[level-membership-for-pulmolory-and-respiratory-category]
Neuromuscular and Other Diseases of the Chest Wall
 Identify pulmonary function test results seen in patients with neuromuscular disease.
Identify pulmonary function test results seen in patients with neuromuscular disease.
 List the potential respiratory complications associated with neuromuscular disease.
List the potential respiratory complications associated with neuromuscular disease.
 Identify the clinical signs and symptoms associated with respiratory muscle weakness.
Identify the clinical signs and symptoms associated with respiratory muscle weakness.
 Describe techniques for monitoring patients with respiratory muscle weakness.
Describe techniques for monitoring patients with respiratory muscle weakness.
 Describe general respiratory care management of patients with respiratory muscle weakness.
Describe general respiratory care management of patients with respiratory muscle weakness.

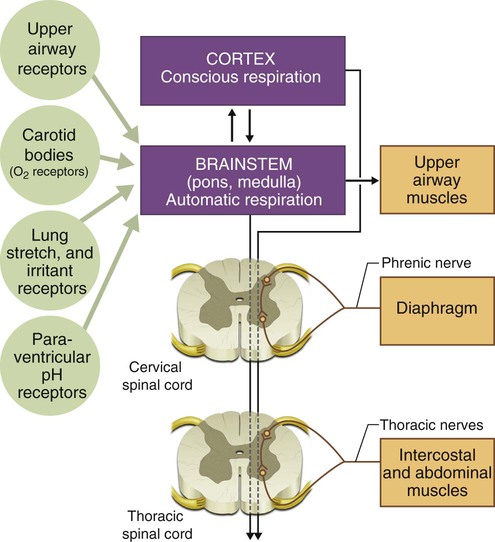
The respiratory system comprises the lungs, which provide an interface between inhaled air and circulating blood, mediating gas exchange; the thoracic cage, which forms the structure of the ventilatory pump; and the muscles of respiration, which are linked to respiratory centers in the brainstem by nerves exiting the spinal column and whose action on the thoracic cage produces movement of air into and out of the lungs. Understanding the interactions between these components is essential to understanding how their dysfunction leads to disease. The neuromuscular organization of the components of the respiratory system is shown in Figure 29-1. Maintenance of normal ventilation depends critically on intact, functional components of the neuromuscular system, which contribute to breathing in three principal ways: (1) regulation of respiratory drive and rate, (2) control of the mechanics of ventilation, and (3) cough and other airway protection. Diseases that affect the brain, nerves, muscles, or thoracic cage can lead to respiratory failure or hypoxemia even if the lungs are normal. The pulmonary consequences of neuromuscular disease include the following:
1. Dysregulation of respiratory drive or rate
2. Loss of strength or control of the mechanics of breathing
• Respiratory failure owing to excessive work of breathing
• Atelectasis leading to hypoxemia
• Secondary effects of chronic hypoxemia (e.g., cor pulmonale, pulmonary hypertension)
3. Loss of strength or control of the muscles that produce cough and protect the airway
Some systemic diseases that affect the neuromuscular system also cause interstitial lung disease, which can lead to considerable respiratory dysfunction (see Chapter 24). Respiratory failure, often associated with pulmonary infection, is a frequent cause of death in patients with neuromuscular disorders.
A thorough understanding of the physiology of ventilation and chest wall mechanics (see Chapters 10 and 18) is needed to help the reader understand how abnormalities of the upper airway, chest wall, diaphragm, and abdominal muscles cause disease. This chapter reviews major disorders of the neuromuscular and skeletal systems that affect breathing. Disorders are grouped according to which functional unit of the neuromuscular system is affected, focusing on pulmonary manifestations of these disease processes (Table 29-1).
TABLE 29-1
Locations at Which Several Neuromuscular Diseases Affect the Respiratory System
| Location | Disease |
| Cortex and upper motor neurons | Stroke, traumatic brain injury |
| Spinal cord | Trauma, transverse myelitis, multiple sclerosis |
| Anterior horn cells (lower motor neurons) | ALS, spinal muscular atrophy, poliomyelitis and postpoliomyelitis |
| Peripheral nerves | GBS, critical illness polyneuropathy, Lyme disease |
| Neuromuscular junction | MG, LES, botulism |
| Muscle | DMD, polymyositis, acid maltase deficiency |
| Interstitial lung disease* | Polymyositis, dermatomyositis, tuberous sclerosis, neurofibromatosis |
*A category of systemic diseases that can affect neuromuscular function.
General Principles Relating To Neuromuscular Weakness Of The Ventilatory Muscles
Pathophysiology and Pulmonary Function Testing
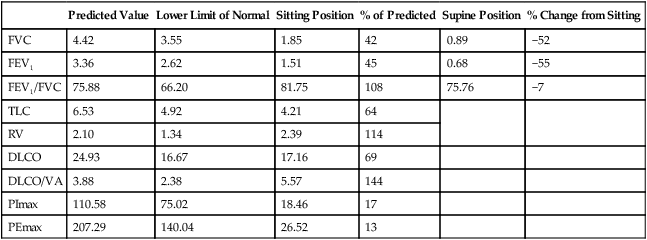
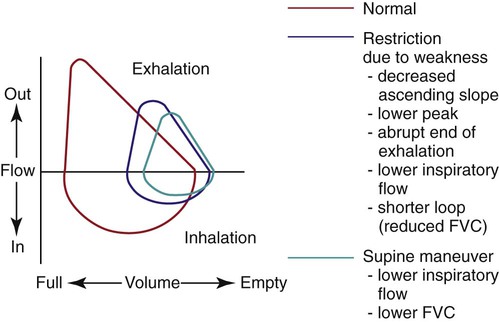
Weakness of the respiratory muscles leads to the inability to generate or maintain normal respiratory pressures. Pulmonary function testing in patients with neuromuscular weakness typically reveals a restrictive ventilatory defect even if the lungs are normal. Vital capacity (VC), forced expiratory volume in 1 second (FEV1), and total lung capacity (TLC) are decreased. Residual volume (RV) is normal or decreased, and diffusing capacity corrected for alveolar volume is normal or near-normal but has been reported to be decreased.1 Comparison of spirometric results obtained with the patient in seated and supine positions can be useful in showing that orthopnea is caused by neuromuscular weakness. A decrease in VC or FEV1 of greater than 20% when a patient moves from the seated to the supine position suggests diaphragmatic weakness (Table 29-2 and Figure 29-2). The inability to generate normal respiratory pressures is reflected in a decreased maximal inspiratory pressure (PImax). Expiratory muscle weakness is characterized by a decreased maximal expiratory pressure (PEmax).
TABLE 29-2
Pulmonary Function Testing Results from a Patient With Profound Diaphragm Weakness
| Predicted Value | Lower Limit of Normal | Sitting Position | % of Predicted | Supine Position | % Change from Sitting | |
| FVC | 4.42 | 3.55 | 1.85 | 42 | 0.89 | −52 |
| FEV1 | 3.36 | 2.62 | 1.51 | 45 | 0.68 | −55 |
| FEV1/FVC | 75.88 | 66.20 | 81.75 | 108 | 75.76 | −7 |
| TLC | 6.53 | 4.92 | 4.21 | 64 | ||
| RV | 2.10 | 1.34 | 2.39 | 114 | ||
| DLCO | 24.93 | 16.67 | 17.16 | 69 | ||
| DLCO/VA | 3.88 | 2.38 | 5.57 | 144 | ||
| PImax | 110.58 | 75.02 | 18.46 | 17 | ||
| PEmax | 207.29 | 140.04 | 26.52 | 13 |
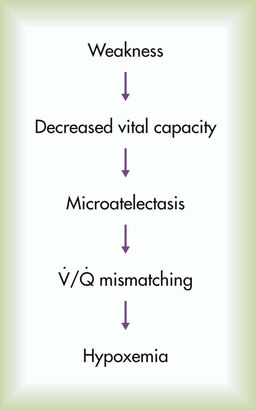
Arterial blood gases in the setting of a rapid, shallow breathing pattern may show a decreased PaCO2, although progressive inspiratory muscle weakness leads to hypoventilation and hypercapnia. Hypoxemia can occur in patients unable to take deep breaths and may be caused by microatelectasis, which leads to ventilation/perfusion mismatching within the lung and a resulting decrease in PaO2 (Figure 29-3). Chronic hypoxemia in this situation may be protective against acute respiratory muscle failure. Experimental data suggest that when hypoxemia is chronic, it may increase diaphragm muscle endurance.2 Hypoventilation that occurs with progressive neuromuscular disease may be a protective mechanism that avoids acute respiratory muscle fatigue. However, when hypoxemia is acute, it potentiates respiratory muscle fatigue, hastening respiratory failure.3
 , Ventilation/perfusion.
