384 |
The Spondyloarthritides |
The spondyloarthritides are a group of overlapping disorders that share certain clinical features and genetic associations. These disorders include ankylosing spondylitis (AS), reactive arthritis, psoriatic arthritis and spondylitis, enteropathic arthritis and spondylitis, juvenile-onset spondyloarthritis (SpA), and undifferentiated SpA. The similarities in clinical manifestations and genetic predisposition suggest that these disorders share pathogenic mechanisms.
ANKYLOSING SPONDYLITIS
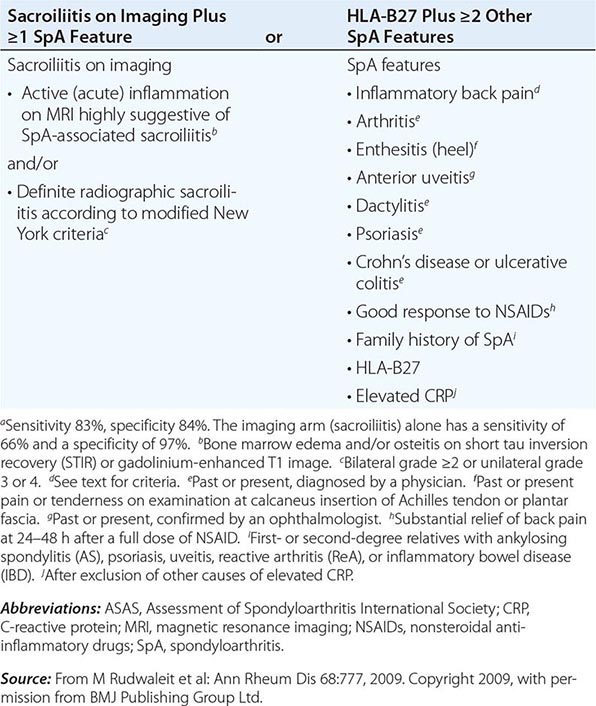
AS is an inflammatory disorder of unknown cause that primarily affects the axial skeleton; peripheral joints and extraarticular structures are also frequently involved. The disease usually begins in the second or third decade; male-to-female prevalence is between 2:1 and 3:1. The term axial spondyloarthritis is coming into common use, supported by criteria formulated in 2009 (Table 384-1). This classification includes both definite AS and early stages that do not yet meet classical criteria for AS, but it probably also includes other conditions with a different natural history.
|
ASAS CRITERIA FOR CLASSIFICATION OF AXIAL SPONDYLOARTHRITIS (TO BE APPLIED FOR PATIENTS WITH BACK PAIN ≥3 MONTHS AND AGE OF ONSET <45 YEARS)a |
EPIDEMIOLOGY
AS shows a striking correlation with the histocompatibility antigen HLA-B27 and occurs worldwide roughly in proportion to the prevalence of B27 (Chap. 373e). In North American whites, the prevalence of B27 is 7%, whereas it is 90% in patients with AS, independent of disease severity.
In population surveys, AS is present in 1–6% of adults inheriting B27, whereas the prevalence is 10–30% among B27+ adult first-degree relatives of AS probands. Concordance rate in identical twins is about 65%. Susceptibility to AS is determined largely by genetic factors, with B27 comprising less than one-half of the genetic component. Genomewide single-nucleotide polymorphism (SNP) analysis has identified over 30 additional susceptibility alleles.
PATHOLOGY
Sacroiliitis is often the earliest manifestation of AS. Knowledge of its pathology comes from both biopsy and autopsy studies that cover a range of disease durations. Synovitis and myxoid marrow represent the earliest changes, followed by pannus and subchondral granulation tissue. Marrow edema, enthesitis, and chondroid differentiation are also found. Macrophages, T cells, plasma cells, and osteoclasts are prevalent. Eventually the eroded joint margins are gradually replaced by fibrocartilage regeneration and then by ossification. The joint may become totally obliterated.
In the spine, the specimens studied have either been surgically resected in advanced disease or taken from autopsies. There is inflammatory granulation tissue in the paravertebral connective tissue at the junction of annulus fibrosus and vertebral bone, and in some cases along the entire outer annulus. The outer annular fibers are eroded and eventually replaced by bone, forming the beginning of a syndesmophyte, which then grows by continued endochondral ossification, ultimately bridging the adjacent vertebral bodies. Ascending progression of this process leads to the “bamboo spine.” Other lesions in the spine include diffuse osteoporosis (loss of trabecular bone despite accretion of periosteal bone), erosion of vertebral bodies at the disk margin, “squaring” or “barreling” of vertebrae, and inflammation and destruction of the disk-bone border. Inflammatory arthritis of the apophyseal (facet) joints is common, with synovitis, inflammation at the bony attachment of the joint capsule, and subchondral bone marrow granulation tissue. Erosion of joint cartilage by pannus is often followed by bony ankylosis. This may precede formation of syndesmophytes bridging the adjacent disks. Bone mineral density is diminished in the spine and proximal femur early in the course of the disease.
Peripheral synovitis in AS shows marked vascularity, which is also evident as tortuous macrovascularity seen during arthroscopy. Lining layer hyperplasia, lymphoid infiltration, and pannus formation are also found. Central cartilaginous erosions caused by proliferation of subchondral granulation tissue are common. It should be emphasized that the characteristics of peripheral arthritis in AS and other forms of SpA are similar, and distinct from those of rheumatoid arthritis.
Inflammation in the fibrocartilaginous enthesis, the region where a tendon, ligament, or joint capsule attaches to bone, is a characteristic lesion in AS and other SpAs, both at axial and peripheral sites. Enthesitis is associated with prominent edema of the adjacent bone marrow and is often characterized by erosive lesions that eventually undergo ossification.
Subclinical intestinal inflammation has been found in the colon or distal ileum in a majority of patients with SpA. The histology is described below under “Enteropathic Arthritis.”
PATHOGENESIS
The pathogenesis of AS is immune-mediated, but there is little direct evidence for antigen-specific autoimmunity, and there is evidence to suggest more of an autoinflammatory pathogenesis. Uncertainty remains regarding the primary site of disease initiation. The dramatic response of the disease to therapeutic blockade of tumor necrosis factor α (TNF-α) indicates that this cytokine plays a central role in the immunopathogenesis of AS. Other genes related to TNF pathways show association with AS, including TNFRSF1A, LTBR, and TBKBP1. More recent evidence strongly implicates the interleukin (IL) 23/IL-17 cytokine pathway in AS pathogenesis. At least five genes in this pathway show association with AS, including IL23R, PTER4, IL12B, CARD9, and TYK2. All of these genes are also associated with inflammatory bowel disease (IBD), and three of them are associated with psoriasis. Serum levels of IL-23 and IL-17 are elevated in AS patients. Mice expressing high levels of IL-23 show spontaneous infiltration in the entheses of CD3+CD4–CD8– cells bearing IL-23 receptors and producing IL-17 and IL-22. This finding suggests the possibility that site-specific innate immune cells may play a critical role in the anatomic specificity of the lesions. Mast cells and, to a lesser extent, neutrophils appear to be the major IL-17-producing cells in peripheral arthritis, whereas neutrophils producing IL-17 are prominent in apophyseal joints. High levels of circulating γδ T cells expressing IL-23 receptors and producing IL-17 have been found in AS patients.
Other associated genes encode other cytokines or cytokine receptors (IL6R, IL1R1, IL1R2, IL7R, IL27), transcription factors involved in the differentiation of immune cells (RUNX3, EOMES, BACH2, NKX2-3, TBX21), or other molecules involved in activation or regulation of immune or inflammatory responses (FCGR2A, ZMIZ1, NOS2, ICOSLG).
The inflamed sacroiliac joint is infiltrated with CD4+ and CD8+ T cells and macrophages and shows high levels of TNF-α, particularly early in the disease. Abundant transforming growth factor β (TGF-β) has been found in more advanced lesions. Peripheral synovitis in AS and the other spondyloarthritides is characterized by neutrophils, macrophages expressing CD68 and CD163, CD4+ and CD8+ T cells, and B cells. There is prominent staining for intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), matrix metalloproteinase 3 (MMP-3), and myeloid-related proteins 8 and 14 (MRP-8 and MRP-14). Unlike rheumatoid arthritis (RA) synovium, citrullinated proteins and cartilage gp39 peptide–major histocompatibility complexes (MHCs) are absent. However, citrullinated proteins can be seen in the circulation.
No specific event or exogenous agent that triggers the onset of disease has been identified, although overlapping features with reactive arthritis and IBD and the involvement of the IL-23/IL-17 pathway suggest that enteric bacteria may play a role, and microdamage from mechanical stress at enthesial sites has also been implicated.
It is firmly established that HLA-B27 plays a direct role in AS pathogenesis, but its precise role at the molecular level remains unresolved. Rats transgenic for HLA-B27 develop arthritis and spondylitis, and this is un-affected by the absence of CD8. It thus appears that classical peptide antigen presentation to CD8+ T cells may not be the primary disease mechanism. However, the association of AS with ERAP1, which strongly influences the MHC class I peptide repertoire, is only found in B27+ patients, and this suggests that peptide binding to B27 is nonetheless important. The pairs of ERAP1 alleles found in AS patients show diminished peptidase activity, compared with those found in healthy controls. The B27 heavy chain has an unusual tendency to misfold, a process that may be proinflammatory. Genetic and functional studies in humans have suggested a role for natural killer (NK) cells in AS, possibly through interaction with B27 heavy chain homodimers. SpA-prone B27 rats show defective dendritic cell function and share with AS patients a characteristic “reverse interferon” gene expression signature in antigen-presenting cells.
New bone formation in AS appears to be largely based on enchondral bone formation and occurs only in the periosteal compartment. It correlates with lack of regulation of the Wnt signaling pathway, which controls the differentiation of mesenchymal cells into osteophytes, by the inhibitors DKK-1 and sclerostin. Indirect evidence and data from animal models also implicate bone morphogenic proteins, hedgehog proteins, and prostaglandin E2. There is sharp controversy as to whether vertebral new bone formation in AS is a sequela of inflammation or whether it arises independently of inflammation. The second hypothesis is based on the observation that syndesmophyte formation is not suppressed by anti-TNF-α therapy that potently suppresses inflammation. TNF-α is also a known inducer of DKK-1, which inhibits bone formation. Recent magnetic resonance imaging (MRI) studies suggest that it is vertebral inflammatory lesions that undergo metaplasia to fat (increased T1-weighted signal) that are the predominant site of subsequent syndesmophytes despite anti-TNF-α therapy, whereas early acute inflammatory lesions resolve. A recent study suggested that the rate of syndesmophyte formation decreases after >4 years of anti-TNF-α therapy.
CLINICAL MANIFESTATIONS
The symptoms of the disease are usually first noticed in late adolescence or early adulthood; the median age in Western countries is approximately 23 years. In 5% of patients, symptoms begin after age 40. The initial symptom is usually dull pain, insidious in onset, felt deep in the lower lumbar or gluteal region, accompanied by low-back morning stiffness of up to a few hours’ duration that improves with activity and returns following inactivity. Within a few months, the pain has usually become persistent and bilateral. Nocturnal exacerbation of pain often forces the patient to rise and move around.
In some patients, bony tenderness (presumably reflecting enthesitis or osteitis) may accompany back pain or stiffness, whereas in others it may be the predominant complaint. Common sites include the costosternal junctions, spinous processes, iliac crests, greater trochanters, ischial tuberosities, tibial tubercles, and heels. Hip and shoulder (“root” joint) arthritis is considered part of the axial disease. Hip arthritis occurs in 25–35% of patients. Shoulder arthritis is much less common. Severe isolated hip arthritis or bony chest pain may be the presenting complaint, and symptomatic hip disease can dominate the clinical picture. Arthritis of peripheral joints other than the hips and shoulders, usually asymmetric, occurs in up to 30% of patients. Neck pain and stiffness from involvement of the cervical spine are usually relatively late manifestations but are occasionally dominant symptoms. Rare patients, particularly in the older age group, present with predominantly constitutional symptoms.
AS often has a juvenile onset in developing countries. Peripheral arthritis and enthesitis usually predominate, with axial symptoms supervening in late adolescence.
Initially, physical findings mirror the inflammatory process. The most specific findings involve loss of spinal mobility, with limitation of anterior and lateral flexion and extension of the lumbar spine and of chest expansion. Limitation of motion is usually out of proportion to the degree of bony ankylosis and is thought to possibly reflect muscle spasm secondary to pain and inflammation. Pain in the sacroiliac joints may be elicited either with direct pressure or with stress on the joints. In addition, there is commonly tenderness upon palpation of the posterior spinous processes and other sites of symptomatic bony tenderness.
The modified Schober test is a useful measure of lumbar spine flexion. The patient stands erect, with heels together, and marks are made on the spine at the lumbosacral junction (identified by a horizontal line between the posterosuperior iliac spines) and 10 cm above. The patient then bends forward maximally with knees fully extended, and the distance between the two marks is measured. This distance increases by ≥5 cm in the case of normal mobility and by <4 cm in the case of decreased mobility. Chest expansion is measured as the difference between maximal inspiration and maximal forced expiration in the fourth intercostal space in males or just below the breasts in females, with the patient’s hands resting on or just behind the head. Normal chest expansion is ≥5 cm. Lateral bending measures the distance the patient’s middle finger travels down the leg with maximal lateral bending. Normal is >10 cm.
Limitation or pain with motion of the hips or shoulders is usually present if these joints are involved. It should be emphasized that early in the course of mild cases, symptoms may be subtle and nonspecific, and the physical examination may be unrevealing.
The course of the disease is extremely variable, ranging from the individual with mild stiffness and normal radiographs to the patient with a totally fused spine and severe bilateral hip arthritis, accompanied by severe peripheral arthritis and extraarticular manifestations. Pain tends to be persistent early in the disease and intermittent later, with alternating exacerbations and quiescent periods. In a typical severe untreated case with progression of the spondylitis to syndesmophyte formation, the patient’s posture undergoes characteristic changes, with obliterated lumbar lordosis, buttock atrophy, and accentuated thoracic kyphosis. There may be a forward stoop of the neck or flexion contractures at the hips, compensated by flexion at the knees. Disease progression can be estimated clinically from loss of height, limitation of chest expansion and spinal flexion, and occiput-to-wall distance. Occasional individuals are encountered with advanced deformities who report having never had significant symptoms.
The factors most predictive of radiographic progression (see below) are the presence of existing syndesmophytes, high inflammatory makers, and smoking. In some but not all studies, onset of AS in adolescence and early hip involvement correlate with a worse prognosis. In women, AS tends to progress less frequently to total spinal ankylosis, although there may be an increased prevalence of isolated cervical ankylosis and peripheral arthritis. In industrialized countries, peripheral arthritis (distal to hips and shoulders) occurs in less than one-half of patients with AS, usually as a late manifestation, whereas in developing countries, the prevalence is much higher, with onset typically early in the disease course. Pregnancy has no consistent effect on AS, with symptoms improving, remaining the same, or deteriorating in one-third of pregnant patients, respectively.
The most serious complication of the spinal disease is spinal fracture, which can occur with even minor trauma to the rigid, osteoporotic spine. The lower cervical spine is most commonly involved. These fractures are often displaced and cause spinal cord injury. A recent survey suggested a >10% lifetime risk of fracture. Occasionally, fracture through a diskovertebral junction and adjacent neural arch, termed pseudoarthrosis, most common in the thoracolumbar spine, can be an unrecognized source of persistent localized pain and/or neurologic dysfunction. Wedging of thoracic vertebrae is common and correlates with accentuated kyphosis.
The most common extraarticular manifestation is acute anterior uveitis, which occurs in up to 40% of patients and can antedate the spondylitis. Attacks are typically unilateral, causing pain, photophobia, and increased lacrimation. These tend to recur, often in the opposite eye. Cataracts and secondary glaucoma are not uncommon sequelae. Up to 60% of patients with AS have inflammation in the colon or ileum. This is usually asymptomatic, but frank IBD occurs in 5–10% of patients with AS (see “Enteropathic Arthritis,” below). About 10% of patients meeting criteria for AS have psoriasis (see “Psoriatic Arthritis,” below). Aortic insufficiency, sometimes leading to congestive heart failure, occurs in a small percentage of patients, occasionally early. Third-degree heart block may occur alone or together with aortic insufficiency. Subclinical pulmonary lesions and cardiac dysfunction may be relatively common. Cauda equina syndrome and upper pulmonary lobe fibrosis are rare late complications. Retroperitoneal fibrosis is a rare associated condition. Prostatitis has been reported to have an increased prevalence. Amyloidosis is rare (Chap. 137).
Several validated measures of disease activity and functional outcome are in widespread use in the study and management of AS, particularly the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) and the Ankylosing Spondylitis Disease Activity Score (ASDAS), both measures of disease activity; the Bath Ankylosing Spondylitis Functional Index (BASFI), a measure of limitation in activities of daily living; and several measures of radiographic changes. The Harris hip score, although not specific for AS, can be helpful. Despite persistence of the disease, most patients remain gainfully employed. Some but not all studies of survival in AS have suggested that AS shortens life span, compared with the general population. Mortality attributable to AS is largely the result of spinal trauma, aortic insufficiency, respiratory failure, amyloid nephropathy, or complications of therapy such as upper gastrointestinal hemorrhage. The impact of anti-TNF therapy on outcome and mortality is not yet known, except for significantly improved work productivity.
LABORATORY FINDINGS
No laboratory test is diagnostic of AS. In most ethnic groups, HLA-B27 is present in 80–90% of patients. Erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) are often, but not always, elevated. Mild anemia may be present. Patients with severe disease may show an elevated alkaline phosphatase level. Elevated serum IgA levels are common. Rheumatoid factor, anti-cyclic citrullinated peptide (CCP), and antinuclear antibodies (ANAs) are largely absent unless caused by a coexistent disease, although ANAs may appear with anti-TNF therapy. Circulating levels of CD8+ T cells tend to be low, and serum matrix metalloproteinase 3 levels correlate with disease activity. Synovial fluid from peripheral joints in AS is nonspecifically inflammatory. In cases with restriction of chest wall motion, decreased vital capacity and increased functional residual capacity are common, but airflow is normal and ventilatory function is usually well maintained.
RADIOGRAPHIC FINDINGS
Radiographically demonstrable sacroiliitis, usually symmetric, is eventually present in AS. The earliest changes by standard radiography are blurring of the cortical margins of the subchondral bone, followed by erosions and sclerosis. Progression of the erosions leads to “pseudowidening” of the joint space; as fibrous and then bony ankylosis supervene, the joints may become obliterated.
In the lumbar spine, progression of the disease leads to straightening, caused by loss of lordosis, and reactive sclerosis, caused by osteitis of the anterior corners of the vertebral bodies with subsequent erosion, leading to “squaring” or even “barreling” of one or more vertebral bodies. Progressive ossification leads to eventual formation of marginal syndesmophytes, visible on plain films as bony bridges connecting successive vertebral bodies anteriorly and laterally.
Years may elapse before unequivocal sacroiliac abnormalities are evident on plain radiographs, and consequently, MRI is being increasingly used in diagnosing AS. Active sacroiliitis is best visualized by dynamic MRI with fat saturation, either T2-weighed turbo spin-echo sequence or short tau inversion recovery (STIR) with high resolution, or T1-weighted images with contrast enhancement. These techniques sensitively identify early intraarticular inflammation, cartilage changes, and underlying bone marrow edema in sacroiliitis (Fig. 384-1). They are also highly sensitive for evaluation of acute and chronic spinal changes (Fig. 384-2).
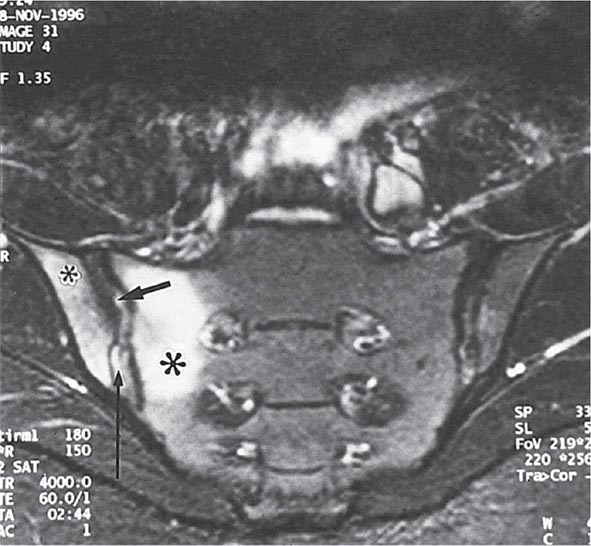
FIGURE 384-1 Early sacroiliitis in a patient with ankylosing spondylitis, indicated by prominent edema in the juxtaarticular bone marrow (asterisks), synovium and joint capsule (thin arrow), and interosseous ligaments (thick arrow) on a short tau inversion recovery (STIR) magnetic resonance image. (From M Bollow et al: Zeitschrift für Rheumatologie 58:61, 1999. Reproduced with permission.)
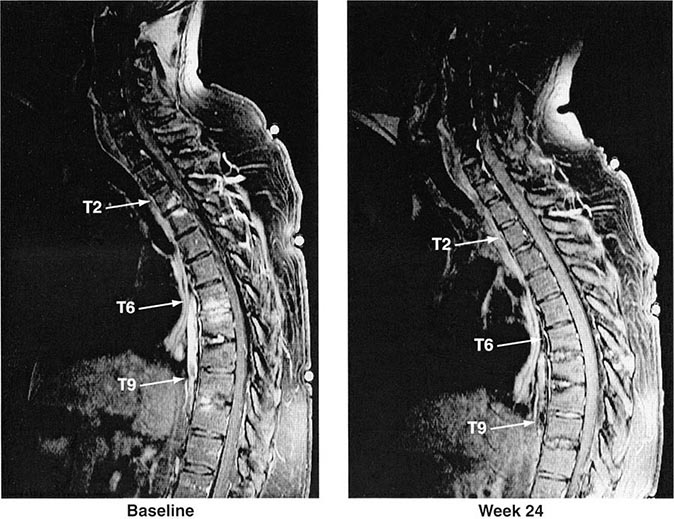
FIGURE 384-2 Spinal inflammation (spondylodiskitis) in a patient with ankylosing spondylitis and its dramatic response to treatment with infliximab. Gadolinium-enhanced T1-weighted magnetic resonance images, with fat saturation, at baseline and after 24 weeks of infliximab therapy. (From J Braun et al: Arthritis Rheum 54:1646, 2006.)
Reduced bone mineral density can be detected by dual-energy x-ray absorptiometry of the femoral neck and the lumbar spine. Use of a lateral projection of the L3 vertebral body can prevent falsely elevated readings related to spinal ossification.
DIAGNOSIS
It is important to establish the diagnosis of early AS before the development of irreversible deformity. This goal presents a challenge for several reasons: (1) Back pain is very common, but AS is much less common; (2) an early presumptive diagnosis often relies on clinical grounds requiring considerable expertise; and (3) young individuals with symptoms of AS often do not seek medical care. The widely used modified New York criteria (1984) are based on the presence of definite radiographic sacroiliitis and are too insensitive in early or mild cases. In 2009, new criteria for axial SpA were proposed by the Assessment of Spondyloarthritis International Society (ASAS) (Table 384-1). They are applicable to individuals with ≥3 months of back pain with age of onset <45 years old. Active inflammation of the sacroiliac joints as determined by dynamic MRI is considered equivalent to definite radiographic sacroiliitis (see below).
AS must be differentiated from numerous other causes of low-back pain, some far more common than AS. To qualify as the criterion for inflammatory back pain of axial SpA (Table 384-1), the chronic (≥3 months) back pain should have four or more of the following characteristic features: (1) age of onset <40 years old; (2) insidious onset; (3) improvement with exercise; (4) no improvement with rest; and (5) pain at night with improvement upon getting up. Other common features of inflammatory back pain include morning stiffness >30 min, awakening from back pain during only the second half of the night, and alternating buttock pain. In clinical decision-making, all of these features are additive. The most common causes of back pain other than AS are primarily mechanical or degenerative rather than primarily inflammatory and tend not to show clustering of these features.
Less-common metabolic, infectious, and malignant causes of back pain must also be differentiated from AS, including infectious spondylitis, spondylodiskitis, and sacroiliitis, and primary or metastatic tumor. Ochronosis can produce a phenotype that is clinically and radiographically similar to AS. Calcification and ossification of paraspinous ligaments occur in diffuse idiopathic skeletal hyperostosis (DISH), which occurs in the middle-aged and elderly and is usually not symptomatic. Ligamentous calcification gives the appearance of “flowing wax” on the anterior bodies of the vertebrae. Intervertebral disk spaces are preserved, and sacroiliac and apophyseal joints appear normal, helping to differentiate DISH from spondylosis and from AS, respectively.
REACTIVE ARTHRITIS
Reactive arthritis (ReA) refers to acute nonpurulent arthritis complicating an infection elsewhere in the body. In recent years, the term has been used primarily to refer to SpA following enteric or urogenital infections.
Other forms of reactive and infection-related arthritis not associated with B27 and showing a spectrum of clinical features different from SpA, such as Lyme disease and rheumatic fever, are discussed in Chaps. 210 and 381.
HISTORIC BACKGROUND
The association of acute arthritis with episodes of diarrhea or urethritis has been recognized for centuries. A large number of cases during World Wars I and II focused attention on the triad of arthritis, urethritis, and conjunctivitis, often with additional mucocutaneous lesions, which became widely known by eponyms that are now of historic interest only.
The identification of bacterial species capable of triggering the clinical syndrome and the finding that many patients possess the B27 antigen led to the unifying concept of ReA as a clinical syndrome triggered by specific etiologic agents in a genetically susceptible host. A similar spectrum of clinical manifestations can be triggered by enteric infection with any of several Shigella, Salmonella, Yersinia, and Campylobacter species; by genital infection with Chlamydia trachomatis; and by other agents as well. The triad of arthritis, urethritis, and conjunctivitis represents a small part of the spectrum of the clinical manifestations of ReA and only a minority of patients present with this “classic triad” of symptoms. Although emerging data suggest that asymptomatic Chlamydia trachomatis infections might trigger ReA, for the purposes of this chapter, the use of the term ReA will be restricted to those cases of SpA in which there is at least presumptive evidence for a related symptomatic antecedent infection. Patients with clinical features of ReA who lack evidence of an antecedent infection will be considered to have undifferentiated spondyloarthritis, discussed below.
EPIDEMIOLOGY
Initial reports may have overestimated the association of ReA with HLA-B27, since 60–85% of patients who developed ReA triggered by Shigella, Yersinia, or Chlamydia were B27-positive. However, other studies demonstrated a lower prevalence of B27 in ReA triggered by Salmonella, with one study suggesting no association in Campylobacter-induced ReA. Several more recent community-based or common-source epidemic studies demonstrated that the prevalence of B27 in ReA was below 50%. The most common age range is 18–40 years, but ReA can occur rarely in children and occasionally in older adults.
