also been case reports of sensory peripheral neuropathy following cytarabine exposure.82
ETOPOSIDE
Etoposide, a topoisomerase II inhibitor used in treatment of hematological, lung, ovarian, and testicular cancers,69 causes very little neurotoxicity, although at very high doses there have been reports of peripheral neuropathy, headache, seizures, and somnolence, in bone marrow transplant recipients and patients with malignant gliomas.83,84
FLUDARABINE
Neurotoxicity with the antimetabolite fludarabine is uncommon, but somnolence, acute encephalopathy, and chronic leukoencephalopathy progressing to coma and death have all been reported.85
IFOSFAMIDE
Central nervous system toxicity occurs in approximately 10% to 20% of patients receiving ifosfamide, who present with personality changes, confusion, hallucinations, stupor, and coma.9 More unusually, patients may experience seizures, myoclonus, cranial nerve palsies, or extrapyramidal symptoms.86,87 Symptoms usually begin 12 to14 hours after initiation of an ifosfamide infusion and spontaneously resolve 2 to 3 days after its cessation, although rarely symptoms may persist or even be fatal. EEG tends to show severe slowing with delta wave activity with or without seizure activity. Patients most at risk are those with impaired renal function, low serum albumin, pelvic tumors, and previous exposure to cisplatin.88 The risk of encephalopathy varies with route of administration. It is more common after oral administration, and is also more frequent with short intravenous infusion durations.89–91 The mechanism of toxicity is unclear but may be related to accumulation of metabolites such as chloracetaldehyde and chloro- ethylamine, which deplete intracellular glutathione and NAD and impair mitochondrial electron transport.92 Methylene blue, 300 mg IV in 6 divided doses, is used in the treatment of ifosfamide-induced encephalopathy and 50 mg IV qds may be given prophylatically.93–96 It is thought to act primarily as an alternative electron acceptor restoring mitochondrial respiratory chain function, but may also oxidate NADH and inhibits plasma monoamine oxidases.
METHOTREXATE
Methotrexate is one of the most widely used cytotoxic antimetabolites in the treatment of hematological, breast, and head/ neck cancers.9 In addition, prophylactic intrathecal use in ALL and high grade non-Hodgkin lymphomas has reduced the incidence of CNS relapse in high risk patients.97 Methotrexate acts by inhibiting dihydrofolate reductase, thus blocking purine and thymidine biosynthesis. Its neurotoxicity is thought to stem from widespread disruption of various metabolic pathways in the brain and can be acute or chronic.
Acute encephalopathy is associated with the administration of high-dose methotrexate (>3 mg/m2) and is characterized by somnolence, confusion, and seizures.98 Other symptoms include emotional lability and alternating hemiparesis, giving rise to the misdiagnosis of a functional disorder. The imaging appearances are characteristic and show symmetrical restricted diffusion on diffusion-weighted imaging, even when the T2-weighted sequences appear normal (Figure 16-1). Patients most at risk are children, those who have received previous cranial radiotherapy, or those receiving concomitant intravenous and intrathecal therapy. Metabolic abnormalities associated with the development of encephalopathy include widespread reduction in glucose utilization and protein synthesis,99 which are reversible by replacing depleted folate stores with the administration of leucovorin.100 Leucovorin is now given routinely following high-dose methotrexate (folinic acid rescue). Other studies have also postulated that changes in adenosine,101 homocysteine,102,103 or biopterin104 levels may also contribute to development of encephalopathy. In one study, aminophylline (2.5 mg/kg IV over 1 hour), given to six encephalopathic patients, caused immediate resolution of symptoms in four and improved symptoms in the remaining two patients.101
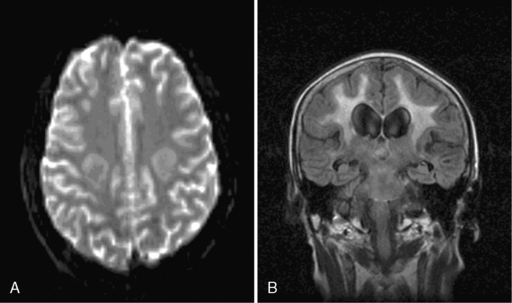
Administration of intrathecal methotrexate alone may result in an aseptic meningitis, with headache, neck stiffness, vomiting, and fever developing 2 to 4 hours after injection and lasting 12 to 72 hours.105 Transverse myelopathy has also been associated with intrathecal methotrexate, but usually in patients who have received repeated administrations. Patients develop progressive back pain, sensory loss, paraplegia, and sphincter disturbance over several days or weeks. Myelin basic protein may be raised in the CSF prior to the development of neurological signs.106 Approximately half the patients recover some function. Histologically, there are areas of focal necrosis associated with axonal swelling and demyelination. Pathogenesis is due to transient inhibition of myelin formation by S-adenosylmethionine107 and choline16 depletion secondary to methotrexate-induced folate deficiency.
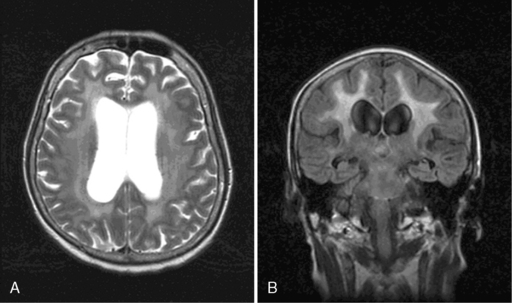
Leukoencephalopathy may develop several months to years after both intravenous and intrathecal methotrexate administration. Patients present with cognitive impairment, gait abnormalities, focal neurologic deficits, and/or seizures, and may progress to coma and death. MRI reveals diffuse atrophy, ventricular dilation, and cortical calcification (Figure 16-2).108 Histologically, there are areas of axonal necrosis with associated myelin loss as well as microangiopathic calcification within the vasculature.19 As with the development of acute encephalopathy, those most at risk appear to be children and recipients of cranial radiotherapy prior to chemotherapy.20 The neurological damage is not reversible, and there is no available treatment. Neurocognitive complications are significantly increased in older patients with primary CNS lymphoma treated with high-dose methotrexate and whole brain radiotherapy.
NITROSOUREAS
The nitrosureas, carmustine (BCNU) and lomustine (CCNU) are DNA-alkylating agents that cross the blood-brain barrier and are therefore commonly used in the treatment of brain tumors, melanoma, and CNS lymphomas.69 At conventional doses neurotoxicity is unusual; however, when used at high doses, particularly in association with intraarterial injection or previous radiotherapy, blindness due to optic neuropathy, encephalopathy, and seizures have been reported.109–112
PLATINUM COMPOUNDS
Platinum-containing compounds such as cisplatin, carboplatin, and oxaliplatin all act by alkylating DNA to produce interstrand and intrastrand breaks, which result in lethal DNA strand breakage during replication.69
The commonest neurological toxicity is a painful sensory peripheral neuropathy, which is dose-related and cumulative. Patients typically present with dysesthesia and decreased proprioception and vibration sense, and may have absent deep tendon reflexes.27,113–125 If treatment is halted at this stage, these symptoms generally resolve completely over several months.126 However, if treatment continues, mild motor neuropathy and severe sensory ataxia may develop, affecting ambulation in some patients. The pathogenesis of the peripheral neuropathy is unknown, but histologically there is axonal swelling and myelin sheath breakdown, predominantly affecting large sensory fibers within the dorsal column.127
Oxaliplatin also causes an acute sensory disturbance in 80% to 90% of patients, characterized by transient distal or perioral paresthesia that starts during drug administration, is frequently triggered by cold and touching cold objects, and resolves completely over several hours to days. It is thought to be caused by disruption of voltage-gated ion channels within neurons. Symptoms can, therefore, be dramatically reduced if patients are advised to keep warm and avoid cold food or drinks.115
Irreversible high-frequency hearing loss associated with tinnitus also occurs frequently with cisplatin exposure and may progress to affect lower frequency ranges if treatment is not terminated.116,128–131 Cisplatin appears to be toxic to the outer hair cells of the cochlea, while the vestibular apparatus is unaffected. Young age and previous cranial radiotherapy increase risk, as does peak plasma cisplatin concentration and cumulative dose. Despite the frequent occurrence of hearing loss, most patients remain asymptomatic, although small losses can be a significant problem for professional singers and musicians.
