[level-membership-for-neurology-category]
Chapter 33D Neuroimaging
Chemical Imaging: Ligands and Pathology Seeking Agents
Neurochemical Targets of Interest
General studies of cerebral glucose metabolism or regional CBF can be found in Chapter 33C. Neurochemical systems of interest that have been well studied using PET—monoamines (particularly dopamine and serotonin), cholinergic systems, opioid and non-opioid peptides, and amino acids—will be addressed in this chapter (Table 33D.1).
Table 33D.1 Neurochemical Tracers and Pathology-Seeking Agents
| MONOAMINES | |
| Dopamine | |
| Vesicular monoamine transporter type 2 | [11C]dihydrotetrabenazine |
| Dopamine transporter | [11C]d-threo-methylphenidate |
| [11C]- and [18F]-fluoropropyl-CIT | |
| [123I]β-CIT | |
| [99mTc]TRODAT | |
| Numerous other tropanes (cocaine analogs) | |
| Dopa decarboxylase | 6-[18F]fluoro-l-dopa |
| D1 receptors | [11C]SCH 23390 |
| D2 receptors | [11C]raclopride (also dopamine release) |
| [11]N-methylspiperone | |
| [18F]benperidol | |
| [11C]FLB 457 (extrastriatal sites) | |
| [11C] or [18F]fallypride (extrastriatal) | |
| Serotonin | |
| Tryptophan hydroxylase/kynurenin | α-[11C]-l-methyltryptophan |
| 5HT transporter | [11C]DASB |
| 5HT1A receptors | [11C]WAY 100635 |
| [18F]MPPF | |
| [18F]FCWAY | |
| 5HT2 receptors | [11C]MDL 100907 |
| [18F]setoperone | |
| [18F]altanserin | |
| CHOLINERGIC | |
| Acetylcholinesterase | [11C]MP4A |
| [11C]PMP | |
| Cholinergic vesicular transporter | [123I]iodobenzovesamicol |
| Muscarinic receptors | [11C]N-methyl-piperidyl benzylate |
| [123I]quinuclidinyl benzylate | |
| Nicotinic receptors | [11C]N-methyl-iodo-epibatidine |
| OPIOID RECEPTORS | |
| µ-Opioid receptor | [11C]carfentanil |
| Non-selective opioid receptor | [11C]diprenorphine |
| AMINO ACID RECEPTORS | |
| GABAA/benzodiazepine receptor | [11C]flumazenil |
| Excitatory amino acid receptors: | |
| NMDA receptors | [18F]fluoroethyl-diarylguanidine |
| [11C]GMOM | |
| mGluR5 receptors | [11C]MPEP |
| [11C]ABP688 | |
| [18F]FE-DABP688 | |
| [18F]SP203 | |
| [18F]FP-ECMO | |
| [18F]PEB | |
| NEUROINFLAMMATION | |
| Peripheral benzodiazepine receptor (microglia) | [11C]PK 11195 |
| BLOOD-BRAIN BARRIER FUNCTION | |
| P-glycoprotein | [11C]verapamil |
| Amyloid and other protein deposition | [11C]Pittsburgh compound B |
| [18F]AV-45 | |
| [18F]BAY94-9172 | |
| [18F]FDDNP |
Monoamines
Dopaminergic function (Fig. 33D.1) can be assessed using 6-[18F]fluoro-l-dopa (FD), an analog of levodopa that is decarboxylated to [18F]fluorodopamine and trapped in synaptic vesicles, or by its false neurotransmitter analog 6-[18F]fluoro-meta-tyrosine (FMT). The membrane dopamine transporter (DAT) can be assessed using either PET or SPECT using a variety of tropane (cocaine-like) analogs labeled with C-11, F-18, iodine-123 (123I), or technetium-99m (99mTc), or with the non-tropane [11C]d-threo-methylphenidate. FD uptake/decarboxylation and expression are subject to changes that may arise as a compensatory mechanism or in response to pharmacological manipulations. In contrast, [11C]dihydrotetrabenazine (DTBZ), which labels the vesicular monoamine transporter type 2 (VMAT2) responsible for packaging monoamines into synaptic vesicles, is theoretically less subject to such influences. VMAT2 is, however, expressed by all monoaminergic neurons and is therefore not specific for dopamine (although dopaminergic nerve terminals represent the majority of VMAT2 binding in the striatum).
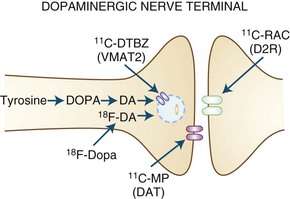
Dopamine receptors can be studied using a variety of C-11- or F-18-labeled ligands for the D2 receptor (some 123I-labeled ligands are available for SPECT as well), with fewer options available for the D1 receptor. Some D2 receptor ligands are susceptible to competition from endogenous dopamine or by pharmacological agents that bind to dopamine receptors. On the one hand, this can lead to problems of interpretation because differences in binding could potentially reflect alterations in receptor occupancy by endogenous neurotransmitter rather than changes in receptor expression. However, this property may also be extremely useful for estimating changes in dopamine release in response to a variety of behavioral (Monchi et al., 2006), pharmacological (Piccini et al., 2003; Tedroff et al., 1996), or physical (Strafella et al., 2003) interventions.
Serotonin (5-hydroxytryptamine [5HT]) nerve terminal function can be studied by the radiolabeled precursor α-[11C]methyl-l-tryptophan (analogous to FD uptake as a measure of dopaminergic integrity) or by agents that bind to the membrane 5HT transporter, of which the most widely accepted example is [11C]DASB. The 5HT2 receptor can be labeled with [11C]MDL 100,907 or [18F]setoperone, but these tracers have suboptimal kinetics (MDL) or selectivity (setoperone). Another option is [18F]altanserin, whose binding characteristics are very similar to those of [3H]MDL 100,907 in vitro (Kristiansen et al., 2005). Binding is relatively insensitive to endogenous 5HT, and interpretation could theoretically be affected by the presence of radiolabeled metabolites that cross the blood-brain barrier (BBB) (Price et al., 2001), but standard graphical analysis appears to be adequate, and changes are seen in Alzheimer disease (AD; decreased) (Marner et al., 2010) and Tourette syndrome (TS) (increased) (Haugbol et al., 2007). Binding of [18F]altanserin correlates with response to tonic heat pain (Kupers et al., 2009), and 5HT1A receptors can be labeled with [11C]WAY 100,635. In the raphe, the latter agent binds to presynaptic somatodendritic autoreceptors, and its binding accordingly gives an indirect measure of serotonergic integrity, whereas binding in other regions is predominantly postsynaptic. Unlike the situation with dopamine receptor binding, changes in the binding of serotonergic ligands cannot routinely be used to assess alterations in the availability of endogenous 5HT.