The attack rate of postenteric ReA generally ranges from 1% to about 30% depending on the study and causative organism, whereas the attack rate of postchlamydial ReA is about 4–8%. The gender ratio in ReA following enteric infection is nearly 1:1, whereas venereally acquired ReA occurs mainly in men. The overall prevalence and incidence of ReA are difficult to assess because of the lack of validated diagnostic criteria, variable prevalence and arthritogenic potential of the triggering infectious agents, and inconstant genetic susceptibility factors in different populations. In Scandinavia, an annual incidence of 10–28:100,000 has been reported. The spondyloarthritides were formerly almost unknown in sub-Saharan Africa. However, ReA and other peripheral SpAs have now become the most common rheumatic diseases in Africans in the wake of the AIDS epidemic, without association to B27, which is very rare in these populations. ReA is often the first manifestation of HIV infection and often remits with disease progression. In contrast, Western white patients with HIV and SpA are usually B27-positive, and the arthritis flares as AIDS advances.
PATHOLOGY
Synovial histology is similar to that of other SpAs. Enthesitis shows increased vascularity and macrophage infiltration of fibrocartilage. Microscopic histopathologic evidence of inflammation mimicking IBD has routinely been demonstrated in the colon and ileum of patients with postenteric ReA and less commonly in postvenereal ReA. The skin lesions of keratoderma blennorrhagica, associated mainly with venereally acquired ReA, are histologically indistinguishable from pustular psoriatic lesions.
ETIOLOGY AND PATHOGENESIS
The bacteria identified as definitive triggers of ReA include several Salmonella spp., Shigella spp., Yersinia enterocolitica, Yersinia pseudotuberculosis, Campylobacter jejuni, and Chlamydia trachomatis. These triggering microbes are gram-negative bacteria with a lipopolysaccharide component to their cell walls. All four Shigella species (Shigella sonnei, Shigella boydii, Shigella flexneri, and Shigella dysenteriae) have been implicated in cases of ReA, with S. flexneri and S. sonnei being the most common. After Salmonella infection, individuals of Caucasian descent may be more likely than those of Asian descent to develop ReA. Children may be less susceptible to ReA caused by Salmonella and Campylobacter. Yersinia species in Europe and Scandinavia may have greater arthritogenic potential than in other parts of the world, and C. trachomatis appears to be a common cause worldwide. The ocular serovars of C. trachomatis appear to be particularly, perhaps uniquely, arthritogenic.
There is also evidence implicating Clostridium difficile, Campylobacter coli, certain toxigenic Escherichia coli, and possibly Ureaplasma urealyticum and Mycoplasma genitalium as potential triggers of ReA. Chlamydia pneumoniae is another trigger of ReA, albeit far less common than C. trachomatis. There have also been numerous isolated reports of acute arthritis preceded by other bacterial, viral, or parasitic infections, and even following intravesicular bacillus Calmette-Guérin (BCG) treatment for bladder cancer.
It has not been determined whether ReA occurs by the same pathogenic mechanism following infection with each of these microorganisms, nor has the mechanism been elucidated in the case of any one of the known bacterial triggers. Most, if not all, of the organisms well established to be triggers share a capacity to attack mucosal surfaces, to invade host cells, and to survive intracellularly. Antigens from Chlamydia, Yersinia, Salmonella, and Shigella have been shown to be present in the synovium and/or synovial fluid leukocytes of patients with ReA for long periods following the acute attack. In ReA triggered by Y. enterocolitica, bacterial lipopolysaccharide (LPS) and heat-shock protein antigens have been found in peripheral blood cells years after the triggering infection. Yersinia DNA and C. trachomatis DNA and RNA have been detected in synovial tissue from ReA patients, suggesting the presence of viable organisms despite uniform failure to culture the organism from these specimens. In C. trachomatis–induced ReA specifically, the bacterial load in synovial tissue of patients with remitting disease is lower than that of active disease, but mRNAs encoding proinflammatory proteins are equal to or higher than those of active disease. The specificity of these findings is unclear, however, since chromosomal bacterial DNA and 16S rRNA from a very wide variety of bacteria have also been found in synovium in other rheumatic diseases, albeit less frequently. In several older studies, synovial T cells that specifically responded to antigens of the inciting organism were reported and characterized as predominantly CD4+ with a TH2 or T regulatory phenotype. More recent work has documented high levels of IL-17 in ReA synovial fluid, but the source has not been identified. HLA-B27 seems to be associated with more severe and chronic forms of the “classic triad” of ReA, but its pathogenic role remains to be determined. HLA-B27 significantly prolongs the intracellular survival of Y. enterocolitica and Salmonella enteritidis in human and mouse cell lines. Prolonged intracellular bacterial survival, promoted by B27, other factors, or both, may permit trafficking of infected leukocytes from the site of primary infection to joints, where an innate and/or adaptive immune response to persistent bacterial antigens may then promote arthritis.
CLINICAL FEATURES
The clinical manifestations of ReA constitute a spectrum that ranges from an isolated, transient monoarthritis or enthesitis to severe multisystem disease. A careful history will usually elicit evidence of an antecedent infection 1–4 weeks before onset of symptoms of the reactive disease, particularly in postenteric ReA. However, in a sizable minority, no clinical or laboratory evidence of an antecedent infection can be found, particularly in the case of postchlamydial ReA. In cases of presumed venereally acquired reactive disease, there is often a history of a recent new sexual partner, even without laboratory evidence of infection.
Constitutional symptoms are common, including fatigue, malaise, fever, and weight loss. The musculoskeletal symptoms are usually acute in onset. Arthritis is usually asymmetric and additive, with involvement of new joints occurring over a few days to 1–2 weeks. The joints of the lower extremities, especially the knee, ankle, and subtalar, metatarsophalangeal, and toe interphalangeal joints, are most commonly involved, but the wrist and fingers can be involved as well. The arthritis is usually quite painful, and tense joint effusions are not uncommon, especially in the knee. Dactylitis, or “sausage digit,” a diffuse swelling of a solitary finger or toe, is a distinctive feature of ReA and other peripheral spondyloarthritides but can be seen in polyarticular gout and sarcoidosis. Tendinitis and fasciitis are particularly characteristic lesions, producing pain at multiple insertion sites (entheses), especially the Achilles insertion, the plantar fascia, and sites along the axial skeleton. Spinal, low-back, and buttock pain are quite common and may be caused by insertional inflammation, muscle spasm, acute sacroiliitis, or, presumably, arthritis in intervertebral joints.
Urogenital lesions may occur throughout the course of the disease. In males, urethritis may be marked or relatively asymptomatic and may be either an accompaniment of the triggering infection or a result of the reactive phase of the disease; interestingly, it occurs in both postvenereal and postenteric ReA. Prostatitis is also common. Similarly, in females, cervicitis or salpingitis may be caused either by the infectious trigger or by the sterile reactive process.
Ocular disease is common, ranging from transient, asymptomatic conjunctivitis to an aggressive anterior uveitis that occasionally proves refractory to treatment and may result in blindness.
Mucocutaneous lesions are frequent. Oral ulcers tend to be superficial, transient, and often asymptomatic. The characteristic skin lesions, keratoderma blennorrhagica, consist of vesicles and/or pustules that become hyperkeratotic, ultimately forming a crust before disappearing. They are most common on the palms and soles but may occur elsewhere as well. In patients with HIV infection, these lesions are often extremely severe and extensive, sometimes dominating the clinical picture (Chap. 226). Lesions may occur on the glans penis, termed circinate balanitis; these consist of vesicles that quickly rupture to form painless superficial erosions, which in circumcised individuals can form crusts similar to those of keratoderma blennorrhagica. Nail changes are common and consist of onycholysis, distal yellowish discoloration, and/or heaped-up hyperkeratosis.
Less-frequent or rare manifestations of ReA include cardiac conduction defects, aortic insufficiency, central or peripheral nervous system lesions, and pleuropulmonary infiltrates.
Arthritis typically persists for 3–5 months, but more chronic courses do occur. Chronic joint symptoms persist in about 15% of patients and in up to 60% of patients in hospital-based series, but these tend to be less severe than in the acute stage. Recurrences of the acute syndrome are also common. Work disability or forced change in occupation is common in those with persistent joint symptoms. Chronic heel pain is often particularly distressing. Low-back pain, sacroiliitis, and frank AS are also common sequelae. In most studies, HLA-B27–positive patients have shown a worse outcome than B27-negative patients. Patients with Yersinia– or Salmonella-induced arthritis have less chronic disease than those whose initial episode follows epidemic shigellosis.
LABORATORY AND RADIOGRAPHIC FINDINGS
The ESR and acute-phase reactants are usually elevated during the acute phase of the disease, often markedly so. Mild anemia may be present. Synovial fluid is nonspecifically inflammatory. In most ethnic groups, 30–50% of the patients are B27-positive. The triggering infection usually does not persist at the site of primary mucosal infection through the time of onset of the reactive disease, but it may be possible to culture the organism, e.g., in the case of Yersinia– or Chlamydia-induced disease. Serologic evidence of exposure to one of the causative organisms with elevation of antibodies is nonspecific and is of questionable utility. Polymerase chain reaction (PCR) for chlamydial DNA in first-voided urine specimens may have high sensitivity in the acute stage but is less useful with chronic disease.
In early or mild disease, radiographic changes may be absent or confined to juxtaarticular osteoporosis. With long-standing persistent disease, radiographic features share those of psoriatic arthritis; marginal erosions and loss of joint space can be seen in affected joints. Periostitis with reactive new bone formation is characteristic, as in all the SpAs. Spurs at the insertion of the plantar fascia are common.
Sacroiliitis and spondylitis may be seen as late sequelae. Sacroiliitis is more commonly asymmetric than in AS, and spondylitis, rather than ascending symmetrically, can begin anywhere along the lumbar spine. The syndesmophytes are described as nonmarginal; they are coarse, asymmetric, and “comma”-shaped, arising from the middle of a vertebral body, a pattern less commonly seen in primary AS. Progression to spinal fusion is uncommon.
DIAGNOSIS
ReA is a clinical diagnosis with no definitively diagnostic laboratory test or radiographic finding. The diagnosis should be entertained in any patient with an acute inflammatory, asymmetric, additive arthritis or tendinitis. The evaluation should include questioning regarding possible triggering events such as an episode of diarrhea or dysuria. On physical examination, attention must be paid to the distribution of the joint and tendon involvement and to possible sites of extraarticular involvement, such as the eyes, mucous membranes, skin, nails, and genitalia. Synovial fluid analysis may be helpful in excluding septic or crystal-induced arthritis. Culture, serology, or molecular methods may help to identify a triggering infection, but they cannot be relied upon.
Although typing for B27 has low negative predictive value in ReA, it may have prognostic significance in terms of severity, chronicity, and the propensity for spondylitis and uveitis. Furthermore, if positive, it can be helpful diagnostically in atypical cases. HIV testing is often indicated and may be necessary in order to select appropriate therapy.
It is important to differentiate ReA from disseminated gonococcal disease (Chap. 181), both of which can be venereally acquired and associated with urethritis. Unlike ReA, gonococcal arthritis and tenosynovitis tend to involve both upper and lower extremities equally, spare the axial skeleton, and be associated with characteristic vesicular skin lesions. A positive gonococcal culture from the urethra or cervix does not exclude a diagnosis of ReA; however, culturing gonococci from blood, skin lesion, or synovium establishes the diagnosis of disseminated gonococcal disease. PCR assay for Neisseria gonorrhoeae and C. trachomatis may be helpful. Occasionally, only a therapeutic trial of antibiotics can distinguish the two.
ReA shares many features in common with psoriatic arthropathy. However, psoriatic arthritis is usually gradual in onset; the arthritis tends to affect primarily the upper extremities; there is less associated periarthritis; and there are usually no associated mouth ulcers, urethritis, or bowel symptoms.
PSORIATIC ARTHRITIS
Psoriatic arthritis (PsA) refers to an inflammatory musculoskeletal disease that has both autoimmune and autoinflammatory features characteristically occurring in individuals with psoriasis.
HISTORIC BACKGROUND
The association between arthritis and psoriasis was noted in the nineteenth century. In the 1960s, on the basis of epidemiologic and clinical studies, it became clear that unlike RA, the arthritis associated with psoriasis was usually seronegative, often involved the distal interphalangeal (DIP) joints of the fingers and the spine and sacroiliac joints, had distinctive radiographic features, and showed considerable familial aggregation. In the 1970s, PsA was included in the broader category of the spondyloarthritides because of features similar to those of AS and ReA.
EPIDEMIOLOGY
Estimates of the prevalence of PsA among individuals with psoriasis range from 5 to 42%. The prevalence of PsA appears to be increasing in parallel with disease awareness; recent data using screening tools suggest that 20% or more of patients with psoriasis have undiagnosed PsA. The duration and severity of psoriasis increase one’s likelihood of developing PsA. In white populations, psoriasis is estimated to have a prevalence of 1–3%. Psoriasis and PsA are less common in other races in the absence of HIV infection, and the prevalence of PsA in individuals with psoriasis may be less common. First-degree relatives of PsA patients have an elevated risk for psoriasis, for PsA itself, and for other forms of SpA. Of patients with psoriasis, up to 30% have an affected first-degree relative. In monozygotic twins, the reported concordance for psoriasis varies from 35 to 72%, and for PsA from 10 to 30%. A variety of HLA associations have been found. The HLA-Cw*0602 gene is directly associated with psoriasis, particularly familial juvenile-onset (type I) psoriasis. HLA-B27 is associated with psoriatic spondylitis (see below). HLA-DR7, -DQ3, and -B57 are associated with PsA because of linkage disequilibrium with Cw6. Other associations with PsA include HLA-B13, -B37, -B38, -B39, -C12, and -DR4. A recent genome-wide scan found association of both psoriasis and PsA with a polymorphism at the HCP5 locus closely linked to HLA-B, and also to IL-23R, IL-12B (chromosome 5q31), IL-13, and several other chromosomal regions. Certain genetic loci are associated with PsA but not psoriasis, e.g., RUNX3 and IL-13.
PATHOLOGY
The inflamed synovium in PsA resembles that of RA, although with somewhat less hyperplasia and cellularity than in RA. As noted with AS above, the synovial vascular pattern in PsA is generally greater and more tortuous than in RA, independent of disease duration. Some studies have indicated a higher tendency to synovial fibrosis in PsA. Unlike RA, PsA shows prominent enthesitis, with histology similar to that of the other spondyloarthritides.
PATHOGENESIS
PsA is almost certainly immune-mediated and probably shares pathogenic mechanisms with psoriasis. PsA synovium is characterized by lining layer hyperplasia; diffuse infiltration with T cells, B cells, macrophages, and NK receptor–expressing cells, with upregulation of leukocyte homing receptors; and neutrophil proliferation with angiogenesis. Clonally expanded T cell subpopulations are frequent and have been demonstrated both in the synovium and the skin. Plasmacytoid dendritic cells are thought to play a key role in psoriasis, and there is some evidence for their participation in PsA. There is abundant synovial overexpression of proinflammatory cytokines, and synovial tissue staining has identified an overexpression of monocyte-derived cytokines, such as myeloid-related protein (S100A8/A9). Interferon γ, TNF-α, and IL-1β, -2, -6, -8, -10, -12, -13, and -15 are found in PsA synovium or synovial fluid. TH17-derived cytokines are important in PsA, given the genetic association with genes in the IL-12/IL-23 axis and the therapeutic response to an antibody to the shared IL-12/23 p40 subunit (see below). TH17 cells have been identified from the dermal extracts of psoriatic lesions and the synovial fluid of PsA patients. The majority of these CD4+ IL-17+ T cells are of memory phenotype (CD4RO[+]CD45RA[–]CD11a[+]). Consistent with the extensive bone remodeling in PsA, patients with PsA have been found to have a marked increase in osteoclastic precursors in peripheral blood and upregulation of receptor activator of nuclear factor κβ ligand (RANKL) in the synovial lining layer. Increased serum levels of TNF-α, RANKL, leptin, and omentin positively correlate with these osteoclastic precursors.
CLINICAL FEATURES
In 60–70% of cases, psoriasis precedes joint disease. In 15–20% of cases, the two manifestations appear within 1 year of each other. In about 15–20% of cases, the arthritis precedes the onset of psoriasis and can present a diagnostic challenge. The frequency in men and women is almost equal, although the frequency of disease patterns differs somewhat in the two sexes. The disease can begin in childhood or late in life but typically begins in the fourth or fifth decade, at an average age of 37 years.
The spectrum of arthropathy associated with psoriasis is quite broad. Many classification schemes have been proposed. In the original scheme of Wright and Moll, five patterns are described: (1) arthritis of the DIP joints; (2) asymmetric oligoarthritis; (3) symmetric polyarthritis similar to RA; (4) axial involvement (spine and sacroiliac joints); and (5) arthritis mutilans, a highly destructive form of disease. These patterns frequently coexist, and the pattern that persists chronically often differs from that of the initial presentation. A simpler scheme in recent use contains three patterns: oligoarthritis, polyarthritis, and axial arthritis.
Nail changes in the fingers or toes occur in up to 90% of patients with PsA, compared with 40% of psoriatic patients without arthritis, and pustular psoriasis is said to be associated with more severe arthritis. Several articular features distinguish PsA from other joint disorders; such hallmark features include dactylitis and enthesitis. Dactylitis occurs in >30%; enthesitis and tenosynovitis are also common and are probably present in most patients, although often not appreciated on physical examination. Shortening of digits because of underlying osteolysis is particularly characteristic of PsA (Fig. 384-3), and there is a much greater tendency than in RA for both fibrous and bony ankylosis of small joints. Rapid ankylosis of one or more proximal interphalangeal (PIP) joints early in the course of disease is not uncommon. Back and neck pain and stiffness are also common in PsA.
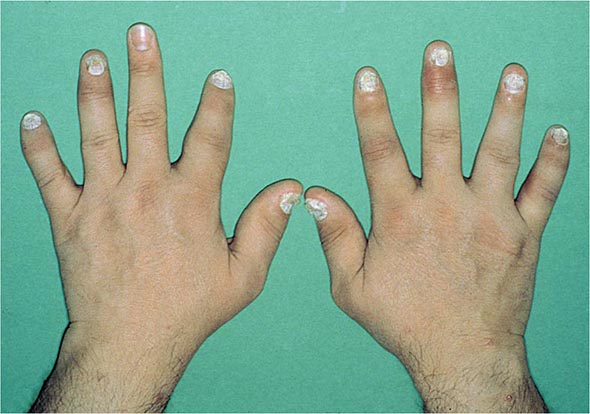
FIGURE 384-3 Characteristic lesions of psoriatic arthritis. Inflammation is prominent in the distal interphalangeal joints (left 5th, 4th, 2nd; right 2nd, 3rd, and 5th) and proximal interphalangeal joints (left 2nd, right 2nd, 4th, and 5th). There is dactylitis in the left 2nd finger and thumb, with pronounced telescoping of the left 2nd finger. Nail dystrophy (hyperkeratosis and onycholysis) affects each of the fingers except the left 3rd finger, the only finger without arthritis. (Courtesy of Donald Raddatz, MD; with permission.)
Arthropathy confined to the DIP joints occurs in about 5% of cases. Accompanying nail changes in the affected digits are almost always present. These joints are also often affected in the other patterns of PsA. Approximately 30% of patients have asymmetric oligoarthritis. This pattern commonly involves a knee or another large joint with a few small joints in the fingers or toes, often with dactylitis. Symmetric polyarthritis occurs in about 40% of PsA patients at presentation. It may be indistinguishable from RA in terms of the joints involved, but other features characteristic of PsA are usually also present. Almost any peripheral joint can be involved. Axial arthropathy without peripheral involvement is found in about 5% of PsA patients. It may be clinically indistinguishable from idiopathic AS, although more neck involvement and less thoracolumbar spinal involvement are characteristic, and nail changes are not found in idiopathic AS. A small percentage of PsA patients have arthritis mutilans, in which there can be widespread shortening of digits (“telescoping”), sometimes coexisting with ankylosis and contractures in other digits.
Six patterns of nail involvement are identified: pitting, horizontal ridging, onycholysis, yellowish discoloration of the nail margins, dystrophic hyperkeratosis, and combinations of these findings. Other extraarticular manifestations of the spondyloarthritides are common. Eye involvement, either conjunctivitis or uveitis, is reported in 7–33% of PsA patients. Unlike the uveitis associated with AS, the uveitis in PsA is more often bilateral, chronic, and/or posterior. Aortic valve insufficiency has been found in <4% of patients, usually after long-standing disease.
Widely varying estimates of clinical outcome have been reported in PsA. At its worst, severe PsA with arthritis mutilans is potentially at least as crippling and ultimately fatal as severe RA. Unlike RA, however, many patients with PsA experience temporary remissions. Overall, erosive disease develops in the majority of patients, progressive disease with deformity and disability is common, and in some large published series, mortality was found to be significantly increased compared with the general population. There appears to be a greater incidence of cardiovascular death in psoriatic disease.
The psoriasis and associated arthropathy seen with HIV infection both tend to be severe and can occur in populations with very little psoriasis in noninfected individuals. Severe enthesopathy, dactylitis, and rapidly progressive joint destruction are seen, but axial involvement is very rare. This condition is prevented by or responds well to antiretroviral therapy.
LABORATORY AND RADIOGRAPHIC FINDINGS
There are no laboratory tests diagnostic of PsA. ESR and CRP are often elevated. A small percentage of patients may have low titers of rheumatoid factor or antinuclear antibodies. About 10% of patients have anti-CCP antibodies. Uric acid may be elevated in the presence of extensive psoriasis. HLA-B27 is found in 50–70% of patients with axial disease, but in ≤20% of patients with only peripheral joint involvement.
The peripheral and axial arthropathies in PsA show a number of radiographic features that distinguish them from RA and AS, respectively. Characteristics of peripheral PsA include DIP involvement, including the classic “pencil-in-cup” deformity; marginal erosions with adjacent bony proliferation (“whiskering”); small-joint ankylosis; osteolysis of phalangeal and metacarpal bone, with telescoping of digits; and periostitis and proliferative new bone at sites of enthesitis. Characteristics of axial PsA include asymmetric sacroiliitis. When compared with idiopathic AS, axial PsA manifests less zygapophyseal joint arthritis; nonmarginal, bulky, “comma”-shaped syndesmophytes that tend to be fewer and less symmetric and delicate than the marginal syndesmophytes of AS; fluffy hyperperiostosis on anterior vertebral bodies; severe cervical spine involvement, with a tendency to atlantoaxial subluxation but relative sparing of the thoracolumbar spine; and paravertebral ossification. Ultrasound and MRI both readily demonstrate enthesitis and tendon sheath effusions that can be difficult to assess on physical examination. A recent MRI study of 68 PsA patients found sacroiliitis in 35%, unrelated to B27 but correlated with restricted spinal movement.
DIAGNOSIS
Classification criteria for PsA were published in 2006 (Classification of Psoriatic Arthritis [CASPAR] criteria) that have been widely accepted (Table 384-2). The sensitivity and specificity of these criteria exceed 90%, and they are useful for early diagnosis. The criteria are based on the history, presence of psoriasis, characteristic peripheral or spinal joint symptoms, signs, and imaging. Diagnosis can be challenging when the arthritis precedes psoriasis, the psoriasis is undiagnosed or obscure, or the joint involvement closely resembles another form of arthritis. A high index of suspicion is needed in any patient with an undiagnosed inflammatory arthropathy. The history should include inquiry about psoriasis in the patient and family members. Patients should be asked to disrobe for the physical examination, and psoriasiform lesions should be sought in the scalp, ears, umbilicus, and gluteal folds in addition to more accessible sites; the finger and toe nails should also be carefully examined. Axial symptoms or signs, dactylitis, enthesitis, ankylosis, the pattern of joint involvement, and characteristic radiographic changes can be helpful clues. The differential diagnosis includes all other forms of arthritis, which can occur coincidentally in individuals with psoriasis. The differential diagnosis of isolated DIP involvement is short. Osteoarthritis (Heberden’s nodes) is usually not inflammatory; gout involving more than one DIP joint often involves other sites and may be accompanied by tophi; the very rare entity multicentric reticulohistiocytosis involves other joints and has characteristic small pearly periungual skin nodules; and the uncommon entity inflammatory osteoarthritis, like the others, lacks the nail changes of PsA. Radiography can be helpful in all of these cases and in distinguishing between psoriatic spondylitis and idiopathic AS. A history of trauma to an affected joint preceding the onset of arthritis is said to occur more frequently in PsA than in other types of arthritis, perhaps reflecting the Koebner phenomenon in which psoriatic skin lesions can arise at sites of the skin trauma.
|
THE CASPAR (CLASSIFICATION CRITERIA FOR PSORIATIC ARTHRITIS) CRITERIAa |
aSpecificity of 99% and sensitivity of 91%. bCurrent psoriasis is assigned 2 points; all other features are assigned 1 point. cPsoriatic skin or scalp disease present at the time of examination, as judged by a rheumatologist or dermatologist. dHistory of psoriasis in a first- or second-degree relative. eOnycholysis, pitting, or hyperkeratosis. fSwelling of an entire digit. gIll-defined ossification near joint margins, excluding osteophyte formation.
Source: From W Taylor et al: Arthritis Rheum, 54:2665, 2006.
UNDIFFERENTIATED AND JUVENILE-ONSET SPONDYLOARTHRITIS
Many patients, usually young adults, present with some features of one or more of the spondyloarthritides discussed above. Until recently, these patients were said to have undifferentiated spondylo- arthritis, or simply spondyloarthritis, as defined by the 1991 European Spondyloarthropathy Study Group criteria. For example, a patient may present with inflammatory synovitis of one knee, Achilles tendinitis, and dactylitis of one digit. Some of these patients may have ReA in which the triggering infection remains clinically silent. In some other cases, the patient subsequently develops IBD or psoriasis, or the process eventually meets criteria for AS. This diagnosis of undifferentiated SpA was also commonly applied to patients with inflammatory back pain, who did meet modified New York criteria for AS. Most of these would now be classified under the new category of axial SpA (Table 384-1).
