Chapter 42 Neuroendocrinology
Neuropeptides, Neurotransmitters, and Neurohormones
One of the features of the neuroendocrine system is that it uses neuropeptides as both neurotransmitters and neurohormones. The term neurotransmitter is applied traditionally to a substance that is released by one neuron and acts on an adjacent neuron in a stimulatory or inhibitory fashion. The effect usually is rapid, brief, and confined to a small area of the neuron surface. In contrast, a hormone is a substance that is released into the bloodstream and travels to a distant site to act over seconds, minutes, or hours to produce its effect over a large area of the cell or over many cells. Neuropeptides can act in either fashion. For example, the neuropeptide, vasopressin, produced by the neurons of the supraoptic and paraventricular nuclei, is released into the bloodstream and has a hormonal action on the collecting ducts in the kidney. Vasopressin is also released within the central nervous system (CNS), where it acts as a neurotransmitter (Landgraf and Neumann, 2004). Similarly, the neuropeptide, substance P, acts as a neurotransmitter in primary sensory neurons that convey pain signals, and more as a neurohormone in the hypothalamus.
Numerous neuropeptides are found in the brain, where they have a wide variety of effects on neuronal function (Table 42.1). Current understanding of all the actions of neuropeptides in the nervous system is far from complete.
Table 42.1 Neuropeptides Found in the Brain and Their Effects on Brain Function*
| Neuropeptide | Central Nervous System Function |
|---|---|
| HYPOTHALAMIC PEPTIDES MODULATING PITUITARY FUNCTION | |
| Corticotropin (ACTH)-releasing hormone (CRH) | Regulation of ACTH secretion |
| Integration of behavioral and biochemical responses to stress | |
| Modulatory effects on learning and memory | |
| Growth hormone–releasing hormone (GHRH) | Regulation of growth hormone secretion |
| Growth hormone release–inhibiting hormone (somatostatin) | Regulation of growth hormone secretion |
| Ghrelin | Regulation of growth hormone secretion |
| Regulation of feeding | |
| Thyrotropin-releasing hormone (TRH) | Regulation of thyroid-stimulating hormone secretion |
| May be involved in depression | |
| Enhances neuromuscular function (in the periphery) | |
| Gonadotropin-releasing hormone (luteinizing hormone–releasing hormone) (GnRH) | Regulates gonadotropin secretionControls sexual receptivity |
| Prolactin-releasing peptide | Stimulates prolactin secretion |
| Neurotensin | Endogenous neuroleptic |
| Regulates mesolimbic, mesocortical, and nigrostriatal dopamine neurons | |
| Thermoregulation | |
| Analgesia | |
| Neuropeptide Y | Satiety (induces obesity) and drinking |
| Sexual behavior | |
| Locomotion | |
| Memory | |
| Orexins (hypocretins) | Stimulates CRH and antidiuretic hormone (ADH) |
| Inhibits GHRH | |
| Stimulates GnRH | |
| May stimulate preovulatory prolactin release | |
| Inhibits TRH release | |
| PITUITARY PEPTIDES | |
| Prolactin | Maternal behavior |
| Mood | |
| Anxiety | |
| Growth hormone | |
| Thyroid-stimulating hormone | |
| Follicle-stimulating hormone | |
| Luteinizing hormone | Elevated levels may promote neurodegeneration |
| Pro-opiomelanocortin | |
| ACTH | |
| ACTH-like intermediate lobe peptides | |
| β-Endorphin | Analgesic mechanisms |
| Feeding | |
| Thermoregulation | |
| Learning and memory | |
| β-Lipotropic hormone | Skin tanning |
| Melanocyte-stimulating hormone (α- and γ-) | Weight loss |
| Skin tanning | |
| Increased sexual desire | |
| Antiinflammatory effect | |
| Important mediator of leptin control on energy homeostasis | |
| Oxytocin | Anxiety and mood |
| Active/passive stress coping | |
| Maternal behavior, aggression | |
| Pair bonding | |
| Vasopressin | Active/passive stress coping |
| Anxiety | |
| Spatial memory | |
| Social discrimination, social interaction | |
| Pair bonding | |
| Neurophysins | |
| BRAIN–GASTROINTESTINAL TRACT PEPTIDES | |
| Vasoactive intestinal polypeptide | Cerebral blood flow |
| Potent antiinflammatory factor | |
| Somatostatin | |
| Insulin | Feeding behavior |
| Modulatory effect on learning and memory | |
| Hunger | |
| Glucagon | Inhibition of feeding |
| Pancreatic polypeptide | |
| Gastrin | |
| Cholecystokinin | Feeding behavior |
| Satiety | |
| Modulates dopamine neuron activity | |
| Facilitation of memory processing (especially under stress) | |
| Tachykinins (e.g., substance P) | Substance P co-localizes with serotonin and is involved in nociception |
| Secretin | Modulates motor and other functions in brain, facilitating GABA |
| Thyrotropin-releasing hormone | |
| Bombesin | Thermoregulation |
| Inhibition of feeding | |
| Modulatory effect on learning and memory | |
| Orexins (hypocretin) | Gastric and gastrointestinal motility and secretion |
| Pancreatic hormone release | |
| Regulation of energy homeostasis | |
| Feeding behavior | |
| Locomotion and muscle tone | |
| Wakefulness/sleep | |
| Galanin | Modulates release of gonadotropin-releasing hormone, prolactin, insulin, glucagons, growth hormone, and somatostatin |
| Affects feeding, sexual behavior, and anxiety | |
| Potent anticonvulsant effects | |
| Leptin | Satiety factor |
| GROWTH FACTORS | |
| Insulin-like growth factors (IGF) 1 and 2 | |
| Nerve growth factors | Axonal plasticity |
| OPIOID FAMILY | |
| Endorphins | Analgesia |
| Enkephalins (met-, leu-) | Analgesic mechanisms |
| Feeding | |
| Temperature control | |
| Learning and memory | |
| Cardiovascular control | |
| Dynorphins | |
| Kytorphin | |
| NEUROPEPTIDES MODULATING IMMUNE FUNCTION | |
| ACTH | |
| Endorphins | |
| Interferons | |
| Neuroleukins | |
| Thymosin | |
| Thymopeptin | |
| OTHER NEUROPEPTIDES | |
| Atrial natriuretic factors | ?Role in cerebral salt wasting |
| Bradykinins | Cerebral blood flow |
| Migraine | |
| OTHER NEUROPEPTIDES | |
| Angiotensin | Hypertension |
| Thirst | |
| Synapsins | |
| Calcitonin gene–related peptide | Migraine and other vascular headaches |
| Calcitonin | |
| Sleep peptides | Regulation of sleep cycles |
| Orexins (hypocretin) | Sleep-wake regulation |
| Narcolepsy | |
| Energy homeostasis | |
| Carnosine | |
| PRECURSOR PEPTIDES | |
| Pro-opiomelanocortin | |
| Proenkephalins (A and B) | |
| Calcitonin gene product | |
| Vasoactive intestinal polypeptide gene product | |
| Proglucagon | |
| Proinsulin |
ACTH, Adrenocorticotropic hormone; GABA, γ-aminobutyric acid.