More rarely, patients may develop Lhermitte sign, optic neuropathy, or cortical blindness secondary to cisplatin-induced nerve demyelination, all of which are capable of total spontaneous recovery if treatment is stopped.9
Cisplatin is the most neurotoxic of the platinum compounds, with 23% of patients developing peripheral neuropathy27,132,133 and up to 81% developing high frequency hearing loss on audiometry,134 while carboplatin is the least with only 1% of patients developing peripheral neuropathy and 19%, asymptomatic hearing loss.27,131,133
There have been many small trials looking at prophylaxis of neuropathy in platinum-treated patients, predominantly with compounds containing thiol groups or heavy metal chelators such as glutathione, antiepileptics, and amifostine. Some of these trials reported some benefit.35–39,41–52 However, study numbers are small and no definite conclusion can be drawn from these studies. Infusional calcium gluconate with magnesium sulfate (1g of each in 250 ml 5% dextrose IV over 20 minutes, pre- and post-chemotherapy) may lessen the severity of established peripheral neuropathy due to oxaliplatin.135
PROCARBAZINE
Procarbazine is a DNA alkylator commonly used in the treatment of Hodgkin lymphoma; it readily crosses the blood brain barrier.69 It causes an acute encephalopathy with confusion and somnolence in 14% to 33% of patients, which is probably secondary to its ability to inhibit monoamine oxidase.109,136,137 Symptoms are therefore worsened by concurrent administration of phenothiazines such as chlorpromazine, which is used frequently as antiemetic prophylaxis. Peripheral neuropathy has also been reported in 2% to 20% of patients receiving procarbazine. Patients typically present with a subjective paresthesia, which usually resolves completely on cessation of treatment.136,137
TAXANES
The taxanes paclitaxol and docetaxol both act by stabilizing microtubule polymerization, thus preventing the completion of successful mitosis. They are widely used in the treatment of breast, lung, and ovarian cancers. Unfortunately, their mechanism of action also leads to microtubule accumulation within axons, interfering with axonal transport and causing neurotoxicity. This is predominantly in the form of a distal peripheral polyneuropathy; patients typically present with painful paresthesia in a glove-and-stocking distribution, associated with reduced vibration sense, absent deep tendon reflexes, and difficulty with activities of daily living.138 Infrequently, there may also be a mild motor neuropathy, proximal muscle weakness, or autonomic neuropathy. Histologically, there is axonal atrophy, lack of axonal sprouting, and secondary demyelination predominantly involving small pain and temperature fibers, although motor and large sensory fibers may also be affected to a lesser extent. Risk of neuropathy is greater with paclitaxol than docetaxol139 and increases with dose, cumulative exposure, diabetes, or other preexisting neuropathy, and multiple sclerosis. Once peripheral neuropathy has developed, the taxane should be stopped and spontaneous recovery will usually occur over several months. Pyridoxine may speed recovery in the case of docetaxol, and amitryptiline or gabapentin may be useful if neuropathic pain is significant. Although no neuroprotective agents can be recommended at present, laboratory studies with nerve growth factor,140 ACTH analogues,141 and phase I clinical trials of glutamate and amifostine25 have produced some encouraging results.
CNS toxicity is rare, reflecting poor penetrance of the blood-brain barrier, although there have been occasional case reports of paclitaxol precipitating a mild encephalopathy. Presentation was with confusion, dysphasia, and nonspecific diffuse slowing on EEG, and recurred on reexposure.142 Transient visual disturbances have also been documented during taxane infusions, which may respond to a slowing of the rate of infusion.
VINCA ALKALOIDS
The vinca alkaloids vincristine, vinblastine, vindesine, and vinorelbine are widely used in the treatment of hematological malignancies, sarcomas, breast cancer, and lung cancer. They act by binding tubulin and thus inhibit microtubule polymerization during mitosis.9 Neurotoxicity, in the form of peripheral or autonomic neuropathy, occurs with all of these drugs, especially with vincristine. It is less frequently reported with vinblastine, vindesine, or vinorelbine. The neuropathy is probably caused by impairment of axonal flow due to inhibition of axonal microtubules, leading to axonal atrophy and adjacent myelin sheath damage. Patients usually present with a predominantly sensory peripheral neuropathy, which may be accompanied by the loss of deep tendon reflexes or, in the severest forms, distal muscle weakness. Constipation also occurs frequently due to reduced bowel motility, requiring treatment with stimulant laxatives. More rarely ocular palsies, voice hoarseness, and autonomic neuropathy (with postural hypotension and bladder atony) may occur. There have been several case reports of vincristine administration unmasking previously undiagnosed Charcot-Marie-Tooth syndrome, and therefore vinca alkaloids are not recommended for patients with hereditary polyneuropathies.143–148 Because of the fact that this toxicity is dose related and cumulative, the dose of vincristine is always capped at 2 mg. Concurrent administration of itraconazole or nifedipine has also been reported to enhance neurotoxicity by inhibiting vincristine degradation within the liver. Following development of significant neuropathy, vinca alkaloid treatment should either be reduced in dose or stopped. Symptomatic recovery usually occurs spontaneously, although this can take several months. There has been one case report of lithium (600 mg od PO) administration149 successfully reversing vincristine-induced peripheral neuropathy, allowing vincristine therapy to continue.
When administered intrathecally, vincristine is usually fatal due an ascending myeloencephalopathy causing ascending paralysis, coma, and death. At postmortem, there is widespread necrosis within the central nervous system, particularly the periventricular regions. Even so, there are several cases worldwide each year of inadvertent intrathecal administration,150–153 usually as a result of a series of procedural errors including mistaking vincristine for an intended intrathecal drug, not checking physician orders, mistaking the route of administration and mislabeling of syringes.154 Stringent regulations have now been introduced in the UK regarding who can administer intrathecal chemotherapy, and where and how this takes place. Once this mistake has been discovered, cerebrospinal fluid exchange should be initiated immediately and continued until as much of the vincristine as possible has been recovered.151,154 Unfortunately, most patients still suffer extensive neurological damage resulting in death despite aggressive cerebrospinal lavage.
Biological Agents
Since the late 1990s, the search for new anticancer agents has switched away from classical cytotoxics that kill cancer cells by inducing DNA damage to agents that modify growth signaling pathways. There are two main types of agents, the -mabs, which are monoclonal antibodies that target either cell surface receptors (e.g., trastuzumab against Her 2 receptors) or circulating ligand (e.g., bevacizumab against VEGF), and the -ibs, which are small drug molecules that target intracellular stages of signaling pathways (e.g., erlotinib which inhibits the tyrosine kinase complexed to the intracellular portion of the epidermal growth factor (EGF) receptor). In general, these drugs have a much gentler side effect profile compared with cytotoxic agents and very little in the way of neurotoxicity has been reported in the literature to date, although there have been isolated case reports of reversible posterior leukoencephalopathy (RPLE) with bevacizumab and sorafenib.155–157 This condition typically presents with headache, cortical blindness, and seizures. MRI imaging reveals vasogenic edema in the posterior cerebral white matter. Although the exact etiology is unknown, RPLE is frequently associated with hypertension, which is a commonly reported side effect of this group of drugs. Concomitant cytotoxic chemotherapy may also be an additional influence on the development of RPLE by causing endothelial damage or vasospasm.