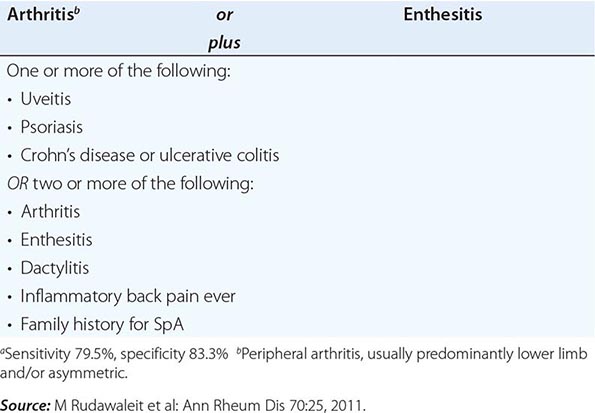
Comparable to the classification criteria for axial symptoms, the ASAS has recently formulated criteria for peripheral SpA. This is intended to exclude patients with axial symptoms and thus to divide the universe of patients with SpA into axial and exclusively peripheral subsets. These criteria are shown in Table 384-3.
|
ASAS CRITERIA FOR PERIPHERAL SPONDYLOARTHRITISa |
Approximately one-half of the patients with undifferentiated SpA are HLA-B27-positive, and thus the absence of B27 is not useful in establishing or excluding the diagnosis. In familial cases, which are much more frequently B27-positive, there is often eventual progression to classical AS.
In juvenile-onset SpA, which begins between ages 7 and 16, most commonly in boys (60–80%), an asymmetric, predominantly lower-extremity oligoarthritis and enthesitis without extraarticular features is the typical mode of presentation. The prevalence of B27 in this condition, which has been termed the seronegative enthesopathy and arthropathy (SEA) syndrome, is approximately 80%. Many, but not all, of these patients go on to develop AS in late adolescence or adulthood.
Management of undifferentiated SpA is similar to that of the other spondyloarthritides. Response to anti-TNF-α therapy has been documented, and this therapy is indicated in severe, persistent cases not responsive to other treatment.
Current pediatric textbooks and journals should be consulted for information on management of juvenile-onset SpA.
ENTEROPATHIC ARTHRITIS
HISTORIC BACKGROUND
A relationship between arthritis and IBD was observed in the 1930s. The relationship was further defined by the epidemiologic studies in the 1950s and 1960s and included in the concept of the spondylo- arthritides in the 1970s.
EPIDEMIOLOGY
Both of the common forms of IBD, ulcerative colitis (UC) and Crohn’s disease (CD) (Chap. 351), are associated with SpA. UC and CD both have an estimated prevalence of 0.05–0.1%, and the incidence of each is thought to have increased in recent decades. AS and peripheral arthritis are both associated with UC and CD. Wide variations have been reported in the estimated frequencies of these associations. In recent series, AS was diagnosed in 1–10%, and peripheral arthritis in 10–50% of patients with IBD. Inflammatory back pain and enthesopathy are common, and many patients have sacroiliitis on imaging studies.
The prevalence of UC or CD in patients with AS is thought to be 5–10%. However, investigation of unselected SpA patients by ileocolonoscopy has revealed that from one-third to two-thirds of patients with AS have subclinical intestinal inflammation that is evident either macroscopically or histologically. These lesions have also been found in patients with undifferentiated SpA or ReA (both enterically and urogenitally acquired).
Both UC and CD have a tendency to familial aggregation, more so for CD. HLA associations have been weak and inconsistent. HLA-B27 is found in up to 70% of patients with IBD and AS, but in ≤15% of patients with IBD and peripheral arthritis or IBD alone. Three alleles of the NOD2/CARD15 gene on chromosome 16 have been found in approximately one-half of patients with CD. These alleles are not associated with the spondyloarthritides per se. However, they are found significantly more often in (1) CD patients with sacroiliitis than in those without sacroiliitis, and (2) SpA patients with chronic inflammatory gut lesions than in those with normal gut histology. These associations are independent of HLA-B27. In addition to NOD2, over 100 other genes have been found to be associated with CD, UC, or both. Around 20 of these are also associated with AS.
PATHOLOGY
Available data for IBD-associated peripheral arthritis suggest a synovial histology similar to other spondyloarthritides. Association with arthropathy does not affect the gut histology of UC or CD (Chap. 351). The subclinical inflammatory lesions in the colon and distal ileum associated with SpA have been classified as either acute or chronic. The former resemble acute bacterial enteritis, with largely intact architecture and neutrophilic infiltration in the lamina propria. The latter resemble the lesions of CD, with distortion of villi and crypts, aphthoid ulceration, and mononuclear cell infiltration in the lamina propria.
PATHOGENESIS
Both IBD and SpA are immune-mediated, but the specific pathogenic mechanisms are poorly understood, and the connection between the two is obscure. The shared genetics evidently reflects shared pathogenic mechanisms. A number of rodent models showing various immune perturbations manifest both IBD and arthritis. Several lines of evidence indicate trafficking of leukocytes between the gut and the joint. Mucosal leukocytes from IBD patients have been shown to bind avidly to synovial vasculature through several different adhesion molecules. Macrophages expressing CD163 are prominent in the inflammatory lesions of both gut and synovium in the spondyloarthritides.
CLINICAL FEATURES
AS associated with IBD is clinically indistinguishable from idiopathic AS. It runs a course independent of the bowel disease, and in some patients, it precedes the onset of IBD, sometimes by many years. Peripheral arthritis may also begin before onset of overt bowel disease. The spectrum of peripheral arthritis includes acute self-limited attacks of oligoarthritis that often coincide with relapses of IBD, and more chronic and symmetric polyarticular arthritis that runs a course independent of IBD activity. The patterns of joint involvement are similar in UC and CD. In general, erosions and deformities are infrequent in IBD-associated peripheral arthritis, and joint surgery is infrequently required. Isolated destructive hip arthritis is a rare complication of CD, apparently distinct from osteonecrosis and septic arthritis. Dactylitis and enthesopathy are occasionally found. In addition to the ∼20% of IBD patients with SpA, a comparable percentage have arthralgias or fibromyalgia symptoms.
Other extraintestinal manifestations of IBD are seen in addition to arthropathy, including uveitis, pyoderma gangrenosum, erythema nodosum, and finger clubbing, all somewhat more commonly in CD than UC. The uveitis shares the features described above for PsA-associated uveitis.
LABORATORY AND RADIOGRAPHIC FINDINGS
Laboratory findings reflect the inflammatory and metabolic manifestations of IBD. Joint fluid is usually at least mildly inflammatory. Of patients with AS and IBD, 30–70% carry the HLA-B27 gene, compared with >85% of patients with AS alone and 50–70% of those with AS and psoriasis. Hence, definite or probable AS in a B27-negative individual in the absence of psoriasis should prompt a search for occult IBD. Radiographic changes in the axial skeleton are the same as in uncomplicated AS. Erosions are uncommon in peripheral arthritis but may occur, particularly in the metatarsophalangeal joints. Isolated destructive hip disease has been described.
DIAGNOSIS
Diarrhea and arthritis are both common conditions that can coexist for a variety of reasons. When etiopathogenically related, ReA and IBD-associated arthritis are the most common causes. Rare causes include celiac disease, blind loop syndromes, and Whipple’s disease. In most cases, diagnosis depends on investigation of the bowel disease.
SAPHO SYNDROME
The syndrome of synovitis, acne, pustulosis, hyperostosis, and osteitis (SAPHO) is characterized by a variety of skin and musculoskeletal manifestations. Dermatologic manifestations include palmoplantar pustulosis, acne conglobata, acne fulminans, and hidradenitis suppurativa. The main musculoskeletal findings are sternoclavicular and spinal hyperostosis, chronic recurrent foci of sterile osteomyelitis, and axial or peripheral arthritis. Cases with one or a few manifestations are probably the rule. The ESR is usually elevated, sometimes dramatically. In some cases, bacteria, most often Propionibacterium acnes, have been cultured from bone biopsy specimens and occasionally other sites. IBD was coexistent in 8% of patients in one large series. B27 is not associated. Either bone scan or computed tomography scan is helpful diagnostically. An MRI report described characteristic vertebral body corner cortical erosions in 12 of 12 patients. High-dose NSAIDs may provide relief from bone pain. A number of uncontrolled series and case reports describe successful therapy with pamidronate or other bisphosphonates. Response to anti-TNF-α therapy has also been observed, although in a few cases this has been associated with a flare of skin manifestations. Successful prolonged antibiotic therapy has also been reported. Recent reports suggest a possible autoinflammatory pathogenesis and successful treatment with the IL-1 receptor antagonist anakinra.
1Azathioprine, methotrexate, sulfasalazine, pamidronate, and thalidomide have not been approved for this purpose by the U.S. Food and Drug Administration at the time of publication.
385 |
The Vasculitis Syndromes |
DEFINITION
Vasculitis is a clinicopathologic process characterized by inflammation of and damage to blood vessels. The vessel lumen is usually compromised, and this is associated with ischemia of the tissues supplied by the involved vessel. A broad and heterogeneous group of syndromes may result from this process, since any type, size, and location of blood vessel may be involved. Vasculitis and its consequences may be the primary or sole manifestation of a disease; alternatively, vasculitis may be a secondary component of another disease. Vasculitis may be confined to a single organ, such as the skin, or it may simultaneously involve several organ systems.
CLASSIFICATION
A major feature of the vasculitic syndromes as a group is the fact that there is a great deal of heterogeneity at the same time as there is considerable overlap among them. This heterogeneity and overlap in addition to a lack of understanding of the pathogenesis of these syndromes have been major impediments to the development of a coherent classification system for these diseases. Table 385-1 lists the major vasculitis syndromes. The distinguishing and overlapping features of these syndromes are discussed below.
|
VASCULITIS SYNDROMES |

PATHOPHYSIOLOGY AND PATHOGENESIS
Generally, most of the vasculitic syndromes are assumed to be mediated at least in part by immunopathogenic mechanisms that occur in response to certain antigenic stimuli. However, evidence supporting this hypothesis is for the most part indirect and may reflect epiphenomena as opposed to true causality. Furthermore, it is unknown why some individuals might develop vasculitis in response to certain antigenic stimuli, whereas others do not. It is likely that a number of factors are involved in the ultimate expression of a vasculitic syndrome. These include the genetic predisposition, environmental exposures, and the regulatory mechanisms associated with immune response to certain antigens. Although immune complex formation, antineutrophil cytoplasmic antibodies (ANCA), and pathogenic T lymphocyte responses (Table 385-2) have been among the prominent hypothesized mechanisms, it is likely that the pathogenesis of individual forms of vasculitis is complex and varied.
|
POTENTIAL MECHANISMS OF VESSEL DAMAGE IN VASCULITIS SYNDROMES |
Source: Adapted from MC Sneller, AS Fauci: Med Clin North Am 81:221, 1997.
PATHOGENIC IMMUNE-COMPLEX FORMATION
Deposition of immune complexes was the first and most widely accepted pathogenic mechanism of vasculitis. However, the causal role of immune complexes has not been clearly established in most of the vasculitic syndromes. Circulating immune complexes need not result in deposition of the complexes in blood vessels with ensuing vasculitis, and many patients with active vasculitis do not have demonstrable circulating or deposited immune complexes. The actual antigen contained in the immune complex has only rarely been identified in vasculitic syndromes. In this regard, hepatitis B antigen has been identified in both the circulating and deposited immune complexes in a subset of patients who have features of a systemic vasculitis, most notably in polyarteritis nodosa (see “Polyarteritis Nodosa”). Cryoglobulinemic vasculitis is strongly associated with hepatitis C virus infection; hepatitis C virions and hepatitis C virus antigen-antibody complexes have been identified in the cryoprecipitates of these patients (see “Cryoglobulinemic Vasculitis”).
The mechanisms of tissue damage in immune complex–mediated vasculitis resemble those described for serum sickness. In this model, antigen-antibody complexes are formed in antigen excess and are deposited in vessel walls whose permeability has been increased by vasoactive amines such as histamine, bradykinin, and leukotrienes released from platelets or from mast cells as a result of IgE-triggered mechanisms. The deposition of complexes results in activation of complement components, particularly C5a, which is strongly chemotactic for neutrophils. These cells then infiltrate the vessel wall, phagocytose the immune complexes, and release their intracytoplasmic enzymes, which damage the vessel wall. As the process becomes subacute or chronic, mononuclear cells infiltrate the vessel wall. The common denominator of the resulting syndrome is compromise of the vessel lumen with ischemic changes in the tissues supplied by the involved vessel. Several variables may explain why only certain types of immune complexes cause vasculitis and why only certain vessels are affected in individual patients. These include the ability of the reticuloendothelial system to clear circulating complexes from the blood, the size and physicochemical properties of immune complexes, the relative degree of turbulence of blood flow, the intravascular hydrostatic pressure in different vessels, and the preexisting integrity of the vessel endothelium.
ANTINEUTROPHIL CYTOPLASMIC ANTIBODIES (ANCA)
ANCA are antibodies directed against certain proteins in the cytoplasmic granules of neutrophils and monocytes. These autoantibodies are present in a high percentage of patients with active granulomatosis with polyangiitis (Wegener’s) and microscopic polyangiitis, and in a lower percentage of patients with eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Because these diseases share the presence of ANCA and small-vessel vasculitis, some investigators have come to refer to them collectively as “ANCA-associated vasculitis.” However, as these diseases possess unique clinical phenotypes in which ANCA may be absent, it remains our opinion that granulomatosis with polyangiitis (Wegener’s), microscopic polyangiitis, and eosinophilic granulomatosis with polyangiitis (Churg-Strauss) should continue to be viewed as separate entities.
There are two major categories of ANCA based on different targets for the antibodies. The terminology of cytoplasmic ANCA (cANCA) refers to the diffuse, granular cytoplasmic staining pattern observed by immunofluorescence microscopy when serum antibodies bind to indicator neutrophils. Proteinase-3, a 29-kDa neutral serine proteinase present in neutrophil azurophilic granules, is the major cANCA antigen. More than 90% of patients with typical active granulomatosis with polyangiitis (Wegener’s) have detectable antibodies to proteinase-3 (see below). The terminology of perinuclear ANCA (pANCA) refers to the more localized perinuclear or nuclear staining pattern of the indicator neutrophils. The major target for pANCA is the enzyme myeloperoxidase; other targets that can produce a pANCA pattern of staining include elastase, cathepsin G, lactoferrin, lysozyme, and bactericidal/permeability-increasing protein. However, only antibodies to myeloperoxidase have been convincingly associated with vasculitis. Antimyeloperoxidase antibodies have been reported to occur in variable percentages of patients with microscopic polyangiitis, eosinophilic granulomatosis with polyangiitis (Churg-Strauss), isolated necrotizing crescentic glomerulonephritis, and granulomatosis with polyangiitis (Wegener’s) (see below). A pANCA pattern of staining that is not due to antimyeloperoxidase antibodies has been associated with nonvasculitic entities such as rheumatic and nonrheumatic autoimmune diseases, inflammatory bowel disease, certain drugs, and infections such as endocarditis and bacterial airway infections in patients with cystic fibrosis.
It is unclear why patients with these vasculitis syndromes develop antibodies to myeloperoxidase or proteinase-3 or what role these antibodies play in disease pathogenesis. There are a number of in vitro observations that suggest possible mechanisms whereby these antibodies can contribute to the pathogenesis of the vasculitis syndromes. Proteinase-3 and myeloperoxidase reside in the azurophilic granules and lysosomes of resting neutrophils and monocytes, where they are apparently inaccessible to serum antibodies. However, when neutrophils or monocytes are primed by tumor necrosis factor α (TNF-α) or interleukin 1 (IL-1), proteinase-3 and myeloperoxidase translocate to the cell membrane, where they can interact with extracellular ANCA. The neutrophils then degranulate and produce reactive oxygen species that can cause tissue damage. Furthermore, ANCA-activated neutrophils can adhere to and kill endothelial cells in vitro. Activation of neutrophils and monocytes by ANCA also induces the release of proinflammatory cytokines such as IL-1 and IL-8. Adoptive transfer experiments in genetically engineered mice provide further evidence for a direct pathogenic role of ANCA in vivo. In contradiction, however, a number of clinical and laboratory observations argue against a primary pathogenic role for ANCA. Patients may have active granulomatosis with polyangiitis (Wegener’s) in the absence of ANCA; the absolute height of the antibody titers does not correlate well with disease activity; and patients with granulomatosis with polyangiitis (Wegener’s) in remission may continue to have high antiproteinase-3 (cANCA) titers for years (see below).
PATHOGENIC T LYMPHOCYTE RESPONSES AND GRANULOMA FORMATION
The histopathologic feature of granulomatous vasculitis has provided evidence to support a role of pathogenic T lymphocyte responses and cell-mediated immune injury. Vascular endothelial cells can express HLA class II molecules following activation by cytokines such as interferon (IFN) γ. This allows these cells to participate in immunologic reactions such as interaction with CD4+ T lymphocytes in a manner similar to antigen-presenting macrophages. Endothelial cells can secrete IL-1, which may activate T lymphocytes and initiate or propagate in situ immunologic processes within the blood vessel. In addition, IL-1 and TNF-α are potent inducers of endothelial-leukocyte adhesion molecule 1 (ELAM-1) and vascular cell adhesion molecule 1 (VCAM-1), which may enhance the adhesion of leukocytes to endothelial cells in the blood vessel wall.
APPROACH TO THE PATIENT:
General Principles of Diagnosis
The diagnosis of vasculitis is often considered in any patient with an unexplained systemic illness. However, there are certain clinical abnormalities that when present alone or in combination should suggest a diagnosis of vasculitis. These include palpable purpura, pulmonary infiltrates and microscopic hematuria, chronic inflammatory sinusitis, mononeuritis multiplex, unexplained ischemic events, and glomerulonephritis with evidence of multisystem disease. A number of nonvasculitic diseases may also produce some or all of these abnormalities. Thus, the first step in the workup of a patient with suspected vasculitis is to exclude other diseases that produce clinical manifestations that can mimic vasculitis (Table 385-3). It is particularly important to exclude infectious diseases with features that overlap those of vasculitis, especially if the patient’s clinical condition is deteriorating rapidly and empirical immunosuppressive treatment is being contemplated.
|
CONDITIONS THAT CAN MIMIC VASCULITIS |
Once diseases that mimic vasculitis have been excluded, the workup should follow a series of progressive steps that establish the diagnosis of vasculitis and determine, where possible, the category of the vasculitis syndrome (Fig. 385-1). This approach is of considerable importance since several of the vasculitis syndromes require aggressive therapy with glucocorticoids and other immunosuppressive agents, whereas other syndromes usually resolve spontaneously and require symptomatic treatment only. The definitive diagnosis of vasculitis is usually made based on biopsy of involved tissue. The yield of “blind” biopsies of organs with no subjective or objective evidence of involvement is very low and should be avoided. When syndromes such as polyarteritis nodosa, Takayasu arteritis, or primary central nervous system (CNS) vasculitis are suspected, arteriogram of organs with suspected involvement should be performed.
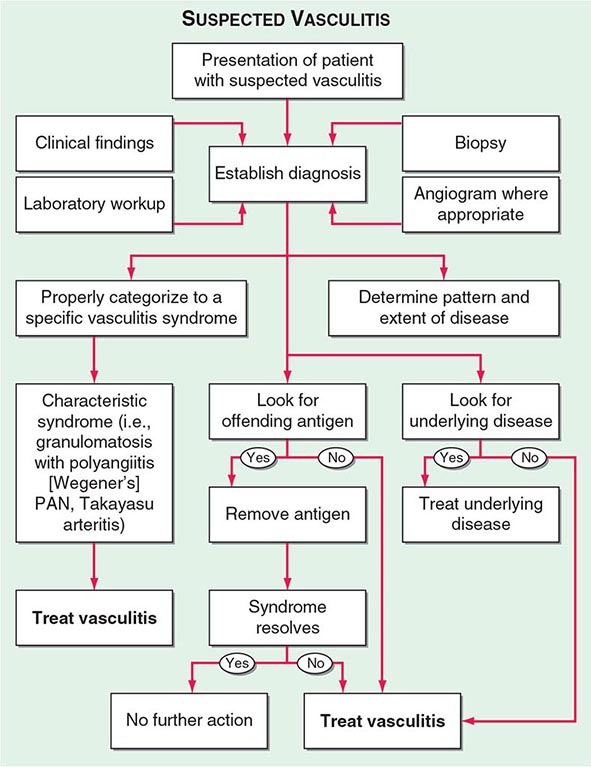
FIGURE 385-1 Algorithm for the approach to a patient with suspected diagnosis of vasculitis. PAN, polyarteritis nodosa.
GENERAL PRINCIPLES OF TREATMENT Once a diagnosis of vasculitis has been established, a decision regarding therapeutic strategy must be made (Fig. 385-1). If an offending antigen that precipitates the vasculitis is recognized, the antigen should be removed where possible. If the vasculitis is associated with an underlying disease such as an infection, neoplasm, or connective tissue disease, the underlying disease should be treated. If the syndrome represents a primary vasculitic disease, treatment should be initiated according to the category of the vasculitis syndrome. Specific therapeutic regimens are discussed below for the individual vasculitis syndromes; however, certain general principles regarding therapy should be considered. Decisions regarding treatment should be based on the use of regimens for which there has been published literature supporting efficacy for that particular vasculitic disease. Since the potential toxic side effects of certain therapeutic regimens may be substantial, the risk-versus-benefit ratio of any therapeutic approach should be weighed carefully. On the one hand, glucocorticoids and/or other immunosuppressive agents should be instituted immediately in diseases where irreversible organ system dysfunction and high morbidity and mortality rates have been clearly established. Granulomatosis with polyangiitis (Wegener’s) is the prototype of a severe systemic vasculitis requiring such a therapeutic approach (see below). On the other hand, when feasible, aggressive therapy should be avoided for vasculitic manifestations that rarely result in irreversible organ system dysfunction and that usually do not respond to such therapy. For example, isolated idiopathic cutaneous vasculitis usually resolves with symptomatic treatment, and prolonged courses of glucocorticoids uncommonly result in clinical benefit. Cytotoxic agents have not proved to be beneficial in idiopathic cutaneous vasculitis, and their toxic side effects generally outweigh any potential beneficial effects. Glucocorticoids should be initiated in those systemic vasculitides that cannot be specifically categorized or for which there is no established standard therapy; or other immunosuppressive therapy should be added in these diseases only if an adequate response does not result or if remission can only be achieved and maintained with an unacceptably toxic regimen of glucocorticoids. When remission is achieved, one should continually attempt to taper glucocorticoids and discontinue when possible. When using other immunosuppressive regimens, one should base the choice of agent upon the available therapeutic data supporting efficacy in that disease, the site and severity of organ involvement, and the toxicity profile of the drug.
Physicians should be thoroughly aware of the toxic side effects of therapeutic agents employed that can include both acute and long-term complications (Table 385-4). Morbidity and mortality can occur as a result of treatment, and strategies to monitor for and prevent toxicity represent an essential part of patient care. Glucocorticoids are an important part of treatment for most vasculitides but are associated with substantial toxicities. Monitoring and prevention of glucocorticoid-induced bone loss are important in all patients. With the use of daily cyclophosphamide, strategies are particularly important and are directed toward minimization of bladder toxicity and prevention of leukopenia. Instructing the patient to take cyclophosphamide all at once in the morning with a large amount of fluid throughout the day in order to maintain a dilute urine can reduce the risk of bladder injury. Bladder cancer can occur several years after discontinuation of cyclophosphamide therapy; therefore, monitoring for bladder cancer should continue indefinitely in patients who have received cyclophosphamide. Bone marrow suppression is an important toxicity of cyclophosphamide and can be observed during glucocorticoid tapering or over time, even after periods of stable measurements. Monitoring of the complete blood count every 1–2 weeks for as long as the patient receives cyclophosphamide can effectively prevent cytopenias. Maintaining the white blood cell (WBC) count at >3000/μL and the neutrophil count at >1500/μL is essential to reduce the risk of life-threatening infections.
|
MAJOR TOXIC SIDE EFFECTS OF DRUGS USED IN THE TREATMENT OF SYSTEMIC VASCULITIS |
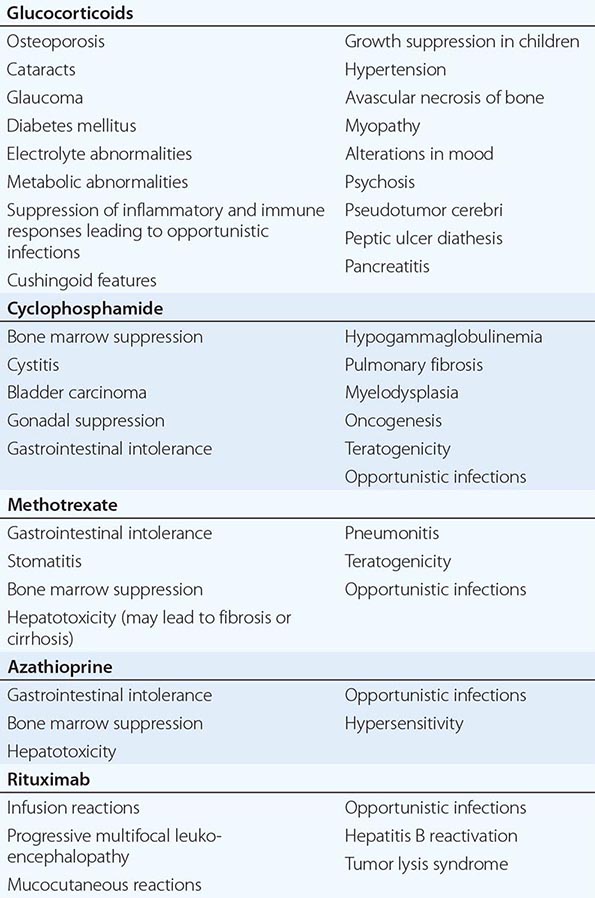
Methotrexate and azathioprine are also associated with bone marrow suppression, and complete blood counts should be obtained every 1–2 weeks for the first 1–2 months after their initiation and once a month thereafter. To lessen toxicity, methotrexate is often given together with folic acid, 1 mg daily, or folinic acid, 5–10 mg once a week 24 h following methotrexate. Prior to initiation of azathioprine, thiopurine methyltransferase (TPMT), an enzyme involved in the metabolism of azathioprine, should be assayed because inadequate levels may result in severe cytopenia.
Infection represents a significant toxicity for all vasculitis patients treated with immunosuppressive therapy. Infections with Pneumocystis jiroveci and certain fungi can be seen even in the face of WBCs that are within normal limits, particularly in patients receiving glucocorticoids. All vasculitis patients who are receiving daily glucocorticoids in combination with another immunosuppressive agent should receive trimethoprim-sulfamethoxazole (TMP-SMX) or another prophylactic therapy to prevent P. jiroveci infection.
Finally, it should be emphasized that each patient is unique and requires individual decision-making. The above outline should serve as a framework to guide therapeutic approaches; however, flexibility should be practiced in order to provide maximal therapeutic efficacy with minimal toxic side effects in each patient.
