[level-membership-for-neurosurgery-category]
Chapter 184 Neurologic Problems of the Spine in Achondroplasia
Achondroplasia is characterized by disproportionately short stature with rhizomelic shortening of the extremities, macrocephaly, midface hypoplasia, and frontal bossing.1,2 This skeletal dysplasia results from defective formation of endochondral bone.3–5 Morbidity in achondroplasia results largely from bony compression of the neuraxis6–12 and respiratory failure.13,14 This chapter focuses on the main indications for neurosurgical interventions for problems attributed to the spine in achondroplasia, namely, cervicomedullary compression and spinal stenosis.
Thoracolumbar stenosis resulting in spinal compression is the commonest complication of achondroplasia, becoming symptomatic in most patients in their 20s or later.15 Thoracolumbar stenosis can be accelerated in infants who develop progressive thoracolumbar kyphosis if bracing is not undertaken before vertebral wedging develops.16 Less common problems in infancy include symptomatic airway obstruction17 and severe cervicomedullary compression secondary to foramen magnum stenosis.18–20 The latter can be sometimes accompanied by swallowing difficulties and central apnea.21
Most individuals with achondroplasia have normal intelligence. Motor milestones are delayed,22 partly because of generalized hypotonia and partly because of the mechanical disadvantage imposed by short limbs. Psychological problems arising from short stature include lack of acceptance by peers and a tendency of adults, including parents and teachers, to treat children with achondroplasia appropriately for their height rather than their age.23 Involvement with other families with children of short stature can improve self-esteem and can assist parents in guiding their achondroplastic children through the difficulties of growing up in a culture that equates stature to status.
Respiratory complications include obstructive sleep apnea secondary to a small upper airway.14 Many people with achondroplasia snore. Many infants sleep with their necks in a hyperextended position, a position that functionally increases the size of the upper airway, relieving intermittent obstruction. However, the hyperextended neck position may exacerbate neurologic sequelae of a small foramen magnum and cervicomedullary compression.24 A small thoracic cage may result in restrictive pulmonary disease in infancy, and respiration can be compromised further by aspiration secondary to gastroesophageal reflux, swallowing dysfunction, or both, resulting in recurrent pneumonia.
Reproductive difficulties have not been conclusively documented,25 but evidence of reduced fertility, frequent fibroid cysts, and early menopause have been reported. Women with achondroplasia must deliver their infants by cesarean section because of cephalopelvic disproportion, and administration of spinal anesthesia is strongly discouraged due to the small size of the canal.
Although life expectancy was formerly thought to be normal for people with achondroplasia, age-specific mortality is increased at all ages, with the highest increase occurring in children.26 Moreover, cardiovascular cases of death are increased in the adult group (25-54 years of age). The increased mortality in childhood is likely related to severe cervicomedullary compression.
Causes and Pathophysiology
Clinical Genetics and Growth Plate Ultrastructures
Achondroplasia is an autosomal dominant disorder; most estimates of its frequency cluster between 1:25,000 and 1:35,000 live births27,28; however, the true frequency may be slightly higher.29 New mutations account for about 80% of children born with achondroplasia.28 As in many autosomal dominant disorders, a positive correlation exists between advanced paternal age and occurrence of new mutations.30 Offspring of couples in which both partners are affected by achondroplasia have a 25% chance of inheriting both parental achondroplasia alleles, resulting in homozygous achondroplasia, which is universally fatal within the first year of life.31 The skeletal features of achondroplasia are highly exaggerated in the homozygous condition, resulting in significantly shorter limbs, a smaller chest size, and a smaller foramen magnum. Death is usually secondary to respiratory complications, sometimes in concordance with foramen magnum stenosis and brain stem compression.32
Achondroplasia results from impaired formation of endochondral bone. A missense mutation, G380R, in the transmembrane domain of fibroblast growth factor receptor 3 has been traced to chromosome 4, at 4p16.3.33,34 The protein is a tyrosine kinase receptor expressed in developing bones. The G380R mutation has been found in most patients.35,36 Several groups used this discovery to develop polymerase chain reaction diagnostic tests. The histochemical features of the endochondral growth plates of achondroplastic bone have been interpreted in several ways. Some researchers suggested that mitotic abnormalities indicate cessation of normal cell function and arrest of cell division of the chondrocytes.37 This impaired formation of bone from cartilage is seen in the growth of the diaphyses of long bones.3 In addition, an enlargement of the epiphyses occurs. Cartilaginous synchondroses in the spine and skull seem to fuse prematurely, and hypertrophy of the spinal articular surface occurs. Cervicomedullary compression is typically a pediatric concern, while spinal stenosis is usually seen in adults.
Cervicomedullary Compression
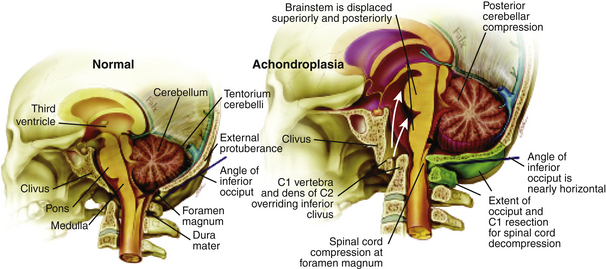
Cervicomedullary compression stems primarily from a reduction in the diameter of the foramen magnum in the sagittal and coronal dimensions that is sometimes more than five standard deviations less than normal.38 The cranial base (chondrocranium) derives from endochondral ossification. In achondroplasia, the base is stunted, shorter, and narrower that normal.39 The basioccipital bone, which forms the anterior border of the foramen magnum, is narrow and angulated. The lateral and posterior parts, consisting of the exoccipitalis bone, are similarly deformed, resulting in the diamond, triangular, or teardrop shape of the achondroplastic foramen magnum. In addition, the articular surfaces of the occipital bone (between the lateral occipital and the basioccipital bones and between the lateral bones and the planum nuchale of the squama) are hypertrophic and can encroach on the neural elements within the foramen. The pathology of the achondroplastic skull is further complicated by the small size of the posterior fossa, resulting from stunting of the endochondrally derived planum nuchale, and the resultant horizontalization of the squamous portions of the occipital bones. This constricted arrangement of the skull base displaces the brain stem upward and the foramen magnum anteriorly, resulting in posterior tilting to the brain stem and further impingement of the neuraxis posteriorly24 (Fig. 184-1).
Spinal Stenosis
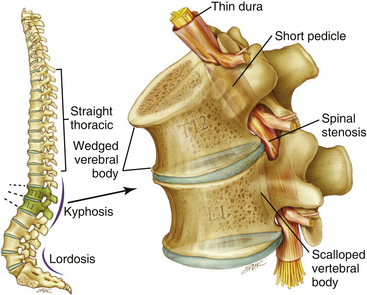
The anatomy of the achondroplastic spine is distinctive in several respects, all of which can contribute to compromise of the spinal cord or nerve roots.40,41 The hypertrophy of epiphyseal articular processes in the long bones is mirrored at the caudal and cephalic surfaces of the vertebral bodies, resulting in a mushroom shape at each end and concomitant scalloping along the posterior surface that is appreciable in a contrast myelogram.42 Abbreviated and thickened pedicles of the vertebral arches result from premature fusion of synchondroses between the laminae and the vertebral bodies3; the laminae are also thickened. Intervertebral discs tend to bulge prominently,42 further aggravating neural encroachment by the enlarged vertebral body articular surfaces. The interpediculate distance decreases in the lumbar region of the spine, resulting in a canal that tapers caudally,43 the opposite of normal (the canal normally widens caudally). The overall picture is one of dramatic stenosis in every dimension of the spine, a stenosis sometimes aggravated by osteoarthritic changes and disc ruptures.44 Consequently, a generalized constriction of spinal neural elements occurs (Fig. 184-2).
