PYROGENS
The term pyrogen (Greek pyro, “fire”) is used to describe any substance that causes fever. Exogenous pyrogens are derived from outside the patient; most are microbial products, microbial toxins, or whole microorganisms (including viruses). The classic example of an exogenous pyrogen is the lipopolysaccharide (endotoxin) produced by all gram-negative bacteria. Pyrogenic products of gram-positive organisms include the enterotoxins of Staphylococcus aureus and the groups A and B streptococcal toxins, also called superantigens. One staphylococcal toxin of clinical importance is that associated with isolates of S. aureus from patients with toxic shock syndrome. These products of staphylococci and streptococci cause fever in experimental animals when injected intravenously at concentrations of 1–10 μg/kg. Endotoxin is a highly pyrogenic molecule in humans: when injected intravenously into volunteers, a dose of 2–3 ng/kg produces fever, leukocytosis, acute-phase proteins, and generalized symptoms of malaise.
PYROGENIC CYTOKINES
Cytokines are small proteins (molecular mass, 10,000–20,000 Da) that regulate immune, inflammatory, and hematopoietic processes. For example, the elevated leukocytosis seen in several infections with an absolute neutrophilia is attributable to the cytokines interleukin (IL) 1 and IL-6. Some cytokines also cause fever; formerly referred to as endogenous pyrogens, they are now called pyrogenic cytokines. The pyrogenic cytokines include IL-1, IL-6, tumor necrosis factor (TNF), and ciliary neurotropic factor, a member of the IL-6 family. Interferons (IFNs), particularly IFN-α, also are pyrogenic cytokines; fever is a prominent side effect of IFN-α used in the treatment of hepatitis. Each pyrogenic cytokine is encoded by a separate gene, and each has been shown to cause fever in laboratory animals and in humans. When injected into humans at low doses (10–100 ng/kg), IL-1 and TNF produce fever; in contrast, for IL-6, a dose of 1–10 μg/kg is required for fever production.
A wide spectrum of bacterial and fungal products induce the synthesis and release of pyrogenic cytokines. However, fever can be a manifestation of disease in the absence of microbial infection. For example, inflammatory processes, trauma, tissue necrosis, and antigen-antibody complexes induce the production of IL-1, TNF, and/or IL-6; individually or in combination, these cytokines trigger the hypothalamus to raise the set point to febrile levels.
ELEVATION OF THE HYPOTHALAMIC SET POINT BY CYTOKINES
During fever, levels of prostaglandin E2 (PGE2) are elevated in hypothalamic tissue and the third cerebral ventricle. The concentrations of PGE2 are highest near the circumventricular vascular organs (organum vasculosum of lamina terminalis)—networks of enlarged capillaries surrounding the hypothalamic regulatory centers. Destruction of these organs reduces the ability of pyrogens to produce fever. Most studies in animals have failed to show, however, that pyrogenic cytokines pass from the circulation into the brain itself. Thus, it appears that both exogenous pyrogens and pyrogenic cytokines interact with the endothelium of these capillaries and that this interaction is the first step in initiating fever—i.e., in raising the set point to febrile levels.
The key events in the production of fever are illustrated in Fig. 23-1. Myeloid and endothelial cells are the primary cell types that produce pyrogenic cytokines. Pyrogenic cytokines such as IL-1, IL-6, and TNF are released from these cells and enter the systemic circulation. Although these circulating cytokines lead to fever by inducing the synthesis of PGE2, they also induce PGE2 in peripheral tissues. The increase in PGE2 in the periphery accounts for the nonspecific myalgias and arthralgias that often accompany fever. It is thought that some systemic PGE2 escapes destruction by the lung and gains access to the hypothalamus via the internal carotid. However, it is the elevation of PGE2 in the brain that starts the process of raising the hypothalamic set point for core temperature.
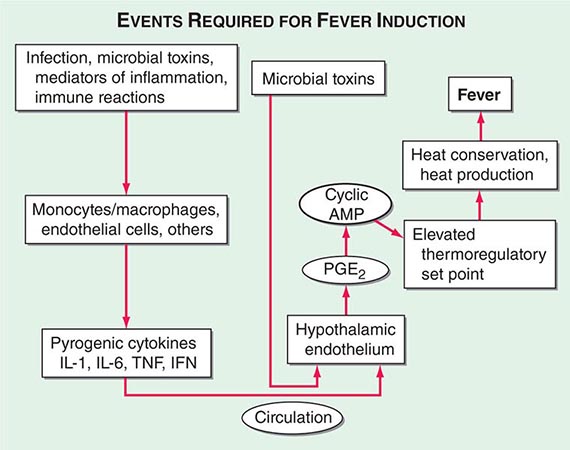
FIGURE 23-1 Chronology of events required for the induction of fever. AMP, adenosine 5′-monophosphate; IFN, interferon; IL, interleukin; PGE2, prostaglandin E2; TNF, tumor necrosis factor.
There are four receptors for PGE2, and each signals the cell in different ways. Of the four receptors, the third (EP-3) is essential for fever: when the gene for this receptor is deleted in mice, no fever follows the injection of IL-1 or endotoxin. Deletion of the other PGE2 receptor genes leaves the fever mechanism intact. Although PGE2 is essential for fever, it is not a neurotransmitter. Rather, the release of PGE2 from the brain side of the hypothalamic endothelium triggers the PGE2 receptor on glial cells, and this stimulation results in the rapid release of cyclic adenosine 5′-monophosphate (cAMP), which is a neurotransmitter. As shown in Fig. 23-1, the release of cAMP from glial cells activates neuronal endings from the thermoregulatory center that extend into the area. The elevation of cAMP is thought to account for changes in the hypothalamic set point either directly or indirectly (by inducing the release of neurotransmitters). Distinct receptors for microbial products are located on the hypothalamic endothelium. These receptors are called Toll-like receptors and are similar in many ways to IL-1 receptors. IL-1 receptors and Toll-like receptors share the same signal-transducing mechanism. Thus, the direct activation of Toll-like receptors or IL-1 receptors results in PGE2 production and fever.
PRODUCTION OF CYTOKINES IN THE CNS
Cytokines produced in the brain may account for the hyperpyrexia of CNS hemorrhage, trauma, or infection. Viral infections of the CNS induce microglial and possibly neuronal production of IL-1, TNF, and IL-6. In experimental animals, the concentration of a cytokine required to cause fever is several orders of magnitude lower with direct injection into the brain substance or brain ventricles than with systemic injection. Therefore, cytokines produced in the CNS can raise the hypothalamic set point, bypassing the circumventricular organs. CNS cytokines likely account for the hyperpyrexia of CNS hemorrhage, trauma, or infection.
24 |
Fever and Rash |
The acutely ill patient with fever and rash may present a diagnostic challenge for physicians. However, the distinctive appearance of an eruption in concert with a clinical syndrome can facilitate a prompt diagnosis and the institution of life-saving therapy or critical infection-control interventions. Representative images of many of the rashes discussed in this chapter are included in Chap. 25e.
For further discussion, see Chaps. 70, 72, and 147.
CLASSIFICATION OF RASH
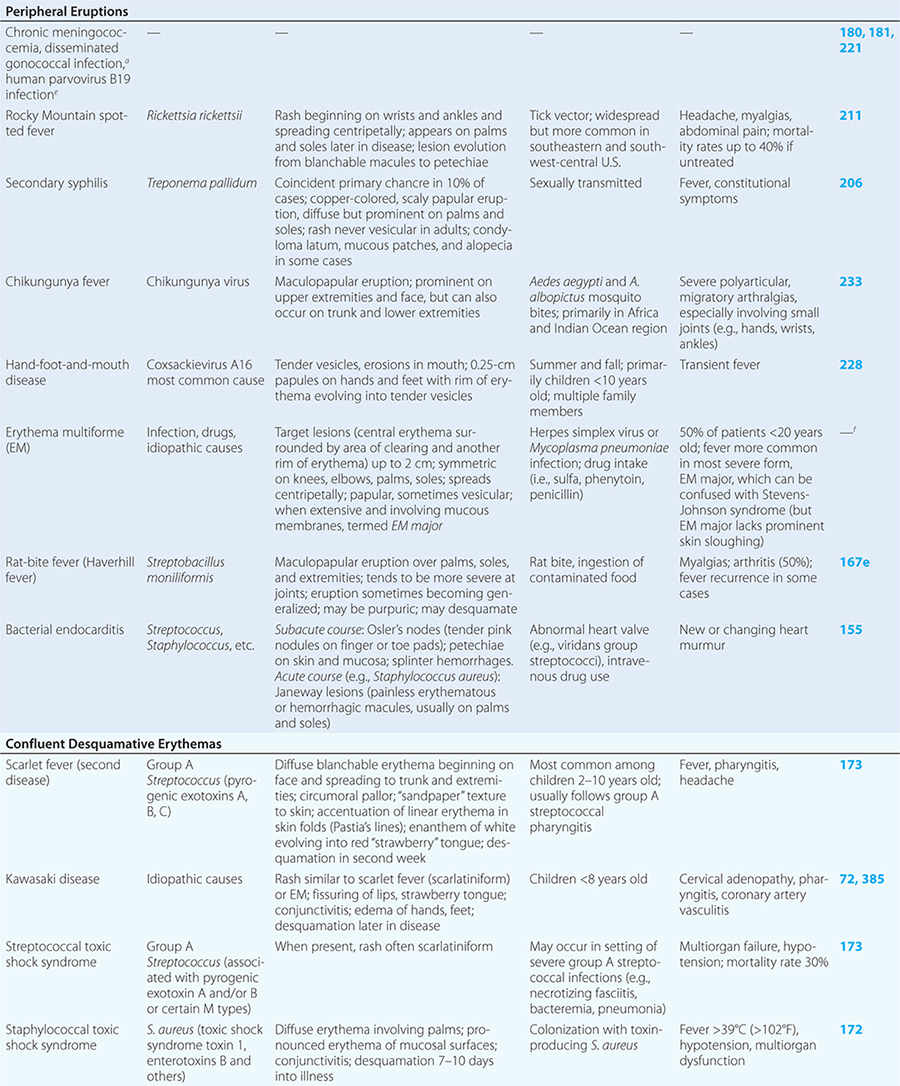
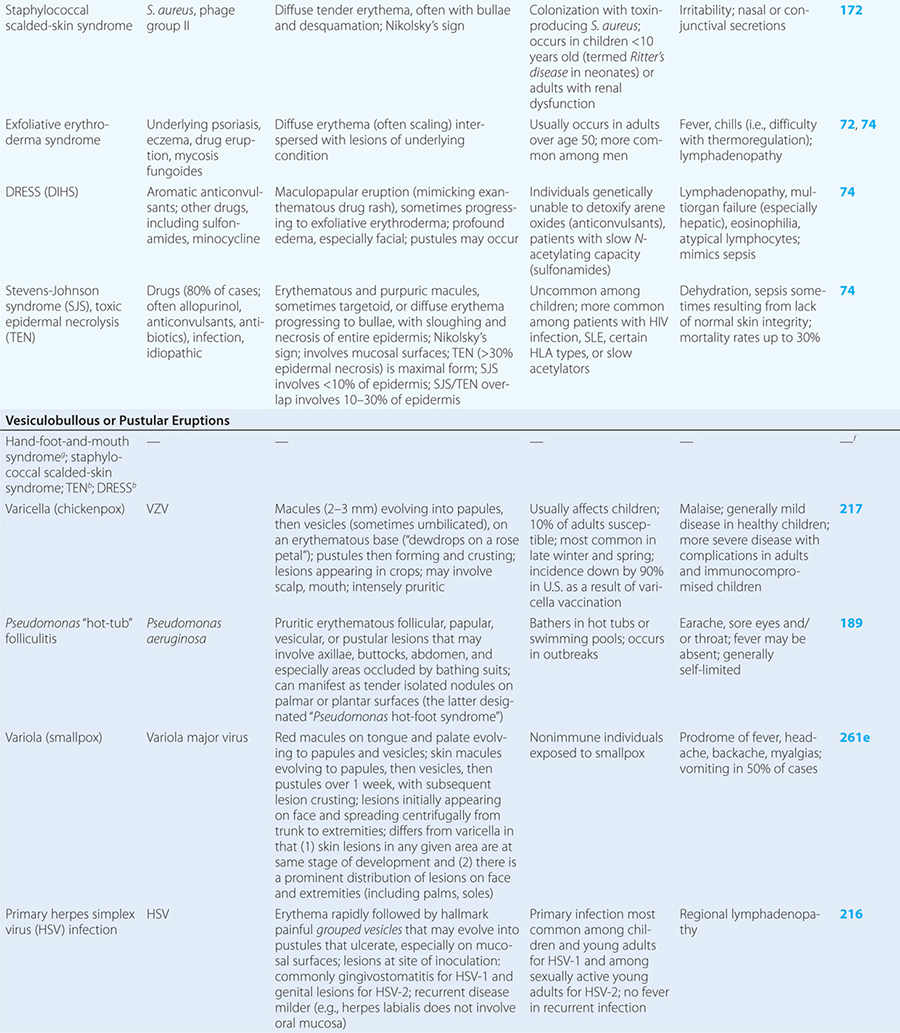
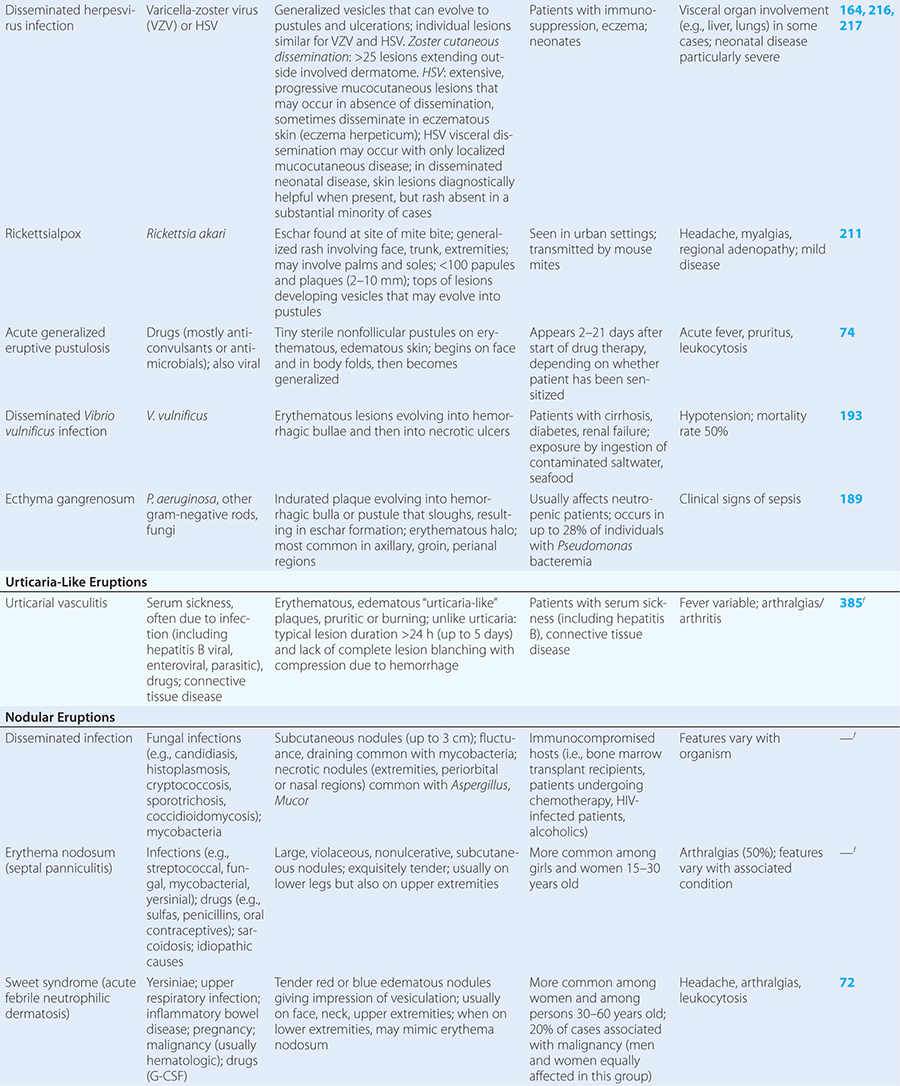
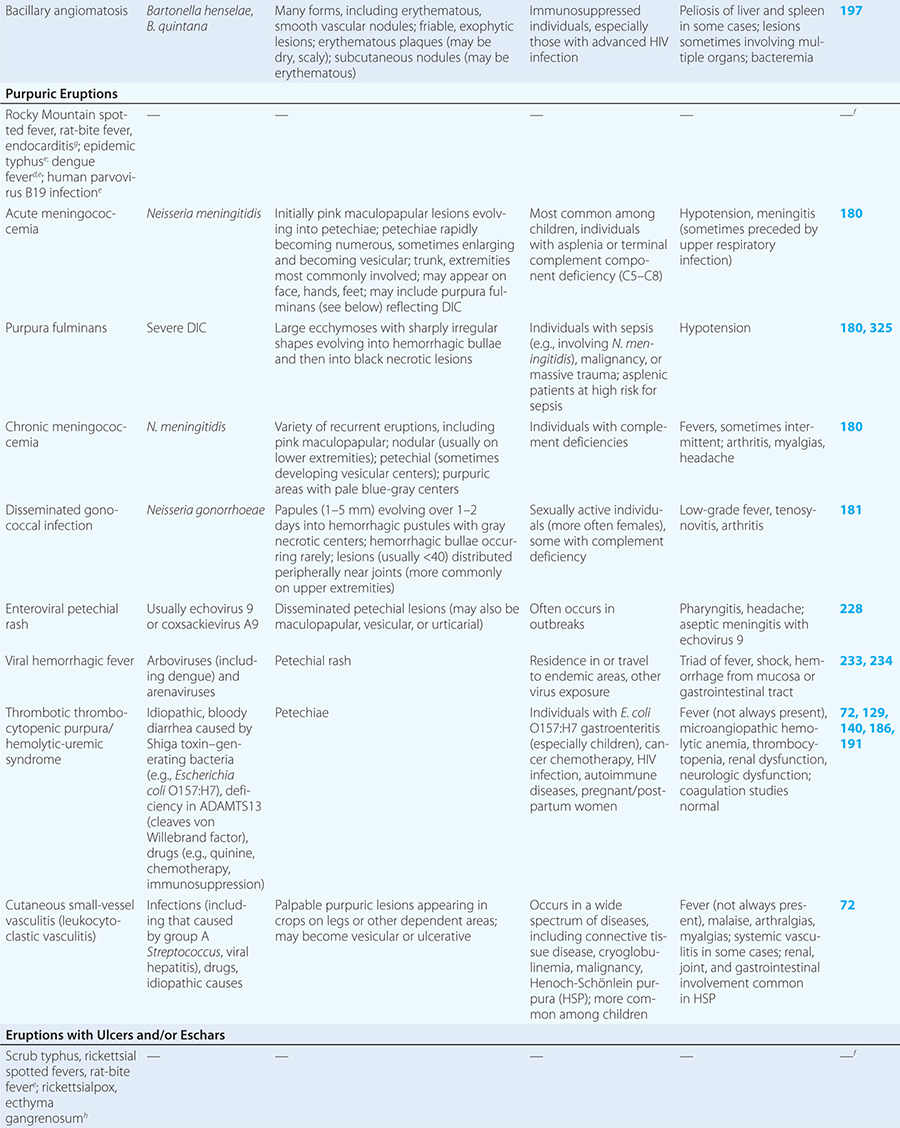
This chapter reviews rashes that reflect systemic disease, but it does not include localized skin eruptions (i.e., cellulitis, impetigo) that may also be associated with fever (Chap. 156). The chapter is not intended to be all-inclusive, but it covers the most important and most common diseases associated with fever and rash. Rashes are classified herein on the basis of lesion morphology and distribution. For practical purposes, this classification system is based on the most typical disease presentations. However, morphology may vary as rashes evolve, and the presentation of diseases with rashes is subject to many variations (Chap. 72). For instance, the classic petechial rash of Rocky Mountain spotted fever (Chap. 211) may initially consist of blanchable erythematous macules distributed peripherally; at times, however, the rash associated with this disease may not be predominantly acral, or no rash may develop at all.
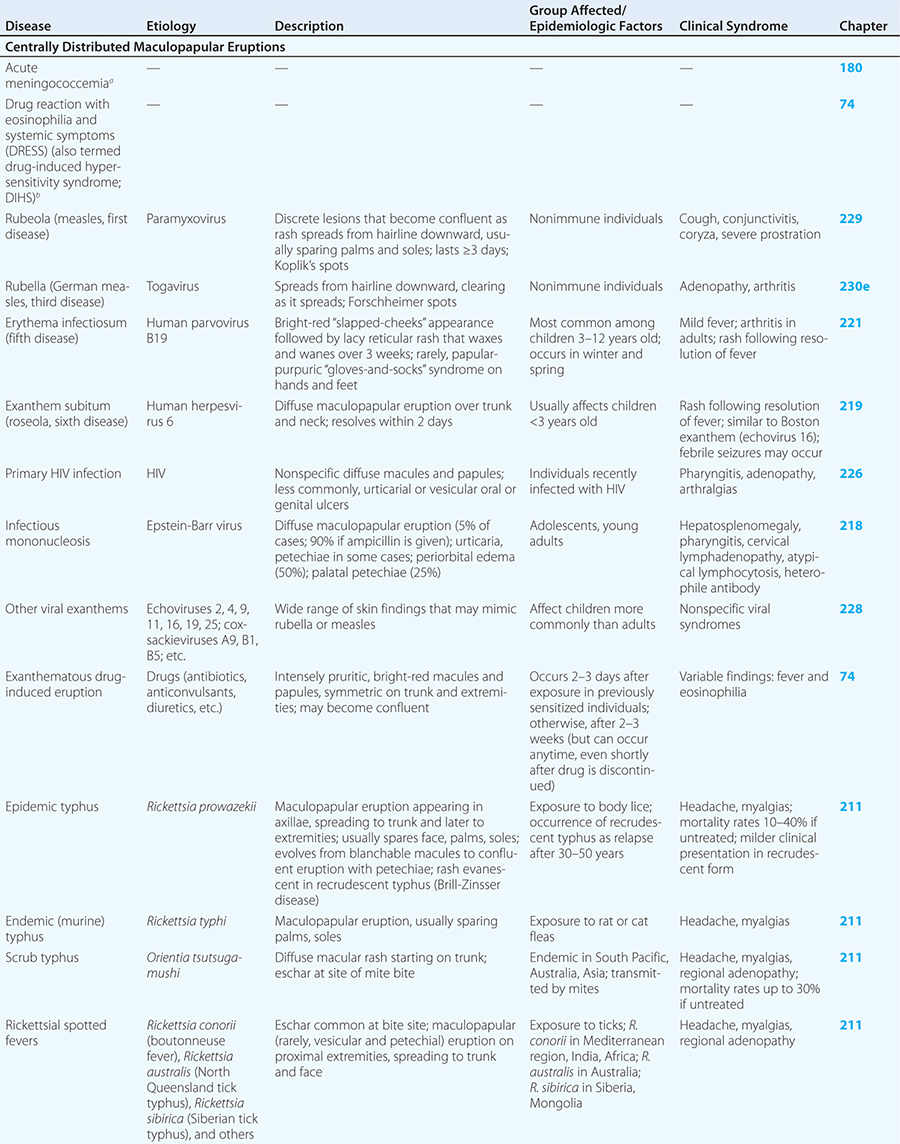
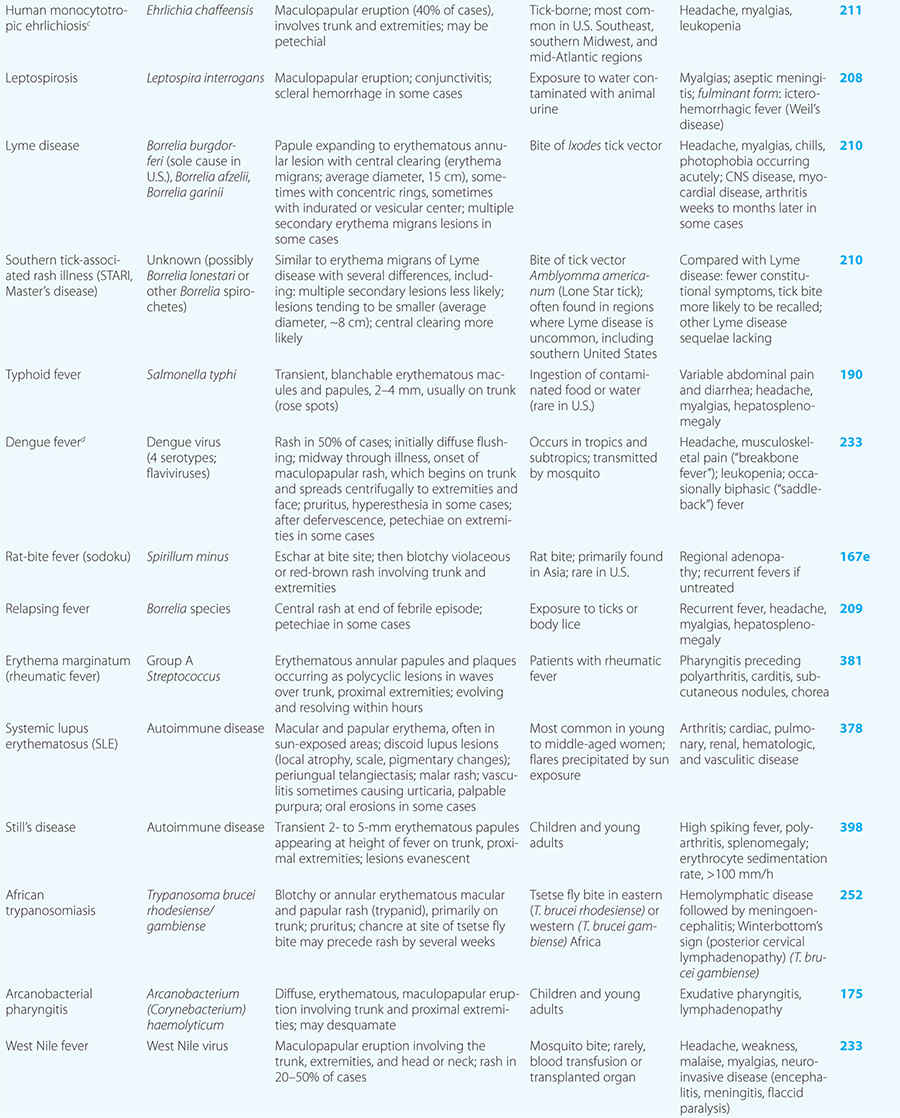
Diseases with fever and rash may be classified by type of eruption: centrally distributed maculopapular, peripheral, confluent desquamative erythematous, vesiculobullous, urticaria-like, nodular, purpuric, ulcerated, or with eschars. Diseases are listed by these categories in Table 24-1, and many are highlighted in the text. However, for a more detailed discussion of each disease associated with a rash, the reader is referred to the chapter dealing with that specific disease. (Reference chapters are cited in the text and listed in Table 24-1.)
|
DISEASES ASSOCIATED WITH FEVER AND RASH |

CENTRALLY DISTRIBUTED MACULOPAPULAR ERUPTIONS
Centrally distributed rashes, in which lesions are primarily truncal, are the most common type of eruption. The rash of rubeola (measles) starts at the hairline 2–3 days into the illness and moves down the body, typically sparing the palms and soles (Chap. 229). It begins as discrete erythematous lesions, which become confluent as the rash spreads. Koplik’s spots (1- to 2-mm white or bluish lesions with an erythematous halo on the buccal mucosa) are pathognomonic for measles and are generally seen during the first 2 days of symptoms. They should not be confused with Fordyce’s spots (ectopic sebaceous glands), which have no erythematous halos and are found in the mouth of healthy individuals. Koplik’s spots may briefly overlap with the measles exanthem.