* This is only a partial list of all of the neuropeptides that have been found in the brain, and not all of the putative functions have been listed.
Neuropeptides and the Immune System
It has been known for many years that stress, acting through the hypothalamic-pituitary-adrenal axis, modulates the function of the immune system (Tsigos and Chrousos, 2002; Wrona, 2006). Certain peptides and their receptors, once thought to be unique to either the immune or the neuroendocrine systems, actually are found in both.
Cytokines—interleukins (IL)-1, -2, -4, and -6 and tumor necrosis factor (TNF)—are synthesized by glial cells in the CNS in response to cell injury. IL-1 and the other cytokines, through their ability to stimulate the synthesis of nerve growth factor, may be important promoters of neuron damage repair. Circulating cytokines have been thought to play a role in the hypothalamus to activate the hypothalamic-pituitary-adrenal axis in response to inflammation elsewhere in the body (see Fever, later in this chapter) and inhibit the pituitary-thyroid and pituitary-gonadal axes in response to systemic disease.
Several other hormones and neuropeptides have modulatory effects on immune function. Similarly, immunocompetent cells contain hormones and neuropeptides that may affect neuroendocrine and brain cells (Table 42.2). Despite speculation about the ability of the psyche to influence immunological function and therefore disease outcome, conclusive evidence suggesting a clinically significant effect remains lacking (Padgett and Glaser, 2003).
Table 42.2 Immunoregulatory Effects of Several Hormones and Peptides
| Hormone or Peptide | Immunocompetent Cell in which Hormone Is Found | Comments |
|---|---|---|
| INHIBITORY | ||
| Glucocorticoids | Inhibit lymphokine synthesis, inflammation | |
| Corticotropin (ACTH) | B lymphocytes | Stimulated by corticotropin-releasing hormone; inhibited by cortisol |
| Macrophage activation, synthesis of IgG and γ-interferon | ||
| Chorionic gonadotropin | T cells | Stimulated by thyrotropin-releasing hormone; inhibited by somatostatin |
| Activity of T cells and natural killer cells | ||
| α-Endorphin | IgG synthesis, T-cell proliferation | |
| Somatostatin | Mononuclear leukocytes, mast cells | T-cell proliferation, inflammatory cascade |
| Vasoactive intestinal peptide | Mononuclear leukocytes, mast cells | T-cell proliferation and migration in Peyer’s patches |
| Potent antiinflammatory effect | ||
| α-Melanocyte-stimulating hormone | Fever, prostaglandin synthesis, secretion of interleukin 2 | |
| Impairs function of antigen-producing cells and T cells | ||
| Antiinflammatory effects | ||
| STIMULATORY | ||
| Estrogens | Lymphocyte proliferation and secretion | |
| Growth hormone | T lymphocytes | Stimulated by growth hormone |
| Thymic growth, lymphocyte reactivity | ||
| Prolactin | Mononuclear cells | Thymic growth, lymphocyte proliferation |
| STIMULATORY | ||
| Thyrotropin (TSH) | T cells | Stimulated by thyrotropin-releasing hormone; inhibited by somatostatin |
| IgG synthesis | ||
| β-Endorphin | Activity of T, B, and natural killer cells | |
| Substance P | Proliferation of T cells and macrophages, inflammatory cascade | |
| Corticotropin (ACTH)-releasing hormone | Lymphocyte and monocyte proliferation and activation | |
| NOT KNOWN TO BE STIMULATORY OR INHIBITORY | ||
| Enkephalins | B lymphocytes | |
| Vasopressin | Thymus | |
| Oxytocin | Thymus | |
| Neurophysin | Thymus | |
ACTH, Adrenocorticotropic hormone; IgG, immunoglobulin G; TSH, thyroid-stimulating hormone.
Modified from Reichlin, S., 1993. Neuroendocrine-immune interactions. N Engl J Med 329, 1246-1253.
Nonendocrine Hypothalamus
Temperature Regulation
The hypothalamus plays a key role in ensuring that body temperature is maintained within narrow limits by balancing the heat gained from metabolic activity and the environment with the heat lost to the environment. A theoretical schema of the mechanisms of hypothalamic temperature regulation is depicted in Fig. 42.1. Although numerous neurotransmitters and peptides alter body temperature, their physiological roles remain unclear.