Evaluation and Diagnosis
Referral Criteria
Primary-care physicians have recognized the need for comprehensive prospective management of achondroplastic patients for sleep, respiratory, and central nervous system disorders.14,17,45 To aid the efforts, we present our recommendations for evaluation of achondroplastic patients for cervicomedullary compression and spinal stenosis. Achondroplastic patients referred to our institution are usually evaluated according to a standardized protocol that involves a multidisciplinary team of neurosurgeons, neurologists, pulmonary and sleep specialists, geneticists, anesthesiologists, neuroradiologists, orthopedic surgeons, and otolaryngologists. Because these patients are at risk for brain stem compression, comprehensive testing is directed toward detection of central and obstructive apnea and cervicomedullary compression, all of which contribute to the risk of sudden death.
Cervicomedullary Compression
Clinical Pathology and Presentation
Cervicomedullary compression warrants early and aggressive treatment. Results of studies suggest such compression is progressive and potentially fatal because it increases the risk of sudden death by central respiratory failure.46,47 This condition has gained increasing attention as a cause of respiratory and neurologic impairment in children with achondroplasia.48–50 In our prospective evaluation of achondroplastic infants, we found radiographic evidence of craniocervical stenosis in 58% of the studied patients, and a diagnosis of cervicomedullary compression was made in 35%.49 These figures are for a selected population and are certainly higher that the proportion in the general population. Nonetheless, they are a strong argument for the careful evaluation and treatment of achondroplastic children. A retrospective study found excess mortality in sudden death resulting from brain stem compression, which was identified as the cause of half of the excess deaths.26 The same study also found a 7.5% risk of sudden death in the first year of life.
Chronic medullary and upper cervical cord compression may exist as a neurologically asymptomatic lesion, exhibiting neither signs of root compression in the arms nor symptoms of cranial nerve impairment.51 Nonetheless, microcystic histopathologic changes, cervical syringomyelia, and necrosis and gliosis have been reported in autopsies of achondroplastic children who died unexpectedly.19,20,47 Presumably, lesions of this type interrupt neural respiratory pathways from the nucleus tractus solitarius to the phrenic nerve nucleus—arresting the muscles of respiration and resulting in sudden death in some cases. We consider infants with a history of sleep apnea or other severe respiratory or neurologic abnormalities to be at increased risk for respiratory complications resulting from occult cervicomedullary compression.49 Some authors have recommended performing sleep and imaging studies on all children with achondroplasia.45 A composite profile of the patient with cervicomedullary compression includes upper or lower extremity paresis, apnea and cyanosis, hyperreflexia or hypertonia, and delay in motor milestones beyond achondroplastic standards. These patients can present a striking contrast to the usual floppy, hypotonic achondroplastic infants.52 More recently, a study indicated that although normal imaging studies may be found in achondroplastic children on magnetic resonance imaging (MRI) in a neutral neck position, flexion can lead to increased intracranial pressure (ICP) due to venous outflow obstruction and complete cerebrospinal fluid (CSF) outflow block.53 The imaging findings have to be correlated with the clinical status of the patient, and the decision for treatment is ultimately guided by the patient’s neurologic status and the surgeon’s experience.
Evaluation
Once the high-risk patient with respiratory or neurologic symptoms or signs has been identified, we advise comprehensive testing. Parents should be carefully interviewed about the health and medical history of the child, with emphasis on respiratory symptoms, and a general physical examination, including chest measurements, should be performed. Respiratory evaluation should include the evaluation of blood pH, a chest radiograph, and overnight polysomnography. Electrocardiograms and echocardiograms should be performed to see whether there is cardiac evidence of chronic oxygen deprivation during sleep. A neurologic examination for signs of brain stem compression, such as hyperreflexia, hypertonia, paresis, asymmetry of movement or strength, or abnormal plantar response, is essential. Brain stem auditory evoked potential and upper extremity somatosensory evoked potential evaluation should be considered as an adjunct, especially in patients with normal results on neurologic examination.54 Imaging studies of the craniocervical junction are necessary, particularly MRI in the sagittal plane. We also strongly recommend MRI flow studies of the CSF at the foramen magnum, using the technique of synchronizing the succession of images with the heartbeat. Some of these procedures have been described elsewhere.49
Indication for Surgery
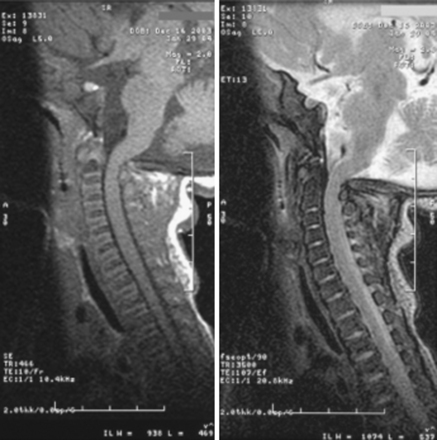
The underlying principle for surgery must be to identify patients who are at risk for neurologic damage or sudden death. We recommend that patients with cervicomedullary compression be identified and treated prophylactically, before abrupt and irreversible changes occur. For the purpose of diagnosis, we define clinically significant cervicomedullary compression to be (1) neurologic evidence of upper cervical myelopathy; (2) evidence of stenosis on imaging studies, including the absence of flow above and below the foramen magnum; and (3) an otherwise unexplained respiratory or developmental abnormality. It is possible to see a patient with brain stem compression and obstructive apnea who nonetheless meets these criteria. Having discovered these indications, the treatment team should also ask whether the patient’s status is stable or deteriorating before undertaking operative decompression and should evaluate the probability for catastrophic deterioration if decompression is not performed (Fig. 184-3).
Operative Management
Craniocervical surgical decompression for cervicomedullary compression in children with achondroplasia55 has been used at several centers with generally good results.12,21,56 Decompression of the cervicomedullary junction has shown to bring about dramatic, sustained improvement in neurologic and respiratory compromise.49,52 The procedure has not received as wide acceptance as it might, however, because its successful performance relies on careful management of the anatomic difficulties presented by achondroplastic patients. Clinical evaluation is frequently difficult for many reasons, some of which are unrelated to neurologic compromise. Long-term follow-up data that would allow a definitive assessment of craniocervical decompression have also been lacking. As with any surgical procedure, detailed prior consultation must be conducted with the parents to inform them of the potential risks and expected benefits for their achondroplastic child.
For decompression, a midline suboccipital incision is made, and the ligaments and musculature are dissected subperiosteally to expose the occiput and the spinous process and laminae of C1 and C2. The arch of C1 is then removed with a high-speed drill and small curettes. The surgeon frequently sees a thick, fibrous band or pannus about the level of C1 that should be left in place during the initial bone drilling to create a protective layer for the underlying dura and spinal cord. Removal of the arch of C2 is sometimes necessary; more caudal areas of compression necessitate even further caudad decompression. The posterior rim of the foramen magnum is thinned gradually with a high-speed drill and removed with small, straight, and angled curettes. Invariably, the bone of the foramen magnum is thickened and oriented more horizontally than usual, severely indenting the underlying dura. The most delicate part of the dissection occurs as the drill approaches the posterior rim of the foramen magnum. Once bone decompression is complete, the fibrous pannus or band is removed as well, often revealing the transverse dural channel that offers dramatic evidence of the extent of the dural restriction; consequently, adequate attention must be paid to the soft tissue aspects of the decompression. We used to perform duraplasty routinely with the placement of a dural patch. However, more recently, this step of the procedure has been only required if there is any persistent dural constriction after the fibrous band has been removed. After a duraplasty is performed, adequate cord pulsation and CSF flow are confirmed. A dural patch can be performed using pericranium, paraspinal fascia, or cadaveric dura. A watertight seal is essential. We do not recommend the placement of a wound drain so as not to potentiate the development of a CSF fistula; however, if the dura is opened, it is wise to place a ventriculostomy, because many patients with achondroplasia have increased ICP and will experience CSF leak and possible pseudomeningocele if the duraplasty is challenged early.