Cholinergic Systems
A number of ligands have been developed for both muscarinic (Asahina et al., 1998; Eckelman, 2006) and nicotinic (Ding et al., 2006; Horti et al., 2010) cholinergic receptors.
Neuropeptides
Opioid peptide receptors have been most widely studied using PET with [11C]diprenorphine (nonselective) or [11C]carfentanil (selective for µ-opioid receptors). Both agents are thought to be susceptible to competition from endogenous opioids. In the case of [11C]carfentanil, this property has been used to demonstrate opioid release in response to pain (Zubieta et al., 2001) and to placebo analgesia (Zubieta et al., 2005). In vivo imaging of opioid receptors has been extensively reviewed in a recent paper (Henriksen and Willoch, 2008).
Amino Acids
There has been relatively limited use of agents to study excitatory amino acid receptors, but recently agents for the mGluR5 (Honer et al., 2007; Kimura et al., 2010; Lucatelli et al., 2009; Mu et al., 2010; Patel et al., 2007; Sanchez-Pernaute et al., 2008; Treyer et al., 2007; Yu, 2007), as well as the N-methyl-d-aspartate (NMDA) (Robins et al., 2010; Waterhouse, 2003) and glycine-binding site of the NMDA receptor (Fuchigami et al., 2009) have been developed.
Assessment of Pathology
Inflammation
The peripheral benzodiazepine receptor (PBR) ligand, [11C]PK 11195, has been used as a marker of microglial activation; [11C]PK 11195 binding is increased in disorders with known inflammatory response such as multiple sclerosis (MS) (Banati et al., 2000), encephalitis (Banati et al., 1999; Cagnin et al., 2001b), and stroke (Gerhard et al., 2000), but changes can also be seen in neurodegenerative disorders, lending support to an inflammatory contribution to these conditions. More recently, other agents with higher affinity for the PBR have been developed. These may presumably have a higher sensitivity for detecting inflammatory changes.
Blood-Brain Barrier Function
The P-glycoprotein (P-gp) is responsible for extrusion of potential toxins across the BBB; [11C]verapamil is a putative substrate for P-gp. Thus, increased [11C]verapamil binding may be seen as evidence of impaired P-gp function. A number of studies have been conducted in Parkinson disease (PD) and related disorders, with somewhat unclear findings (see Clinical Studies).
Clinical Studies
Parkinson Disease
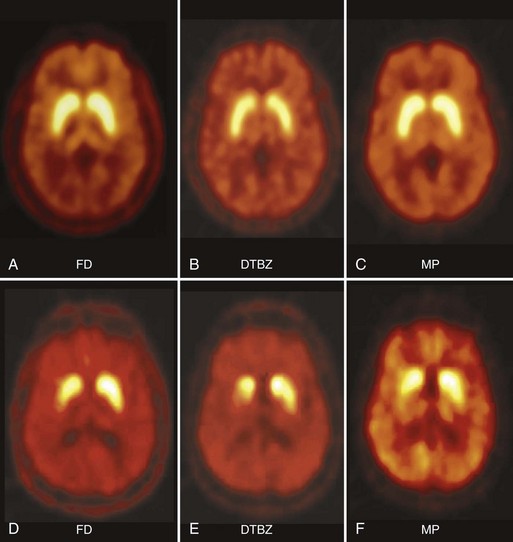
In PD, FD uptake, DAT binding, and VMAT2 binding are all reduced in a similar pattern, with a rostral-caudal gradient in which the posterior striatum is maximally affected and the caudate nucleus is relatively spared (Fig. 33D.2). The degree of abnormality is typically asymmetrical, in keeping with clinical findings, but even patients with clinically unilateral disease have evidence of bilateral striatal dopamine denervation on PET or SPECT (Marek et al., 1996). With disease progression, uptake of all tracers declines according to an exponential function. The rostral-caudal gradient of involvement is maintained throughout the course of the illness, but the asymmetry between sides lessens over time (Nandhagopal et al., 2009). Because the symptoms of PD do not become manifest until loss of approximately 50% of nigral neurons or 80% of striatal dopamine, imaging may be used to detect preclinical abnormalities in individuals at high risk of developing parkinsonism, including persons exposed to the selective nigral toxin, N-methyl-4-phenyl-1,2,3,6-tetrahydropridine (MPTP) (Calne et al., 1985); twins of persons with PD (Piccini et al., 1999); family members from pedigrees with dominantly inherited PD (Adams et al., 2005; Nandhagopal et al., 2008); and individuals with REM sleep behavior disorder (Albin et al. 2000). Interestingly, family members from kindreds with recessively inherited PD who carry heterozygous mutations also demonstrate imaging evidence of dopamine denervation (Hilker et al., 2001; Khan et al., 2002; Khan et al., 2005). The significance of the latter observation is unclear. While imaging can be used to assess disease progression, there have been numerous examples of discordance between PET or SPECT findings and clinical observations, particularly in studies designed to assess the effects of potential disease-modifying or cell-based therapies (Fahn et al., 2004; Marek et al., 2002; Olanow et al., 2003; Whone et al., 2003). This has led to caution with respect to the use of imaging as a surrogate marker in such studies (Brooks et al., 2003; Ravina et al., 2005).
By using displacement of [11C]raclopride as a measure of dopamine release, it can be shown that PD patients who go on to develop fluctuations in response to levodopa therapy have a relatively large but poorly sustained increase in synaptic dopamine following levodopa, compared to those who have a stable response to medication, in whom dopamine release is lower in magnitude but more sustained. These differences are evident even at a time when both groups still have a stable response to medication (de la Fuente-Fernandez et al., 2001a). Levodopa-induced dopamine release increases with disease progression and is also increased in patients with medication-induced dyskinesias (de la Fuente-Fernandez et al., 2004). A similar approach has been used to demonstrate increased levodopa-derived dopamine release in the ventral (but not dorsal) striatum of patients with the dopamine dysequilibrium syndrome (Evans et al., 2006). In this situation, dopamine release correlates with how much subjects want the drug as opposed to how much they enjoy the effects. Dopamine release is also increased during performance of a gambling task with monetary reward, and this effect is enhanced in PD patients with pathological gambling (Steeves et al., 2009).