GRANULOMATOSIS WITH POLYANGIITIS (WEGENER’S)
DEFINITION
Granulomatosis with polyangiitis (Wegener’s) is a distinct clinicopathologic entity characterized by granulomatous vasculitis of the upper and lower respiratory tracts together with glomerulonephritis. In addition, variable degrees of disseminated vasculitis involving both small arteries and veins may occur.
INCIDENCE AND PREVALENCE
Granulomatosis with polyangiitis (Wegener’s) is an uncommon disease with an estimated prevalence of 3 per 100,000. It is extremely rare in blacks compared with whites; the male-to-female ratio is 1:1. The disease can be seen at any age; ∼15% of patients are <19 years of age, but only rarely does the disease occur before adolescence; the mean age of onset is ∼40 years.
PATHOLOGY AND PATHOGENESIS
The histopathologic hallmarks of granulomatosis with polyangiitis (Wegener’s) are necrotizing vasculitis of small arteries and veins together with granuloma formation, which may be either intravascular or extravascular (Fig. 385-2). Lung involvement typically appears as multiple, bilateral, nodular cavitary infiltrates (Fig. 385-3), which on biopsy almost invariably reveal the typical necrotizing granulomatous vasculitis. Upper airway lesions, particularly those in the sinuses and nasopharynx, typically reveal inflammation, necrosis, and granuloma formation, with or without vasculitis.
FIGURE 385-2 Lung histology in granulomatosis with polyangiitis (Wegener’s). This area of geographic necrosis has a serpiginous border of histiocytes and giant cells surrounding a central necrotic zone. Vasculitis is also present with neutrophils and lymphocytes infiltrating the wall of a small arteriole (upper right). (Courtesy of William D. Travis, MD; with permission.)
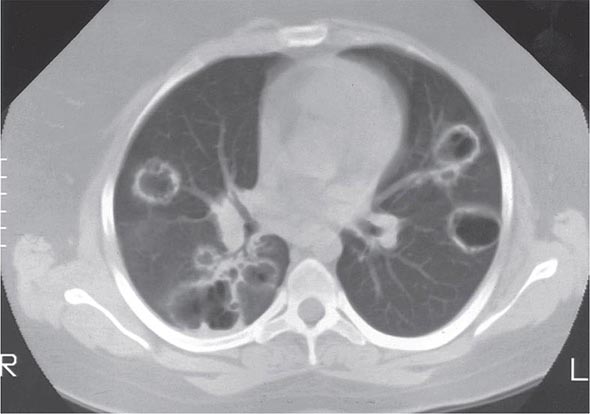
FIGURE 385-3 Computed tomography scan of a patient with granulomatosis with polyangiitis (Wegener’s). The patient developed multiple, bilateral, and cavitary infiltrates.
In its earliest form, renal involvement is characterized by a focal and segmental glomerulitis that may evolve into a rapidly progressive crescentic glomerulonephritis. Granuloma formation is only rarely seen on renal biopsy. In contrast to other forms of glomerulonephritis, evidence of immune complex deposition is not found in the renal lesion of granulomatosis with polyangiitis (Wegener’s). In addition to the classic triad of disease of the upper and lower respiratory tracts and kidney, virtually any organ can be involved with vasculitis, granuloma, or both.
The immunopathogenesis of this disease is unclear, although the involvement of upper airways and lungs with granulomatous vasculitis suggests an aberrant cell-mediated immune response to an exogenous or even endogenous antigen that enters through or resides in the upper airway. Chronic nasal carriage of Staphylococcus aureus has been reported to be associated with a higher relapse rate of granulomatosis with polyangiitis (Wegener’s); however, there is no evidence for a role of this organism in the pathogenesis of the disease.
Peripheral blood mononuclear cells obtained from patients with granulomatosis with polyangiitis (Wegener’s) manifest increased secretion of IFN-γ but not of IL-4, IL-5, or IL-10 compared to normal controls. In addition, TNF-α production from peripheral blood mononuclear cells and CD4+ T cells is elevated. Furthermore, monocytes from patients with granulomatosis with polyangiitis (Wegener’s) produce increased amounts of IL-12. These findings indicate an unbalanced TH1-type T cell cytokine pattern in this disease that may have pathogenic and perhaps ultimately therapeutic implications.
A high percentage of patients with granulomatosis with polyangiitis (Wegener’s) develop ANCA, and these autoantibodies may play a role in the pathogenesis of this disease (see above).
CLINICAL AND LABORATORY MANIFESTATIONS
Involvement of the upper airways occurs in 95% of patients with granulomatosis with polyangiitis (Wegener’s). Patients often present with severe upper respiratory tract findings such as paranasal sinus pain and drainage and purulent or bloody nasal discharge, with or without nasal mucosal ulceration (Table 385-5). Nasal septal perforation may follow, leading to saddle nose deformity. Serous otitis media may occur as a result of eustachian tube blockage. Subglottic tracheal stenosis resulting from active disease or scarring occurs in ∼16% of patients and may result in severe airway obstruction.
|
GRANULOMATOSIS WITH POLYANGIITIS (WEGENER’S): FREQUENCY OF CLINICAL MANIFESTATIONS IN 158 PATIENTS STUDIED AT THE NATIONAL INSTITUTES OF HEALTH |
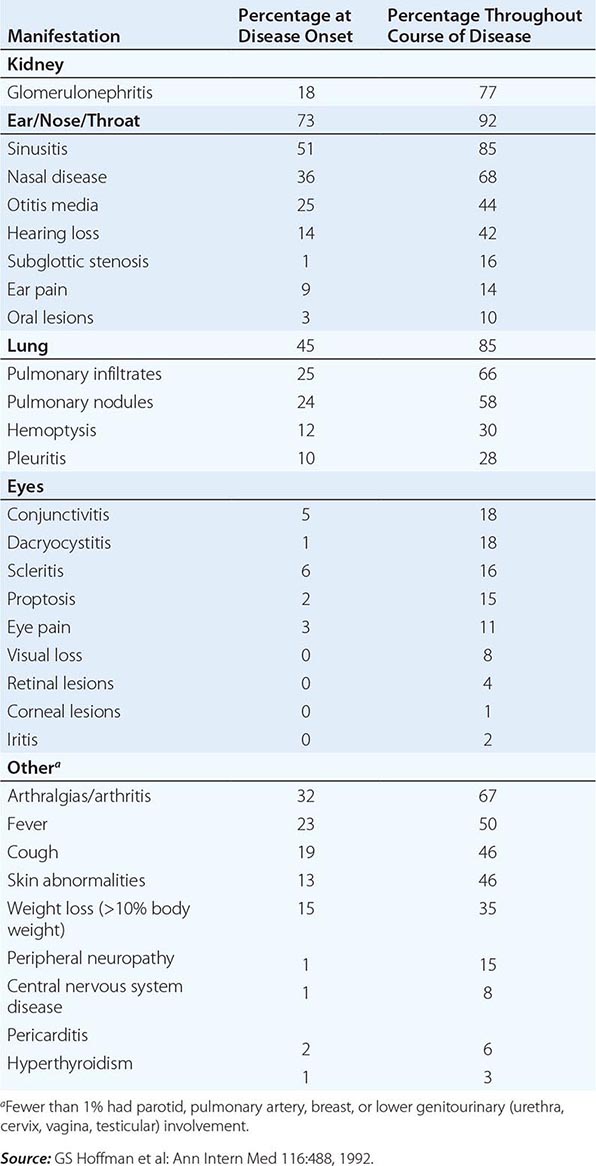
Pulmonary involvement may be manifested as asymptomatic infiltrates or may be clinically expressed as cough, hemoptysis, dyspnea, and chest discomfort. It is present in 85–90% of patients. Endobronchial disease, either in its active form or as a result of fibrous scarring, may lead to obstruction with atelectasis.
Eye involvement (52% of patients) may range from a mild conjunctivitis to dacryocystitis, episcleritis, scleritis, granulomatous sclerouveitis, ciliary vessel vasculitis, and retroorbital mass lesions leading to proptosis.
Skin lesions (46% of patients) appear as papules, vesicles, palpable purpura, ulcers, or subcutaneous nodules; biopsy reveals vasculitis, granuloma, or both. Cardiac involvement (8% of patients) manifests as pericarditis, coronary vasculitis, or, rarely, cardiomyopathy. Nervous system manifestations (23% of patients) include cranial neuritis, mononeuritis multiplex, or, rarely, cerebral vasculitis and/or granuloma.
Renal disease (77% of patients) generally dominates the clinical picture and, if left untreated, accounts directly or indirectly for most of the mortality rate in this disease. Although it may smolder in some cases as a mild glomerulitis with proteinuria, hematuria, and red blood cell casts, it is clear that once clinically detectable renal functional impairment occurs, rapidly progressive renal failure usually ensues unless appropriate treatment is instituted.
While the disease is active, most patients have nonspecific symptoms and signs such as malaise, weakness, arthralgias, anorexia, and weight loss. Fever may indicate activity of the underlying disease but more often reflects secondary infection, usually of the upper airway.
Characteristic laboratory findings include a markedly elevated erythrocyte sedimentation rate (ESR), mild anemia and leukocytosis, mild hypergammaglobulinemia (particularly of the IgA class), and mildly elevated rheumatoid factor. Thrombocytosis may be seen as an acute-phase reactant. Approximately 90% of patients with active granulomatosis with polyangiitis (Wegener’s) have a positive antiproteinase-3 ANCA. However, in the absence of active disease, the sensitivity drops to ∼60–70%. A small percentage of patients with granulomatosis with polyangiitis (Wegener’s) may have antimyeloperoxidase rather than antiproteinase-3 antibodies, and up to 20% may lack ANCA.
Patients with granulomatosis with polyangiitis (Wegener’s) have been found to have an increased incidence of venous thrombotic events. Although routine anticoagulation for all patients is not recommended, a heightened awareness for any clinical features suggestive of deep venous thrombosis or pulmonary emboli is warranted.
DIAGNOSIS
The diagnosis of granulomatosis with polyangiitis (Wegener’s) is made by the demonstration of necrotizing granulomatous vasculitis on tissue biopsy in a patient with compatible clinical features. Pulmonary tissue offers the highest diagnostic yield, almost invariably revealing granulomatous vasculitis. Biopsy of upper airway tissue usually reveals granulomatous inflammation with necrosis but may not show vasculitis. Renal biopsy can confirm the presence of pauci-immune glomerulonephritis.
The specificity of a positive antiproteinase-3 ANCA for granulomatosis with polyangiitis (Wegener’s) is very high, especially if active glomerulonephritis is present. However, the presence of ANCA should be adjunctive and, with rare exceptions, should not substitute for a tissue diagnosis. False-positive ANCA titers have been reported in certain infectious and neoplastic diseases.
In its typical presentation, the clinicopathologic complex of granulomatosis with polyangiitis (Wegener’s) usually provides ready differentiation from other disorders. However, if all the typical features are not present at once, it needs to be differentiated from the other vasculitides, antiglomerular basement membrane disease (Goodpasture’s syndrome) (Chap. 338), relapsing polychondritis (Chap. 389), tumors of the upper airway or lung, and infectious diseases such as histoplasmosis (Chap. 236), mucocutaneous leishmaniasis (Chap. 251), and rhinoscleroma (Chap. 44) as well as noninfectious granulomatous diseases.
Of particular note is the differentiation from other midline destructive diseases. These diseases lead to extreme tissue destruction and mutilation localized to the midline upper airway structures including the sinuses; erosion through the skin of the face commonly occurs, a feature that is extremely rare in granulomatosis with polyangiitis (Wegener’s). Although blood vessels may be involved in the intense inflammatory reaction and necrosis, primary vasculitis is not seen. Upper airway neoplasms and specifically extranodal natural killer (NK)/T cell lymphoma (nasal type) are important causes of midline destructive disease. These lesions are diagnosed based on histology, which reveals polymorphous atypical lymphoid cells with an NK cell immunophenotype, typically Epstein-Barr virus (Chap. 134). Such cases are treated based on their degree of dissemination, and localized lesions have responded to irradiation. Upper airway lesions should never be irradiated in granulomatosis with polyangiitis (Wegener’s). Cocaine-induced tissue injury can be another important mimic of granulomatosis with polyangiitis (Wegener’s) in patients who present with isolated midline destructive disease. ANCA that target human neutrophil elastase can be found in patients with cocaine-induced midline destructive lesions and can complicate the differentiation from granulomatosis with polyangiitis (Wegener’s). This has been further confounded by the high frequency of levamisole adulteration of cocaine, which can result in cutaneous infarction and serologic changes that may mimic vasculitis. Granulocytopenia is a common finding in levamisole-induced disease that would not be associated with granulomatosis with polyangiitis (Wegener’s).
Granulomatosis with polyangiitis (Wegener’s) must also be differentiated from lymphomatoid granulomatosis, which is an Epstein-Barr virus–positive B cell proliferation that is associated with an exuberant T cell reaction. Lymphomatoid granulomatosis is characterized by lung, skin, CNS, and kidney involvement in which atypical lymphocytoid and plasmacytoid cells infiltrate nonlymphoid tissue in an angioinvasive manner. In this regard, it clearly differs from granulomatosis with polyangiitis (Wegener’s) in that it is not an inflammatory vasculitis in the classic sense but an angiocentric perivascular infiltration of atypical mononuclear cells. Up to 50% of patients may develop a true malignant lymphoma.
MICROSCOPIC POLYANGIITIS
DEFINITION
The term microscopic polyarteritis was introduced into the literature by Davson in 1948 in recognition of the presence of glomerulonephritis in patients with polyarteritis nodosa. In 1992, the Chapel Hill Consensus Conference on the Nomenclature of Systemic Vasculitis adopted the term microscopic polyangiitis to connote a necrotizing vasculitis with few or no immune complexes affecting small vessels (capillaries, venules, or arterioles). Glomerulonephritis is very common in microscopic polyangiitis, and pulmonary capillaritis often occurs. The absence of granulomatous inflammation in microscopic polyangiitis is said to differentiate it from granulomatosis with polyangiitis (Wegener’s).
INCIDENCE AND PREVALENCE
The incidence of microscopic polyangiitis has not yet been reliably established due to its previous inclusion as part of polyarteritis nodosa. The mean age of onset is ∼57 years of age, and males are slightly more frequently affected than females.
PATHOLOGY AND PATHOGENESIS
The vasculitis seen in microscopic polyangiitis has a predilection to involve capillaries and venules in addition to small and medium-sized arteries. Immunohistochemical staining reveals a paucity of immunoglobulin deposition in the vascular lesion of microscopic polyangiitis, suggesting that immune-complex formation does not play a role in the pathogenesis of this syndrome. The renal lesion seen in microscopic polyangiitis is identical to that of granulomatosis with polyangiitis (Wegener’s). Like granulomatosis with polyangiitis (Wegener’s), microscopic polyangiitis is highly associated with the presence of ANCA, which may play a role in pathogenesis of this syndrome (see above).
CLINICAL AND LABORATORY MANIFESTATIONS
Because of its predilection to involve the small vessels, microscopic polyangiitis and granulomatosis with polyangiitis (Wegener’s) share similar clinical features. Disease onset may be gradual, with initial symptoms of fever, weight loss, and musculoskeletal pain; however, it is often acute. Glomerulonephritis occurs in at least 79% of patients and can be rapidly progressive, leading to renal failure. Hemoptysis may be the first symptom of alveolar hemorrhage, which occurs in 12% of patients. Other manifestations include mononeuritis multiplex and gastrointestinal tract and cutaneous vasculitis. Upper airway disease and pulmonary nodules are not typically found in microscopic polyangiitis and, if present, suggest granulomatosis with polyangiitis (Wegener’s).
Features of inflammation may be seen, including an elevated ESR, anemia, leukocytosis, and thrombocytosis. ANCA are present in 75% of patients with microscopic polyangiitis, with antimyeloperoxidase antibodies being the predominant ANCA associated with this disease.
DIAGNOSIS
The diagnosis is based on histologic evidence of vasculitis or pauci-immune glomerulonephritis in a patient with compatible clinical features of multisystem disease. Although microscopic polyangiitis is strongly ANCA-associated, no studies have as yet established the sensitivity and specificity of ANCA in this disease.
EOSINOPHILIC GRANULOMATOSIS WITH POLYANGIITIS (CHURG-STRAUSS)
DEFINITION
Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) was described in 1951 by Churg and Strauss and is characterized by asthma, peripheral and tissue eosinophilia, extravascular granuloma formation, and vasculitis of multiple organ systems.
INCIDENCE AND PREVALENCE
Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) is an uncommon disease with an estimated annual incidence of 1–3 per million. The disease can occur at any age with the possible exception of infants. The mean age of onset is 48 years, with a female-to-male ratio of 1.2:1.
PATHOLOGY AND PATHOGENESIS
The necrotizing vasculitis of eosinophilic granulomatosis with polyangiitis (Churg-Strauss) involves small and medium-sized muscular arteries, capillaries, veins, and venules. A characteristic histopathologic feature of eosinophilic granulomatosis with polyangiitis (Churg-Strauss) is granulomatous reactions that may be present in the tissues or even within the walls of the vessels themselves. These are usually associated with infiltration of the tissues with eosinophils. This process can occur in any organ in the body; lung involvement is predominant, with skin, cardiovascular system, kidney, peripheral nervous system, and gastrointestinal tract also commonly involved. Although the precise pathogenesis of this disease is uncertain, its strong association with asthma and its clinicopathologic manifestations, including eosinophilia, granuloma, and vasculitis, point to aberrant immunologic phenomena.
CLINICAL AND LABORATORY MANIFESTATIONS
Patients with eosinophilic granulomatosis with polyangiitis (Churg-Strauss) often exhibit nonspecific manifestations such as fever, malaise, anorexia, and weight loss, which are characteristic of a multisystem disease. The pulmonary findings in eosinophilic granulomatosis with polyangiitis (Churg-Strauss) clearly dominate the clinical picture with severe asthmatic attacks and the presence of pulmonary infiltrates. Mononeuritis multiplex is the second most common manifestation and occurs in up to 72% of patients. Allergic rhinitis and sinusitis develop in up to 61% of patients and are often observed early in the course of disease. Clinically recognizable heart disease occurs in ∼14% of patients and is an important cause of mortality. Skin lesions occur in ∼51% of patients and include purpura in addition to cutaneous and subcutaneous nodules. The renal disease in eosinophilic granulomatosis with polyangiitis (Churg-Strauss) is less common and generally less severe than that of granulomatosis with polyangiitis and microscopic polyangiitis.
The characteristic laboratory finding in virtually all patients with eosinophilic granulomatosis with polyangiitis (Churg-Strauss) is a striking eosinophilia, which reaches levels >1000 cells/μL in >80% of patients. Evidence of inflammation as evidenced by elevated ESR, fibrinogen, or α2-globulins can be found in 81% of patients. The other laboratory findings reflect the organ systems involved. Approximately 48% of patients with eosinophilic granulomatosis with polyangiitis (Churg-Strauss) have circulating ANCA that is usually antimyeloperoxidase.
DIAGNOSIS
Although the diagnosis of eosinophilic granulomatosis with polyangiitis (Churg-Strauss) is optimally made by biopsy in a patient with the characteristic clinical manifestations (see above), histologic confirmation can be challenging because the pathognomonic features often do not occur simultaneously. In order to be diagnosed with eosinophilic granulomatosis with polyangiitis (Churg-Strauss), a patient should have evidence of asthma, peripheral blood eosinophilia, and clinical features consistent with vasculitis.
POLYARTERITIS NODOSA
DEFINITION
Polyarteritis nodosa was described in 1866 by Kussmaul and Maier. It is a multisystem, necrotizing vasculitis of small and medium-sized muscular arteries in which involvement of the renal and visceral arteries is characteristic. Polyarteritis nodosa does not involve pulmonary arteries, although bronchial vessels may be involved; granulomas, significant eosinophilia, and an allergic diathesis are not observed.
INCIDENCE AND PREVALENCE
It is difficult to establish an accurate incidence of polyarteritis nodosa because previous reports have included polyarteritis nodosa and microscopic polyangiitis as well as other related vasculitides. Polyarteritis nodosa, as currently defined, is felt to be a very uncommon disease.
PATHOLOGY AND PATHOGENESIS
The vascular lesion in polyarteritis nodosa is a necrotizing inflammation of small and medium-sized muscular arteries. The lesions are segmental and tend to involve bifurcations and branchings of arteries. They may spread circumferentially to involve adjacent veins. However, involvement of venules is not seen in polyarteritis nodosa and, if present, suggests microscopic polyangiitis (see below). In the acute stages of disease, polymorphonuclear neutrophils infiltrate all layers of the vessel wall and perivascular areas, which results in intimal proliferation and degeneration of the vessel wall. Mononuclear cells infiltrate the area as the lesions progress to the subacute and chronic stages. Fibrinoid necrosis of the vessels ensues with compromise of the lumen, thrombosis, infarction of the tissues supplied by the involved vessel, and, in some cases, hemorrhage. As the lesions heal, there is collagen deposition, which may lead to further occlusion of the vessel lumen. Aneurysmal dilations up to 1 cm in size along the involved arteries are characteristic of polyarteritis nodosa. Granulomas and substantial eosinophilia with eosinophilic tissue infiltrations are not characteristically found and suggest eosinophilic granulomatosis with polyangiitis (Churg-Strauss) (see above).
Multiple organ systems are involved, and the clinicopathologic findings reflect the degree and location of vessel involvement and the resulting ischemic changes. As mentioned above, pulmonary arteries are not involved in polyarteritis nodosa, and bronchial artery involvement is uncommon. The pathology in the kidney in classic polyarteritis nodosa is that of arteritis without glomerulonephritis. In patients with significant hypertension, typical pathologic features of glomerulosclerosis may be seen. In addition, pathologic sequelae of hypertension may be found elsewhere in the body.
The presence of a polyarteritis nodosa–like vasculitis in patients with hepatitis B together with the isolation of circulating immune complexes composed of hepatitis B antigen and immunoglobulin, and the demonstration by immunofluorescence of hepatitis B antigen, IgM, and complement in the blood vessel walls, strongly suggest the role of immunologic phenomena in the pathogenesis of this disease. A polyarteritis nodosa–like vasculitis has also been reported in patients with hepatitis C. Hairy cell leukemia can be associated with polyarteritis nodosa; the pathogenic mechanisms of this association are unclear.
CLINICAL AND LABORATORY MANIFESTATIONS
Nonspecific signs and symptoms are the hallmarks of polyarteritis nodosa. Fever, weight loss, and malaise are present in over one-half of cases. Patients usually present with vague symptoms such as weakness, malaise, headache, abdominal pain, and myalgias that can rapidly progress to a fulminant illness. Specific complaints related to the vascular involvement within a particular organ system may also dominate the presenting clinical picture as well as the entire course of the illness (Table 385-6). In polyarteritis nodosa, renal involvement most commonly manifests as hypertension, renal insufficiency, or hemorrhage due to microaneurysms.
|
CLINICAL MANIFESTATIONS RELATED TO ORGAN SYSTEM INVOLVEMENT IN POLYARTERITIS NODOSA |
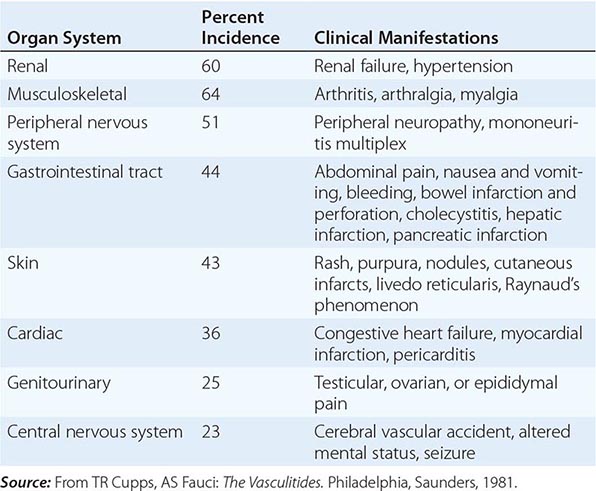
There are no diagnostic serologic tests for polyarteritis nodosa. In >75% of patients, the leukocyte count is elevated with a predominance of neutrophils. Eosinophilia is seen only rarely and, when present at high levels, suggests the diagnosis of eosinophilic granulomatosis with polyangiitis (Churg-Strauss). The anemia of chronic disease may be seen, and an elevated ESR is almost always present. Other common laboratory findings reflect the particular organ involved. Hypergammaglobulinemia may be present, and all patients should be screened for hepatitis B and C. Antibodies against myeloperoxidase or proteinase-3 (ANCA) are rarely found in patients with polyarteritis nodosa.
DIAGNOSIS
The diagnosis of polyarteritis nodosa is based on the demonstration of characteristic findings of vasculitis on biopsy material of involved organs. In the absence of easily accessible tissue for biopsy, the arteriographic demonstration of involved vessels, particularly in the form of aneurysms of small and medium-sized arteries in the renal, hepatic, and visceral vasculature, is sufficient to make the diagnosis. This should consist of a catheter-directed dye arteriogram because magnetic resonance and computed tomography arteriograms do not have sufficient resolution at the current time to visualize the vessels affected in polyarteritis nodosa. Aneurysms of vessels are not pathognomonic of polyarteritis nodosa; furthermore, aneurysms need not always be present, and arteriographic findings may be limited to stenotic segments and obliteration of vessels. Biopsy of symptomatic organs such as nodular skin lesions, painful testes, and nerve/muscle provides the highest diagnostic yields.
GIANT CELL ARTERITIS AND POLYMYALGIA RHEUMATICA
DEFINITION
Giant cell arteritis, historically referred to as temporal arteritis, is an inflammation of medium- and large-sized arteries. It characteristically involves one or more branches of the carotid artery, particularly the temporal artery. However, it is a systemic disease that can involve arteries in multiple locations, particularly the aorta and its main branches.
Giant cell arteritis is closely associated with polymyalgia rheumatica, which is characterized by stiffness, aching, and pain in the muscles of the neck, shoulders, lower back, hips, and thighs. Most commonly, polymyalgia rheumatica occurs in isolation, but it may be seen in 40–50% of patients with giant cell arteritis. In addition, ∼10–20% of patients who initially present with features of isolated polymyalgia rheumatica later go on to develop giant cell arteritis. This strong clinical association together with data from pathophysiologic studies has increasingly supported that giant cell arteritis and polymyalgia rheumatica represent differing clinical spectrums of a single disease process.