Rubella (German measles) also spreads from the hairline downward; unlike that of measles, however, the rash of rubella tends to clear from originally affected areas as it migrates, and it may be pruritic (Chap. 230e). Forchheimer spots (palatal petechiae) may develop but are nonspecific because they also develop in infectious mononucleosis (Chap. 218) and scarlet fever (Chap. 173). Postauricular and suboccipital adenopathy and arthritis are common among adults with rubella. Exposure of pregnant women to ill individuals should be avoided, as rubella causes severe congenital abnormalities. Numerous strains of enteroviruses (Chap. 228), primarily echoviruses and coxsackieviruses, cause nonspecific syndromes of fever and eruptions that may mimic rubella or measles. Patients with infectious mononucleosis caused by Epstein-Barr virus (Chap. 218) or with primary HIV infection (Chap. 226) may exhibit pharyngitis, lymphadenopathy, and a nonspecific maculopapular exanthem.
The rash of erythema infectiosum (fifth disease), which is caused by human parvovirus B19, primarily affects children 3–12 years old; it develops after fever has resolved as a bright blanchable erythema on the cheeks (“slapped cheeks”) with perioral pallor (Chap. 221). A more diffuse rash (often pruritic) appears the next day on the trunk and extremities and then rapidly develops into a lacy reticular eruption that may wax and wane (especially with temperature change) over 3 weeks. Adults with fifth disease often have arthritis, and fetal hydrops can develop in association with this condition in pregnant women.
Exanthem subitum (roseola) is caused by human herpesvirus 6 and is most common among children <3 years of age (Chap. 219). As in erythema infectiosum, the rash usually appears after fever has subsided. It consists of 2- to 3-mm rose-pink macules and papules that coalesce only rarely, occur initially on the trunk and sometimes on the extremities (sparing the face), and fade within 2 days.
Although drug reactions have many manifestations, including urticaria, exanthematous drug-induced eruptions (Chap. 74) are most common and are often difficult to distinguish from viral exanthems. Eruptions elicited by drugs are usually more intensely erythematous and pruritic than viral exanthems, but this distinction is not reliable. A history of new medications and an absence of prostration may help to distinguish a drug-related rash from an eruption of another etiology. Rashes may persist for up to two weeks after administration of the offending agent is discontinued. Certain populations are more prone than others to drug rashes. Of HIV-infected patients, 50–60% develop a rash in response to sulfa drugs; 90% of patients with mononucleosis due to Epstein-Barr virus develop a rash when given ampicillin.
Rickettsial illnesses (Chap. 211) should be considered in the evaluation of individuals with centrally distributed maculopapular eruptions. The usual setting for epidemic typhus is a site of war or natural disaster in which people are exposed to body lice. Endemic typhus or leptospirosis (the latter caused by a spirochete) (Chap. 208) may be seen in urban environments where rodents proliferate. Outside the United States, other rickettsial diseases cause a spotted-fever syndrome and should be considered in residents of or travelers to endemic areas. Similarly, typhoid fever, a nonrickettsial disease caused by Salmonella typhi (Chap. 190), is usually acquired during travel outside the United States. Dengue fever, caused by a mosquito-transmitted flavivirus, occurs in tropical and subtropical regions of the world (Chap. 233).
Some centrally distributed maculopapular eruptions have distinctive features. Erythema migrans, the rash of Lyme disease (Chap. 210), typically manifests as single or multiple annular plaques. Untreated erythema migrans lesions usually fade within a month but may persist for more than a year. Southern tick-associated rash illness (STARI) (Chap. 210) has an erythema migrans–like rash but is less severe than Lyme disease and often occurs in regions where Lyme is not endemic. Erythema marginatum, the rash of acute rheumatic fever (Chap. 381), has a distinctive pattern of enlarging and shifting transient annular lesions.
Collagen vascular diseases may cause fever and rash. Patients with systemic lupus erythematosus (Chap. 378) typically develop a sharply defined, erythematous eruption in a butterfly distribution on the cheeks (malar rash) as well as many other skin manifestations. Still’s disease (Chap. 398) presents as an evanescent, salmon-colored rash on the trunk and proximal extremities that coincides with fever spikes.
PERIPHERAL ERUPTIONS
These rashes are alike in that they are most prominent peripherally or begin in peripheral (acral) areas before spreading centripetally. Early diagnosis and therapy are critical in Rocky Mountain spotted fever (Chap. 211) because of its grave prognosis if untreated. Lesions evolve from macular to petechial, start on the wrists and ankles, spread centripetally, and appear on the palms and soles only later in the disease. The rash of secondary syphilis (Chap. 206), which may be generalized but is prominent on the palms and soles, should be considered in the differential diagnosis of pityriasis rosea, especially in sexually active patients. Chikungunya fever (Chap. 233), which is transmitted by mosquito bite in Africa and the Indian Ocean region, is associated with a maculopapular eruption and severe polyarticular small-joint arthralgias. Hand-foot-and-mouth disease (Chap. 228), most commonly caused by coxsackievirus A16, is distinguished by tender vesicles distributed peripherally and in the mouth; outbreaks commonly occur within families. The classic target lesions of erythema multiforme appear symmetrically on the elbows, knees, palms, soles, and face. In severe cases, these lesions spread diffusely and involve mucosal surfaces. Lesions may develop on the hands and feet in endocarditis (Chap. 155).
CONFLUENT DESQUAMATIVE ERYTHEMAS
These eruptions consist of diffuse erythema frequently followed by desquamation. The eruptions caused by group A Streptococcus or Staphylococcus aureus are toxin-mediated. Scarlet fever (Chap. 173) usually follows pharyngitis; patients have a facial flush, a “strawberry” tongue, and accentuated petechiae in body folds (Pastia’s lines). Kawasaki disease (Chaps. 72 and 385) presents in the pediatric population as fissuring of the lips, a strawberry tongue, conjunctivitis, adenopathy, and sometimes cardiac abnormalities. Streptococcal toxic shock syndrome (Chap. 173) manifests with hypotension, multiorgan failure, and, often, a severe group A streptococcal infection (e.g., necrotizing fasciitis). Staphylococcal toxic shock syndrome (Chap. 172) also presents with hypotension and multiorgan failure, but usually only S. aureus colonization—not a severe S. aureus infection—is documented. Staphylococcal scalded-skin syndrome (Chap. 172) is seen primarily in children and in immunocompromised adults. Generalized erythema is often evident during the prodrome of fever and malaise; profound tenderness of the skin is distinctive. In the exfoliative stage, the skin can be induced to form bullae with light lateral pressure (Nikolsky’s sign). In a mild form, a scarlatiniform eruption mimics scarlet fever, but the patient does not exhibit a strawberry tongue or circumoral pallor. In contrast to the staphylococcal scalded-skin syndrome, in which the cleavage plane is superficial in the epidermis, toxic epidermal necrolysis (Chap. 74), a maximal variant of Stevens-Johnson syndrome, involves sloughing of the entire epidermis, resulting in severe disease. Exfoliative erythroderma syndrome (Chaps. 72 and 74) is a serious reaction associated with systemic toxicity that is often due to eczema, psoriasis, a drug reaction, or mycosis fungoides. Drug rash with eosinophilia and systemic symptoms (DRESS), often due to antiepileptic and antibiotic agents (Chap. 74), initially appears similar to an exanthematous drug reaction but may progress to exfoliative erythroderma; it is accompanied by multi-organ failure and has an associated mortality rate of ~10%.
VESICULOBULLOUS OR PUSTULAR ERUPTIONS
Varicella (Chap. 217) is highly contagious, often occurring in winter or spring. At any point in time, within a given region of the body, varicella lesions are in different stages of development. In immunocompromised hosts, varicella vesicles may lack the characteristic erythematous base or may appear hemorrhagic. Lesions of Pseudomonas “hot-tub” folliculitis (Chap. 189) are also pruritic and may appear similar to those of varicella. However, hot-tub folliculitis generally occurs in outbreaks after bathing in hot tubs or swimming pools, and lesions occur in regions occluded by bathing suits. Lesions of variola (smallpox) (Chap. 261e) also appear similar to those of varicella but are all at the same stage of development in a given region of the body. Variola lesions are most prominent on the face and extremities, while varicella lesions are most prominent on the trunk. Herpes simplex virus infection (Chap. 216) is characterized by hallmark grouped vesicles on an erythematous base. Primary herpes infection is accompanied by fever and toxicity, while recurrent disease is milder. Rickettsialpox (Chap. 211) is often documented in urban settings and is characterized by vesicles followed by pustules. It can be distinguished from varicella by an eschar at the site of the mouse-mite bite and the papule/plaque base of each vesicle. Acute generalized eruptive pustulosis should be considered in individuals who are acutely febrile and are taking new medications, especially anticonvulsant or antimicrobial agents (Chap. 74). Disseminated Vibrio vulnificus infection (Chap. 193) or ecthyma gangrenosum due to Pseudomonas aeruginosa (Chap. 189) should be considered in immunosuppressed individuals with sepsis and hemorrhagic bullae.
URTICARIA-LIKE ERUPTIONS
Individuals with classic urticaria (“hives”) usually have a hypersensitivity reaction without associated fever. In the presence of fever, urticaria-like eruptions are most often due to urticarial vasculitis (Chap. 385). Unlike individual lesions of classic urticaria, which last up to 24 h, these lesions may last 3–5 days. Etiologies include serum sickness (often induced by drugs such as penicillins, sulfas, salicylates, or barbiturates), connective-tissue disease (e.g., systemic lupus erythematosus or Sjögren’s syndrome), and infection (e.g., with hepatitis B virus, enteroviruses, or parasites). Malignancy, especially lymphoma, may be associated with fever and chronic urticaria (Chap. 72).
NODULAR ERUPTIONS
In immunocompromised hosts, nodular lesions often represent disseminated infection. Patients with disseminated candidiasis (often due to Candida tropicalis) may have a triad of fever, myalgias, and eruptive nodules (Chap. 240). Disseminated cryptococcosis lesions (Chap. 239) may resemble molluscum contagiosum (Chap. 220e). Necrosis of nodules should raise the suspicion of aspergillosis (Chap. 241) or mucormycosis (Chap. 242). Erythema nodosum presents with exquisitely tender nodules on the lower extremities. Sweet syndrome (Chap. 72) should be considered in individuals with multiple nodules and plaques, often so edematous that they give the appearance of vesicles or bullae. Sweet syndrome may occur in individuals with infection, inflammatory bowel disease, or malignancy and can also be induced by drugs.
PURPURIC ERUPTIONS
Acute meningococcemia (Chap. 180) classically presents in children as a petechial eruption, but initial lesions may appear as blanchable macules or urticaria. Rocky Mountain spotted fever should be considered in the differential diagnosis of acute meningococcemia. Echovirus 9 infection (Chap. 228) may mimic acute meningococcemia; patients should be treated as if they have bacterial sepsis because prompt differentiation of these conditions may be impossible. Large ecchymotic areas of purpura fulminans (Chaps. 180 and 325) reflect severe underlying disseminated intravascular coagulation, which may be due to infectious or noninfectious causes. The lesions of chronic meningococcemia (Chap. 180) may have a variety of morphologies, including petechial. Purpuric nodules may develop on the legs and resemble erythema nodosum but lack its exquisite tenderness. Lesions of disseminated gonococcemia (Chap. 181) are distinctive, sparse, countable hemorrhagic pustules, usually located near joints. The lesions of chronic meningococcemia and those of gonococcemia may be indistinguishable in terms of appearance and distribution. Viral hemorrhagic fever (Chaps. 233 and 234) should be considered in patients with an appropriate travel history and a petechial rash. Thrombotic thrombocytopenic purpura (Chaps. 72, 129, and 140) and hemolytic-uremic syndrome (Chaps. 140, 186, and 191) are closely related and are noninfectious causes of fever and petechiae. Cutaneous small-vessel vasculitis (leukocytoclastic vasculitis) typically manifests as palpable purpura and has a wide variety of causes (Chap. 72).
ERUPTIONS WITH ULCERS OR ESCHARS
The presence of an ulcer or eschar in the setting of a more widespread eruption can provide an important diagnostic clue. For example, the presence of an eschar may suggest the diagnosis of scrub typhus or rickettsialpox (Chap. 211) in the appropriate setting. In other illnesses (e.g., anthrax) (Chap. 261e), an ulcer or eschar may be the only skin manifestation.
25e |
Atlas of Rashes Associated with Fever |
Given the extremely broad differential diagnosis, the presentation of a patient with fever and rash often poses a thorny diagnostic challenge for even the most astute and experienced clinician. Rapid narrowing of the differential by prompt recognition of a rash’s key features can result in appropriate and sometimes life-saving therapy. This atlas presents high-quality images of a variety of rashes that have an infectious etiology and are commonly associated with fever.
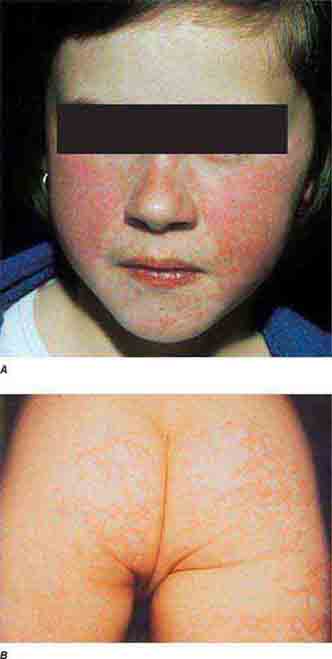
FIGURE 25e-1 A. Erythema leading to “slapped cheeks” appearance in erythema infectiosum (fifth disease) caused by parvovirus B19. B. Lacy reticular rash of erythema infectiosum. (Panel A reprinted from K Wolff, RA Johnson: Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology, 6th ed. New York, McGraw-Hill, 2009.)
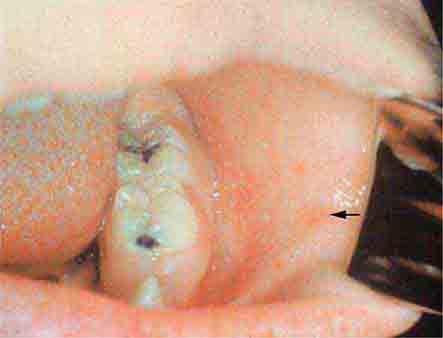
FIGURE 25e-2 Koplik’s spots, which manifest as white or bluish lesions with an erythematous halo on the buccal mucosa, usually occur in the first 2 days of measles symptoms and may briefly overlap the measles exanthem. The presence of the erythematous halo (arrow indicates one example) differentiates Koplik’s spots from Fordyce’s spots (ectopic sebaceous glands), which occur in the mouths of healthy individuals. (Courtesy of the Centers for Disease Control and Prevention.)
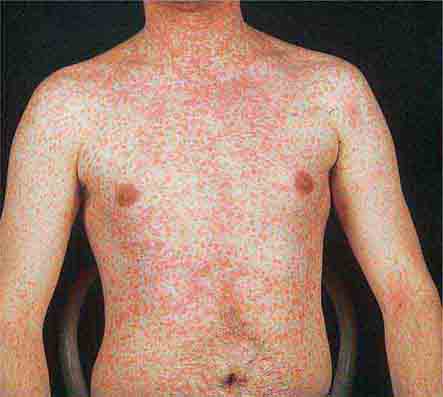
FIGURE 25e-3 In measles, discrete erythematous lesions become confluent on the face and neck over 2–3 days as the rash spreads downward to the trunk and arms, where lesions remain discrete. (Reprinted from K Wolff, RA Johnson: Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology, 5th ed. New York, McGraw-Hill, 2005.)
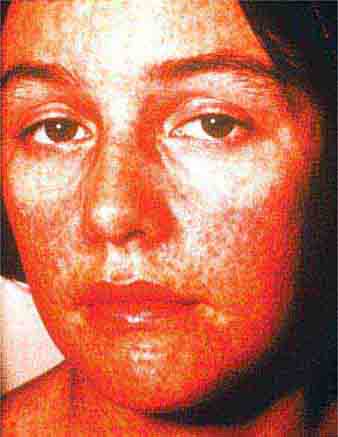
FIGURE 25e-4 In rubella, an erythematous exanthem spreads from the hairline downward and clears as it spreads. (Courtesy of Stephen E. Gellis, MD; with permission.)
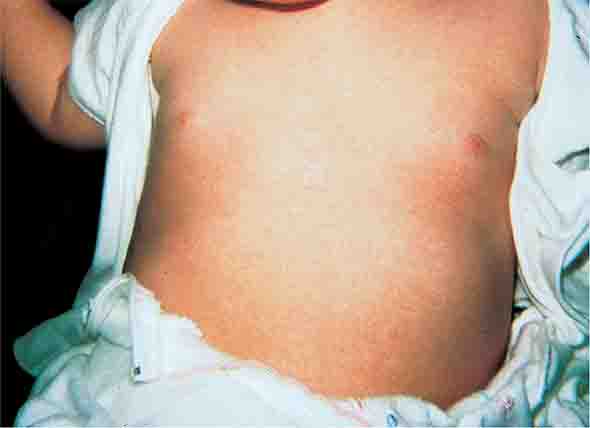
FIGURE 25e-5 Exanthem subitum (roseola) occurs most commonly in young children. A diffuse maculopapular exanthem follows resolution of fever. (Courtesy of Stephen E. Gellis, MD; with permission.)
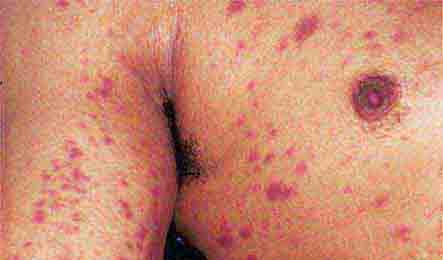
FIGURE 25e-6 Erythematous macules and papules are apparent on the trunk and arm of this patient with primary HIV infection. (Reprinted from K Wolff, RA Johnson: Color Atlas and Synopsis of Clinical Dermatology, 5th ed. New York, McGraw-Hill, 2005.)
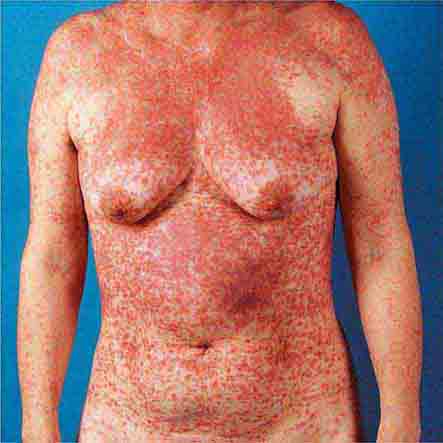
FIGURE 25e-7 This exanthematous, drug-induced eruption consists of brightly erythematous macules and papules, some of which are confluent, distributed symmetrically on the trunk and extremities. Ampicillin caused this rash. (Reprinted from K Wolff, RA Johnson: Color Atlas and Synopsis of Clinical Dermatology, 5th ed. New York, McGraw-Hill, 2005.)
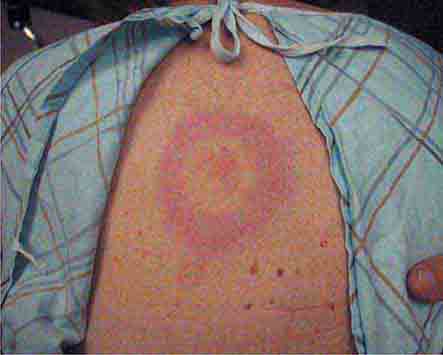
FIGURE 25e-8 Erythema migrans is the early cutaneous manifestation of Lyme disease and is characterized by erythematous annular patches, often with a central erythematous focus at the tick-bite site. (Reprinted from RP Usatine et al: Color Atlas of Family Medicine, 2nd ed. New York, McGraw-Hill, 2013. Courtesy of Thomas Corson, MD.)
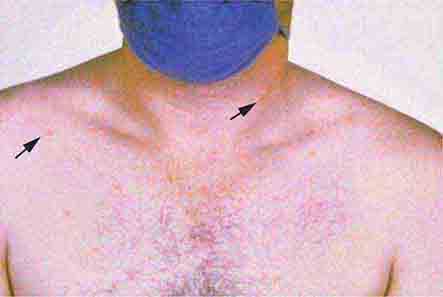
FIGURE 25e-9 Rose spots are evident as erythematous macules on the trunk of this patient with typhoid fever. (Courtesy of the Centers for Disease Control and Prevention.)
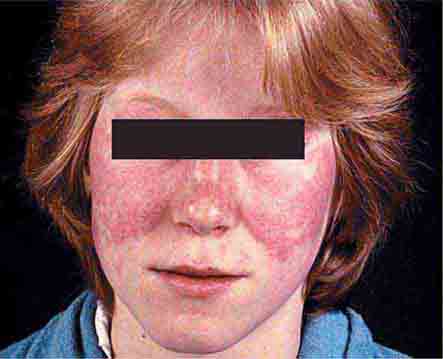
FIGURE 25e-10 Systemic lupus erythematosus showing prominent malar erythema and minimal scaling. Involvement of other sun-exposed sites is also common. (Reprinted from K Wolff, RA Johnson: Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology, 6th ed. New York, McGraw-Hill, 2009.)
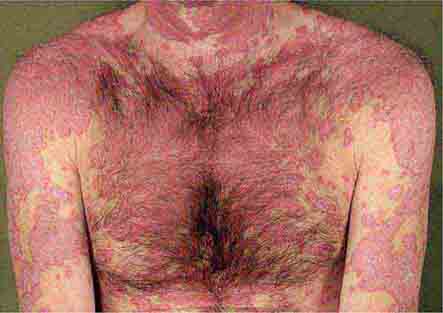
FIGURE 25e-11 Subacute lupus erythematosus on the upper chest, with brightly erythematous and slightly edematous coalescent papules and plaques. (Reprinted from K Wolff, RA Johnson: Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology, 6th ed. New York, McGraw-Hill, 2009.)
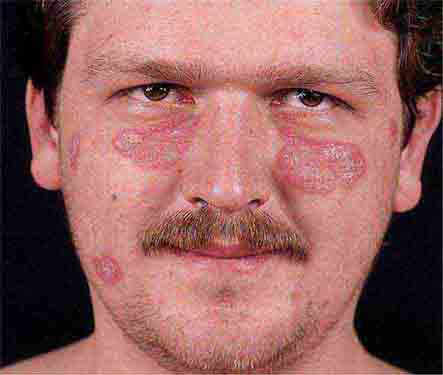
FIGURE 25e-12 Chronic discoid lupus erythematosus. Violaceous, hyperpigmented, atrophic plaques, often with evidence of follicular plugging (which may result in scarring), are characteristic of this cutaneous form of lupus. (Reprinted from K Wolff, RA Johnson, AP Saavedra: Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology, 7th ed. New York, McGraw-Hill, 2013.)
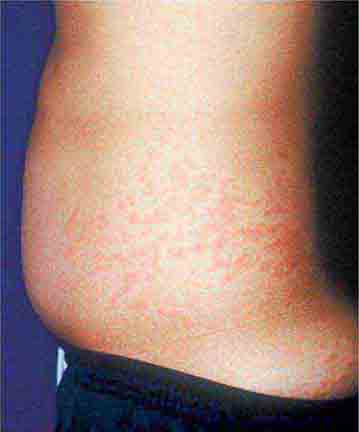
FIGURE 25e-13 The rash of Still’s disease typically exhibits evanescent, erythematous papules that appear at the height of fever on the trunk and proximal extremities. (Courtesy of Stephen E. Gellis, MD; with permission.)
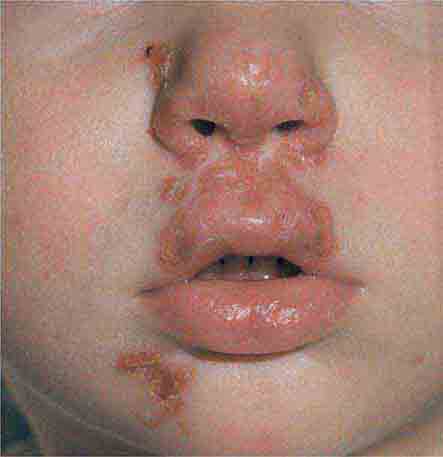
FIGURE 25e-14 Impetigo is a superficial group A streptococcal or Staphylococcus aureus infection consisting of honey-colored crusts and erythematous weeping erosions. (Reprinted from K Wolff, RA Johnson: Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology, 6th ed. New York, McGraw-Hill, 2009.)
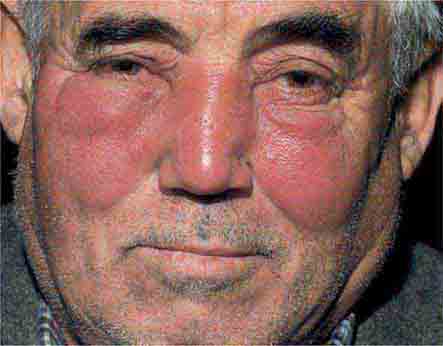
FIGURE 25e-15 Erysipelas is a group A streptococcal infection of the superficial dermis and consists of well-demarcated, erythematous, edematous, warm plaques. (Reprinted from K Wolff, RA Johnson, AP Saavedra: Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology, 7th ed. New York, McGraw-Hill, 2013.)
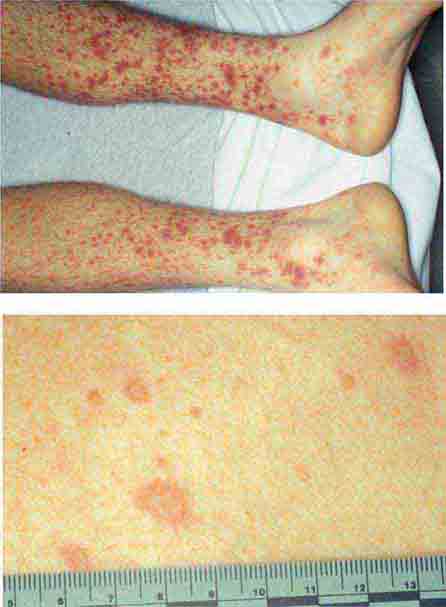
FIGURE 25e-16 Top: Petechial lesions of Rocky Mountain spotted fever on the lower legs and soles of a young, otherwise healthy patient. Bottom: Close-up of lesions from the same patient. (Courtesy of Lindsey Baden, MD; with permission.)
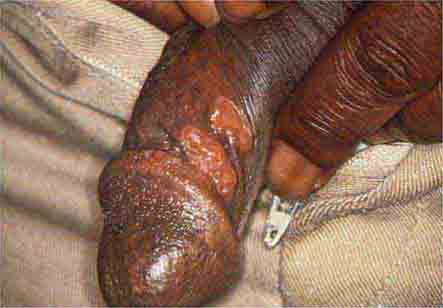
FIGURE 25e-17 Primary syphilis with firm, nontender chancres. (Courtesy of M. Rein and the Centers for Disease Control and Prevention.)
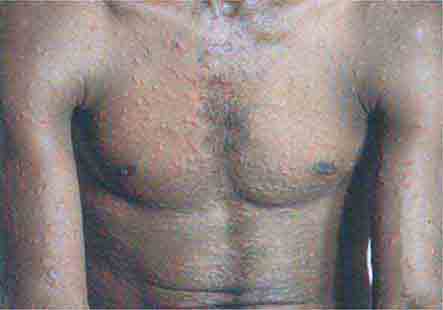
FIGURE 25e-18 Secondary syphilis, demonstrating the papulosquamous truncal eruption.