The same technique has been used to demonstrate dopamine release underlying the placebo effect in PD (de la Fuente-Fernandez et al., 2001b). This finding, initially demonstrated using placebo medication, has been confirmed with sham repetitive transcranial magnetic stimulation, which also induces ventral striatal dopamine release (Strafella et al., 2006).
PET has been used to investigate depression in PD, and the results are somewhat surprising. Using the selective 5HT transporter ligand, [11C]DASB, Guttman and colleagues demonstrated widespread reductions in 5HT transporter binding in PD compared to healthy controls, in keeping with loss of serotonergic fibers (Guttman et al., 2007). In PD patients with depression, however, 5HT transporter binding was increased, particularly in dorsolateral and prefrontal cortex (Boileau et al., 2008); 5HT transporter binding correlated with clinical ratings of depression. Although somewhat surprising, this finding is reminiscent of those in major depression, where 5HT transporter binding is increased in those subjects with negativistic dysfunctional attitude (Meyer et al., 2004).
Dementia in PD (PDD) is associated with marked reductions in cholinergic activity (Bohnen et al., 2003; Hilker et al., 2005) greater than those seen in AD (see later discussion). The pathology of PDD is mixed but will often include cortical Lewy body deposition (with or without evidence of AD pathology), so there has been considerable interest in whether agents that bind to aberrantly folded protein can be used to image dementia with Lewy bodies (DLB) or PDD. Most studies to date have suggested that [11C]PiB binding is increased in DLB but not in PDD. This may be seen as somewhat surprising, as many investigators consider these to represent variations of the same disorder. It is possible that patients with PDD who demonstrate [11C]PiB uptake in fact have concurrent AD pathology, as suggested by a relationship to ApoE4 allele and CSF Aβ42 levels (Maetzler et al., 2009), as well as recent postmortem (Burack et al., 2010) and in vitro (Fodero-Tavoletti et al., 2007) studies.
As is the case for other neurodegenerative disorders, there has been great interest in the possibility of an inflammatory component to the pathogenesis and progression of PD. Using the peripheral benzodiazepine ligand, [11C]PK 11195, as a marker of microglial activation, Ouchi et al. demonstrated increased binding in the substantia nigra of PD patients that correlated with dopaminergic nerve loss and with clinical measures of disease severity (Ouchi et al., 2005). In contrast, Gerhard et al. found more widespread increases in [11C]PK 11195 binding that did not correlate with either FD uptake or clinical measures of disease progression (Gerhard et al., 2006). Quantitation with this tracer is difficult, and results vary according to the analytical model employed; furthermore, binding is apparently not reduced in response to treatment with celecoxib (Bartels et al., 2010).
Another tantalizing possibility for pathogenic mechanisms in PD was raised by the observation that [11C]verapamil binding was increased in the midbrain of patients with PD (Kortekaas et al., 2005). In the initial report, this was interpreted by the authors as reflecting impairment of BBB P-gp function, resulting in reduced extrusion of toxins. Similar abnormalities have been reported in the basal ganglia of patients with multiple system atrophy (MSA) and progressive supranuclear palsy (PSP) (Bartels et al., 2008b). However, the pathogenic significance of this finding in PD is questionable, as it is not seen in patients with early PD (Bartels et al., 2008a).
Alzheimer Disease
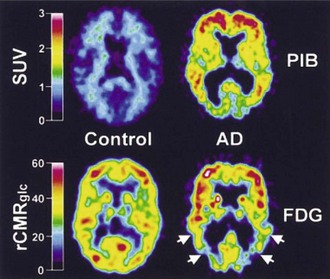
Both [11C]PiB and [18F]FDDNP bind to β-amyloid, and their uptake is increased in cortical and subcortical regions in AD (Klunk et al., 2004; Shoghi-Jadid et al., 2002) (Fig. 33D.3). Since [18F]FDDNP binds to neurofibrillary tangles as well as amyloid plaques, one might anticipate some differences between the uptake of the two ligands. This does appear to be the case, with voxel-based analysis revealing preferential uptake of [18F]FDDNP in the hippocampus (Shin et al., 2010). Similarly, global [11C]PiB uptake is inversely associated with CSF Aβ1-42 levels, while [18F]FDDNP binding correlates with CSF τ (Tolboom et al., 2009a). Increased [18F]FDDNP binding seems to correlate better with impairment of episodic memory, while [11C]PiB binding may be associated with more widespread cognitive deficits (Tolboom et al., 2009b). Binding of [11C]PiB does not necessarily correlate with the Mini-Mental State Examination, even where glucose metabolism does (Jagust et al., 2009). Both agents reveal increased uptake in subjects with minimal cognitive impairment (MCI) (Small et al., 2006), although there is some variability reported in the ability of [18F]FDDNP to differentiate between control, MCI, and AD (Tolboom et al., 2009c). In healthy aging, [11C]PiB binding is increased, particularly in carriers of the ApoE-ε4 allele (Rowe et al., 2010). Perhaps not surprisingly, binding is also increased in healthy adults with a family history of late-onset AD (Mosconi et al., 2010). Increased binding in subjects with minimal cognitive impairment (Okello et al., 2009) or indeed in apparently healthy controls (Morris et al., 2009) is associated with a significant risk of progression to dementia. While [11C]PiB binding appears to be sensitive to the effects of therapeutic interventions such as monoclonal anti-amyloid antibodies, placebo-treated patients showed ongoing accumulation (Rinne et al., 2010).
Clearly these agents may be helpful in differentiating AD from other disorders resulting in dementia. Thus [11C]PiB binding may distinguish between AD and frontotemporal lobar dementia (FTLD). A significant proportion of patients with clinical evidence of FTLD may display increased [11C]PiB binding, and it is as yet unclear whether this represents false positivity, misdiagnosis, or concurrent AD (Rabinovici et al., 2007). Binding of [18F]FDDNP is increased in the cerebellum, neocortex, and subcortical structures of patients with the prion amyloid disorder, Gerstmann-Sträussler-Scheinker disease and in caudate and thalamus of some asymptomatic mutation carriers (Kepe et al., 2010).