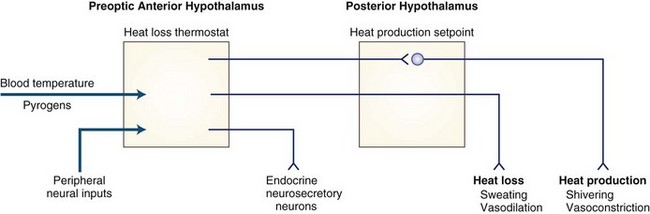
Fever
Classical teaching has been that inflammatory cells in the periphery (primarily monocytes) released cytokines in response to infection and inflammation. These cytokines were thought to act on the hypothalamus to induce production of prostaglandin E2 (PGE2) and cause elevation of the body temperature set point. The body then used its normal physiological mechanisms of vasoconstriction, vasodilation, sweating, and shivering to maintain this new higher set point (i.e., fever). This view of the mechanism by which bacterial infections cause fever probably is incorrect. Bacterial endotoxic lipopolysaccharide (LPS) does appear to work in the periphery to cause macrophages to release a variety of factors; however, the initial signal to the brain probably travels by vagal afferents to the preoptic anterior hypothalamus via norepinephrine, which activates the cyclo-oxygenase isoenzyme, COX-2, to generate and release PGE2. The slower or second febrile increase in PGE2 is due to COX-2 activation by IL-1β produced locally in the brain, not to circulating factors (Blatteis et al., 2000). Interference with prostaglandins is probably how drugs such as acetylsalicylic acid and acetaminophen act to treat fever.
Drug-Induced Hyperthermia
Drug-induced hyperthermic syndromes include anticholinergic poisoning, sympathomimetic poisoning, malignant hyperthermia, neuroleptic malignant syndrome, and serotonin syndrome. In malignant hyperthermia, a syndrome associated most often with the use of various general anesthetic agents and muscle relaxants, an inherited defect leads to excessive release of calcium from sarcoplasmic reticulum, stimulating severe muscle contraction (see Chapters 64 and 79). The neuroleptic malignant syndrome (NMS) is characterized by diffuse muscular rigidity, akinesia, and fever accompanied by a decreased consciousness level and evidence of autonomic dysfunction—namely, labile blood pressure, tachyarrhythmias, excessive sweating, and incontinence. NMS can be associated with administration of major tranquilizers (primarily those that work by blocking dopamine receptors), with rapid withdrawal from dopaminergic agents, including entacapone, and less commonly with administration of tricyclic antidepressants. It appears to result from an alteration of temperature control mechanisms in the hypothalamus. As part of treatment, withdrawal of all neuroleptics is mandatory. In addition to general supportive measures and cooling, the use of bromocriptine (2.5 to 10 mg 4 times daily, increasing by 7.5 mg daily in divided doses to a maximum of 60 mg daily) is helpful. Hypotension, psychosis, and nausea are possible side effects. An alternative is dantrolene (50 to 200 mg/day orally, or 2 to 3 mg/kg/day intravenously (IV), to a maximum of 10 mg/kg/day). The serotonin syndrome is characterized by mental status changes, neuromuscular symptoms, autonomic dysfunction, and gastrointestinal dysfunction. In addition to tremor and rigidity (seen also in NMS), other features may include shivering, myoclonus, and hyperreflexia. Nausea, vomiting, and diarrhea are common in serotonin syndrome but are uncommon in NMS. Treatment entails withdrawal from the offending drug and general supportive care.
Appetite
Given free access to food and water, most animals maintain their body weight within narrow limits. With a change in energy intake/expenditure (a change in the size or number of individual meals that is not balanced by an equal and opposite change in energy use), the animal experiences a change in weight. One possible model of nutrient balance is depicted in Fig. 42.2. The four components of energy balance are (1) the afferent system, (2) the CNS processing unit, (3) the efferent system, and (4) the absorption of food from the gut and its metabolism in the liver. Defects at any point in these systems may lead to weight loss or weight gain (Wynne et al., 2005).
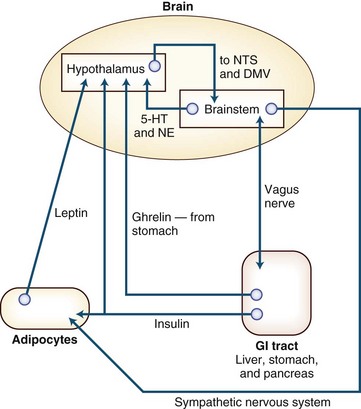
Fig. 42.2 Sympathetic nervous system. In the regulation of energy balance, the brain is the central processing unit. It receives afferent neuronal signals from the vagus nerve, via the brainstem and hormonal signals—ghrelin (from the stomach), insulin (from the pancreas), and leptin (from adipocytes). The brainstem also has input to the hypothalamus via norepinephrine (NE) (from the locus ceruleus) and serotonin, or 5-hydroxytryptamine (5-HT) (from the raphe nuclei). These afferent signals are interpreted both in the brainstem (in the nucleus of the tractus solitarius) and in the hypothalamus (in the ventromedial nucleus). The ventromedial nucleus of the hypothalamus communicates with the lateral hypothalamic area (LHA) and the paraventricular nucleus (PVN) by means of pro-opiomelanocortin-cocaine/amphetamine-regulated transcript (POMC-CART), an anorexigenic peptide (the release of which is stimulated by insulin and leptin), and by neuropeptide Y/agouti-related protein (NPY/AGRP), an orexigenic peptide (the release of which is stimulated by ghrelin and inhibited by leptin and insulin)—or through the parasympathetic nervous system. Output from the LHA and PVN is either via the sympathetic nervous system, which leads to energy expenditure through physical activity, activation of β2-adrenergic receptors, and uncoupling proteins in the adipocyte to cause energy release through lipolysis, or through the parasympathetic nervous system. Output via the vagus nerve leads to increased insulin secretion, which causes adipogenesis and energy storage. For a more complete explanation of this control of energy balance, the interested reader is directed to the article by Lustig (2001). DMV, Dorsal motor nucleus of the vagus; GI, gastrointestinal; NTS, nuclear tractus solitarius.