Outcome
An early series of 15 patients with cervicomedullary compression were treated with decompression and showed significant symptomatic improvement.52 The mortality rate was zero, and the main complication was CSF leak. These results were then confirmed by a larger study involving 43 pediatric patients with heterozygous achondroplasia and foramen magnum stenosis who underwent 45 cervicomedullary decompressions at our institution over an 11-year period.57 Complete resolution or partial improvement in the preoperative symptoms was observed in all patients. There was no mortality, and the surgical morbidity rate was low, consisting of CSF leak in patients in whom the dura mater was opened (both accidental and intentional for decompression and with patch placement). This problem was successfully managed in all cases with local measures (wound oversewing) or CSF diversion. A comparative study of the quality of life using the Medical Outcome Study 36-Item Short-Form General Health Survey between patients who underwent decompression for symptomatic craniomedullary compression and asymptomatic patients with achondroplasia was done at our institution, examining a total 122 patients58: 56 individuals (46%) had craniomedullary decompression, and 66 patients (54%) did not. The quality of life of patients who had undergone surgery was found to be comparable to those treated conservatively (controlled for age and sex), indicating that once neurologic symptoms become severe, surgical decompression can provide the same level of quality of life as that of an achondroplastic patient without craniomedullary compression.
Concerns have been expressed about the morbidity accompanying the relatively high number of patients with cervicomedullary compression who undergo decompression and require subsequent shunt placement.59 In our opinion, this hydrocephalus is not a complication of the operation but rather is discovered as a result of the violation of the dural layer, a situation that allows a more sensitive picture of the ICP dynamics than is otherwise possible. This view received additional support from research that suggests a strong predisposition to hydrocephalus in achondroplastic children and a higher-than-normal ICP as a result of the bony anatomy. In light of these findings, the morbidity and mortality of our reported craniocervical decompression are not only low for a procedure of this delicacy but also low relative to the general morbidity and mortality of achondroplastic disease. This argument is strengthened by a large number of the patients we diagnose having a combination of severe respiratory and neurologic disease; after their procedures, they go on to make developmental strides at a rapid pace, with resolution of their symptoms. Although we believe that our treatment of achondroplastic hydrocephalus is unconventional, our understanding of it rests on some unproven assumptions. First, the current understanding of ICP dynamics is neither comprehensive nor general. Despite evidence strongly favoring the theory that raised ICP in an achondroplastic patient is secondary to venous hypertension, this view has not been conclusively demonstrated. However, the balance of the evidence, both published and experiential, favors this view, and we will continue to use it as a working theory until better understanding of ICP in general, and hydrocephalus in achondroplasia in particular, are available.
Spinal Stenosis
Clinical Pathology and Presentation
Spinal stenosis is the most common complication of achondroplasia, usually becoming symptomatic in the third decade or later. The anatomy of the achondroplastic spine is distinctive in several aspects, all of which contribute to spinal cord compromise and nerve root compression. The superior and inferior articular facets of the vertebral bodies are susceptible to hypertrophy, resulting in a mushroom shape that is clearly evident on a contrast myelogram.42 Abbreviated and thickened pedicles of the vertebral arches result from premature fusion. Intervertebral discs tend to bulge prominently, further aggravating neural encroachment by the enlarged vertebral body articular surfaces. The interpediculate distance decreases in the lumbar region of the spine, resulting in a canal that tapers caudally, the opposite of normal (the canal normally widens caudally). The overall picture is one of dramatic stenosis in every dimension of the spine.
Although the problems relating to cervicomedullary compression are frequently identified in infancy and childhood, neurologic problems below the foramen magnum often present in late adolescence and adulthood, perhaps as a result of postural or degenerative changes superimposed on congenital stenosis. In one series of patients treated at our institution, however, 35% became symptomatic before age 15.60 Estimates of the incidence of symptomatic spinal stenosis range from 37.5% to 89%, suggesting that a significant proportion of patients eventually have this problem.8,61–63 Because the achondroplastic spinal canal tends to have severe congenital constriction, more intensive early screening might reveal substantial numbers of young achondroplastic patients with occult symptoms of spinal stenosis.
Although symptomatic stenosis can warrant neurosurgical intervention, it is generally possible to distinguish between the neurosurgical and the orthopedic aspects of the management of the achondroplastic spine, inasmuch as some neurologic complaints requiring surgical intervention are secondary results of orthopedic deterioration.64,65 The hypotonia that the achondroplastic infant typically exhibits suggests that muscular tone may be insufficient for adequate protection of pediatric skeletal structures in weight-bearing postures.66 Achondroplastic children are developmentally delayed in supporting their heads independently, as well as in upright sitting and walking; thus, in our opinion, parents should not encourage early sitting because of the potential for aggravation of thoracolumbar kyphosis in this posture.22 Sitting and standing postures affect the curvature of the spine, and in achondroplastic children, muscular weakness, short vertebral pedicles, and lax spinal ligaments complicate these mechanics.67 Attention has also been drawn to the dynamic effect of a small chest and a globulus abdomen in the formation of a progressive kyphosis.68 Moreover, delayed standing predisposes to development of a gibbus, with wedging of one or more vertebral elements. These wedged deformities are both debilitating and preventable. Because surgical repair has risks, effort is well spent on prevention. The prophylactic use of orthopedic bracing can be applied to cases in which formation of a wedged gibbus seems likely.55,64,65,67 Parents should also be urged not to use infant carriers, strollers, or baby seats that exaggerate the thoracolumbar kyphosis.
In the adult, compromise can result from such abnormalities as hyperlordosis, minor disk bulging, hypertrophic osteoarthritis, or ligamentous hypertrophy.47 The presence of a thoracolumbar kyphosis is also positively correlated with symptomatic spinal stenosis.68 Although lower back pain is a common complaint among achondroplastic patients, those with severe stenosis can develop symptomatic neurogenic claudication. Prolonged walking produces first paresthesia and later weakness of the lower extremities, which is usually bilateral. These symptoms are promptly relieved by rest, squatting, and leaning forward, which straighten the lordosis and increase the transverse diameter of the lumbosacral canal.67 With progressive stenosis, the distance walked before claudication ensues decreases, making this symptom a useful clinical parameter. Bowel and bladder function are also compromised in many cases of severe stenosis (Table 184-1).
Table 184-1 Baseline Characteristics of Pediatric Patients with Achondroplasia Before Decompressive Surgery for Symptomatic Spinal Stenosis
| Variable | No. of Cases/Value |
|---|---|
| No. of patients | 44 |
| Sex (%) | |
| Boys | 25 (57) |
| Girls | 19 (43) |
| Mean age at surgery (yrs) | 12.7 (range 5–21) |
| Mean duration of preoperative symptoms (mos) | 9.2 (range 1–54) |
| History of CMD (%) | 27 (61) |
| Preoperative kyphosis (%) | 22 (50) |
| Preoperative scoliosis (%) | 14 (32) |
| Hydrocephalus (%) | 9 (20) |
| Presenting symptoms (%) | |
| Pain | 42 (95.4) |
| Neurogenic claudication/cauda equina∗ | 40 (91) |
| Radicular pain only | 1 (2.3) |
| Low-back pain only | 1 (2.3) |
| Myelopathy | 2 (4.5) |
∗ Presence of urinary and/or bowel incontinence occurred in the context of neurogenic claudication in 11 (25%) of 44 patients.
(Sciubba et al. “Spinal stenosis surgery in pediatric patients with achondroplasia.” J Neurosurg. 2007 May;106(5 Suppl):372-378.)
