Chapter 102 Neurological emergencies in children
Neurological emergencies are the most common life-threatening emergencies in children. In developed societies after the first year of life, the leading cause of death in childhood is injury, particularly traumatic brain injury. There is a range of conditions affecting the brain, spinal cord and peripheral nervous system that require prompt recognition, resuscitation and definitive management. The pathophysiology, clinical features, treatment and outcome of these acute neurological emergencies are influenced by several important differences between adults and children. These differences include response to injury, developmental maturity and capacity for growth and recovery.
PATHOPHYSIOLOGY OF BRAIN INJURIES IN CHILDREN
Features of brain injury particular to the paediatric patient are described below.
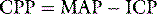
BONE DEVELOPMENT
The skull bones in the first year of life are thin with open sutures and open fontanelles. Beyond 2 years, the skull sutures close and the cranial vault thickens. In young children there tends to be less bony protection from high impact trauma, while the non-rigid skull may expand to partially decompress expanding lesions.1
UNDIAGNOSED COMA
An ordered approach to diagnosis and treatment is required for a child with depressed conscious state of unknown origin. This approach must consider common life-threatening and rare treatable diseases (Table 102.1).
| Structural | Metabolic |
|---|---|
| Trauma | Post-ictal state |
| Accidental | Infection |
| Inflicted | Meningitis |
| Hydrocephalus | Encephalitis |
| Haemorrhage | Drugs and toxins |
| AVM | Hypoxia–ischaemia |
| Aneurysms | Circulatory shock |
| Tumour | Biochemical |
| Tumour | Hypoglycaemia |
| Cerebral abscess | Electrolyte disorders |
| Sodium/water | |
| Calcium | |
| Acid–base disturbance | |
| Hyperthermia | |
| Hepatic failure | |
| Haemolytic–uraemic syndrome | |
| Inborn errors of metabolism | |
| Reye’s syndrome |
INITIAL MANAGEMENT
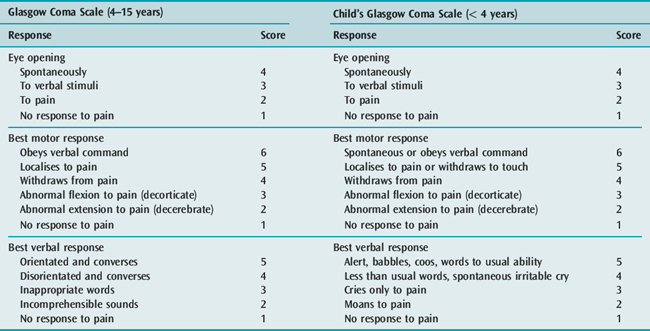
Concurrent with the initial assessment and resuscitation, relevant details of the present and past history should be obtained. A detailed neurological and general physical examination should be performed. It is important to document accurately the conscious state so that changes over time, particularly deterioration, can be easily recognised. The Glasgow Coma Scale (GCS) is appropriate for this purpose. The responses of children change with development and therefore the GCS requires modification for paediatric use (Table 102.2). After completing the clinical assessment, the likely diagnosis is often apparent and appropriate investigation and treatment can commence. Multiple factors may compound to produce coma. For example, a child with severe gastroenteritis may have hyperthermia, hyponatraemic dehydration, metabolic acidosis and hypovolaemic shock.
CONTROLLED VENTILATION
Indications for ventilating a comatose child are:
Once ventilation is initiated the stomach should be drained with a gastric tube and blood pressure checked every 5 minutes. Raised ICP should be considered in any case of rapidly progressive coma. Intracranial hypertension should be managed with moderate hyperventilation and intravenous mannitol (0.25 g/kg). Hypertonic saline given as 0.5 ml/kg of 20% solution (3.4 mmol/ml) can also rapidly reduce ICP.2 Once stability is achieved, adequate sedation and analgesia is required. Muscle relaxants may be necessary to facilitate ventilation and prevent straining; however, their use precludes further neurological assessment and therefore, if long-acting muscle relaxants are continued, ICP monitoring is advisable. Hyperventilation is a short-term manoeuvre and following the early resuscitation phase, gradual return to a low-normal PaCO2 should be the aim. This is best achieved with end tidal CO2 and ICP monitoring. Particular attention should be paid to restoring intravascular volume and maintaining an adequate CPP.
ADDITIONAL INVESTIGATIONS
Additional investigations include arterial blood gas analysis, serum electrolytes, glucose, urea and creatinine, liver function tests, serum ammonia, serum and CSF lactate and pyruvate, and urine analysis. Appropriate screening of blood and urine will exclude common poisons and drug intoxications.