, Ventilation/perfusion. Rule of Thumb
Rule of ThumbClinical Signs and Symptoms
In the early stages of neuromuscular disease, patients with respiratory muscle weakness initially report exertional dyspnea and fatigue. As the disease process progresses, patients may complain of orthopnea or symptoms of cor pulmonale. These symptoms occur because the muscles involved with respiration can no longer generate or maintain normal ventilation. The response to hypoxemia and respiratory drive, measured with airway occlusion pressure (P0.1), is preserved in most patients with neuromuscular weakness.4 Because these patients do not have the strength to take deep breaths, they increase minute ventilation by increasing respiratory rate and adopting a rapid, shallow breathing pattern, which uses less respiratory muscle strength but provides less efficient ventilation. Patients with poor inspiratory muscle function (especially diaphragm weakness) may have marked orthopnea and prefer to sleep in a seated position. They may also experience a decline in the volume and power of voice or voice quality. Progressive muscle weakness can progress to the point where adequate ventilation is no longer maintained, and hypercapnia occurs.
 Problem
Problem Rule of Thumb
Rule of ThumbMonitoring and Assessing Patients With Muscle Weakness for Respiratory Insufficiency
Monitoring the ventilatory function of a patient with neuromuscular weakness can involve repeated measurement of inspiratory pressure, VC, and arterial blood gas values. Depending on the condition, the need for mechanical support can be signaled by reaching either a critical value on testing or an overall clinical condition that does not allow unassisted ventilation to continue. Additional measurements that have been suggested to be more indicative of early respiratory insufficiency, at least in amyotrophic lateral sclerosis (ALS), include maximal sniff nasal inspiratory force5 and nocturnal oximetry.6
At least two caveats to this general approach should be mentioned. First, patients with myasthenia gravis having an acute myasthenic crisis may have normal test results within minutes of acute ventilatory failure because of the nature of the disorder.7 Second, the need to protect the upper airway from secretions and aspiration may not be clearly reflected in results of pulmonary function tests, which evaluate only the mechanical function of the ventilatory pump. Neuromuscular weakness may not manifest uniformly in all muscle groups. Ventilation may be only moderately reduced in patients with gross oropharyngeal dysfunction leading to aspiration. Patients with ventilatory weakness can have a high risk of acute respiratory failure if upper respiratory tract infection or pneumonia develops. In these patients, inability to clear secretions can increase the work of breathing; the results are muscle fatigue, hypoventilation, and acute respiratory failure.
 Problem
ProblemManagement of Respiratory Muscle Weakness
Respiratory insufficiency and failure to clear secretions are the major consequences of inspiratory and expiratory muscle weakness. Treatment of these patients involves consideration of mechanical ventilation via face mask or other noninvasive interfaces or via tracheostomy. Although often overlooked, therapies to augment secretion clearance and assist with cough are important in these patients (see Chapters 39 and 40). Used together, these interventions can decrease hospitalizations secondary to respiratory complications in patients with neuromuscular disease.8 These measures may also be useful in delaying or preventing the need for intubation or tracheostomy, although severe bulbar muscle weakness may limit a patient’s ability to avoid invasive ventilatory support.9 In addition to these interventions, general rehabilitation focusing on aerobic conditioning, muscle strengthening, and respiratory muscle training can often delay the need for ventilatory support and improve the overall quality of life for patients with muscle weakness.10
Noninvasive ventilation is being used increasingly for short-term and intermittent ventilatory support of patients with neuromuscular disease.11 Acute deterioration, such as during a pneumonia, and surgical procedures such as gastrostomy tube insertion are situations where noninvasive ventilation is safe and effective if used carefully.9,12 If a patient needs long-term ventilatory support on an intermittent basis, such as at night only, noninvasive ventilation may be appropriate.13 Decisions to begin mechanical ventilation in some patients with neuromuscular weakness have varied greatly among physicians14 and may be motivated by many different patient factors.15 No uniform guidelines exist for the use of long-term mechanical ventilation for patients with neuromuscular weakness. However, it is considered a standard option for patients who have reached the point of respiratory failure. Starting a patient on long-term mechanical ventilation requires careful planning and consideration of various issues related to respiratory care in alternative settings. These issues have been addressed in consensus statements16,17 and are discussed in Chapter 51.
Diaphragm pacing in patients with spinal cord injury has been described for some time using direct stimulation of an intact phrenic nerve to contract the diaphragm and produce negative intrathoracic pressure and inspiration.18 This technique usually requires a thoracotomy, with its associated risks and high cost, and carries some risk of phrenic nerve injury. These objections have led to the development of an alternative system for diaphragm pacing using direct pacing of the diaphragm employing intramuscular electrodes implanted laparoscopically.19 When the electrodes can be placed in the diaphragm muscle at an electrophysiologically mapped motor point, the result is often elimination of the need for mechanical ventilation in patients with spinal cord injury and delay in the need to start mechanical ventilation in patients with ALS and other neuromuscular diseases.20
Mini Clini
Care of a Patient With Neuromuscular Weakness
 Problem
ProblemNoninvasive positive pressure ventilation (NIPPV) may be a reasonable first choice in the care of this patient. The patient’s mental status and bulbar function are intact. He has no significant problems with secretions. (Important factors for successful application of NIPPV are discussed in Chapter 45.) Use of a nasal mask with a biphasic positive airway pressure unit may be instituted and titrated to patient tolerance. (Most pressure or volume-cycled ventilators can be used to deliver NIPPV and invasive ventilation.) A time-cycled backup rate can be set on some ventilators to facilitate ventilation of patients who may inadequately trigger the ventilator. If the ability to clear secretions becomes compromised, cough augmentation strategies should be implemented. As long as bulbar function—swallowing and secretion management—are maintained, noninvasive ventilation is a reasonable choice for ventilatory support.
 Rule of Thumb
Rule of ThumbSpecific Neuromuscular Diseases
Disorders of the Muscle (Myopathic Disease)
Primary muscle disease can decrease the ability of a normal neural impulse to generate effective muscle contraction. Some commonly recognized myopathies include Duchenne muscular dystrophy (DMD), myotonic dystrophy, and polymyositis. Table 29-3 presents a more complete list of myopathic diseases associated with ventilatory dysfunction.
TABLE 29-3
Myopathic Diseases With Associated Respiratory Dysfunction
| Muscular Dystrophies | Myopathies |
| DMD | Congenital myopathies |
| Myotonic dystrophy | Nemaline rod myopathy |
| Facioscapulohumeral muscular dystrophy | Centronuclear myopathy |
| Limb-girdle dystrophy | Metabolic myopathies |
| Oculopharyngeal dystrophy | Acid maltase deficiency |
| Mitochondrial myopathies (Kearns-Sayre syndrome) | |
| Inflammatory myopathies | |
| Polymyositis | |
| Dermatomyositis | |
| Hypothyroid-related and hyperthyroid-related myopathies | |
| Endocrine myopathies | |
| Steroid-induced myopathies (including critical illness myopathy) | |
| Miscellaneous myopathies | |
| Electrolyte disorders (e.g., hypophosphatemia, hypokalemia) | |
| Rhabdomyolysis | |
| Periodic paralysis | |
| Postneuromuscular blockade myopathy |
Duchenne Muscular Dystrophy and Becker Muscular Dystrophy
Duchenne muscular dystrophy (DMD) is a genetic muscle-wasting disorder caused by mutations in the dystrophin gene.21 Because it is an X-linked recessive disorder, it affects mostly males. The diagnosis is made when a dystrophin mutation is found in DNA from peripheral white blood cells or when dystrophin is found to be absent or abnormal in muscle biopsy tissue.