One obvious disadvantage of [11C]-labeled agents is their requirement for proximity to a cyclotron. When an [18F]-labeled agent is used, the longer half-life permits a somewhat greater degree of flexibility. Other β-amyloid–selective agents with favorable kinetic properties are [18F]AV-45 (flobetapir) (Wong et al., 2010) and [18F]BAY94-9172 (Rowe et al., 2008); like [11C]PiB, these agents demonstrate widespread increased binding in patients with AD compared to healthy controls.
Binding of [11C]PK 11195 suggests increased microglial activation in entorhinal, temporoparietal, and cingulate cortex of patients with AD (Cagnin et al., 2001a).
Epilepsy
There is abundant evidence for altered opioid transmission in experimental models of seizures. Combined PET studies using both the δ-opioid antagonist, [11C]N-methylnaltrindole, and the µ-opioid agonist, [11C]carfentanil, in patients with temporal lobe epilepsy (TLE) revealed increases in the binding of both ligands (associated with reduced glucose metabolism), although with somewhat different distributions (Madar et al., 1997). In contrast, binding of the nonselective opioid ligand, [11C]diprenorphine, was reduced during reading-induced seizures, compared to the baseline state, suggestive of opioid release in patients with reading epilepsy (Koepp et al., 1998), although [11C]diprenorphine binding increased in the fusiform gyrus and temporal pole following seizures in patients with TLE (Hammers et al., 2007).
The GABAA/benzodiazepine receptor has been studied using [11C]flumazenil. In patients with mesial temporal epilepsy and hippocampal sclerosis, binding is reduced in the affected hippocampus (with or without concurrent reductions in binding in the amygdala), but variable increases or decreases may be seen in neocortical regions (Hammers et al., 2001). Focal abnormalities (increases, decreases, or both concurrently) may be seen in many patients with cortical epilepsy. In some patients, these abnormalities are seen in periventricular regions, possibly representing neuronal migration abnormalities (Hammers et al., 2003).
Multiple lines of evidence suggest dysfunction in serotonergic mechanisms in patients with epilepsy. Uptake of the 5HT precursor, α-[11C]-methyl-l-tryptophan (AMT), is increased in the hippocampus of patients with TLE and preserved hippocampal volume (Natsume et al., 2003), and focal cortical increases of this tracer in children with intractable epilepsy are often associated with epileptogenic cortical developmental abnormalities (Juhasz et al., 2003). Increases in AMT uptake are smaller in extent and have a lower sensitivity but greater specificity compared with areas of altered glucose metabolism. Decreases in 5HT1A binding have been demonstrated in epileptogenic regions of TLE patients using the antagonist ligands, [18F]MPPF (Merlet et al., 2004) and [18F]FCWAY (Liew et al., 2009).
More recent studies also suggest abnormalities in dopamine transmission. Uptake of 6-[18F]-fluoro-l-dopa was reduced in patients with multiple types of epilepsy (Bouilleret et al., 2005). Striatal and thalamic binding of [11C]raclopride is increased in patients with Unverricht-Lundborg myoclonic epilepsy (Korja et al., 2007), also in keeping with a striatal dopaminergic deficit. This is further supported by reduced dopamine transporter binding, which is restricted to the midbrain in juvenile myoclonic epilepsy but found in the putamen in patients with generalized tonic-clonic epilepsy (Ciumas et al., 2010). In patients with mesial TLE, dopamine D2/D3 receptor binding (as measured by [18F]fallypride) is reduced at the pole and in lateral aspects of the epileptogenic temporal lobe, possibly corresponding to the “irritative zone” (Werhahn et al., 2006).
Adams J.R., van Netten H., Schulzer M., et al. PET in LRRK2 mutations: comparison to sporadic Parkinson’s disease and evidence for presymptomatic compensation. Brain. 2005;128:2777-2785.
Albin R.L., Koeppe R.A., Chervin R.D., et al. Decreased striatal dopaminergic innervation in REM sleep behavior disorder. Neurology. 2000;55:1410-1412.
Asahina M., Suhara T., Shinotoh H., et al. Brain muscarinic receptors in progressive supranuclear palsy and Parkinson’s disease: a positron emission tomographic study. J Neurol Neurosurg Psychiatry. 1998;65:155-163.
Banati R.B., Goerres G.W., Myers R., et al. [11C](R)-PK11195 positron emission tomography imaging of activated microglia in vivo in Rasmussen’s encephalitis. Neurology. 1999;53:2199-2203.
Banati R.B., Newcombe J., Gunn R.N., et al. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain. 2000;123(Pt 11):2321-2337.
Bartels A.L., van Berckel B.N., Lubberink M., et al. Blood-brain barrier P-glycoprotein function is not impaired in early Parkinson’s disease. Parkinsonism Relat Disord. 2008.
Bartels A.L., Willemsen A.T., Doorduin J., et al. [11C]-PK11195 PET: quantification of neuroinflammation and a monitor of anti-inflammatory treatment in Parkinson’s disease? Parkinsonism Relat Disord. 2010;16:57-59.
Bartels A.L., Willemsen A.T., Kortekaas R., et al. Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J. Neural Transm. 2008;115:1001-1009.
Bohnen N.I., Kaufer D.I., Ivanco L.S., et al. Cortical cholinergic function is more severely affected in parkinsonian dementia than in Alzheimer disease: an in vivo positron emission tomographic study. Arch Neurol. 2003;60:1745-1748.
Boileau I., Warsh J.J., Guttman M., et al. Elevated serotonin transporter binding in depressed patients with Parkinson’s disease: a preliminary PET study with [11C]DASB. Mov Disord. 2008;23:1776-1780.
Bouilleret V., Semah F., Biraben A., et al. Involvement of the basal ganglia in refractory epilepsy: an 18F-fluoro-l-dopa PET study using 2 methods of analysis. J Nucl Med. 2005;46:540-547.