STATUS EPILEPTICUS
Convulsive status epilepticus (CSE) is usually defined as a continuous convulsion lasting 30 minutes or longer or repeated convulsions lasting 30 minutes or longer without recovery of consciousness between convulsions.3 The common causes of CSE in children are:
PATHOPHYSIOLOGY
Many physiological changes occur during prolonged seizures. There is an initial phase of compensation lasting less than 30 minutes. Following a period of transition there is a phase of decompensation commencing between 30 and 60 minutes and evolving over hours. Physiological changes during the compensated phase include tachycardia, hypertension, increased catecholamine release and increased cardiac output. Changes within the brain include increased cerebral blood flow and increased cerebral utilisation of glucose and oxygen. After 30–60 minutes the mechanisms for homeostatic compensation fail. During the decompensated phase there may be falling blood pressure and cardiac output, hypoglycaemia, hypoxia, acidosis, electrolyte disturbance and rhabdomyolysis. The cerebral physiology is characterised by failing autoregulation and reduced cerebral blood flow and oxygen and glucose utilisation. Over hours a deficit in brain energy develops and this is associated with the development of brain damage.4
EXCITATORY AMINO ACIDS AND BRAIN INJURY
Mesial temporal sclerosis is the most common acquired brain lesion following CSE. There is evidence that the accumulation of a number of excitatory and inhibitory amino acids have a role in the pathophysiology of neuronal injury. In particular, glutamate accumulation and stimulation of NMDA receptors (N-methyl-D-aspartate) leads to an influx of intracellular calcium, which triggers a number of cytotoxic events and ultimately cell death.5
MANAGEMENT
The initial management is as for other neurological emergencies with attention to airway and oxygenation. Most seizures in childhood cease spontaneously in a short time, but if they persist for 5 minutes or continue after presentation to an emergency department, they should be stopped to avoid metabolic and ischaemic neuronal damage. Hypoglycaemia should be excluded or detected early. If intravenous access cannot be obtained rapidly, drugs can be administered intramuscularly, intranasally, rectally or via the intraosseous (i.o.) route. If benzodiazepines are administered within 20 minutes of a seizure commencing, the rate of seizure control is higher than if they are administered after 30 minutes.6 This justifies early prehospital administration of benzodiazepines to children with active seizures at the time of ambulance arrival.7 Specific drug treatment includes the following.
BENZODIAZEPINES
Diazepam, midazolam and lorazepam are the most useful agents.
Lorazepam (0.05–0.1 mg/kg i.v.) may have advantages over diazepam as it is as effective, has a longer half-life, and causes less respiratory depression.8,9
Midazolam and clonazepam are also effective. When i.v. access is not available, intramuscular or intranasal administration of midazolam is an alternative to rectal diazepam.10–12 In children presenting to an emergency department still convulsing, 0.2 mg/kg i.m. of midazolam has been reported to control 83% of seizures within 5 minutes of administration and 93% of seizures within 10 minutes.13 Midazolam has also been used by constant i.v. infusion (1–8 μg/kg per min) to control refractory CSE in children.14–16
PHENYTOIN
Phenytoin should be commenced if benzodiazepines are not effective. It is given as 20 mg/kg i.v. over 30 minutes, which is followed by a maintenance dose (4 mg/kg 8-hourly past the neonatal period). It causes minimal sedation or respiratory depression, but is not suitable in neonates and in patients who are already on maintenance doses. As it is given slowly, phenytoin will not have rapid effects and if generalised CSE persists, consideration should be given to the use of thiopental (see below). Fosphenytoin, a prodrug of phenytoin, can be administered i.m. or i.v., but has a less irritant effect when given i.v.17 Its place in the management of CSE in children is not yet clear.18
PARALDEHYDE
In many centres rectal paraldehyde (0.4 ml/kg mixed with an equal quantity of olive oil) is frequently used in the management of CSE.18 One advantage of paraldehyde is that, like diazepam, it can be administered rectally if i.v. access is difficult to obtain.
OUTCOME
The outcome of CSE is dependent on the aetiology. Neurologically normal children in whom CSE is precipitated by fever are considered to have a good prognosis with mortality reported between 0 and 2%.19 The incidence of neurological deficits or cognitive impairment in this group is also very low.5 In acute symptomatic CSE (where CSE is a symptom of an acute neurological process such as infection or trauma), mortality is 12–16% and the incidence of new neurological dysfunction is more than 20%.19 In this setting, however, it is very difficult to tease out the extent to which prolonged seizures contribute to neurological sequelae.
BACTERIAL MENINGITIS
INVESTIGATIONS
Lumbar puncture for CSF microscopy, culture and bacterial antigen testing is required for definitive diagnosis of BM and to guide antibiotic therapy. While an LP can be performed safely in the majority of children with BM, LP may precipitate brainstem herniation if raised ICP is present.20 Identifying children at risk of this complication is difficult, so it is generally recommended that empiric antibiotic therapy be commenced and LP deferred if any of the following clinical features are present:
A normal CT scan does not exclude the possibility of elevated ICP.20 Therefore, the decision to defer an LP should be based on clinical rather than radiological signs. In addition to signs of increased ICP, other indications for deferring the LP include cardiorespiratory instability and severe coagulopathy.