Progressive scoliosis is associated with DMD and can contribute further to respiratory insufficiency. Many patients undergo spine fusion surgery, and often the procedure allows greater comfort and ease in maintaining an upright posture. Although no randomized trials have proven any benefit of surgery on pulmonary function,22 observational data suggest that the rate of respiratory decline is slower after fusion surgery.23
Obstructive sleep apnea has been described in a significant proportion of patients with DMD, prompting some authors to recommend formal polysomnography in patients with symptoms of obstructive sleep apnea or at the stage of becoming wheelchair-bound.24 Institution of positive pressure ventilation (PPV) is a decision most patients face at some point in the disease. The point at which to begin different stages of ventilatory support depends on both test results and clinical condition. Nocturnal PPV is usually indicated when FVC reaches 30% of predicted with signs of hypoventilation.25,26 It can be instituted in response to oxygen desaturation during sleep, which is common in patients with increased disability and scoliosis. Nocturnal ventilation seems to improve daytime ventilatory function in patients with DMD,27 presumably through the prevention of fatigue of the affected respiratory muscles. Despite this improvement, results of studies of early “prophylactic” institution of PPV in the care of patients with DMD have shown that early PPV failed to delay the need for formal ventilatory support.28 However, long-term inspiratory muscle training using resistive loading was shown in at least one study to improve PImax in patients with DMD and VC values greater than 27%,29 theoretically delaying the need for ventilatory support. Although there are concerns that pulmonary rehabilitation could produce deleterious effects through overloading weak respiratory muscles, such concerns have not been confirmed in studies in Becker muscular dystrophy.30
Myotonic Dystrophy
Myotonic dystrophy is the most common form of muscular dystrophy in adults, with an estimated frequency of 1 in 8000 persons.31 Myotonia, or delayed muscle relaxation, is the hallmark of this neuromuscular disorder but does not clearly occur in respiratory muscles or directly cause respiratory insufficiency.32 This autosomal dominant disorder causes progressive muscle weakness, abnormalities of the cardiac conduction system, endocrine dysfunction, and cataracts. There are two main types of myotonic dystrophy, both caused by an expansion of a repeated DNA sequence on chromosome 19.33
Respiratory dysfunction in myotonic dystrophy is common, usually occurring late in the course of disease, and can include respiratory muscle weakness, obstructive sleep apnea, central sleep apnea, and bulbar muscle dysfunction leading to aspiration. Sleep-related disorders are particularly common, even at an early age.34
Patients with myotonic dystrophy can be very sensitive to anesthesia and respiratory depressants. Both respiratory failure and prolonged neuromuscular blockade have been reported in patients with myotonic dystrophy given usual doses of these agents. For this reason, prolonged perioperative monitoring after surgery is important.35,36
Polymyositis
Polymyositis, dermatomyositis, and inclusion body myositis are inflammatory myopathies of unknown cause. Respiratory compromise is rare in inclusion body myositis but can be seen in both polymyositis and dermatomyositis. Clinical respiratory muscle weakness is uncommon but can lead to respiratory weakness or failure within weeks to months in the setting of rapidly progressing disease. Diagnosis of these diseases is based on clinical findings of myalgia, elevated muscle enzyme levels (creatine phosphokinase or aldolase), and compatible electromyographic or muscle biopsy results. Diagnostic criteria may apply to any inflammatory myopathy, and specific findings or antibody identification may be needed to differentiate the various diseases (Table 29-4).37,38
TABLE 29-4
Diagnostic Criteria for Inflammatory Myopathies
| Criterion | Polymyositis | Dermatomyositis | Inclusion Body Myositis |
| Symmetric proximal muscle weakness on physical examination | Yes | Yes | May be asymmetric and more distal weakness |
| Elevation of serum muscle enzymes (creatine kinase, aldolase, glutamate oxaloacetate, pyruvate transaminases, and lactate dehydrogenase) | Yes | Yes | Yes, lower levels than in polymyositis or dermatomyositis |
| Electromyographic triad of (1) short, small polyphasic potentials, (2) fibrillations, (3) high-frequency repetitive discharges | Yes | Yes | Yes |
| Muscle biopsy showing mononuclear inflammation, phagocytosis, necrosis, degeneration, and regeneration | Yes | Yes | Yes, may have fatty infiltration |
| Skin findings: Gottron sign and papules; heliotrope rash | No | Common | No |
| Anti–Jo-1 antibody (or other antisynthetase antibodies) | 30%-50%, may indicate antisynthetase syndrome | No | |
| Interstitial lung disease | 86% in patients with antisynthetase syndrome | No | |
Ventilatory insufficiency and failure caused by these inflammatory myopathies are unusual but tend to parallel the development of limb muscle weakness when they occur. In rare reported instances, diaphragmatic function is decreased disproportionately to the degree of limb weakness.39
Corticosteroids are the mainstay of initial management of polymyositis and dermatomyositis, although other immunosuppressive and cytotoxic regimens are used to limit long-term steroid exposure; 35% to 40% of patients with inflammatory myopathy have interstitial lung disease associated with myopathy. This lung disease appears as diffuse interstitial infiltrates that may be caused by various lung processes, but the most common (56%) is nonspecific interstitial pneumonia.40 Various antisynthetase antibodies (e.g., Jo-1) have been identified that are associated with polymyositis and dermatomyositis,41–43 although the role of these antibodies in the pathogenesis of these diseases is unclear. Pulmonary vasculitis can occur with polymyositis and dermatomyositis and can lead to oxygen exchange abnormalities and pulmonary hypertension.
Critical Illness Myopathy
Critical illness myopathy is a heterogeneous entity that occurs commonly in intensive care units (ICUs) and in which patients develop flaccid weakness of proximal muscles. Patients are often very difficult to wean from mechanical ventilation. Risk factors for developing this myopathy include use of corticosteroids (likely dose-dependent); use of paralytic agents; hyperglycemia; hyperthyroidism; and possibly systemic inflammatory response syndrome, with or without sepsis.44,45 Weakness improves on its own in most cases but may take weeks or months to resolve. There is no specific therapy, and prevention by avoiding the aforementioned risk factors is the best approach.
Disorders of the Neuromuscular Junction
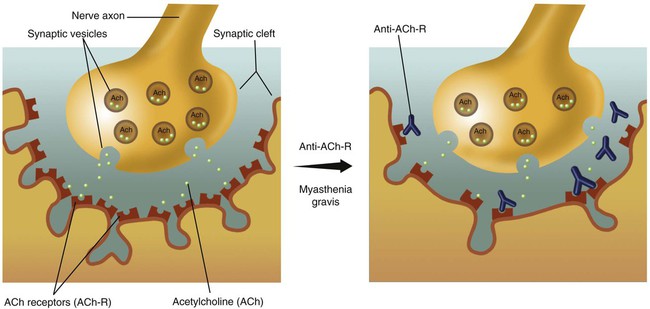
Disorders of the neuromuscular junction decrease conduction of nervous system impulses to the peripheral muscles, resulting in muscle weakness. Different clinical syndromes are caused by defects in different molecules or components of the neuromuscular junction, which is represented schematically in Figure 29-4. Disorders of the neuromuscular junction include the following:
Myasthenia Gravis
Myasthenia gravis (MG) is characterized by intermittent muscular weakness, which worsens on repetitive stimulation and improves with administration of anticholinesterase medications, such as edrophonium (Tensilon) or neostigmine. Most cases of MG arise from production of antibodies directed against the acetylcholine receptor (ACh-R). The antibodies inactivate the ACh-R and block transmission of electrical impulses from the nerve to the muscle.46
Approximately 20% of patients with MG do not exhibit such antibodies but may have antibodies to alternative targets, such as muscle-specific kinase.47 Abnormalities of the thymus gland are common in MG. Approximately 10% of patients with MG have a neoplastic growth within the thymus called thymoma, which can be malignant but usually is not. Patients without thymoma typically have some degree of thymic hyperplasia. Congenital or fetal myasthenic syndromes are caused by either autoantibodies or inherited defects in the ACh-R.