Brooks D.J., Frey K.A., Marek K.L., et al. Assessment of neuroimaging techniques as biomarkers of the progression of Parkinson’s disease. Exp Neurol. 2003;184(Suppl 1)):S68-S79.
Burack M.A., Hartlein J., Flores H.P., et al. In vivo amyloid imaging in autopsy-confirmed Parkinson disease with dementia. Neurology. 2010;74:77-84.
Cagnin A., Brooks D.J., Kennedy A.M., et al. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358:461-467.
Cagnin A., Myers R., Gunn R.N., et al. In vivo visualization of activated glia by [11C] (R)-PK11195-PET following herpes encephalitis reveals projected neuronal damage beyond the primary focal lesion. Brain. 2001;124:2014-2027.
Calne D.B., Langston J.W., Martin W.R., et al. Positron emission tomography after MPTP: observations relating to the cause of Parkinson’s disease. Nature. 1985;317:246-248.
Ciumas C., Wahlin T.B., Espino C., et al. The dopamine system in idiopathic generalized epilepsies: identification of syndrome-related changes. Neuroimage. 2010;51:606-615.
de la Fuente-Fernandez R., Lu J.Q., Sossi V., et al. Biochemical variations in the synaptic level of dopamine precede motor fluctuations in Parkinson’s disease: PET evidence of increased dopamine turnover. Ann Neurol. 2001;49:298-303.
de la Fuente-Fernandez R., Ruth T.J., Sossi V., et al. Expectation and dopamine release: mechanism of the placebo effect in Parkinson’s disease. Science. 2001;293:1164-1166.
de la Fuente-Fernandez R., Sossi V., Huang Z., et al. Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: implications for dyskinesias. Brain. 2004;127:2747-2754.
Ding Y.S., Kil K.E., Lin K.S., et al. A novel nicotinic acetylcholine receptor antagonist radioligand for PET studies. Bioorg Med Chem Lett. 2006;16:1049-1053.
Eckelman W.C. Imaging of muscarinic receptors in the central nervous system. Curr Pharm Des. 2006;12:3901-3913.
Evans A.H., Pavese N., Lawrence A.D., et al. Compulsive drug use linked to sensitized ventral striatal dopamine transmission. Ann Neurol. 2006;59:852-858.
Fahn S., Oakes D., Shoulson I., et al. Levodopa and the progression of Parkinson’s disease. N Engl J Med. 2004;351:2498-2508.
Fodero-Tavoletti M.T., Smith D.P., McLean C.A., et al. In vitro characterization of Pittsburgh Compound-B binding to Lewy bodies. J Neurosci. 2007;27:10365-10371.
Fuchigami T., Haradahira T., Fujimoto N., et al. Development of N-[11C]methylamino 4-hydroxy-2(1H)-quinolone derivatives as PET radioligands for the glycine-binding site of NMDA receptors. Bioorg Med Chem. 2009;17:5665-5675.
Gerhard A., Neumaier B., Elitok E., et al. In vivo imaging of activated microglia using [11C]PK11195 and positron emission tomography in patients after ischemic stroke. Neuroreport. 2000;11:2957-2960.
Gerhard A., Pavese N., Hotton G., et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis. 2006;21:404-412.
Guttman M., Boileau I., Warsh J., et al. Brain serotonin transporter binding in non-depressed patients with Parkinson’s disease. Eur J Neurol. 2007;14:523-528.
Hammers A., Asselin M.C., Hinz R., et al. Upregulation of opioid receptor binding following spontaneous epileptic seizures. Brain. 2007;130:1009-1016.
Hammers A., Koepp M.J., Labbe C., et al. Neocortical abnormalities of [11C]-flumazenil PET in mesial temporal lobe epilepsy. Neurology. 2001;56:897-906.
Hammers A., Koepp M.J., Richardson M.P., et al. Grey and white matter flumazenil binding in neocortical epilepsy with normal MRI. A PET study of 44 patients. Brain. 2003;126:1300-1318.
Haugbol S., Pinborg L.H., Regeur L., et al. Cerebral 5-HT2A receptor binding is increased in patients with Tourette’s syndrome. Int J Neuropsychopharmacol. 2007;10:245-252.
Henriksen G., Willoch F. Imaging of opioid receptors in the central nervous system. Brain. 2008;131:1171-1196.
Hilker R., Klein C., Ghaemi M., et al. Positron emission tomographic analysis of the nigrostriatal dopaminergic system in familial parkinsonism associated with mutations in the parkin gene. Ann Neurol. 2001;49:367-376.
Hilker R., Thomas A.V., Klein J.C., et al. Dementia in Parkinson disease: functional imaging of cholinergic and dopaminergic pathways. Neurology. 2005;65:1716-1722.
Honer M., Stoffel A., Kessler L.J., et al. Radiolabeling and in vitro and in vivo evaluation of [18F]-FE-DABP688 as a PET radioligand for the metabotropic glutamate receptor subtype 5. Nucl. Med. Biol. 2007;34:973-980.
Horti A.G., Gao Y., Kuwabara H., Dannals R.F. Development of radioligands with optimized imaging properties for quantification of nicotinic acetylcholine receptors by positron emission tomography. Life Sci. 2010;86:575-584.
Jagust W.J., Landau S.M., Shaw L.M., et al. Relationships between biomarkers in aging and dementia. Neurology. 2009;73:1193-1199.
Juhasz C., Chugani D.C., Muzik O., et al. Alpha-methyl-l-tryptophan PET detects epileptogenic cortex in children with intractable epilepsy. Neurology. 2003;60:960-968.
Kepe V, Ghetti B, Farlow M.R., et al. PET of brain prion protein amyloid in Gerstmann-Straussler-Scheinker disease. Brain Pathol. 2010;20:419-430.
Khan N.L., Scherfler C., Graham E., et al. Dopaminergic dysfunction in unrelated, asymptomatic carriers of a single parkin mutation. Neurology. 2005;64:134-136.
Khan N.L., Valente E.M., Bentivoglio A.R., et al. Clinical and subclinical dopaminergic dysfunction in PARK6-linked parkinsonism: an 18F-dopa PET study. Ann. Neurol. 2002;52:849-853.
Kimura Y., Simeon F.G., Hatazawa J., et al. Biodistribution and radiation dosimetry of a positron emission tomographic ligand, (18)F-SP203, to image metabotropic glutamate subtype 5 receptors in humans. Eur J Nucl Med Mol. Imaging. 2010;37:1943-1949.