Destruction of the ventromedial hypothalamus, both in animals and in humans, leads to obesity. Lesions in the paraventricular nucleus have a similar effect. Overeating (hyperphagia) is only one of the mechanisms that produces hypothalamic obesity; more efficient handling of calories by the eater is probably another important factor. Hypothalamic lesions also can cause weight loss. Lesions in the dorsomedial nucleus lead to a reduction in body weight and fat stores, as do lesions in the lateral hypothalamus. Studies in the decerebrate rat suggest that oral motor and meal-size responses are dependent on centers in the caudal brainstem, on which the hypothalamus has only a modulatory effect (Grill and Kaplan, 2002).
When α-melanocyte-stimulating hormone (α-MSH) binds to its receptor in the hypothalamus, it causes satiety. Up to 5% of obese children have been found to have an abnormality of the α-MSH receptor, MC4R, as a cause of their obesity (Lustig, 2001).
The orexins (hypocretins) are neuropeptides that play a role in energy balance and arousal (Ferguson and Samson, 2003). Narcolepsy is caused by failure of orexin-mediated signaling. Orexins are found in the hypothalamus, where they regulate sleep/wake cycles (Baumann and Bassetti, 2005), and in the GI tract, where they excite secretomotor neurons and modulate gastric and intestinal motility and secretion (Kirchgessner, 2002).
Anorexia nervosa and bulimia nervosa are clinical eating disorders of unknown etiology seen primarily in young women and girls. Anorexia nervosa is characterized by reduced caloric intake and increased physical activity associated with weight loss, a distorted body image, and a fear of gaining weight. Bulimia nervosa is characterized by episodic gorging followed by self-induced vomiting or laxative and diuretic abuse or dieting and exercise to reduce weight. Initially these syndromes were considered to be neuroendocrine in origin, then for many years, they were assumed to be purely psychiatric. Although malnutrition produces changes in neuroendocrine function, disturbances in corticotropin-releasing hormone (CRH), opioids, neuropeptide Y, vasopressin, oxytocin, cholecystokinin (CCK), and leptin as well as the monoamines—serotonin, dopamine, and norepinephrine—have been found in patients with eating disorders (Barbarich et al., 2003). These neurotransmitters play a role not only in appetite but in mood and impulse control. The abnormalities have been found to persist, in some instances, long after recovery. Furthermore, patients with anorexia nervosa seem to have an increased total daily energy expenditure because of their increased physical activity. All of this suggests a complex interaction between the psyche and the endocrine system as a cause for these syndromes.
Emotion and Libido
Libido, like other feelings, requires the participation of both hypothalamic and extrahypothalamic sites. In most instances of hypothalamic disease, loss of libido is caused by impaired release of gonadotropin-releasing hormone (GnRH) and a subsequent decrease in testicular testosterone in men. In women, libido is related more to adrenal androgens, and in menstruating women to ovarian androgens. Adrenal androgen levels may be low in women with corticotropin (i.e., adrenocorticotropic hormone [ACTH]) deficiency and secondary adrenal insufficiency. Hypersexuality associated with hypothalamic disease is rare and may occur with or without a subjective increase in libido. The melanocortins (ACTH and α-, β-, and γ-MSH) may play a role in the motivational aspects of sexual behavior, as well as having a sildenafil-like effect on penile and vaginal blood flow, albeit through a central rather than a peripheral mechanism (Raffin-Sanson et al., 2003).
Current understanding of the human hypothalamus in relation to normal development, sexual differentiation, aging, and some degenerative neurological disorders is gradually expanding. The sexually dimorphic nucleus, or intermediate nucleus, of the preoptic area is twice the volume in male subjects compared to that in female subjects—although this finding is disputed by some investigators. Two other cell groups in the preoptic anterior hypothalamus are larger in males than in females. The size does not differ between homosexual and heterosexual men. Although the shape of the suprachiasmatic nucleus differs in male and female subjects, the vasopressin cell number and volume are similar in men and women. Homosexual men seem to have a larger suprachiasmatic nucleus containing twice as many cells as in heterosexual men (Swaab et al., 2001). The site of the central nucleus of the bed nucleus of the stria terminalis is perhaps involved in gender identity in transsexuals. At present, the significance of these observations is uncertain.
It has been known for some time that oxytocin, the classical posterior pituitary hormone, works in the brain to promote mother-infant bonding. Now, it has been shown that it can promote more appropriate social behavior and affect in patients with high-functioning autism or with Asperger syndrome (Andari et al., 2010).
Endocrine Hypothalamus: The Hypothalamic-Pituitary Unit
Functional Anatomy
The hypothalamus, despite its small size, is the region of brain with the highest concentrations of neurotransmitters and neuropeptides. Beginning with the pioneering work of Ernst and Berta Scharrer and Geoffrey Harris in the 1940s, the hypothalamus has been assigned a central role in regulating anterior pituitary function. In addition to the identified hypophysiotropic hormones (Table 42.3), other peptides with putative regulatory functions are found in high concentration in the hypothalamus: neurotensin, substance P, cholecystokinin, neuropeptide Y, vasoactive intestinal polypeptide, and the opioid peptides. The hypothalamus also is rich in acetylcholine, norepinephrine, dopamine, serotonin, histamine, and γ-aminobutyric acid (GABA). In many neurons, these neurotransmitters co-localize with peptides, although this co-localization and presumptive co-release have uncertain physiological significance. In patients with nonfunctioning pituitary or parapituitary tumors, symptoms produced by compression of neural structures adjacent to the pituitary gland are a common presentation. Understanding these symptoms requires knowledge of the anatomy of the region (Fig. 42.3).