INCIDENCE AND PREVALENCE
Giant cell arteritis occurs almost exclusively in individuals >50 years. It is more common in women than in men and is rare in blacks. The incidence of giant cell arteritis varies widely in different studies and in different geographic regions. A high incidence has been found in Scandinavia and in regions of the United States with large Scandinavian populations, compared to a lower incidence in southern Europe. The annual incidence rates in individuals ≥50 years range from 6.9 to 32.8 per 100,000 population. Familial aggregation has been reported, as has an association with HLA-DR4. In addition, genetic linkage studies have demonstrated an association of giant cell arteritis with alleles at the HLA-DRB1 locus, particularly HLA-DRB1*04 variants. In Olmsted County, Minnesota, the annual incidence of polymyalgia rheumatica in individuals ≥50 years is 58.7 per 100,000 population.
PATHOLOGY AND PATHOGENESIS
Although the temporal artery is most frequently involved in giant cell arteritis, patients often have a systemic vasculitis of multiple medium- and large-sized arteries, which may go undetected. Histopathologically, the disease is a panarteritis with inflammatory mononuclear cell infiltrates within the vessel wall with frequent giant cell formation. There is proliferation of the intima and fragmentation of the internal elastic lamina. Pathophysiologic findings in organs result from the ischemia related to the involved vessels.
Experimental data support that giant cell arteritis is an antigen-driven disease in which activated T lymphocytes, macrophages, and dendritic cells play a critical role in the disease pathogenesis. Sequence analysis of the T cell receptor of tissue-infiltrating T cells in lesions of giant cell arteritis indicates restricted clonal expansion, suggesting the presence of an antigen residing in the arterial wall. Giant cell arteritis is believed to be initiated in the adventitia where CD4+ T cells enter through the vasa vasorum, become activated, and orchestrate macrophage differentiation. T cells recruited to vasculitic lesions in patients with giant cell arteritis produce predominantly IL-2 and IFN-γ, and the latter has been suggested to be involved in the progression to overt arteritis. Recent data demonstrate that at least two separate lineages of CD4 T cells–-IFN-γ-producing TH1 cells and IL-17-producing TH17 cells—participate in vascular inflammation and may have differing levels of responsiveness to glucocorticoids.
CLINICAL AND LABORATORY MANIFESTATIONS
Giant cell arteritis is most commonly characterized clinically by the complex of fever, anemia, high ESR, and headaches in a patient over the age of 50 years. Other phenotypic manifestations include features of systemic inflammation including malaise, fatigue, anorexia, weight loss, sweats, arthralgias, polymyalgia rheumatica, or large-vessel disease.
In patients with involvement of the cranial arteries, headache is the predominant symptom and may be associated with a tender, thickened, or nodular artery, which may pulsate early in the disease but may become occluded later. Scalp pain and claudication of the jaw and tongue may occur. A well-recognized and dreaded complication of giant cell arteritis, particularly in untreated patients, is ischemic optic neuropathy, which may lead to serious visual symptoms, even sudden blindness in some patients. However, most patients have complaints relating to the head or eyes before visual loss. Attention to such symptoms with institution of appropriate therapy (see below) will usually avoid this complication. Other cranial ischemic complications include strokes and scalp or tongue infarction.
Up to one-third of patients can have large-vessel disease that can be the primary presentation of giant cell arteritis or can emerge at a later point in patients who have had previous cranial arteritis features or polymyalgia rheumatica. Manifestations of large-vessel disease can include subclavian artery stenosis that can present as arm claudication or aortic aneurysms involving the thoracic and to a lesser degree the abdominal aorta, which carry risks of rupture or dissection.
Characteristic laboratory findings in addition to the elevated ESR include a normochromic or slightly hypochromic anemia. Liver function abnormalities are common, particularly increased alkaline phosphatase levels. Increased levels of IgG and complement have been reported. Levels of enzymes indicative of muscle damage such as serum creatine kinase are not elevated.
DIAGNOSIS
The diagnosis of giant cell arteritis and its associated clinicopathologic syndrome can often be suggested clinically by the demonstration of the complex of fever, anemia, and high ESR with or without symptoms of polymyalgia rheumatica in a patient >50 years. The diagnosis is confirmed by biopsy of the temporal artery. Since involvement of the vessel may be segmental, positive yield is increased by obtaining a biopsy segment of 3–5 cm together with serial sectioning of biopsy specimens. Ultrasonography of the temporal artery has been reported to be helpful in diagnosis. A temporal artery biopsy should be obtained as quickly as possible in the setting of ocular signs and symptoms, and under these circumstances, therapy should not be delayed pending a biopsy. In this regard, it has been reported that temporal artery biopsies may show vasculitis even after ∼14 days of glucocorticoid therapy. A dramatic clinical response to a trial of glucocorticoid therapy can further support the diagnosis.
Large-vessel disease may be suggested by symptoms and findings on physical examination such as diminished pulses or bruits. It is confirmed by vascular imaging, most commonly through magnetic resonance or computed tomography.
Isolated polymyalgia rheumatica is a clinical diagnosis made by the presence of typical symptoms of stiffness, aching, and pain in the muscles of the hip and shoulder girdle, an increased ESR, the absence of clinical features suggestive of giant cell arteritis, and a prompt therapeutic response to low-dose prednisone.
TAKAYASU ARTERITIS
DEFINITION
Takayasu arteritis is an inflammatory and stenotic disease of medium- and large-sized arteries characterized by a strong predilection for the aortic arch and its branches.
INCIDENCE AND PREVALENCE
Takayasu arteritis is an uncommon disease with an estimated annual incidence rate of 1.2–2.6 cases per million. It is most prevalent in adolescent girls and young women. Although it is more common in Asia, it is neither racially nor geographically restricted.
PATHOLOGY AND PATHOGENESIS
The disease involves medium- and large-sized arteries, with a strong predilection for the aortic arch and its branches; the pulmonary artery may also be involved. The most commonly affected arteries seen by arteriography are listed in Table 385-7. The involvement of the major branches of the aorta is much more marked at their origin than distally. The disease is a panarteritis with inflammatory mononuclear cell infiltrates and occasionally giant cells. There are marked intimal proliferation and fibrosis, scarring and vascularization of the media, and disruption and degeneration of the elastic lamina. Narrowing of the lumen occurs with or without thrombosis. The vasa vasorum are frequently involved. Pathologic changes in various organs reflect the compromise of blood flow through the involved vessels.
|
FREQUENCY OF ARTERIOGRAPHIC ABNORMALITIES AND POTENTIAL CLINICAL MANIFESTATIONS OF ARTERIAL INVOLVEMENT IN TAKAYASU ARTERITIS |
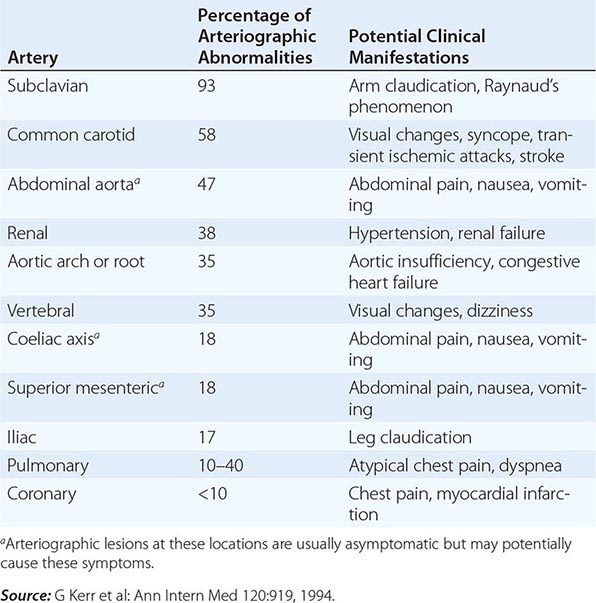
Immunopathogenic mechanisms, the precise nature of which is uncertain, are suspected in this disease. As with several of the vasculitis syndromes, circulating immune complexes have been demonstrated, but their pathogenic significance is unclear.
CLINICAL AND LABORATORY MANIFESTATIONS
Takayasu arteritis is a systemic disease with generalized as well as vascular symptoms. The generalized symptoms include malaise, fever, night sweats, arthralgias, anorexia, and weight loss, which may occur months before vessel involvement is apparent. These symptoms may merge into those related to vascular compromise and organ ischemia. Pulses are commonly absent in the involved vessels, particularly the subclavian artery. The frequency of arteriographic abnormalities and the potentially associated clinical manifestations are listed in Table 385-7. Hypertension occurs in 32–93% of patients and contributes to renal, cardiac, and cerebral injury.
Characteristic laboratory findings include an elevated ESR, mild anemia, and elevated immunoglobulin levels.
DIAGNOSIS
The diagnosis of Takayasu arteritis should be suspected strongly in a young woman who develops a decrease or absence of peripheral pulses, discrepancies in blood pressure, and arterial bruits. The diagnosis is confirmed by the characteristic pattern on arteriography, which includes irregular vessel walls, stenosis, poststenotic dilation, aneurysm formation, occlusion, and evidence of increased collateral circulation. Complete aortic arteriography by catheter-directed dye arteriography or magnetic resonance arteriography should be obtained in order to fully delineate the distribution and degree of arterial disease. Histopathologic demonstration of vessel wall inflammation that is predominantly lymphocytic with granuloma formation and giant cells involving the media and adventitia adds confirmatory data; however, tissue is rarely readily available for examination. IgG4-related disease is a potential cause of aortitis and periaortitis that is histologically differentiated from Takayasu arteritis by a dense lymphoplasmacytic infiltrate rich in IgG4-positive plasma cells, a storiform pattern of fibrosis, and obliterative phlebitis.
IgA VASCULITIS (HENOCH-SCHÖNLEIN)
DEFINITION
IgA vasculitis (Henoch-Schönlein) is a small-vessel vasculitis characterized by palpable purpura (most commonly distributed over the buttocks and lower extremities), arthralgias, gastrointestinal signs and symptoms, and glomerulonephritis.
INCIDENCE AND PREVALENCE
IgA vasculitis (Henoch-Schönlein) is usually seen in children; most patients range in age from 4 to 7 years; however, the disease may also be seen in infants and adults. It is not a rare disease; in one series it accounted for between 5 and 24 admissions per year at a pediatric hospital. The male-to-female ratio is 1.5:1. A seasonal variation with a peak incidence in spring has been noted.
PATHOLOGY AND PATHOGENESIS
The presumptive pathogenic mechanism for IgA (Henoch-Schönlein) vasculitis is immune-complex deposition. A number of inciting antigens have been suggested including upper respiratory tract infections, various drugs, foods, insect bites, and immunizations. IgA is the antibody class most often seen in the immune complexes and has been demonstrated in the renal biopsies of these patients.
CLINICAL AND LABORATORY MANIFESTATIONS
In pediatric patients, palpable purpura is seen in virtually all patients; most patients develop polyarthralgias in the absence of frank arthritis. Gastrointestinal involvement, which is seen in almost 70% of pediatric patients, is characterized by colicky abdominal pain usually associated with nausea, vomiting, diarrhea, or constipation and is frequently accompanied by the passage of blood and mucus per rectum; bowel intussusception may occur. Renal involvement occurs in 10–50% of patients and is usually characterized by mild glomerulonephritis leading to proteinuria and microscopic hematuria, with red blood cell casts in the majority of patients (Chap. 338); it usually resolves spontaneously without therapy. Rarely, a progressive glomerulonephritis will develop. In adults, presenting symptoms are most frequently related to the skin and joints, while initial complaints related to the gut are less common. Although certain studies have found that renal disease is more frequent and more severe in adults, this has not been a consistent finding. However, the course of renal disease in adults may be more insidious and thus requires close follow-up. Myocardial involvement can occur in adults but is rare in children.
Laboratory studies generally show a mild leukocytosis, a normal platelet count, and occasionally eosinophilia. Serum complement components are normal, and IgA levels are elevated in about one-half of patients.
DIAGNOSIS
The diagnosis of IgA vasculitis (Henoch-Schönlein) is based on clinical signs and symptoms. Skin biopsy specimen can be useful in confirming leukocytoclastic vasculitis with IgA and C3 deposition by immunofluorescence. Renal biopsy is rarely needed for diagnosis but may provide prognostic information in some patients.
CRYOGLOBULINEMIC VASCULITIS
DEFINITION
Cryoglobulins are cold-precipitable monoclonal or polyclonal immunoglobulins. Cryoglobulinemia may be associated with a systemic vasculitis characterized by palpable purpura, arthralgias, weakness, neuropathy, and glomerulonephritis. Although this can be observed in association with a variety of underlying disorders including multiple myeloma, lymphoproliferative disorders, connective tissue diseases, infection, and liver disease, in many instances it appears to be idiopathic. Because of the apparent absence of an underlying disease and the presence of cryoprecipitate containing oligoclonal/polyclonal immunoglobulins, this entity was referred to as essential mixed cryoglobulinemia. Since the discovery of hepatitis C, it has been established that the vast majority of patients who were considered to have essential mixed cryoglobulinemia have cryoglobulinemic vasculitis related to hepatitis C infection.
INCIDENCE AND PREVALENCE
The incidence of cryoglobulinemic vasculitis has not been established. It has been estimated, however, that 5% of patients with chronic hepatitis C will develop cryoglobulinemic vasculitis.
PATHOLOGY AND PATHOGENESIS
Skin biopsies in cryoglobulinemic vasculitis reveal an inflammatory infiltrate surrounding and involving blood vessel walls, with fibrinoid necrosis, endothelial cell hyperplasia, and hemorrhage. Deposition of immunoglobulin and complement is common. Abnormalities of uninvolved skin including basement membrane alterations and deposits in vessel walls may be found. Membranoproliferative glomerulonephritis is responsible for 80% of all renal lesions in cryoglobulinemic vasculitis.
The association between hepatitis C and cryoglobulinemic vasculitis has been supported by the high frequency of documented hepatitis C infection, the presence of hepatitis C RNA and anti–hepatitis C antibodies in serum cryoprecipitates, evidence of hepatitis C antigens in vasculitic skin lesions, and the effectiveness of antiviral therapy (see below). Current evidence suggests that in the majority of cases, cryoglobulinemic vasculitis occurs when an aberrant immune response to hepatitis C infection leads to the formation of immune complexes consisting of hepatitis C antigens, polyclonal hepatitis C–specific IgG, and monoclonal IgM rheumatoid factor. The deposition of these immune complexes in blood vessel walls triggers an inflammatory cascade that results in cryoglobulinemic vasculitis.
CLINICAL AND LABORATORY MANIFESTATIONS
The most common clinical manifestations of cryoglobulinemic vasculitis are cutaneous vasculitis, arthritis, peripheral neuropathy, and glomerulonephritis. Renal disease develops in 10–30% of patients. Life-threatening rapidly progressive glomerulonephritis or vasculitis of the CNS, gastrointestinal tract, or heart occurs infrequently.
The presence of circulating cryoprecipitates is the fundamental finding in cryoglobulinemic vasculitis. Rheumatoid factor is almost always found and may be a useful clue to the disease when cryoglobulins are not detected. Hypocomplementemia occurs in 90% of patients. An elevated ESR and anemia occur frequently. Evidence for hepatitis C infection must be sought in all patients by testing for hepatitis C antibodies and hepatitis C RNA.
SINGLE-ORGAN VASCULITIS
The potential for vasculitis to affect single organs has become increasingly recognized. This has been defined as vasculitis in arteries or veins of any size in a single organ that has no features that indicate that it is a limited expression of a systemic vasculitis. Examples include isolated aortitis, testicular vasculitis, vasculitis of the breast, isolated cutaneous vasculitis, and primary CNS vasculitis. In some instances, this may be discovered at the time of surgery such as orchiectomy for a testicular mass where there is concern for neoplasm that is found instead to be vasculitis. Some patients originally diagnosed with single-organ vasculitis may later develop additional manifestations of a more systemic disease. In instances where there is no evidence of systemic vasculitis and the affected organ has been removed in its entirety, the patient may be followed closely without immunosuppressive therapy. In other instances, such as primary CNS vasculitis or some patients with isolated cutaneous vasculitis, medical intervention is warranted.
IDIOPATHIC CUTANEOUS VASCULITIS
DEFINITION
The term cutaneous vasculitis is defined broadly as inflammation of the blood vessels of the dermis. Due to its heterogeneity, cutaneous vasculitis has been described by a variety of terms including hypersensitivity vasculitis and cutaneous leukocytoclastic angiitis. However, cutaneous vasculitis is not one specific disease but a manifestation that can be seen in a variety of settings. In >70% of cases, cutaneous vasculitis occurs either as part of a primary systemic vasculitis or as a secondary vasculitis related to an inciting agent or an underlying disease (see “Secondary Vasculitis,” below). In the remaining 30% of cases, cutaneous vasculitis occurs idiopathically.
INCIDENCE AND PREVALENCE
Cutaneous vasculitis represents the most commonly encountered vasculitis in clinical practice. The exact incidence of idiopathic cutaneous vasculitis has not been determined due to the predilection for cutaneous vasculitis to be associated with an underlying process and the variability of its clinical course.
PATHOLOGY AND PATHOGENESIS
The typical histopathologic feature of cutaneous vasculitis is the presence of vasculitis of small vessels. Postcapillary venules are the most commonly involved vessels; capillaries and arterioles may be involved less frequently. This vasculitis is characterized by a leukocytoclasis, a term that refers to the nuclear debris remaining from the neutrophils that have infiltrated in and around the vessels during the acute stages. In the subacute or chronic stages, mononuclear cells predominate; in certain subgroups, eosinophilic infiltration is seen. Erythrocytes often extravasate from the involved vessels, leading to palpable purpura. Cutaneous arteritis can also occur, which involves slightly larger-sized vessels within the dermis.
CLINICAL AND LABORATORY MANIFESTATIONS
The hallmark of idiopathic cutaneous vasculitis is the predominance of skin involvement. Skin lesions may appear typically as palpable purpura; however, other cutaneous manifestations of the vasculitis may occur, including macules, papules, vesicles, bullae, subcutaneous nodules, ulcers, and recurrent or chronic urticaria. The skin lesions may be pruritic or even quite painful, with a burning or stinging sensation. Lesions most commonly occur in the lower extremities in ambulatory patients or in the sacral area in bedridden patients due to the effects of hydrostatic forces on the postcapillary venules. Edema may accompany certain lesions, and hyperpigmentation often occurs in areas of recurrent or chronic lesions.
There are no specific laboratory tests diagnostic of idiopathic cutaneous vasculitis. A mild leukocytosis with or without eosinophilia is characteristic, as is an elevated ESR. Laboratory studies should be aimed toward ruling out features to suggest an underlying disease or a systemic vasculitis.
DIAGNOSIS
The diagnosis of cutaneous vasculitis is made by the demonstration of vasculitis on biopsy. An important diagnostic principle in patients with cutaneous vasculitis is to search for an etiology of the vasculitis—be it an exogenous agent, such as a drug or an infection, or an endogenous condition, such as an underlying disease (Fig. 385-1). In addition, a careful physical and laboratory examination should be performed to rule out the possibility of systemic vasculitis. This should start with the least invasive diagnostic approach and proceed to the more invasive only if clinically indicated.
PRIMARY CENTRAL NERVOUS SYSTEM (CNS) VASCULITIS
Primary CNS vasculitis is an uncommon clinicopathologic entity characterized by vasculitis restricted to the vessels of the CNS without other apparent systemic vasculitis. The inflammatory process is usually composed of mononuclear cell infiltrates with or without granuloma formation.
Patients may present with headaches, altered mental function, and focal neurologic defects. Systemic symptoms are generally absent. Devastating neurologic abnormalities may occur depending on the extent of vessel involvement. The diagnosis can be suggested by abnormal magnetic resonance imaging of the brain, an abnormal lumbar puncture, and/or demonstration of characteristic vessel abnormalities on arteriography (Fig. 385-4), but it is confirmed by biopsy of the brain parenchyma and leptomeninges. In the absence of a brain biopsy, care should be taken not to misinterpret as true primary vasculitis arteriographic abnormalities that might actually be related to another cause. An important entity in the differential diagnosis is reversible cerebral vasoconstrictive syndrome, which typically presents with “thunderclap” headache and is associated with arteriographic abnormalities that mimic primary CNS vasculitis that are reversible. Other diagnostic considerations include infection, atherosclerosis, emboli, connective tissue disease, sarcoidosis, malignancy, and drug-associated causes. The prognosis of granulomatous primary CNS vasculitis is poor; however, some reports indicate that glucocorticoid therapy, alone or together with cyclophosphamide administered as described above, has induced clinical remissions.
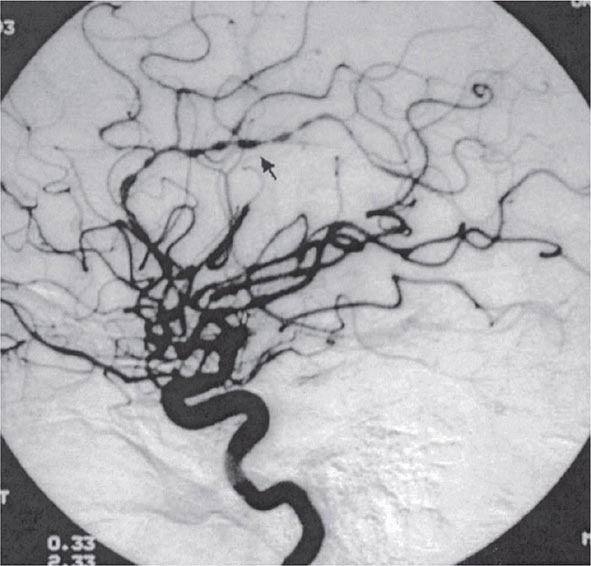
FIGURE 385-4 Cerebral arteriogram from a 32-year-old male with primary central nervous system vasculitis. Dramatic beading (arrow) typical of vasculitis is seen.
BEHÇET’S DISEASE
Behçet’s disease is a clinicopathologic entity characterized by recurrent episodes of oral and genital ulcers, iritis, and cutaneous lesions. The underlying pathologic process is a leukocytoclastic venulitis, although vessels of any size and in any organ can be involved. This disorder is described in detail in Chap. 387.
COGAN’S SYNDROME
Cogan’s syndrome is characterized by interstitial keratitis together with vestibuloauditory symptoms. It may be associated with a systemic vasculitis, particularly aortitis with involvement of the aortic valve. Glucocorticoids are the mainstay of treatment. Initiation of treatment as early as possible after the onset of hearing loss improves the likelihood of a favorable outcome.
KAWASAKI DISEASE
Kawasaki disease is an acute, febrile, multisystem disease of children. Some 80% of cases occur prior to the age of 5, with the peak incidence occurring at ≤2 years. It is characterized by nonsuppurative cervical adenitis and changes in the skin and mucous membranes such as edema; congested conjunctivae; erythema of the oral cavity, lips, and palms; and desquamation of the skin of the fingertips. Although the disease is generally benign and self-limited, it is associated with coronary artery aneurysms in ∼25% of cases, with an overall case fatality rate of 0.5–2.8%. These complications usually occur between the third and fourth weeks of illness during the convalescent stage. Vasculitis of the coronary arteries is seen in almost all the fatal cases that have been autopsied. There is typical intimal proliferation and infiltration of the vessel wall with mononuclear cells. Beadlike aneurysms and thromboses may be seen along the artery. Other manifestations include pericarditis, myocarditis, myocardial ischemia and infarction, and cardiomegaly.
Apart from the up to 2.8% of patients who develop fatal complications, the prognosis of this disease for uneventful recovery is excellent. High-dose IV γ-globulin (2 g/kg as a single infusion over 10 h) together with aspirin (100 mg/kg per day for 14 days followed by 3–5 mg/kg per day for several weeks) have been shown to be effective in reducing the prevalence of coronary artery abnormalities when administered early in the course of the disease. Surgery may be necessary for Kawasaki disease patients who have giant coronary artery aneurysms or other coronary complications. Surgical treatment most commonly includes thromboendarterectomy, thrombus clearing, aneurysmal reconstruction, and coronary artery bypass grafting.
POLYANGIITIS OVERLAP SYNDROMES
Some patients with systemic vasculitis manifest clinicopathologic characteristics that do not fit precisely into any specific disease but have overlapping features of different vasculitides. Active systemic vasculitis in such settings has the same potential for causing irreversible organ system damage as when it occurs in one of the defined syndromes listed in Table 385-1. The diagnostic and therapeutic considerations as well as the prognosis for these patients depend on the sites and severity of active vasculitis. Patients with vasculitis that could potentially cause irreversible damage to a major organ system should be treated as described under “Granulomatosis with Polyangiitis (Wegener’s).”
SECONDARY VASCULITIS
DRUG-INDUCED VASCULITIS
Vasculitis associated with drug reactions usually presents as palpable purpura that may be generalized or limited to the lower extremities or other dependent areas; however, urticarial lesions, ulcers, and hemorrhagic blisters may also occur (Chap. 74). Signs and symptoms may be limited to the skin, although systemic manifestations such as fever, malaise, and polyarthralgias may occur. Although the skin is the predominant organ involved, systemic vasculitis may result from drug reactions. Drugs that have been implicated in vasculitis include allopurinol, thiazides, gold, sulfonamides, phenytoin, and penicillin (Chap. 74).
An increasing number of drugs have been reported to cause vasculitis associated with antimyeloperoxidase ANCA. Of these, the best evidence of causality exists for hydralazine and propylthiouracil. The clinical manifestations in ANCA-positive drug-induced vasculitis can range from cutaneous lesions to glomerulonephritis and pulmonary hemorrhage. Outside of drug discontinuation, treatment should be based on the severity of the vasculitis. Patients with immediately life-threatening small-vessel vasculitis should initially be treated with glucocorticoids and cyclophosphamide as described for granulomatosis with polyangiitis (Wegener’s). Following clinical improvement, consideration may be given for tapering such agents along a more rapid schedule.
SERUM SICKNESS AND SERUM SICKNESS–LIKE REACTIONS
These reactions are characterized by the occurrence of fever, urticaria, polyarthralgias, and lymphadenopathy 7–10 days after primary exposure and 2–4 days after secondary exposure to a heterologous protein (classic serum sickness) or a nonprotein drug such as penicillin or sulfa (serum sickness–like reaction). Most of the manifestations are not due to a vasculitis; however, occasional patients will have typical cutaneous venulitis that may progress rarely to a systemic vasculitis.
VASCULITIS ASSOCIATED WITH OTHER UNDERLYING DISEASES
Certain infections may directly trigger an inflammatory vasculitic process. For example, rickettsias can invade and proliferate in the endothelial cells of small blood vessels causing a vasculitis (Chap. 211). In addition, the inflammatory response around blood vessels associated with certain systemic fungal diseases such as histoplasmosis (Chap. 236) may mimic a primary vasculitic process. A leukocytoclastic vasculitis predominantly involving the skin with occasional involvement of other organ systems may be a minor component of many other infections. These include subacute bacterial endocarditis, Epstein-Barr virus infection, HIV infection, and a number of other infections.