MG typically occurs earlier in life in women and later in men. As the population has aged, more patients are diagnosed with MG later in life, and now there are more men affected than women.48 This disorder may be associated with other autoimmune diseases, such as endocrinopathies, rheumatoid arthritis, ulcerative colitis, sarcoidosis, and pernicious anemia.
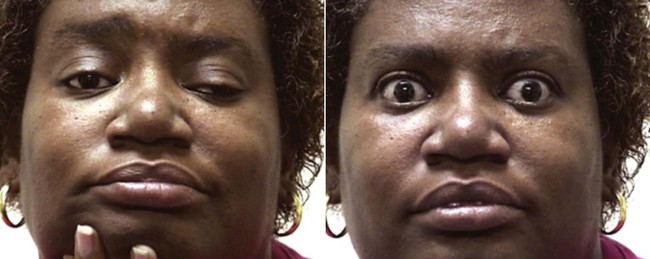
MG is characterized by progressive loss of muscular function, which may affect only the eye muscles (ocular myasthenia) or may be more widespread. The initial symptom is diplopia (double vision) or ptosis (a drooping eyelid) in greater than 65% of patients (Figure 29-5).49 The patient typically reports weakness of the affected muscles that may vary through the day or progress, especially with repetitive use. The diagnosis of MG is supported by the detection of anti–ACh-R antibodies in the blood, a characteristic fading of nerve impulses with repeated nerve stimulation testing during electromyography, and improvement of strength or symptoms in response to an anticholinesterase inhibitor drug (edrophonium) (Figure 29-6).

Treatment of MG is generally effective, although it is largely empiric because clinical trials are rare. Long-term management includes thymectomy50 and administration of anticholinesterases (edrophonium, neostigmine, pyridostigmine) with or without corticosteroids or other immunosuppressants, such as azathioprine or cyclosporine.49 For patients in acute myasthenic crisis, who often are in respiratory failure on mechanical ventilation, circulating antibodies can be removed by plasmapheresis, which results in clinical improvement51,52 usually after five or six treatments and has been used to facilitate weaning from mechanical ventilation in the care of patients with myasthenic crisis.53 Intravenous IgG has also been used and can improve muscle strength and hasten recovery from respiratory failure. Neither apheresis nor intravenous IgG is generally used for long-term management of MG. The effect of both treatments is temporary but may last several months.49 Clinical trials comparing these treatments have shown a slight efficacy advantage of plasmapheresis but at the cost of a higher complication rate.54,55
The pulmonary complications of MG depend on the magnitude and location of the affected muscle groups and tend to occur in patients most severely disabled with the disease. Upper airway obstruction, exertional dyspnea, and overt ventilatory failure all are reported in MG. Pulmonary function testing of MG patients who have respiratory muscle weakness shows decreased TLC, VC, PImax, and PEmax similar to other neuromuscular disorders, with PImax and PEmax being more sensitive markers of early respiratory muscle weakness.56
Myasthenic crisis is an acute event in MG and is characterized either by respiratory failure or by inability to maintain a patent airway. Myasthenic crisis can occur acutely in response to worsening of disease, intercurrent infection, or surgery or when excess anticholinesterase inhibitors have been given. Endotracheal intubation and mechanical ventilation are required immediately and may be prolonged.57
Lambert-Eaton Syndrome
Another syndrome of neuromuscular weakness arising from a disorder at the neuromuscular junction is Lambert-Eaton syndrome (LES). More than 50% of cases of LES are associated with cancer. Of these cancer-related cases, greater than 80% are associated with small cell carcinoma of the lung.58 The mean age at presentation is approximately 60 years, although LES can occur in all age groups. Autoantibodies against voltage-gated calcium channels at the nerve terminals impair the release of acetylcholine and can lead to both muscular weakness and autonomic insufficiency.59 These autoantibodies can be detected in a patient’s serum, which confirms the diagnosis.60 The clinical diagnosis of LES is supported by results of nerve conduction studies. Increasing muscle strength with repetitive stimuli is a characteristic feature of LES, which differentiates it from MG. In contrast, MG is characterized by progressive fatigue of muscular contraction with repetitive stimulation.
Patients with LES usually present with tiredness or weakness of proximal muscle groups out of proportion to findings on clinical examination. Although patients are subject to respiratory complications because of their increased sensitivity to the effects of anesthesia, respiratory failure is rare. The clinical course of LES tends to be one of relative stability with less fluctuation than MG. Management of LES includes treatment of the underlying malignancy when present. If no malignancy is found, surveillance for lung cancer is recommended every 6 months, and LES is managed symptomatically with immunosuppressive medication or acetylcholinesterase inhibitors.60
Disorders of the Nerves
The peripheral nerves may be affected by toxic agents, inflammatory processes, vascular disorders, malignant diseases, and metabolic or nutritional imbalances. Hundreds of conditions have been associated with neuropathies leading to respiratory muscle dysfunction. Representative conditions are listed in Box 29-1.
Guillain-Barré Syndrome
Guillain-Barré syndrome (GBS) is acute inflammatory demyelinating polyneuropathy and is the most common peripheral neuropathy causing respiratory insufficiency. GBS is characterized by paralysis and hyporeflexia with or without sensory symptoms. GBS is typically a self-limited disease, but overall mortality ranges from 3% to 6%.61 Before modern mechanical ventilation techniques, mortality in this condition was greater than 33%.62 Most patients recover their respiratory muscle strength completely, but the proportion of patients with significant disability 1 year after onset of the disease can be 20%.63 Autonomic nervous system problems, such as hypotension, flushing, bronchorrhea, dermatographia, and bradycardia, are common. Two-thirds of patients with GBS report a triggering event, such as respiratory or gastrointestinal infection, immunization, or surgery, 1 to 4 weeks before the onset of symptoms. Other reported triggers include pregnancy and malignancy.62
GBS is a demyelinating process widely believed to be caused by autoantibodies directed against the myelin constituting the nerve sheath. The diagnosis of GBS is based on a combination of clinical, laboratory, and electrophysiologic data (Table 29-5).64 Cerebrospinal fluid protein levels are elevated with minimal cellularity after approximately 1 week of illness. Nerve conduction studies show slowing of conduction with preserved amplitude, which is typical of demyelination. Approximately one-third of all patients with GBS have respiratory muscle compromise. Although the diaphragm is typically affected later in the course of GBS, cases of respiratory failure in the absence of substantial peripheral weakness have been reported. The need for mechanical ventilatory support for patients with GBS increases with age.62
TABLE 29-5
Diagnostic Criteria for Guillain-Barré Syndrome
| Required for Diagnosis | Supportive of Diagnosis |
| Progressive weakness of both legs and arms | Symptoms progress over days to weeks |
| Areflexia | Symmetry of weakness |
| Cranial nerve involvement (facial palsies) | |
| Improvement begins 2-4 wk after progression stops | |
| Absence of fever at onset of symptoms | |
| Laboratory features not suggestive of alternate diagnosis | |
| Cerebrospinal fluid with high protein, low cell count | |
| Nerve conduction slowing or block with normal amplitude |
Treatment strategies that have improved outcome in GBS include intravenous immunoglobulin infusions and plasmapheresis. Both treatments are equally effective in hastening recovery of muscle strength. The benefit is greatest when treatment is started within 2 weeks of symptom onset. There is no additional benefit of combining the two therapies. Corticosteroids have no beneficial role in GBS.65