Klunk W.E., Engler H., Nordberg A., et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306-319.
Koepp M.J., Richardson M.P., Brooks D.J., et al. Focal cortical release of endogenous opioids during reading-induced seizures. Lancet. 1998;352:952-955.
Korja M., Kaasinen V., Lamusuo S., et al. Substantial thalamostriatal dopaminergic defect in Unverricht-Lundborg disease. Epilepsia. 2007;48:1768-1773.
Kortekaas R., Leenders K.L., van Oostrom J.C., et al. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol. 2005;57:176-179.
Kristiansen H., Elfving B., Plenge P., et al. Binding characteristics of the 5-HT2A receptor antagonists altanserin and MDL 100907. Synapse. 2005;58:249-257.
Kupers R., Frokjaer V.G., Naert A., et al. A PET [18F]altanserin study of 5-HT2A receptor binding in the human brain and responses to painful heat stimulation. Neuroimage. 2009;44:1001-1007.
Liew C.J., Lim Y.M., Bonwetsch R., et al. 18F-FCWAY and 18F-FDG PET in MRI-negative temporal lobe epilepsy. Epilepsia. 2009;50:234-239.
Lucatelli C., Honer M., Salazar J.F., et al. Synthesis, radiolabeling, in vitro and in vivo evaluation of [18F]-FPECMO as a positron emission tomography radioligand for imaging the metabotropic glutamate receptor subtype 5. Nucl Med Biol. 2009;36:613-622.
Madar I., Lesser R.P., Krauss G., et al. Imaging of delta- and mu-opioid receptors in temporal lobe epilepsy by positron emission tomography. Ann Neurol. 1997;41:358-367.
Maetzler W., Liepelt I., Reimold M., et al. Cortical PIB binding in Lewy body disease is associated with Alzheimer-like characteristics. Neurobiol Dis. 2009;34:107-112.
Marek K., Seibyl J., Shoulson I., et al. Dopamine transporter brain imaging to assess the effects of pramipexole vs levodopa on Parkinson disease progression. JAMA. 2002;287:1653-1661.
Marek K.L., Seibyl J.P., Zoghbi S.S., et al. [123I]beta-CIT/SPECT imaging demonstrates bilateral loss of dopamine transporters in hemi-Parkinson’s disease. Neurology. 1996;46:231-237.
Marner L., Frokjaer V.G., Kalbitzer J., et al. Loss of serotonin 2A receptors exceeds loss of serotonergic projections in early Alzheimer’s disease: a combined [(11)C]DASB and [(18)F]altanserin-PET study. Neurobiol Aging. 2010.
Merlet I., Ostrowsky K., Costes N., et al. 5-HT1A receptor binding and intracerebral activity in temporal lobe epilepsy: an [18F]MPPF-PET study. Brain. 2004;127:900-913.
Meyer J.H., Houle S., Sagrati S., et al. Brain serotonin transporter binding potential measured with carbon 11-labeled DASB positron emission tomography: effects of major depressive episodes and severity of dysfunctional attitudes. Arch Gen Psychiatry. 2004;61:1271-1279.
Monchi O., Ko J.H., Strafella A.P. Striatal dopamine release during performance of executive functions: A [(11)C] raclopride PET study. Neuroimage. 2006;33:907-912.
Morris J.C., Roe C.M., Grant E.A., et al. Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch Neurol. 2009;66:1469-1475.
Mosconi L., Rinne J.O., Tsui W.H., et al. Increased fibrillar amyloid-β burden in normal individuals with a family history of late-onset Alzheimer’s. Proc Natl Acad Sci U.S.A. 2010;107:5949-5954.
Mu L., Schubiger P.A., Ametamey S.M. Radioligands for the PET imaging of metabotropic glutamate receptor subtype 5 (mGluR5). Curr Top Med Chem. 2010.
Nandhagopal R., Kuramoto L., Schulzer M., et al. Longitudinal progression of sporadic Parkinson’s disease: a multi-tracer positron emission tomography study. Brain. 2009;132:2970-2979.
Nandhagopal R., Mak E., Schulzer M., et al. Progression of dopaminergic dysfunction in a LRRK2 kindred: a multitracer PET study. Neurology. 2008;71:1790-1795.
Natsume J., Kumakura Y., Bernasconi N., et al. Alpha-[11C] methyl-l-tryptophan and glucose metabolism in patients with temporal lobe epilepsy. Neurology. 2003;60:756-761.
Okello A., Koivunen J., Edison P., et al. Conversion of amyloid positive and negative MCI to AD over 3 years: an 11C-PIB PET study. Neurology. 2009;73:754-760.
Olanow C.W., Goetz C.G., Kordower J.H., et al. A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann Neurol. 2003;54:403-414.
Ouchi Y., Yoshikawa E., Sekine Y., et al. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann Neurol. 2005;57:168-175.
Patel S., Hamill T.G., Connolly B., et al. Species differences in mGluR5 binding sites in mammalian central nervous system determined using in vitro binding with [18F]F-PEB. Nucl Med Biol. 2007;34:1009-1017.
Piccini P., Burn D.J., Ceravolo R., et al. The role of inheritance in sporadic Parkinson’s disease: evidence from a longitudinal study of dopaminergic function in twins. Ann Neurol. 1999;45:577-582.
Piccini P., Pavese N., Brooks D.J. Endogenous dopamine release after pharmacological challenges in Parkinson’s disease. Ann Neurol. 2003;53:647-653.
Price J.C., Lopresti B.J., Meltzer C.C., et al. Analyses of [(18)F]altanserin bolus injection PET data. II: consideration of radiolabeled metabolites in humans. Synapse. 2001;41:11-21.
Rabinovici G.D., Furst A.J., O’Neil J.P., et al. 11C-PIB PET imaging in Alzheimer disease and frontotemporal lobar degeneration. Neurology. 2007;68:1205-1212.
Ravina B., Eidelberg D., Ahlskog J.E., et al. The role of radiotracer imaging in Parkinson disease. Neurology. 2005;64:208-215.
Rinne J.O., Brooks D.J., Rossor M.N., et al. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;9:363-372.