Vasculitis can be associated with certain malignancies, particularly lymphoid or reticuloendothelial neoplasms. Leukocytoclastic venulitis confined to the skin is the most common finding; however, widespread systemic vasculitis may occur. Of particular note is the association of hairy cell leukemia (Chap. 134) with polyarteritis nodosa.
A number of connective tissue diseases have vasculitis as a secondary manifestation of the underlying primary process. Foremost among these are systemic lupus erythematosus (Chap. 378), rheumatoid arthritis (Chap. 380), inflammatory myositis (Chap. 388), relapsing polychondritis (Chap. 389), and Sjögren’s syndrome (Chap. 383). The most common form of vasculitis in these conditions is the small-vessel venulitis isolated to the skin. However, certain patients may develop a fulminant systemic necrotizing vasculitis.
Secondary vasculitis has also been observed in association with ulcerative colitis, congenital deficiencies of various complement components, sarcoidosis, primary biliary cirrhosis, ≥1-antitrypsin deficiency, and intestinal bypass surgery.
386e |
Atlas of the Vasculitic Syndromes |
Diagnosis of the vasculitic syndromes is usually based on characteristic histologic or arteriographic findings in a patient who has clinically compatible features. The images provided in this atlas highlight some of the characteristic histologic and radiographic findings that may be seen in the vasculitic diseases. These images demonstrate the importance that tissue histology may have in securing the diagnosis of vasculitis, the utility of diagnostic imaging in the vasculitic diseases, and the improvements in the care of vasculitis patients that have resulted from radiologic innovations.
Tissue biopsies represent vital information in many patients with a suspected vasculitic syndrome, not only in confirming the presence of vasculitis and other characteristic histologic features, but also in ruling out other diseases that can have similar clinical presentations. The determination of where biopsies should be performed is based on the presence of clinical disease in an affected organ, the likelihood of a positive diagnostic yield from data contained in the published literature, and the risk of performing a biopsy in an affected site. Common sites where biopsies may be performed include the lung, kidney, and skin. Other sites such as sural nerve, brain, testicle, and gastrointestinal tissues may also demonstrate features of vasculitis and be appropriate locations for biopsy when clinically affected.
Surgical biopsies of radiographically abnormal pulmonary parenchyma have a diagnostic yield of 90% in patients with granulomatosis with polyangiitis (Wegener’s) and play an important role in ruling out infection or malignancy. The yield of lung biopsies is highly associated with amount of tissue that can be obtained, and transbronchial biopsies, while less invasive, have a yield of only 7%. Lung biopsies also play an important role in microscopic polyangiitis, eosinophilic granulomatosis with polyangiitis (Churg-Strauss), and any vasculitic disease where an immunosuppressed patient has pulmonary disease that is suspected to be an infection.
Kidney biopsy findings of a focal, segmental, crescentic, necrotizing glomerulonephritis with few to no immune complexes (pauci-immune glomerulonephritis) are characteristic in patients with granulomatosis with polyangiitis (Wegener’s), microscopic polyangiitis, or eosinophilic granulomatosis with polyangiitis (Churg-Strauss), who have active renal disease. These findings not only distinguish these entities from other causes of glomerulonephritis, but can also confirm the presence of active glomerulonephritis that requires treatment. As a result, renal biopsies can also be helpful to guide management decisions in these diseases when an established patient has worsening renal function and an inactive or equivocal urine sediment. Cryoglobulinemic vasculitis and IgA vasculitis (Henoch-Schönlein) are other vasculitides where renal involvement may occur and where biopsy may be important in diagnosis or prognosis.
Biopsies of the skin are commonly performed and are well tolerated. Because not all purpuric or ulcerative lesions are due to vasculitis, skin biopsy plays an important role to confirm the presence of vasculitis as the cause of the manifestation. Cutaneous vasculitis represents the most common vasculitic feature that affects people and can be seen in a broad spectrum of settings including infections, medications, malignancies, and connective tissue diseases. As a result, for systemic vasculitides that will require aggressive immunosuppressive treatment, a skin biopsy may not represent sufficient evidence to secure the diagnosis.
Diagnostic imaging represents a critical assessment tool in patients who are known or suspected to have a systemic vasculitic disease. Imaging contributes unique information about the patient that, when taken together with the history, physical examination, and laboratory determinations, can guide the differential diagnosis and the subsequent assessment or treatment plan. A diverse range of imaging techniques is used in the assessment of vasculitis including plain radiography, ultrasonography, computed tomography (CT), magnetic resonance imaging (MRI), positron emission tomography, and catheter-directed dye arteriography. These procedures have specific utilities that can allow differing perspectives on the spectrum and severity of vasculitis.
For vasculitic diseases that involve the large- or medium-sized blood vessels, arteriography provides information regarding blood vessel stenoses or aneurysms that can support the diagnosis. Catheter-directed dye arteriography provides information on central blood pressure and offers the most precise detail regarding vessel lumen dimensions but carries risks related to dye exposure and the invasive nature of the procedure. Advancements in magnetic resonance (MR) and CT arteriography have brought about noninvasive options to view the lumen and vessel wall, thus enhancing the ability to perform serial studies for patient monitoring in large-vessel vasculitis. However, in patients suspected to have a medium-vessel vasculitis such as polyarteritis nodosa, catheter-directed dye arteriography should still be performed because MR and CT arteriograms do not currently have sufficient resolution to visualize arteries of this size.
Although vasculitis involving the small blood vessels cannot be directly visualized, diagnostic imaging plays an essential role in detecting tissue injury that occurs as result of blood vessel and tissue inflammation. In granulomatosis with polyangiitis (Wegener’s), 80% of patients may have pulmonary involvement during their disease course. Chest imaging should be obtained whenever active disease is suspected, because up to one-third of patients with radiographic abnormalities are asymptomatic. Pulmonary imaging is also important to detect complications of vasculitis therapy such as opportunistic pneumonias and medication-related pneumonitis.
FIGURE 386e-1 Bilateral nodular infiltrates seen on computed tomography of the chest in a 40-year-old woman with granulomatosis with polyangiitis (Wegener’s).
FIGURE 386e-2 Computed tomography of the chest in two patients with granulomatosis with polyangiitis (Wegener’s) demonstrating (A) single and (B) multiple cavitary lung lesions.
FIGURE 386e-3 Bilateral ground-glass infiltrates due to alveolar hemorrhage from pulmonary capillaritis as seen in the same patient by (A) chest radiograph and (B) computed tomography. This manifestation can occur in granulomatosis with polyangiitis (Wegener’s) or microscopic polyangiitis.
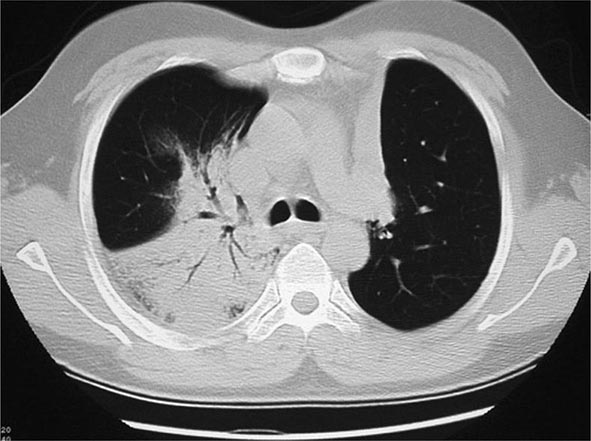
FIGURE 386e-4 Computed tomography of the chest demonstrating a dense infiltrate with air bronchograms involving a segment of the right upper lobe due to bacterial pneumonia in an immunosuppressed patient with granulomatosis with polyangiitis (Wegener’s). Collapse of the left upper lobe secondary to endobronchial stenosis from granulomatosis with polyangiitis (Wegener’s) also is seen on this image.
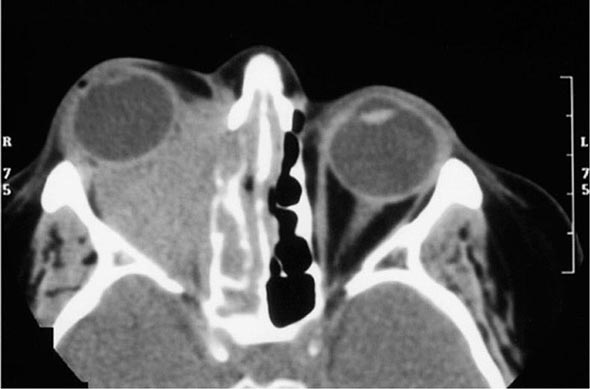
FIGURE 386e-5 Computed tomography of the orbits in a patient with granulomatosis with polyangiitis (Wegener’s) who presented with right-eye proptosis. The image demonstrates inflammatory tissue extending from the ethmoid sinus through the lamina papyracea and filling the orbital space.
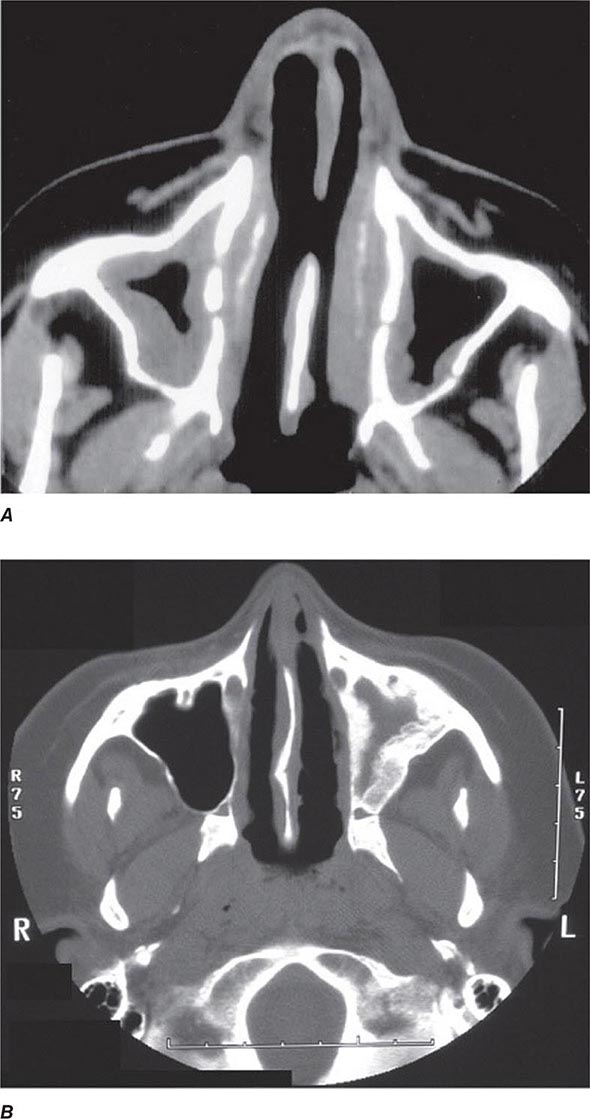
FIGURE 386e-6 Computed tomography of the sinuses in two patients with granulomatosis with polyangiitis (Wegener’s). (A) Mucosal thickening of the bilateral maxillary sinuses and a perforation of the nasal septum. (B) Osteitis with obliteration of the left maxillary sinus in a patient with long-standing sinus disease.
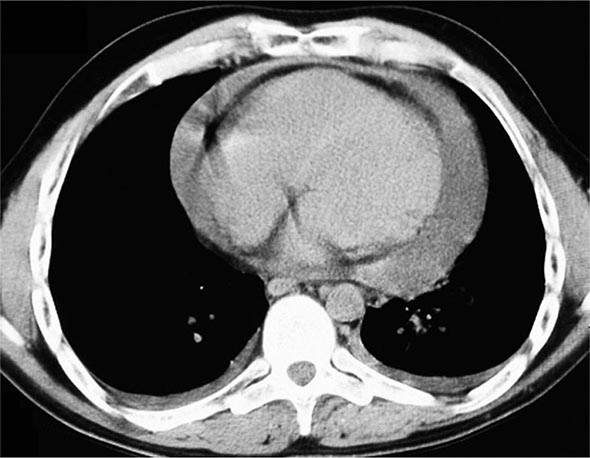
FIGURE 386e-7 Computed tomography of the chest demonstrating a large pericardial effusion in a patient with eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Cardiac involvement is an important cause of morbidity and mortality in eosinophilic granulomatosis with polyangiitis and can include myocarditis, endocarditis, and pericarditis.
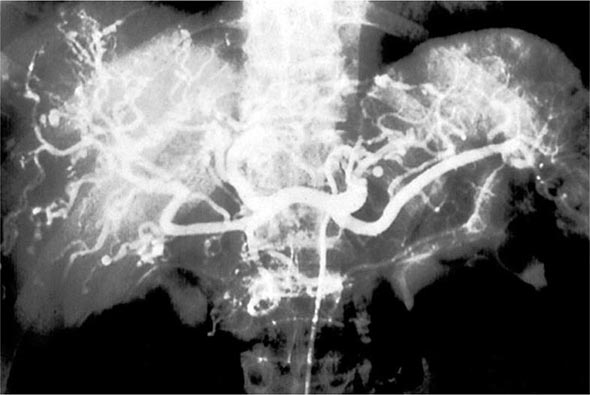
FIGURE 386e-8 Arteriogram of a 40-year-old man with polyarteritis nodosa demonstrating microaneurysms in the hepatic circulation.
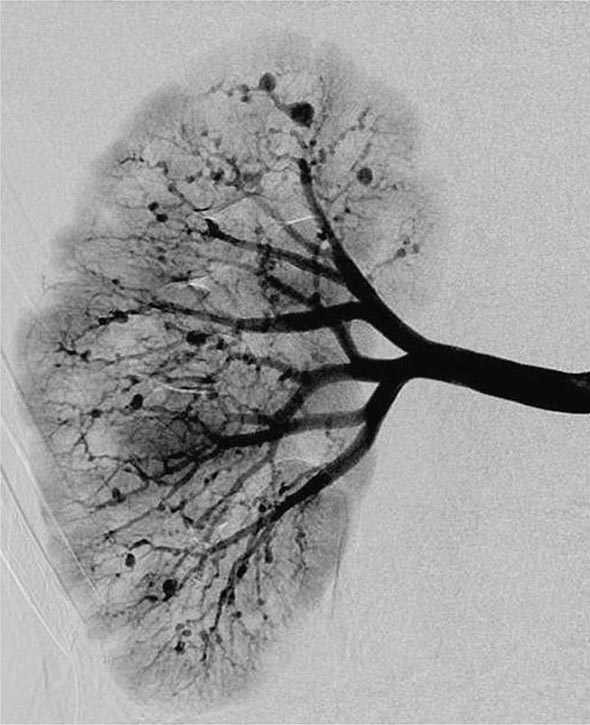
FIGURE 386e-9 Arteriogram of a 19-year-old man with polyarteritis nodosa demonstrating multiple microaneurysms in the renal circulation. The patient presented with headache and severe hypertension that was due to medium-vessel vasculitis affecting the kidney.
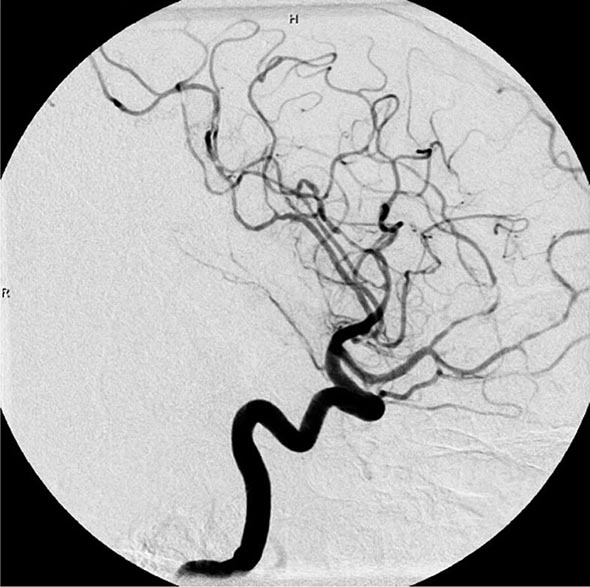
FIGURE 386e-10 Cerebral arteriogram demonstrating beading along branches of the internal carotid artery in a patient with primary central nervous system vasculitis.
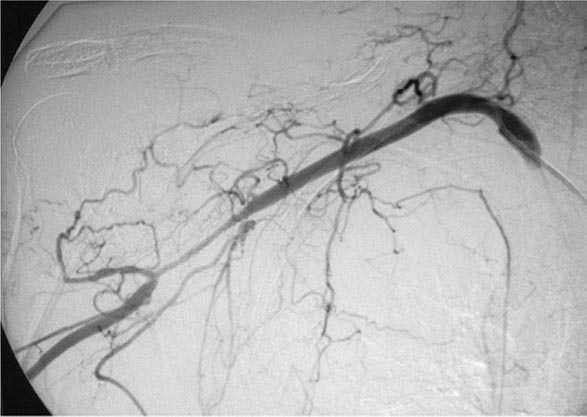
FIGURE 386e-11 Upper-extremity arteriogram demonstrating a long stenotic lesion of the axillary artery in a 75-year-old female with giant cell arteritis.
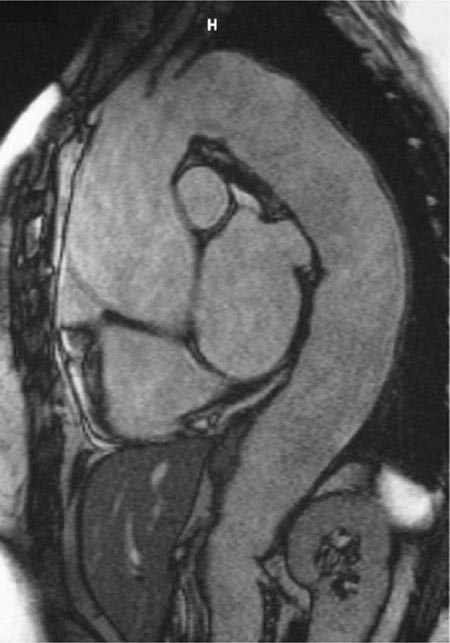
FIGURE 386e-12 Magnetic resonance imaging demonstrating extensive aneurysmal disease of the thoracic aorta in an 80-year-old female. The patient had been diagnosed with biopsy-proven giant cell arteritis 10 years prior to presenting with this aneurysm.
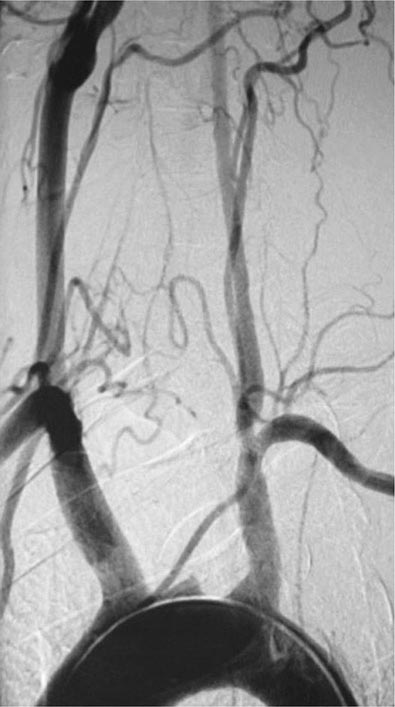
FIGURE 386e-13 Arteriogram of the aortic arch demonstrating complete occlusion of the left common carotid artery just after its origin from the aorta. This 20-year-old female presented with syncope and was subsequently diagnosed with Takayasu arteritis.
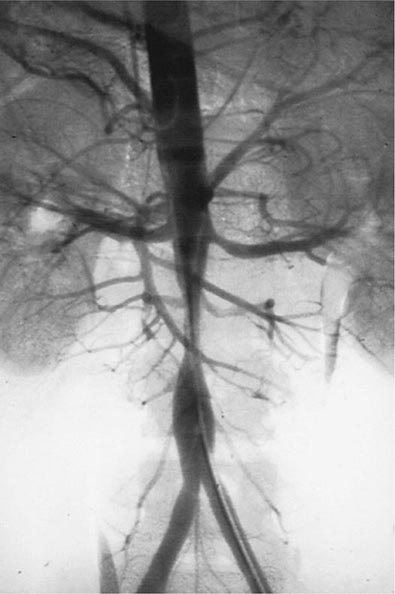
FIGURE 386e-14 Arteriogram demonstrating stenosis of the abdominal aorta in a 25-year-old female with Takayasu arteritis.
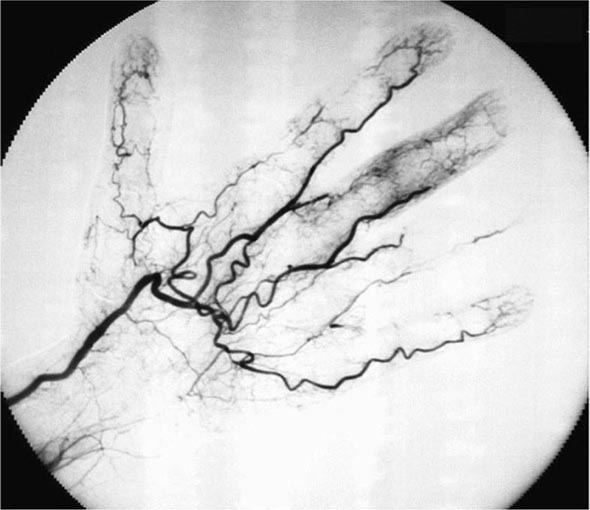
FIGURE 386e-15 Arteriogram of the hand demonstrating arterial skip lesions and vessel cutoffs in a patient with cryoglobulinemic vasculitis due to multiple myeloma.
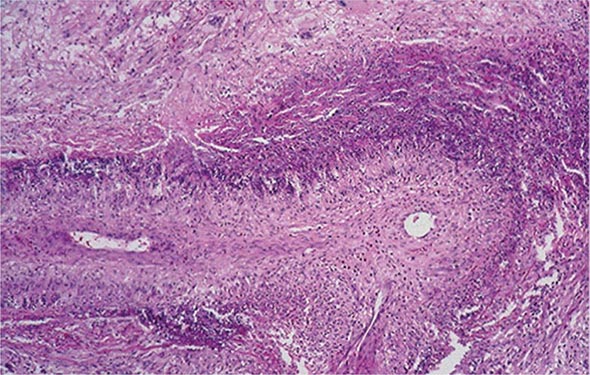
FIGURE 386e-16 Lung histology in granulomatosis with polyangiitis (Wegener’s). This lung biopsy shows areas of geographic necrosis with a border of histiocytes and giant cells. There is also vasculitis with neutrophils, lymphocytes, and giant cells infiltrating the wall of an artery.
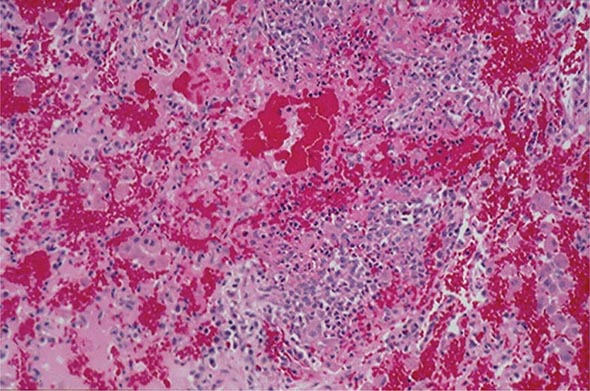
FIGURE 386e-17 Lung histology in microscopic polyangiitis. This lung biopsy demonstrates hemorrhage in the alveolar spaces due to capillaritis in a patient with microscopic polyangiitis. Similar findings can also be seen in granulomatosis with polyangiitis (Wegener’s) and less commonly in eosinophilic granulomatosis with polyangiitis (Churg-Strauss).
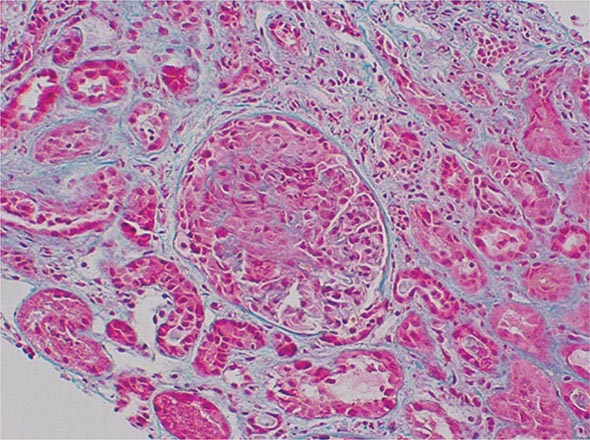
FIGURE 386e-18 Kidney biopsy in granulomatosis with polyangiitis (Wegener’s). This renal biopsy shows a crescentic and necrotizing glomerulonephritis. These findings were focal and segmental with normal and scarred glomeruli also being found in the biopsy. By immunofluorescence and electron microscopy, no immune deposits were present, indicative of a pauci-immune glomerulonephritis. Similar findings can also be seen in microscopic polyangiitis and eosinophilic granulomatosis with polyangiitis (Churg-Strauss).
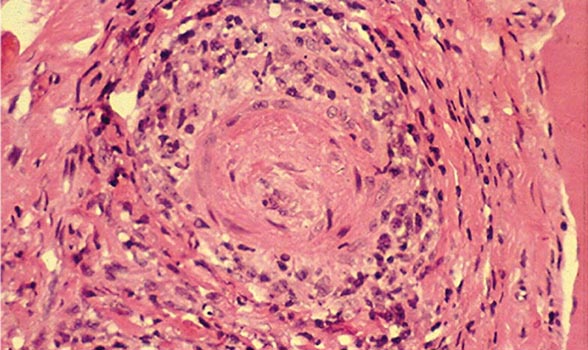
FIGURE 386e-19 Sural nerve biopsy in polyarteritis nodosa. This sural nerve biopsy was performed in a patient with polyarteritis nodosa, who had presented with a mononeuritis multiplex. Neutrophils are seen infiltrating all layers of this medium-sized vessel, which resulted in vessel occlusion and nerve infraction.
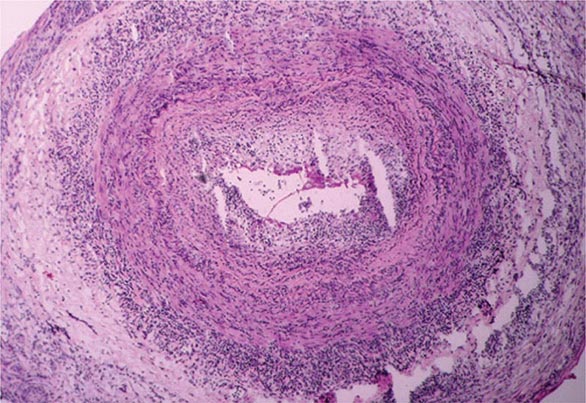
FIGURE 386e-20 Temporal artery biopsy in giant cell arteritis. This temporal artery biopsy demonstrates a panmural infiltration of mononuclear cells and lymphocytes that are particularly seen in the media and adventitia. Scattered giant cells are also present.