Bortezomib, a proteosome inhibitor used in patients with multiple myeloma, causes a peripheral sensory neuropathy in one third of patients.158 On examination, affected patients typically have reduced ankle reflexes, decreased vibration sensation, and impaired heel-toe gait. Symptoms generally improve with dose reduction or discontinuation of therapy. More rarely, it has been associated with life-threatening motor neuropathy.159
Thalidomide is also currently widely used in myeloma because of its immunomodulatory and antiangiogenic effects. Unfortunately, up to 50% of patients develop a distal sensory peripheral neuropathy and a smaller number, a motor neuropathy. Patients may benefit from dose reductions when these complications occur. Newer thalidomide analogues such as lenalidomide seem to have a more favorable toxicity profile with reduced incidences of neuropathy (<5%).160
1. C.A. Meyers. How chemotherapy damages the central nervous system. J Biol. 2008;7:11.
2. J.S. Wefel, et al. The cognitive sequelae of standard dose adjuvant chemotherapy in women with breast cancer: results of a prospective, randomised longitudinal trial. Cancer. 2004;100:2292-2299.
3. T.A. Ahles, et al. Neuropsychological impact of standard dose chemotherapy in long-term survivors of breast cancer and lymphoma. J Clin Oncol. 2002;20:485-493.
4. S.B. Schagen, et al. Cognitive deficits after post-operative chemotherapy for breast cancer. Cancer. 1999;85:640-650.
5. R. Han, et al. Systemmic 5-fluorouracil treatment causes a syndrome of delayed myelin destruction in the central nervous system. J Biol. 2008;7:12.
6. M. Gilbert. Neurologic complications. In: M.D. Abeloff, editor. Clinical Oncology. Philadelphia: Churchill Livingstone, 2000.
7. R. Korinthenberg, et al. On the origin of EEG-slowing and encephalopathy during induction treatment of acute lymphoblastic leukemia. Med Pediatr Oncol. 2002;39(6):566-572.
8. W.J. Baker, G.L. RoyerJr, R.B. Weiss. Cytarabine and neurologic toxicity. J Clin Oncol. 1991;9(4):679-693.
9. C.C. Verstappen, et al. Neurotoxic complications of chemotherapy in patients with cancer: clinical signs and optimal management. Drugs. 2003;63(15):1549-1563.
10. M. Saumoy, et al. Progressive multifocal leukoencephalopathy in chronic lymphocytic leukemia after treatment with fludarabine. Leuk Lymphoma. 2002;43(2):433-436.
11. S.M. Choi, et al. 5-fluorouracil-induced leukoencephalopathy in patients with breast cancer. J Korean Med Sci. 2001;16(3):328-334.
12. J. Honkaniemi, et al. Reversible posterior leukoencephalopathy after combination chemotherapy. Neuroradiology. 2000;42(12):895-899.
13. H. Gonzalez, et al. Progressive multifocal leukoencephalitis (PML) in three patients treated with standard-dose fludarabine (FAMP). Hematol Cell Ther. 1999;41(4):183-186.
14. D.L. Hoffman, et al. Encephalopathy, myelopathy, optic neuropathy, and anosmia associated with intravenous cytosine arabinoside. Clin Neuropharmacol. 1993;16(3):258-262.
15. L.H. Leung, et al. White-matter diffusion anisotropy after chemo-irradiation: a statistical parametric mapping study and histogram analysis. Neuroimage. 2004;21(1):261-268.
16. A. Davidson, et al. Proton magnetic resonance spectroscopy ((1)H-MRS) of the brain following high-dose methotrexate treatment for childhood cancer. Med Pediatr Oncol. 2000;35(1):28-34.
17. R.D. Rao, et al. Methotrexate induced seizures associated with acute reversible magnetic resonance imaging (MRI) changes in a patient with acute lymphoblastic leukemia. Leuk Lymphoma. 2002;43(6):1333-1336.
18. D.J. Vaughn, et al. High-dose cytarabine neurotoxicity: MR findings during the acute phase. AJNR Am J Neuroradiol. 1993;14(4):1014-1016.
19. H.M. Liu, et al. Methotrexate encephalopathy. A neuropathologic study. Hum Pathol. 1978;9(6):635-648.
20. J.Y. Blay, et al. High-dose methotrexate for the treatment of primary cerebral lymphomas: analysis of survival and late neurologic toxicity in a retrospective series. J Clin Oncol. 1998;16(3):864-871.
21. C. Barbieux, et al. (Acute cerebellar syndrome after treatment with 5-fluorouracil). Bull Cancer. 1996;83(1):77-80.
22. N.A. Pirzada, I.I. Ali, R.M. Dafer. Fluorouracil-induced neurotoxicity. Ann Pharmacother. 2000;34(1):35-38.
23. P.S. Hodgson, et al. The neurotoxicity of drugs given intrathecally (spinal). Anesth Analg. 1999;88(4):797-809.
24. A.W. Clark, et al. Paraplegia following intrathecal chemotherapy: neuropathologic findings and elevation of myelin basic protein. Cancer. 1982;50(1):42-47.
25. H. Makino. Treatment and care of neurotoxicity from taxane anticancer agents. Breast Cancer. 2004;11(1):100-104.
26. N.A. Othieno-Abinya, L.O. Nyabola. Experience with vincristine—associated neurotoxicity. East Afr Med J. 2001;78(7):376-378.
27. P.H. Hilkens, et al. Peripheral neuropathy induced by combination chemotherapy of docetaxel and cisplatin. Br J Cancer. 1997;75(3):417-422.
28. D. Cella, et al. Measuring the side effects of taxane therapy in oncology: the functional assesment of cancer therapy-taxane (FACT-taxane). Cancer. 2003;98(4):822-831.
29. J.H. Uhm, W.K. Yung. Neurologic Complications of Cancer Therapy. Curr Treat Options Neurol. 1999;1(5):428-437.
30. G. Cavaletti, et al. Grading of chemotherapy-induced peripheral neurotoxicity using the Total Neuropathy Scale. Neurology. 2003;61(9):1297-1300.
31. S. Quasthoff, H.P. Hartung. Chemotherapy-induced peripheral neuropathy. J Neurol. 2002;249(1):9-17.
32. U. Schlegel, et al. Neurologic sequelae of treatment of primary CNS lymphomas. J Neurooncol. 1999;43(3):277-286.
33. M.J. Taphoorn. Neurocognitive sequelae in the treatment of low-grade gliomas. Semin Oncol. 2003;30(6 Suppl. 19):45-48.
34. C.A. Pardo, J.C. McArthur, J.W. Griffin. HIV neuropathy: insights in the pathology of HIV peripheral nerve disease. J Peripher Nerv Syst. 2001;6(1):21-27.
35. L. Aloe, et al. Evidence that nerve growth factor promotes the recovery of peripheral neuropathy induced in mice by cisplatin: behavioral, structural and biochemical analysis. Auton Neurosci. 2000;86(1–2):84-93.
36. C. Bokemeyer, et al. Silibinin protects against cisplatin-induced nephrotoxicity without compromising cisplatin or ifosfamide anti-tumour activity. Br J Cancer. 1996;74(12):2036-2041.
37. R.L. Capizzi. Protection of normal tissues from the cytotoxic effects of chemotherapy by amifostine (Ethyol): clinical experiences. Semin Oncol. 1994;21(5 Suppl. 11):8-15.
38. G. Cavaletti, et al. Neuroprotectant drugs in cisplatin neurotoxicity. Anticancer Res. 1996;16(5B):3149-3159.
39. S. Cascinu, et al. Neuroprotective effect of reduced glutathione on oxaliplatin-based chemotherapy in advanced colorectal cancer: a randomized, double-blind, placebo-controlled trial. J Clin Oncol. 2002;20(16):3478-3483.
40. J. Cassidy, et al. Clinical trials of nimodipine as a potential neuroprotector in ovarian cancer patients treated with cisplatin. Cancer Chemother Pharmacol. 1998;41(2):161-166.
41. F. Eckel, et al. (Prevention of oxaliplatin-induced neuropathy by carbamazepine. A pilot study). Dtsch Med Wochenschr. 2002;127(3):78-82.
42. J.A. Foster-Nora, R. Siden. Amifostine for protection from antineoplastic drug toxicity. Am J Health Syst Pharm. 1997;54(7):787-800.
43. O. Kanat, et al. Protective effect of amifostine against toxicity of paclitaxel and carboplatin in non-small cell lung cancer: a single center randomized study. Med Oncol. 2003;20(3):237-245.