Patients with dyspnea, orthopnea, or impaired ability to maintain a patent airway should receive spirometry every 4 to 6 hours for documentation of function and assessment of the need for endotracheal intubation. Patients with poor upper airway control, weak cough, or large amounts of secretions should be considered for endotracheal intubation even though their VC is greater than 20 ml/kg. The increased work of breathing imposed by mucous plugging or atelectasis can hasten decompensation. A small subgroup may need mechanical ventilation for 1 year or more. Weaning of patients with GBS from mechanical ventilation is predicted by VC greater than 18 ml/kg,68 transdiaphragmatic pressure greater than 31 cm H2O, or a PImax stronger than −30 cm H2O.69
 Rule of Thumb
Rule of Thumb
Patients with GBS whose VC becomes less than 20 ml/kg or declines more than 30% from baseline or whose maximal inspiratory pressure is less than −30 cm H2O and maximal expiratory pressure is less than 40 cm H2O are at risk of respiratory failure and may need ventilatory support. Patients who meet the criteria of this “20-30-40 rule” should be observed in an ICU.66
 Rule of Thumb
Rule of Thumb
Patients with GBS should be intubated for mechanical ventilatory support when VC decreases to 12 to 15 ml/kg or sooner if they have bulbar dysfunction with difficulty managing oral secretions or when PaO2 values are less than 70 mm Hg while breathing room air.62 As with all patients requiring intubation, as soon as it is clear that intubation will exceed 2 weeks’ duration, a tracheostomy should be considered.67
Phrenic Nerve Damage and Diaphragmatic Paralysis
Each hemidiaphragm is supplied by its own phrenic nerve. The phrenic nerves emerge from the spinal cord at level C3-5 and descend through the mediastinum along the great vessels of the chest and pericardium. Damage to or interruption of either phrenic nerve leads to paralysis of the ipsilateral hemidiaphragm. Bilateral interruption is seen in high spinal cord injury and causes complete diaphragmatic paralysis. Unilateral diaphragmatic paralysis can be seen in various disease processes. Reversible unilateral diaphragmatic paralysis is a rare complication of acute pneumonia, but it can occur in 10% of patients who undergo cardiac surgery with cardiopulmonary bypass, usually secondary to cold cardioplegia or traction on the nerve during surgery or ischemic nerve injury.70
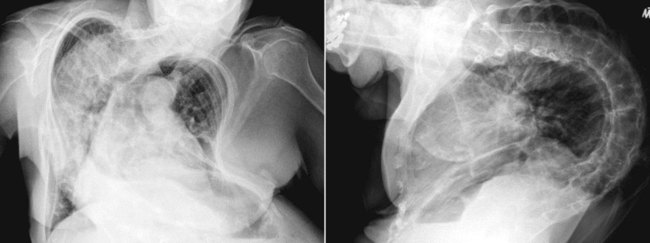
Patients with unilateral diaphragmatic paralysis may have a 15% to 20% reduction in VC and TLC in the upright position and a further reduction while supine. If they have no other diseases, patients with unilateral diaphragmatic paralysis may have no symptoms. Athletes, musicians, and others who use their lungs more fully are more likely to notice the decreased ventilatory capacity caused by unilateral diaphragm weakness. Diaphragmatic paralysis is diagnosed most often with chest radiography. The paralyzed side retains its contour but is displaced upward (Figure 29-7, A). At fluoroscopy, the paralyzed hemidiaphragm paradoxically rises into the thorax during a sudden forceful inspiration (sniff test). This paradoxical motion dampens the effect of the normal diaphragm on the opposite side.
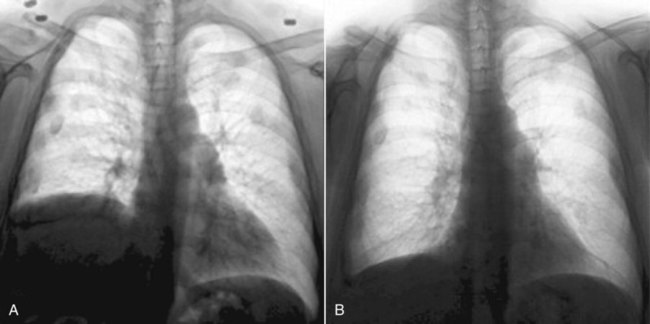
For patients with unilateral diaphragm paralysis, surgical plication of the weak side can move the diaphragm downward to a more normal position (Figure 29-7, B) and minimize paradoxical motion and improve overall lung function.71,72 Historically, results of diaphragm plication procedures have been disappointing, but newer laparoscopic techniques are more promising.73 Appropriate patient selection is crucial to avoid operating on patients whose diaphragms would recover their strength in time and patients whose weakness is due to primary neuromuscular disorders, which would not be improved by plication.
 Rule of Thumb
Rule of ThumbCritical Illness Polyneuropathy
In contrast to critical illness myopathy, discussed previously, critical illness polyneuropathy is associated almost entirely with severe sepsis in the ICU. Patients develop muscle weakness and atrophy, loss of deep tendon reflexes, and loss of peripheral sensation to pinprick and touch. Cranial nerve function is usually spared. The mechanism of nerve damage is unknown but, similar to other complications of sepsis, may be related to ischemia caused by thrombosis or underperfusion of the microcirculation, or both, in this case of the affected nerves.45
Disorders of the Spinal Cord
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS), or Lou Gehrig disease, is a neuromuscular disease characterized by progressive degeneration of both upper and lower motor neurons; early in the disease, degeneration of either upper or lower motor neurons may predominate. Approximately 5% to 10% of cases are familial; others are sporadic. The male-to-female ratio for ALS is approximately 1.2 : 1, and the peak incidence is between 65 years old and 75 years old, although cases have been reported in patients younger than 30 years old. The prognosis of ALS is poor, with a mean survival from diagnosis of 3 years. By 5 years, 80% of patients have died, and 90% have died by 10 years.74 Medical treatment of ALS is disappointing. There is no cure, and therapies to halt or slow progression of the disease are few and ineffective. One approved therapy for ALS is riluzole, an antiglutamate agent. Randomized trials have shown modest improvements in median survival of 4.2 months.75 The cost of the medication is prohibitive for many patients, especially in light of the modest results usually seen.
Muscle weakness secondary to ALS usually begins in a localized muscle group and spreads out geographically from there. It most often begins in the arms but may originate in the bulbar muscles (i.e., muscles supplied by nerves in the upper spinal cord, such as the nerves controlling swallowing and speaking) 25% of the time. In 1% to 2% of cases, ALS manifests initially as isolated respiratory muscle dysfunction in an otherwise relatively intact patient. Respiratory involvement eventually occurs in all patients with ALS, and pulmonary complications are the most frequent cause of death. Muscle weakness is associated with fasciculations, or involuntary quivering of the affected muscles. If muscle fasciculation does not develop in a patient with presumed ALS soon after weakness, another diagnosis should be considered.76
 Rule of Thumb
Rule of ThumbThe prevention of respiratory complications and assessment of the need for ventilatory assistance are central in caring for patients with advancing ALS. Helpful therapeutic interventions include (1) modification of food consistency or placement of feeding tubes in patients with marked bulbar dysfunction, (2) clearing of secretions with assisted cough techniques or postural drainage, and (3) ventilatory assistance with positive or negative pressure devices. Noninvasive techniques are a viable option for many patients and have been shown to slow the rate of pulmonary decline, improve symptoms, and prolong survival.77 When these techniques no longer suffice, many patients with ALS choose not to receive invasive mechanical ventilation and opt for palliative management at that point. However, many patients do desire invasive ventilatory support, and 90% of such patients in one study reported satisfaction with their decision and would choose tracheostomy and ventilation again in the same situation.78 Although overall survival with ALS is poor, prolonged survival of ventilated patients has been reported.
 Rule of Thumb
Rule of Thumb
The timing and type of ventilatory intervention (noninvasive or invasive) in the care of patients with ALS are the subject of much discussion. General guidelines for considering ventilatory assistance79 include the following: VC less than 50% predicted, orthopnea, maximal sniff nasal inspiratory force less than −40 cm H2O, PImax less than (i.e., more negative than) −60 cm H2O, and abnormal nocturnal oximetry.