Table 42.3 Hypothalamic Peptides Controlling Anterior Pituitary Hormone Release
| Pituitary Hormone | Hypothalamic Factor |
|---|---|
| Growth hormone | Growth hormone–releasing hormone (GHRH) |
| Growth hormone release–inhibiting hormone (somatostatin) | |
| Prolactin | Prolactin-releasing factor(s) (PRF) |
| Prolactin release–inhibiting factor: dopamine and possibly the precursor of gonadotropin-releasing hormone (GnRH) | |
| Thyrotropin | Thyrotropin-releasing hormone (TRH) |
| Thyrotropin release–inhibiting factor: somatostatin can do this but has not been confirmed to do so physiologically | |
| Pro-opiomelanocortin is cleaved to form corticotropin (ACTH) | Corticotropin-releasing hormone (CRH) |
| Luteinizing hormone (LH) and follicle-stimulating hormone (FSH) | Gonadotropin-releasing hormone (GnRH) |
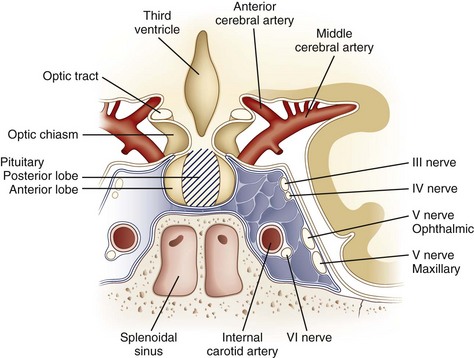
Expansion of pituitary tumors out of the sella tends to lead to compression of the anterior and inferior crossing fibers of the optic chiasm (see Chapters 14 and 36). These fibers subserve vision in the superior temporal quadrants. Therefore, pituitary adenomas typically cause bitemporal superior quadrantanopias. Lesions such as craniopharyngiomas that impinge on the posterior and superior fibers of the optic chiasm tend to manifest with bitemporal inferior quadrantanopias. Nevertheless, owing to the variability of the positioning of the optic chiasm and the tendency for tumors to be asymmetrical in their growth, parasellar lesions result in a wide variety of field defects.
Blood Supply
The superior and inferior hypophysial arteries are the pituitary’s major source of blood (Fig. 42.4). The posterior pituitary gland is supplied principally by the inferior hypophysial arteries and is drained by the inferior hypophysial veins. The superior hypophysial artery forms a primary capillary plexus in the median eminence of the hypothalamus. From here, blood flows into the long hypophysial portal veins, which carry it to the anterior pituitary. Although some blood from the anterior pituitary drains into the cavernous sinus, some drains into the posterior pituitary, and some returns to the median eminence by way of the long portal veins, which are capable of bidirectional blood flow. This vascular anatomy provides a potential mechanism for the important feedback loops necessary for regulation of hypothalamic-pituitary function.
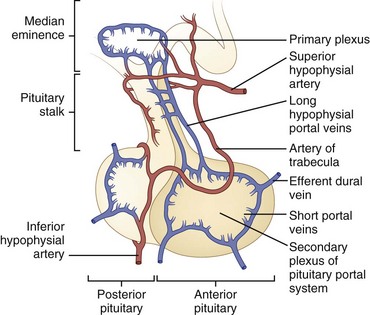
Anterior Pituitary
Hypothalamic Control of Anterior Pituitary Secretion
The hypothalamus produces hypophysiotropic substances that control the secretion of anterior pituitary hormones. Five neuropeptides and one neurotransmitter (dopamine) are known to be important physiological regulators of pituitary function (see Table 42.3). In addition, several neurotransmitters affect pituitary hormone release, although their physiological role remains uncertain. Since their discovery in 1998, the orexins (hypocretins) have been found to regulate virtually all the hypothalamic-pituitary axes as well as participate in the coordination of anterior pituitary function with sleep, arousal, and general metabolism. This is well reviewed in a recent article by López, Tena-Sempere and Diéguez (2010).
Abnormalities of Anterior Pituitary Function
Hypofunction
The causes of pituitary insufficiency are summarized in Box 42.1. In general, the symptoms of hypopituitarism (Table 42.4) are those of the secondary failure of end-organ function. Because the associated changes usually develop slowly and some autonomous end-organ function remains, the symptoms often are less severe than those that occur with primary end-organ disease. The term Simmonds disease is applied to panhypopituitarism. When this syndrome develops in the postpartum period after an episode of pituitary infarction, it is called Sheehan syndrome.
Table 42.4 Clinical Syndromes of Anterior Pituitary Dysfunction
| Hormone | Excess Secretion | Deficient Secretion |
|---|---|---|
| Growth hormone | In children: Gigantism | In children: Growth failure and tendency to hypoglycemia |
| In adults: Acromegaly | ||
| Prolactin | In children: Delayed puberty | |
| In adults: | In adults: | |
| Women: Amenorrhea, galactorrhea, and infertility | Women: Inability to breast-feed and possible infertility | |
| Men: Impotence, infertility, and (rarely) galactorrhea | ||
| Luteinizing hormone and follicle-stimulating hormone | In children: Precocious pubertyIn adults: Infertility, hypogonadism, polycystic ovary syndrome | In children: Delayed pubertyIn adults: Amenorrhea, infertility, erectile dysfunction |
| Thyrotropin (TSH) | Hyperthyroidism | Hypothyroidism |
| Hyperprolactinemia (due to excessive TRH stimulation) | ||
| Pro-opiomelanocortin | Cushing disease, Nelson syndrome | Hypoadrenalism; glucocorticoids affected more severely than mineralocorticoids |
Hyperfunction
Hyperprolactinemia
Probably the most common abnormality of pituitary function encountered by the neuroendocrinologist is hyperprolactinemia (Wand, 2003); causes are summarized in Box 42.2. Prolactin levels in excess of 200 ng/mL (normal, <25ng/mL) are almost always due to excessive production of the hormone by a pituitary adenoma. In premenopausal women, the development of amenorrhea secondary to direct inhibition of LH and FSH by prolactin leads to early investigation and diagnosis of tumors at the microadenoma (<10 mm in diameter) stage. In men, the insidious onset of erectile dysfunction and reduced libido usually means that these tumors are found late, often only after they have produced signs and symptoms of optic nerve compression. Galactorrhea is a common accompaniment of elevated prolactin in women and a rare finding in men.