Evaluation
In more advanced cases of stenosis, neurologic abnormalities, such as weakness of the lower extremities (particularly dorsiflexors of the toes and feet) and hypoesthesia, often at the truncal level, persist at rest. Occasionally, a partial Brown-Séquard syndrome is seen. Spasticity and hyperreflexia of the legs typically indicate compression of the thoracic cord but may indicate coexistent cervical compression. The results of neurologic examinations of patients with claudication often remain otherwise normal, unless a superimposed disc problem is present. The thoracolumbar spine must be evaluated in all symptomatic achondroplastic patients, even in the absence of neurologic findings. If the patient has urinary incontinence or hesitancy, such an investigation is performed as an emergency.
Indication for Surgery
We recommend decompressive laminectomies for achondroplastic patients whose ambulation is severely limited by claudication, who show significant weakness (other than mild extensor hallucis longus weakness) at rest, or who have urinary incontinence or urgency that is attributable to compression of the cauda equina or spinal cord. There is a high incidence of urologic complications after laminectomy and a high correlation between preoperative urologic abnormalities and postoperative complaints.60 We recommend thorough urologic testing as part of the preoperative evaluation for patients undergoing laminectomy.
Operative Management
Decompression of the achondroplastic spinal canal is difficult because of the extent and severity of the stenosis. The quantitative magnitude of this stenosis has been well documented.69,70 Poor postoperative results were relatively common for spinal decompression in achondroplastic patients.15,63,71 Before the era of CT and MRI, the degree and extent of spinal decompression were often not appreciated with conventional myelography because of the lack of adequate contrast medium diffusion.6,42 Insertion of bulky instruments under the laminae during conventional techniques also frequently traumatized neural tissue. Other sources of poor results included postoperative instability resulting from overly wide laminectomies.61,62,71
The following procedure has been used at our institution with good results.60 Adequate anatomic delineation is obtained by MRI and intrathecally enhanced CT, but myelography is sometimes useful as an adjunct. Based on the results of these studies, the surgeon can devise an operative plan for adequate decompression that includes at least three segments above the level of demonstrated block and three segments below (or to S2). The incision is midline, and dissection is carried subperiosteally to expose the spinous processes, laminae, and facet joints over the extent of the area to be decompressed. When adequate exposure has been achieved, the laminae immediately medial to the facet joints are gradually thinned with a high-speed drill, forming a groove approximately parallel to the longitudinal axis of the spinal column. The drill head is pointed toward the facet at an angle of about 45 degrees to the laminal surface; this angle—rather than a 90-degree angle—offers the surgeon the control necessary to avoid accidental perforation of the laminae and the dura. The groove is deepened until the dura can be seen through the thinned laminal mantle. Drilling is then concluded in this area and is continued on the contralateral side in a similar fashion. An opening is made at the caudad part of the groove on the first side, a thin surgical punch is inserted into the epidural space, and the laminectomy is carried along the groove. This technique minimizes dural tearing, preserves the facet joints, and protects the neural structures from injury. If the facet joints are violated on both sides, spinal fusion is necessary.
A different viewpoint concerning the ideal strategy for spinal laminectomy recommends wide decompression with foraminotomies and mandatory undermining of the facets.64 The rationale for this strategy is that decompression is lateral, as well as longitudinal, in the achondroplastic spinal canal. Our experience, however, does not bear this out, despite the hypothesized impact of small lateral recesses in the achondroplastic vertebral foramen. The primary compressive dimension in the achondroplastic spine is cephalocaudal, not lateral, and undermining the facets was not necessary. Moreover, the stabilization problems encountered with wide laminectomies can be more debilitating than the initial disease. There is no reason every spinal level could not be subjected to the laminectomy we described without the need for concomitant spinal stabilization in adults. Therefore, in light of the results obtained at our institution with spinal decompressions, we believe that a narrow, extensive laminectomy is the spinal decompression of choice for adult achondroplastic patients. Some surgeons have adopted this operative technique for their nonachondroplastic patients as well. The goal is adequate decompression of neural elements, not simply enlargement of the bony canals.
Removal of the spinous processes and detachment of the paraspinal soft tissue create a large, deep void, particularly at the lumbosacral junction. To minimize the risk of pseudomeningocele formation, an overlapping closure using paraspinal muscle was developed, whereby muscle masses are brought in to fill the dead space.70 First, a paraspinal muscle encased in fascia is partially detached from the iliac spine and the lumbosacral laminae by use of a split-thickness incision, if necessary, to mobilize the required tissue, which is then reflected around its pedicle. The edge of the flap is brought down to the opposite lateral end of the lamina and is attached with heavy sutures to the inferior part of the paraspinal muscle mass on that side. The superior part of the muscle mass on that side is then retracted over the first flap, completing the muscle closure and collapsing the void. As with craniocervical decompression, somatosensory evoked potentials are monitored intraoperatively. Postoperative care is generally routine, but because of the high incidence of urologic complications, the nursing staff should be advised to help the patient be prepared for this possibility. In the pediatric population, wide laminectomy and fusion are preferred, because the immature spinal canal has a greater propensity toward instability and deformation.
Outcome
In 2006, Ain et al. examined the risk of postlaminectomy kyphosis in the thoracolumbar region in achondroplastic children with stenosis. They wanted to evaluate the need for concurrent fusion at multiple levels in a retrospective chart review of 10 patients.72 The average age of the 6 male and 4 female patients at surgery was 9.2 years (range 6-16 years). Decompression consisted of multilevel5–8 thoracolumbar laminectomies. More than 50% of each medial facet was preserved bilaterally to maintain spinal stability. All of those patients developed postoperative kyphosis ranging from 78 to 135 degrees (mean 94 degrees), and they subsequently underwent spinal fusions with instrumentation, performed from 10 months to 2.6 years after the decompressions, to stabilize the kyphoses. The authors concluded that in achondroplastic children with symptomatic stenosis, there is a high risk for postlaminectomy thoracolumbar kyphosis, and concurrent spinal fusion is indicated in patients undergoing at least five levels of laminectomy.
This conclusion was further supported by another retrospective study at our institution, indicating that fusion procedures are recommended in patients with a large decompression overlying a thoracolumbar kyphosis to avoid progressive postoperative deformity.73 The study also correlated a high risk of developing symptomatic stenosis prior to adolescence to patients with a history of cervicomedullary compression.
In a spine that has been previously decompressed, restenosis may occur because of accelerated facet hypertrophy, bony overgrowth, and scarring. This acceleration of facet hypertrophy may represent instability in the previously operated achondroplastic spine or some exaggerated response to normal motion that results from the genetic defect in this disease. Many reports document the efficacy of decompressive therapy in the treatment of achondroplastic spinal stenosis. In several of these series, reoperation was often necessary for achondroplastic spinal restenosis.74
Outcome assessment revealed that six of the patients in the series (75%) had postoperative improvement in their strength. Bladder symptoms disappeared in two patients and remained unchanged in one patient. In summary, our retrospective review suggests that, despite its technical challenges, redecompression of the spinal canal can be successful in alleviating the majority of achondroplastic patients’ symptoms (Table 184-2).
Table 184-2 Characteristics of Surgical Procedures in 44 Patients with Achondroplasia
| Variable | No. of Cases/Value |
|---|---|
| Total no. of procedures | 60 |
| Initial procedures | 49 |
| Revision procedures | 11 |
| Location of initial procedure (%) | |
| Thoracolumbar | 32 (65.3) |
| Lumbar | 10 (20.4) |
| Cervical | 4 (8) |
| Cervicothoracic | 2 (4) |
| Thoracic | 1 (2) |
| Fusion procedures with internal fixation | 43 |
| Nonfusion procedures | 17 |
| Mean follow up (mos)∗ | 34 (range 8–93) |
| Reasons for revision surgery | |
| Progressive deformity in nonfused spine | 5 |
| Decompression of junctional stenosis | 5 |
| Repeat decompression at same levels | 1 |
| Complications (%) | 7 (11.6) |
| Durotomy | 4 |
| Wound breakdown/infection | 2 |
| Instrumentation revision | 1 |
Nine of 44 patients had limited follow up because of moving out of state/country; not included in mean follow up.