44. C. Lersch, et al. Prevention of oxaliplatin-induced peripheral sensory neuropathy by carba-mazepine in patients with advanced colorectal cancer. Clin Colorectal Cancer. 2002;2(1):54-58.
45. M. Links, C. Lewis. Chemoprotectants: a review of their clinical pharmacology and therapeutic efficacy. Drugs. 1999;57(3):293-308.
46. M. Orditura, et al. Amifostine: A selective cytoprotective agent of normal tissues from chemo-radiotherapy induced toxicity (Review). Oncol Rep. 1999;6(6):1357-1362.
47. A. Pace, et al. Neuroprotective effect of vitamin E supplementation in patients treated with cisplatin chemotherapy. J Clin Oncol. 2003;21(5):927-931.
48. O. Rick, et al. Assessment of amifostine as protection from chemotherapy-induced toxicities after conventional-dose and high-dose chemotherapy in patients with germ cell tumor. Ann Oncol. 2001;12(8):1151-1155.
49. J.F. Smyth, et al. Glutathione reduces the toxicity and improves quality of life of women diagnosed with ovarian cancer treated with cisplatin: results of a double-blind, randomised trial. Ann Oncol. 1997;8(6):569-573.
50. C.M. Spencer, K.L. Goa. Amifostine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential as a radioprotector and cytotoxic chemoprotector. Drugs. 1995;50(6):1001-1031.
51. Y. Sumiyoshi, et al. (Glutathione chemoprotection therapy against CDDP-induced neurotoxicity in patients with invasive bladder cancer). Gan To Kagaku Ryoho. 1996;23(11):1506-1508.
52. C.S. Viele, B.C. Holmes. Amifostine: drug profile and nursing implications of the first pancytoprotectant. Oncol Nurs Forum. 1998;25(3):515-523.
53. J. Hildebrand. Neurological complications of cancer chemotherapy. Curr Opin Oncol. 2006;18(4):321-324.
54. H.A. Bygrave, et al. Neurological complications of 5-fluorouracil chemotherapy: case report and review of the literature. Clin Oncol (R Coll Radiol). 1998;10(5):334-336.
55. D.H. Moore, W.C. FowlerJr, L.S. Crumpler. 5-Fluorouracil neurotoxicity. Gynecol Oncol. 1990;36(1):152-154.
56. S.S. Ki, et al. A case of neurotoxicity following 5-fluorouracil-based chemotherapy. Korean J Intern Med. 2002;17(1):73-77.
57. N. Shehata, A. Pater, S.C. Tang. Prolonged severe 5-fluorouracil-associated neurotoxicity in a patient with dihydropyrimidine dehydrogenase deficiency. Cancer Invest. 1999;17(3):201-205.
58. C.H. Takimoto, et al. Severe neurotoxicity following 5-fluorouracil-based chemotherapy in a patient with dihydropyrimidine dehydrogenase deficiency. Clin Cancer Res. 1996;2(3):477-481.
59. G.B. Morrison. Dihydropyrimidine dehydrogenase deficiency: a pharmacogenetic defect causing severe adverse reactions to 5-fluorouracil-based chemotherapy. Oncol Nurs Forum. 1997;24(1):83-88.
60. C.H. Hsu, et al. Weekly 24-hour infusion of high-dose 5-fluorouracil and leucovorin in the treatment of advanced gastric cancers. An effective and low-toxic regimen for patients with poor general condition. Oncology. 1997;54(4):275-280.
61. A.T. Figueredo, et al. Disabling encephalopathy during 5-fluorouracil and levamisole adjuvant therapy for resected colorectal cancer: a report of two cases. Cancer Invest. 1995;13(6):608-611.
62. C.L. Murray, et al. Multifocal inflammatory leukoencephalopathy after fluorouracil and levamisole therapy for colon cancer. AJNR Am J Neuroradiol. 1997;18(8):1591-1592.
63. P. Saletti, et al. Two cases of neurotoxicity possibly related to 5-fluorouracil and FA administration. Ann Oncol. 1996;7(2):213-214.
64. W.S. Wang, et al. Weekly 24-hour infusion of high-dose 5-fluorouracil and leucovorin in patients with advanced colorectal cancer: Taiwan experience. Jpn J Clin Oncol. 1998;28(1):16-19.
65. H.W. van Laarhoven, et al. 5-FU-induced peripheral neuropathy: a rare complication of a well-known drug. Anticancer Res. 2003;23(1B):647-648.
66. M. Ray, R.K. Marwaha, A. Trehan. Chemotherapy related fatal neurotoxicity during induction in acute lymphoblastic leukemia. Indian J Pediatr. 2002;69(2):185-187.
67. W.M. Feinberg, M.R. Swenson. Cerebrovascular complications of L-asparaginase therapy. Neurology. 1988;38(1):127-133.
68. J.C. Holland. Psychiatric symptoms assocated with L-asparaginase therapy. J Psychiatr Res. 1974;10:174.
69. M.C. Perry, C.M. Anderson, R.C. Donehower. Chemotherapy. In: M.D. Abeloff, editor. Clinical Oncology. Philadelphia: Churchill Livingstone, 2000.
70. M.J. Webberley, J.A. Murray. Life-threatening acute hyponatraemia induced by low dose cyclophosphamide and indomethacin. Postgrad Med J. 1989;65(770):950-952.
71. Y. Tsujita, et al. (Syndrome of inappropriate secretion of antidiuretic hormone and neurotoxicity induced by vincristine and alkylating agents during chemotherapy for malignant lymphoma of thyroid gland). Gan To Kagaku Ryoho. 1998;25(5):757-760.
72. R.A. DeFronzo, et al. Proceedings: Cyclophosphamide and the kidney. Cancer. 1974;33(2):483-491.
73. G. Kende, et al. Blurring of vision: a previously undescribed complication of cyclophosphamide therapy. Cancer. 1979;44(1):69-71.
74. C.K. Tashima. Immediate cerebral symptoms during rapid intravenous administration of cyclophosphamide (NSC-26271). Cancer Chemother Rep. 1975;59(2 Pt 1):441-442.
75. G. Schiller, M. Lee. Long-term outcome of high-dose cytarabine-based consolidation chemotherapy for older patients with acute myelogenous leukemia. Leuk Lymphoma. 1997;25(1–2):111-119.
76. G.A. Smith, et al. High-dose cytarabine dose modification reduces the incidence of neurotoxicity in patients with renal insufficiency. J Clin Oncol. 1997;15(2):833-839.
77. D.P. Martin, T.L. Wallace, E.M. JohnsonJr. Cytosine arabinoside kills postmitotic neurons in a fashion resembling trophic factor deprivation: evidence that a deoxycytidine-dependent process may be required for nerve growth factor signal transduction. J Neurosci. 1990;10(1):184-193.
78. H.M. Geller, et al. Oxidative stress mediates neuronal DNA damage and apoptosis in response to cytosine arabinoside. J Neurochem. 2001;78(2):265-275.
79. J. Garcia-Tena, et al. Intrathecal chemotherapy-related myeloencephalopathy in a young child with acute lymphoblastic leukemia. Pediatr Hematol Oncol. 1995;12(4):377-385.
80. A. Ozon, et al. Acute ascending myelitis and encephalopathy after intrathecal cytosine arabinoside and methotrexate in an adolescent boy with acute lymphoblastic leukemia. Brain and Development. 1994;16(3):246-248.
81. A. Ruggiero, et al. Intrathecal chemotherapy with antineoplastic agents in children. Paediatr Drugs. 2001;3(4):237-246.
82. A. Borgeat, B. De Muralt, M. Stalder. Peripheral neuropathy associated with high-dose Ara-C therapy. Cancer. 1986;58(4):852-854.
83. K.R. Imrie, et al. Peripheral neuropathy following high-dose etoposide and autologous bone marrow transplantation. Bone Marrow Transplant. 1994;13(1):77-79.