Gigantism and Acromegaly
Presence of excessive amounts of circulating GH before closure of the epiphyses leads to gigantism. If the epiphyses have closed, only tissue still capable of responding to GH will grow, leading to the clinical syndrome of acromegaly; its clinical features are summarized in Box 42.3. Of particular note for the neurologist and the neurosurgeon is the frequent complaint of headache and symptoms related to carpal tunnel syndrome. It is not uncommon to find patients with acromegaly in whom surgery for carpal tunnel release was performed 3 to 5 years before diagnosis of their disease.
Cushing Disease and Nelson Syndrome
The term Cushing syndrome refers to the clinical picture resulting from exposure to excessive corticosteroids, either endogenous or exogenous. If the clinical manifestations are caused by excessive production of ACTH from the pituitary, the condition is referred to as Cushing disease. Common clinical features of Cushing disease are listed in Box 42.4. The syndrome of hyperpigmentation and local compression of parapituitary structures that occurs in approximately 10% of patients with Cushing disease who have been treated with bilateral adrenalectomy is called Nelson syndrome. Given the generally good results from surgery on the pituitary gland in Cushing disease, Nelson syndrome is now quite uncommon.
Box 42.4 Common Clinical Features of Cushing’s Disease
The diagnosis of Cushing syndrome, although simple in theory, often is quite difficult in practice (Findling and Raff, 2005). It is also often difficult for tests to distinguish between true Cushing syndrome and so-called pseudo-Cushing syndrome due to alcoholism, depression, and eating disorders.
As a screening test, the most sensitive and specific screening tool is an 11 pm salivary cortisol determination. Unfortunately, this test may not be readily available in all clinical centers, and 24-hour urine collections for urinary free cortisol are still used. The sensitivity and specificity of the 24-hour collection can be increased by doing two collections on consecutive days. For years now, the dexamethasone suppression test has been pivotal in the diagnosis of Cushing disease. For this test, 0.5 mg of dexamethasone is given every 6 hours for 8 doses; during the second 24 hours of administration, the normal response is suppression of cortisol production, as reflected by reduced urinary levels of 17-ketogenic steroids or urinary free cortisol. Patients with Cushing disease usually show a similar suppression only when the dose of dexamethasone is increased to 2 mg every 6 hours for 8 doses. The formal dexamethasone suppression test is cumbersome, requiring 6 consecutive days of collection of urine for urinary free cortisol levels. Various modifications of this test may be useful and less cumbersome. More detailed discussion can be found in the literature, both for screening (Findling and Raff, 2005) and for diagnosis (Lindsay and Nieman, 2005) of Cushing syndrome.
Pituitary Tumors and Pituitary Hyperplasia
Pituitary tumors account for approximately 15% of all intracranial tumors. Although most are benign, they can be locally invasive. Only rarely is true malignancy evidenced by metastases. The old classification of pituitary adenomas into chromophobe, acidophil, and basophil has been supplanted by a more functional classification based on findings of immunological and electron microscopic examinations (Table 42.5). Hyperplasia of various cellular elements of the pituitary is relatively rare and usually only seen in cases of ectopic hypothalamic-releasing hormone production (Ironside, 2003).
| Tumor | Frequency (%) |
|---|---|
| Growth hormone cell adenoma | 14.0 |
| Prolactin cell adenoma | 27.2 |
| Growth hormone–prolactin cell adenoma | 8.4 |
| Corticotroph cell adenoma | 8.1 |
| Thyrotroph cell adenoma | 1.0 |
| Gonadotroph cell adenoma | 6.4 |
| Clinically nonfunctioning adenoma | 31.2 |
| Plurihormonal adenoma | 3.7 |
Modified with permission from Thapar, K., Kovacs, K., Muller, P.J., 1995. Clinical-pathological correlations of pituitary tumours. Clin Endocrinol Metab 9, 243-270.
Levy and Lightman (2003) suggest that although some pituitary tumors may be due to genetic abnormalities, it is possible that a majority of pituitary tumors are not “tumors” in the usual sense but rather represent an overresponse to normal trophic factors that has failed to normalize once the growth stimulus has returned to normal.
Posterior Pituitary
Physiology
Oxytocin
Oxytocin, like vasopressin, is synthesized in magnocellular neurons of the supraoptic and paraventricular nuclei as a large precursor molecule that is cleaved into oxytocin and a specific neurophysin. Many of the physiological stimuli of vasopressin also result in oxytocin release, and although in supraphysiological doses, oxytocin does have ADH-like properties, its physiological role in these circumstances remains obscure. The only specific stimulus that causes the release of oxytocin but not vasopressin is suckling. Oxytocin’s role in normal lactation and parturition in the human remains to be defined clearly. Oxytocin receptors are found in the limbic system, particularly in the amygdala, and for this reason oxytocin has been implicated in emotion. Lim and Young (2006) have shown that in mammals it plays a role in a variety of complex social behaviors as well as helping regulate the response to stress.
Diabetes Insipidus
Diabetes insipidus (DI) is a clinical syndrome characterized by severe thirst, polydipsia, and polyuria. Central DI must be distinguished from nephrogenic DI (an inability of the kidney to respond to ADH) and from compulsive water drinking (Baylis, 1995). Distinguishing among these entities normally is done using a water deprivation test. A urine osmolality of greater than 750 mmol/L after water deprivation excludes the diagnosis of DI. Central DI is characterized by an increase in osmolality to greater than 750 mmol/L after administration of desamino-d-arginine-vasopressin (DDAVP). In nephrogenic DI, little change in osmolality occurs after DDAVP administration. The polyuria induced by chronic compulsive water drinking may produce a renal tubular concentrating defect because of medullary washout—that is, the loss of sodium and other solutes from around the loops of Henle. This can make it difficult to differentiate partial DI of central or renal cause from polydipsia. Treatment by gradual fluid restriction, with or without DDAVP, can be used to reverse the medullary washout, thereby increasing the sensitivity of the test.