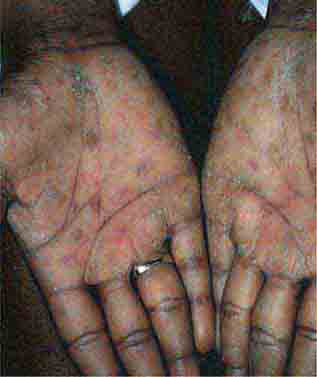
FIGURE 25e-19 Secondary syphilis commonly affects the palms and soles with scaling, firm, red-brown papules.
FIGURE 25e-20 Condylomata lata are moist, somewhat verrucous intertriginous plaques seen in secondary syphilis.
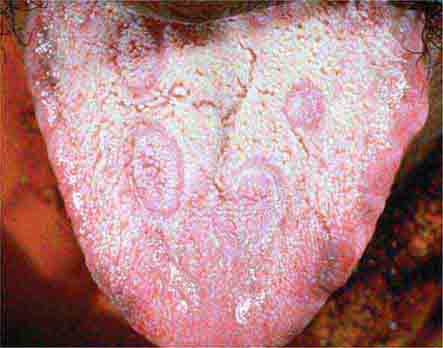
FIGURE 25e-21 Mucous patches on the tongue of a patient with secondary syphilis. (Courtesy of Ron Roddy; with permission.)
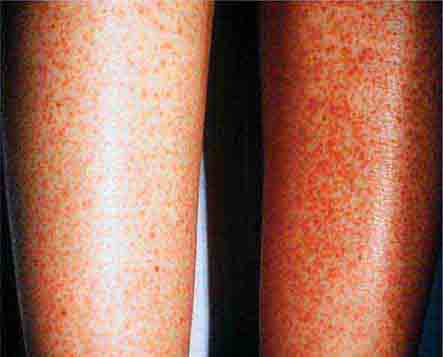
FIGURE 25e-22 Petechial lesions in a patient with atypical measles. (Courtesy of Stephen E. Gellis, MD; with permission.)
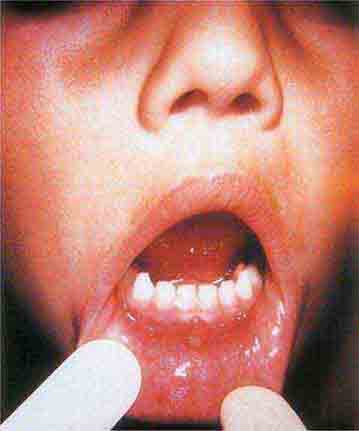
FIGURE 25e-23 Tender vesicles and erosions in the mouth of a patient with hand-foot-and-mouth disease. (Courtesy of Stephen E. Gellis, MD; with permission.)
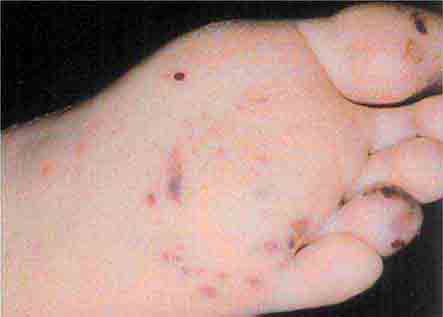
FIGURE 25e-24 Septic emboli with hemorrhage and infarction due to acute Staphylococcus aureus endocarditis. (Courtesy of Lindsey Baden, MD; with permission.)
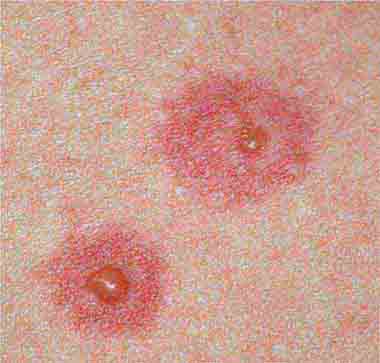
FIGURE 25e-25 Erythema multiforme is characterized by erythematous plaques with a target or iris morphology, sometimes with a vesicle in the center. It usually represents a hypersensitivity reaction to infections (especially herpes simplex virus or Mycoplasma pneumoniae) or drugs. (Reprinted from K Wolff, RA Johnson: Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology, 6th ed. New York, McGraw-Hill, 2009.)
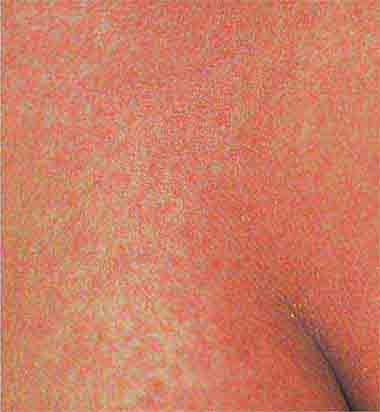
FIGURE 25e-26 Scarlet fever exanthem. Finely punctuated erythema has become confluent (scarlatiniform); accentuation of linear erythema in body folds (Pastia’s lines) is seen here. (Reprinted from K Wolff, RA Johnson: Color Atlas and Synopsis of Clinical Dermatology, 6th ed. New York, McGraw-Hill, 2009.)
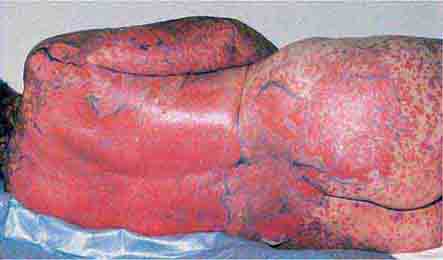
FIGURE 25e-27 Erythema progressing to bullae with resulting sloughing of the entire thickness of the epidermis occurs in toxic epidermal necrolysis. This reaction was due to a sulfonamide. (Reprinted from K Wolff, RA Johnson: Color Atlas and Synopsis of Clinical Dermatology, 5th ed. New York, McGraw-Hill, 2005.)
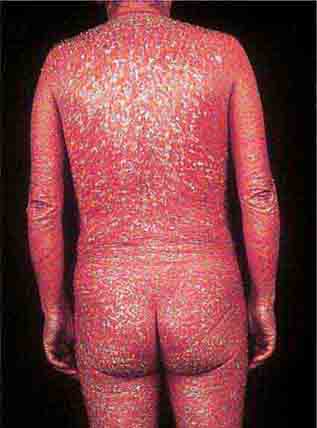
FIGURE 25e-28 Diffuse erythema and scaling are present in this patient with psoriasis and the exfoliative erythroderma syndrome. (Reprinted from K Wolff, RA Johnson: Color Atlas and Synopsis of Clinical Dermatology, 6th ed. New York, McGraw-Hill, 2009.)
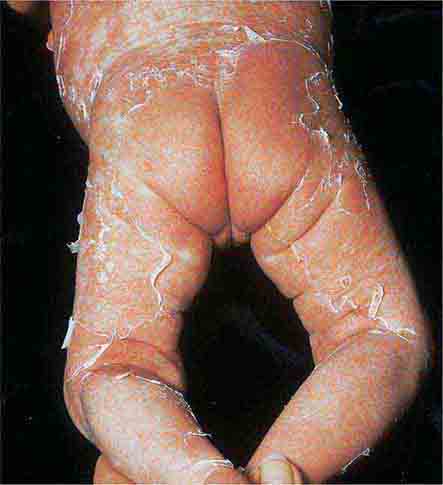
FIGURE 25e-29 This infant with staphylococcal scalded skin syndrome demonstrates generalized desquamation. (Reprinted from K Wolff, RA Johnson: Color Atlas and Synopsis of Clinical Dermatology, 6th ed. New York, McGraw-Hill, 2009.)
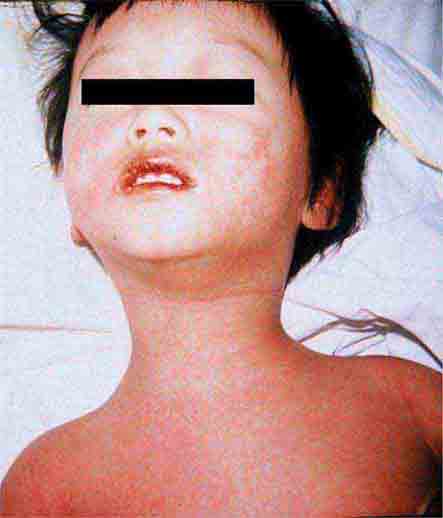
FIGURE 25e-30 Fissuring of the lips and an erythematous exanthem are evident in this patient with Kawasaki disease. (Courtesy of Stephen E. Gellis, MD; with permission.)
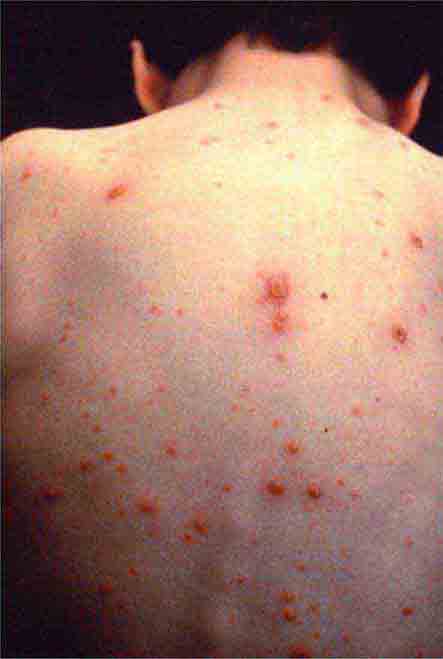
FIGURE 25e-31 Numerous varicella lesions at various stages of evolution: vesicles on an erythematous base and umbilicated vesicles, which then develop into crusting lesions. (Courtesy of the Centers for Disease Control and Prevention.)
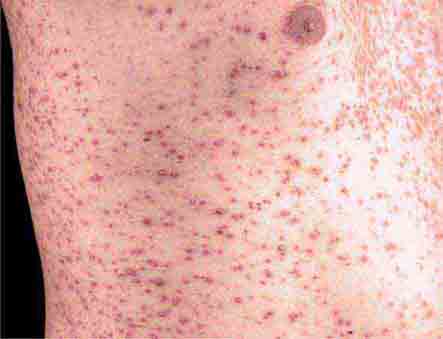
FIGURE 25e-32 Lesions of disseminated zoster at different stages of evolution, including pustules and crusting, similar to varicella. Note nongrouping of lesions, in contrast to herpes simplex or zoster. (Reprinted from K Wolff, RA Johnson, AP Saavedra: Color Atlas and Synopsis of Clinical Dermatology, 7th ed. New York, McGraw-Hill, 2013.)
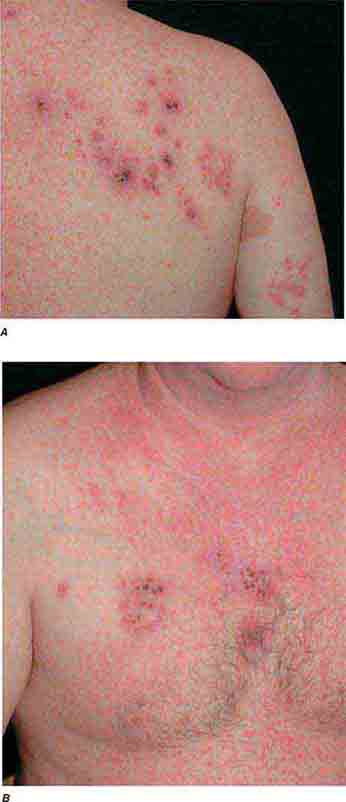
FIGURE 25e-33 Herpes zoster is seen in this patient taking prednisone. Grouped vesicles and crusted lesions are seen in the T2 dermatome on the back and arm (A) and on the right side of the chest (B). (Reprinted from K Wolff, RA Johnson: Color Atlas and Synopsis of Clinical Dermatology, 6th ed. New York, McGraw-Hill, 2009.)
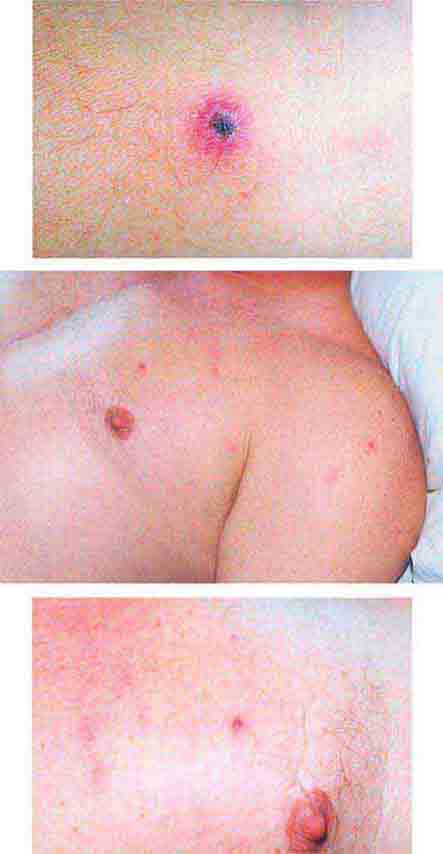
FIGURE 25e-34 Top: Eschar at the site of the mite bite in a patient with rickettsialpox. Middle: Papulovesicular lesions on the trunk of the same patient. Bottom: Close-up of lesions from the same patient. (Reprinted from A Krusell et al: Emerg Infect Dis 8:727, 2002.)
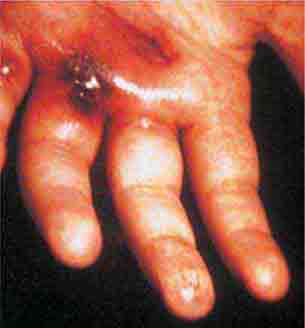
FIGURE 25e-35 Ecthyma gangrenosum in a neutropenic patient with Pseudomonas aeruginosa bacteremia.
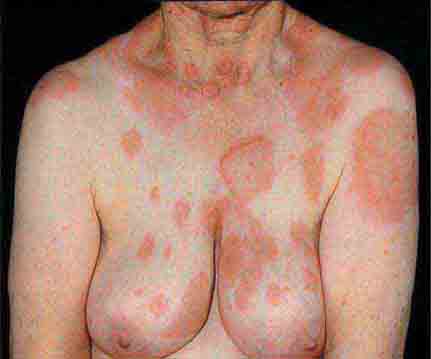
FIGURE 25e-36 Urticaria showing characteristic discrete and confluent, edematous, erythematous papules and plaques. (Reprinted from K Wolff, RA Johnson, AP Saavedra: Color Atlas and Synopsis of Clinical Dermatology, 7th ed. New York, McGraw-Hill, 2013.)
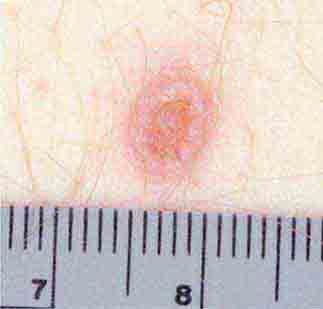
FIGURE 25e-37 Disseminated cryptococcal infection. A liver transplant recipient developed six cutaneous lesions similar to the one shown. Biopsy and serum antigen testing demonstrated Cryptococcus. Important features of the lesion include a benign-appearing fleshy papule with central umbilication resembling molluscum contagiosum. (Courtesy of Lindsey Baden, MD; with permission.)
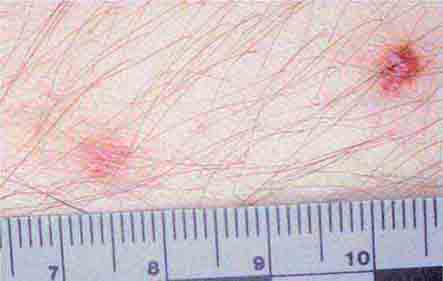
FIGURE 25e-38 Disseminated candidiasis. Tender, erythematous, nodular lesions developed in a neutropenic patient with leukemia who was undergoing induction chemotherapy. (Courtesy of Lindsey Baden, MD; with permission.)
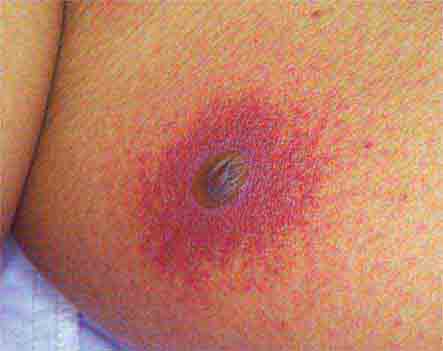
FIGURE 25e-39 Disseminated Aspergillus infection. Multiple necrotic lesions developed in this neutropenic patient undergoing hematopoietic stem cell transplantation. The lesion in the photograph is on the inner thigh and is several centimeters in diameter. Biopsy demonstrated infarction caused by Aspergillus fumigatus. (Courtesy of Lindsey Baden, MD; with permission.)
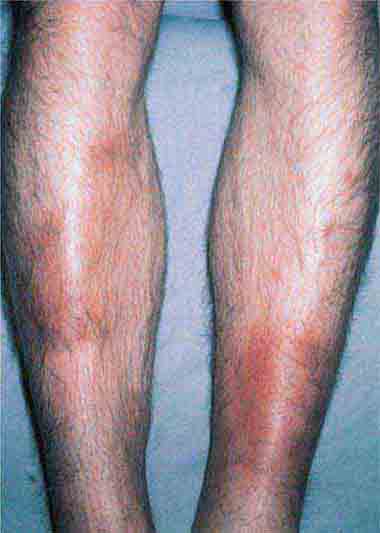
FIGURE 25e-40 Erythema nodosum is a panniculitis characterized by tender, deep-seated nodules and plaques usually located on the lower extremities. (Courtesy of Robert Swerlick, MD; with permission.)
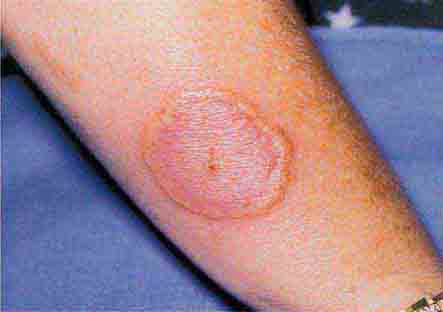
FIGURE 25e-41 Sweet syndrome is an erythematous indurated plaque with a pseudovesicular border. (Courtesy of Robert Swerlick, MD; with permission.)
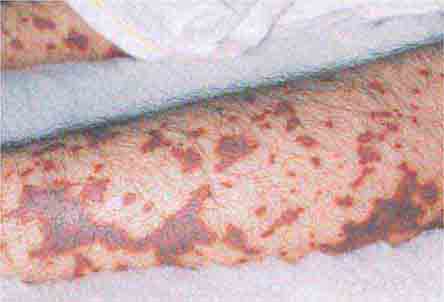
FIGURE 25e-42 Fulminant meningococcemia with extensive angular purpuric patches. (Courtesy of Stephen E. Gellis, MD; with permission.)
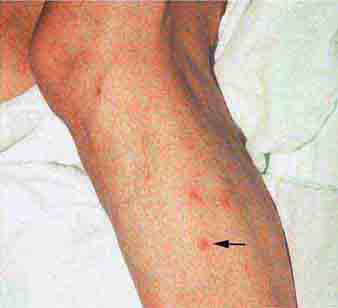
FIGURE 25e-43 Erythematous papular lesions are seen on the leg of this patient with chronic meningococcemia (arrow indicates a lesion).
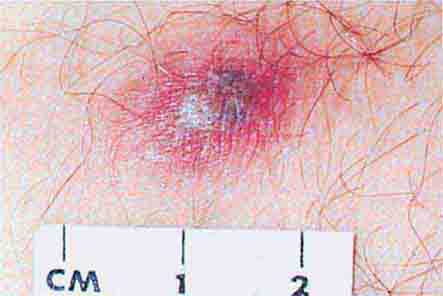
FIGURE 25e-44 Disseminated gonococcemia in the skin is seen as hemorrhagic papules and pustules with purpuric centers in a centrifugal distribution. (Courtesy of Daniel M. Musher, MD; with permission.)
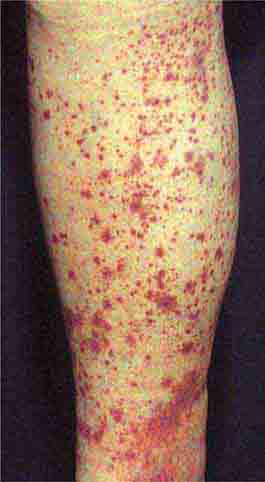
FIGURE 25e-45 Palpable purpuric papules on the lower leg are seen in this patient with cutaneous small-vessel hypersensitivity vasculitis. (Reprinted from K Wolff, RA Johnson: Color Atlas and Synopsis of Clinical Dermatology, 6th ed. New York, McGraw-Hill, 2009.)
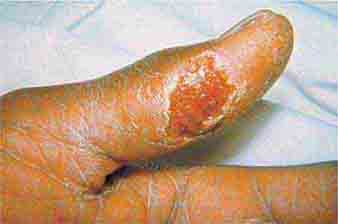
FIGURE 25e-46 The thumb of a patient with a necrotic ulcer of tularemia. (Courtesy of the Centers for Disease Control and Prevention.)
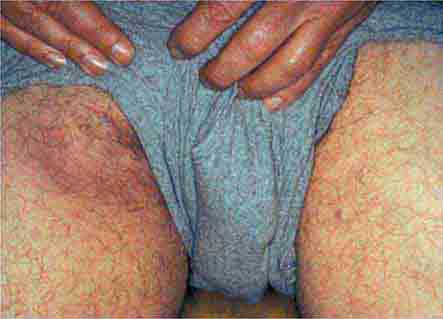
FIGURE 25e-47 This 50-year-old man developed high fever and massive inguinal lymphadenopathy after a small ulcer healed on his foot. Tularemia was diagnosed. (Courtesy of Lindsey Baden, MD; with permission.)
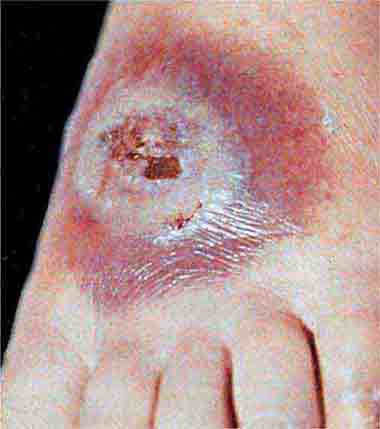
FIGURE 25e-48 This painful trypanosomal chancre developed at the site of a tsetse fly bite on the dorsum of the foot. Trypanosoma brucei was diagnosed from an aspirate of the ulcer. (Courtesy of Edward T. Ryan, MD. N Engl J Med 346:2069, 2002; with permission.)
FIGURE 25e-49 Drug reaction with eosinophilia and systemic symptoms/drug-induced hypersensitivity syndrome (DRESS/DIHS). This patient developed a progressive eruption exhibiting early desquamation after taking phenobarbital. There was also associated lymphadenopathy and hepatomegaly. (Courtesy of Peter Lio, MD; with permission.)
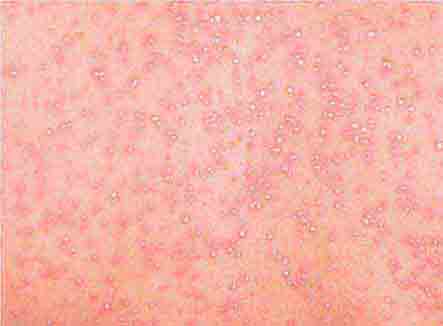
FIGURE 25e-50 Many small, nonfollicular pustules are seen against a background of erythema in this patient with acute generalized eruptive pustulosis (AGEP). The rash began in body folds and progressed to cover the trunk and face. (Reprinted from K Wolff, RA Johnson: Color Atlas and Synopsis of Clinical Dermatology, 6th ed. New York, McGraw-Hill, 2009.)
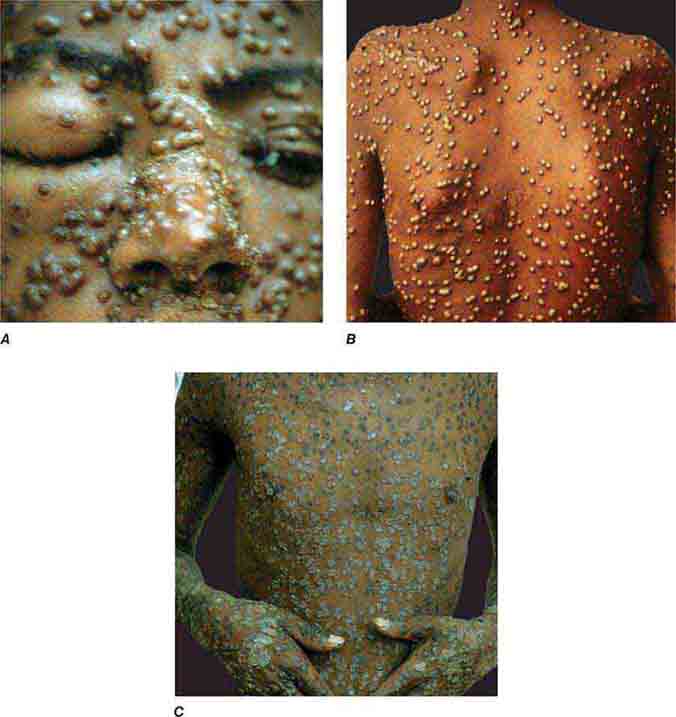
FIGURE 25e-51 Smallpox is shown with many pustules on the face, becoming confluent (A), and on the trunk (B). Pustules are all in the same stage of development. C. Crusting, healing lesions are noted on the trunk, arms, and hands. (Reprinted from K Wolff, RA Johnson: Color Atlas and Synopsis of Clinical Dermatology, 6th ed. New York, McGraw-Hill, 2009.)
26 |
Fever of Unknown Origin |
DEFINITION
Clinicians commonly refer to any febrile illness without an initially obvious etiology as fever of unknown origin (FUO). Most febrile illnesses either resolve before a diagnosis can be made or develop distinguishing characteristics that lead to a diagnosis. The term FUO should be reserved for prolonged febrile illnesses without an established etiology despite intensive evaluation and diagnostic testing. This chapter focuses on classic FUO in the adult patient.
FUO was originally defined by Petersdorf and Beeson in 1961 as an illness of >3 weeks’ duration with fever of ≥38.3°C (101°F) on two occasions and an uncertain diagnosis despite 1 week of inpatient evaluation. Nowadays, most patients with FUO are hospitalized if their clinical condition requires it, but not for diagnostic purposes only; thus the in-hospital evaluation requirement has been eliminated from the definition. The definition of FUO has been further modified by the exclusion of immunocompromised patients, whose workup requires an entirely different diagnostic and therapeutic approach. For the optimal comparison of patients with FUO in different geographic areas, it has been proposed that the quantitative criterion (diagnosis uncertain after 1 week of evaluation) be changed to a qualitative criterion that requires the performance of a specific list of investigations. Accordingly, FUO is now defined as:
1. Fever >38.3°C (101°F) on at least two occasions
2. Illness duration of ≥3 weeks
3. No known immunocompromised state
4. Diagnosis that remains uncertain after a thorough history-taking, physical examination, and the following obligatory investigations: determination of erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) level; platelet count; leukocyte count and differential; measurement of levels of hemoglobin, electrolytes, creatinine, total protein, alkaline phosphatase, alanine aminotransferase, aspartate aminotransferase, lactate dehydrogenase, creatine kinase, ferritin, antinuclear antibodies, and rheumatoid factor; protein electrophoresis; urinalysis; blood cultures (n = 3); urine culture; chest x-ray; abdominal ultrasonography; and tuberculin skin test (TST).
ETIOLOGY AND EPIDEMIOLOGY
The range of FUO etiologies has evolved over time as a result of changes in the spectrum of diseases causing FUO, the widespread use of antibiotics, and the availability of new diagnostic techniques. The proportion of cases caused by intraabdominal abscesses and tumors, for example, has decreased because of earlier detection by CT and ultrasound. In addition, infective endocarditis is a less frequent cause because blood culture and echocardiographic techniques have improved. Conversely, some diagnoses, such as acute HIV infection, were unknown four decades ago.