84. R.S. Leff, et al. Acute neurologic dysfunction after high-dose etoposide therapy for malignant glioma. Cancer. 1988;62(1):32-35.
85. H.G. Chun, et al. Central nervous system toxicity of fludarabine phosphate. Cancer Treat Rep. 1986;70(10):1225-1228.
86. T. Meyer, A.C. Ludolph, C. Munch. Ifosfamide encephalopathy presenting with asterixis. J Neurol Sci. 2002;199(1–2):85-88.
87. A. Primavera, D. Audenino, L. Cocito. Ifosfamide encephalopathy and nonconvulsive status epilepticus. Can J Neurol Sci. 2002;29(2):180-183.
88. J.P. Curtin, et al. Ifosfamide-induced neurotoxicity. Gynecol Oncol. 1991;42(3):193-196. discussion 191–2
89. A. Comandone, et al. Two episodes of ifosfamide-related neurotoxicity in the same patient following different schedules and doses of the drug. A case report. Tumori. 2000;86(6):483-486.
90. L. Carlson, et al. Toxicity, pharmacokinetics, and in vitro hemodialysis clearance of ifosfamide and metabolites in an anephric pediatric patient with Wilms’ tumor. Cancer Chemother Pharmacol. 1998;41(2):140-146.
91. H.J. Keizer, et al. Ifosfamide treatment as a 10-day continuous intravenous infusion. J Cancer Res Clin Oncol. 1995;121(5):297-302.
92. C. Sood, P.J. O’Brien. 2-Chloroacetaldehyde-induced cerebral glutathione depletion and neurotoxicity. Br J Cancer Suppl. 1996;27:S287-S293.
93. A.R. Turner, C.D. Duong, D.J. Good. Methylene blue for the treatment and prophylaxis of ifosfamide-induced encephalopathy. Clin Oncol (R Coll Radiol). 2003;15(7):435-439.
94. A.B. Raj, S.J. Bertolone, N. Jaffe. Methylene blue reversal of ifosfamide-related encephalopathy. J Pediatr Hematol Oncol. 2004;26(2):116.
95. A. Kupfer, C. Aeschlimann, T. Cerny. Methylene blue and the neurotoxic mechanisms of ifosfamide encephalopathy. Eur J Clin Pharmacol. 1996;50(4):249-252.
96. A. Kupfer, et al. Prophylaxis and reversal of ifosfamide encephalopathy with methylene-blue. Lancet. 1994;343(8900):763-764.
97. J.M. Hill, et al. A comparative study of the long term psychosocial functioning of childhood acute lymphoblastic leukemia survivors treated by intrathecal methotrexate with or without cranial radiation. Cancer. 1998;82(1):208-218.
98. R.W. Walker, et al. Transient cerebral dysfunction secondary to high-dose methotrexate. J Clin Oncol. 1986;4(12):1845-1850.
99. P.C. Phillips, et al. Acute high-dose methotrexate neurotoxicity in the rat. Ann Neurol. 1986;20(5):583-589.
100. P.C. Phillips, et al. High-dose leucovorin reverses acute high-dose methotrexate neurotoxicity in the rat. Ann Neurol. 1989;25(4):365-372.
101. J.C. Bernini, et al. Aminophylline for methotrexate-induced neurotoxicity. Lancet. 1995;345(8949):544-547.
102. S. Kishi, et al. Homocysteine, pharmacogenetics, and neurotoxicity in children with leukemia. J Clin Oncol. 2003;21(16):3084-3091.
103. C.T. Quinn, et al. Elevation of homocysteine and excitatory amino acid neurotransmitters in the CSF of children who receive methotrexate for the treatment of cancer. J Clin Oncol. 1997;15(8):2800-2806.
104. F. Millot, et al. Changes of cerebral biopterin and biogenic amine metabolism in leukemic children receiving 5 g/m2 intravenous methotrexate. Pediatr Res. 1995;37(2):151-154.
105. M.G. Mott, P. Stevenson, C.B. Wood. Methotrexate meningitis. Lancet. 1972;2(7778):656.
106. S.E. Bates, et al. Ascending myelopathy after chemotherapy for central nervous system acute lymphoblastic leukemia: correlation with cerebrospinal fluid myelin basic protein. Med Pediatr Oncol. 1985;13(1):4-8.
107. R. Surtees, J. Clelland, I. Hann. Demyelination and single-carbon transfer pathway metabolites during the treatment of acute lymphoblastic leukemia: CSF studies. J Clin Oncol. 1998;16(4):1505-1511.
108. H.H. Lien, et al. Osteogenic sarcoma: MR signal abnormalities of the brain in asymptomatic patients treated with high-dose methotrexate. Radiology. 1991;179(2):547-550.
109. T.J. Postma, et al. Neurotoxicity of combination chemotherapy with procarbazine, CCNU and vincristine (PCV) for recurrent glioma. J Neurooncol. 1998;38(1):69-75.
110. B.J. Shingleton, et al. Ocular toxicity associated with high-dose carmustine. Arch Ophthalmol. 1982;100(11):1766-1772.
111. M.K. Rosenblum, et al. Fatal necrotizing encephalopathy complicating treatment of malignant gliomas with intra-arterial BCNU and irradiation: a pathological study. J Neurooncol. 1989;7(3):269-281.
112. W.B. Wilson, G.M. Perez, B.K. Kleinschmidt-Demasters. Sudden onset of blindness in patients treated with oral CCNU and low-dose cranial irradiation. Cancer. 1987;59(5):901-907.
113. E. Donzelli, et al. Neurotoxicity of platinum compounds: comparison of the effects of cisplatin and oxaliplatin on the human neuroblastoma cell line SH-SY5Y. J Neurooncol. 2004;67(1–2):65-73.
114. P. Gent, K. Massey. An overview of chemotherapy-induced peripheral sensory neuropathy, focusing on oxaliplatin. Int J Palliat Nurs. 2001;7(7):354-359.
115. A. Grothey. Oxaliplatin-safety profile: neurotoxicity. Semin Oncol. 2003;30(4 Suppl. 15):5-13.
116. G. Cavaletti, et al. Neurotoxicity and ototoxicity of cisplatin plus paclitaxel in comparison to cisplatin plus cyclophosphamide in patients with epithelial ovarian cancer. J Clin Oncol. 1997;15(1):199-206.
117. S.D. Fossa, et al. Clinical and biochemical long-term toxicity after postoperative cisplatin-based chemotherapy in patients with low-stage testicular cancer. Oncology. 1995;52(4):300-305.
118. G.M. Higa, T.C. Wise, E.B. Crowell. Severe, disabling neurologic toxicity following cisplatin retreatment. Ann Pharmacother. 1995;29(2):134-137.
119. F. Keime-Guibert, M. Napolitano, J.Y. Delattre. Neurological complications of radiotherapy and chemotherapy. J Neurol. 1998;245(11):695-708.
120. V. Lorusso, et al. Carboplatin plus ifosfamide as salvage treatment of epithelial ovarian cancer: a pilot study. J Clin Oncol. 1993;11(10):1952-1956.
121. J.P. Neijt, B. Lund. Paclitaxel with carboplatin for the treatment of ovarian cancer. Semin Oncol. 1996;23(6 Suppl. 15):2-4.
122. P.M. Petersen, S.W. Hansen. The course of long-term toxicity in patients treated with cisplatin-based chemotherapy for non-seminomatous germ-cell cancer. Ann Oncol. 1999;10(12):1475-1483.
123. K.K. Fu, E.F. Kai, C.K. Leung. Cisplatin neuropathy: a prospective clinical and electrophysiological study in Chinese patients with ovarian carcinoma. J Clin Pharm Ther. 1995;20(3):167-172.
124. L. Troy, et al. Cisplatin-based therapy: a neurological and neuropsychological review. Psychooncology. 2000;9(1):29-39.
125. M. von Schlippe, C.J. Fowler, S.J. Harland. Cisplatin neurotoxicity in the treatment of metastatic germ cell tumour: time course and prognosis. Br J Cancer. 2001;85(6):823-826.
