Neurofibromatosis, type 1
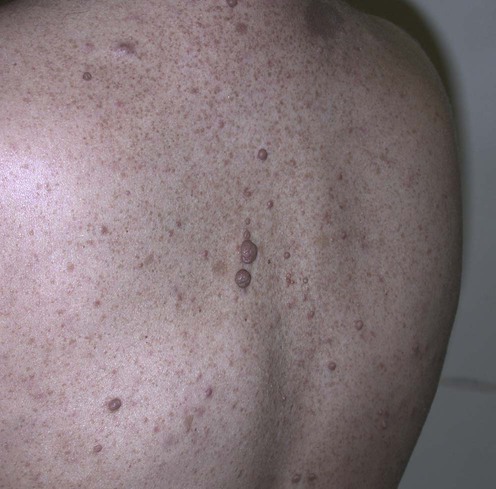
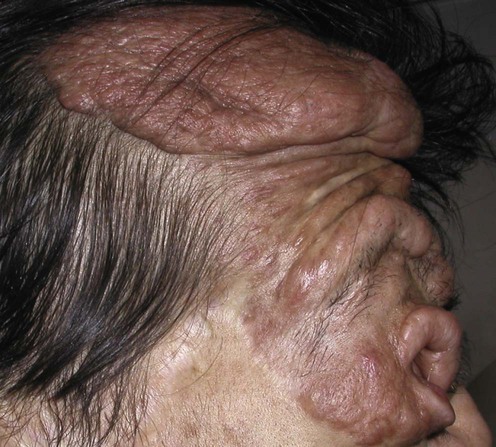
Specific investigations
 Annual complete cutaneous examinations, particularly in those patients with large plexiform lesions
Annual complete cutaneous examinations, particularly in those patients with large plexiform lesions

 Skin and eye exams for first-degree relatives
Skin and eye exams for first-degree relatives

 Regular blood pressure checks to screen for renal artery stenosis or pheochromocytoma
Regular blood pressure checks to screen for renal artery stenosis or pheochromocytoma

 MRI or PET scan, with and without contrast, for deep or changing plexiform neurofibromas
MRI or PET scan, with and without contrast, for deep or changing plexiform neurofibromas
 Biopsy or excision of changing or suspicious lesions
Biopsy or excision of changing or suspicious lesions


Neurofibromatosis 1.
Friedman JM. In: GeneReviews at GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle 1997–2008. Updated May 3, 2012. Available at http://www.genetests.org. Accessed July 7, 2012.
This is one of the most comprehensive reviews of NF1 which is frequently updated.
First-line therapies
 Surgical excision
Surgical excision Laser therapy
Laser therapySecond-line therapies
 Ketotifen
Ketotifen Chemotherapy
Chemotherapy
 Imatinib
Imatinib mTOR inhibitor (rapamycin)
mTOR inhibitor (rapamycin) VEGFR/c-Kit inhibitor (cediranib)
VEGFR/c-Kit inhibitor (cediranib) Multi-kinase inhibitor (sorafenib)
Multi-kinase inhibitor (sorafenib) Tamoxifen
Tamoxifen Antisense morpholino oligomers
Antisense morpholino oligomers Statins (simvistatin, lovastin)
Statins (simvistatin, lovastin) Farnesyl transferase inhibitor (tipifarnib)
Farnesyl transferase inhibitor (tipifarnib) Pirfenidone
Pirfenidone Radiofrequency
Radiofrequency