Robins E.G., Zhao Y., Khan I., et al. Synthesis and in vitro evaluation of (18)F-labelled S-fluoroalkyl diarylguanidines: novel high-affinity NMDA receptor antagonists for imaging with PET. Bioorg Med Chem Lett. 2010;20:1749-1751.
Rowe C.C., Ackerman U., Browne W., et al. Imaging of amyloid beta in Alzheimer’s disease with 18F-BAY94-9172, a novel PET tracer: proof of mechanism. Lancet Neurol. 2008;7:129-135.
Rowe C.C., Ellis K.A., Rimajova M., et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging. 2010.
Sanchez-Pernaute R., Wang J.Q., Kuruppu D., et al. Enhanced binding of metabotropic glutamate receptor type 5 (mGluR5) PET tracers in the brain of parkinsonian primates. Neuroimage. 2008;42:248-251.
Shin J., Lee S.Y., Kim S.J., et al. Voxel-based analysis of Alzheimer’s disease PET imaging using a triplet of radiotracers: PIB, FDDNP, and FDG. Neuroimage. 2010;52:488-496.
Shoghi-Jadid K., Small G.W., Agdeppa E.D., et al. Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry. 2002;10:24-35.
Small G.W., Kepe V., Ercoli L.M., et al. PET of brain amyloid and tau in mild cognitive impairment. N Engl J Med. 2006;355:2652-2663.
Steeves T.D., Miyasaki J., Zurowski M., et al. Increased striatal dopamine release in Parkinsonian patients with pathological gambling: a [11C] raclopride PET study. Brain. 2009;132:1376-1385.
Strafella A.P., Ko J.H., Monchi O. Therapeutic application of transcranial magnetic stimulation in Parkinson’s disease: the contribution of expectation. Neuroimage. 2006;31:1666-1672.
Strafella A.P., Paus T., Fraraccio M., et al. Striatal dopamine release induced by repetitive transcranial magnetic stimulation of the human motor cortex. Brain. 2003;126:2609-2615.
Tedroff J., Pedersen M., Aquilonius S-M., et al. Levodopa-induced changes in synaptic dopamine in patients with Parkinson’s disease as measured by [11C]raclopride displacement and PET. Neurology. 1996;46:1430-1436.
Tolboom N., van der Flier W.M., Yaqub M., et al. Relationship of cerebrospinal fluid markers to 11C-PiB and 18F-FDDNP binding. J Nucl Med. 2009;50:1464-1470.
Tolboom N., van der Flier W.M., Yaqub M., et al. Differential association of [11C]PIB and [18F]FDDNP binding with cognitive impairment. Neurology. 2009;73:2079-2085.
Tolboom N., Yaqub M., van der Flier W.M., et al. Detection of Alzheimer pathology in vivo using both 11C-PIB and 18F-FDDNP PET. J Nucl Med. 2009;50:191-197.
Treyer V., Streffer J., Wyss M.T., et al. Evaluation of the metabotropic glutamate receptor subtype 5 using PET and 11C-ABP688: assessment of methods. J. Nucl. Med. 2007;48:1207-1215.
Waterhouse R.N. Imaging the PCP site of the NMDA ion channel. Nucl Med Biol. 2003;30:869-878.
Werhahn K.J., Landvogt C., Klimpe S., et al. Decreased dopamine D2/D3-receptor binding in temporal lobe epilepsy: an [18F]fallypride PET study. Epilepsia. 2006;47:1392-1396.
Whone A.L., Watts R.L., Stoessl A.J., et al. Slower progression of Parkinson’s disease with ropinirole versus levodopa: The REAL-PET. study. Ann Neurol. 2003;54:93-101.
Wong D.F., Rosenberg P.B., Zhou Y., et al. In vivo imaging of amyloid deposition in Alzheimer disease using the radioligand 18F-AV-45 (flobetapir F-18). J Nucl Med. 2010;51:913-920.
Yu M. Recent developments of the PET imaging agents for metabotropic glutamate receptor subtype 5. Curr Top Med Chem. 2007;7:1800-1805.
Zubieta J.K., Bueller J.A., Jackson L.R., et al. Placebo effects mediated by endogenous opioid activity on mu-opioid receptors. J Neurosci. 2005;25:7754-7762.
Zubieta J.K., Smith Y.R., Bueller J.A., et al. Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science. 2001;293:311-315.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 33D Neuroimaging
Chemical Imaging: Ligands and Pathology Seeking Agents
Neurochemical Targets of Interest
General studies of cerebral glucose metabolism or regional CBF can be found in Chapter 33C. Neurochemical systems of interest that have been well studied using PET—monoamines (particularly dopamine and serotonin), cholinergic systems, opioid and non-opioid peptides, and amino acids—will be addressed in this chapter (Table 33D.1).