126. J. Cassidy, J.L. Misset. Oxaliplatin-related side effects: characteristics and management. Semin Oncol. 2002;29(5 Suppl. 15):11-20.
127. R.W. Gregg, et al. Cisplatin neurotoxicity: the relationship between dosage, time, and platinum concentration in neurologic tissues, and morphologic evidence of toxicity. J Clin Oncol. 1992;10(5):795-803.
128. F. Salvinelli, et al. Bilateral irreversible hearing loss associated with the combination of carboplatin and paclitaxel chemotherapy: a unusual side effect. J Exp Clin Cancer Res. 2003;22(1):155-158.
129. C. Kollmannsberger, et al. Late toxicity following curative treatment of testicular cancer. Semin Surg Oncol. 1999;17(4):275-281.
130. E.A. Calhoun, et al. Perceptions of cisplatin-related toxicity among ovarian cancer patients and gynecologic oncologists. Gynecol Oncol. 1998;71(3):369-375.
131. C. Bokemeyer, et al. Evaluation of long-term toxicity after chemotherapy for testicular cancer. J Clin Oncol. 1996;14(11):2923-2932.
132. U.B. Chaudhary, J.R. Haldas. Long-term complications of chemotherapy for germ cell tumours. Drugs. 2003;63(15):1565-1577.
133. M. Perol, et al. Multicenter randomized trial comparing cisplatin-mitomycin-vinorelbine versus cisplatin-mitomycin-vindesine in advanced non-small cell lung cancer. ‘Groupe Francais de Pneumo-Cancerologie’. Lung Cancer. 1996;14(1):119-134.
134. R.J. van der Hulst, W.A. Dreschler, N.A. Urbanus. High frequency audiometry in prospective clinical research of ototoxicity due to platinum derivatives. Ann Otol Rhinol Laryngol. 1988;97(2 Pt 1):133-137.
135. L. Gamelin, M. Boisdron-Celle, R. Delva. Prevention of oxaliplatin related neurotoxicity by calcium and magnesium infusions: A retrospective study of 161 patints receiving oxaliplatin combined with 5-fluorouracil and leucovorin for advanced colorectal cancer. Clin Cancer Res. 2004;10:4055-4061.
136. S.D. Spivack. Drugs 5 years later: procarbazine. Ann Int Med. 1974;81(6):795-800.
137. D.C. Stolinsky, et al. Clinical experience with procarbazine in Hodgkin’s disease, reticulum cell sarcoma, and lymphosarcoma. Cancer. 1970;26(5):984-990.
138. K. Kuroi, K. Shimozuma. Neurotoxicity of taxanes: symptoms and quality of life assessment. Breast Cancer. 2004;11(1):92-99.
139. J.P. Guastalla3rd, V. Dieras. The taxanes: toxicity and quality of life considerations in advanced ovarian cancer. Br J Cancer. 2003;89(Suppl. 3):S16-S22.
140. S.C. Apfel, et al. Nerve growth factor prevents toxic neuropathy in mice. Ann Neurol. 1991;29(1):87-90.
141. F.P. Hamers, et al. The ACTH-(4–9) analog, ORG 2766, prevents taxol-induced neuropathy in rats. Eur J Pharmacol. 1993;233(1):177-178.
142. J.R. Perry, E. Warner. Transient encephalopathy after paclitaxel (Taxol) infusion. Neurology. 1996;46(6):1596-1599.
143. R. Naumann, et al. Early recognition of hereditary motor and sensory neuropathy type 1 can avoid life-threatening vincristine neurotoxicity. Br J Haematol. 2001;115(2):323-325.
144. A.R. Chauvenet, et al. Vincristine-induced neuropathy as the initial presentation of charcot-marie-tooth disease in acute lymphoblastic leukemia: a Pediatric Oncology Group study. J Pediatr Hematol Oncol. 2003;25(4):316-320.
145. A.D. Trobaugh-Lotrario, A.A. Smith, L.F. Odom. Vincristine neurotoxicity in the presence of hereditary neuropathy. Med Pediatr Oncol. 2003;40(1):39-43.
146. S. Uno, et al. (Acute vincristine neurotoxicity in a non-Hodgkin’s lymphoma patient with Charcot-Marie-Tooth disease). Rinsho Ketsueki. 1999;40(5):414-419.
147. Y. Neumann, et al. Vincristine treatment triggering the expression of asymptomatic Charcot-Marie-Tooth disease. Med Pediatr Oncol. 1996;26(4):280-283.
148. A.M. Orejana-Garcia, J. Pascual-Huerta, A. Perez-Melero. Charcot-Marie-Tooth disease and vincristine. J Am Podiatr Med Assoc. 2003;93(3):229-233.
149. M. Petrini, et al. Is lithium able to reverse neurological damage induced by vinca alkaloids? (Short communication). J Neural Transm. 1999;106(5–6):569-575.
150. A. Al Ferayan, et al. Cerebrospinal fluid lavage in the treatment of inadvertent intrathecal vincristine injection. Childs Nerv Syst. 1999;15(2–3):87-89.
151. A. Alcaraz, et al. Intrathecal vincristine: fatal myeloencephalopathy despite cerebrospinal fluid perfusion. J Toxicol Clin Toxicol. 2002;40(5):557-561.
152. E.K. Kwack, et al. Neural toxicity induced by accidental intrathecal vincristine administration. J Korean Med Sci. 1999;14(6):688-692.
153. W.J. Meggs, R.S. Hoffman. Fatality resulting from intraventricular vincristine administration. J Toxicol Clin Toxicol. 1998;36(3):243-246.
154. C.V. Fernandez, et al. Intrathecal vincristine: an analysis of reasons for recurrent fatal chemotherapeutic error with recommendations for prevention. J Pediatr Hematol Oncol. 1998;20(6):587-590.
155. P. Glusker, L. Recht, B. Lane. Reversible posterior leukoencephalopathy syndrome and bevacizumab. N Engl J Med. 2006;354(9):980-982. discussion 980–2
156. S. Peter, et al. Reversible posterior leukoencephalopathy syndrome and intravenous bevacizumab. Clin Experiment Ophthalmol. 2008;36(1):94-96.
157. C. Vaughn, L. Zhang, D. Schiff. Reversible posterior leukoencephalopathy syndrome in cancer. Curr Oncol Rep. 2008;10(1):86-91.
158. A. Badros, et al. Neurotoxicity of bortezomib therapy in multiple myeloma: a single-center experience and review of the literature. Cancer. 2007;110(5):1042-1049.
159. S. Gupta, et al. Life-threatening motor neurotoxicity in association with bortezomib. Haematologica. 2006;91(7):1001.
160. G. Kannarkat, E.E. Lasher, D. Schiff. Neurologic complications of chemotherapy agents. Curr Opin Neurol. 2007;20(6):719-725.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
16 Neurotoxicity of Chemotherapy
Types of Neurological Damage
COGNITIVE DYSFUNCTION
Cancer patients have frequently recognized decreased cognitive function (“chemo-brain”) during chemotherapy, which, in the past, was attributed by their physicians to stress or depression. Patients report problems with memory retrieval, learning, and concentration, which may persist after treatment has finished or never fully resolve.1 The incidence of acute problems during treatment ranges from 15% to 70%, with 50% of patients in one study identifying persistent problems a year after treatment.2 Cross-sectional studies also suggest persistent cognitive dysfunction in 20% to 30% of patients 2 to 10 years posttreatment.3,4
Mechanisms for this functional decline are not fully understood, but recent work by Han et al.5 on 5-fluorouracil neurotoxicity in mice suggest extensive myelin damage and persistent suppression of both oligodendrocyte and progenitor cell proliferation in the subventricular zone, hippocampus, and corpus callosum.