(Sciubba et al. Spinal stenosis surgery in pediatric patients with achondroplasia. J Neurosurg. 2007 May;106(5 Suppl):372-378.)
Allanson J.E., Hall J.G. Obstetric and gynecologic problems in women with chondrodystrophies. Obstet Gynecol. 1986;67(1):74-78.
Bergstrom K., Laurent U., Lundberg P.O. Neurological symptoms in achondroplasia. Acta Neurol Scand. 1971;47(1):59-70.
Cohen M.E., Rosenthal A.D., Matson D.D. Neurological abnormalities in achondroplastic children. J Pediatr. 1967;71(3):367-376.
Cohen M.M.Jr., Walker G.F., Phillips C. A morphometric analysis of the craniofacial configuration in achondroplasia. J Craniofac Genet Dev Biol Suppl. 1985;1:139-165.
Duvoisin R.C., Yahr M.D. Compressive spinal cord and root syndromes in achondroplastic dwarfs. Neurology. 1962;12:202-207.
Epstein J.A., Malis L.I. Compression of spinal cord and cauda equina in achondroplastic dwarfs. Neurology. 1955;5(12):875-881.
Eulert J. Scoliosis and kyphosis in dwarfing conditions. Arch Orthop Trauma Surg. 1983;102(1):45-47.
Fremion A.S., Garg B.P., Kalsbeck J. Apnea as the sole manifestation of cord compression in achondroplasia. J Pediatr. 1984;104(3):398-401.
Galanski M., Herrmann R., Knoche U. Neurological complications and myelographic features of achondroplasia. Neuroradiology. 1978;17(1):59-63.
Goldstein S.J., Shprintzen R.J., Wu R.H.K., et al. Achondroplasia and obstructive sleep apnea: correction of apnea and abnormal sleep-entrained growth hormone release by tracheostomy. Birth Defects Orig Artic Ser. 1985;21(2):93-101.
Hancock D.O., Philips D.G. Spinal compression in achondroplasia. Paraplegia. 1965;3(1):23-33.
Hecht J.T., Butler I.J., Scott C.I.Jr. Long-term neurological sequelae in achondroplasia. Eur J Pediatr. 1984;143(1):58-60.
Hurko O., Pyeritz R., Uematsu S. Neurological considerations in achondroplasia. Basic Life Sci. 1988;48:153-162.
Lutter L.D., Langer L.O. Neurological symptoms in achondroplastic dwarfs: surgical treatment. J Bone Joint Surg Am. 1977;59(1):87-92.
Nelson F.W., Hecht J.T., Horton W.A., et al. Neurological basis of respiratory complications in achondroplasia. Ann Neurol. 1988;24(1):89-93.
Nelson M.A. Spinal stenosis in achondroplasia. Proc R Soc Med. 1972;65(11):1028-1029.
Oberklaid F., Danks D.M., Jensen F., et al. Achondroplasia and hypochondroplasia. Comments on frequency, mutation rate, and radiological features in skull and spine. J Med Genet. 1979;16(2):140-146.
Scott C.I.Jr. Medical and social adaptation in dwarfing conditions. Birth Defects Orig Artic Ser. 1977;13(3C):29-43.
Spillane J.D. Three cases of achondroplasia with neurological complications. J Neurol Neurosurg Psychiatry. 1952;15(4):246-252.
Stokes D.C., Phillips J.A., Leonard C.O., et al. Respiratory complications of achondroplasia. J Pediatr. 1983;102(4):534-541.
Thomas I.T., Frias J.L., Williams J.L., et al. Magnetic resonance imaging in the assessment of medullary compression in achondroplasia. Am J Dis Child. 1988;142(9):989-992.
Todorov A.B., Scott C.l.Jr., Warren A.E., et al. Developmental screening tests in achondroplastic children. Am J Med Genet. 1981;9(1):19-23.
Vogl A., Osborne R.L. Lesions of the spinal cord (transverse myelopathy) in achondroplasia. Arch Neurol Psychiatry. 1949;61(6):644-662.
Yamada H., Nakamura S., Tajima M., et al. Neurological manifestations of pediatric achondroplasia. J Neurosurg. 1981;54(1):49-57.
Yang S.S., Corbett D.P., Brough A.J., et al. Upper cervical myelopathy in achondroplasia. Am J Clin Pathol. 1977;68(1):68-72.
1. Cohen M.M.Jr., Walker G.F., Phillips C. A morphometric analysis of the craniofacial configuration in achondroplasia. J Craniofac Genet Dev Biol Suppl. 1985;1:139-165.
2. Oberklaid F., et al. Achondroplasia and hypochondroplasia. Comments on frequency, mutation rate, and radiological features in skull and spine. J Med Genet. 1979;16(2):140-146.
3. Duvoisin R.C., Yahr M.D. Compressive spinal cord and root syndromes in achondroplastic dwarfs. Neurology. 1962;12:202-207.
4. Vogl A., Osborne R.L. Lesions of the spinal cord (transverse myelopathy) in achondroplasia. Arch Neurol Psychiatry. 1949;61(6):644-662.
5. Spillane J.D. Three cases of achondroplasia with neurological complications. J Neurol Neurosurg Psychiatry. 1952;15(4):246-252.
6. Epstein J.A., Malis L.I. Compression of spinal cord and cauda equina in achondroplastic dwarfs. Neurology. 1955;5(12):875-881.
7. Bergstrom K., Laurent U., Lundberg P.O. Neurological symptoms in achondroplasia. Acta Neurol Scand. 1971;47(1):59-70.
8. Nelson M.A. Spinal stenosis in achondroplasia. Proc R Soc Med. 1972;65(11):1028-1029.
9. Cohen M.E., Rosenthal A.D., Matson D.D. Neurological abnormalities in achondroplastic children. J Pediatr. 1967;71(3):367-376.
10. Galanski M., Herrmann R., Knoche U. Neurological complications and myelographic features of achondroplasia. Neuroradiology. 1978;17(1):59-63.
11. Hancock D.O., Philips D.G. Spinal compression in achondroplasia. Paraplegia. 1965;3(1):23-33.
12. Yamada H., Nakamura S., Tajima M., et al. Neurological manifestations of pediatric achondroplasia. J Neurosurg. 1981;54(1):49-57.
13. Nelson F.W., Hecht J.T., Horton W.A., et al. Neurological basis of respiratory complications in achondroplasia. Ann Neurol. 1988;24(1):89-93.
14. Stokes D.C., Phillips J.A., Leonard C.O., et al. Respiratory complications of achondroplasia. J Pediatr. 1983;102(4):534-541.
15. Lutter L.D., Langer L.O. Neurological symptoms in achondroplastic dwarfs: surgical treatment. J Bone Joint Surg Am. 1977;59(1):87-92.
16. Eulert J. Scoliosis and kyphosis in dwarfing conditions. Arch Orthop Trauma Surg. 1983;102(1):45-47.
17. Goldstein S.J., Shpritzen R.J., Wu R.H.K., et al. Achondroplasia and obstructive sleep apnea: correction of apnea and abnormal sleep-entrained growth hormone release by tracheostomy. Birth Defects Orig Artic Ser. 1985;21(2):93-101.
18. Hecht J.T., Butler I.J., Scott C.I.Jr. Long-term neurological sequelae in achondroplasia. Eur J Pediatr. 1984;143(1):58-60.
19. Thomas I.T., Frias J.L., Williams J.L., et al. Magnetic resonance imaging in the assessment of medullary compression in achondroplasia. Am J Dis Child. 1988;142(9):989-992.
20. Yang S.S., Corbett D.P., Brough A.J., et al. Upper cervical myelopathy in achondroplasia. Am J Clin Pathol. 1977;68(1):68-72.
21. Fremion A.S., Garg B.P., Kalsbeck J. Apnea as the sole manifestation of cord compression in achondroplasia. J Pediatr. 1984;104(3):398-401.