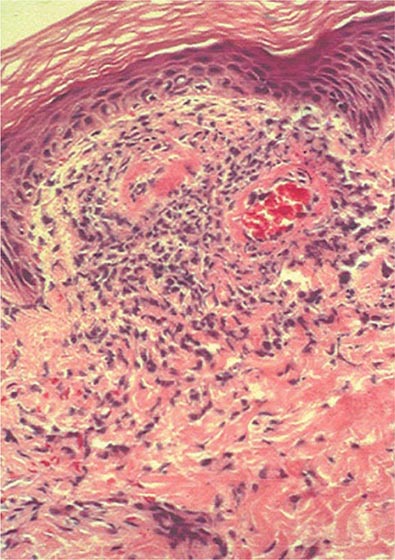
FIGURE 386e-21 Cutaneous vasculitis. This skin biopsy reveals two arterioles beneath the dermis with a neutrophilic inflammatory infiltrate in and around the vessel wall with leukocytoclasis (nuclear debris). While such features are diagnostic of vasculitis, they can be seen in a variety of settings and are not specific for any single disease.
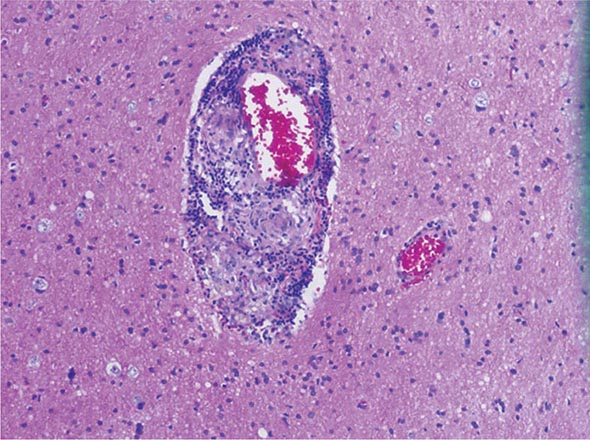
FIGURE 386e-22 Granulomatous primary central nervous system vasculitis. This brain biopsy demonstrates a medium-sized artery with granulomatous inflammation present within the vessel wall indicative of a granulomatous vasculitis. This patient presented with progressive headache, clinical and radiographic features of a stroke, and had arteriographic features consistent with vasculitis. Because no evidence of vasculitis could be found outside of the brain, this was consistent with granulomatous primary central nervous system vasculitis.
387 |
Behçet’s Syndrome |
DEFINITION, INCIDENCE, AND PREVALENCE
Behçet’s syndrome is a multisystem disorder presenting with recurrent oral and genital ulcerations as well as ocular involvement. The diagnosis is clinical and based on internationally agreed diagnostic criteria (Table 387-1).
|
DIAGNOSTIC CRITERIA OF BEHÇET’S SYNDROME |
The syndrome affects young males and females from the Mediterranean region, the Middle East, and the Far East, suggesting a link with the ancient Silk Route. Males and females are affected equally, but males often have more severe disease. Blacks are very infrequently affected.
PATHOGENESIS
The etiology and pathogenesis of this syndrome remain obscure. The disease appears to be in the crossroads of autoinflammatory and autoimmune disorders. The main pathologic lesion is systemic perivasculitis with early neutrophil infiltration and endothelial swelling. In some patients, diffuse inflammatory disease, involving all layers of large vessels and resulting to formation of pseudoaneurysms, suggests vasculitis of vasa vasorum. Apart from activated neutrophils, increased numbers of infiltrating TH1, TH17, cytotoxic CD8+, and γδ T cells are observed, suggesting a link between innate and adaptive autoreactive immune response. Circulating autoantibodies against α-enolase of endothelial cells, selenium binding protein, and anti-Saccharomyces cerevisiae antibodies have been observed, but their pathogenic role remains unclear. A recent genome-wide association study confirmed the known association of Behçet’s syndrome with HLA-B*51 and identified a second, independent association within the major histocompatibility complex (MHC) class I region. In addition, an association with interleukin (IL) 10 and the IL-23R–IL-12RB2 locus was also observed. Interestingly, the disease-associated IL-10 variant was correlated with diminished mRNA expression and low protein production.
CLINICAL FEATURES
The recurrent aphthous ulcerations are a sine qua non for the diagnosis. The ulcers are usually painful, are shallow or deep with a central yellowish necrotic base, appear singly or in crops, and are located anywhere in the oral cavity. Small ulcers, less than 10 mm in diameter, are seen in 85% of patients, whereas large or herpetiform lesions are less frequent. The ulcers persist for 1–2 weeks and subside without leaving scars. The genital ulcers are less common but more specific, are painful, do not affect the glans penis or urethra, and produce scrotal scars.
Skin involvement is observed in 80% of patients and includes folliculitis, erythema nodosum, an acne-like exanthem, and, infrequently, vasculitis, Sweet syndrome, and pyoderma gangrenosum. Nonspecific skin inflammatory reactivity to any scratches or intradermal saline injection (pathergy test) is a common and specific manifestation.
Eye involvement with scarring and bilateral panuveitis is the most dreaded complication, since it occasionally progresses rapidly to blindness. The eye disease, occurring in 50% of patients, is usually present at the onset but may also develop within the first few years. In addition to iritis, posterior uveitis, retinal vessel occlusions, and optic neuritis can be seen in some patients with the syndrome.
Nondeforming arthritis or arthralgias are seen in 50% of patients and affect the knees and ankles.
Superficial or deep peripheral vein thrombosis is seen in 30% of patients. Pulmonary emboli are a rare complication. The superior vena cava is obstructed occasionally, producing a dramatic clinical picture. Arterial involvement occurs in less than 5% of patients and presents with aortitis or peripheral arterial aneurysm and arterial thrombosis. Pulmonary artery vasculitis presenting with dyspnea, cough, chest pain, hemoptysis, and infiltrates on chest roentgenograms has been reported in 5% of patients and should be differentiated from thromboembolic disease since it warrants anti-inflammatory and not thrombolytic therapy.
Neurologic involvement (5–10%) appears mainly in the parenchymal form (80%); it is associated with brainstem involvement and has a serious prognosis (central nervous system [CNS]-Behçet’s syndrome). IL-6 is persistently raised in cerebrospinal fluid of these patients. Cerebral venous thrombosis is most frequently observed in the superior sagittal and transverse sinuses and is associated with headache and increased intracranial pressure. Magnetic resonance imaging (MRI) and/or proton magnetic resonance spectroscopy (MRS) are very sensitive and should be employed if CNS-Behçet’s syndrome is suspected.
Gastrointestinal involvement is seen more frequently in patients from Japan and consists of mucosal ulcerations of the gut, resembling Crohn’s disease.
Epididymitis is seen in 5% of patients, whereas amyloidosis of AA type and glomerulonephritis are uncommon.
Laboratory findings are mainly nonspecific indices of inflammation, such as leukocytosis and elevated erythrocyte sedimentation rate, as well as C-reactive protein levels.
388 |
Polymyositis, Dermatomyositis, and Inclusion Body Myositis |
The inflammatory myopathies represent the largest group of acquired and potentially treatable causes of skeletal muscle weakness. They are classified into three major groups: polymyositis (PM), dermatomyositis (DM), and inclusion body myositis (IBM).
CLINICAL FEATURES
The prevalence of the inflammatory myopathies is estimated at 1 in 100,000. PM as a stand-alone entity is a rare disease. DM affects both children and adults and women more often than men. IBM is three times more frequent in men than in women, more common in whites than blacks, and is most likely to affect persons age >50 years.
These disorders present as progressive and symmetric muscle weakness except for IBM, which can have an asymmetric pattern. Patients usually report increasing difficulty with everyday tasks requiring the use of proximal muscles, such as getting up from a chair, climbing steps, stepping onto a curb, lifting objects, or combing hair. Finemotor movements that depend on the strength of distal muscles, such as buttoning a shirt, sewing, knitting, or writing, are affected only late in the course of PM and DM, but fairly early in IBM. Falling is common in IBM because of early involvement of the quadriceps muscle, with buckling of the knees. Ocular muscles are spared, even in advanced, untreated cases; if these muscles are affected, the diagnosis of inflammatory myopathy should be questioned. Facial muscles are unaffected in PM and DM, but mild facial muscle weakness is common in patients with IBM. In all forms of inflammatory myopathy, pharyngeal and neck-flexor muscles are often involved, causing dysphagia or difficulty in holding up the head (head drop). In advanced and rarely in acute cases, respiratory muscles may also be affected. Severe weakness, if untreated, is almost always associated with muscle wasting. Sensation remains normal. The tendon reflexes are preserved but may be absent in severely weakened or atrophied muscles, especially in IBM, where atrophy of the quadriceps and the distal muscles is common. Myalgia and muscle tenderness may occur in a small number of patients, usually early in the disease, and particularly in DM associated with connective tissue disorders. Weakness in PM and DM progresses subacutely over a period of weeks or months and rarely acutely; by contrast, IBM progresses very slowly, over years, simulating a late-life muscular dystrophy (Chap. 462e) or slowly progressive motor neuron disorder (Chap. 452).
SPECIFIC FEATURES
|
FEATURES ASSOCIATED WITH INFLAMMATORY MYOPATHIES |
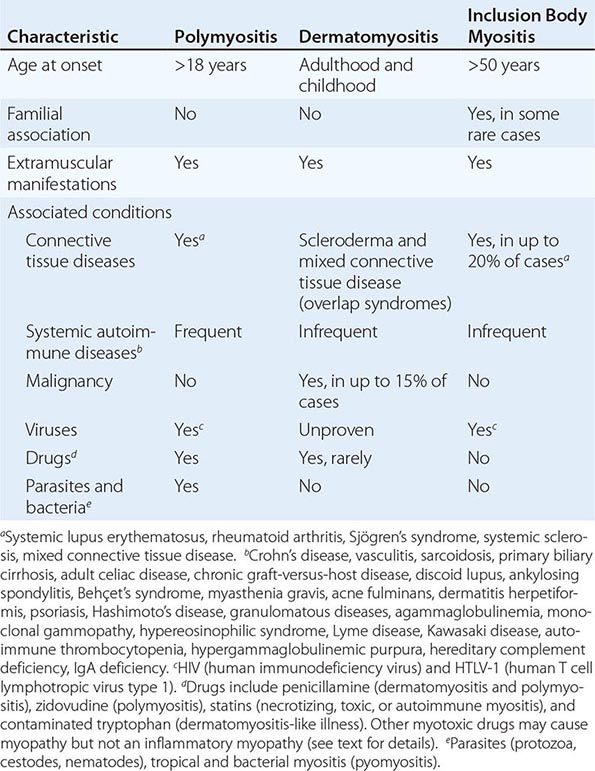
Polymyositis The actual onset of PM is often not easily determined, and patients typically delay seeking medical advice for several weeks or even months. This is in contrast to DM, in which the rash facilitates early recognition (see below). PM mimics many other myopathies and is a diagnosis of exclusion. It is a subacute inflammatory myopathy affecting adults, and rarely children, who do not have any of the following: rash, involvement of the extraocular and facial muscles, family history of a neuromuscular disease, history of exposure to myotoxic drugs or toxins, endocrinopathy, neurogenic disease, muscular dystrophy, biochemical muscle disorder (deficiency of a muscle enzyme), or IBM as excluded by muscle biopsy analysis (see below). As an isolated entity, PM is a rare (and overdiagnosed) disorder; more commonly, PM occurs in association with a systemic autoimmune or connective tissue disease or with a known viral or bacterial infection. Drugs, especially D-penicillamine, statins, or zidovudine (AZT), may also trigger an inflammatory myopathy similar to PM.
Dermatomyositis DM is a distinctive entity identified by a characteristic rash accompanying, or more often preceding, muscle weakness. The rash may consist of a blue-purple discoloration on the upper eyelids with edema (heliotrope rash; see Fig. 73-3), a flat red rash on the face and upper trunk, and erythema of the knuckles with a raised violaceous scaly eruption (Gottron’s sign; see Fig. 73-4). The erythematous rash can also occur on other body surfaces, including the knees, elbows, malleoli, neck and anterior chest (often in a V sign), or back and shoulders (shawl sign), and may worsen after sun exposure. In some patients, the rash is pruritic, especially on the scalp, chest, and back. Dilated capillary loops at the base of the fingernails are also characteristic. The cuticles may be irregular, thickened, and distorted, and the lateral and palmar areas of the fingers may become rough and cracked, with irregular, “dirty” horizontal lines, resembling mechanic’s hands. The weakness can be mild, moderate, or severe enough to lead to quadriparesis. At times, the muscle strength appears normal, hence the term dermatomyositis sine myositis. When muscle biopsy is performed in such cases, however, significant perivascular and perimysial inflammation is often seen.
DM usually occurs alone but may overlap with scleroderma and mixed connective tissue disease. Fasciitis and thickening of the skin, similar to that seen in chronic cases of DM, have occurred in patients with the eosinophilia-myalgia syndrome associated with the ingestion of contaminated L-tryptophan.
Inclusion Body Myositis In patients ≥50 years of age, IBM is the most common of the inflammatory myopathies. It is often misdiagnosed as PM and is suspected only later when a patient with presumed PM does not respond to therapy. Weakness and atrophy of the distal muscles, especially foot extensors and deep finger flexors, occur in almost all cases of IBM and may be a clue to early diagnosis. Some patients present with falls because their knees collapse due to early quadriceps weakness. Others present with weakness in the small muscles of the hands, especially finger flexors, and complain of inability to hold objects such as golf clubs or perform tasks such as turning keys or tying knots. On occasion, the weakness and accompanying atrophy can be asymmetric and selectively involve the quadriceps, iliopsoas, triceps, biceps, and finger flexors, resembling a lower motor neuron disease. Dysphagia is common, occurring in up to 60% of IBM patients, and may lead to episodes of choking. Sensory examination is generally normal; some patients have mildly diminished vibratory sensation at the ankles that presumably is age-related. The pattern of distal weakness, which superficially resembles motor neuron or peripheral nerve disease, results from the myopathic process affecting distal muscles selectively. Disease progression is slow but steady, and most patients require an assistive device such as cane, walker, or wheelchair within several years of onset.
In at least 20% of cases, IBM is associated with systemic autoimmune or connective tissue diseases. Familial aggregation of typical IBM may occur; such cases have been designated as familial inflammatory IBM. This disorder is distinct from hereditary inclusion body myopathy (h-IBM), which describes a heterogeneous group of recessive, and less frequently dominant, inherited syndromes; the h-IBMs are noninflammatory myopathies. A subset of h-IBM that spares the quadriceps muscles has emerged as a distinct entity. This disorder, originally described in Iranian Jews and now seen in many ethnic groups, is linked to chromosome 9p1 and results from mutations in the UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) gene.
ASSOCIATED CLINICAL FINDINGS
Extramuscular Manifestations These may be present to a varying degree in patients with PM or DM, and include:
1. Systemic symptoms, such as fever, malaise, weight loss, arthralgia, and Raynaud’s phenomenon, especially when inflammatory myopathy is associated with a connective tissue disorder.
2. Joint contractures, mostly in DM and especially in children.
3. Dysphagia and gastrointestinal symptoms, due to involvement of oropharyngeal striated muscles and upper esophagus, especially in DM and IBM.
4. Cardiac disturbances, including atrioventricular conduction defects, tachyarrhythmias, dilated cardiomyopathy, a low ejection fraction, and congestive heart failure, which may rarely occur either from the disease itself or from hypertension associated with long-term use of glucocorticoids.
5. Pulmonary dysfunction, due to weakness of the thoracic muscles, interstitial lung disease, or drug-induced pneumonitis (e.g., from methotrexate), which may cause dyspnea, nonproductive cough, and aspiration pneumonia. Interstitial lung disease may precede myopathy or occur early in the disease and develops in up to 10% of patients with PM or DM, most of whom have antibodies to t-RNA synthetases, as described below.
6. Subcutaneous calcifications, in DM, sometimes extruding on the skin and causing ulcerations and infections.
7. Arthralgias, synovitis, or deforming arthropathy with subluxation in the interphalangeal joints, which can occur in some patients with DM and PM who have Jo-1 antibodies (see below).
Association with Malignancies Although all the inflammatory myopathies can have a chance association with malignant lesions, especially in older age groups, the incidence of malignant conditions appears to be specifically increased only in patients with DM and not in those with PM or IBM. The most common tumors associated with DM are ovarian cancer, breast cancer, melanoma, colon cancer, and non-Hodgkin’s lymphoma. The extent of the search that should be conducted for an occult neoplasm in adults with DM depends on the clinical circumstances. Tumors in these patients are usually uncovered by abnormal findings in the medical history and physical examination and not through an extensive blind search. The weight of evidence argues against performing expensive, invasive, and nondirected tumor searches. A complete annual physical examination with pelvic, breast (mammogram, if indicated), and rectal examinations (with colonoscopy according to age and family history); urinalysis; complete blood count; blood chemistry tests; and a chest film should suffice in most cases. In Asians, nasopharyngeal cancer is common, and a careful examination of ears, nose, and throat is indicated. If malignancy is clinically suspected, screening with a whole-body positron emission tomography (PET) scan should be considered.
Overlap Syndromes These describe the association of inflammatory myopathies with connective tissue diseases. A well-characterized overlap syndrome occurs in patients with DM who also have manifestations of systemic sclerosis or mixed connective tissue disease, such as sclerotic thickening of the dermis, contractures, esophageal hypomotility, microangiopathy, and calcium deposits (Table 388-1). By contrast, signs of rheumatoid arthritis, systemic lupus erythematosus, or Sjögren’s syndrome are very rare in patients with DM. Patients with the overlap syndrome of DM and systemic sclerosis may have a specific antinuclear antibody, the anti-PM/Scl, directed against a nucleolar-protein complex.
PATHOGENESIS
An autoimmune etiology of the inflammatory myopathies is indirectly supported by an association with other autoimmune or connective tissue diseases; the presence of various autoantibodies; an association with specific major histocompatibility complex (MHC) genes; demonstration of T cell–mediated myocytotoxicity or complement-mediated microangiopathy; and a response to immunotherapy.
Autoantibodies and Immunogenetics Various autoantibodies against nuclear antigens (antinuclear antibodies) and cytoplasmic antigens are found in up to 30% of patients with inflammatory myopathies. The antibodies to cytoplasmic antigens are directed against ribonucleoproteins involved in protein synthesis (antisynthetases) or translational transport (anti-signal-recognition particles). The antibody directed against the histidyl-transfer RNA synthetase, called anti-Jo-1, accounts for 75% of all the antisynthetases and is clinically useful because up to 80% of patients with anti-Jo-1 antibodies have interstitial lung disease. Some patients with the anti-Jo-1 antibody also have Raynaud’s phenomenon, nonerosive arthritis, and the MHC molecules DR3 and DRw52. DR3 haplotypes (molecular designation DRB1*0301, DQB1*0201) occur in up to 75% of patients with PM and IBM, whereas in juvenile DM, there is an increased frequency of DQA1*0501 (Chap. 373e). Antibodies against the cytosolic 5′-nucleotidase 1A, an enzyme abundantly expressed in muscle and thought to be involved in DNA degradation and repair, have been detected in one-third of IBM patients. Although the pathogenic significance of these antibodies is still unknown, they highlight the presence of an immune response, as discussed below.
Immunopathologic Mechanisms In DM, humoral immune mechanisms are implicated, resulting in a microangiopathy and muscle ischemia (Fig. 388-1). Endomysial inflammatory infiltrates are composed of B cells located in proximity to CD4 T cells, plasmacytoid dendritic cells, and macrophages; there is a relative absence of lymphocytic invasion of nonnecrotic muscle fibers. Activation of the complement C5b-9 membranolytic attack complex is thought to be a critical early event that triggers release of proinflammatory cytokines and chemokines, induces expression of vascular cell adhesion molecule (VCAM) 1 and intercellular adhesion molecule (ICAM) 1 on endothelial cells, and facilitates migration of activated lymphoid cells to the perimysial and endomysial spaces. Necrosis of the endothelial cells, reduced numbers of endomysial capillaries, ischemia, and muscle-fiber destruction resembling microinfarcts occur. The remaining capillaries often have dilated lumens in response to the ischemic process. Larger intramuscular blood vessels may also be affected in the same pattern. Residual perifascicular atrophy reflects the endofascicular hypoperfusion that is prominent in the periphery of muscle fascicles. Increased expression of type I interferon–inducible proteins is also noted in these regions.
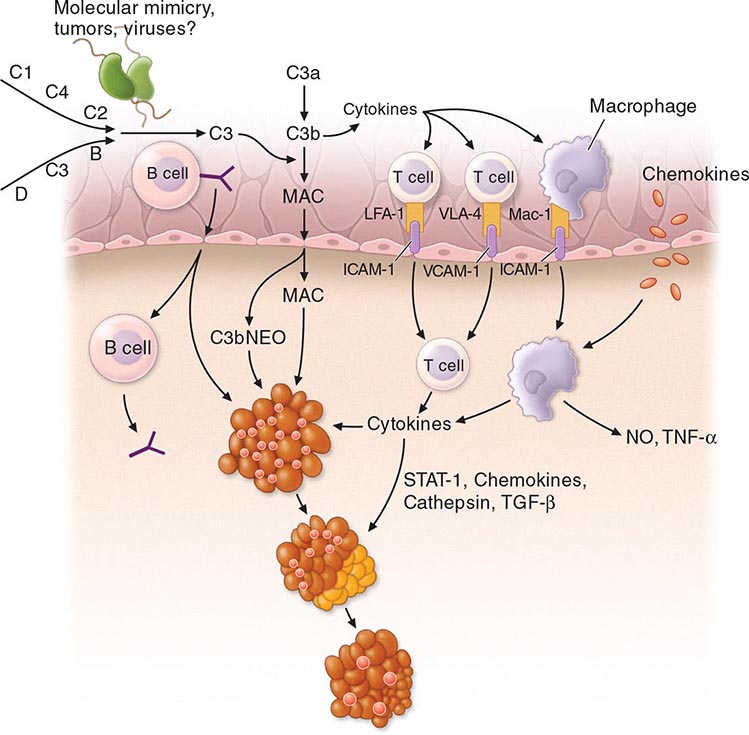
FIGURE 388-1 Immunopathogenesis of dermatomyositis. Activation of complement, possibly by autoantibodies (Y), against endothelial cells and formation of C3 via the classic or alternative pathway. Activated C3 leads to formation of C3b, C3bNEO, and membrane attack complexes (MAC), which are deposited in and around the endothelial cell wall of the endomysial capillaries. Deposition of MAC leads to destruction of capillaries, ischemia, or microinfarcts, most prominent in the periphery of the fascicles, and perifascicular atrophy. B cells, plasmacytoid dendritic cells, CD4 T cells, and macrophages traffic from the circulation to the muscle. Endothelial expression of vascular cell adhesion molecule (VCAM) and intercellular adhesion molecule (ICAM) is induced by cytokines released by the mononuclear cells. Integrins, specifically very late activation antigen (VLA)-4 and lymphocyte function–associated antigen (LFA)-1, bind VCAM and ICAM and promote T cell and macrophage infiltration of muscle through the endothelial cell wall.
By contrast, in PM and IBM, a mechanism of T cell–mediated cytotoxicity is likely. CD8 T cells, along with macrophages, initially surround and eventually invade and destroy healthy, nonnecrotic muscle fibers that aberrantly express class I MHC molecules. MHC-I expression, absent from the sarcolemma of normal muscle fibers, is probably induced by cytokines secreted by activated T cells and macrophages. The CD8/MHC-I complex is characteristic of PM and IBM; its detection can aid in confirming the histologic diagnosis of PM, as discussed below. The cytotoxic CD8 T cells contain perforin and granzyme granules directed toward the surface of the muscle fibers and capable of inducing myonecrosis. Analysis of T cell receptor molecules expressed by the infiltrating CD8 cells has revealed clonal expansion and conserved sequences in the antigen-binding region, both suggesting an antigen-driven T cell response. Whether the putative antigens are endogenous (e.g., muscle) or exogenous (e.g., viral) sequences is unknown. Viruses have not been identified within the muscle fibers. Co-stimulatory molecules and their counterreceptors, which are fundamental for T cell activation and antigen recognition, are strongly upregulated in PM and IBM. As noted above, the possibility that B cells and the humoral immune system might also play a role in IBM is suggested by the identification of anti-cytosolic 5′-nucleotidase 1A antibodies in some patients. Key molecules involved in T cell–mediated cytotoxicity are depicted in Fig. 388-2.
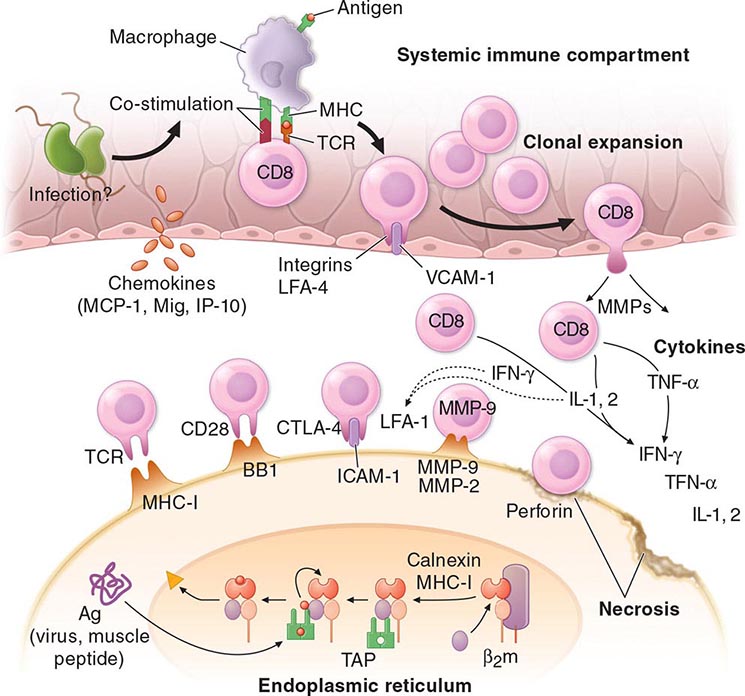
FIGURE 388-2 Cell-mediated mechanisms of muscle damage in polymyositis (PM) and inclusion body myositis (IBM). Antigen-specific CD8 cells are expanded in the periphery, cross the endothelial barrier, and bind directly to muscle fibers via T cell receptor (TCR) molecules that recognize aberrantly expressed major histocompatibility complex (MHC)-I. Engagement of co-stimulatory molecules (BB1 and ICOSL) with their ligands (CD28, CTLA-4, and ICOS), along with ICAM-1/LFA-1, stabilize the CD8–muscle fiber interaction. Metalloproteinases (MMPs) facilitate the migration of T cells and their attachment to the muscle surface. Muscle fiber necrosis occurs via perforin granules released by the autoaggressive T cells. A direct myocytotoxic effect exerted by the cytokines interferon (IFN) γ, interleukin (IL) 1, or tumor necrosis factor (TNF) α may also play a role. Death of the muscle fiber is mediated by necrosis. MHC class I molecules consist of a heavy chain and a light chain (β2 microglobulin [β2m]) complexed with an antigenic peptide that is transported into the endoplasmic reticulum by TAP proteins (Chap. 373e).