ACUTE ENCEPHALOPATHY
Acute encephalopathy is a common problem in oncology patients; it has a wide range of precipitating factors including metabolic derangements, hypoxia, brain metastases, meningeal carcinomatosis, infection, paraneoplastic phenomena, and drugs.6 Presenting symptoms typically include lethargy, confusion, somnolence, seizures, or coma. The diagnosis of drug-induced encephalopathy is typically one of exclusion. A careful drug history should be taken, including recent administration of narcotic analgesia and antiemetic cover, used frequently for cancer patients undergoing chemotherapy and/or for symptom control. CNS infections should also be excluded, particularly in immunocompromised and neutropenic patients. Investigations should include urea/electrolytes, liver function, serum glucose, calcium magnesium, viral serology, and CSF examination. If focal neurological deficit is present, CT or MRI imaging may be helpful and, in any case, should be undertaken prior to a lumbar puncture. EEG is particularly helpful if seizures occur, and will typically show generalized slowing with delta wave activity.7 Encephalopathy due to cytotoxic drug exposure is generally self-limiting and recovers spontaneously. The only specific therapy is the use of methylene blue in ifosfamide-induced encephalopathy, which should be considered in any patients undergoing ifosfamide chemotherapy. Risk factors include extremes of age, dose/ schedule, previous cranial radiotherapy, and renal or hepatic dysfunction.8,9
LEUKOENCEPHALOPATHY
Leukoencephalopathy may follow on from acute encephalopathy, but may also be the first indication of neurotoxicity several months to years after administration of cytotoxic drugs. Patients present with cognitive deficits, which may progress to dementia, coma, and death.10–14 MRI imaging shows widespread changes throughout the white matter,15–18 and histologically, there is axonal swelling, demyelination, and neuronal death.19 Those most at risk are patients treated with methotrexate or cytarabine, particularly if given intrathecally or if cranial radiotherapy preceded cytotoxic administration.9 Elderly patients with primary central nervous system lymphoma undergoing treatment with high-dose methotrexate and whole-brain radiotherapy (WBRT) are at particularly high risk of developing this complication.20 There is no specific therapy that can halt the progressive decline, and overall the prognosis is poor. Elderly patients with primary CNS lymphoma should be informed about these risks; it should also be considered by the treating oncologists whether these patients can be treated with reduced-dose WBRT or avoid WBRT altogether after-high dose methotrexate.
CEREBELLAR DYSFUNCTION
Cerebellar signs in an oncology patient are usually due to direct spread of cancer, particularly if it is asymmetric. In rarer situations, this can be due to paraneoplastic syndromes, which tend to have a subacute onset and may be associated with the presence of antineuronal antibodies.6 Care should also be taken to distinguish cerebellar ataxia from sensory ataxia due to a severe sensory neuropathy. Cytarabine and 5-fluorouracil are the cytotoxics most likely to cause cerebellar dysfunction including truncal ataxia, gait disturbance, and ataxia.8,14,21,22 Acutely, the MRI tends to be normal, but subsequent scans may show chronic atrophy due to irreversible Purkinje cell loss.15–18
SPINAL CORD TOXICITY
Spinal cord toxicity can occur following intrathecal administration of certain cytotoxics in acute leukemias, lymphomas, and brain tumors. Intrathecal chemotherapy is administered either as part of a lumbar puncture procedure or into the ventricles, via an Ommaya reservoir.6 The drugs most commonly used are cytarabine, methotrexate, and hydrocortisone; they may be given singly, sequentially, or together as “triple therapy.” Symptoms usually arise after multiple cycles of therapy and include both spinal cord and nerve root signs. Loss of neurological function may progress upwards.23 Histologically, there are focal areas of necrosis, particularly at the periphery of the spinal cord, associated with axonal swelling and demyelination. Myelin basic protein levels in the CSF may be elevated.24 Typically, only half of those affected will show any sign of recovery.
PERIPHERAL NEUROPATHY
Peripheral neuropathies are the most common neurological complications in patients receiving chemotherapy, especially with regimens containing taxanes (taxol, docataxel), platinum (cisplatin, carboplatin, oxaliplatin), and vinca alkaloids (vincristine). The neuropathy tends to be predominantly sensory in nature, with a glove and stocking distribution. Generally, symptoms are self-limiting, but in some patients the symptoms persist. The effect is cumulative,6 and patients frequently complain of acute subjective paresthesia of the extremities 2 to 3 days after chemotherapy. With subsequent treatment cycles, symptoms may progress to permanent paresthesia, with decreased sensation to pinprick, light touch, and vibration on formal testing.9,25–29 At this stage, chemotherapy should be stopped or the dose reduced, as continuing can lead to difficulty with activities of daily living. The neuropathy is largely reversible over several months but many patients may be left with some degree of paresthesia. In rare instances, neuropathy may be paraneoplastic in origin. Oxaliplatin is unusual in that it causes acute cold dysesthesias, as well as pharyngolaryngospasm, which usually starts shortly after administration of chemotherapy and then resolves.
Grading Toxicity
There is no universal grading system for the evaluation of patients with neurological toxicity although two neurotoxicity scoring systems are frequently used: NCI-CTC 3 (National Cancer Institute Common Toxicity Criteria version 3) and ECOG (Eastern Cooperative Oncology Group).30 The NCI-CTCAE scores a variety of symptoms from ataxia to motor neuropathy on a scale of 0 to 5, with 0 being normal, 1 mild self-limiting, 2 moderate, 3 severe undesirable, 4 life-threatening, and 5 death induced by adverse event. The full version can be downloaded from www.fda.gov. The ECOG criteria are organized in a similar manner with a scale from 0 to 4 and can be viewed at www.ecog.org. Generally patients with a toxicity score of 1 to 2 can continue with their treatment unmodified, while those with a score of 3 or 4 require dose modification or cessation of treatment.
Prevention
The patients most at risk are those receiving a high cumulative dose or intensive schedule, particularly if there is a preexisting condition such as diabetes mellitus, hereditary neuropathy, or multiple sclerosis.31 Previous radiotherapy may also increase the risk of developing neurotoxicity if patients are subsequently treated with cisplatin or methotrexate.32,33
Patients with HIV-related malignancies are also at increased risk of cytotoxic-induced neuropathy, since both HIV and the drugs used to treat it (highly active antiretroviral therapy, or HAART) can cause neurological damage independently.34 Distal sensory neuropathy is the commonest form of HIV-associated neuropathy and can be difficult to distinguish from that caused by specific nucleoside antiretrovirals. In addition, compounded neurological toxicities frequently occur because of reduced clearance of vincristine, vinblastine, and taxane; these complications can be reduced by changing these patients’ antiretroviral medications. More rarely, patients can develop inflammatory demyelinating polyneuropathy, mononeuritis multiplex, or neuronal damage due to opportunistic infections such as CMV and HZV.
Currently there are few therapies able to prevent neurological toxicity preemptively. Infusions of methylene blue are used prophylatically in patients receiving ifosfamide who have previously developed acute encephalopathy. A number of trials have investigated the benefits of agents such as calcium-magnesium infusions, carbamazepine, gabapentin, amifostine, and glutathione.7,35–52 However, the trials are small studies and no specific prophylaxis can be routinely recommended.
Treatment of Neurotoxicity
Apart from dose reduction or discontinuing the drugs implicated in the development of neurotoxicity, there is very little in the way of specific pharmacological management to reverse their side effects. Methylene blue is used for ifosfamide-induced encephalopathy and infusional calcium with magnesium may lessen the severity of established peripheral neuropathy due to oxaliplatin. However, considerable input from the multidisciplinary team is required in the holistic care of the patient,53 including analgesia for neuropathic pain, maximization of mobility (with occupational and physiotherapist involvement where there is loss of balance, sensory loss, or muscle weakness), and good nursing care to manage bowel or bladder dysfunction, protection of pressure areas in immobilized patients, and maintenance of a safe environment if patients are confused.