22. Todorov A.B., Scott J.R., Warren A.E., et al. Developmental screening tests in achondroplastic children. Am J Med Genet. 1981;9(1):19-23.
23. Scott C.I.Jr. Medical and social adaptation in dwarfing conditions. Birth Defects Orig Artic Ser. 1977;13(3C):29-43.
24. Hurko O., Pyeritz R., Uematsu S. Neurological considerations in achondroplasia. Basic Life Sci. 1988;48:153-162.
25. Allanson J.E., Hall J.G. Obstetric and gynecologic problems in women with chondrodystrophies. Obstet Gynecol. 1986;67(1):74-78.
26. Hecht J.T., Francomano C.A., Horton W.A., Annegers J.F. Mortality in achondroplasia. Am J Hum Genet. 1987;41(3):454-464.
27. Camera G., Mastroiacovo P. Birth prevalence and mutation rate of achondroplasia in the Italian Multicentre Monitoring System for Birth Defects. Basic Life Sci. 1988;48:11-15.
28. Gardner R.J. A new estimate of the achondroplasia mutation rate. Clin Genet. 1977;11(1):31-38.
29. Opitz J.M. Premutation in achondroplasia. Basic Life Sci. 1988;48:17-25.
30. Murdoch J.L., Walker B.A., Hall J.C., et al. Achondroplasia: a genetic and statistical survey. Ann Hum Genet. 1970;33(3):227-244.
31. Pauli R.M., Conroy M.M., Langer L.O.Jr., et al. Homozygous achondroplasia with survival beyond infancy. Am J Med Genet. 1983;16(4):459-473.
32. Moskowitz N., Carson B., Kopits S., et al. Foramen magnum decompression in an infant with homozygous achondroplasia. Case report. J Neurosurg. 1989;70(1):126-128.
33. Bellus G.A., Hufferton T.M., Ortiz de Luna R.I., et al. Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am J Hum Genet. 1995;56(2):368-373.
34. Bellus G.A., McIntosh I., Smith E.A., et al. A recurrent mutation in the tyrosine kinase domain of fibroblast growth factor receptor 3 causes hypochondroplasia. Nat Genet. 1995;10(3):357-359.
35. Francomano C.A. The genetic basis of dwarfism. N Engl J Med. 1995;332(1):58-59.
36. McKusick V.A., Amberger J.S., Francomano C.A. Progress in medical genetics: map-based gene discovery and the molecular pathology of skeletal dysplasias. Am J Med Genet. 1996;63(1):98-105.
37. Ippolito E., et al. Histochemical and ultrastructural study of the growth plate in achondroplasia. Basic Life Sci. 1988;48:61-71.
38. Marin-Padilla M., Marin-Padilla T.M. Developmental abnormalities of the occipital bone in human chondrodystrophies (achondroplasia and thanatophoric dwarfism). Birth Defects Orig Artic Ser. 1977;13(3D):7-23.
39. Hecht J.T., Nelson F.W., Butler I.J., et al. Computerized tomography of the foramen magnum: achondroplastic values compared to normal standards. Am J Med Genet. 1985;20(2):355-360.
40. Alexander E.Jr. Significance of the small lumbar spinal canal: cauda equina compression syndromes due to spondylosis. 5. Achondroplasia. J Neurosurg. 1969;31(5):513-519.
41. Bailey J.A.2nd. Orthopaedic aspects of achondroplasia. J Bone Joint Surg Am. 1970;52(7):1285-1301.
42. Vogl A. The fate of the achondroplastic dwarf (neurologic complications of achondroplasia). Exp Med Surg. 1962;20:108-117.
43. Langer L.O.Jr., Baumann P.A., Gorlin R.J. Achondroplasia. Am J Roentgenol Radium Ther Nucl Med. 1967;100(1):12-26.
44. Schreiber F., Rosenthal H. Paraplegia from ruptured lumbar discs in achondroplastic dwarfs. J Neurosurg. 1952;9(6):648-651.
45. Thomas I.T., Frias J.L. The prospective management of cervicomedullary compression in achondroplasia. Birth Defects Orig Artic Ser. 1989;25(4):83-90.
46. Pauli R.M., Horton V.K., Glinski L.P., et al. Apnea and sudden unexpected death in infants with achondroplasia. J Pediatr. 1984;104(3):342-348.
47. Bland J.D., Emery J.L. Unexpected death of children with achondroplasia after the perinatal period. Dev Med Child Neurol. 1982;24(4):489-492.
48. Mador M.J., Tobin M.J. Apneustic breathing. A characteristic feature of brainstem compression in achondroplasia? Chest. 1990;97(4):877-883.
49. Reid C.S., Pyeritz R.E., Kopits S.E., et al. Cervicomedullary compression in young patients with achondroplasia: value of comprehensive neurologic and respiratory evaluation. J Pediatr. 1987;110(4):522-530.
50. Wang H., Rosenbaum A.E., Reid C.S., et al. Pediatric patients with achondroplasia: CT evaluation of the craniocervical junction. Radiology. 1987;164(2):515-519.
51. Blom S., Ekbom K.A. Early clinical signs of meningiomas of the foramen magnum: a new syndrome. J Neurosurg. 1962;19:661-664.
52. Aryanpur J., Hurko O., Francomano C., et al. Craniocervical decompression for cervicomedullary compression in pediatric patients with achondroplasia. J Neurosurg. 1990;73(3):375-382.
53. Danielpour M., Wilcox W.R., Alanay Y., et al. Dynamic cervicomedullary cord compression and alterations in cerebrospinal fluid dynamics in children with achondroplasia. Report of four cases. J Neurosurg. 2007;107(suppl 6):504-507.
54. Nelson F.W., Goldie W.E., Hecht J.T., et al. Short-latency somatosensory evoked potentials in the management of patients with achondroplasia. Neurology. 1984;34(8):1053-1058.
55. Carson B., Winfield J., Wang H., et al. Surgical management of cervicomedullary compression in achondroplastic patients. Basic Life Sci. 1988;48:207-214.
56. Luyendijk W., Matricali B., Thomeer R.T. Basilar impression in an achondroplastic dwarf: causative role in tetraparesis. Acta Neurochir (Wien). 1978;41(1-3):243-253.
57. Bagley C.A., Pendrik J.A., Bookland M.J., et al. Cervicomedullary decompression for foramen magnum stenosis in achondroplasia. J Neurosurg. 2006;104(suppl 3):166-172.
58. Ho N.C., Guarnieri M., Brant L.J., et al. Living with achondroplasia: quality of life evaluation following cervico-medullary decompression. Am J Med Genet A. 2004;131(2):163-167.
59. Wassman E.R.Jr., Rimoin D.L. Cervicomedullary compression with achondroplasia. J Pediatr. 1988;113(2):411.
60. Streeten E., Uematsu S., Hurko O., et al. Extended laminectomy for spinal stenosis in achondroplasia. Basic Life Sci. 1988;48:261-273.
61. Bethem D., Winter R.D., Lutter L., et al. Spinal disorders of dwarfism. Review of the literature and report of eighty cases. J Bone Joint Surg Am. 1981;63(9):1412-1425.
62. Wynne-Davies R., Walsh W.K., Gormley J. Achondroplasia and hypochondroplasia. Clinical variation and spinal stenosis. J Bone Joint Surg Br. 1981;63B(4):508-515.
63. Morgan D.F., Young R.F. Spinal neurological complications of achondroplasia. Results of surgical treatment. J Neurosurg. 1980;52(4):463-472.
64. Lonstein J.E. Treatment of kyphosis and lumbar stenosis in achondroplasia. Basic Life Sci. 1988;48:283-292.
65. O’Brien J.P., Mehdian H. Relevant principles in the management of spinal disorders in achondroplasia. Basic Life Sci. 1988;48:293-298.
66. Siebens A.A., Hungerford D.S., Kirby N.A. Curves of the achondroplastic spine: a new hypothesis. Johns Hopkins Med J. 1978;142(6):205-210.