Mini Clini
Respiratory Care of a Patient with Amyotrophic Lateral Sclerosis
 Problem
ProblemReduced lung compliance owing to atelectasis or retained secretions can greatly increase the work of breathing for weakened muscles and worsen gas exchange. Maneuvers to recruit collapsed alveoli and augment his cough to clear secretions can counteract these factors. These maneuvers include chest physiotherapy and mechanical insufflation and exsufflation devices. Increased secretion production is another factor in many patients’ illness, which can be managed with medications such as atropine or amitriptyline. In some cases, salivary glands are treated with low-dose radiation. Patients with bulbar dysfunction may lose additional respiratory muscle strength as a result of poor nutrition; placement of a percutaneous endoscopic gastrostomy tube for nutrition may avoid this as eating becomes more difficult. Keeping these additional aspects in mind allows a care provider to address them along with the primary weakness, and it is this care in many cases that prolongs a patient’s ability to breathe independently and function as he or she would like.80
 Rule of Thumb
Rule of Thumb Rule of Thumb
Rule of ThumbSpinal Cord Trauma
Approximately 12,000 new spinal cord injuries occur in the United States each year, and 55% involve the cervical spine. The causes are varied, but the most common causes are motor vehicle accidents, falls, violence, and sports. Many patients with a spine injury have other injuries associated with their trauma, including 25% to 50% with traumatic head injury81; 5% to 10% of these spinal cord injuries cause quadriplegia. Complete cord injury is associated with absent motor and sensory function below the level of injury, and the patient’s condition rarely improves. Patients with incomplete injury have residual function and tend to improve to varying degrees.
 Problem
ProblemDisorders of the Brain
Traumatic brain injury, stroke, hemorrhage, and infection can lead to abnormality of respiration through various mechanisms. The motor cortex contains voluntary centers for control of the upper airway and pharynx (see Chapter 14). The pons and medulla, located in the brainstem, contain (1) chemoreceptors for automatic control of ventilation in response to increasing pH and hypercapnia and (2) centers that generate and modify patterns of automatic ventilation in response to visceral and chemical afferent information (see Figure 29-1). Both stroke and traumatic brain injury can lead to disordered patterns of breathing, which are listed in Table 29-6, and abnormalities in the lungs themselves, such as neurogenic pulmonary edema. This section describes the clinical entities of stroke and traumatic brain injury and their effects on the respiratory system.
TABLE 29-6
Abnormal Respiratory Patterns Associated With Stroke
| Cheyne-Stokes respiration | Common abnormal pattern characterized by crescendo-decrescendo breathing force and tidal volume; it is not specific for stroke and is more commonly due to cardiopulmonary disease |
| Periodic breathing | Similar to Cheyne-Stokes but with complete central apneas in between periods of crescendo-decrescendo breathing; it is seen in 25% of acute strokes, especially in patients with subarachnoid hemorrhage |
| Gasping | Very short inspiration, often involving contraction of accessory muscles, with long expiratory phase; it often heralds impending respiratory failure |
| Ataxic breathing | Irregularly irregular respiratory rate and tidal volumes; it nearly always means a medullary stroke or lesion; it is not the sign of a poor prognosis |
| Apneustic breathing | Very long inspiratory phase (several seconds), then brief, rapid exhalation followed by a respiratory pause; it is usually associated with bulbar dysfunction with difficulty protecting airway from secretions, so usually requires intubation, and may be difficult to wean from ventilator |
| Central neurogenic hyperventilation | Rapid deep breaths resulting in hypocapnia; other causes of hyperventilation must be ruled out, but if so, this pattern signifies a poor prognosis |
| Apnea | Unusual in strokes except in brain death; prognosis is generally poor |
| Ondine curse | Apnea during sleep with normal respiration while awake; it is treated with mechanical ventilation during sleep only; some patients improve and no longer need ventilatory support |
Stroke
Strokes in the cerebral cortex can produce decreased chest wall and diaphragmatic movement. Infarction in this area usually does not lead to significant alteration of ventilation. However, the patient may have significant impairment of speech and movement, including impairment of muscles that affect upper airway tone and control secretions. Swallowing is also frequently a problem, leading to aspiration or poor nutrition. Chronic changes in pharyngeal muscle tone can also lead to obstructive sleep apnea. Rarely, localized strokes lead to profound alterations of the respiratory system. These alterations often result from strokes in the midbrain and brainstem or from subarachnoid hemorrhage. They often resolve or improve after the acute time frame of the stroke (see Table 29-6).
Therapy for stroke has evolved considerably. Previous therapy for thrombotic stroke was largely supportive. At the present time, early use of thrombolytic therapy to dissolve the clot and restore circulation and function has been shown to improve function and survival, particularly if the thrombolytic agent is given less than 3 hours after the onset of symptoms.85,86 More recent trials have also shown benefit in a more restricted patient population when thrombolytic therapy was given in the 3- to 4.5-hour time frame.87,88 Physical therapy and occupational therapy continue to be important components for optimizing function in the setting of residual deficit after stroke. Patients with substantial impairment of speech and swallowing may be at risk of aspiration pneumonia. As in many catastrophic illnesses, stroke presents a complex set of problems, and patients do best when managed using a multidisciplinary approach in a center with high volumes of similar patients and well-developed expertise.89
Disorders of the Thoracic Cage
Kyphoscoliosis
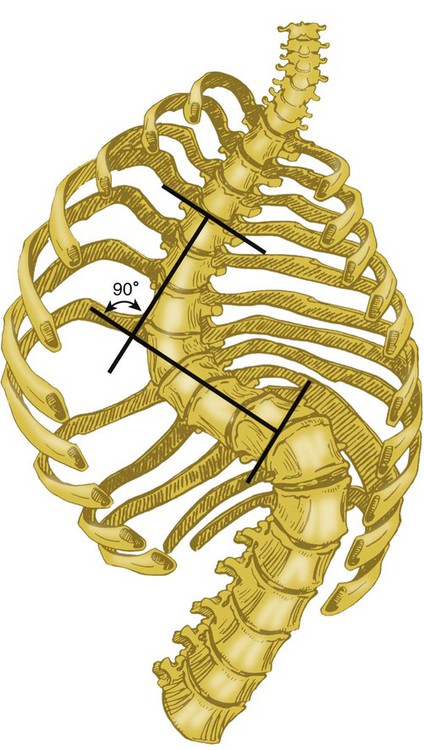
Kyphosis is posterior angulation of the thoracic cage. Scoliosis is lateral curvature of the spine (Figure 29-8). These two deformities often occur together (called kyphoscoliosis) as a result of the compensatory effects of the spine in response to the primary lateral curve in scoliosis.
Scoliosis is typically noticed during childhood and progresses during adolescence, although idiopathic adult kyphoscoliosis has been reported. The degree of scoliosis is measured by the Cobb angle, which is determined by the intersection of lines drawn between the upper and lower limbs of the primary curve in scoliosis (Figure 29-9). Severe kyphoscoliosis (Cobb angle >90 to 100 degrees) can lead to hypoventilation, hypercapnia, and, if untreated, complications of pulmonary hypertension. However, the degree of pulmonary dysfunction cannot be predicted from the Cobb angle alone.90,91 Respiratory dysfunction is probably multifactorial in most patients. Compliance of the chest wall and lung is decreased in patients with significant kyphoscoliosis. The result is a restrictive ventilatory defect with decreased TLC and VC in pulmonary function testing. Maximal transdiaphragmatic pressure also is decreased, a sign of impaired diaphragmatic function in the pathogenesis of respiratory dysfunction in severe kyphoscoliosis.