Cytotoxic Agents and Their Neurological Toxicities (See Table 16-1)
5-FLUOROURACIL
5-fluorouracil (5-FU), a pyrimidine analogue, is widely used in the treatment of gastrointestinal malignancies; it is administered either as a short intravenous bolus or as a prolonged continuous infusion. Neurotoxicity is rare, but may include acute cerebellar dysfunction in 3% to 7% of patients, causing gait ataxia, nystagmus, and scanning speech.54,55 Symptoms tend to resolve spontaneously within a few days of treatment cessation although administration of thiamine may be helpful.22 Patients may also develop acute confusion in the absence of cerebellar signs, which may recur on reexposure to 5-FU.56 The main risk factor for development of neurotoxicity is deficiency of the enzyme dihydropyrimidine dehydrogenase which metabolizes 5-FU.57–59
A subacute form of leukoencephalopathy occurs in approximately 2% of patients receiving 5-FU in combination with levamisole (an immunomodulatory agent).60–64 Presentation is with focal neurological abnormalities and cognitive impairment that may be mistaken for metastatic disease. MRI shows patchy abnormalities within the white matter that enhance with gadolinium. Histologically, these patches contain an intense inflammatory infiltration with demyelination but axonal sparing. It is thought that the levamisole disrupts the blood-brain barrier potentiating 5-FU’s access to the CNS. Although treatment with steroids has been advocated, this syndrome is frequently self-limiting and patients usually recover completely over the course of several weeks without specific therapy. In some patients, thymidine has also been used successfully.58 Rarely, a peripheral sensory neuropathy has also been reported.65 Because many patients received adjuvant 5-FU based treatments for a variety of cancers, including breast, gastrointestinal, and bowel cancers, patients should be monitored for possible long-term neurocognitive damage.5
ASPARAGINASE
Asparaginase (either as the L- or pegylated formulation) is a component of remission-induction therapy used to treat acute lymphoblastic leukemia (ALL). It may cause cerebrovascular accidents during the first few weeks of its administration.66 This is due to depletion of plasma proteins involved in coagulation, such as fibrinogen and antithrombin III. Thrombosis typically occurs within the dural sinuses and cerebral veins, leading to secondary hemorrhage or infarction.67 Patients present with a range of symptoms from mild headache to coma and death, which are caused by rapid increases in intracranial pressure. Once diagnosed, the asparaginase should be stopped and the patient anticoagulated unless hemorrhage is present.
Asparaginase acts to cleave asparagine, an essential amino acid required by rapidly proliferating cells (hence its antimitotic action) and also as a neurotransmitter. Therefore depletion of asparagine during treatment has also been associated with the development of neuropsychiatric symptoms such as depression and hallucinations.68
CYCLOPHOSPHAMIDE
Cyclophosphamide is a prodrug, which alkylates DNA after hepatic activation.69 It is used in a wide range of malignancies, including lymphoma, breast cancer, and testicular cancer. When given in high doses, it may lead to inappropriate secretion of ADH (SIADH), and hence a secondary metabolic encephalopathy may occur, with confusion, seizures, or coma.70–72 There have been a few case reports of cyclophosphamide also being associated with blurred vision, dizziness, and confusion in the absence of SIADH.73,74
CYTARABINE
Cytarabine is an analogue of adenosine, causing chain termination during DNA synthesis; it is one of the most effective cytotoxic drugs in the treatment of acute leukemia. It is used either at a conventional dose of 100 to 200 mg/m2/day or at high doses of 2 to 6 g/m2/day.69 Neurotoxicity is particularly common at the higher dose levels, affecting 16% to 50% of patients;8 it predominantly affects the central nervous system. The risk of neurotoxicity is increased by age, dose/ schedule (particularly cumulative dose), renal or hepatic impairment, and the concurrent use of neurotropic antiemetics such as phenothiazines.8,75,76
The mechanism of toxicity is not well-understood, but it appears that cytarabine directly causes neuronal death, possibly by the inhibition of cytidine-dependent neurotropic signal transduction,77 although it has also been shown to stimulate the production of reactive oxygen species that may also damage DNA directly by inducing strand breaks.78
Acute cerebellar dysfunction is the commonest central neurotoxicity, occurring in approximately 14% of patients; they typically present with dysarthria, nystagmus, gait ataxia, and confusion. Onset usually occurs during administration of a multi-day regimen, particularly above a cumulative dose of 36 g/m2 and generally resolves rapidly once cytarabine is withdrawn.8,9,14 However, toxicity may be permanent once more than 8% to 20% of Purkinje cells have been lost.8 Acute encephalopathy is also common, presenting with somnolence or seizures. MRI scanning reveals diffuse high-intensity lesions within the central matter on T2-weighting that may be reversible.18 The encephalopathy should rapidly resolve entirely on stopping cytarabine; however, damage may be permanent and progress to leukoencephalopathy in a minority of patients, usually those with preexisting organ dysfunction or neurological problems.8 Less commonly, optic neuropathy, anosmia, and an incompletely reversible myelopathy have been reported.14 As with methotrexate, the intrathecal administration of cytarabine may cause ascending myelitis.8,79–81 There have also been case reports of sensory peripheral neuropathy following cytarabine exposure.82
ETOPOSIDE
Etoposide, a topoisomerase II inhibitor used in treatment of hematological, lung, ovarian, and testicular cancers,69 causes very little neurotoxicity, although at very high doses there have been reports of peripheral neuropathy, headache, seizures, and somnolence, in bone marrow transplant recipients and patients with malignant gliomas.83,84
FLUDARABINE
Neurotoxicity with the antimetabolite fludarabine is uncommon, but somnolence, acute encephalopathy, and chronic leukoencephalopathy progressing to coma and death have all been reported.85
IFOSFAMIDE
Central nervous system toxicity occurs in approximately 10% to 20% of patients receiving ifosfamide, who present with personality changes, confusion, hallucinations, stupor, and coma.9 More unusually, patients may experience seizures, myoclonus, cranial nerve palsies, or extrapyramidal symptoms.86,87 Symptoms usually begin 12 to14 hours after initiation of an ifosfamide infusion and spontaneously resolve 2 to 3 days after its cessation, although rarely symptoms may persist or even be fatal. EEG tends to show severe slowing with delta wave activity with or without seizure activity. Patients most at risk are those with impaired renal function, low serum albumin, pelvic tumors, and previous exposure to cisplatin.88 The risk of encephalopathy varies with route of administration. It is more common after oral administration, and is also more frequent with short intravenous infusion durations.89–91 The mechanism of toxicity is unclear but may be related to accumulation of metabolites such as chloracetaldehyde and chloro- ethylamine, which deplete intracellular glutathione and NAD and impair mitochondrial electron transport.92 Methylene blue, 300 mg IV in 6 divided doses, is used in the treatment of ifosfamide-induced encephalopathy and 50 mg IV qds may be given prophylatically.93–96 It is thought to act primarily as an alternative electron acceptor restoring mitochondrial respiratory chain function, but may also oxidate NADH and inhibits plasma monoamine oxidases.
METHOTREXATE
Methotrexate is one of the most widely used cytotoxic antimetabolites in the treatment of hematological, breast, and head/ neck cancers.9 In addition, prophylactic intrathecal use in ALL and high grade non-Hodgkin lymphomas has reduced the incidence of CNS relapse in high risk patients.97 Methotrexate acts by inhibiting dihydrofolate reductase, thus blocking purine and thymidine biosynthesis. Its neurotoxicity is thought to stem from widespread disruption of various metabolic pathways in the brain and can be acute or chronic.
Acute encephalopathy is associated with the administration of high-dose methotrexate (>3 mg/m2) and is characterized by somnolence, confusion, and seizures.98 Other symptoms include emotional lability and alternating hemiparesis, giving rise to the misdiagnosis of a functional disorder. The imaging appearances are characteristic and show symmetrical restricted diffusion on diffusion-weighted imaging, even when the T2-weighted sequences appear normal (Figure 16-1). Patients most at risk are children, those who have received previous cranial radiotherapy, or those receiving concomitant intravenous and intrathecal therapy. Metabolic abnormalities associated with the development of encephalopathy include widespread reduction in glucose utilization and protein synthesis,99 which are reversible by replacing depleted folate stores with the administration of leucovorin.100 Leucovorin is now given routinely following high-dose methotrexate (folinic acid rescue). Other studies have also postulated that changes in adenosine,101 homocysteine,102,103 or biopterin104
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