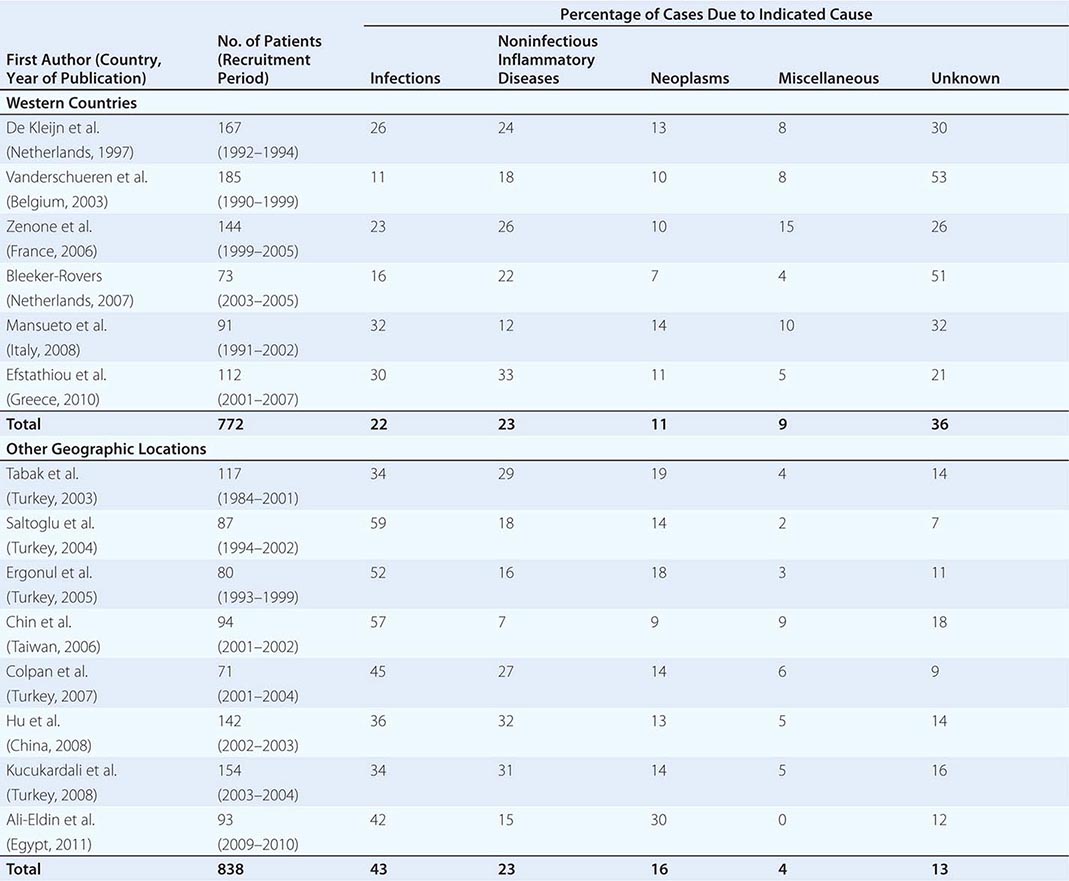
![]() Table 26-1 summarizes the findings of several large studies on FUO conducted over the past 20 years. In general, infection accounts for about 20–25% of cases of FUO in Western countries; next in frequency are neoplasms and noninfectious inflammatory diseases (NIIDs), the latter including “collagen or rheumatic diseases,” vasculitis syndromes, and granulomatous disorders. In geographic areas outside the West, infections are a much more common cause of FUO (43% vs 22%), while the proportions of cases due to NIIDs and neoplasms are similar. Up to 50% of cases caused by infections in patients with FUO outside Western nations are due to tuberculosis, which is a less common cause in the United States and Western Europe. The number of FUO patients diagnosed with NIIDs probably will not decrease in the near future, as fever may precede more typical manifestations or serologic evidence by months in these diseases. Moreover, many NIIDs can be diagnosed only after prolonged observation and exclusion of other diseases.
Table 26-1 summarizes the findings of several large studies on FUO conducted over the past 20 years. In general, infection accounts for about 20–25% of cases of FUO in Western countries; next in frequency are neoplasms and noninfectious inflammatory diseases (NIIDs), the latter including “collagen or rheumatic diseases,” vasculitis syndromes, and granulomatous disorders. In geographic areas outside the West, infections are a much more common cause of FUO (43% vs 22%), while the proportions of cases due to NIIDs and neoplasms are similar. Up to 50% of cases caused by infections in patients with FUO outside Western nations are due to tuberculosis, which is a less common cause in the United States and Western Europe. The number of FUO patients diagnosed with NIIDs probably will not decrease in the near future, as fever may precede more typical manifestations or serologic evidence by months in these diseases. Moreover, many NIIDs can be diagnosed only after prolonged observation and exclusion of other diseases.
|
ETIOLOGY OF FEVER OF UNKNOWN ORIGIN (FUO) OVER THE PAST 20 YEARS: FINDINGS FROM LARGE FUO STUDIES |
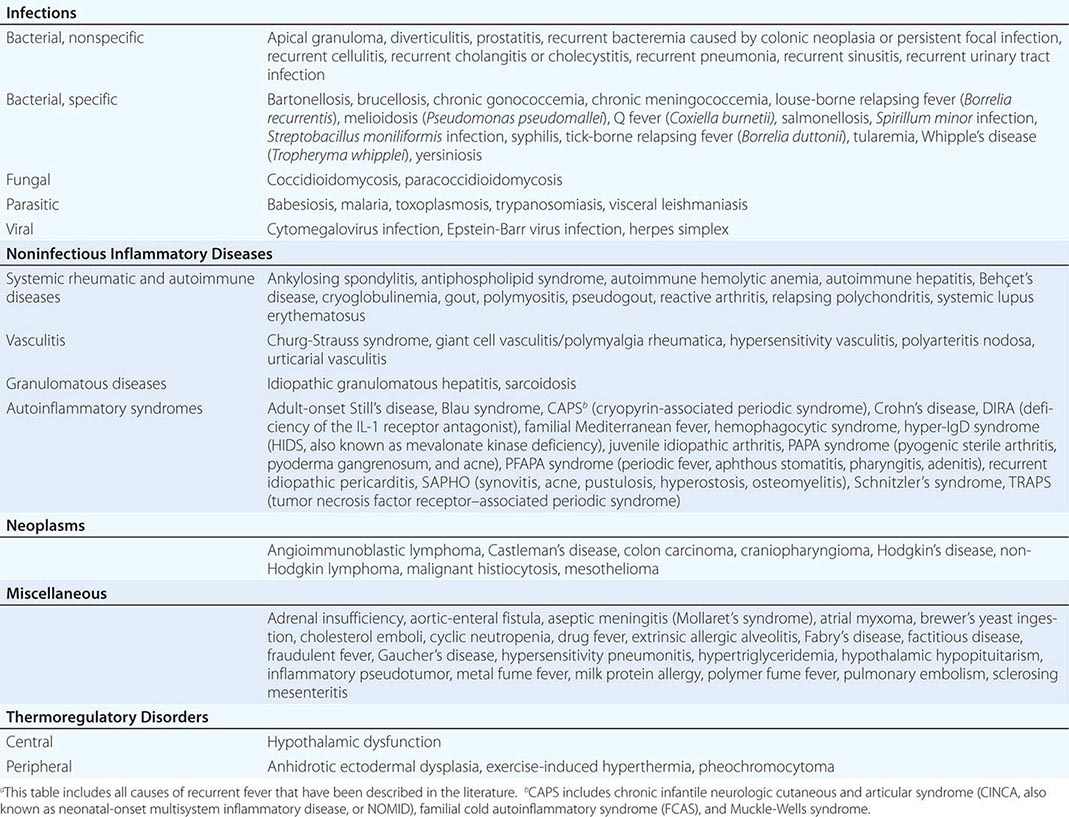
In the West, the percentage of undiagnosed cases of FUO has increased in more recent studies. An important factor contributing to the seemingly high diagnostic failure rate is that a diagnosis is more often being established before 3 weeks have elapsed, given that patients with fever tend to seek medical advice earlier and better diagnostic techniques, such as CT and MRI, are widely available; therefore, only the cases that are more difficult to diagnose continue to meet the criteria for FUO. Furthermore, most patients who have FUO without a diagnosis currently do well, and thus a less aggressive diagnostic approach may be used in clinically stable patients once diseases with immediate therapeutic or prognostic consequences have been ruled out to a reasonable extent. This factor may be especially relevant to patients with recurrent fever who are asymptomatic in between febrile episodes. In patients with recurrent fever (defined as repeated episodes of fever interspersed with fever-free intervals of at least 2 weeks and apparent remission of the underlying disease), the chance of attaining an etiologic diagnosis is <50%.
DIFFERENTIAL DIAGNOSIS
The differential diagnosis for FUO is extensive, but it is important to remember that FUO is far more often caused by an atypical presentation of a rather common disease than by a very rare disease. Table 26-2 presents an overview of possible causes of FUO. An atypical presentation of endocarditis, diverticulitis, vertebral osteomyelitis, and extrapulmonary tuberculosis are the more common infectious disease diagnoses. Q fever and Whipple’s disease are quite rare but should always be kept in mind as a cause of FUO since the presenting symptoms can be nonspecific. Serologic testing for Q fever, which results from exposure to animals or animal products, should be performed when the patient lives in a rural area or has a history of heart valve disease, an aortic aneurysm, or a vascular prosthesis. In patients with unexplained symptoms localized to the central nervous system (CNS), gastrointestinal tract, or joints, polymerase chain reaction (PCR) testing for Tropheryma whipplei should be performed. Travel to or (former) residence in tropical countries or the American Southwest should lead to consideration of infectious diseases such as malaria, leishmaniasis, histoplasmosis, or coccidioidomycosis. Fever with signs of endocarditis and negative blood culture results poses a special problem. Culture-negative endocarditis may be due to difficult-to-culture bacteria such as nutritionally variant bacteria, HACEK organisms (Haemophilus parainfluenzae, H. paraphrophilus, Aggregatibacter species [actinomycetemcomitans, aphrophilus], Cardiobacterium species [hominis, valvarum], Eikenella corrodens, and Kingella kingae; discussed below), Coxiella burnetii (as indicated above), T. whipplei, and Bartonella species. Marantic endocarditis is a sterile thrombotic disease that occurs as a paraneoplastic phenomenon, especially with adenocarcinomas. Sterile endocarditis is also seen in the context of systemic lupus erythematosus and antiphospholipid syndrome.
|
ALL REPORTED CAUSES OF FUOa |
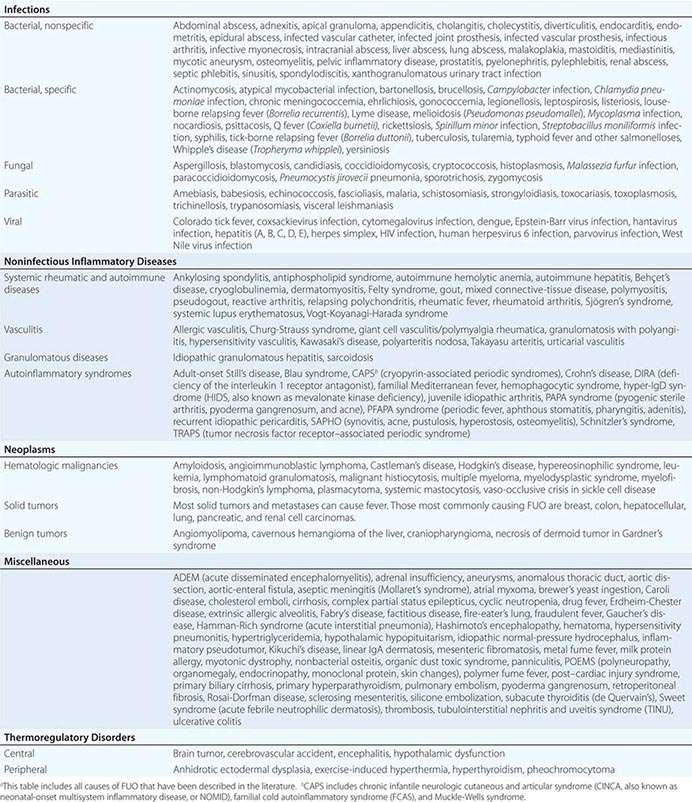
Of the NIIDs, large-vessel vasculitis, polymyalgia rheumatica, sarcoidosis, familial Mediterranean fever, and adult-onset Still’s disease are rather common diagnoses in patients with FUO. The hereditary autoinflammatory syndromes are very rare and usually present in young patients. Schnitzler’s syndrome, which can present at any age, is uncommon but can often be diagnosed easily in a patient with FUO who presents with urticaria, bone pain, and monoclonal gammopathy.
Although most tumors can present with fever, malignant lymphoma is by far the most common diagnosis of FUO among the neoplasms. Sometimes the fever even precedes lymphadenopathy detectable by physical examination.
Apart from drug-induced fever and exercise-induced hyperthermia, none of the miscellaneous causes of fever is found very frequently in patients with FUO. Virtually all drugs can cause fever, even that commencing after long-term use. Drug-induced fever, including DRESS (drug reaction with eosinophilia and systemic symptoms; Fig. 25e-49), is often accompanied by eosinophilia and also by lymphadenopathy, which can be extensive. More common causes of drug-induced fever are allopurinol, carbamazepine, lamotrigine, phenytoin, sulfasalazine, furosemide, antimicrobial drugs (especially sulfonamides, minocycline, vancomycin, β-lactam antibiotics, and isoniazid), some cardiovascular drugs (e.g., quinidine), and some antiretroviral drugs (e.g., nevirapine). Exercise-induced hyperthermia (Chap. 479e) is characterized by an elevated body temperature that is associated with moderate to strenuous exercise lasting from half an hour up to several hours without an increase in CRP level or ESR; typically these patients sweat during the temperature elevation. Factitious fever (fever artificially induced by the patient—for example, by IV injection of contaminated water) should be considered in all patients but is more common among young women in health care professions. In fraudulent fever, the patient is normothermic but manipulates the thermometer. Simultaneous measurements at different body sites (rectum, ear, mouth) should rapidly identify this diagnosis. Another clue to fraudulent fever is a dissociation between pulse rate and temperature.
Previous studies of FUO have shown that a diagnosis is more likely in elderly patients than in younger age groups. In many cases, FUO in the elderly results from an atypical manifestation of a common disease, among which giant cell arteritis and polymyalgia rheumatica are most frequently involved. Tuberculosis is the most common infectious disease associated with FUO in elderly patients, occurring much more often than in younger patients. As many of these diseases are treatable, it is well worth pursuing the cause of fever in elderly patients.
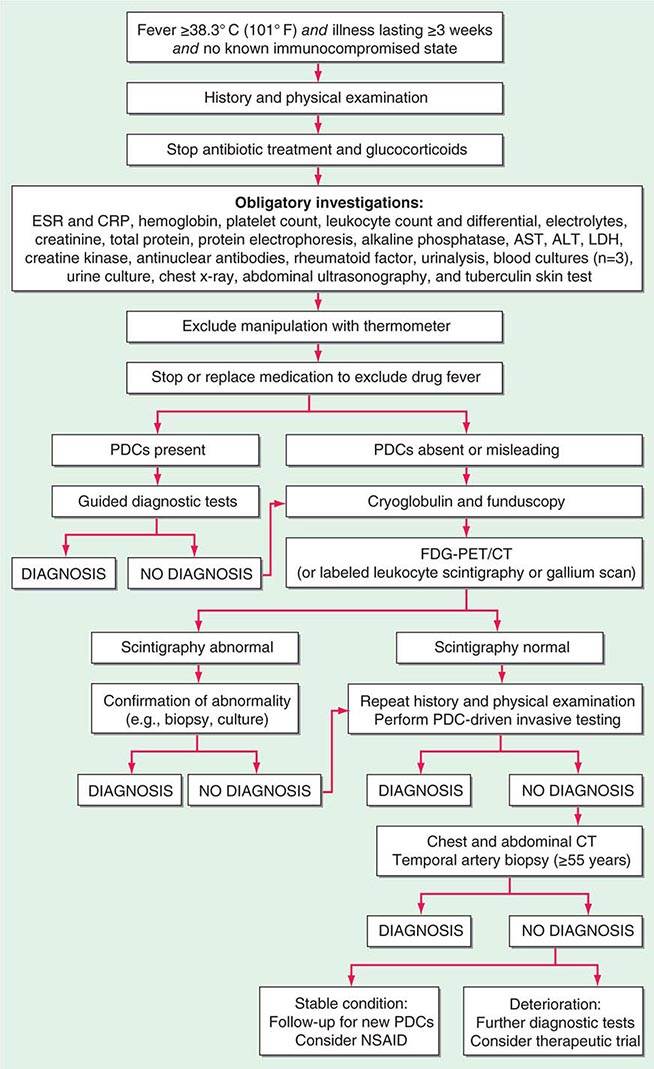
FIGURE 26-1 Structured approach to patients with FUO. ALT, alanine aminotransferase; AST, aspartate aminotransferase; CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; FDG-PET/CT, 18F-fluorodeoxyglucose positron emission tomography combined with low-dose computed tomography; LDH, lactate dehydrogenase; PDCs, potentially diagnostic clues (all localizing signs, symptoms, and abnormalities potentially pointing toward a diagnosis); NSAID, nonsteroidal anti-inflammatory drug.
|
ALL REPORTED CAUSES OF RECURRENT FEVERa |
PROGNOSIS
FUO-related mortality rates have continuously declined over recent decades. The majority of fevers are caused by treatable diseases, and the risk of death related to FUO is, of course, dependent on the underlying disease. In a study by our group (Table 26-1), none of 37 FUO patients without a diagnosis died during a follow-up period of at least 6 months; 4 of 36 patients with a diagnosis died during follow-up due to infection (n = 1) or malignancy (n = 3). Other studies have also shown that malignancy accounts for most FUO-related deaths. Non-Hodgkin’s lymphoma carries a disproportionately high death toll. In nonmalignant FUO, fatality rates are very low. The good outcome in patients without a diagnosis confirms that potentially lethal occult diseases are very unusual and that empirical therapy with antibiotics, antituberculous agents, or glucocorticoids is rarely required in stable patients. In less affluent regions, infectious diseases are still a major cause of FUO, and outcomes may be different.
SECTION 3 |
NERVOUS SYSTEM DYSFUNCTION |
27 |
Syncope |
Syncope is a transient, self-limited loss of consciousness due to acute global impairment of cerebral blood flow. The onset is rapid, duration brief, and recovery spontaneous and complete. Other causes of transient loss of consciousness need to be distinguished from syncope; these include seizures, vertebrobasilar ischemia, hypoxemia, and hypoglycemia. A syncopal prodrome (presyncope) is common, although loss of consciousness may occur without any warning symptoms. Typical presyncopal symptoms include dizziness, lightheadedness or faintness, weakness, fatigue, and visual and auditory disturbances. The causes of syncope can be divided into three general categories: (1) neurally mediated syncope (also called reflex or vasovagal syncope), (2) orthostatic hypotension, and (3) cardiac syncope.
Neurally mediated syncope comprises a heterogeneous group of functional disorders that are characterized by a transient change in the reflexes responsible for maintaining cardiovascular homeostasis. Episodic vasodilation (or loss of vasoconstrictor tone) and bradycardia occur in varying combinations, resulting in temporary failure of blood pressure control. In contrast, in patients with orthostatic hypotension due to autonomic failure, these cardiovascular homeostatic reflexes are chronically impaired. Cardiac syncope may be due to arrhythmias or structural cardiac diseases that cause a decrease in cardiac output. The clinical features, underlying pathophysiologic mechanisms, therapeutic interventions, and prognoses differ markedly among these three causes.
EPIDEMIOLOGY AND NATURAL HISTORY
Syncope is a common presenting problem, accounting for approximately 3% of all emergency room visits and 1% of all hospital admissions. The annual cost for syncope-related hospitalization in the United States is ~$2.4 billion. Syncope has a lifetime cumulative incidence of up to 35% in the general population. The peak incidence in the young occurs between ages 10 and 30 years, with a median peak around 15 years. Neurally mediated syncope is the etiology in the vast majority of these cases. In elderly adults, there is a sharp rise in the incidence of syncope after 70 years.
In population-based studies, neurally mediated syncope is the most common cause of syncope. The incidence is slightly higher in females than males. In young subjects, there is often a family history in first-degree relatives. Cardiovascular disease due to structural disease or arrhythmias is the next most common cause in most series, particularly in emergency room settings and in older patients. Orthostatic hypotension also increases in prevalence with age because of the reduced baroreflex responsiveness, decreased cardiac compliance, and attenuation of the vestibulosympathetic reflex associated with aging. In the elderly, orthostatic hypotension is substantially more common in institutionalized (54–68%) than community-dwelling (6%) individuals, an observation most likely explained by the greater prevalence of predisposing neurologic disorders, physiologic impairment, and vasoactive medication use among institutionalized patients.
The prognosis after a single syncopal event for all age groups is generally benign. In particular, syncope of noncardiac and unexplained origin in younger individuals has an excellent prognosis; life expectancy is unaffected. By contrast, syncope due to a cardiac cause, either structural heart disease or primary arrhythmic disease, is associated with an increased risk of sudden cardiac death and mortality from other causes. Similarly, mortality rate is increased in individuals with syncope due to orthostatic hypotension related to age and the associated comorbid conditions (Table 27-1).
|
HIGH-RISK FEATURES INDICATING HOSPITALIZATION OR INTENSIVE EVALUATION OF SYNCOPE |
PATHOPHYSIOLOGY
The upright posture imposes a unique physiologic stress upon humans; most, although not all, syncopal episodes occur from a standing position. Standing results in pooling of 500–1000 mL of blood in the lower extremities and splanchnic circulation. There is a decrease in venous return to the heart and reduced ventricular filling that result in diminished cardiac output and blood pressure. These hemodynamic changes provoke a compensatory reflex response, initiated by the baroreceptors in the carotid sinus and aortic arch, resulting in increased sympathetic outflow and decreased vagal nerve activity (Fig. 27-1). The reflex increases peripheral resistance, venous return to the heart, and cardiac output and thus limits the fall in blood pressure. If this response fails, as is the case chronically in orthostatic hypotension and transiently in neurally mediated syncope, cerebral hypoperfusion occurs.
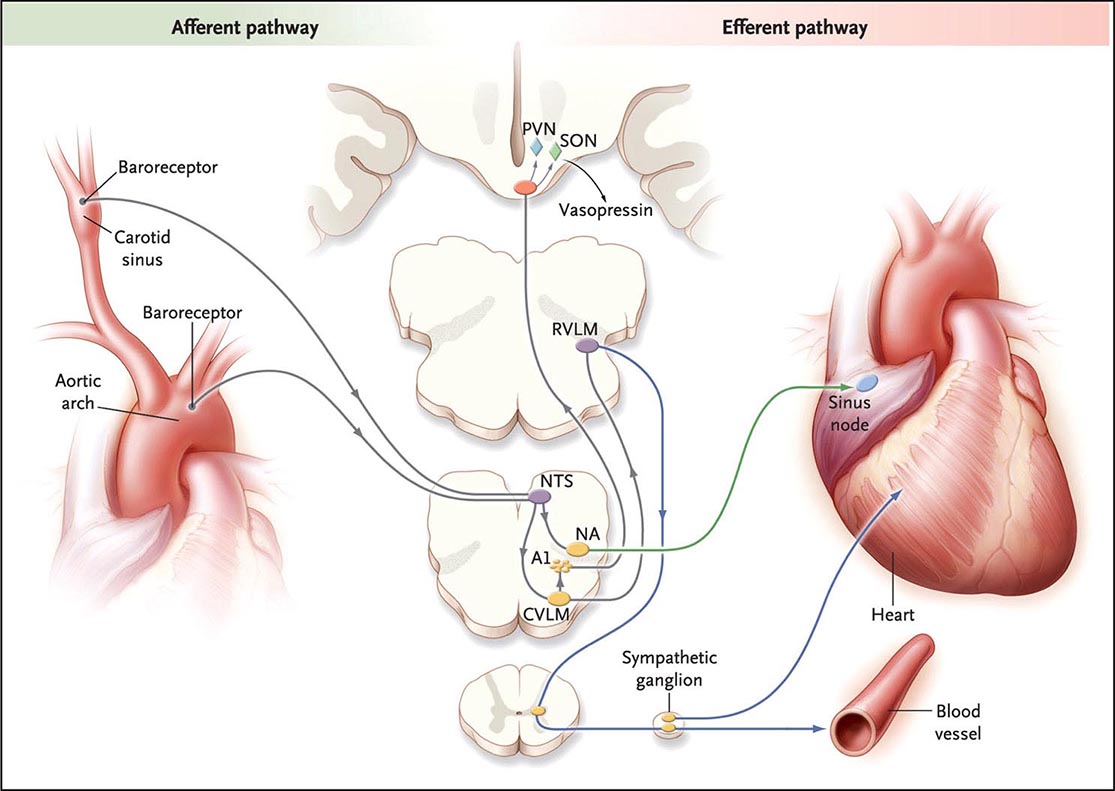
FIGURE 27-1 The baroreflex. A decrease in arterial pressure unloads the baroreceptors—the terminals of afferent fibers of the glossopharyngeal and vagus nerves—that are situated in the carotid sinus and aortic arch. This leads to a reduction in the afferent impulses that are relayed from these mechanoreceptors through the glossopharyngeal and vagus nerves to the nucleus of the tractus solitarius (NTS) in the dorsomedial medulla. The reduced baroreceptor afferent activity produces a decrease in vagal nerve input to the sinus node that is mediated via connections of the NTS to the nucleus ambiguus (NA). There is an increase in sympathetic efferent activity that is mediated by the NTS projections to the caudal ventrolateral medulla (CVLM) (an excitatory pathway) and from there to the rostral ventrolateral medulla (RVLM) (an inhibitory pathway). The activation of RVLM presympathetic neurons in response to hypotension is thus predominantly due to disinhibition. In response to a sustained fall in blood pressure, vasopressin release is mediated by projections from the A1 noradrenergic cell group in the ventrolateral medulla. This projection activates vasopressin-synthesizing neurons in the magnocellular portion of the paraventricular nucleus (PVN) and the supraoptic nucleus (SON) of the hypothalamus. Blue denotes sympathetic neurons, and green denotes parasympathetic neurons. (From R Freeman: N Engl J Med 358:615, 2008.)
Syncope is a consequence of global cerebral hypoperfusion and thus represents a failure of cerebral blood flow autoregulatory mechanisms. Myogenic factors, local metabolites, and to a lesser extent autonomic neurovascular control are responsible for the autoregulation of cerebral blood flow (Chap. 330). The latency of the autoregulatory response is 5–10 s. Typically cerebral blood flow ranges from 50 to 60 mL/min per 100 g brain tissue and remains relatively constant over perfusion pressures ranging from 50 to 150 mmHg. Cessation of blood flow for 6–8 s will result in loss of consciousness, while impairment of consciousness ensues when blood flow decreases to 25 mL/min per 100 g brain tissue.
From the clinical standpoint, a fall in systemic systolic blood pressure to ~50 mmHg or lower will result in syncope. A decrease in cardiac output and/or systemic vascular resistance—the determinants of blood pressure—thus underlies the pathophysiology of syncope. Common causes of impaired cardiac output include decreased effective circulating blood volume; increased thoracic pressure; massive pulmonary embolus; cardiac brady- and tachyarrhythmias; valvular heart disease; and myocardial dysfunction. Systemic vascular resistance may be decreased by central and peripheral autonomic nervous system diseases, sympatholytic medications, and transiently during neurally mediated syncope. Increased cerebral vascular resistance, most frequently due to hypocarbia induced by hyperventilation, may also contribute to the pathophysiology of syncope.