If LP is deferred, alternative methods of establishing a bacterial diagnosis include blood culture and bacterial antigen testing in urine and polymerase chain reaction (PCR) testing of blood.21,22
MANAGEMENT
ANTIBIOTIC THERAPY
Empiric broad-spectrum antibiotics should be selected based on likely pathogens and local resistance patterns. A common protocol for BM is to use ampicillin plus cefotaxime for the first month of life and to use a third-generation cephalosporin (cefotaxime or ceftriaxone) after the first month.23 In regions where penicillin and cephalosporin-resistant Pneumococcus occurs, vancomycin should be added to the initial empiric antibiotics until the causative organism is identified and the antibiotic sensitivities are known. When cephalosporin-resistant pneumococci are found to be the causative organism in meningitis, both a third-generation cephalosporin and vancomycin should be continued as vancomycin penetrates into CSF poorly and therefore should not be used as a single antibiotic. The addition of rifampicin should be considered.24
ADJUVANT THERAPY
Although a number of adjuvant therapies have been investigated experimentally, the only one that is commonly used clinically is dexamethasone. If dexamethasone is used it should ideally be given before the first dose of antibiotics and continued for 48 hours (0.4 mg/kg 12-hourly).25 There is evidence that dexamethasone reduces the incidence of neurological sequelae and sensineural deafness; however, the beneficial effects are greatest in Hib meningitis.26,27 Now that immunisation has changed the epidemiology and antibiotic-resistant pneumococcal strains are more common, it is possible that the relationship between risk and benefit of dexamethasone therapy has changed. However, current recommendations support the use of dexamethasone for children more than 6 weeks of age with bacterial meningitis.24,28
FLUID THERAPY
Although there is consensus that hypovolaemia should be treated rapidly and aggressively, fluid therapy after the initial resuscitation is controversial. It is important to consider both the tonicity of i.v. fluids as well as the rate of fluid administration. Although dextrose saline solutions with low sodium content (0.18–0.3% saline) are isotonic with plasma when first administered, as the dextrose crosses the blood–brain barrier or is metabolised, the net effect is that the cerebral circulation is exposed to hypotonic fluids, which potentially exacerbates cerebral oedema. Therefore, the i.v. fluid used in BM should be 0.45% or 0.9% saline in 5% dextrose.29,30
Restriction of maintenance fluids in BM has been commonly practised. Hyponatraemia is common and if it is assumed that this is secondary to the syndrome of inappropriate antidiuretic hormone secretion (SIADH) then fluid restriction is logical. Recent evidence, however, suggests that ADH is appropriately elevated in response to hypovolaemia31 and that hyponatraemic patients with BM tend to be more dehydrated than normonatraemic patients.32 Compared to fluid restriction, fluid therapy aimed at providing maintenance plus the fluid deficit has been reported to result in a more rapid correction of sodium and ADH.31,33
CHEMOPROPHYLAXIS FOR CONTACTS
Prophylaxis is required for every household member in cases of meningococcal infection and in cases of Hib infection when there is another child in the household under 5 years who is unimmunised. Rifampicin (10 mg/kg to a maximum of 600 mg b.d.) for 2 days in meningococcal disease or 4 days in Hib disease is appropriate. Neonates require 10 mg/kg per day. Pregnant women can be effectively managed with a single intramuscular dose of ceftriaxone (250 mg). Ciprofloxacin (15 mg/kg to a maximum of 750 mg) is also effective and has the advantage of single dose therapy.34
OUTCOME
Bacterial meningitis is associated with significant mortality and morbidity. Overall mortality for BM in childhood is 5–10%; however, in children requiring mechanical ventilation a mortality rate of 30% has been reported, with major neurosequelae occurring in 33% of survivors.35
PREVENTION
Immunisation has significantly reduced the incidence of Hib meningitis in developed countries,36,37 while initial trials of heptavalent pneumococcal vaccine have shown a reduction in invasive pneumococcal infection in children.38 Meningococcal serogroup C conjugate vaccines have reduced the incidence of severe meningococcal disease in infants in the UK and Australia.39,40
ENCEPHALITIS
Presenting symptoms of encephalitis include seizures, focal neurological deficits in the setting of an acute febrile illness, confusion and coma. Meningeal irritation may not be obvious. CSF analysis may show a pleocytosis and in the early phase this can consist predominantly of neutrophils. As mentioned, herpes is the most important diagnosis to make, because it is treatable. Electroencephalography (EEG), CT and magnetic resonance imaging (MRI) are helpful in making the diagnosis. MRI is more sensitive than CT for detecting signs of encephalitis, particularly during the early stages of the illness. PCR on CSF may also aid rapid diagnosis.41,42 Aciclovir used early improves outcome and should be commenced when the diagnosis is suspected.
The enteroviruses are important causes of encephalitis in children. In addition, they cause other acute neurological illnesses including acute flaccid paralysis due to transverse myelitis and Guillain–Barré syndrome.43 Pleconaril is a promising new antiviral agent that appears to have clinical benefit in enteroviral infections, including CNS disease.44 Outcome of viral encephalitis is worse in infancy than in older age groups.45
NON-TRAUMATIC INTRACRANIAL HAEMORRHAGE
Non-traumatic intracranial haemorrhage (ICH) is uncommon in children. Arteriovenous malformations (AVM) are a more common cause of haemorrhage in children than aneurysms46 (Table 102.3). The presenting features are similar to those seen in adults, and include sudden severe headache, altered conscious state and seizures. Diagnosis can usually be made by CT scan. Raised ICP, if present, is managed in the usual manner. A mass lesion or acute obstructive hydrocephalus requires neurosurgical assessment.
Table 102.3 Aetiology of spontaneous intracranial haemorrhage in children
Following diagnosis of ICH further investigations may be required to clarify the underlying cause. These include coagulation profile and platelet count. Angiographic images can be obtained with CT, MRA (magnetic resonance angiography) or digital subtraction. In some cases definitive cerebral angiography may be required to define the underlying vascular malformation or tumour. Definitive surgery or endovascular treatment of an AVM can be planned once the underlying lesion has been defined. A period of close observation is required as children may be at greater risk of rebleeding from an AVM than adults.47 The efficacy of therapies (e.g. calcium channel blockers) used to prevent vasospasm in adults has not been studied in children with aneurysmal haemorrhage. Sequelae of ICH include hemiparesis, aphasia, seizures and hydrocephalus.
HYPOXIC–ISCHAEMIC ENCEPHALOPATHY
AETIOLOGY
The most common causes of hypoxic–ischaemic encephalopathy outside the neonatal period are:
PATHOLOGY
Following restoration of cerebral blood flow there is a period of relative hyperaemia followed by relative hypoperfusion. Animal studies suggest that the blood flow during this phase of postischaemic hypoperfusion is determined by the metabolic needs of the brain.48 Cytotoxic cerebral oedema may subsequently develop, but significant elevation of ICP is unusual unless ischaemia and damage are profound.
MANAGEMENT
The principles of therapy are similar to those for other brain injuries. It is mandatory to provide rapid cardiopulmonary resuscitation (CPR) and prevent secondary insults. In cases of out-of-hospital cardiac arrest, full resuscitation must be attempted while the history is sought. Postresuscitation care is important for optimising outcome. Comatose patients with hyper- or hypotonia and a Glasgow Coma Scale score (GCS) < 8 are probably best managed by mechanical ventilation, sedation and paralysis for at least 1–2 days although benefits are not proven. Ventilation should be targeted at normocapnia, and hyperventilation avoided due to the risk of further cerebral ischaemia.49 Haemodynamic disturbance may develop because of primary cardiac dysfunction or hypovolaemia secondary to fluid loss from capillary leak syndrome. Circulating volume should be restored and inotropic agents considered to improve the state of the circulation. The dose and choice of vasoactive agent should be individualised based on haemodynamic monitoring.