The Role of Nonimmune Factors in IBM In IBM, the presence of Congo red–positive amyloid deposits within some vacuolated muscle fibers and abnormal mitochondria with cytochrome oxidase–negative fibers suggest that, in addition to the autoimmune component, there is also a degenerative process. Similar to Alzheimer’s disease, the intracellular amyloid deposits in IBM are immunoreactive against amyloid precursor protein (APP), β-amyloid, chymotrypsin, apolipoprotein E, presenilin, ubiquitin, and phosphorylated tau, but it is unclear whether these deposits, which are also seen in other vacuolar myopathies, are directly pathogenic or represent secondary phenomena. The same is true for the mitochondrial abnormalities, which may also be secondary to the effects of aging or a bystander effect of upregulated cytokines. Expression of cytokines and upregulation of MHC class I by the muscle fibers may cause an endoplasmic reticulum stress response resulting in intracellular accumulation of stressor molecules or misfolded glycoproteins and activation of nuclear factor κB (NF-κB), leading to further cytokine activation.
Association with Viral Infections and the Role of Retroviruses Several viruses, including coxsackieviruses, influenza, paramyxoviruses, mumps, cytomegalovirus, and Epstein-Barr virus, have been indirectly associated with myositis. For the coxsackieviruses, an autoimmune myositis triggered by molecular mimicry has been proposed because of structural homology between histidyl-transfer RNA synthetase that is the target of the Jo-1 antibody (see above) and genomic RNA of an animal picornavirus, the encephalomyocarditis virus. Sensitive polymerase chain reaction (PCR) studies, however, have repeatedly failed to confirm the presence of such viruses in muscle biopsies.
The best evidence of a viral connection in PM and IBM is with the retroviruses. Some individuals infected with HIV or with human T cell lymphotropic virus 1 (HTLV-1) develop PM or IBM; a similar disorder has been described in nonhuman primates infected with the simian immunodeficiency virus. The inflammatory myopathy may occur as the initial manifestation of a retroviral infection, or myositis may develop later in the disease course. Retroviral antigens have been detected only in occasional endomysial macrophages and not within the muscle fibers themselves, suggesting that persistent infection and viral replication within the muscle does not occur. Histologic findings are identical to retroviral-negative PM or IBM. The infiltrating T cells in the muscle have a clonal bias and a number of them are retroviral-specific. This disorder should be distinguished from a toxic myopathy related to long-term therapy with AZT, characterized by fatigue, myalgia, mild muscle weakness, and mild elevation of creatine kinase (CK). AZT-induced myopathy, which generally improves when the drug is discontinued, is a mitochondrial disorder characterized histologically by “ragged-red” fibers. AZT inhibits γ-DNA polymerase, an enzyme found solely in the mitochondrial matrix.
GLOBAL ISSUES
![]() Inadequate data exist with respect to possible differences in the prevalence of the inflammatory myopathies in various parts of the world. PM appears to be reported more often in Asia and southern Europe, whereas IBM seems to be more frequently recognized in North America, northern Europe, and Australia. Whether this represents differences in diagnostic methods and disease awareness or true disease prevalence remains unclear. Pyomyositis and parasitic myositis are clearly more common in the tropics, whereas HIV-associated PM and IBM are more commonly encountered in areas endemic for HIV. In patients from Asia, nasopharyngeal cancer appears to be a malignancy more commonly associated with DM, necessitating the need to specifically search for these tumors in this population.
Inadequate data exist with respect to possible differences in the prevalence of the inflammatory myopathies in various parts of the world. PM appears to be reported more often in Asia and southern Europe, whereas IBM seems to be more frequently recognized in North America, northern Europe, and Australia. Whether this represents differences in diagnostic methods and disease awareness or true disease prevalence remains unclear. Pyomyositis and parasitic myositis are clearly more common in the tropics, whereas HIV-associated PM and IBM are more commonly encountered in areas endemic for HIV. In patients from Asia, nasopharyngeal cancer appears to be a malignancy more commonly associated with DM, necessitating the need to specifically search for these tumors in this population.
DIFFERENTIAL DIAGNOSIS
The clinical picture of the typical skin rash and proximal or diffuse muscle weakness has few causes other than DM. However, proximal muscle weakness without skin involvement can be due to many conditions other than PM or IBM.
Subacute or Chronic Progressive Muscle Weakness This may be due to denervating conditions such as the spinal muscular atrophies or amyotrophic lateral sclerosis (Chap. 452). In addition to the muscle weakness, upper motor neuron signs in the latter and signs of denervation detected by electromyography (EMG) aid in the diagnosis. The muscular dystrophies (Chap. 462e) may be additional considerations; however, these disorders usually develop over years rather than weeks or months and rarely present after the age of 30 years. It may be difficult, even with a muscle biopsy, to distinguish chronic PM from a rapidly advancing muscular dystrophy. This is particularly true of facioscapulohumeral muscular dystrophy, dysferlin myopathy, and the dystrophinopathies where inflammatory cell infiltration is often found early in the disease. Such doubtful cases should always be given an adequate trial of glucocorticoid therapy and undergo genetic testing to exclude muscular dystrophy. Identification of the MHC/CD8 lesion by muscle biopsy is helpful to identify cases of PM. Some metabolic myopathies, including glycogen storage disease due to myophosphorylase or acid maltase deficiency, lipid storage myopathies due to carnitine deficiency, and mitochondrial diseases produce weakness that is often associated with other characteristic clinical signs; diagnosis rests upon histochemical and biochemical studies of the muscle biopsy. The endocrine myopathies such as those due to hypercorticosteroidism, hyper- and hypothyroidism, and hyper- and hypoparathyroidism require the appropriate laboratory investigations for diagnosis. Muscle wasting in patients with an underlying neoplasm may be due to disuse, cachexia, or rarely a paraneoplastic neuromyopathy (Chap. 122).
Diseases of the neuromuscular junction, including myasthenia gravis or the Lambert-Eaton myasthenic syndrome, cause fatiguing weakness that also affects ocular and other cranial muscles (Chap. 461). Repetitive nerve stimulation and single-fiber EMG studies aid in diagnosis.
Acute Muscle Weakness This may be caused by an acute neuropathy such as Guillain-Barré syndrome (Chap. 460), transverse myelitis (Chap. 456), a neurotoxin (Chap. 462e), or a neurotropic viral infection such as poliomyelitis or West Nile virus (Chap. 164). When acute weakness is associated with very high levels of serum CK (often in the thousands), painful muscle cramps, rhabdomyolysis, and myoglobinuria, it may be due to a necrotizing autoimmune myositis, as discussed below, a viral infection or a metabolic disorder such as myophosphorylase deficiency, or carnitine palmitoyltransferase deficiency (Chap. 462e). Several animal parasites, including protozoa (Toxoplasma, Trypanosoma), cestodes (cysticerci), and nematodes (trichinae), may produce a focal or diffuse inflammatory myopathy known as parasitic polymyositis. Staphylococcus aureus, Yersinia, Streptococcus, or anaerobic bacteria may produce a suppurative myositis, known as tropical polymyositis, or pyomyositis. Pyomyositis, previously rare in the west, is now occasionally seen in AIDS patients. Other bacteria, such as Borrelia burgdorferi (Lyme disease) and Legionella pneumophila (Legionnaire’s disease), may infrequently cause myositis.
Patients with periodic paralysis experience recurrent episodes of acute muscle weakness without pain, always beginning in childhood. Chronic alcoholics may develop painful myopathy with myoglobinuria after a bout of heavy drinking. Acute painless muscle weakness with myoglobinuria may occur with prolonged hypokalemia, or hypophosphatemia and hypomagnesemia, usually in chronic alcoholics or in patients on nasogastric suction receiving parenteral hyperalimentation.
Myofasciitis This distinctive inflammatory disorder affecting muscle and fascia presents as diffuse myalgias, skin induration, fatigue, and mild muscle weakness; mild elevations of serum CK are usually present. The most common form is eosinophilic myofasciitis characterized by peripheral blood eosinophilia and eosinophilic infiltrates in the endomysial tissue. In some patients, the eosinophilic myositis/fasciitis occurs in the context of parasitic infections, vasculitis, mixed connective tissue disease, hypereosinophilic syndrome, or toxic exposures (e.g., toxic oil syndrome, contaminated L-tryptophan) or with mutations in the calpain gene. A distinct subset of myofasciitis is characterized by pronounced infiltration of the connective tissue around the muscle by sheets of periodic acid–Schiff-positive macrophages and occasional CD8 T cells (macrophagic myofasciitis or inflammatory myositis with abundant macrophages [IMAM]). A focal form of this disorder, limited to sites of previous vaccinations, administered months or years earlier, has been linked to an aluminum-containing substrate in vaccines. This disorder, which to date has not been observed outside of France, responds to glucocorticoid therapy, and the overall prognosis seems favorable.
Necrotizing Autoimmune Myositis This is an increasingly recognized entity that has distinct features, even though it is often labeled as PM. It presents as an acute or subacute onset of symmetric muscle weakness; CK is typically extremely high. The weakness can be severe. Coexisting interstitial lung disease and cardiomyopathy may be present. The disorder may develop after a viral infection, in association with cancer, or in patients taking statins when the myopathy continues to worsen after statin withdrawal. Some patients have antibodies against signal recognition particle (SRP) or against the 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), a 100-kDa protein considered the pharmacologic target of statins. The muscle biopsy demonstrates necrotic fibers infiltrated by macrophages but only rare, if any, T cell infiltrates. Muscle MHC-I expression is only slightly and focally upregulated. The capillaries may be swollen with hyalinization, thickening of the capillary wall, and deposition of complement. Most patients respond to immunotherapy, but some are resistant.
Hyperacute Necrotizing Fasciitis/Myositis (Flesh-Eating Disease) This a fulminant infectious disease, seen most often in the tropics or in conditions with poor hygiene, characterized by widespread necrosis of the superficial fascia and muscle of a limb; if the scrotum, perineum, and abdominal wall are affected, the condition is referred to as Fournier’s gangrene. It may be caused by group A β-hemolytic Streptococcus, methicillin-sensitive S. aureus, Pseudomonas aeruginosa, Vibrio vulnificus, clostridial species (gas gangrene; Chap. 179), or polymicrobial infection with anaerobes and facultative bacteria (Chap. 201); toxins from these bacteria may act as superantigens (Chap. 372e). The port of bacterial entry is usually a trivial cut or skin abrasion, and the source is contact with carriers of the organism. Individuals with diabetes mellitus, immunodeficiency states, or systemic illnesses such as liver failure are most susceptible. Systemic varicella is a predisposing factor in children.
The disease presents with swelling, pain, and redness in the involved area followed by a rapid tissue necrosis of fascia and muscle that progresses at an estimated rate of 3 cm/h. Emergency debridement, antibiotics, IV immunoglobulin (IVIg), and even hyperbaric oxygen have been recommended. In progressive or advanced cases, amputation of the affected limb may be necessary to avoid a fatal outcome.
Drug-Induced Myopathies D-Penicillamine, procainamide, and statins may produce a true myositis resembling PM or necrotizing myositis. A DM-like illness has been associated with the contaminated preparations of L-tryptophan. As noted above, AZT causes a mitochondrial myopathy. Other drugs may elicit a toxic noninflammatory myopathy that is histologically different from DM, PM, or IBM. These include cholesterol-lowering agents such as clofibrate, lovastatin, simvastatin, or pravastatin, especially when combined with cyclosporine, amiodarone, or gemfibrozil. Mild statin-induced myopathic symptoms (such as myalgia, fatigue, or asymptomatic elevations of CK) are self-limited and usually improve after discontinuation of the drug. In rare patients, however, muscle weakness continues to progress even after the statin is withdrawn; in these cases, a diagnostic muscle biopsy is indicated and search for antibodies to HMGCR is suggested; if histologic evidence of PM or necrotizing myositis is present, immunotherapy should be initiated. Rhabdomyolysis and myoglobinuria have been rarely associated with amphotericin B, ε-aminocaproic acid, fenfluramine, heroin, and phencyclidine. The use of amiodarone, chloroquine, colchicine, carbimazole, emetine, etretinate, and ipecac syrup; chronic laxative or licorice use resulting in hypokalemia; and glucocorticoid or growth hormone administration have also been associated with myopathic muscle weakness. Some neuromuscular blocking agents such as pancuronium, in combination with glucocorticoids, may cause an acute critical illness myopathy. A careful drug history is essential for diagnosis of these drug-induced myopathies, which do not require immunosuppressive therapy except when an autoimmune myopathy has been triggered, as noted above.
“Weakness” due to Muscle Pain and Muscle Tenderness A number of conditions including polymyalgia rheumatica (Chap. 385) and arthritic disorders of adjacent joints may enter into the differential diagnosis of inflammatory myopathy, even though they do not cause myositis. The muscle biopsy is either normal or discloses type II muscle fiber atrophy. Patients with fibrositis and fibromyalgia (Chap. 396) complain of focal or diffuse muscle tenderness, fatigue, and aching, which is sometimes poorly differentiated from joint pain. Some patients, however, have muscle tenderness, painful muscles on movement, and signs suggestive of a collagen vascular disorder, such as an increased erythrocyte sedimentation rate, C-reactive protein, antinuclear antibody, or rheumatoid factor, along with modest elevation of the serum CK and aldolase. They demonstrate a “break-away” pattern of weakness with difficulty sustaining effort but not true muscle weakness. The muscle biopsy is usually normal or nonspecific. Many such patients show some response to nonsteroidal anti-inflammatory agents or glucocorticoids, although most continue to have indolent complaints. An indolent fasciitis in the setting of an ill-defined connective tissue disorder may be at times present, and these patients should not be labeled as having a psychosomatic disorder. Chronic fatigue syndrome, which may follow a viral infection, can present with debilitating fatigue, sore throat, painful lymphadenopathy, myalgia, arthralgia, sleep disorder, and headache (Chap. 464e). These patients do not have muscle weakness, and the muscle biopsy is normal.
DIAGNOSIS
The clinically suspected diagnosis of PM, DM, IBM, or necrotizing myositis is confirmed by analysis of serum muscle enzymes, EMG findings, and muscle biopsy (Table 388-2).
|
CRITERIA FOR DIAGNOSIS OF INFLAMMATORY MYOPATHIES |
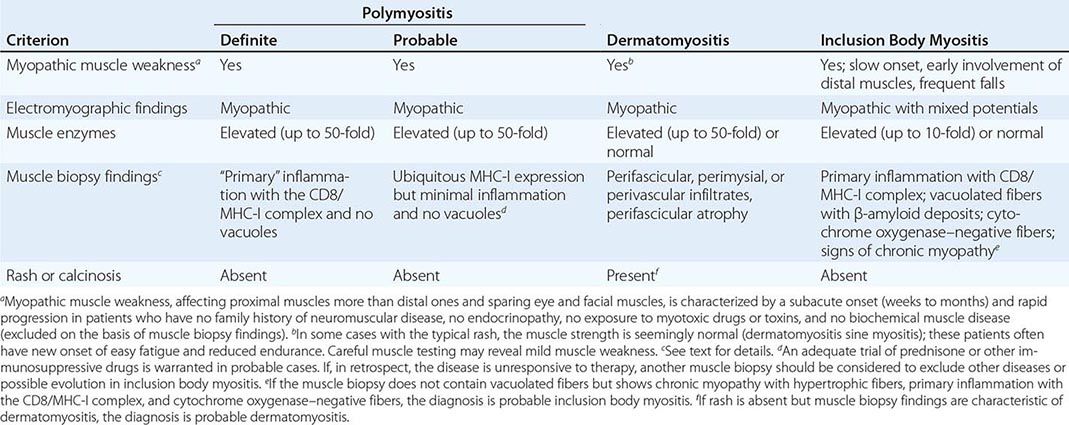
The most sensitive enzyme is CK, which in active disease can be elevated as much as 50-fold. Although the CK level usually parallels disease activity, it can be normal in some patients with active IBM or DM, especially when associated with a connective tissue disease. The CK is always elevated in patients with active PM. Along with the CK, the serum glutamic-oxaloacetic and glutamate pyruvate transaminases, lactate dehydrogenase, and aldolase may be elevated.
Needle EMG shows myopathic potentials characterized by short-duration, low-amplitude polyphasic units on voluntary activation and increased spontaneous activity with fibrillations, complex repetitive discharges, and positive sharp waves. Mixed potentials (polyphasic units of short and long duration) indicating a chronic process and muscle fiber regeneration are often present in IBM. These EMG findings are not diagnostic of an inflammatory myopathy but are useful to identify the presence of active or chronic myopathy and to exclude neurogenic disorders.
Magnetic resonance imaging (MRI) is not routinely used for the diagnosis of PM, DM, or IBM. However, it may provide information or guide the location of the muscle biopsy in certain clinical settings.
Muscle biopsy—despite occasional variability in demonstrating all of the typical pathologic findings—is the most sensitive and specific test for establishing the diagnosis of inflammatory myopathy and for excluding other neuromuscular diseases. Inflammation is the histologic hallmark for these diseases; however, additional features are characteristic of each subtype (Figs. 388-3, 388-4, and 388-5).
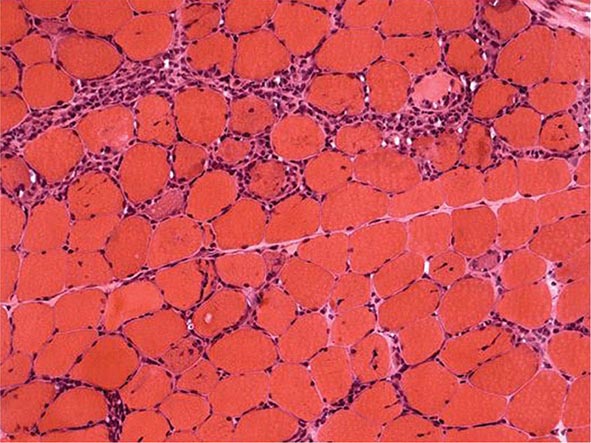
FIGURE 388-3 Cross-section of a muscle biopsy from a patient with polymyositis demonstrates scattered inflammatory foci with lymphocytes invading or surrounding muscle fibers. Note lack of chronic myopathic features (increased connective tissue, atrophic or hypertrophic fibers) as seen in inclusion body myositis.
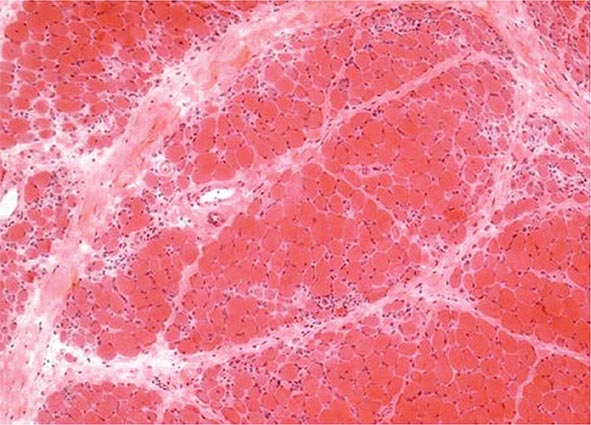
FIGURE 388-4 Cross-section of a muscle biopsy from a patient with dermatomyositis demonstrates atrophy of the fibers at the periphery of the fascicle (perifascicular atrophy).
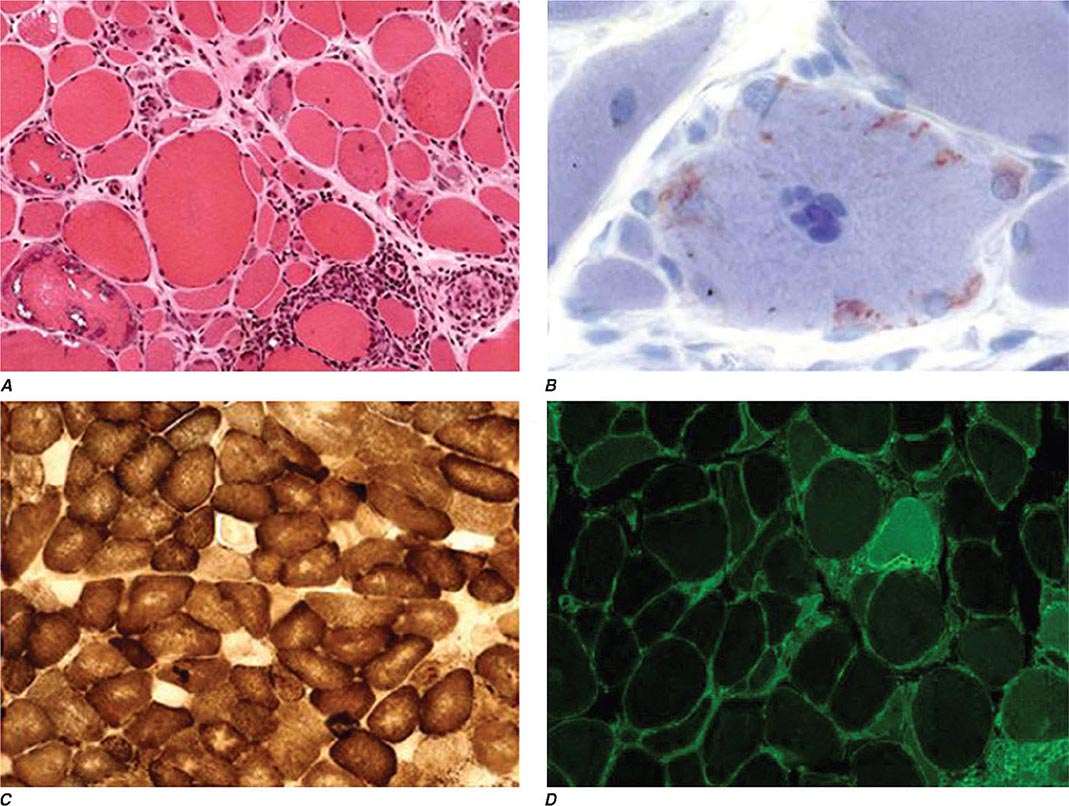
FIGURE 388-5 Cross-sections of a muscle biopsy from a patient with inclusion body myositis demonstrate the typical features of vacuoles with lymphocytic infiltrates surrounding nonvacuolated or necrotic fibers (A), tiny endomysial deposits of amyloid visualized with crystal violet (B), cytochrome oxidase–negative fibers, indicative of mitochondrial dysfunction (C), and ubiquitous major histocompatibility complex class I expression at the periphery of all fibers (D).
In PM the inflammation is primary, a term used to indicate that the inflammation is not reactive and the T cell infiltrates, located primarily within the muscle fascicles (endomysially), surround individual, healthy muscle fibers and result in phagocytosis and necrosis (Fig. 388-3). The MHC-I molecule is ubiquitously expressed on the sarcolemma, even in fibers not invaded by CD8+ cells. The CD8/MHC-I lesion is characteristic and useful to confirm or establish the diagnosis and to exclude disorders with secondary, nonspecific, inflammation, such as in some muscular dystrophies. When the disease is chronic, connective tissue is increased and may react positively with alkaline phosphatase. In necrotizing myositis, there are abundant necrotic fibers invaded or surrounded by macrophages, but no lymphocytic infiltrates or MHC-I expression beyond the necrotic fibers.
In DM the endomysial inflammation is predominantly perivascular or in the interfascicular septae and around—rather than within—the muscle fascicles (Fig. 388-4). The intramuscular blood vessels show endothelial hyperplasia with tubuloreticular profiles, fibrin thrombi, and obliteration of capillaries. The muscle fibers undergo necrosis, degeneration, and phagocytosis, often in groups involving a portion of a muscle fasciculus in a wedge-like shape or at the periphery of the fascicle, due to microinfarcts within the muscle. This results in perifascicular atrophy, characterized by 2–10 layers of atrophic fibers at the periphery of the fascicles. The presence of perifascicular atrophy is diagnostic of DM, even in the absence of inflammation.
In IBM (Fig. 388-5), there is endomysial inflammation with T cells invading MHC-I-expressing nonvacuolated muscle fibers; basophilic granular deposits distributed around the edge of slit-like vacuoles (rimmed vacuoles); loss of fibers, replaced by fat and connective tissue, hypertrophic fibers, and angulated or round fibers; rare eosinophilic cytoplasmic inclusions; abnormal mitochondria characterized by the presence of ragged-red fibers or cytochrome oxidase–negative fibers; and amyloid deposits within or next to the vacuoles best visualized with crystal violet or Congo-red staining viewed with fluorescent optics. Electron microscopy demonstrates filamentous inclusions in the vicinity of the rimmed vacuoles. In at least 15% of patients with the typical clinical phenotype of IBM, there is brisk inflammation in the muscle biopsy but no vacuoles or amyloid deposits, leading to an erroneous diagnosis of PM. Such patients are often referred to as having “clinical IBM.” Close clinicopathologic correlations are therefore essential; if uncertain, a repeat muscle biopsy from another site is often helpful.
PROGNOSIS
The 5-year survival rate for treated patients with PM and DM is ~95%, and the 10-year survival rate is 84%; death is usually due to pulmonary, cardiac, or other systemic complications. The prognosis is worse for patients who are severely affected at presentation, when initial treatment is delayed, and in cases with severe dysphagia or respiratory difficulties. Older patients and those with associated cancer also have a worse prognosis. DM responds more favorably to therapy than PM and thus has a better prognosis. Most patients improve with therapy, and many make a full functional recovery, which is often sustained with maintenance therapy. Up to 30% may be left with some residual muscle weakness. Relapses may occur at any time.
IBM has the least favorable prognosis of the inflammatory myopathies. Most patients will require the use of an assistive device such as a cane, walker, or wheelchair within 5–10 years of onset. In general, the older the age of onset in IBM, the more rapidly progressive is the course.