Two patterns of electroencephalographic (EEG) changes occur in syncopal subjects. The first is a “slow-flat-slow” pattern (Fig. 27-2) in which normal background activity is replaced with high-amplitude slow delta waves. This is followed by sudden flattening of the EEG—a cessation or attenuation of cortical activity—followed by the return of slow waves, and then normal activity. A second pattern, the “slow pattern,” is characterized by increasing and decreasing slow wave activity only. The EEG flattening that occurs in the slow-flat-slow pattern is a marker of more severe cerebral hypoperfusion. Despite the presence of myoclonic movements and other motor activity during some syncopal events, EEG seizure discharges are not detected.
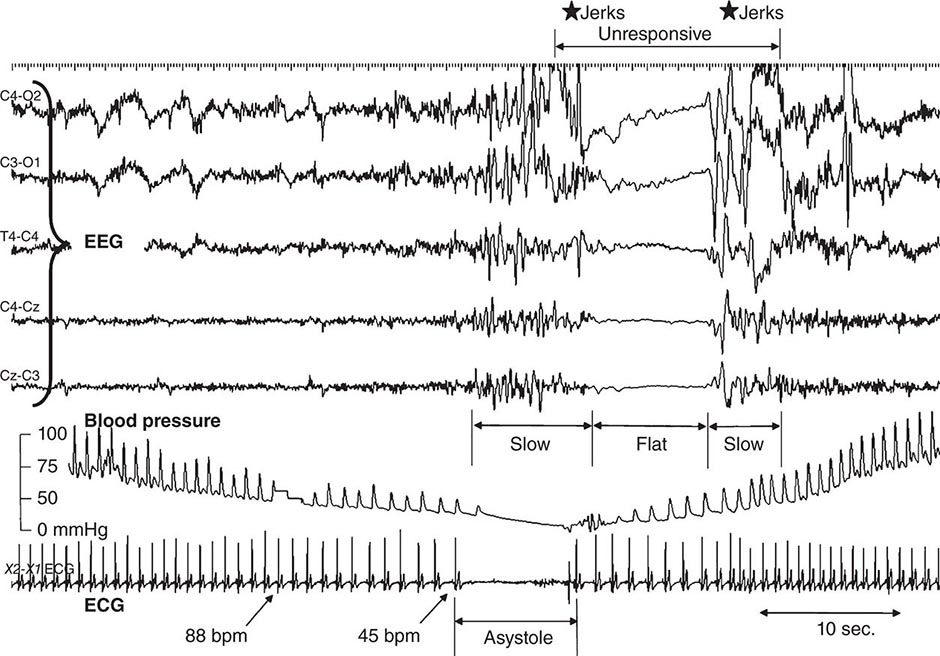
FIGURE 27-2 The electroencephalogram (EEG) in vasovagal syncope. A 1-min segment of a tilt-table test with typical vasovagal syncope demonstrating the “slow-flat-slow” EEG pattern. Finger beat-to-beat blood pressure, electrocardiogram (ECG), and selected EEG channels are shown. EEG slowing starts when systolic blood pressure drops to ~50 mmHg; heart rate is then approximately 45 beats/min (bpm). Asystole occurred, lasting about 8 s. The EEG flattens for a similar period, but with a delay. A transient loss of consciousness, lasting 14 s, was observed. There were muscle jerks just before and just after the flat period of the EEG. (Figure reproduced with permission from W Wieling et al: Brain 132:2630, 2009.)
CLASSIFICATION
NEURALLY MEDIATED SYNCOPE
Neurally mediated (reflex; vasovagal) syncope is the final pathway of a complex central and peripheral nervous system reflex arc. There is a sudden, transient change in autonomic efferent activity with increased parasympathetic outflow, plus sympathoinhibition (the vasodepressor response), resulting in bradycardia, vasodilation, and/or reduced vasoconstrictor tone. The resulting fall in systemic blood pressure can then reduce cerebral blood flow to below the compensatory limits of autoregulation (Fig. 27-3). In order to elicit neutrally mediated syncope, a functioning autonomic nervous system is necessary, in contrast to syncope resulting from autonomic failure (discussed below).
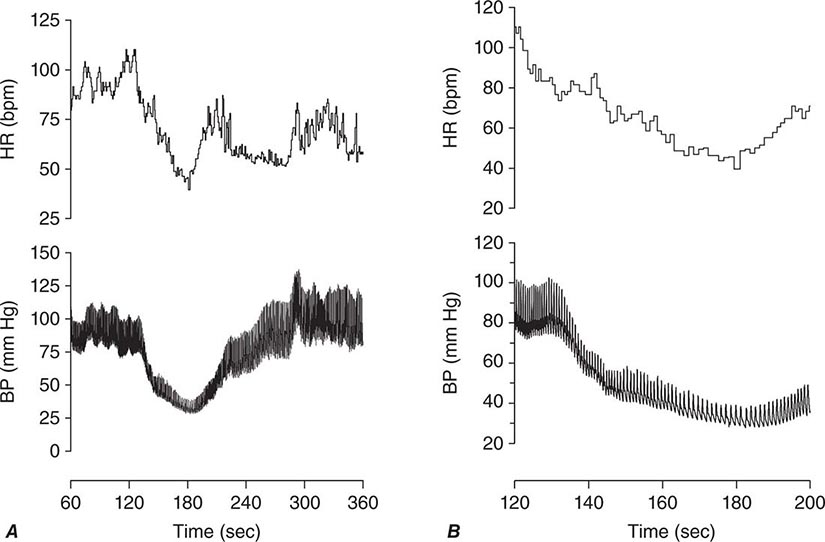
FIGURE 27-3 A. The paroxysmal hypotensive-bradycardic response that is characteristic of neurally mediated syncope. Noninvasive beat-to-beat blood pressure and heart rate are shown over 5 min (from 60 to 360 s) of an upright tilt on a tilt table. B. The same tracing expanded to show 80 s of the episode (from 80 to 200 s). BP, blood pressure; bpm, beats per minute; HR, heart rate.
Multiple triggers of the afferent limb of the reflex arc can result in neutrally mediated syncope. In some situations, these can be clearly defined, e.g., the carotid sinus, the gastrointestinal tract, or the bladder. Often, however, the trigger is less easily recognized and the cause is multifactorial. Under these circumstances, it is likely that different afferent pathways converge on the central autonomic network within the medulla that integrates the neural impulses and mediates the vasodepressor-bradycardic response.
Classification of Neurally Mediated Syncope Neurally mediated syncope may be subdivided based on the afferent pathway and provocative trigger. Vasovagal syncope (the common faint) is provoked by intense emotion, pain, and/or orthostatic stress, whereas the situational reflex syncopes have specific localized stimuli that provoke the reflex vasodilation and bradycardia that leads to syncope. The underlying mechanisms have been identified and pathophysiology delineated for most of these situational reflex syncopes. The afferent trigger may originate in the pulmonary system, gastrointestinal system, urogenital system, heart, and carotid artery (Table 27-2). Hyperventilation leading to hypocarbia and cerebral vasoconstriction, and raised intrathoracic pressure that impairs venous return to the heart, play a central role in many of the situational reflex syncopes. The afferent pathway of the reflex arc differs among these disorders, but the efferent response via the vagus and sympathetic pathways is similar.
|
CAUSES OF SYNCOPE |
aHyperventilation for ~1 minute, followed by sudden chest compression. bHyperventilation (~20 breaths) in a squatting position, rapid rise to standing, then Valsalva.
Alternately, neurally mediated syncope may be subdivided based on the predominant efferent pathway. Vasodepressor syncope describes syncope predominantly due to efferent, sympathetic, vasoconstrictor failure; cardioinhibitory syncope describes syncope predominantly associated with bradycardia or asystole due to increased vagal outflow; and mixed syncope describes syncope in which there are both vagal and sympathetic reflex changes.
Features of Neurally Mediated Syncope In addition to symptoms of orthostatic intolerance such as dizziness, lightheadedness, and fatigue, premonitory features of autonomic activation may be present in patients with neurally mediated syncope. These include diaphoresis, pallor, palpitations, nausea, hyperventilation, and yawning. During the syncopal event, proximal and distal myoclonus (typically arrhythmic and multifocal) may occur, raising the possibility of epilepsy. The eyes typically remain open and usually deviate upward. Pupils are usually dilated. Roving eye movements may occur. Grunting, moaning, snorting, and stertorous breathing may be present. Urinary incontinence may occur. Fecal incontinence is very rare. Postictal confusion is also rare, although visual and auditory hallucinations and near death and out-of-body experiences are sometimes reported.
Although some predisposing factors and provocative stimuli are well established (for example, motionless upright posture, warm ambient temperature, intravascular volume depletion, alcohol ingestion, hypoxemia, anemia, pain, the sight of blood, venipuncture, and intense emotion), the underlying basis for the widely different thresholds for syncope among individuals exposed to the same provocative stimulus is not known. A genetic basis for neurally mediated syncope may exist; several studies have reported an increased incidence of syncope in first-degree relatives of fainters, but no gene or genetic marker has been identified, and environmental, social, and cultural factors have not been excluded by these studies.
ORTHOSTATIC HYPOTENSION
Orthostatic hypotension, defined as a reduction in systolic blood pressure of at least 20 mmHg or diastolic blood pressure of at least 10 mmHg within 3 min of standing or head-up tilt on a tilt table, is a manifestation of sympathetic vasoconstrictor (autonomic) failure (Fig. 27-4). In many (but not all) cases, there is no compensatory increase in heart rate despite hypotension; with partial autonomic failure, heart rate may increase to some degree but is insufficient to maintain cardiac output. A variant of orthostatic hypotension is “delayed” orthostatic hypotension, which occurs beyond 3 min of standing; this may reflect a mild or early form of sympathetic adrenergic dysfunction. In some cases, orthostatic hypotension occurs within 15 s of standing (so-called “initial” orthostatic hypotension), a finding that may reflect a transient mismatch between cardiac output and peripheral vascular resistance and does not represent autonomic failure.
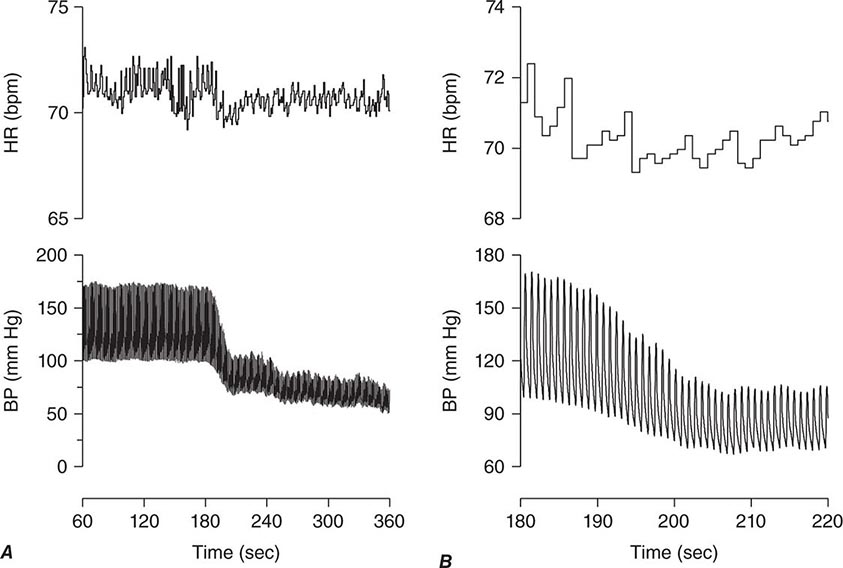
FIGURE 27-4 A. The gradual fall in blood pressure without a compensatory heart rate increase that is characteristic of orthostatic hypotension due to autonomic failure. Blood pressure and heart rate are shown over 5 min (from 60 to 360 s) of an upright tilt on a tilt table. B. The same tracing expanded to show 40 s of the episode (from 180 to 220 s). BP, blood pressure; bpm, beats per minute; HR, heart rate.
Characteristic symptoms of orthostatic hypotension include light-headedness, dizziness, and presyncope (near-faintness) occurring in response to sudden postural change. However, symptoms may be absent or nonspecific, such as generalized weakness, fatigue, cognitive slowing, leg buckling, or headache. Visual blurring may occur, likely due to retinal or occipital lobe ischemia. Neck pain, typically in the suboccipital, posterior cervical, and shoulder region (the “coat-hanger headache”), most likely due to neck muscle ischemia, may be the only symptom. Patients may report orthostatic dyspnea (thought to reflect ventilation-perfusion mismatch due to inadequate perfusion of ventilated lung apices) or angina (attributed to impaired myocardial perfusion even with normal coronary arteries). Symptoms may be exacerbated by exertion, prolonged standing, increased ambient temperature, or meals. Syncope is usually preceded by warning symptoms, but may occur suddenly, suggesting the possibility of a seizure or cardiac cause.
Supine hypertension is common in patients with orthostatic hypotension due to autonomic failure, affecting over 50% of patients in some series. Orthostatic hypotension may present after initiation of therapy for hypertension, and supine hypertension may follow treatment of orthostatic hypotension. However, in other cases, the association of the two conditions is unrelated to therapy; it may in part be explained by baroreflex dysfunction in the presence of residual sympathetic outflow, particularly in patients with central autonomic degeneration.
Causes of Neurogenic Orthostatic Hypotension Causes of neurogenic orthostatic hypotension include central and peripheral autonomic nervous system dysfunction (Chap. 454). Autonomic dysfunction of other organ systems (including the bladder, bowels, sexual organs, and sudomotor system) of varying severity frequently accompanies orthostatic hypotension in these disorders (Table 27-2).
The primary autonomic degenerative disorders are multiple system atrophy (the Shy-Drager syndrome; Chap. 454), Parkinson’s disease (Chap. 449), dementia with Lewy bodies (Chap. 448), and pure autonomic failure (Chap. 454). These are often grouped together as “synucleinopathies” due to the presence of alpha-synuclein, a small protein that precipitates predominantly in the cytoplasm of neurons in the Lewy body disorders (Parkinson’s disease, dementia with Lewy bodies, and pure autonomic failure) and in the glia in multiple system atrophy.
Peripheral autonomic dysfunction may also accompany small-fiber peripheral neuropathies such as those seen in diabetes, amyloid, immune-mediated neuropathies, hereditary sensory and autonomic neuropathies (HSAN; particularly HSAN type III, familial dysautonomia), and inflammatory neuropathies (Chaps. 459 and 460). Less frequently, orthostatic hypotension is associated with the peripheral neuropathies that accompany vitamin B12 deficiency, neurotoxic exposure, HIV and other infections, and porphyria.
Patients with autonomic failure and the elderly are susceptible to falls in blood pressure associated with meals. The magnitude of the blood pressure fall is exacerbated by large meals, meals high in carbohydrate, and alcohol intake. The mechanism of postprandial syncope is not fully elucidated.
Orthostatic hypotension is often iatrogenic. Drugs from several classes may lower peripheral resistance (e.g., alpha-adrenoreceptor antagonists used to treat hypertension and prostatic hypertrophy; antihypertensive agents of several classes; nitrates and other vasodilators; tricyclic agents and phenothiazines). Iatrogenic volume depletion due to diuresis and volume depletion due to medical causes (hemorrhage, vomiting, diarrhea, or decreased fluid intake) may also result in decreased effective circulatory volume, orthostatic hypotension, and syncope.
CARDIAC SYNCOPE
Cardiac (or cardiovascular) syncope is caused by arrhythmias and structural heart disease. These may occur in combination because structural disease renders the heart more vulnerable to abnormal electrical activity.
Arrhythmias Bradyarrhythmias that cause syncope include those due to severe sinus node dysfunction (e.g., sinus arrest or sinoatrial block) and atrioventricular (AV) block (e.g., Mobitz type II, high-grade, and complete AV block). The bradyarrhythmias due to sinus node dysfunction are often associated with an atrial tachyarrhythmia, a disorder known as the tachycardia-bradycardia syndrome. A prolonged pause following the termination of a tachycardic episode is a frequent cause of syncope in patients with the tachycardia-bradycardia syndrome. Medications of several classes may also cause bradyarrhythmias of sufficient severity to cause syncope. Syncope due to bradycardia or asystole is referred to as a Stokes-Adams attack.
Ventricular tachyarrhythmias frequently cause syncope. The likelihood of syncope with ventricular tachycardia is in part dependent on the ventricular rate; rates below 200 beats/min are less likely to cause syncope. The compromised hemodynamic function during ventricular tachycardia is caused by ineffective ventricular contraction, reduced diastolic filling due to abbreviated filling periods, loss of AV synchrony, and concurrent myocardial ischemia.
Several disorders associated with cardiac electrophysiologic instability and arrhythmogenesis are due to mutations in ion channel subunit genes. These include the long QT syndrome, Brugada syndrome, and catecholaminergic polymorphic ventricular tachycardia. The long QT syndrome is a genetically heterogeneous disorder associated with prolonged cardiac repolarization and a predisposition to ventricular arrhythmias. Syncope and sudden death in patients with long QT syndrome result from a unique polymorphic ventricular tachycardia called torsades des pointes that degenerates into ventricular fibrillation. The long QT syndrome has been linked to genes encoding K+ channel α-subunits, K+ channel β-subunits, voltage-gated Na+ channel, and a scaffolding protein, ankyrin B (ANK2). Brugada syndrome is characterized by idiopathic ventricular fibrillation in association with right ventricular electrocardiogram (ECG) abnormalities without structural heart disease. This disorder is also genetically heterogeneous, although it is most frequently linked to mutations in the Na+ channel α-subunit, SCN5A. Catecholaminergic polymorphic tachycardia is an inherited, genetically heterogeneous disorder associated with exercise- or stress-induced ventricular arrhythmias, syncope, or sudden death. Acquired QT interval prolongation, most commonly due to drugs, may also result in ventricular arrhythmias and syncope. These disorders are discussed in detail in Chap. 277.
Structural Disease Structural heart disease (e.g., valvular disease, myocardial ischemia, hypertrophic and other cardiomyopathies, cardiac masses such as atrial myxoma, and pericardial effusions) may lead to syncope by compromising cardiac output. Structural disease may also contribute to other pathophysiologic mechanisms of syncope. For example, cardiac structural disease may predispose to arrhythmogenesis; aggressive treatment of cardiac failure with diuretics and/or vasodilators may lead to orthostatic hypotension; and inappropriate reflex vasodilation may occur with structural disorders such as aortic stenosis and hypertrophic cardiomyopathy, possibly provoked by increased ventricular contractility.
28 |
Dizziness and Vertigo |
Dizziness is an imprecise symptom used to describe a variety of sensations that include vertigo, light-headedness, faintness, and imbalance. When used to describe a sense of spinning or other motion, dizziness is designated as vertigo. Vertigo may be physiologic, occurring during or after a sustained head rotation, or it may be pathologic, due to vestibular dysfunction. The term light-headedness is commonly applied to presyncopal sensations due to brain hypoperfusion but also may refer to disequilibrium and imbalance. A challenge to diagnosis is that patients often have difficulty distinguishing among these various symptoms, and the words they choose do not reliably indicate the underlying etiology.
There are a number of potential causes of dizziness. Vascular disorders cause presyncopal dizziness as a result of cardiac dysrhythmia, orthostatic hypotension, medication effects, or other causes. Such presyncopal sensations vary in duration; they may increase in severity until loss of consciousness occurs, or they may resolve before loss of consciousness if the cerebral ischemia is corrected. Faintness and syncope, which are discussed in detail in Chap. 27, should always be considered when one is evaluating patients with brief episodes of dizziness or dizziness that occurs with upright posture.
Vestibular causes of dizziness (vertigo or imbalance) may be due to peripheral lesions that affect the labyrinths or vestibular nerves or to involvement of the central vestibular pathways. They may be paroxysmal or due to a fixed unilateral or bilateral vestibular deficit. Acute unilateral lesions cause vertigo due to a sudden imbalance in vestibular inputs from the two labyrinths. Bilateral lesions cause imbalance and instability of vision (oscillopsia) when the head moves. Other causes of dizziness include nonvestibular imbalance and gait disorders (e.g., loss of proprioception from sensory neuropathy, parkinsonism) and anxiety.
When evaluating patients with dizziness, questions to consider include the following: (1) Is it dangerous (e.g., arrhythmia, transient ischemic attack/stroke)? (2) Is it vestibular? (3) If vestibular, is it peripheral or central? A careful history and examination often provide sufficient information to answer these questions and determine whether additional studies or referral to a specialist is necessary.
DIFFERENTIAL DIAGNOSIS AND TREATMENT
Treatment of vestibular symptoms should be driven by the underlying diagnosis. Simply treating dizziness with vestibular suppressant medications is often not helpful and may make the symptoms worse and prolong recovery. The diagnostic and specific treatment approaches for the most commonly encountered vestibular disorders are discussed below.
ACUTE PROLONGED VERTIGO (VESTIBULAR NEURITIS)
An acute unilateral vestibular lesion causes constant vertigo, nausea, vomiting, oscillopsia (motion of the visual scene), and imbalance. These symptoms are due to a sudden asymmetry of inputs from the two labyrinths or in their central connections, simulating a continuous rotation of the head. Unlike BPPV, continuous vertigo persists even when the head remains still.
When a patient presents with an acute vestibular syndrome, the most important question is whether the lesion is central (e.g., a cerebellar or brainstem infarct or hemorrhage), which may be life-threatening, or peripheral, affecting the vestibular nerve or labyrinth (vestibular neuritis). Attention should be given to any symptoms or signs that point to central dysfunction (diplopia, weakness or numbness, dysarthria). The pattern of spontaneous nystagmus, if present, may be helpful (Table 28-1). If the head impulse test is normal, an acute peripheral vestibular lesion is unlikely. A central lesion cannot always be excluded with certainty based on symptoms and examination alone; thus, older patients with vascular risk factors who present with an acute vestibular syndrome should be evaluated for the possibility of stroke even when there are no specific findings that indicate a central lesion.
Most patients with vestibular neuritis recover spontaneously, but glucocorticoids can improve outcome if administered within 3 days of symptom onset. Antiviral medications are of no proven benefit and are not typically given unless there is evidence to suggest herpes zoster oticus (Ramsay Hunt syndrome). Vestibular suppressant medications may reduce acute symptoms but should be avoided after the first several days because they may impede central compensation and recovery. Patients should be encouraged to resume a normal level of activity as soon as possible, and directed vestibular rehabilitation therapy may accelerate improvement.
BENIGN PAROXYSMAL POSITIONAL VERTIGO
BPPV is a common cause of recurrent vertigo. Episodes are brief (<1 min and typically 15–20 s) and are always provoked by changes in head position relative to gravity, such as lying down, rolling over in bed, rising from a supine position, and extending the head to look upward. The attacks are caused by free-floating otoconia (calcium carbonate crystals) that have been dislodged from the utricular macula and have moved into one of the semicircular canals, usually the posterior canal. When head position changes, gravity causes the otoconia to move within the canal, producing vertigo and nystagmus. With posterior canal BPPV, the nystagmus beats upward and torsionally (the upper poles of the eyes beat toward the affected lower ear). Less commonly, the otoconia enter the horizontal canal, resulting in a horizontal nystagmus when the patient is lying with either ear down. Superior (also called anterior) canal involvement is rare. BPPV is treated with repositioning maneuvers that use gravity to remove the otoconia from the semicircular canal. For posterior canal BPPV, the Epley maneuver (Fig. 28-1) is the most commonly used procedure. For more refractory cases of BPPV, patients can be taught a variant of this maneuver that they can perform alone at home. A demonstration of the Epley maneuver is available online (http://www.dizziness-and-balance.com/disorders/bppv/bppv.html).
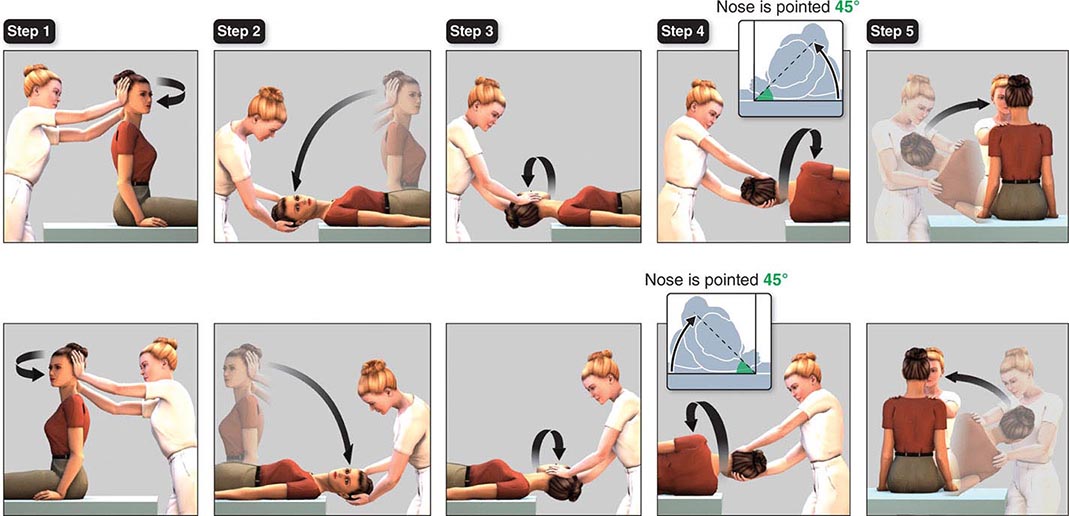
FIGURE 28-1 Modified Epley maneuver for treatment of benign paroxysmal positional vertigo of the right (top panels) and left (bottom panels) posterior semicircular canals. Step 1. With the patient seated, turn the head 45 degrees toward the affected ear. Step 2. Keeping the head turned, lower the patient to the head-hanging position and hold for at least 30 s and until nystagmus disappears. Step 3. Without lifting the head, turn it 90 degrees toward the other side. Hold for another 30 s. Step 4. Rotate the patient onto her side while turning the head another 90 degrees, so that the nose is pointed down 45 degrees. Hold again for 30 s. Step 5. Have the patient sit up on the side of the table. After a brief rest, the maneuver should be repeated to confirm successful treatment. (Figure adapted from http://www.dizziness-and-balance.com/disorders/bppv/movies/Epley-480×640.avi.)
VESTIBULAR MIGRAINE
Vestibular symptoms occur frequently in migraineurs, sometimes as a headache aura but usually independent of headache. The duration of vertigo may be from minutes to hours, and some patients also experience more prolonged periods of disequilibrium (lasting days to weeks). Motion sensitivity and sensitivity to visual motion (e.g., movies) are common in patients with vestibular migraine. Although data from controlled studies are generally lacking, vestibular migraine typically is treated with medications that are used for prophylaxis of migraine headaches. Antiemetics may be helpful to relieve symptoms at the time of an attack.
MÉNIÈRE’S DISEASE
Attacks of Ménière’s disease consist of vertigo and hearing loss, as well as pain, pressure, and/or fullness in the affected ear. The low-frequency hearing loss and aural symptoms are key features that distinguish Ménière’s disease from other peripheral vestibulopathies and from vestibular migraine. Audiometry at the time of an attack shows a characteristic asymmetric low-frequency hearing loss; hearing commonly improves between attacks, although permanent hearing loss may eventually occur. Ménière’s disease is thought to be due to excess fluid (endolymph) in the inner ear; hence the term endolymphatic hydrops. Patients suspected of having Ménière’s disease should be referred to an otolaryngologist for further evaluation. Diuretics and sodium restriction are the initial treatments. If attacks persist, injections of gentamicin into the middle ear are typically the next line of therapy. Full ablative procedures (vestibular nerve section, labyrinthectomy) are seldom required.