Following cardiac arrest children are usually hypothermic. Fever commonly develops during the subsequent hours and is associated with worse outcome:50 fever should therefore be anticipated and prevented. Further to preventing fever, there is evidence that therapeutic hypothermia of 32–34°C improves outcome. There are two trials in adults following cardiac arrest51,52 and two trials in newborns following birth asphyxia53,54 demonstrating improved neurological outcome if hypothermia is used for between 12 and 72 hours. Although there are no paediatric trials, therapeutic hypothermia should be considered as a treatment option in children who are comatose following hypoxic–ischaemic events. Hyperglycaemia has been associated with a worse prognosis and, although it may simply be a marker of injury severity, active treatment has been advocated. The role of ICP measurement is limited as intracranial hypertension usually only occurs in the setting of severe injury and poor outcome.55
PROGNOSIS
The major determinants of recovery are:
In immersion injuries in young children, full recovery may be possible despite prolonged ischaemia if sufficient rapid cerebral cooling has occurred. In these cases the onset of ischaemia may be delayed by bradycardia with preferential cerebral flow (the ‘diving reflex’). In general, survival from out-of-hospital cardiac arrest is unlikely, even with expert CPR, if asystole is present on arrival at hospital.56 The rare exception is the hypothermic child who presents following immersion; prolonged CPR may be justified in selected cases when profound hypothermia was induced rapidly. If cardiac output is present on arrival at hospital, with either flexion or extension to pain, recovery is likely. Normothermic patients, who present apnoeic, flaccid and unresponsive to pain, are likely to die or have serious neurological deficits. This group is also prone to further deterioration several days after the insult, with progression of cerebral oedema.55 Coma persisting for more than 24 hours is a predictor of poor prognosis and minimal long-term improvement is likely in this group.57 Residual neurological deficits present at the end of the first week are less likely to improve following ischaemic injury than following traumatic brain injury.
A number of ancillary tests have been investigated as predictors of neurological outcome. Somatosensory-evoked potentials (SEPs) performed at the bedside are the most useful aid to prediction. One report of 109 children with severe brain injury concluded that, with appropriate patient selection, the positive predictive value for poor outcome of bilaterally absent SEPs is 100% (95% CI, 92–100%).58
GUILLAIN–BARRé SYNDROME
CLINICAL FEATURES
Guillain–Barré syndrome (GBS) is the most common cause of acute motor paralysis in children. Although most patients develop typical ascending, symmetrical areflexic weakness, GBS may present insidiously with apparent lethargy or loss of motor milestones in the young child. There may also be rapid progression and admission criteria to the intensive care unit (ICU) include respiratory failure, bulbar palsy, severe autonomic disturbance or rapidly progressive weakness. Sensory loss is usually minimal and transient. Pain in the back and legs, possibly neurogenic in origin, is common and may be the presenting feature.59 This pain may be severe and is often difficult to control. Papilloedema and encephalopathy occasionally occur.60 The complications of deep venous thrombosis and thromboembolism are not common problems in young children but may occur in adolescents.
INVESTIGATIONS
An LP should be done ideally before treatment with intravenous immunoglobulin (IVIG) is commenced. CSF typically shows a white cell count < 10 × 106, while CSF protein is usually raised but may be normal during the first week of illness. Nerve conduction studies may be normal if performed early. Neurophysiological criteria have been used to classify GBS subtypes according to either demylinating or axonal and whether or not both sensory and motor nerves are involved.61 Anti-ganglioside antibodies are raised in some but not all patients.62
MANAGEMENT
Plasma exchange (PE) and IVIG are effective therapies. The indications for either are rapid progression and respiratory insufficiency or weakness to the point of being unable to walk unassisted. The strongest evidence for these therapies is from adult trials.63–65 Although large trials adequately powered to separately test the efficacy of PE and IVIG in children have not been performed, a number of small retrospective studies have described local experiences with these immune therapies in children with mixed results.66–70 As IVIG has significant potential technical advantages over PE, IVIG is generally the first-line therapy in children. Indications for IVIG are based on the degree of functional impairment and time from onset of symptoms. The indications are not influenced by the clinical or neurophysiological subtype or the results of antibody screening. There is no evidence to support sequential treatment using both PE and IVIG.71
PROGNOSIS
The prognosis in acute GBS may be better for children than adults. Full recovery is likely if the time from maximal deficit to onset of recovery is less than 18 days. Complete recovery, despite a longer plateau phase, has been reported, however.72 Good recovery can occur in patients who have required ventilation and the need for ventilation may not be a poor prognostic factor in children.66 Those presenting with a subacute course are at risk of relapses and permanent motor deficits.
METABOLIC ENCEPHALOPATHY
Approximately 0.1% of babies have an inborn error of metabolism. Acute encephalopathy is one of the many ways neurometabolic diseases present in childhood.73,74 In general, acute presentations occur in the neonatal period and early infancy. Symptoms are often vague and include lethargy, poor feeding and vomiting. Older infants and children more commonly present with a chronic encephalopathy, with features that may include seizures, long-tract signs, visual impairment and loss of milestones.