Etiology
Approximately a third of patients with central DI have no demonstrable disease of the hypothalamic-posterior pituitary unit. The remaining patients have damage to the supraoptic-hypophysial-portal pathway from trauma, surgery, tumors, inflammatory lesions (which may be granulomatous or infectious), or vascular lesions. In patients with polyuria, the urine should be examined to ensure that a solute diuresis, as with hyperglycemia, has not occurred, or that a type of nephrogenic DI has not been induced by hypokalemia, hypercalcemia, or lithium carbonate therapy. In the acute care setting on a neurosurgical ward, administration of mannitol may be one of the more common causes of polyuria. The investigation of DI has been well summarized by Diederich and colleagues (2001).
Syndrome of Inappropriate Antidiuretic Hormone Secretion
Etiology and Pathophysiology
The initial clue to the diagnosis of SIADH is the low serum sodium (Reynolds et al., 2006). Measured serum osmolality also must be low to exclude the artifactual hyponatremia that occurs with hyperlipidemia and hyperproteinemia, in which the sodium concentration in the plasma water actually is normal. The urine osmolality in SIADH is not always above the serum osmolality, but the urine is less than maximally dilute, which excludes the dilutional hyponatremia of water intoxication. Causes of SIADH are listed in Box 42.5.
Clinical Features
The clinical features of SIADH are nonspecific and are related to the hypo-osmolality of the body fluids. The more rapidly this condition develops, the more symptomatic the patient. Serum sodium less than 115 mmol/L almost always is associated with confusion or obtundation, and seizures can occur. With milder hyponatremia, the symptoms may be nonspecific, including fatigue, general malaise, loss of appetite, and some clouding of consciousness (Bhardwaj, 2006).
Treatment
For asymptomatic or mildly symptomatic SIADH-induced hyponatremia, the mainstay of treatment is restriction of fluid. Intake should be reduced to insensible losses (approximately 800 mL/day). Obtundation by itself does not necessitate more aggressive treatment of hyponatremia. In patients with severe hyponatremia complicated by seizures, however, a more rapid partial correction can be undertaken. Diuresis is induced with furosemide (1 mg/kg IV), and urinary losses are replaced with 3% sodium chloride through a central line at a rate of 0.1 mL/kg/min, to which appropriate amounts of potassium are added (to counter urinary losses). Considerable controversy exists over the rate at which serum sodium can be raised safely. Some clinicians suggest that active correction be by no more than 20 to 25 mmol/L during the first 48 hours, to a level no higher than 130 mmol/L, and that the rate of replacement is not important. Patients must be monitored carefully to avoid acute elevation to hypernatremic or even normonatremic levels, and to avoid a change of more than 25 mmol/L in 48 hours, which seems to be dangerous and can cause the syndrome of osmotic demyelination, one form of which is central pontine myelinolysis (Tisdall et al., 2006).
Approach to the Patient with Hypothalamic-Pituitary Dysfunction
History and Physical Examination
Because of the proximity of the optic nerves to the hypothalamic-pituitary unit, a careful examination of the visual fields is essential (see Chapters 14 and 36).
Endocrinological Investigation
No single endocrine test can provide all the answers about pituitary function. Conclusions are based on a synthesis of evidence gained from clinical examination, endocrine tests, and MRI. Endocrine testing is used to determine residual pituitary function after surgery or radiotherapy. The return of biochemical markers of abnormal pituitary secretion or their failure to resolve can be used to gauge the success of surgery and aid in differentiating scar tissue from tumor recurrence on postoperative MRI scans. Table 42.6 summarizes some of the more common pituitary tests and their use.
| Test | Comments |
|---|---|
| Insulin hypoglycemia (Regular insulin, 0.1 unit/kg of body weight IV, fasting) | Adequate hypoglycemia is associated with a rise in growth hormone, ACTH, and to a lesser extent, prolactin. It probably is the most physiological stressor of the hypothalamic-pituitary-adrenal axis. The test should not be used in patients with epilepsy or unstable angina. |
| Gonadotropin-releasing hormone (2 µg/kg IV to a maximum of 100 µg) | Stimulates LH and FSH release directly at the pituitary. May be used to test LH and FSH reserve. Cannot reliably distinguish between pituitary and long-standing hypothalamic problem. |
| Thyrotropin-releasing hormone (7 µg/kg IV to a maximum of 400 µg) | Stimulates release of thyroid-stimulating hormone and prolactin from pituitary. Failure of prolactin to respond to thyrotropin-stimulating hormone is very suggestive of autonomous secretion by an adenoma, but exceptions do occur. |
| Metyrapone test (750 mg every 4 hours for 6 doses; collect 24-hr urine the day before, day of, and day after the test) | Metyrapone blocks the production of cortisol in the adrenal gland, resulting in increased ACTH secretion. This test is an alternative to insulin hypoglycemia as a test of the hypothalamic-pituitary-adrenal axis. |
| l-Dopa (500 mg PO) | l-Dopa can be used to stimulate growth hormone release (probably by increasing growth hormone–releasing hormone). It is a less potent stimulus than insulin-induced hypoglycemia but can be used as an alternative. |
| Corticotropin-releasing hormone (CRH) (1 µg/kg or 100 µg IV) | Stimulates release of cortisol and ACTH. ACTH is sampled at −5, 0, 15, and 30 minutes and cortisol at −5, 0, 30, and 45 minutes. |
ACTH, Adrenocorticotropic hormone (corticotropin); FSH, follicle-stimulating hormone; IV, intravenously; LH, luteinizing hormone; PO, orally.