VESTIBULAR SCHWANNOMA
Vestibular schwannomas (sometimes termed acoustic neuromas) and other tumors at the cerebellopontine angle cause slowly progressive unilateral sensorineural hearing loss and vestibular hypofunction. These patients typically do not have vertigo, because the gradual vestibular deficit is compensated centrally as it develops. The diagnosis often is not made until there is sufficient hearing loss to be noticed. The examination will show a deficient response to the head impulse test when the head is rotated toward the affected side. As noted above, patients with unexplained unilateral sensorineural hearing loss or vestibular hypofunction require MRI of the internal auditory canals to look for a schwannoma.
BILATERAL VESTIBULAR HYPOFUNCTION
Patients with bilateral loss of vestibular function also typically do not have vertigo, because vestibular function is lost on both sides simultaneously, and there is no asymmetry of vestibular input. Symptoms include loss of balance, particularly in the dark, where vestibular input is most critical, and oscillopsia during head movement, such as while walking or riding in a car. Bilateral vestibular hypofunction may be (1) idiopathic and progressive, (2) part of a neurodegenerative disorder, or (3) iatrogenic, due to medication ototoxicity (most commonly gentamicin or other aminoglycoside antibiotics). Other causes include bilateral vestibular schwannomas (neurofibromatosis type 2), autoimmune disease, superficial siderosis, and meningeal-based infection or tumor. It also may occur in patients with peripheral polyneuropathy; in these patients, both vestibular loss and impaired proprioception may contribute to poor balance. Finally, unilateral processes such as vestibular neuritis and Ménière’s disease may involve both ears sequentially, resulting in bilateral vestibulopathy.
Examination findings include diminished dynamic visual acuity (see above) due to loss of stable vision when the head is moving, abnormal head impulse responses in both directions, and a Romberg sign. Responses to caloric testing are reduced. Patients with bilateral vestibular hypofunction should be referred for vestibular rehabilitation therapy. Vestibular suppressant medications should not be used, as they will increase the imbalance. Evaluation by a neurologist is important not only to confirm the diagnosis but also to consider any other associated neurologic abnormalities that may clarify the etiology.
CENTRAL VESTIBULAR DISORDERS
Central lesions causing vertigo typically involve vestibular pathways in the brainstem and/or cerebellum. They may be due to discrete lesions, such as from ischemic or hemorrhagic stroke (Chap. 446), demyelination (Chap. 458), or tumors (Chap. 118), or they may be due to neurodegenerative conditions that include the vestibulocerebellum (Chap. 448). Subacute cerebellar degeneration may be due to immune, including paraneoplastic, processes (Chaps. 122 and 450). Table 28-1 outlines important features of the history and examination that help to identify central vestibular disorders. Acute central vertigo is a medical emergency, due to the possibility of life-threatening stroke or hemorrhage. All patients with suspected central vestibular disorders should undergo brain MRI, and the patient should be referred for full neurologic evaluation.
PSYCHOSOMATIC DIZZINESS/VERTIGO
Psychological factors play an important role in chronic dizziness. First, dizziness may be a somatic manifestation of a psychiatric condition such as major depression, anxiety, or panic disorder (Chap. 465e). Second, patients may develop anxiety and autonomic symptoms as a consequence or comorbidity of an independent vestibular disorder. One particular form of this has been termed variously phobic postural vertigo, psychophysiologic vertigo, or chronic subjective dizziness. These patients have a chronic feeling (months or longer) of dizziness and disequilibrium, an increased sensitivity to self-motion and visual motion (e.g., movies), and a particular intensification of symptoms when moving through complex visual environments such as supermarkets (visual vertigo). Although there may be a past history of an acute vestibular disorder (e.g., vestibular neuritis), the neurootologic examination and vestibular testing are normal or indicative of a compensated vestibular deficit, indicating that the ongoing subjective dizziness cannot be explained by a primary vestibular disorder. Anxiety disorders are particularly common in patients with chronic dizziness and contribute substantially to the morbidity. Thus, treatment with antianxiety medications (selective serotonin reuptake inhibitors [SSRIs]) and cognitive-behavioral therapy may be helpful. Vestibular rehabilitation therapy is also sometimes beneficial. Vestibular suppressant medications generally should be avoided. This condition should be suspected when the patient states, “My dizziness is so bad, I’m afraid to leave my house” (agoraphobia).
|
TREATMENT |
VERTIGO |
Table 28-2 provides a list of commonly used medications for suppression of vertigo. As noted, these medications should be reserved for short-term control of active vertigo, such as during the first few days of acute vestibular neuritis, or for acute attacks of Ménière’s disease. They are less helpful for chronic dizziness and, as previously stated, may hinder central compensation. An exception is that benzodiazepines may attenuate psychosomatic dizziness and the associated anxiety, although SSRIs are generally preferable in such patients.
|
TREATMENT OF VERTIGO |
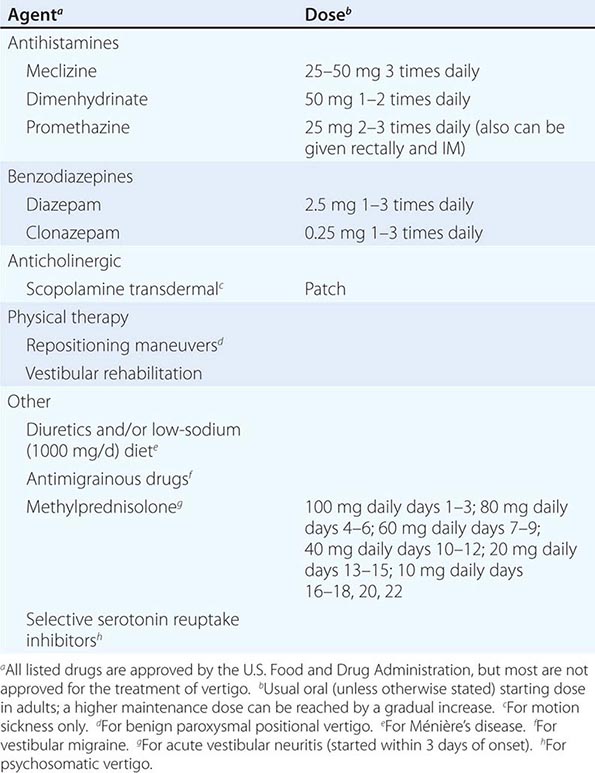
Vestibular rehabilitation therapy promotes central adaptation processes that compensate for vestibular loss and also may help habituate motion sensitivity and other symptoms of psychosomatic dizziness. The general approach is to use a graded series of exercises that progressively challenge gaze stabilization and balance.
29 |
Fatigue |
Fatigue is one of the most common symptoms in clinical medicine. It is a prominent manifestation of a number of systemic, neurologic, and psychiatric syndromes, although a precise cause will not be identified in a substantial minority of patients. Fatigue refers to an inherently subjective human experience of physical and mental weariness, sluggishness, and exhaustion. In the context of clinical medicine, fatigue is most typically and practically defined as difficulty initiating or maintaining voluntary mental or physical activity. Nearly everyone who has ever been ill with a self-limited infection has experienced this near-universal symptomatology, and fatigue is usually brought to medical attention only when it is either of unclear cause or the severity is out of proportion with what would be expected for the associated trigger. Fatigue should be distinguished from muscle weakness, a reduction of neuromuscular power (Chap. 30); most patients complaining of fatigue are not truly weak when direct muscle power is tested. By definition, fatigue is also distinct from somnolence and dyspnea on exertion, although patients may use the word fatigue to describe those two symptoms. The task facing clinicians when a patient presents with fatigue is to identify an underlying cause if one exists and to develop a therapeutic alliance, the goal of which is to spare patients expensive and fruitless diagnostic workups and steer them toward effective therapy.
EPIDEMIOLOGY AND GLOBAL CONSIDERATIONS
![]() Variability in the definitions of fatigue and the survey instruments used in different studies makes it difficult to arrive at precise figures about the global burden of fatigue. The point prevalence of fatigue was 6.7% and the lifetime prevalence was 25% in a large National Institute of Mental Health survey of the U.S. general population. In primary care clinics in Europe and the United States, between 10 and 25% of patients surveyed endorsed symptoms of prolonged (present for >1 month) or chronic (present for >6 months) fatigue, but fatigue was the primary reason for seeking medical attention in only a minority of patients. In a community survey of women in India, 12% reported chronic fatigue. By contrast, the prevalence of chronic fatigue syndrome, as defined by the U.S. Centers for Disease Control and Prevention, is low (Chap. 464e).
Variability in the definitions of fatigue and the survey instruments used in different studies makes it difficult to arrive at precise figures about the global burden of fatigue. The point prevalence of fatigue was 6.7% and the lifetime prevalence was 25% in a large National Institute of Mental Health survey of the U.S. general population. In primary care clinics in Europe and the United States, between 10 and 25% of patients surveyed endorsed symptoms of prolonged (present for >1 month) or chronic (present for >6 months) fatigue, but fatigue was the primary reason for seeking medical attention in only a minority of patients. In a community survey of women in India, 12% reported chronic fatigue. By contrast, the prevalence of chronic fatigue syndrome, as defined by the U.S. Centers for Disease Control and Prevention, is low (Chap. 464e).
DIFFERENTIAL DIAGNOSIS
Psychiatric Disease Fatigue is a common somatic manifestation of many major psychiatric syndromes, including depression, anxiety, and somatoform disorders. Psychiatric symptoms are reported in more than three-quarters of patients with unexplained chronic fatigue. Even in patients with systemic or neurologic syndromes in which fatigue is independently recognized as a manifestation of disease, comorbid psychiatric symptoms or disease may still be an important source of interaction.
Neurologic Disease Patients complaining of fatigue often say they feel weak, but upon careful examination, objective muscle weakness is rarely discernible. If found, muscle weakness must then be localized to the central nervous system, peripheral nervous system, neuromuscular junction, or muscle and the appropriate follow-up studies obtained (Chap. 30). Fatigability of muscle power is a cardinal manifestation of some neuromuscular disorders such as myasthenia gravis and can be distinguished from fatigue by finding clinically apparent diminution of the amount of force that a muscle generates upon repeated contraction (Chap. 461). Fatigue is one of the most common and bothersome symptoms reported in multiple sclerosis (MS) (Chap. 458), affecting nearly 90% of patients; fatigue in MS can persist between MS attacks and does not necessarily correlate with magnetic resonance imaging (MRI) disease activity. Fatigue is also increasingly identified as a troublesome feature of many other neurodegenerative diseases, including Parkinson’s disease, central dysautonomias, and amyotrophic lateral sclerosis. Poststroke fatigue is a well-described but poorly understood entity with a widely varying prevalence. Episodic fatigue can be a premonitory symptom of migraine. Fatigue is also a frequent result of traumatic brain injury, often occurring in association with depression and sleep disorders.
Sleep Disorders Obstructive sleep apnea is an important cause of excessive daytime sleepiness in association with fatigue and should be investigated using overnight polysomnography, particularly in those with prominent snoring, obesity, or other predictors of obstructive sleep apnea (Chap. 319). Whether the cumulative sleep deprivation that is common in modern society contributes to clinically apparent fatigue is not known (Chap. 38).
Endocrine Disorders Fatigue, sometimes in association with true muscle weakness, can be a heralding symptom of hypothyroidism, particularly in the context of hair loss, dry skin, cold intolerance, constipation, and weight gain. Fatigue in association with heat intolerance, sweating, and palpitations is typical of hyperthyroidism. Adrenal insufficiency can also manifest with unexplained fatigue as a primary or prominent symptom, often in association with anorexia, weight loss, nausea, myalgias, and arthralgias; hyponatremia and hyperkalemia may be present at time of diagnosis. Mild hypercalcemia can cause fatigue, which may be relatively vague, whereas severe hypercalcemia can lead to lethargy, stupor, and coma. Both hypoglycemia and hyperglycemia can cause lethargy, often in association with confusion; chronic diabetes, particularly type 1 diabetes, is also associated with fatigue independent of glucose levels. Fatigue may also accompany Cushing’s disease, hypoaldosteronism, and hypogonadism.
Liver and Kidney Disease Both chronic liver failure and chronic kidney disease can cause fatigue. Over 80% of hemodialysis patients complain of fatigue, which makes this one of the most common patient-reported symptoms in chronic kidney disease.
Obesity Obesity is associated with fatigue and sleepiness independent of the presence of obstructive sleep apnea. Obese patients undergoing bariatric surgery experience improvement in daytime sleepiness sooner than would be expected if the improvement were solely the result of weight loss and resolution of sleep apnea. A number of other factors common in obese patients are likely contributors as well, including depression, physical inactivity, and diabetes.
Malnutrition Although fatigue can be a presenting feature of malnutrition, nutritional status may also be an important comorbidity and contributor to fatigue in other chronic illnesses, including cancer-associated fatigue.
Infection Both acute and chronic infections commonly lead to fatigue as part of the broader infectious syndrome. Evaluation for undiagnosed infection as the cause of unexplained fatigue, and particularly prolonged or chronic fatigue, should be guided by the history, physical examination, and infectious risk factors, with particular attention to risk for tuberculosis, HIV, chronic hepatitis B and C, and endocarditis. Infectious mononucleosis may cause prolonged fatigue that persists for weeks to months following the acute illness, but infection with the Epstein-Barr virus is only very rarely the cause of unexplained chronic fatigue.
Drugs Many medications, drug use, drug withdrawal, and chronic alcohol use can all lead to fatigue. Medications that are more likely to be causative in this context include antidepressants, antipsychotics, anxiolytics, opiates, antispasticity agents, antiseizure agents, and beta blockers.
Cardiovascular and Pulmonary Fatigue is one of the most taxing patient-reported symptoms of congestive heart failure and chronic obstructive pulmonary disease and negatively affects quality of life.
Malignancy Fatigue, particularly in association with unexplained unintended weight loss, can be a sign of occult malignancy, but this is only rarely identified as causative in patients with unexplained chronic fatigue in the absence of other telltale signs or symptoms. Cancer-related fatigue is experienced by 40% of patients at time of diagnosis and greater than 80% of patients later in the disease course.
Hematologic Chronic or progressive anemia may present with fatigue, sometimes in association with exertional tachycardia and breathlessness. Anemia may also contribute to fatigue in chronic illness. Low serum ferritin in the absence of anemia may also cause fatigue that is reversible with iron replacement.
Systemic Inflammatory/Rheumatologic Disorders Fatigue is a prominent complaint in many chronic inflammatory disorders, including systemic lupus erythematosus, polymyalgia rheumatica, rheumatoid arthritis, inflammatory bowel disease, antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis, sarcoidosis, and Sjögren’s syndrome, but is not usually an isolated symptom.
Pregnancy Fatigue is very commonly reported by women during all stages of pregnancy and postpartum.
Disorders of Unclear Cause Chronic fatigue syndrome (Chap. 464e) and fibromyalgia (Chap. 396) incorporate chronic fatigue as part of the syndromic definition when present in association with a number of other inclusion and exclusion criteria, as discussed in detail in their respective chapters. The pathophysiology of each is unknown. Idiopathic chronic fatigue is used to describe the syndrome of unexplained chronic fatigue in the absence of enough additional clinical features to meet the diagnostic criteria for chronic fatigue syndrome.
PROGNOSIS
Acute fatigue significant enough to require medical evaluation is more likely to lead to an identifiable medical, neurologic, or psychiatric cause than unexplained chronic fatigue. Evaluation of unexplained chronic fatigue most commonly leads to diagnosis of a psychiatric condition or remains unexplained. Identification of a previously undiagnosed serious or life-threatening culprit etiology is rare on longitudinal follow-up in patients with unexplained chronic fatigue. Complete resolution of unexplained chronic fatigue is uncommon, at least over the short term, but multidisciplinary treatment approaches can lead to symptomatic improvements that can substantially improve quality of life.
30 |
Neurologic Causes of Weakness and Paralysis |
Normal motor function involves integrated muscle activity that is modulated by the activity of the cerebral cortex, basal ganglia, cerebellum, red nucleus, brainstem reticular formation, lateral vestibular nucleus, and spinal cord. Motor system dysfunction leads to weakness or paralysis, discussed in this chapter, or to ataxia (Chap. 450) or abnormal movements (Chap. 449). Weakness is a reduction in the power that can be exerted by one or more muscles. It must be distinguished from increased fatigability (i.e., the inability to sustain the performance of an activity that should be normal for a person of the same age, sex, and size), limitation in function due to pain or articular stiffness, or impaired motor activity because severe proprioceptive sensory loss prevents adequate feedback information about the direction and power of movements. It is also distinct from bradykinesia (in which increased time is required for full power to be exerted) and apraxia, a disorder of planning and initiating a skilled or learned movement unrelated to a significant motor or sensory deficit (Chap. 36).
Paralysis or the suffix “-plegia” indicates weakness so severe that a muscle cannot be contracted at all, whereas paresis refers to less severe weakness. The prefix “hemi-” refers to one-half of the body, “para-” to both legs, and “quadri-” to all four limbs.
The distribution of weakness helps to localize the underlying lesion. Weakness from involvement of upper motor neurons occurs particularly in the extensors and abductors of the upper limb and the flexors of the lower limb. Lower motor neuron weakness depends on whether involvement is at the level of the anterior horn cells, nerve root, limb plexus, or peripheral nerve—only muscles supplied by the affected structure are weak. Myopathic weakness is generally most marked in proximal muscles. Weakness from impaired neuromuscular transmission has no specific pattern of involvement.
Weakness often is accompanied by other neurologic abnormalities that help indicate the site of the responsible lesion (Table 30-1).
|
SIGNS THAT DISTINGUISH THE ORIGIN OF WEAKNESS |
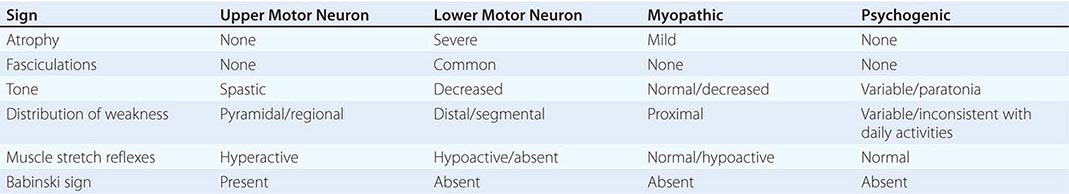
Tone is the resistance of a muscle to passive stretch. Increased tone may be of several types. Spasticity is the increase in tone associated with disease of upper motor neurons. It is velocity-dependent, has a sudden release after reaching a maximum (the “clasp-knife” phenomenon), and predominantly affects the antigravity muscles (i.e., upper-limb flexors and lower-limb extensors). Rigidity is hypertonia that is present throughout the range of motion (a “lead pipe” or “plastic” stiffness) and affects flexors and extensors equally; it sometimes has a cogwheel quality that is enhanced by voluntary movement of the contralateral limb (reinforcement). Rigidity occurs with certain extrapyramidal disorders, such as Parkinson’s disease. Paratonia (or gegenhalten) is increased tone that varies irregularly in a manner seemingly related to the degree of relaxation, is present throughout the range of motion, and affects flexors and extensors equally; it usually results from disease of the frontal lobes. Weakness with decreased tone (flaccidity) or normal tone occurs with disorders of motor units. A motor unit consists of a single lower motor neuron and all the muscle fibers that it innervates.
Muscle bulk generally is not affected by upper motor neuron lesions, although mild disuse atrophy eventually may occur. By contrast, atrophy is often conspicuous when a lower motor neuron lesion is responsible for weakness and also may occur with advanced muscle disease.
Muscle stretch (tendon) reflexes are usually increased with upper motor neuron lesions, but may be decreased or absent for a variable period immediately after onset of an acute lesion. Hyperreflexia is usually—but not invariably—accompanied by loss of cutaneous reflexes (such as superficial abdominals; Chap. 437) and, in particular, by an extensor plantar (Babinski) response. The muscle stretch reflexes are depressed with lower motor neuron lesions directly involving specific reflex arcs. They generally are preserved in patients with myopathic weakness except in advanced stages, when they sometimes are attenuated. In disorders of the neuromuscular junction, reflex responses may be affected by preceding voluntary activity of affected muscles; such activity may lead to enhancement of initially depressed reflexes in Lambert-Eaton myasthenic syndrome and, conversely, to depression of initially normal reflexes in myasthenia gravis (Chap. 461).
The distinction of neuropathic (lower motor neuron) from myopathic weakness is sometimes difficult clinically, although distal weakness is likely to be neuropathic, and symmetric proximal weakness myopathic. Fasciculations (visible or palpable twitch within a muscle due to the spontaneous discharge of a motor unit) and early atrophy indicate that weakness is neuropathic.
PATHOGENESIS
Upper Motor Neuron Weakness Lesions of the upper motor neurons or their descending axons to the spinal cord (Fig. 30-1) produce weakness through decreased activation of lower motor neurons. In general, distal muscle groups are affected more severely than proximal ones, and axial movements are spared unless the lesion is severe and bilateral. Spasticity is typical but may not be present acutely. Rapid repetitive movements are slowed and coarse, but normal rhythmicity is maintained. With corticobulbar involvement, weakness occurs in the lower face and tongue; extraocular, upper facial, pharyngeal, and jaw muscles are typically spared. Bilateral corticobulbar lesions produce a pseudobulbar palsy: dysarthria, dysphagia, dysphonia, and emotional lability accompany bilateral facial weakness and a brisk jaw jerk. Electromyogram (EMG) (Chap. 442e) shows that with weakness of the upper motor neuron type, motor units have a diminished maximal discharge frequency.
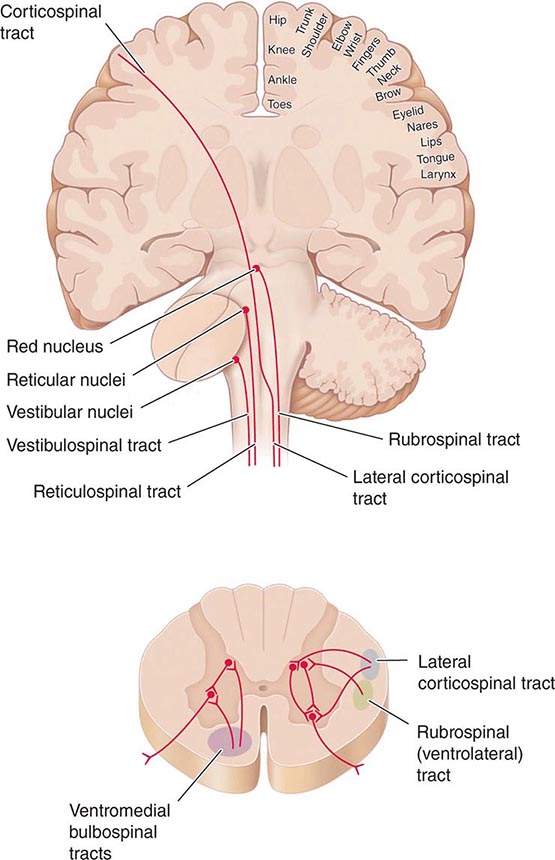
FIGURE 30-1 The corticospinal and bulbospinal upper motor neuron pathways. Upper motor neurons have their cell bodies in layer V of the primary motor cortex (the precentral gyrus, or Brodmann’s area 4) and in the premotor and supplemental motor cortex (area 6). The upper motor neurons in the primary motor cortex are somatotopically organized (right side of figure).
Axons of the upper motor neurons descend through the subcortical white matter and the posterior limb of the internal capsule. Axons of the pyramidal or corticospinal system descend through the brainstem in the cerebral peduncle of the midbrain, the basis pontis, and the medullary pyramids. At the cervicomedullary junction, most corticospinal axons decussate into the contralateral corticospinal tract of the lateral spinal cord, but 10–30% remain ipsilateral in the anterior spinal cord. Corticospinal neurons synapse on premotor interneurons, but some—especially in the cervical enlargement and those connecting with motor neurons to distal limb muscles—make direct monosynaptic connections with lower motor neurons. They innervate most densely the lower motor neurons of hand muscles and are involved in the execution of learned, fine movements. Corticobulbar neurons are similar to corticospinal neurons but innervate brainstem motor nuclei.
Bulbospinal upper motor neurons influence strength and tone but are not part of the pyramidal system. The descending ventromedial bulbospinal pathways originate in the tectum of the midbrain (tectospinal pathway), the vestibular nuclei (vestibulospinal pathway), and the reticular formation (reticulospinal pathway). These pathways influence axial and proximal muscles and are involved in the maintenance of posture and integrated movements of the limbs and trunk. The descending ventrolateral bulbospinal pathways, which originate predominantly in the red nucleus (rubrospinal pathway), facilitate distal limb muscles. The bulbospinal system sometimes is referred to as the extrapyramidal upper motor neuron system. In all figures, nerve cell bodies and axon terminals are shown, respectively, as closed circles and forks.
Lower Motor Neuron Weakness This pattern results from disorders of lower motor neurons in the brainstem motor nuclei and the anterior horn of the spinal cord or from dysfunction of the axons of these neurons as they pass to skeletal muscle (Fig. 30-2). Weakness is due to a decrease in the number of muscle fibers that can be activated through a loss of α motor neurons or disruption of their connections to muscle. Loss of γ motor neurons does not cause weakness but decreases tension on the muscle spindles, which decreases muscle tone and attenuates the stretch reflexes. An absent stretch reflex suggests involvement of spindle afferent fibers.
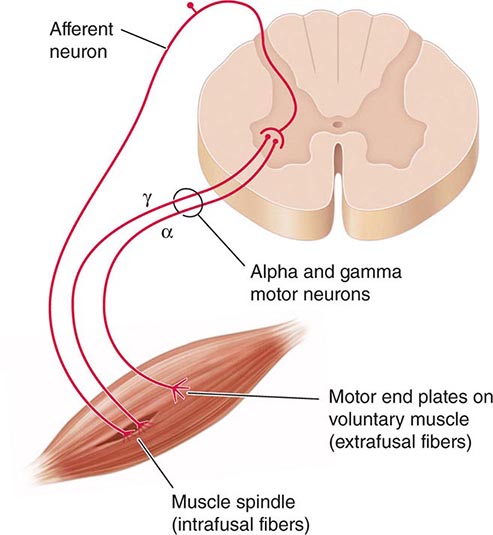
FIGURE 30-2 Lower motor neurons are divided into α and γ types. The larger α motor neurons are more numerous and innervate the extrafusal muscle fibers of the motor unit. Loss of α motor neurons or disruption of their axons produces lower motor neuron weakness. The smaller, less numerous γ motor neurons innervate the intrafusal muscle fibers of the muscle spindle and contribute to normal tone and stretch reflexes. The α motor neuron receives direct excitatory input from corticomotoneurons and primary muscle spindle afferents. The α and γ motor neurons also receive excitatory input from other descending upper motor neuron pathways, segmental sensory inputs, and interneurons. The α motor neurons receive direct inhibition from Renshaw cell interneurons, and other interneurons indirectly inhibit the α and γ motor neurons.
A muscle stretch (tendon) reflex requires the function of all the illustrated structures. A tap on a tendon stretches muscle spindles (which are tonically activated by γ motor neurons) and activates the primary spindle afferent neurons. These neurons stimulate the α motor neurons in the spinal cord, producing a brief muscle contraction, which is the familiar tendon reflex.
When a motor unit becomes diseased, especially in anterior horn cell diseases, it may discharge spontaneously, producing fasciculations. When α motor neurons or their axons degenerate, the denervated muscle fibers also may discharge spontaneously. These single muscle fiber discharges, or fibrillation potentials, cannot be seen but can be recorded with EMG. Weakness leads to delayed or reduced recruitment of motor units, with fewer than normal activated at a particular discharge frequency.
Neuromuscular Junction Weakness Disorders of the neuromuscular junctions produce weakness of variable degree and distribution. The number of muscle fibers that are activated varies over time, depending on the state of rest of the neuromuscular junctions. Strength is influenced by preceding activity of the affected muscle. In myasthenia gravis, for example, sustained or repeated contractions of affected muscle decline in strength despite continuing effort (Chap. 461). Thus, fatigable weakness is suggestive of disorders of the neuromuscular junction, which cause functional loss of muscle fibers due to failure of their activation.