Table 33D.1 Neurochemical Tracers and Pathology-Seeking Agents
| MONOAMINES | |
| Dopamine | |
| Vesicular monoamine transporter type 2 | [11C]dihydrotetrabenazine |
| Dopamine transporter | [11C]d-threo-methylphenidate |
| [11C]- and [18F]-fluoropropyl-CIT | |
| [123I]β-CIT | |
| [99mTc]TRODAT | |
| Numerous other tropanes (cocaine analogs) | |
| Dopa decarboxylase | 6-[18F]fluoro-l-dopa |
| D1 receptors | [11C]SCH 23390 |
| D2 receptors | [11C]raclopride (also dopamine release) |
| [11]N-methylspiperone | |
| [18F]benperidol | |
| [11C]FLB 457 (extrastriatal sites) | |
| [11C] or [18F]fallypride (extrastriatal) | |
| Serotonin | |
| Tryptophan hydroxylase/kynurenin | α-[11C]-l-methyltryptophan |
| 5HT transporter | [11C]DASB |
| 5HT1A receptors | [11C]WAY 100635 |
| [18F]MPPF | |
| [18F]FCWAY | |
| 5HT2 receptors | [11C]MDL 100907 |
| [18F]setoperone | |
| [18F]altanserin | |
| CHOLINERGIC | |
| Acetylcholinesterase | [11C]MP4A |
| [11C]PMP | |
| Cholinergic vesicular transporter | [123I]iodobenzovesamicol |
| Muscarinic receptors | [11C]N-methyl-piperidyl benzylate |
| [123I]quinuclidinyl benzylate | |
| Nicotinic receptors | [11C]N-methyl-iodo-epibatidine |
| OPIOID RECEPTORS | |
| µ-Opioid receptor | [11C]carfentanil |
| Non-selective opioid receptor | [11C]diprenorphine |
| AMINO ACID RECEPTORS | |
| GABAA/benzodiazepine receptor | [11C]flumazenil |
| Excitatory amino acid receptors: | |
| NMDA receptors | [18F]fluoroethyl-diarylguanidine |
| [11C]GMOM | |
| mGluR5 receptors | [11C]MPEP |
| [11C]ABP688 | |
| [18F]FE-DABP688 | |
| [18F]SP203 | |
| [18F]FP-ECMO | |
| [18F]PEB | |
| NEUROINFLAMMATION | |
| Peripheral benzodiazepine receptor (microglia) | [11C]PK 11195 |
| BLOOD-BRAIN BARRIER FUNCTION | |
| P-glycoprotein | [11C]verapamil |
| Amyloid and other protein deposition | [11C]Pittsburgh compound B |
| [18F]AV-45 | |
| [18F]BAY94-9172 | |
| [18F]FDDNP |
Monoamines
Dopaminergic function (Fig. 33D.1) can be assessed using 6-[18F]fluoro-l-dopa (FD), an analog of levodopa that is decarboxylated to [18F]fluorodopamine and trapped in synaptic vesicles, or by its false neurotransmitter analog 6-[18F]fluoro-meta-tyrosine (FMT). The membrane dopamine transporter (DAT) can be assessed using either PET or SPECT using a variety of tropane (cocaine-like) analogs labeled with C-11, F-18, iodine-123 (123I), or technetium-99m (99mTc), or with the non-tropane [11C]d-threo-methylphenidate. FD uptake/decarboxylation and expression are subject to changes that may arise as a compensatory mechanism or in response to pharmacological manipulations. In contrast, [11C]dihydrotetrabenazine (DTBZ), which labels the vesicular monoamine transporter type 2 (VMAT2) responsible for packaging monoamines into synaptic vesicles, is theoretically less subject to such influences. VMAT2 is, however, expressed by all monoaminergic neurons and is therefore not specific for dopamine (although dopaminergic nerve terminals represent the majority of VMAT2 binding in the striatum).
Dopamine receptors can be studied using a variety of C-11- or F-18-labeled ligands for the D2 receptor (some 123I-labeled ligands are available for SPECT as well), with fewer options available for the D1 receptor. Some D2 receptor ligands are susceptible to competition from endogenous dopamine or by pharmacological agents that bind to dopamine receptors. On the one hand, this can lead to problems of interpretation because differences in binding could potentially reflect alterations in receptor occupancy by endogenous neurotransmitter rather than changes in receptor expression. However, this property may also be extremely useful for estimating changes in dopamine release in response to a variety of behavioral (Monchi et al., 2006), pharmacological (Piccini et al., 2003; Tedroff et al., 1996), or physical (Strafella et al., 2003) interventions.
Serotonin (5-hydroxytryptamine [5HT]) nerve terminal function can be studied by the radiolabeled precursor α-[11C]methyl-l-tryptophan (analogous to FD uptake as a measure of dopaminergic integrity) or by agents that bind to the membrane 5HT transporter, of which the most widely accepted example is [11C]DASB. The 5HT2 receptor can be labeled with [11C]MDL 100,907 or [18F]setoperone, but these tracers have suboptimal kinetics (MDL) or selectivity (setoperone). Another option is [18F]altanserin, whose binding characteristics are very similar to those of [3H]MDL 100,907 in vitro (Kristiansen et al., 2005). Binding is relatively insensitive to endogenous 5HT, and interpretation could theoretically be affected by the presence of radiolabeled metabolites that cross the blood-brain barrier (BBB) (Price et al., 2001), but standard graphical analysis appears to be adequate, and changes are seen in Alzheimer disease (AD; decreased) (Marner et al., 2010) and Tourette syndrome (TS) (increased) (Haugbol et al., 2007). Binding of [18F]altanserin correlates with response to tonic heat pain (Kupers et al., 2009), and 5HT1A receptors can be labeled with [11C]WAY 100,635. In the raphe, the latter agent binds to presynaptic somatodendritic autoreceptors, and its binding accordingly gives an indirect measure of serotonergic integrity, whereas binding in other regions is predominantly postsynaptic. Unlike the situation with dopamine receptor binding, changes in the binding of serotonergic ligands cannot routinely be used to assess alterations in the availability of endogenous 5HT.
Cholinergic Systems
A number of ligands have been developed for both muscarinic (Asahina et al., 1998; Eckelman, 2006) and nicotinic (Ding et al., 2006; Horti et al., 2010) cholinergic receptors.
Neuropeptides
Opioid peptide receptors have been most widely studied using PET with [11C]diprenorphine (nonselective) or [11C]carfentanil (selective for µ-opioid receptors). Both agents are thought to be susceptible to competition from endogenous opioids. In the case of [11C]carfentanil, this property has been used to demonstrate opioid release in response to pain (Zubieta et al., 2001) and to placebo analgesia (Zubieta et al., 2005). In vivo imaging of opioid receptors has been extensively reviewed in a recent paper (Henriksen and Willoch, 2008).
Amino Acids
There has been relatively limited use of agents to study excitatory amino acid receptors, but recently agents for the mGluR5 (Honer et al., 2007; Kimura et al., 2010; Lucatelli et al., 2009; Mu et al., 2010; Patel et al., 2007; Sanchez-Pernaute et al., 2008; Treyer et al., 2007; Yu, 2007), as well as the N-methyl-d-aspartate (NMDA) (Robins et al., 2010; Waterhouse, 2003) and glycine-binding site of the NMDA receptor (Fuchigami et al., 2009) have been developed.
Assessment of Pathology
Inflammation
The peripheral benzodiazepine receptor (PBR) ligand, [11C]PK 11195, has been used as a marker of microglial activation; [11C]PK 11195 binding is increased in disorders with known inflammatory response such as multiple sclerosis (MS) (Banati et al., 2000), encephalitis (Banati et al., 1999; Cagnin et al., 2001b
[/not-level-membership-for-neurology-category]