Treatment of Pituitary Tumors
Medical Management
Acromegaly
Until the development of a long-acting somatostatin analog (octreotide), the outcome of medical therapy for acromegaly, like that for Cushing disease, was disappointing. Bromocriptine in doses of 20 to 60 mg/day could reduce GH levels and cause tumor shrinkage, but it seldom normalized GH levels. Octreotide can normalize GH levels when given by subcutaneous injection every 8 hours or when used in a long-acting form given once monthly. The analog can be combined with bromocriptine if necessary to achieve maximal suppression and tumor shrinkage. For those patients who fail to respond to long-acting octreotide, pegvisomant (a growth hormone receptor blocker) can be added to the regimen to normalize growth hormone and IGF-1 levels (Cook et al., 2004). Most patients treated with pegvisomant will require continuing therapy with octreotide to prevent enlargement of the pituitary tumor when pegvisomant is used alone.
Neuroendocrine Tumors
Pheochromocytomas
Tumor localization usually can be achieved by CT scanning of the adrenals. If a wider search is necessary, MRI may be more helpful. Radiolabeled metaiodobenzyl guanidine (123I-MIBG), an iodinated guanethidine derivative, is taken up by chromaffin tissue, and its use with single-photon emission computed tomography (SPECT) can be helpful in localizing nonadrenal tumors and metastases. Some centers have been using indium 111–labeled pentetreotide, an analog of somatostatin, to localize somatostatin receptors on these tumors; 6-18F-fluorodopamine and [11C]hydroxyephedrine PET scanning also are complementary techniques for tumor localization (Eriksson et al., 2005).
Andari E., Duhamel J.-R., Zalla T., et al. Proc Natl Acad Sci USA. 2010;107:4389-4394.
Barbarich N.C., Kaye W.H., Jimerson D. Neurotransmitter and imaging studies in anorexia nervosa: new targets for treatment. Curr Drug Targets CNS Neurol Disord. 2003;2:61-72.
Baumann C., Bassetti C.I. Hypocretins (orexins) and sleep-wake disorders. Lancet Neurol. 2005;4:673-682.
Baylis P.H. Investigation of suspected hypothalamic diabetes insipidus. Clin Endocrinol. 1995;43:507-510.
Bhardwaj A. Neurological impact of vasopressin dysregulation and hyponatremia. Ann Neurol. 2006;59:229-236.
Blatteis C.M., Sheic E., Li S. Pyrogen sensing and signaling: old views and new concepts. Clin Infect Dis. 2000;31(Suppl. 5):S168-S177.
Cook D.M., Ezzat S., Katznelson L., et al. AACE Acromegaly Guidelines Task Force. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the diagnosis and treatment of acromegaly. Endocr Pract. 2004;10:213-225.
Diederich S., Eckmanns T., Exner P., et al. Differential diagnosis of polyuric/polydipsic syndromes with the aid of urinary vasopressin measurement in adults. Clin Endocrinol. 2001;54:665-671.
Eriksson B., Oriefors H., Oberg K., et al. Developments in PET for the detection of endocrine tumours. Best Pract Res Clin Endocrinol Metab. 2005;19:311-324.
Ferguson A.V., Samson W.K. The orexin/hypocretin system: a critical regulator of neuroendocrine and autonomic function. Front Neuroendocrinol. 2003;24:141-150.
Findling J.W., Raff H. Screening and diagnosis of Cushing’s syndrome. Endocrinol Metab Clin North Am. 2005;34:385-402.
Grill H.J., Kaplan J.M. The neuroanatomical basis for control of energy balance. Front Neuroendocrinol. 2002;23:2-40.
Ironside J.W. Best Practice No. 172: pituitary gland pathology. J Clin Pathol. 2003;56:561-568.
Kirchgessner A.L. Orexins in the brain-gut axis. Endocr Rev. 2002;23:1-15.
Landgraf R., Neumann I.D. Vasopressin and oxytocin release within the brain: a dynamic concept of multiple and variable modes of neuropeptide communication. Front Neuroendocrinol. 2004;25:150-176.
Levy A. Pituitary disease: presentation, diagnosis, and management. J Neurol Neurosurg Psychiatry. 2004;75:47-52.
Levy A., Lightman S. Molecular defects in the pathogenesis of pituitary tumours. Front Neuroendocrinol. 2003;24:94-127.
Lim M.M., Young L.J. Neuropeptidergic regulation of affiliative behavior and social bonding in animals. Hum Behav. 2006;50:506-517.
Lindsay J.R., Nieman L.K. Differential diagnosis and imaging in Cushing’s syndrome. Endocrinol Metab Clin North Am. 2005;34:403-421.
López M., Tena-Sempere M., Diéguez C. Cross-talk between orexins (hypocretins) and the neuroendocrine axes (hypothalamic-pituitary axes). Front Neuroendocrinol. 2010;31:113-127.
Lustig R.H. The neuroendocrinology of obesity. Endocrinol Metab Clin North Am. 2001;30:765-785.
Padgett D.A., Glaser R. How stress influences the immune response. Trends Immunol. 2003;24:444-448.
Raffin-Sanson M.L., de Keyzer Y., Bertagna X. Proopiomelanocortin, a polypeptide precursor with multiple functions: from physiology to pathological conditions. Eur J Endocrinol. 2003;149:79-90.
Reynolds R.M., Padfield P.L., Seckl J.R. Disorders of sodium balance. BMJ. 2006;332:702-705.
Swaab D.F., Chung W.C.J., Fruijver F.P.M., et al. Structural and functional sex differences in the human hypothalamus. Horm Behav. 2001;40:93-98.
Tisdall M., Crocker M., Watkiss J., et al. Disturbances of sodium in critically ill adult neurologic patients. J Neurosurg Anesthesiol. 2006;18:57-63.
Tsigos C., Chrousos G.P. Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. Front Neuroendocrinol. 2002;53:865-871.
Wand G.S. Diagnosis and management of hyperprolactinemia. The Endocrinologist. 2003;13:52-57.
Wrona D. Neural-immune interactions: an integrative view of the bi-directional relationship between the brain and immune systems. J Neuroimmunol. 2006;172:38-58.
Wynne K., Stanley S., McGowan B., et al. Appetite control. J Endocrinol. 2005;184:291-318.