Myopathic Weakness Myopathic weakness is produced by a decrease in the number or contractile force of muscle fibers activated within motor units. With muscular dystrophies, inflammatory myopathies, or myopathies with muscle fiber necrosis, the number of muscle fibers is reduced within many motor units. On EMG, the size of each motor unit action potential is decreased, and motor units must be recruited more rapidly than normal to produce the desired power. Some myopathies produce weakness through loss of contractile force of muscle fibers or through relatively selective involvement of type II (fast) fibers. These myopathies may not affect the size of individual motor unit action potentials and are detected by a discrepancy between the electrical activity and force of a muscle.
Psychogenic Weakness Weakness may occur without a recognizable organic basis. It tends to be variable, inconsistent, and with a pattern of distribution that cannot be explained on a neuroanatomic basis. On formal testing, antagonists may contract when the patient is supposedly activating the agonist muscle. The severity of weakness is out of keeping with the patient’s daily activities.
Hemiparesis Hemiparesis results from an upper motor neuron lesion above the midcervical spinal cord; most such lesions are above the foramen magnum. The presence of other neurologic deficits helps localize the lesion. Thus, language disorders, for example, point to a cortical lesion. Homonymous visual field defects reflect either a cortical or a subcortical hemispheric lesion. A “pure motor” hemiparesis of the face, arm, and leg often is due to a small, discrete lesion in the posterior limb of the internal capsule, cerebral peduncle, or upper pons. Some brainstem lesions produce “crossed paralyses,” consisting of ipsilateral cranial nerve signs and contralateral hemiparesis (Chap. 446). The absence of cranial nerve signs or facial weakness suggests that a hemiparesis is due to a lesion in the high cervical spinal cord, especially if associated with the Brown-Séquard syndrome (Chap. 456).
Acute or episodic hemiparesis usually results from focal structural lesions, particularly rapidly expanding lesions, or an inflammatory process. Subacute hemiparesis that evolves over days or weeks may relate to subdural hematoma, infectious or inflammatory disorders (e.g., cerebral abscess, fungal granuloma or meningitis, parasitic infection, multiple sclerosis, sarcoidosis), or primary and metastatic neoplasms. AIDS may present with subacute hemiparesis due to toxoplasmosis or primary central nervous system (CNS) lymphoma. Chronic hemiparesis that evolves over months usually is due to a neoplasm or vascular malformation, a chronic subdural hematoma, or a degenerative disease.
Investigation of hemiparesis (Fig. 30-3) of acute origin starts with a computed tomography (CT) scan of the brain and laboratory studies. If the CT is normal, or in subacute or chronic cases of hemiparesis, magnetic resonance imaging (MRI) of the brain and/or cervical spine (including the foramen magnum) is performed, depending on the clinical accompaniments.
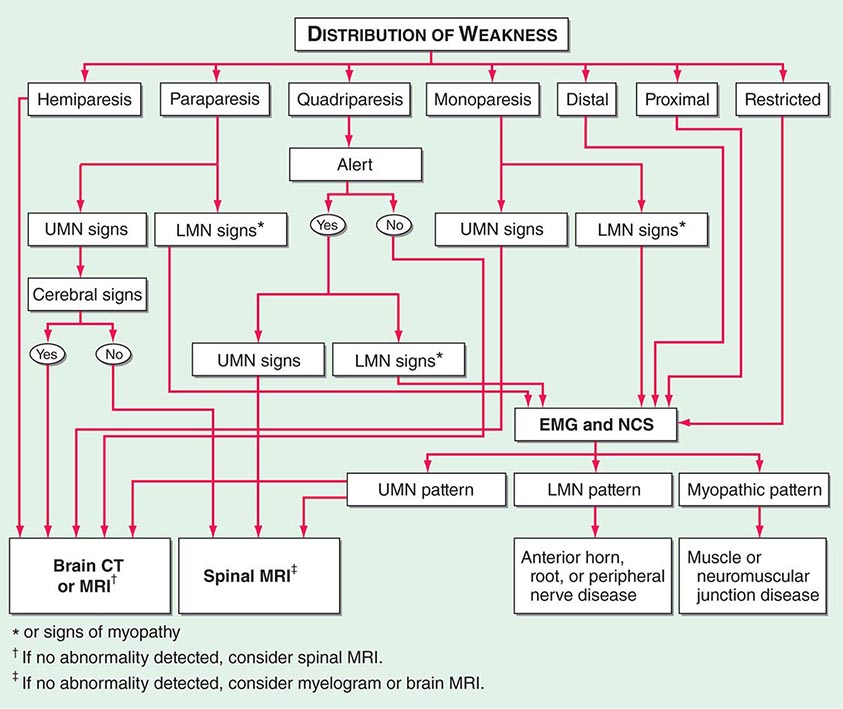
FIGURE 30-3 An algorithm for the initial workup of a patient with weakness. CT, computed tomography; EMG, electromyography; LMN, lower motor neuron; MRI, magnetic resonance imaging; NCS, nerve conduction studies; UMN, upper motor neuron.
Paraparesis Acute paraparesis is caused most commonly by an intraspinal lesion, but its spinal origin may not be recognized initially if the legs are flaccid and areflexic. Usually, however, there is sensory loss in the legs with an upper level on the trunk, a dissociated sensory loss suggestive of a central cord syndrome (Chap. 456), or hyperreflexia in the legs with normal reflexes in the arms. Imaging the spinal cord (Fig. 30-3) may reveal compressive lesions, infarction (proprioception usually is spared), arteriovenous fistulas or other vascular anomalies, or transverse myelitis (Chap. 456).
Diseases of the cerebral hemispheres that produce acute paraparesis include anterior cerebral artery ischemia (shoulder shrug also is affected), superior sagittal sinus or cortical venous thrombosis, and acute hydrocephalus.
Paraparesis may result from a cauda equina syndrome, for example, after trauma to the low back, a midline disk herniation, or an intraspinal tumor; although the sphincters are often affected, hip flexion often is spared, as is sensation over the anterolateral thighs. Rarely, paraparesis is caused by a rapidly evolving anterior horn cell disease (such as poliovirus or West Nile virus infection), peripheral neuropathy (such as Guillain-Barré syndrome; Chap. 460), or myopathy (Chap. 462e).
Subacute or chronic spastic paraparesis is caused by upper motor neuron disease. When associated with lower-limb sensory loss and sphincter involvement, a chronic spinal cord disorder should be considered (Chap. 456). If hemispheric signs are present, a parasagittal meningioma or chronic hydrocephalus is likely. The absence of spasticity in a long-standing paraparesis suggests a lower motor neuron or myopathic etiology.
Investigations typically begin with spinal MRI, but when upper motor neuron signs are associated with drowsiness, confusion, seizures, or other hemispheric signs, brain MRI should also be performed, sometimes as the initial investigation. Electrophysiologic studies are diagnostically helpful when clinical findings suggest an underlying neuromuscular disorder.
Quadriparesis or Generalized Weakness Generalized weakness may be due to disorders of the CNS or the motor unit. Although the terms often are used interchangeably, quadriparesis is commonly used when an upper motor neuron cause is suspected, and generalized weakness is used when a disease of the motor units is likely. Weakness from CNS disorders usually is associated with changes in consciousness or cognition and accompanied by spasticity, hyperreflexia, and sensory disturbances. Most neuromuscular causes of generalized weakness are associated with normal mental function, hypotonia, and hypoactive muscle stretch reflexes. The major causes of intermittent weakness are listed in Table 30-2. A patient with generalized fatigability without objective weakness may have the chronic fatigue syndrome (Chap. 464e).
|
CAUSES OF EPISODIC GENERALIZED WEAKNESS |
ACUTE QUADRIPARESIS Quadriparesis with onset over minutes may result from disorders of upper motor neurons (such as from anoxia, hypotension, brainstem or cervical cord ischemia, trauma, and systemic metabolic abnormalities) or muscle (electrolyte disturbances, certain inborn errors of muscle energy metabolism, toxins, and periodic paralyses). Onset over hours to weeks may, in addition to these disorders, be due to lower motor neuron disorders such as Guillain-Barré syndrome (Chap. 460).
In obtunded patients, evaluation begins with a CT scan of the brain. If upper motor neuron signs are present but the patient is alert, the initial test is usually an MRI of the cervical cord. If weakness is lower motor neuron, myopathic, or uncertain in origin, the clinical approach begins with blood studies to determine the level of muscle enzymes and electrolytes and with EMG and nerve conduction studies.
SUBACUTE OR CHRONIC QUADRIPARESIS Quadriparesis due to upper motor neuron disease may develop over weeks to years from chronic myelopathies, multiple sclerosis, brain or spinal tumors, chronic subdural hematomas, and various metabolic, toxic, and infectious disorders. It may also result from lower motor neuron disease, a chronic neuropathy (in which weakness is often most profound distally), or myopathic weakness (typically proximal).
When quadriparesis develops acutely in obtunded patients, evaluation begins with a CT scan of the brain. If upper motor neuron signs have developed acutely but the patient is alert, the initial test is usually an MRI of the cervical cord. When onset has been gradual, disorders of the cerebral hemispheres, brainstem, and cervical spinal cord can usually be distinguished clinically, and imaging is directed first at the clinically suspected site of pathology. If weakness is lower motor neuron, myopathic, or uncertain in origin, laboratory studies to determine the levels of muscle enzymes and electrolytes, and EMG and nerve conduction studies help to localize the pathologic process.
Monoparesis Monoparesis usually is due to lower motor neuron disease, with or without associated sensory involvement. Upper motor neuron weakness occasionally presents as a monoparesis of distal and nonantigravity muscles. Myopathic weakness rarely is limited to one limb.
ACUTE MONOPARESIS If weakness is predominantly distal and of upper motor neuron type and is not associated with sensory impairment or pain, focal cortical ischemia is likely (Chap. 446); diagnostic possibilities are similar to those for acute hemiparesis. Sensory loss and pain usually accompany acute lower motor neuron weakness; the weakness commonly localizes to a single nerve root or peripheral nerve, but occasionally reflects plexus involvement. If lower motor neuron weakness is likely, evaluation begins with EMG and nerve conduction studies.
SUBACUTE OR CHRONIC MONOPARESIS Weakness and atrophy that develop over weeks or months are usually of lower motor neuron origin. When associated with sensory symptoms, a peripheral cause (nerve, root, or plexus) is likely; otherwise, anterior horn cell disease should be considered. In either case, an electrodiagnostic study is indicated. If weakness is of the upper motor neuron type, a discrete cortical (precentral gyrus) or cord lesion may be responsible, and appropriate imaging is performed.
Distal Weakness Involvement of two or more limbs distally suggests lower motor neuron or peripheral nerve disease. Acute distal lower-limb weakness results occasionally from an acute toxic polyneuropathy or cauda equina syndrome. Distal symmetric weakness usually develops over weeks, months, or years and, when associated with numbness, is due to peripheral neuropathy (Chap. 459). Anterior horn cell disease may begin distally but is typically asymmetric and without accompanying numbness (Chap. 452). Rarely, myopathies present with distal weakness (Chap. 462e). Electrodiagnostic studies help localize the disorder (Fig. 30-3).
Proximal Weakness Myopathy often produces symmetric weakness of the pelvic or shoulder girdle muscles (Chap. 462e). Diseases of the neuromuscular junction, such as myasthenia gravis (Chap. 461), may present with symmetric proximal weakness often associated with ptosis, diplopia, or bulbar weakness and fluctuating in severity during the day. In anterior horn cell disease, proximal weakness is usually asymmetric, but it may be symmetric if familial. Numbness does not occur with any of these diseases. The evaluation usually begins with determination of the serum creatine kinase level and electrophysiologic studies.
Weakness in a Restricted Distribution Weakness may not fit any of these patterns, being limited, for example, to the extraocular, hemifacial, bulbar, or respiratory muscles. If it is unilateral, restricted weakness usually is due to lower motor neuron or peripheral nerve disease, such as in a facial palsy. Weakness of part of a limb is commonly due to a peripheral nerve lesion such as an entrapment neuropathy. Relatively symmetric weakness of extraocular or bulbar muscles frequently is due to a myopathy (Chap. 462e) or neuromuscular junction disorder (Chap. 461). Bilateral facial palsy with areflexia suggests Guillain-Barré syndrome (Chap. 460). Worsening of relatively symmetric weakness with fatigue is characteristic of neuromuscular junction disorders. Asymmetric bulbar weakness usually is due to motor neuron disease. Weakness limited to respiratory muscles is uncommon and usually is due to motor neuron disease, myasthenia gravis, or polymyositis/dermatomyositis (Chap. 388).
31 |
Numbness, Tingling, and Sensory Loss |
Normal somatic sensation reflects a continuous monitoring process, little of which reaches consciousness under ordinary conditions. By contrast, disordered sensation, particularly when experienced as painful, is alarming and dominates the patient’s attention. Physicians should be able to recognize abnormal sensations by how they are described, know their type and likely site of origin, and understand their implications. Pain is considered separately in Chap. 18.
POSITIVE AND NEGATIVE SYMPTOMS
Abnormal sensory symptoms can be divided into two categories: positive and negative. The prototypical positive symptom is tingling (pins and needles); other positive sensory phenomena include itch and altered sensations that are described as pricking, bandlike, lightninglike shooting feelings (lancinations), aching, knifelike, twisting, drawing, pulling, tightening, burning, searing, electrical, or raw feelings. Such symptoms are often painful.
Positive phenomena usually result from trains of impulses generated at sites of lowered threshold or heightened excitability along a peripheral or central sensory pathway. The nature and severity of the abnormal sensation depend on the number, rate, timing, and distribution of ectopic impulses and the type and function of nervous tissue in which they arise. Because positive phenomena represent excessive activity in sensory pathways, they are not necessarily associated with a sensory deficit (loss) on examination.
Negative phenomena represent loss of sensory function and are characterized by diminished or absent feeling that often is experienced as numbness and by abnormal findings on sensory examination. In disorders affecting peripheral sensation, at least one-half the afferent axons innervating a particular site are probably lost or functionless before a sensory deficit can be demonstrated by clinical examination. If the rate of loss is slow, however, lack of cutaneous feeling may be unnoticed by the patient and difficult to demonstrate on examination, even though few sensory fibers are functioning; if it is rapid, both positive and negative phenomena are usually conspicuous. Subclinical degrees of sensory dysfunction may be revealed by sensory nerve conduction studies or somatosensory evoked potentials (Chap. 442e).
Whereas sensory symptoms may be either positive or negative, sensory signs on examination are always a measure of negative phenomena.
TERMINOLOGY
Paresthesias and dysesthesias are general terms used to denote positive sensory symptoms. The term paresthesias typically refers to tingling or pins-and-needles sensations but may include a wide variety of other abnormal sensations, except pain; it sometimes implies that the abnormal sensations are perceived spontaneously. The more general term dysesthesias denotes all types of abnormal sensations, including painful ones, regardless of whether a stimulus is evident.
Another set of terms refers to sensory abnormalities found on examination. Hypesthesia or hypoesthesia refers to a reduction of cutaneous sensation to a specific type of testing such as pressure, light touch, and warm or cold stimuli; anesthesia, to a complete absence of skin sensation to the same stimuli plus pinprick; and hypalgesia or analgesia, to reduced or absent pain perception (nociception). Hyperesthesia means pain or increased sensitivity in response to touch. Similarly, allodynia describes the situation in which a nonpainful stimulus, once perceived, is experienced as painful, even excruciating. An example is elicitation of a painful sensation by application of a vibrating tuning fork. Hyperalgesia denotes severe pain in response to a mildly noxious stimulus, and hyperpathia, a broad term, encompasses all the phenomena described by hyperesthesia, allodynia, and hyperalgesia. With hyperpathia, the threshold for a sensory stimulus is increased and perception is delayed, but once felt, it is unduly painful.
Disorders of deep sensation arising from muscle spindles, tendons, and joints affect proprioception (position sense). Manifestations include imbalance (particularly with eyes closed or in the dark), clumsiness of precision movements, and unsteadiness of gait, which are referred to collectively as sensory ataxia. Other findings on examination usually, but not invariably, include reduced or absent joint position and vibratory sensibility and absent deep tendon reflexes in the affected limbs. The Romberg sign is positive, which means that the patient sways markedly or topples when asked to stand with feet close together and eyes closed. In severe states of deafferentation involving deep sensation, the patient cannot walk or stand unaided or even sit unsupported. Continuous involuntary movements (pseudoathetosis) of the outstretched hands and fingers occur, particularly with eyes closed.
ANATOMY OF SENSATION
Cutaneous receptors are classified by the type of stimulus that optimally excites them. They consist of naked nerve endings (nociceptors, which respond to tissue-damaging stimuli, and thermoreceptors, which respond to noninjurious thermal stimuli) and encapsulated terminals (several types of mechanoreceptor, activated by physical deformation of the skin). Each type of receptor has its own set of sensitivities to specific stimuli, size and distinctness of receptive fields, and adaptational qualities.
Afferent fibers in peripheral nerve trunks traverse the dorsal roots and enter the dorsal horn of the spinal cord (Fig. 31-1). From there, the polysynaptic projections of the smaller fibers (unmyelinated and small myelinated), which subserve mainly nociception, itch, temperature sensibility, and touch, cross and ascend in the opposite anterior and lateral columns of the spinal cord, through the brainstem, to the ventral posterolateral (VPL) nucleus of the thalamus and ultimately project to the postcentral gyrus of the parietal cortex (Chap. 18). This is the spinothalamic pathway or anterolateral system. The larger fibers, which subserve tactile and position sense and kinesthesia, project rostrally in the posterior and posterolateral columns on the same side of the spinal cord and make their first synapse in the gracile or cuneate nucleus of the lower medulla. Axons of second-order neurons decussate and ascend in the medial lemniscus located medially in the medulla and in the tegmentum of the pons and midbrain and synapse in the VPL nucleus; third-order neurons project to parietal cortex as well as to other cortical areas. This large-fiber system is referred to as the posterior column–medial lemniscal pathway (lemniscal, for short). Although the fiber types and functions that make up the spinothalamic and lemniscal systems are relatively well known, many other fibers, particularly those associated with touch, pressure, and position sense, ascend in a diffusely distributed pattern both ipsilaterally and contralaterally in the anterolateral quadrants of the spinal cord. This explains why a complete lesion of the posterior columns of the spinal cord may be associated with little sensory deficit on examination.
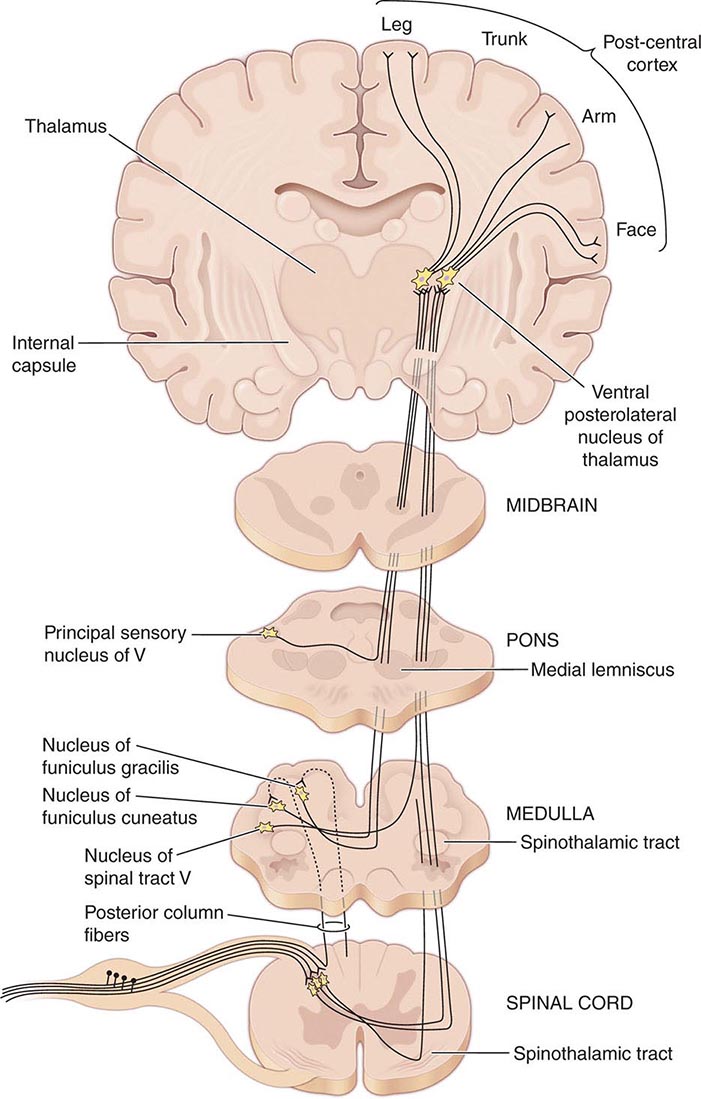
FIGURE 31-1 The main somatosensory pathways. The spinothalamic tract (pain, thermal sense) and the posterior column–lemniscal system (touch, pressure, joint position) are shown. Offshoots from the ascending anterolateral fasciculus (spinothalamic tract) to nuclei in the medulla, pons, and mesencephalon and nuclear terminations of the tract are indicated. (From AH Ropper, MA Samuels: Adams and Victor’s Principles of Neurology, 9th ed. New York, McGraw-Hill, 2009.)
Nerve conduction studies and nerve biopsy are important means of investigating the peripheral nervous system, but they do not evaluate the function or structure of cutaneous receptors and free nerve endings or of unmyelinated or thinly myelinated nerve fibers in the nerve trunks. Skin biopsy can be used to evaluate these structures in the dermis and epidermis.
CLINICAL EXAMINATION OF SENSATION
The main components of the sensory examination are tests of primary sensation (pain, touch, vibration, joint position, and thermal sensation) (Table 31-1). The examiner must depend on patient responses, and this complicates interpretation. Further, examination may be limited in some patients. In a stuporous patient, for example, sensory examination is reduced to observing the briskness of withdrawal in response to a pinch or another noxious stimulus. Comparison of responses on the two sides of the body is essential. In an alert but uncooperative patient, it may not be possible to examine cutaneous sensation, but some idea of proprioceptive function may be gained by noting the patient’s best performance of movements requiring balance and precision.
|
TESTING PRIMARY SENSATION |

In patients with sensory complaints, testing should begin in the center of the affected region and proceed radially until sensation is perceived as normal. The distribution of any abnormality is defined and compared to root and peripheral nerve territories (Figs. 31-2 and 31-3). Some patients present with sensory symptoms that do not fit an anatomic localization and are accompanied by either no abnormalities or gross inconsistencies on examination. The examiner should consider whether the sensory symptoms are a disguised request for help with psychologic or situational problems. Sensory examination of a patient who has no neurologic complaints can be brief and consist of pinprick, touch, and vibration testing in the hands and feet plus evaluation of stance and gait, including the Romberg maneuver (Chap. 438). Evaluation of stance and gait also tests the integrity of motor and cerebellar systems.
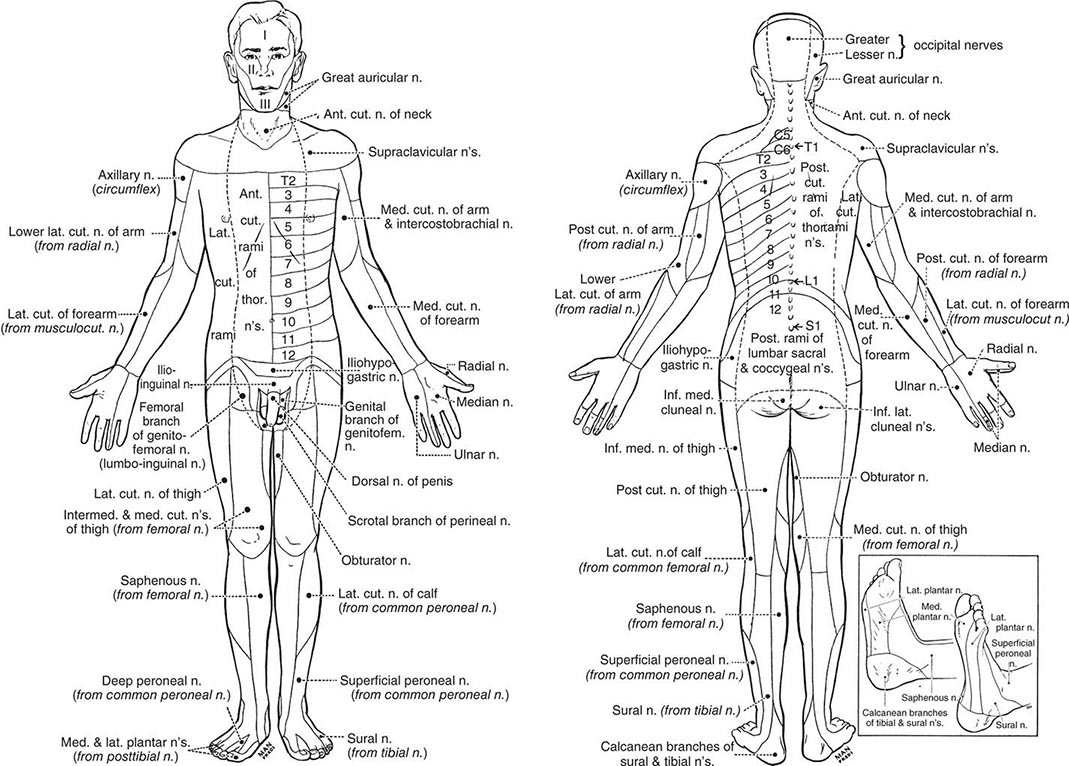
FIGURE 31-2 The cutaneous fields of peripheral nerves. (Reproduced by permission from W Haymaker, B Woodhall: Peripheral Nerve Injuries, 2nd ed. Philadelphia, Saunders, 1953.)
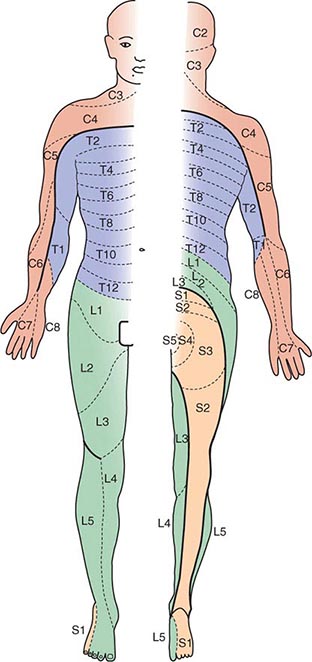
FIGURE 31-3 Distribution of the sensory spinal roots on the surface of the body (dermatomes). (From D Sinclair: Mechanisms of Cutaneous Sensation. Oxford, UK, Oxford University Press, 1981; with permission from Dr. David Sinclair.)
Primary Sensation The sense of pain usually is tested with a clean pin, which is then discarded. The patient is asked to close the eyes and focus on the pricking or unpleasant quality of the stimulus, not just the pressure or touch sensation elicited. Areas of hypalgesia should be mapped by proceeding radially from the most hypalgesic site. Temperature sensation to both hot and cold is best tested with small containers filled with water of the desired temperature. An alternative way to test cold sensation is to touch a metal object, such as a tuning fork at room temperature, to the skin. For testing warm temperatures, the tuning fork or another metal object may be held under warm water of the desired temperature and then used. The appreciation of both cold and warmth should be tested because different receptors respond to each. Touch usually is tested with a wisp of cotton or a fine camel hair brush, minimizing pressure on the skin. In general, it is better to avoid testing touch on hairy skin because of the profusion of the sensory endings that surround each hair follicle. The patient is tested with the eyes closed and should indicate as soon as the stimulus is perceived, indicating its location.