APPROACH TO DIAGNOSIS
Lactic acidosis can be further assessed with measurement of lactate:pyruvate ratio and urine biochemical analysis may provide further diagnostic information (Table 102.4).
| Biochemical marker | Disorder |
|---|---|
| Hypoglycaemia | Organic acidurias |
| Methylmalonic/propionic acidaemia | |
| Reye’s syndrome | |
| Fat oxidation defects | |
| MCAD | |
| Ketoacidosis | Diabetes mellitus |
| Organic acidurias | |
| Lactic acidosis | Mitochondrial encephalomyopathies |
| Respiratory chain disorders | |
| Pyruvate carboxylase deficiency | |
| Hyperammonaemia (2–3 × normal) | Reye’s syndrome |
| Urea cycle defects | |
| Fat oxidation defects | |
| MCAD | |
| Organic acidurias |
MCAD, medium-chain acyl CoA dehydrogenase.
ASSESSMENT
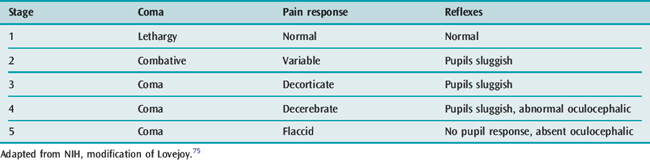
Metabolic coma can be staged using Lovejoy’s classification for Reye’s syndrome (Table 102.5).75
MANAGEMENT
Initial management is largely supportive. Metabolic derangements such as acidosis and hypoglycaemia should be corrected. Acute intracranial hypertension should be managed with mechanical ventilation while the role of ICP monitoring is controversial. Hyperammonaemia should be managed with limited protein intake initially. In certain circumstances increasing ammonia metabolism using agents such as arginine and sodium benzoate is helpful. In some settings providing calories intravenously in the form of glucose and fat may help limit the extent of catabolism and thereby help to curtail the metabolic crisis. Dialysis is used to control severe hyperammonaemia and acidosis. Continuous venovenous haemodiafiltration is considered more effective than peritoneal dialysis.76
REYE’S SYNDROME AND REYE-LIKE ILLNESS
A wide range of conditions has been described that closely mimic typical Reye’s syndrome. Ten percent of cases originally described as being Reye’s syndrome were subsequently found to be due to an inherited metabolic disorder.77 A careful approach is mandatory to ensure detection of inherited disorders.
The introduction of neonatal screening programmes using tandem mass spectrometry will see many inherited metabolic diseases diagnosed in the neonatal period, often prior to the onset of symptoms. This technology is particularly useful for detecting fatty acid oxidation defects (e.g. medium-chain acyl CoA dehydrogenase deficiency, or MCAD) and disorders of amino acid metabolism (e.g. methylmalonic acidaemia).78
SPINAL INJURY
INCIDENCE
Paediatric spinal trauma is relatively rare, comprising < 5% of all spinal injuries. Approximately 5% of children with severe head trauma will have a cervical spine injury. However, a high proportion of children who die following motor vehicle trauma, particularly those who suffer immediate cardiorespiratory arrest, or who die before arrival at hospital, have disruption of the spinal cord above C3, particularly at the cervicomedullary junction.79,80
PATHOPHYSIOLOGY
The patterns of spinal injury in children differ from those seen in adults in many ways. Spinal cord injury without radiographic abnormality (SCIWORA) occurs almost exclusively in children (20–60% of spinal cord injuries). It is associated with a high incidence of complete neurological deficit. Spinal injury in the first decade occurs most commonly in the first two cervical segments, with atlanto-axial rotatory subluxation, bony or ligamentous injuries, or SCIWORA and severe cord injury. Although not as common as upper cervical injuries, lower cervical injuries below C4 do occur in young children,81 while injury may occur at more than one level. Atlanto-axial rotatory subluxation is rarely associated with neurological deficit and ligamentous injury is more likely than high cervical fractures to be associated with permanent neurological deficit.82 As the bony spine matures, the pattern of injury becomes more adult-like with lower cervical and thoracic injuries seen in the second decade.83,84
CLINICAL FEATURES
INVESTIGATIONS
Resuscitation, including emergency intubation, should not be delayed to perform X-rays. The entire spinal column should, however, be X-rayed subsequently to demonstrate the presence of subluxation, fractures or dislocations. MRI is useful in both the acute setting and in assessment at later stages, and is now accepted as the standard for identifying haemorrhage, contusion or compression of the cord. CT produces better definition of bony injuries, and may also show cord involvement by haematomas, bone fragments and foreign bodies. SCIWORA was defined in an era before MRI was routinely available. In the majority of children with SCIWORA, MRI will also be normal; however, if intramedullary lesions are demonstrated with MRI, permanent neurological deficits are more likely.85,86 Somatosensory-evoked potentials may be useful to evaluate the integrity of the spinal cord, particularly in the comatose patient.
MANAGEMENT
The use of steroids is controversial and there are no studies specific to children. Two randomised controlled trials in adults demonstrated that high-dose methylprednisolone administered within 8 hours of the injury (30 mg/kg initially followed by 5.4 mg/kg per hour for the following 24–48 hours) improved outcome.87,88 These studies have significant limitations and the results have not been universally accepted.89–91
The incidence of venous thromboembolism after spinal injury in adolescents approaches that in adults and therefore patients in this age group should receive standard prophylaxis. Venous thromboembolism is rare in prepubertal children (approximately 1%), and the risk–benefit ratio of routine prophylaxis in this age group is unknown.92 There should be early consultation with a specialised spinal injuries unit. Optimal rehabilitation requires a team of orthopaedic and neurosurgeons, rehabilitation specialists, nurses, physiotherapists, occupational therapists, psychiatrists, social workers and schoolteachers.