67. Siebens A.A., Kirby N., Hungerford D. Orthotic correction of sitting abnormality in achondroplastic children. Basic Life Sci. 1988;48:313-317.
68. Kopits S.E. Thoracolumbar kyphosis and lumbosacral hyperlordosis in achondroplastic children. Basic Life Sci. 1988;48:241-255.
69. Lutter L.D., Longstein J.E., Winter R.B., et al. Anatomy of the achondroplastic lumbar canal. Clin Orthop Relat Res, 1977;126:139-142
70. Uematsu S., Wang H., Hurko O., et al. The subarachnoid fluid space in achondroplastic spinal stenosis: the surgical implication. Basic Life Sci. 1988;48:275-281.
71. Shikata J., Yamamuro T., Iida H., et al. Surgical treatment of achondroplastic dwarfs with paraplegia. Surg Neurol. 1988;29(2):125-130.
72. Ain M.C., Shirley E.D., Pirouzmanesh A., et al. Postlaminectomy kyphosis in the skeletally immature achondroplast. Spine (Phila Pa 1976). 2006;31(2):197-201.
73. Sciubba D.M., Noggle J.C., Marupudi N.I., et al. Spinal stenosis surgery in pediatric patients with achondroplasia. J Neurosurg. 2007;106(suppl 5):372-378.
74. Ain M.C., Elmaci I., Hurko O., et al. Reoperation for spinal restenosis in achondroplasia. J Spinal Disord. 2000;13(2):168-173.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
Chapter 184 Neurologic Problems of the Spine in Achondroplasia
Achondroplasia is characterized by disproportionately short stature with rhizomelic shortening of the extremities, macrocephaly, midface hypoplasia, and frontal bossing.1,2 This skeletal dysplasia results from defective formation of endochondral bone.3–5 Morbidity in achondroplasia results largely from bony compression of the neuraxis6–12 and respiratory failure.13,14 This chapter focuses on the main indications for neurosurgical interventions for problems attributed to the spine in achondroplasia, namely, cervicomedullary compression and spinal stenosis.
Thoracolumbar stenosis resulting in spinal compression is the commonest complication of achondroplasia, becoming symptomatic in most patients in their 20s or later.15 Thoracolumbar stenosis can be accelerated in infants who develop progressive thoracolumbar kyphosis if bracing is not undertaken before vertebral wedging develops.16 Less common problems in infancy include symptomatic airway obstruction17 and severe cervicomedullary compression secondary to foramen magnum stenosis.18–20 The latter can be sometimes accompanied by swallowing difficulties and central apnea.21
Most individuals with achondroplasia have normal intelligence. Motor milestones are delayed,22 partly because of generalized hypotonia and partly because of the mechanical disadvantage imposed by short limbs. Psychological problems arising from short stature include lack of acceptance by peers and a tendency of adults, including parents and teachers, to treat children with achondroplasia appropriately for their height rather than their age.23 Involvement with other families with children of short stature can improve self-esteem and can assist parents in guiding their achondroplastic children through the difficulties of growing up in a culture that equates stature to status.
Respiratory complications include obstructive sleep apnea secondary to a small upper airway.14 Many people with achondroplasia snore. Many infants sleep with their necks in a hyperextended position, a position that functionally increases the size of the upper airway, relieving intermittent obstruction. However, the hyperextended neck position may exacerbate neurologic sequelae of a small foramen magnum and cervicomedullary compression.24 A small thoracic cage may result in restrictive pulmonary disease in infancy, and respiration can be compromised further by aspiration secondary to gastroesophageal reflux, swallowing dysfunction, or both, resulting in recurrent pneumonia.
Reproductive difficulties have not been conclusively documented,25 but evidence of reduced fertility, frequent fibroid cysts, and early menopause have been reported. Women with achondroplasia must deliver their infants by cesarean section because of cephalopelvic disproportion, and administration of spinal anesthesia is strongly discouraged due to the small size of the canal.
Although life expectancy was formerly thought to be normal for people with achondroplasia, age-specific mortality is increased at all ages, with the highest increase occurring in children.26 Moreover, cardiovascular cases of death are increased in the adult group (25-54 years of age). The increased mortality in childhood is likely related to severe cervicomedullary compression.
Causes and Pathophysiology
Clinical Genetics and Growth Plate Ultrastructures
Achondroplasia is an autosomal dominant disorder; most estimates of its frequency cluster between 1:25,000 and 1:35,000 live births27,28; however, the true frequency may be slightly higher.29 New mutations account for about 80% of children born with achondroplasia.28 As in many autosomal dominant disorders, a positive correlation exists between advanced paternal age and occurrence of new mutations.30 Offspring of couples in which both partners are affected by achondroplasia have a 25% chance of inheriting both parental achondroplasia alleles, resulting in homozygous achondroplasia, which is universally fatal within the first year of life.31 The skeletal features of achondroplasia are highly exaggerated in the homozygous condition, resulting in significantly shorter limbs, a smaller chest size, and a smaller foramen magnum. Death is usually secondary to respiratory complications, sometimes in concordance with foramen magnum stenosis and brain stem compression.32
Achondroplasia results from impaired formation of endochondral bone. A missense mutation, G380R, in the transmembrane domain of fibroblast growth factor receptor 3 has been traced to chromosome 4, at 4p16.3.33,34 The protein is a tyrosine kinase receptor expressed in developing bones. The G380R mutation has been found in most patients.35,36 Several groups used this discovery to develop polymerase chain reaction diagnostic tests. The histochemical features of the endochondral growth plates of achondroplastic bone have been interpreted in several ways. Some researchers suggested that mitotic abnormalities indicate cessation of normal cell function and arrest of cell division of the chondrocytes.37 This impaired formation of bone from cartilage is seen in the growth of the diaphyses of long bones.3 In addition, an enlargement of the epiphyses occurs. Cartilaginous synchondroses in the spine and skull seem to fuse prematurely, and hypertrophy of the spinal articular surface occurs. Cervicomedullary compression is typically a pediatric concern, while spinal stenosis is usually seen in adults.
Cervicomedullary Compression
Cervicomedullary compression stems primarily from a reduction in the diameter of the foramen magnum in the sagittal and coronal dimensions that is sometimes more than five standard deviations less than normal.38 The cranial base (chondrocranium) derives from endochondral ossification. In achondroplasia, the base is stunted, shorter, and narrower that normal.39 The basioccipital bone, which forms the anterior border of the foramen magnum, is narrow and angulated. The lateral and posterior parts, consisting of the exoccipitalis bone, are similarly deformed, resulting in the diamond, triangular, or teardrop shape of the achondroplastic foramen magnum. In addition, the articular surfaces of the occipital bone (between the lateral occipital and the basioccipital bones and between the lateral bones and the planum nuchale of the squama) are hypertrophic and can encroach on the neural elements within the foramen. The pathology of the achondroplastic skull is further complicated by the small size of the posterior fossa, resulting from stunting of the endochondrally derived planum nuchale, and the resultant horizontalization of the squamous portions of the occipital bones. This constricted arrangement of the skull base displaces the brain stem upward and the foramen magnum anteriorly, resulting in posterior tilting to the brain stem and further impingement of the neuraxis posteriorly24 (Fig. 184-1).
Spinal Stenosis
The anatomy of the achondroplastic spine is distinctive in several respects, all of which can contribute to compromise of the spinal cord or nerve roots.40,41 The hypertrophy of epiphyseal articular processes in the long bones is mirrored at the caudal and cephalic surfaces of the vertebral bodies, resulting in a mushroom shape at each end and concomitant scalloping along the posterior surface that is appreciable in a contrast myelogram.42 Abbreviated and thickened pedicles of the vertebral arches result from premature fusion of synchondroses between the laminae and the vertebral bodies3; the laminae are also thickened. Intervertebral discs tend to bulge prominently,42 further aggravating neural encroachment by the enlarged vertebral body articular surfaces. The interpediculate distance decreases in the lumbar region of the spine, resulting in a canal that tapers caudally,43 the opposite of normal (the canal normally widens caudally). The overall picture is one of dramatic stenosis in every dimension of the spine, a stenosis sometimes aggravated by osteoarthritic changes and disc ruptures.44 Consequently, a generalized constriction of spinal neural elements occurs (Fig. 184-2).