Joint position testing is a measure of proprioception. With the patient’s eyes closed, joint position is tested in the distal interphalangeal joint of the great toe and fingers. The digit is held by its sides, distal to the joint being tested, and moved passively while more proximal joints are stabilized—the patient indicates the change in position or direction of movement. If errors are made, more proximal joints are tested. A test of proximal joint position sense, primarily at the shoulder, is performed by asking the patient to bring the two index fingers together with arms extended and eyes closed. Normal individuals can do this accurately, with errors of 1 cm or less.
The sense of vibration is tested with an oscillating tuning fork that vibrates at 128 Hz. Vibration is tested over bony points, beginning distally; in the feet, it is tested over the dorsal surface of the distal phalanx of the big toes and at the malleoli of the ankles, and in the hands, it is tested dorsally at the distal phalanx of the fingers. If abnormalities are found, more proximal sites should be examined. Vibratory thresholds at the same site in the patient and the examiner may be compared for control purposes.
Quantitative Sensory Testing Effective sensory testing devices are commercially available. Quantitative sensory testing is particularly useful for serial evaluation of cutaneous sensation in clinical trials. Threshold testing for touch and vibratory and thermal sensation is the most widely used application.
Cortical Sensation The most commonly used tests of cortical function are two-point discrimination, touch localization, and bilateral simultaneous stimulation and tests for graphesthesia and stereognosis. Abnormalities of these sensory tests, in the presence of normal primary sensation in an alert cooperative patient, signify a lesion of the parietal cortex or thalamocortical projections. If primary sensation is altered, these cortical discriminative functions usually will be abnormal also. Comparisons should always be made between analogous sites on the two sides of the body because the deficit with a specific parietal lesion is likely to be unilateral.
Two-point discrimination is tested with special calipers, the points of which may be set from 2 mm to several centimeters apart and then applied simultaneously to the test site. On the fingertips, a normal individual can distinguish about a 3-mm separation of points.
Touch localization is performed by light pressure for an instant with the examiner’s fingertip or a wisp of cotton wool; the patient, whose eyes are closed, is required to identify the site of touch. Bilateral simultaneous stimulation at analogous sites (e.g., the dorsum of both hands) can be carried out to determine whether the perception of touch is extinguished consistently on one side (extinction or neglect). Graphesthesia refers to the capacity to recognize, with eyes closed, letters or numbers drawn by the examiner’s fingertip on the palm of the hand. Once again, interside comparison is of prime importance. Inability to recognize numbers or letters is termed agraphesthesia.
Stereognosis refers to the ability to identify common objects by palpation, recognizing their shape, texture, and size. Common standard objects such as keys, paper clips, and coins are best used. Patients with normal stereognosis should be able to distinguish a dime from a penny and a nickel from a quarter without looking. Patients should feel the object with only one hand at a time. If they are unable to identify it in one hand, it should be placed in the other for comparison. Individuals who are unable to identify common objects and coins in one hand but can do so in the other are said to have astereognosis of the abnormal hand.
LOCALIZATION OF SENSORY ABNORMALITIES
Sensory symptoms and signs can result from lesions at many different levels of the nervous system from the parietal cortex to the peripheral sensory receptor. Noting their distribution and nature is the most important way to localize their source. Their extent, configuration, symmetry, quality, and severity are the key observations.
Dysesthesias without sensory findings by examination may be difficult to interpret. To illustrate, tingling dysesthesias in an acral distribution (hands and feet) can be systemic in origin, e.g., secondary to hyperventilation, or induced by a medication such as acetazolamide. Distal dysesthesias can also be an early event in an evolving polyneuropathy or may herald a myelopathy, such as from vitamin B12 deficiency. Sometimes distal dysesthesias have no definable basis. In contrast, dysesthesias that correspond in distribution to that of a particular peripheral nerve structure denote a lesion at that site. For instance, dysesthesias restricted to the fifth digit and the adjacent one-half of the fourth finger on one hand reliably point to disorder of the ulnar nerve, most commonly at the elbow.
Nerve and Root In focal nerve trunk lesions, sensory abnormalities are readily mapped and generally have discrete boundaries (Figs. 31-2 and 31-3). Root (“radicular”) lesions frequently are accompanied by deep, aching pain along the course of the related nerve trunk. With compression of a fifth lumbar (L5) or first sacral (S1) root, as from a ruptured intervertebral disk, sciatica (radicular pain relating to the sciatic nerve trunk) is a common manifestation (Chap. 22). With a lesion affecting a single root, sensory deficits may be minimal or absent because adjacent root territories overlap extensively.
Isolated mononeuropathies may cause symptoms beyond the territory supplied by the affected nerve, but abnormalities on examination typically are confined to appropriate anatomic boundaries. In multiple mononeuropathies, symptoms and signs occur in discrete territories supplied by different individual nerves and—as more nerves are affected—may simulate a polyneuropathy if deficits become confluent. With polyneuropathies, sensory deficits are generally graded, distal, and symmetric in distribution (Chap. 459). Dysesthesias, followed by numbness, begin in the toes and ascend symmetrically. When dysesthesias reach the knees, they usually also have appeared in the fingertips. The process is nerve length–dependent, and the deficit is often described as “stocking-glove” in type. Involvement of both hands and feet also occurs with lesions of the upper cervical cord or the brainstem, but an upper level of the sensory disturbance may then be found on the trunk and other evidence of a central lesion may be present, such as sphincter involvement or signs of an upper motor neuron lesion (Chap. 30). Although most polyneuropathies are pansensory and affect all modalities of sensation, selective sensory dysfunction according to nerve fiber size may occur. Small-fiber polyneuropathies are characterized by burning, painful dysesthesias with reduced pinprick and thermal sensation but with sparing of proprioception, motor function, and deep tendon reflexes. Touch is involved variably; when it is spared, the sensory pattern is referred to as exhibiting sensory dissociation. Sensory dissociation may occur also with spinal cord lesions as well as small-fiber neuropathies. Large-fiber polyneuropathies are characterized by vibration and position sense deficits, imbalance, absent tendon reflexes, and variable motor dysfunction but preservation of most cutaneous sensation. Dysesthesias, if present at all, tend to be tingling or bandlike in quality.
Sensory neuronopathy (or ganglionopathy) is characterized by widespread but asymmetric sensory loss occurring in a non-length-dependent manner so that it may occur proximally or distally and in the arms, legs, or both. Pain and numbness progress to sensory ataxia and impairment of all sensory modalities with time. This condition is usually paraneoplastic or idiopathic in origin (Chaps. 122 and 459) or related to an autoimmune disease, particularly Sjögren’s syndrome.
Spinal Cord (See also Chap. 456) If the spinal cord is transected, all sensation is lost below the level of transection. Bladder and bowel function also are lost, as is motor function. Lateral hemisection of the spinal cord produces the Brown-Séquard syndrome, with absent pain and temperature sensation contralaterally and loss of proprioceptive sensation and power ipsilaterally below the lesion (see Figs. 31-1 and 456-1).
Numbness or paresthesias in both feet may arise from a spinal cord lesion; this is especially likely when the upper level of the sensory loss extends to the trunk. When all extremities are affected, the lesion is probably in the cervical region or brainstem unless a peripheral neuropathy is responsible. The presence of upper motor neuron signs (Chap. 30) supports a central lesion; a hyperesthetic band on the trunk may suggest the level of involvement.
A dissociated sensory loss can reflect spinothalamic tract involvement in the spinal cord, especially if the deficit is unilateral and has an upper level on the torso. Bilateral spinothalamic tract involvement occurs with lesions affecting the center of the spinal cord, such as in syringomyelia. There is a dissociated sensory loss with impairment of pinprick and temperature appreciation but relative preservation of light touch, position sense, and vibration appreciation.
Dysfunction of the posterior columns in the spinal cord or of the posterior root entry zone may lead to a bandlike sensation around the trunk or a feeling of tight pressure in one or more limbs. Flexion of the neck sometimes leads to an electric shock–like sensation that radiates down the back and into the legs (Lhermitte’s sign) in patients with a cervical lesion affecting the posterior columns, such as from multiple sclerosis, cervical spondylosis, or recent irradiation to the cervical region.
Brainstem Crossed patterns of sensory disturbance, in which one side of the face and the opposite side of the body are affected, localize to the lateral medulla. Here a small lesion may damage both the ipsilateral descending trigeminal tract and the ascending spinothalamic fibers subserving the opposite arm, leg, and hemitorso (see “Lateral medullary syndrome” in Fig. 446-10). A lesion in the tegmentum of the pons and midbrain, where the lemniscal and spinothalamic tracts merge, causes pansensory loss contralaterally.
Thalamus Hemisensory disturbance with tingling numbness from head to foot is often thalamic in origin but also can arise from the anterior parietal region. If abrupt in onset, the lesion is likely to be due to a small stroke (lacunar infarction), particularly if localized to the thalamus. Occasionally, with lesions affecting the VPL nucleus or adjacent white matter, a syndrome of thalamic pain, also called Déjerine-Roussy syndrome, may ensue. The persistent, unrelenting unilateral pain often is described in dramatic terms.
Cortex With lesions of the parietal lobe involving either the cortex or the subjacent white matter, the most prominent symptoms are contralateral hemineglect, hemi-inattention, and a tendency not to use the affected hand and arm. On cortical sensory testing (e.g., two-point discrimination, graphesthesia), abnormalities are often found but primary sensation is usually intact. Anterior parietal infarction may present as a pseudothalamic syndrome with contralateral loss of primary sensation from head to toe. Dysesthesias or a sense of numbness and, rarely, a painful state may also occur.
Focal Sensory Seizures These seizures generally are due to lesions in the area of the postcentral or precentral gyrus. The principal symptom of focal sensory seizures is tingling, but additional, more complex sensations may occur, such as a rushing feeling, a sense of warmth, or a sense of movement without detectable motion. Symptoms typically are unilateral; commonly begin in the arm or hand, face, or foot; and often spread in a manner that reflects the cortical representation of different bodily parts, as in a Jacksonian march. Their duration is variable; seizures may be transient, lasting only for seconds, or persist for an hour or more. Focal motor features may supervene, often becoming generalized with loss of consciousness and tonic-clonic jerking.
ACKNOWLEDGMENT
Arthur Asbury authored or co-authored this chapter in earlier editions of this book.
32 |
Gait and Balance Disorders |
PREVALENCE, MORBIDITY, AND MORTALITY
Gait and balance problems are common in the elderly and contribute to the risk of falls and injury. Gait disorders have been described in 15% of individuals older than 65. By age 80 one person in four will use a mechanical aid to assist with ambulation. Among those 85 and older, the prevalence of gait abnormality approaches 40%. In epidemiologic studies, gait disorders are consistently identified as a major risk factor for falls and injury.
A substantial number of older persons report insecure balance and experience falls and fear of falling. Prospective studies indicate that 30% of those older than 65 fall each year. The proportion is even higher in frail elderly and nursing home patients. Each year, 8% of individuals older than 75 suffer a serious fall-related injury. Hip fractures result in hospitalization, can lead to nursing home admission, and are associated with an increased mortality risk in the subsequent year. For each person who is physically disabled, there are others whose functional independence is limited by anxiety and fear of falling. Nearly one in five elderly individuals voluntarily restricts his or her activity because of fear of falling. With loss of ambulation, the quality of life diminishes, and rates of morbidity and mortality increase.
ANATOMY AND PHYSIOLOGY
An upright bipedal gait depends on the successful integration of postural control and locomotion. These functions are widely distributed in the central nervous system. The biomechanics of bipedal walking are complex, and the performance is easily compromised by a neurologic deficit at any level. Command and control centers in the brainstem, cerebellum, and forebrain modify the action of spinal pattern generators to promote stepping. While a form of “fictive locomotion” can be elicited from quadrupedal animals after spinal transection, this capacity is limited in primates. Step generation in primates is dependent on locomotor centers in the pontine tegmentum, midbrain, and subthalamic region. Locomotor synergies are executed through the reticular formation and descending pathways in the ventromedial spinal cord. Cerebral control provides a goal and purpose for walking and is involved in avoidance of obstacles and adaptation of locomotor programs to context and terrain.
Postural control requires the maintenance of the center of mass over the base of support through the gait cycle. Unconscious postural adjustments maintain standing balance: long latency responses are measurable in the leg muscles, beginning 110 milliseconds after a perturbation. Forward motion of the center of mass provides propulsive force for stepping, but failure to maintain the center of mass within stability limits results in falls. The anatomic substrate for dynamic balance has not been well defined, but the vestibular nucleus and midline cerebellum contribute to balance control in animals. Patients with damage to these structures have impaired balance while standing and walking.
Standing balance depends on good-quality sensory information about the position of the body center with respect to the environment, support surface, and gravitational forces. Sensory information for postural control is primarily generated by the visual system, the vestibular system, and proprioceptive receptors in the muscle spindles and joints. A healthy redundancy of sensory afferent information is generally available, but loss of two of the three pathways is sufficient to compromise standing balance. Balance disorders in older individuals sometimes result from multiple insults in the peripheral sensory systems (e.g., visual loss, vestibular deficit, peripheral neuropathy) that critically degrade the quality of afferent information needed for balance stability.
Older patients with cognitive impairment from neurodegenerative diseases appear to be particularly prone to falls and injury. There is a growing body of literature on the use of attentional resources to manage gait and balance. Walking is generally considered to be unconscious and automatic, but the ability to walk while attending to a cognitive task (dual-task walking) may be compromised in frail elderly individuals with a history of falls. Older patients with deficits in executive function may have particular difficulty in managing the attentional resources needed for dynamic balance when distracted.
DISORDERS OF GAIT
Disorders of gait may be attributed to frailty, fatigue, arthritis, and orthopedic deformity, but neurologic causes are disabling and important to address. The heterogeneity of gait disorders observed in clinical practice reflects the large network of neural systems involved in the task. Walking is vulnerable to neurologic disease at every level. Gait disorders have been classified descriptively on the basis of abnormal physiology and biomechanics. One problem with this approach is that many failing gaits look fundamentally similar. This overlap reflects common patterns of adaptation to threatened balance stability and declining performance. The gait disorder observed clinically must be viewed as the product of a neurologic deficit and a functional adaptation. Unique features of the failing gait are often overwhelmed by the adaptive response. Some common patterns of abnormal gait are summarized next. Gait disorders can also be classified by etiology (Table 32-1).
|
ETIOLOGY OF GAIT DISORDERS |
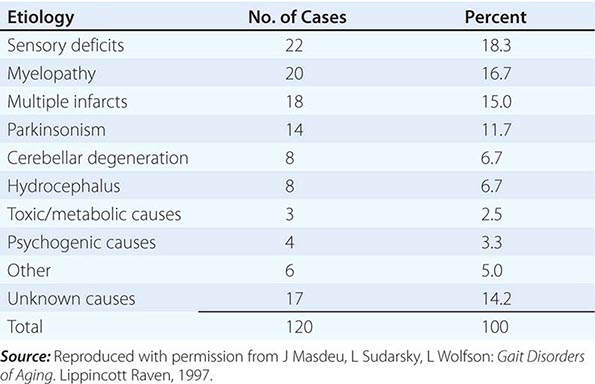
CAUTIOUS GAIT
The term cautious gait is used to describe the patient who walks with an abbreviated stride and lowered center of mass, as if walking on a slippery surface. This disorder is both common and nonspecific. It is, in essence, an adaptation to a perceived postural threat. There may be an associated fear of falling. This disorder can be observed in more than one-third of older patients with gait impairment. Physical therapy often improves walking to the degree that follow-up observation may reveal a more specific underlying disorder.
STIFF-LEGGED GAIT
Spastic gait is characterized by stiffness in the legs, an imbalance of muscle tone, and a tendency to circumduct and scuff the feet. The disorder reflects compromise of corticospinal command and overactivity of spinal reflexes. The patient may walk on the toes. In extreme instances, the legs cross due to increased tone in the adductors. Upper motor neuron signs are present on physical examination. Shoes often reflect an uneven pattern of wear across the outside. The disorder may be cerebral or spinal in origin.
Myelopathy from cervical spondylosis is a common cause of spastic or spastic-ataxic gait in the elderly. Demyelinating disease and trauma are the leading causes of myelopathy in younger patients. In chronic progressive myelopathy of unknown cause, a workup with laboratory and imaging tests may establish a diagnosis. A family history should suggest hereditary spastic paraplegia (Chap. 452); genetic testing is now available for some of the common mutations responsible for this disorder. Tropical spastic paraparesis related to the retrovirus human T-cell lymphotropic virus 1 (HTLV-1) is endemic in parts of the Caribbean and South America. A structural lesion, such as a tumor or a spinal vascular malformation, should be excluded with appropriate testing. Spinal cord disorders are discussed in detail in Chap. 456.
With cerebral spasticity, asymmetry is common, the upper extremities are usually involved, and dysarthria is often an associated feature. Common causes include vascular disease (stroke), multiple sclerosis, and perinatal injury to the nervous system (cerebral palsy).
Other stiff-legged gaits include dystonia (Chap. 449) and stiff-person syndrome (Chap. 122). Dystonia is a disorder characterized by sustained muscle contractions resulting in repetitive twisting movements and abnormal posture. It often has a genetic basis. Dystonic spasms can produce plantar flexion and inversion of the feet, sometimes with torsion of the trunk. In autoimmune stiff-person syndrome, exaggerated lordosis of the lumbar spine and overactivation of antagonist muscles restrict trunk and lower-limb movement and result in a wooden or fixed posture.
PARKINSONISM AND FREEZING GAIT
Parkinson’s disease (Chap. 449) is common, affecting 1% of the population >55 years of age. The stooped posture and shuffling gait are characteristic and distinctive features. Patients sometimes accelerate (festinate) with walking, display retropulsion, or exhibit a tendency to turn en bloc. A National Institutes of Health workshop defined freezing of gait as “brief, episodic absence of forward progression of the feet, despite the intention to walk.” Gait freezing occurs in 26% of Parkinson’s patients by the end of 5 years and develops in most such patients eventually. Postural instability and falling occur as the disease progresses; some falls are precipitated by freezing of gait.
Freezing of gait is even more common in some Parkinson’s-related neurodegenerative disorders, such as progressive supranuclear palsy, multiple-system atrophy, and corticobasal degeneration. Patients with these disorders frequently present with axial stiffness, postural instability, and a shuffling, freezing gait while lacking the characteristic pill-rolling tremor of Parkinson’s disease. Falls within the first year suggest the possibility of progressive supranuclear palsy.
Hyperkinetic movement disorders also produce characteristic and recognizable disturbances in gait. In Huntington’s disease (Chap. 449), the unpredictable occurrence of choreic movements gives the gait a dancing quality. Tardive dyskinesia is the cause of many odd, stereotypic gait disorders seen in patients chronically exposed to antipsychotics and other drugs that block the D2 dopamine receptor.
FRONTAL GAIT DISORDER
Frontal gait disorder, sometimes known as gait apraxia, is common in the elderly and has a variety of causes. The term is used to describe a shuffling, freezing gait with imbalance and other signs of higher cerebral dysfunction. Typical features include a wide base of support, a short stride, shuffling along the floor, and difficulty with starts and turns. Many patients exhibit a difficulty with gait initiation that is descriptively characterized as the “slipping clutch” syndrome or gait ignition failure. The term lower-body parkinsonism is also used to describe such patients. Strength is generally preserved, and patients are able to make stepping movements when not standing and maintaining their balance at the same time. This disorder is best considered a higher-level motor control disorder, as opposed to an apraxia (Chap. 36).
The most common cause of frontal gait disorder is vascular disease, particularly subcortical small-vessel disease. Lesions are frequently found in the deep frontal white matter and centrum ovale. Gait disorder may be the salient feature in hypertensive patients with ischemic lesions of the deep-hemisphere white matter (Binswanger’s disease). The clinical syndrome includes mental changes (variable in degree), dysarthria, pseudobulbar affect (emotional disinhibition), increased tone, and hyperreflexia in the lower limbs.
Communicating hydrocephalus in adults also presents with a gait disorder of this type. Other features of the diagnostic triad (mental changes, incontinence) may be absent in the initial stages. MRI demonstrates ventricular enlargement, an enlarged flow void about the aqueduct, and a variable degree of periventricular white-matter change. A lumbar puncture or dynamic test is necessary to confirm hydrocephalus.
CEREBELLAR GAIT ATAXIA
Disorders of the cerebellum have a dramatic impact on gait and balance. Cerebellar gait ataxia is characterized by a wide base of support, lateral instability of the trunk, erratic foot placement, and decompensation of balance when attempting to walk on a narrow base. Difficulty maintaining balance when turning is often an early feature. Patients are unable to walk tandem heel to toe and display truncal sway in narrow-based or tandem stance. They show considerable variation in their tendency to fall in daily life.
Causes of cerebellar ataxia in older patients include stroke, trauma, tumor, and neurodegenerative disease such as multiple-system atrophy (Chaps. 449 and 454) and various forms of hereditary cerebellar degeneration (Chap. 450). A short expansion at the site of the fragile × mutation (fragile × pre-mutation) has been associated with gait ataxia in older men. Alcoholic cerebellar degeneration can be screened by history and often confirmed by MRI. In patients with ataxia, MRI demonstrates the extent and topography of cerebellar atrophy.
SENSORY ATAXIA
As reviewed earlier in this chapter, balance depends on high-quality afferent information from the visual and the vestibular systems and proprioception. When this information is lost or degraded, balance during locomotion is impaired and instability results. The sensory ataxia of tabetic neurosyphilis is a classic example. The contemporary equivalent is the patient with neuropathy affecting large fibers. Vitamin B12 deficiency is a treatable cause of large-fiber sensory loss in the spinal cord and peripheral nervous system. Joint position and vibration sense are diminished in the lower limbs. The stance in such patients is destabilized by eye closure; they often look down at their feet when walking and do poorly in the dark. Table 32-2 compares sensory ataxia with cerebellar ataxia and frontal gait disorder. Some frail older patients exhibit a syndrome of imbalance from the combined effect of multiple sensory deficits. Such patients have disturbances in proprioception, vision, and vestibular sense that impair postural support.
|
FEATURES OF CEREBELLAR ATAXIA, SENSORY ATAXIA, AND FRONTAL GAIT DISORDERS |
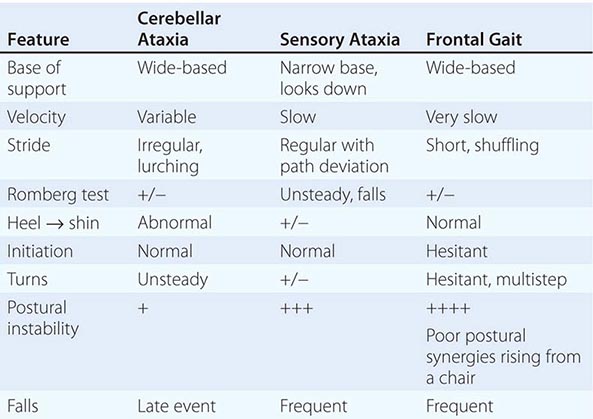
NEUROMUSCULAR DISEASE
Patients with neuromuscular disease often have an abnormal gait, occasionally as a presenting feature. With distal weakness (peripheral neuropathy), the step height is increased to compensate for footdrop, and the sole of the foot may slap on the floor during weight acceptance. Neuropathy may be associated with a degree of sensory imbalance, as described earlier. Patients with myopathy or muscular dystrophy more typically exhibit proximal weakness. Weakness of the hip girdle may result in some degree of excess pelvic sway during locomotion.
TOXIC AND METABOLIC DISORDERS
Alcohol intoxication is the most common cause of acute walking difficulty. Chronic toxicity from medications and metabolic disturbances can impair motor function and gait. Mental status changes may be found, and examination may reveal asterixis or myoclonus. Static equilibrium is disturbed, and such patients are easily thrown off balance. Disequilibrium is particularly evident in patients with chronic renal disease and those with hepatic failure, in whom asterixis may impair postural support. Sedative drugs, especially neuroleptics and long-acting benzodiazepines, affect postural control and increase the risk for falls. These disorders are especially important to recognize because they are often treatable.
PSYCHOGENIC GAIT DISORDER
Psychogenic disorders are common in neurologic practice, and the presentation often involves gait. Some patients with extreme anxiety or phobia walk with exaggerated caution with abduction of the arms, as if walking on ice. This inappropriately overcautious gait differs in degree from the gait of the patient who is insecure and making adjustments for imbalance. Depressed patients exhibit primarily slowness, a manifestation of psychomotor retardation, and lack of purpose in their stride. Hysterical gait disorders are among the most spectacular encountered. Odd gyrations of posture with wastage of muscular energy (astasia–abasia), extreme slow motion, and dramatic fluctuations over time may be observed in patients with somatoform disorders and conversion reactions.
DISORDERS OF BALANCE
DEFINITION, ETIOLOGY, AND MANIFESTATIONS
Balance is the ability to maintain equilibrium—a state in which opposing physical forces cancel one another out. In physiology, this term is taken to mean the ability to control the center of mass with respect to gravity and the support surface. In reality, people are not consciously aware of their center of mass, but everyone (particularly gymnasts, figure skaters, and platform divers, for example) move so as to manage it. Disorders of balance present as difficulty maintaining posture while standing and walking and as a subjective sense of disequilibrium, which is a form of dizziness.
The cerebellum and vestibular system organize antigravity responses needed to maintain an upright posture. These responses are physiologically complex, and the anatomic representation they entail is not well understood. Failure, resulting in disequilibrium, can occur at several levels: cerebellar, vestibular, somatosensory, and higher-level disequilibrium.
Patients with cerebellar ataxia do not generally complain of dizziness, though balance is visibly impaired. Neurologic examination reveals a variety of cerebellar signs. Postural compensation may prevent falls early on, but falls are inevitable with disease progression. The progression of neurodegenerative ataxia is often measured by the number of years to loss of stable ambulation.
Vestibular disorders (Chap. 28) have symptoms and signs that fall into three categories: (1) vertigo (the subjective inappropriate perception or illusion of movement); (2) nystagmus (involuntary eye movements); and (3) impaired standing balance. Not every patient has all manifestations. Patients with vestibular deficits related to ototoxic drugs may lack vertigo or obvious nystagmus, but their balance is impaired on standing and walking, and they cannot navigate in the dark. Laboratory testing is available to investigate vestibular deficits.
Somatosensory deficits also produce imbalance and falls. There is often a subjective sense of insecure balance and fear of falling. Postural control is compromised by eye closure (Romberg’s sign); these patients also have difficulty navigating in the dark. A dramatic example is provided by the patient with autoimmune subacute sensory neuropathy, which is sometimes a paraneoplastic disorder (Chap. 122). Compensatory strategies enable such patients to walk in the virtual absence of proprioception, but the task requires active visual monitoring.
Patients with higher-level disorders of equilibrium have difficulty maintaining balance in daily life and may present with falls. Their awareness of balance impairment may be reduced. Patients taking sedating medications are in this category. In prospective studies, dementia and sedating medications substantially increase the risk for falls.
FALLS
Falls are common in the elderly; 30% of people older than 65 who are living in the community fall each year. Modest changes in balance function have been described in fit older individuals as a result of normal aging. Subtle deficits in sensory systems, attention, and motor reaction time contribute to the risk, and environmental hazards abound. Many falls by older adults are episodes of tripping or slipping, often designated mechanical falls. A fall is not a neurologic problem per se, but there are events for which neurologic evaluation is appropriate. It is important to distinguish falls associated with loss of consciousness (syncope, seizure), which require appropriate evaluation and intervention (Chaps. 27 and 445). In most prospective studies, a small subset of individuals experience a large number of fall events. These individuals with recurrent falls often have gait and balance issues that need to be addressed.
Fall Patterns: The Event Description The history of a fall is often problematic or incomplete, and the underlying mechanism or cause may be difficult to establish in retrospect. The patient and family may have limited information about what triggered the fall. Injuries can complicate the physical examination. While there is no standard nosology of falls, some common clinical patterns may emerge and provide a clue.