PROGNOSIS
The prognosis of all spinal injuries in children may be better than in adults. In one series of 113 children with spinal column injuries, 55 (48%) had no neurological deficit, and 38 (34%) had an incomplete deficit. Of these, 23 (20%) made a complete recovery and 11 (10%) improved.79 The remaining 20 (18%) children had a complete cord injury and, of these, 4 improved and 3 died. In a smaller series, 44% had neurological deficits, SCIWORA was seen in 21% and 11% of injuries were immediate, complete and permanent. Of the 18 children with SCIWORA, 4 had a permanent, complete deficit.82
1 Mann KS, Chan KH, Yue CP. Skull fractures in children: their assessment in relation to developmental skull changes and acute intracranial hematomas. Childs Nerv Syst. 1986;2:258-261.
2 Suarez JI, Qureshi A, Bhardwaj A, et al. Treatment of refractory intracranial hypertension with 23.4% saline. Crit Care Med. 1998;26:1118-1122.
3 Hanhan UA, Fiallos MR, Orlowski JP. Status epilepticus. Pediatr Clin North Am. 2001;48:683-694.
4 Lothman E. The biochemical basis and pathophysiology of status epilepticus. Neurology. 1990;40(S2):13-23.
5 Scott RC, Surtees RAH, Neville BGR. Status epilepticus: pathophysiology, epidemiology, and outcomes. Arch Dis Child. 1998;79:73-77.
6 Lewena S, Young S. When benzodiazepines fail: how effective is second line therapy for status epilepticus in children? Emerg Med Australas. 2006;18:45-50.
7 Dieckmann RA. Is the time overdue for an international reporting standard for convulsive paediatric status epilepticus? Emerg Med Australas. 2006;18:1-3.
8 Chiulli DA, Terndrup TE, Kanter RK. The influence of diazepam or lorazepam on the frequency of endotracheal intubation in childhood status epilepticus. J Emerg Med. 1991;9:13-17.
9 Appleton RE, Sweeney A, Choonara I, et al. Lorazepam vs. diazepam in the treatment of epileptic seizures and status epilepticus. Dev Med Child Neurol. 1995;37:682-688.
10 Chamberlain JM, Altieri MA, Futterman C, et al. A prospective, randomized study comparing intramuscular midazolam with intravenous diazepam for the treatment of seizures in children. Pediatr Emerg Care. 1997;13:92-94.
11 Mahmoudian T, Zadeh MM. Comparison of intranasal midazolam with intravenous diazepam for treating acute seizures in children. Epilepsy Behav. 2004;5:253-255.
12 McIntyre J, Robertson S, Norris E, et al. Safety and efficacy of buccal midazolam versus rectal diazepam for emergency treatment of seizures in children: a randomised controlled trial. Lancet. 2005;366:205-210.
13 Lahat E, Aladjem M, Eshel G, et al. Midazolam in treatment of epileptic seizures. Pediatr Neurol. 1992;8:215-216.
14 Rivera R, Segnini M, Baltodano A, et al. Midazolam in the treatment of status epilepticus in children. Crit Care Med. 1993;21:991-994.
15 Koul RL, Raj Aithala G, Chacko A, et al. Continuous midazolam infusion as treatment of status epilepticus. Arch Dis Child. 1997;76:445-448.
16 Yoshikawa H, Yamazaki S, Abe T, et al. Midazolam as a first-line agent for status epilepticus in children. Brain Dev. 2000;22:239-242.
17 Wheless JW. Paediatric use of intravenous and intramuscular phenytoin: lessons learned. J Child Neurol. 1998;13:S11-14.
18 Appleton R, Choonara I, Martland T, et al. The Status Epilepticus Working Party. The treatment of convulsive status epilepticus in children. Arch Dis Child. 2000;83:415-419.
19 Raspall-Chaure M, Chin RF, Neville BG, et al. Outcome of paediatric convulsive status epilepticus: a systematic review. Lancet Neurol. 2006;5:769-779.
20 Rennick G, Shann F, de Campo J. Cerebral herniation during bacterial meningitis in children. BMJ. 1993;306:953-955.
21 Seward RJ, Towner KJ. Evaluation of a PCR-immunoassay technique for detection of Neisseria meningitidis in cerebrospinal fluid and peripheral blood. J Med Microbiol. 2000;49:451-456.
22 Newcombe J, Cartwright K, Palmer WH, et al. PCR of peripheral blood for diagnosis of meningococcal disease. J Clin Microbiol. 1996;34:1637-1640.
23 Feigin RD, Pearlman E. Bacterial meningitis beyond the neonatal period. In: Feigin RD, Cherry JD, editors. Textbook of Pediatric Infectious Diseases. Philadelphia: WB Saunders; 1998:400-429.
24 Chavez-Bueno S, McCracken GH. Bacterial meningitis in children. Pediatr Clin North Am. 2005;52:795-810.
25 Odio CM, Faingezicht I, Paris M, et al. The beneficial effects of early dexamethasone administration in infants and children with bacterial meningitis. N Engl J Med. 1991;324:1525-1531.
26 McCracken GH, Label MH. Dexamethasone treatment for bacterial meningitis in infants and children. Am J Dis Child. 1989;143:287-289.
27 Schaad UB, Lips U, Gnehm HE, et al. Dexamethasone therapy for bacterial meningitis. Lancet. 1993;342:457-461.
28 McIntyre PB, Berkey CS, King SM, et al. Dexamethasone as adjunctive therapy in bacterial meningitis. A meta-analysis of randomised clinical trial since 1988. JAMA. 1997;278:925-931.
29 Duke T, Molyneux EM. Intravenous fluids for seriously ill children: time to reconsider. Lancet. 2003;362:1320-1323.
30 Duke T. Fluid management of bacterial meningitis in developing countries. Arch Dis Child. 1998;79:181-185.
31 Powell KR, Sugarman LI, Eskenazi AE, et al. Normalisation of plasma arginine vasopressin concentrations when children with meningitis are given maintenance plus replacement fluid therapy. J Pediatr. 1990;117:515-522.