Evaluation and Diagnosis
Referral Criteria
Primary-care physicians have recognized the need for comprehensive prospective management of achondroplastic patients for sleep, respiratory, and central nervous system disorders.14,17,45 To aid the efforts, we present our recommendations for evaluation of achondroplastic patients for cervicomedullary compression and spinal stenosis. Achondroplastic patients referred to our institution are usually evaluated according to a standardized protocol that involves a multidisciplinary team of neurosurgeons, neurologists, pulmonary and sleep specialists, geneticists, anesthesiologists, neuroradiologists, orthopedic surgeons, and otolaryngologists. Because these patients are at risk for brain stem compression, comprehensive testing is directed toward detection of central and obstructive apnea and cervicomedullary compression, all of which contribute to the risk of sudden death.
Cervicomedullary Compression
Clinical Pathology and Presentation
Cervicomedullary compression warrants early and aggressive treatment. Results of studies suggest such compression is progressive and potentially fatal because it increases the risk of sudden death by central respiratory failure.46,47 This condition has gained increasing attention as a cause of respiratory and neurologic impairment in children with achondroplasia.48–50 In our prospective evaluation of achondroplastic infants, we found radiographic evidence of craniocervical stenosis in 58% of the studied patients, and a diagnosis of cervicomedullary compression was made in 35%.49 These figures are for a selected population and are certainly higher that the proportion in the general population. Nonetheless, they are a strong argument for the careful evaluation and treatment of achondroplastic children. A retrospective study found excess mortality in sudden death resulting from brain stem compression, which was identified as the cause of half of the excess deaths.26 The same study also found a 7.5% risk of sudden death in the first year of life.
Chronic medullary and upper cervical cord compression may exist as a neurologically asymptomatic lesion, exhibiting neither signs of root compression in the arms nor symptoms of cranial nerve impairment.51 Nonetheless, microcystic histopathologic changes, cervical syringomyelia, and necrosis and gliosis have been reported in autopsies of achondroplastic children who died unexpectedly.19,20,47 Presumably, lesions of this type interrupt neural respiratory pathways from the nucleus tractus solitarius to the phrenic nerve nucleus—arresting the muscles of respiration and resulting in sudden death in some cases. We consider infants with a history of sleep apnea or other severe respiratory or neurologic abnormalities to be at increased risk for respiratory complications resulting from occult cervicomedullary compression.49 Some authors have recommended performing sleep and imaging studies on all children with achondroplasia.45 A composite profile of the patient with cervicomedullary compression includes upper or lower extremity paresis, apnea and cyanosis, hyperreflexia or hypertonia, and delay in motor milestones beyond achondroplastic standards. These patients can present a striking contrast to the usual floppy, hypotonic achondroplastic infants.52 More recently, a study indicated that although normal imaging studies may be found in achondroplastic children on magnetic resonance imaging (MRI) in a neutral neck position, flexion can lead to increased intracranial pressure (ICP) due to venous outflow obstruction and complete cerebrospinal fluid (CSF) outflow block.53 The imaging findings have to be correlated with the clinical status of the patient, and the decision for treatment is ultimately guided by the patient’s neurologic status and the surgeon’s experience.
Evaluation
Once the high-risk patient with respiratory or neurologic symptoms or signs has been identified, we advise comprehensive testing. Parents should be carefully interviewed about the health and medical history of the child, with emphasis on respiratory symptoms, and a general physical examination, including chest measurements, should be performed. Respiratory evaluation should include the evaluation of blood pH, a chest radiograph, and overnight polysomnography. Electrocardiograms and echocardiograms should be performed to see whether there is cardiac evidence of chronic oxygen deprivation during sleep. A neurologic examination for signs of brain stem compression, such as hyperreflexia, hypertonia, paresis, asymmetry of movement or strength, or abnormal plantar response, is essential. Brain stem auditory evoked potential and upper extremity somatosensory evoked potential evaluation should be considered as an adjunct, especially in patients with normal results on neurologic examination.54 Imaging studies of the craniocervical junction are necessary, particularly MRI in the sagittal plane. We also strongly recommend MRI flow studies of the CSF at the foramen magnum, using the technique of synchronizing the succession of images with the heartbeat. Some of these procedures have been described elsewhere.49
Indication for Surgery
The underlying principle for surgery must be to identify patients who are at risk for neurologic damage or sudden death. We recommend that patients with cervicomedullary compression be identified and treated prophylactically, before abrupt and irreversible changes occur. For the purpose of diagnosis, we define clinically significant cervicomedullary compression to be (1) neurologic evidence of upper cervical myelopathy; (2) evidence of stenosis on imaging studies, including the absence of flow above and below the foramen magnum; and (3) an otherwise unexplained respiratory or developmental abnormality. It is possible to see a patient with brain stem compression and obstructive apnea who nonetheless meets these criteria. Having discovered these indications, the treatment team should also ask whether the patient’s status is stable or deteriorating before undertaking operative decompression and should evaluate the probability for catastrophic deterioration if decompression is not performed (Fig. 184-3).
Operative Management
Craniocervical surgical decompression for cervicomedullary compression in children with achondroplasia55 has been used at several centers with generally good results.12,21,56 Decompression of the cervicomedullary junction has shown to bring about dramatic, sustained improvement in neurologic and respiratory compromise.49,52 The procedure has not received as wide acceptance as it might, however, because its successful performance relies on careful management of the anatomic difficulties presented by achondroplastic patients. Clinical evaluation is frequently difficult for many reasons, some of which are unrelated to neurologic compromise. Long-term follow-up data that would allow a definitive assessment of craniocervical decompression have also been lacking. As with any surgical procedure, detailed prior consultation must be conducted with the parents to inform them of the potential risks and expected benefits for their achondroplastic child.
For decompression, a midline suboccipital incision is made, and the ligaments and musculature are dissected subperiosteally to expose the occiput and the spinous process and laminae of C1 and C2. The arch of C1 is then removed with a high-speed drill and small curettes. The surgeon frequently sees a thick, fibrous band or pannus about the level of C1 that should be left in place during the initial bone drilling to create a protective layer for the underlying dura and spinal cord. Removal of the arch of C2 is sometimes necessary; more caudal areas of compression necessitate even further caudad decompression. The posterior rim of the foramen magnum is thinned gradually with a high-speed drill and removed with small, straight, and angled curettes. Invariably, the bone of the foramen magnum is thickened and oriented more horizontally than usual, severely indenting the underlying dura. The most delicate part of the dissection occurs as the drill approaches the posterior rim of the foramen magnum. Once bone decompression is complete, the fibrous pannus or band is removed as well, often revealing the transverse dural channel that offers dramatic evidence of the extent of the dural restriction; consequently, adequate attention must be paid to the soft tissue aspects of the decompression. We used to perform duraplasty routinely with the placement of a dural patch. However, more recently, this step of the procedure has been only required if there is any persistent dural constriction after the fibrous band has been removed. After a duraplasty is performed, adequate cord pulsation and CSF flow are confirmed. A dural patch can be performed using pericranium, paraspinal fascia, or cadaveric dura. A watertight seal is essential. We do not recommend the placement of a wound drain so as not to potentiate the development of a CSF fistula; however, if the dura is opened, it is wise to place a ventriculostomy, because many patients with achondroplasia have increased ICP and will experience CSF leak and possible pseudomeningocele if the duraplasty is challenged early.
[/not-level-membership-for-neurosurgery-category]