Anterior or posterior spinal fixation can stabilize kyphoscoliosis and restore the thoracic curvature to a condition close to normal. Fixation prevents complications secondary to progressive curvature, loss of compliance, and subsequent ventilatory dysfunction. Few options are available to restore pulmonary function to older patients with established kyphoscoliosis. Surgery to correct the deformity can be undertaken, but this treatment generally does not improve pulmonary function.92 Better long-term results are seen when surgery or brace therapy to correct the angulation are undertaken in adolescence.93
Both noninvasive and invasive ventilation are used in some patients with severe kyphoscoliosis, with improvement in blood gas values, respiratory muscle strength, symptoms of dyspnea,94 and exercise capacity.95 Both negative pressure96 and positive pressure97 ventilation have been reported to stabilize respiratory function in patients with severe kyphoscoliosis.
Flail Chest
Flail chest is defined in different ways but occurs as a result of multiple rib fractures that cause a portion of the chest wall to become free-floating. The destabilized segment of the thoracic cage exhibits paradoxical motion during the respiratory cycle, bowing out with expiration and collapsing inward during a spontaneous breath. The movement is associated with a decreased pressure gradient to drive inspiration and expiration and can result in respiratory failure. Flail chest frequently is accompanied by other pulmonary injuries as a result of the mechanism of injury and the force required to fracture multiple ribs. Pulmonary contusion, hemothorax, and pneumothorax are frequently associated with flail chest and often necessitate urgent or emergency treatment in the trauma patient.98 Flail chest is managed with analgesia and positive pressure ventilation.
Ankylosing Spondylitis
Ankylosing spondylitis is a rheumatologic disease that affects the spine and thoracic cage. Chronic joint inflammation ultimately leads to fusion of the vertebral bodies and the costovertebral joints, typically leading to severe kyphosis and a dramatic decrease in thoracic cage compliance. Because diaphragmatic movement is retained, TLC and VC are only slightly reduced. The most severe respiratory consequence is parenchymal lung disease, which occurs in about 10% of patients with ankylosing spondylitis, in the form of apical fibrocystic changes that can decrease gas exchange and often provide a location for superinfection, especially fungal infection.99
[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]
Neuromuscular and Other Diseases of the Chest Wall
Identify pulmonary function test results seen in patients with neuromuscular disease.
List the potential respiratory complications associated with neuromuscular disease.
Identify the clinical signs and symptoms associated with respiratory muscle weakness.
Describe techniques for monitoring patients with respiratory muscle weakness.
Describe general respiratory care management of patients with respiratory muscle weakness.
The respiratory system comprises the lungs, which provide an interface between inhaled air and circulating blood, mediating gas exchange; the thoracic cage, which forms the structure of the ventilatory pump; and the muscles of respiration, which are linked to respiratory centers in the brainstem by nerves exiting the spinal column and whose action on the thoracic cage produces movement of air into and out of the lungs. Understanding the interactions between these components is essential to understanding how their dysfunction leads to disease. The neuromuscular organization of the components of the respiratory system is shown in Figure 29-1. Maintenance of normal ventilation depends critically on intact, functional components of the neuromuscular system, which contribute to breathing in three principal ways: (1) regulation of respiratory drive and rate, (2) control of the mechanics of ventilation, and (3) cough and other airway protection. Diseases that affect the brain, nerves, muscles, or thoracic cage can lead to respiratory failure or hypoxemia even if the lungs are normal. The pulmonary consequences of neuromuscular disease include the following:
1. Dysregulation of respiratory drive or rate
2. Loss of strength or control of the mechanics of breathing
• Respiratory failure owing to excessive work of breathing
• Atelectasis leading to hypoxemia
• Secondary effects of chronic hypoxemia (e.g., cor pulmonale, pulmonary hypertension)
3. Loss of strength or control of the muscles that produce cough and protect the airway
Some systemic diseases that affect the neuromuscular system also cause interstitial lung disease, which can lead to considerable respiratory dysfunction (see Chapter 24). Respiratory failure, often associated with pulmonary infection, is a frequent cause of death in patients with neuromuscular disorders.
A thorough understanding of the physiology of ventilation and chest wall mechanics (see Chapters 10 and 18) is needed to help the reader understand how abnormalities of the upper airway, chest wall, diaphragm, and abdominal muscles cause disease. This chapter reviews major disorders of the neuromuscular and skeletal systems that affect breathing. Disorders are grouped according to which functional unit of the neuromuscular system is affected, focusing on pulmonary manifestations of these disease processes (Table 29-1).
TABLE 29-1
Locations at Which Several Neuromuscular Diseases Affect the Respiratory System
| Location | Disease |
| Cortex and upper motor neurons | Stroke, traumatic brain injury |
| Spinal cord | Trauma, transverse myelitis, multiple sclerosis |
| Anterior horn cells (lower motor neurons) | ALS, spinal muscular atrophy, poliomyelitis and postpoliomyelitis |
| Peripheral nerves | GBS, critical illness polyneuropathy, Lyme disease |
| Neuromuscular junction | MG, LES, botulism |
| Muscle | DMD, polymyositis, acid maltase deficiency |
| Interstitial lung disease* | Polymyositis, dermatomyositis, tuberous sclerosis, neurofibromatosis |
*A category of systemic diseases that can affect neuromuscular function.
General Principles Relating To Neuromuscular Weakness Of The Ventilatory Muscles
Pathophysiology and Pulmonary Function Testing
Weakness of the respiratory muscles leads to the inability to generate or maintain normal respiratory pressures. Pulmonary function testing in patients with neuromuscular weakness typically reveals a restrictive ventilatory defect even if the lungs are normal. Vital capacity (VC), forced expiratory volume in 1 second (FEV1), and total lung capacity (TLC) are decreased. Residual volume (RV) is normal or decreased, and diffusing capacity corrected for alveolar volume is normal or near-normal but has been reported to be decreased.1 Comparison of spirometric results obtained with the patient in seated and supine positions can be useful in showing that orthopnea is caused by neuromuscular weakness. A decrease in VC or FEV1 of greater than 20% when a patient moves from the seated to the supine position suggests diaphragmatic weakness (Table 29-2 and Figure 29-2). The inability to generate normal respiratory pressures is reflected in a decreased maximal inspiratory pressure (PImax). Expiratory muscle weakness is characterized by a decreased maximal expiratory pressure (PEmax).
TABLE 29-2
Pulmonary Function Testing Results from a Patient With Profound Diaphragm Weakness
| Predicted Value | Lower Limit of Normal | Sitting Position | % of Predicted | Supine Position | % Change from Sitting | |
| FVC | 4.42 | 3.55 | 1.85 | 42 | 0.89 | −52 |
| FEV1 | 3.36 | 2.62 | 1.51 | 45 | 0.68 | −55 |
| FEV1/FVC | 75.88 | 66.20 | 81.75 | 108 | 75.76 | −7 |
| TLC | 6.53 | 4.92 | 4.21 | 64 | ||
| RV | 2.10 | 1.34 | 2.39 | 114 | ||
| DLCO | 24.93 | 16.67 | 17.16 | 69 | ||
| DLCO/VA | 3.88 | 2.38 | 5.57 | 144 | ||
| PImax | 110.58 | 75.02 | 18.46 | 17 | ||
| PEmax | 207.29 | 140.04 | 26.52 | 13 |
Arterial blood gases in the setting of a rapid, shallow breathing pattern may show a decreased PaCO2, although progressive inspiratory muscle weakness leads to hypoventilation and hypercapnia. Hypoxemia can occur in patients unable to take deep breaths and may be caused by microatelectasis, which leads to ventilation/perfusion mismatching within the lung and a resulting decrease in PaO2 (Figure 29-3). Chronic hypoxemia in this situation may be protective against acute respiratory muscle failure. Experimental data suggest that when hypoxemia is chronic, it may increase diaphragm muscle endurance.2 Hypoventilation that occurs with progressive neuromuscular disease may be a protective mechanism that avoids acute respiratory muscle fatigue. However, when hypoxemia is acute, it potentiates respiratory muscle fatigue, hastening respiratory failure.3
Clinical Signs and Symptoms
In the early stages of neuromuscular disease, patients with respiratory muscle weakness initially report exertional dyspnea and fatigue. As the disease process progresses, patients may complain of orthopnea or symptoms of cor pulmonale. These symptoms occur because the muscles involved with respiration can no longer generate or maintain normal ventilation. The response to hypoxemia and respiratory drive, measured with airway occlusion pressure (P0.1), is preserved in most patients with neuromuscular weakness.4 Because these patients do not have the strength to take deep breaths, they increase minute ventilation by increasing respiratory rate and adopting a rapid, shallow breathing pattern, which uses less respiratory muscle strength but provides less efficient ventilation. Patients with poor inspiratory muscle function (especially diaphragm weakness) may have marked orthopnea and prefer to sleep in a seated position. They may also experience a decline in the volume and power of voice or voice quality. Progressive muscle weakness can progress to the point where adequate ventilation is no longer maintained, and hypercapnia occurs.