32 Bianchetti MG, Thyssen HR, Laux-End R, et al. Evidence for fluid volume depletion in hyponatraemic patients with bacterial meningitis. Acta Paediatr. 1996;85:1163-1166.
33 Singhi SC, Singhi PD, Srinivas B, et al. Fluid restriction does not improve the outcome of acute meningitis. Pediatr Infect Dis. 1995;14:495-503.
34 Cuevas LE, Kazembe P, Mughogho GK, et al. Eradication of nasopharyngeal carriage of Neisseria meningitidis in children and adults in rural Africa: a comparison of ciprofloxacin and rifampicin. J Infect Dis. 1995;171:728-731.
35 Madagame ET, Havens PL, Bresnahan JM, et al. Survival and functional outcome of children requiring mechanical ventilation during therapy for acute bacterial meningitis. Crit Care Med. 1995;23:1279-1283.
36 Mclntyre PB, Chey T, Smith WT. The impact of vaccination against invasive Haemophilus influenzae type b disease in the Sydney region. Med J Aust. 1995;162:245-248.
37 Adams WG, Deaver KA, Cochi SL, et al. Decline of childhood Haemophilus influenzae type b (Hib) disease in the Hib vaccine era. JAMA. 1993;269:221-226.
38 Whitney CG, Farley MM, Hadler J, et al. Decline in invasive pneumococcal disease after the introduction of protein–polysaccharide conjugate vaccine. N Engl J Med. 2003;348:1737-1746.
39 Balmer P, Borrow R, Miller E. Impact of meningococcal C conjugate vaccine in the UK. J Med Microbiol. 2002;51:717-722.
40 Booy R, Jelfs J, El Bashir H, et al. Impact of meningococcal C conjugate vaccine use in Australia. Med J Aust. 2007;186:108-109.
41 Aurelius E, Johansson B, Skoldenberg B, et al. Rapid diagnosis of herpes simplex virus encephalitis by nested polymerase chain reaction assay of cerebrospinal fluid. Lancet. 1991;337:189-192.
42 Troendle-Atkins J, Demmler GJ, Buffone GJ. Rapid diagnosis of herpes simplex encephalitis by using polymerase chain reaction. J Paediatr. 1993;123:376-380.
43 McMinn P, Stratov I, Nagarajan L, et al. Neurological manifestations of enterovirus 71 infection in children during an outbreak of hand, foot, and mouth disease in Western Australia. Clin Infect Dis. 2001;32:236-242.
44 Rotbart HA, Webster AD. Treatment of potentially life-threatening enterovirus infections with Pleconaril. Clin Infect Dis. 2001;32:228-235.
45 Koskiniemi M, Vaheri A. Effect of measles, mumps, rubella vaccination on pattern of encephalitis in children. Lancet. 1989;1:31-34.
46 Al-Jarallah A, Al-Rifai MT, Riela AR, et al. Nontraumatic brain hemorrhage in children: etiology and presentation. J Child Neurol. 2000;15:284-289.
47 Stein BM, Wolpert SM. Arteriovenous malformation of the brain II: current concepts and treatment. Arch Neurol. 1980;37:69-75.
48 Michenfelder JD, Milde JH. Post-ischaemic canine cerebral blood flow appears to be determined by cerebral metabolic needs. J Cereb Blood Flow Metab. 1990;10:71-76.
49 International Liaison Committee on Resuscitation. International consensus on cardiopulmonary resuscitation and emergency cardiovascular care science with treatment recommendations. Part 6: Paediatric basic and advanced life support. Resuscitation. 2005;67:271-291.
50 Zeiner A, Holzer M, Sterz F, et al. Hyperthermia after cardiac arrest is associated with an unfavorable neurologic outcome. Arch Intern Med. 2001;161:2007-2012.
51 Bernard SA, Gray TW, Buist MD, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557-563.
52 Hypothermia after Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346:549-556.
53 Shankaran S, Laptook AR, Ehrenkranz RA, et al. Whole-body hypothermia for neonates with hypoxic–ischemic encephalopathy. N Engl J Med. 2005;353:1574-1584.
54 Gluckman PD, Wyatt JS, Azzopardi D, et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet. 2005;365:663-670.
55 Sarnaik AP, Preston G, Lieh-Lai M, et al. Intracranial perfusion pressure in near-drowning. Crit Care Med. 1985;13:224-227.
56 Schindler MB, Cox PN, Jarvis A, et al. Factors influencing outcome from out of hospital paediatric cardiopulmonary arrest. Anaesth Intensive Care. 1995;23:381-382.
57 Kriel RL, Krach LE, Luxenberg MG, et al. Outcome of severe anoxic/ischaemic brain injury in children. Paediatr Neurol. 1994;10:207-212.
58 Beca J, Cox PN, Taylor MJ, et al. Somatosensory evoked potentials for prediction of outcome in acute severe brain injury. J Pediatr. 1995;126:44-49.
59 Manners PJ, Murray KJ. Guillain–Barré syndrome presenting with severe musculoskeletal pain. Acta Paediatr. 1992;81:1049-1051.
60 Cole GF, Matthew DJ. Prognosis in severe Guillain–Barré syndrome. Arch Dis Child. 1987;62:288-291.
61 Hadden RD, Cornblath DR, Hughes RA, et al. Electrophysiological classification of Guillain–Barré syndrome: clinical associations and outcome. Ann Neurol. 1998;44:780-788.
62 Pritchard J. What’s new in Guillain–Barré syndrome? Pract Neurol. 2006;6:208-217.
63 Van der Meche FGA, Schmitz PIM, Dutch Guillain–Barré Study Group. A randomised trial comparing intravenous immune globulin and plasma exchange in Guillain–Barré syndrome. N Engl J Med. 1992;326:1123-1129.