Monitoring and Assessing Patients With Muscle Weakness for Respiratory Insufficiency
Monitoring the ventilatory function of a patient with neuromuscular weakness can involve repeated measurement of inspiratory pressure, VC, and arterial blood gas values. Depending on the condition, the need for mechanical support can be signaled by reaching either a critical value on testing or an overall clinical condition that does not allow unassisted ventilation to continue. Additional measurements that have been suggested to be more indicative of early respiratory insufficiency, at least in amyotrophic lateral sclerosis (ALS), include maximal sniff nasal inspiratory force5 and nocturnal oximetry.6
At least two caveats to this general approach should be mentioned. First, patients with myasthenia gravis having an acute myasthenic crisis may have normal test results within minutes of acute ventilatory failure because of the nature of the disorder.7 Second, the need to protect the upper airway from secretions and aspiration may not be clearly reflected in results of pulmonary function tests, which evaluate only the mechanical function of the ventilatory pump. Neuromuscular weakness may not manifest uniformly in all muscle groups. Ventilation may be only moderately reduced in patients with gross oropharyngeal dysfunction leading to aspiration. Patients with ventilatory weakness can have a high risk of acute respiratory failure if upper respiratory tract infection or pneumonia develops. In these patients, inability to clear secretions can increase the work of breathing; the results are muscle fatigue, hypoventilation, and acute respiratory failure.
Management of Respiratory Muscle Weakness
Respiratory insufficiency and failure to clear secretions are the major consequences of inspiratory and expiratory muscle weakness. Treatment of these patients involves consideration of mechanical ventilation via face mask or other noninvasive interfaces or via tracheostomy. Although often overlooked, therapies to augment secretion clearance and assist with cough are important in these patients (see Chapters 39 and 40). Used together, these interventions can decrease hospitalizations secondary to respiratory complications in patients with neuromuscular disease.8 These measures may also be useful in delaying or preventing the need for intubation or tracheostomy, although severe bulbar muscle weakness may limit a patient’s ability to avoid invasive ventilatory support.9 In addition to these interventions, general rehabilitation focusing on aerobic conditioning, muscle strengthening, and respiratory muscle training can often delay the need for ventilatory support and improve the overall quality of life for patients with muscle weakness.10
Noninvasive ventilation is being used increasingly for short-term and intermittent ventilatory support of patients with neuromuscular disease.11 Acute deterioration, such as during a pneumonia, and surgical procedures such as gastrostomy tube insertion are situations where noninvasive ventilation is safe and effective if used carefully.9,12 If a patient needs long-term ventilatory support on an intermittent basis, such as at night only, noninvasive ventilation may be appropriate.13 Decisions to begin mechanical ventilation in some patients with neuromuscular weakness have varied greatly among physicians14 and may be motivated by many different patient factors.15 No uniform guidelines exist for the use of long-term mechanical ventilation for patients with neuromuscular weakness. However, it is considered a standard option for patients who have reached the point of respiratory failure. Starting a patient on long-term mechanical ventilation requires careful planning and consideration of various issues related to respiratory care in alternative settings. These issues have been addressed in consensus statements16,17 and are discussed in Chapter 51.
Diaphragm pacing in patients with spinal cord injury has been described for some time using direct stimulation of an intact phrenic nerve to contract the diaphragm and produce negative intrathoracic pressure and inspiration.18 This technique usually requires a thoracotomy, with its associated risks and high cost, and carries some risk of phrenic nerve injury. These objections have led to the development of an alternative system for diaphragm pacing using direct pacing of the diaphragm employing intramuscular electrodes implanted laparoscopically.19 When the electrodes can be placed in the diaphragm muscle at an electrophysiologically mapped motor point, the result is often elimination of the need for mechanical ventilation in patients with spinal cord injury and delay in the need to start mechanical ventilation in patients with ALS and other neuromuscular diseases.20
Mini Clini
Care of a Patient With Neuromuscular Weakness
Noninvasive positive pressure ventilation (NIPPV) may be a reasonable first choice in the care of this patient. The patient’s mental status and bulbar function are intact. He has no significant problems with secretions. (Important factors for successful application of NIPPV are discussed in Chapter 45.) Use of a nasal mask with a biphasic positive airway pressure unit may be instituted and titrated to patient tolerance. (Most pressure or volume-cycled ventilators can be used to deliver NIPPV and invasive ventilation.) A time-cycled backup rate can be set on some ventilators to facilitate ventilation of patients who may inadequately trigger the ventilator. If the ability to clear secretions becomes compromised, cough augmentation strategies should be implemented. As long as bulbar function—swallowing and secretion management—are maintained, noninvasive ventilation is a reasonable choice for ventilatory support.
Specific Neuromuscular Diseases
Disorders of the Muscle (Myopathic Disease)
Primary muscle disease can decrease the ability of a normal neural impulse to generate effective muscle contraction. Some commonly recognized myopathies include Duchenne muscular dystrophy (DMD), myotonic dystrophy, and polymyositis. Table 29-3 presents a more complete list of myopathic diseases associated with ventilatory dysfunction.
TABLE 29-3
Myopathic Diseases With Associated Respiratory Dysfunction
| Muscular Dystrophies | Myopathies |
| DMD | Congenital myopathies |
| Myotonic dystrophy | Nemaline rod myopathy |
| Facioscapulohumeral muscular dystrophy | Centronuclear myopathy |
| Limb-girdle dystrophy | Metabolic myopathies |
| Oculopharyngeal dystrophy | Acid maltase deficiency |
| Mitochondrial myopathies (Kearns-Sayre syndrome) | |
| Inflammatory myopathies | |
| Polymyositis | |
| Dermatomyositis | |
| Hypothyroid-related and hyperthyroid-related myopathies | |
| Endocrine myopathies | |
| Steroid-induced myopathies (including critical illness myopathy) | |
| Miscellaneous myopathies | |
| Electrolyte disorders (e.g., hypophosphatemia, hypokalemia) | |
| Rhabdomyolysis | |
| Periodic paralysis | |
| Postneuromuscular blockade myopathy |
Duchenne Muscular Dystrophy and Becker Muscular Dystrophy
Duchenne muscular dystrophy (DMD) is a genetic muscle-wasting disorder caused by mutations in the dystrophin gene.21 Because it is an X-linked recessive disorder, it affects mostly males. The diagnosis is made when a dystrophin mutation is found in DNA from peripheral white blood cells or when dystrophin is found to be absent or abnormal in muscle biopsy tissue.
Progressive scoliosis is associated with DMD and can contribute further to respiratory insufficiency. Many patients undergo spine fusion surgery, and often the procedure allows greater comfort and ease in maintaining an upright posture. Although no randomized trials have proven any benefit of surgery on pulmonary function,22 observational data suggest that the rate of respiratory decline is slower after fusion surgery.23
Obstructive sleep apnea has been described in a significant proportion of patients with DMD, prompting some authors to recommend formal polysomnography in patients with symptoms of obstructive sleep apnea or at the stage of becoming wheelchair-bound.24 Institution of positive pressure ventilation (PPV) is a decision most patients face at some point in the disease. The point at which to begin different stages of ventilatory support depends on both test results and clinical condition. Nocturnal PPV is usually indicated when FVC reaches 30% of predicted with signs of hypoventilation.25,26
[/not-level-membership-for-pulmolory-and-respiratory-category]