64 Hughes RAC, Raphaël J-C, Swan AV, et al. Intravenous immunoglobulin for Guillain–Barré syndrome. Cochrane Database Syst Rev. (Issue 1):2006. Art. No. CD002063
65 Raphaël JC, Chevret S, Hughes RAC, et al. Plasma exchange for Guillain–Barré syndrome. Cochrane Database Syst Rev. (Issue 2):2002. Art. No. CD001798
66 Jansen PW, Perkin RM, Ashwal S. Guillain–Barré syndrome in childhood: natural course and efficacy of plasmapheresis. Pediatr Neurol. 1993;9:16-20.
67 Lamont PJ, Johnston HM, Berdoukas VA. Plasmapheresis in children with Guillain–Barré syndrome. Neurology. 1991;41:1928-1931.
68 Shahar E, Leiderman M. Outcome of severe Guillain–Barré syndrome in children: comparison between untreated cases versus gamma-globulin therapy. Clin Neuropharmacol. 2003;26:84-87.
69 Ortiz-Corredor F, Pena-Preciado M. Use of immunoglobulin in severe childhood Guillain–Barré syndrome. Acta Neurol Scand. 2007;115:289-293.
70 Vajsar J, Sloane A, Wood E, et al. Plasmapheresis vs intravenous immunoglobulin treatment in childhood Guillain–Barré syndrome. Arch Pediatr Adolesc Med. 1994;148:1210-1212.
71 Hughes RA, Wijdicks EF, Barohn R, et al. Practice parameter: immunotherapy for Guillain–Barré syndrome: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2003;61:736-740.
72 Briscoe DM, McMeniman LB, O’Donohoe NV. Prognosis in Guillain–Barré syndrome. Arch Dis Child. 1987;62:733-735.
73 Neville BGR. Paediatric neurology. In: Walton J, editor. Brain’s Diseases of the Nervous System. Oxford: Oxford Medical Publications; 1993:453-477.
74 Chaves-Carballo E. Detection of inherited neuro-metabolic disorders. A practical clinical approach. Pediatr Clin North Am. 1992;39:801-819.
75 National Institutes of Health Conference on Reye’s Syndrome. Diagnosis and treatment of Reye’s syndrome. JAMA. 1981;246:2441.
76 Schaffer F, Straube Oh J, et al. Dialysis in neonates with inborn errors of metabolism. Nephrol Dial Transplant. 1999;14:918-919.
77 Green A, Hall S. Investigation of metabolic disorders resembling Reye’s syndrome. Arch Dis Child. 1992;67:1313-1317.
78 Andresen BS, Dobrowolski SF, O’Reilly L, et al. Medium-chain acyl CoA dehydrogenase deficiency (MCAD) mutations identified by MS/MS-based screening of newborns differ from those observed in patients with clinical symptoms: identification and characterization of a new, prevalent mutation that results in mild MCAD deficiency. Am J Hum Genet. 2001;68:1408-1418.
79 Hadley MN, Zabramski MD, Browner CM, et al. Paediatric spinal trauma. Review of 122 cases of spinal cord and vertebral column injuries. J Neurosurg. 1988;68:18-24.
80 Swan PK, Bohn DJ, Sides CA, et al. Cervical spine damage associated with severe head injury in the paediatric patient: implications for airway management. Anaesth Intensive Care. 1987;15:115-116.
81 Givens TG, Polley KA, Smith GF, et al. Pediatric cervical spine injury: a three year experience. J Trauma. 1996;41:310-314.
82 Ruge JR, Sinson GP, McLone DG, et al. Pediatric spinal injury: the very young. J Neurosurg. 1988;68:25-30.
83 Birney TJ, Hanley EN. Traumatic cervical spine injuries in childhood and adolescence. Spine. 1989;14:1277-1282.
84 McCall T, Fassett D, Brockmeyer D. Cervical spine trauma in children: a review. Neurosurg Focus. 2006;20:E5.
85 Dare AO, Dias MS, Li V. Magnetic resonance imaging correlation in pediatric spinal cord injury without radiographic abnormality. J Neurosurg. 2002;97(S1):33-39.
86 Liao CC, Lui TN, Chen LR, et al. Spinal cord injury without radiological abnormality in preschool-aged children: correlation of magnetic resonance imaging findings with neurological outcomes. J Neurosurg. 2005;103:17-23.
87 Bracken MB, Shepard MJ, Collins WF, et al. A randomized controlled trial of methylprednisolone or naloxone in the treatment of acute spinal cord injury: result of the Second National Acute Spinal Cord Injury Study. N Engl J Med. 1990;322:1405-1411.
88 Bracken MB, Shepard MJ, Holford TR, et al. Administration of methylprednisolone for 24 or 48 hours or tirilazad mesylate for 48 hours in the treatment of acute spinal cord injury: results of the third National Acute Spinal Cord Injury randomized controlled trial. JAMA. 1997;277:1597-1604.
89 Short DJ, El Masry WS, Jones PW. High-dose methyl-prednisone in the management of acute spinal cord injury – a systematic review from a clinical perspective. Spinal Cord. 2000;38:273-286.
90 Hurlbert RJ. The role of steroids in acute spinal cord injury: an evidence-based analysis. Spine. 2001;26:S39-46.
91 Sayer FT, Kronvall E, Nilsson OG. Methylprednisolone treatment in acute spinal cord injury: the myth challenged through a structured analysis of published literature. Spine J. 2006;6:335-343.
92 Jones T, Ugalde V, Franks P, et al. Venous thromboembolism after spinal cord injury: incidence, time course, and associated risk factors in 16,240 adults and children. Arch Phys Med Rehabil. 2005;86:2240-2247.