Chapter 65 Neurocutaneous Syndromes
Tuberous Sclerosis
Tuberous sclerosis complex (TSC) is a disorder of cellular differentiation and proliferation that can affect the brain, skin, kidneys, heart, and other organs. Many clinical features of TSC result from hamartomas, but true neoplasms also occur, particularly in the kidney and brain. Abnormal neuronal migration plays a major additional role in neurological dysfunction (Roach and Sparagana, 2004).
Population-based studies suggest a prevalence of 1 per 6000 to 9000 individuals. However, because of the striking variability of clinical expression, establishing the diagnosis of TSC can be difficult in individuals with subtle findings, and the true prevalence may be considerably higher. Cutaneous findings are usually the first clue that a patient has TSC, but other features may lead to the diagnosis (Box 65.1). In infants, cardiac involvement and seizures frequently are presenting signs, whereas dermatological, pulmonary, or renal involvement may lead to diagnosis in older individuals.
Box 65.1
Diagnostic Criteria for Tuberous Sclerosis Complex
Major Features
Minor Features
1. Multiple randomly distributed pits in dental enamel
2. Hamartomatous rectal polyps‡
4. Cerebral white-matter radial migration lines*,†,i
Definite tuberous sclerosis complex: either two major features or one major feature plus two minor features.
Probable tuberous sclerosis complex: one major plus one minor feature.
Possible tuberous sclerosis complex: either one major feature or two or more minor features.
Reprinted with permission from Roach, E.S., Gomez, M.R., Northrup, H., 1998. Tuberous sclerosis consensus conference: revised clinical diagnostic criteria. J Child Neurol 13, 624-628.
* When cerebral cortical dysplasias and cerebral white-matter migration tracts occur together, they should be counted as one rather than two features of tuberous sclerosis.
† When both lymphangiomyomatosis and renal angiomyolipomas are present, other features of tuberous sclerosis should be present before a definite diagnosis is assigned.
‡ Histological confirmation is suggested.
§ Radiographical confirmation is sufficient.
i One panel member felt strongly that three or more radial migration lines should constitute a major sign.
The inheritance of TSC is as an autosomal dominant trait with variable penetrance. The estimated spontaneous mutation rate for TSC varies from 66% to 86%, depending in part on the completeness of investigation of the extended family. Two genes are responsible for TSC: TSC1, coding for hamartin at chromosome 9q34.3; and TSC2, coding for tuberin adjacent to the gene for adult polycystic kidney disease at chromosome 16p13.3. The clinical features of TSC1 and TSC2 overlap, since the two gene products form a single functional unit that is an upstream modulator in the mTOR (mammalian target of rapamycin) signaling pathway. Both gene products down-regulate small G-protein Ras-homologue enriched in brain (RHEB) activity in this pathway. However, genotype-phenotype studies indicate that individuals with a TSC2 mutation tend to have more severe disease, and the frequency of TSC2 mutations is greater among individuals with spontaneous mutations (Sancak et al., 2005). Multiple mutation types exist in different regions of each gene, and even individuals with identical genetic mutations can have different phenotypes. Molecular diagnostic testing—including prenatal testing—has been available since the early 2000s, and a disease-causing mutation is identified in 85% meeting clinical criteria. Individuals with no mutation identified may have distinctive clinical features. Large genomic deletions and rearrangements are more common in the TSC2 gene compared to TSC1, and more mutations have been identified for TSC2 versus TSC1. TSC2 mutations appear more commonly than TSC1 in patients with subependymal nodules, mental retardation, renal angiomyolipomas, and retinal phakomas. Mental retardation and other neuropsychiatric involvement are more likely in individuals with TSC2 versus TSC1 mutation (Au et al., 2007).
Cutaneous Features
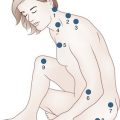
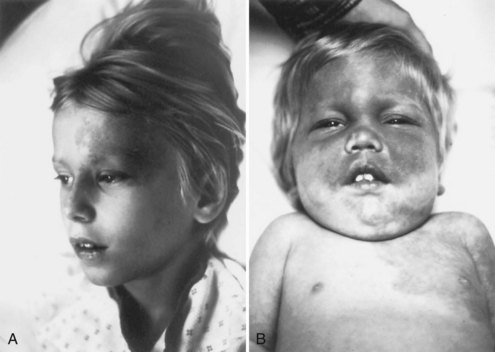
The cutaneous lesions of TSC include hypomelanotic macules, the shagreen patch, ungual fibromas, and facial angiofibromas. Hypomelanotic macules (ash leaf spots) occur in up to 90% of affected individuals (Fig. 65.1). The lesions usually are present at birth but may be seen in the newborn only with an ultraviolet light. Other pigmentary abnormalities include confetti lesions (areas with stippled hypopigmentation, typically on the extremities) and poliosis (a white patch or forelock) of the scalp, hair, or eyelids (Fig. 65.2, D). Hypomelanotic macules are common in normal individuals (Table 65.1), but three or more hypomelanotic papules is a major diagnostic criterion for TSC.
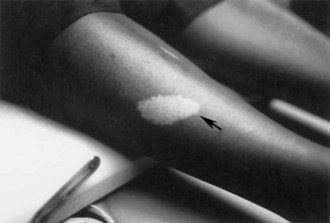
Fig. 65.1 A hypomelanotic macule (ash leaf spot) (arrow) from the leg of a patient with tuberous sclerosis.
(Reprinted with permission from Weiner, D.M., Ewalt, D.H., Roach, E.S., et al., 1998. The tuberous sclerosis complex: a comprehensive review. J Am Coll Surg 187, 548-561.)
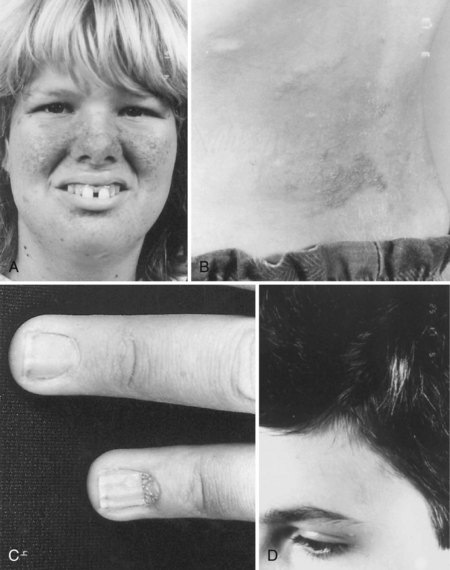
Table 65.1 Frequency of Lesions in Individuals with Tuberous Sclerosis Versus Other Individuals
| Lesion | Tuberous Sclerosis Complex | Other Individuals |
|---|---|---|
| Hypomelanotic macules | Occur in over 95% of tuberous sclerosis (TSC) patients, often with many lesions11 | Occur in up to 5% of the population (but usually fewer than three lesions per person)12 |
| Facial angiofibromas | Eventually seen in 75% but less often in children11 | Seen in individuals with multiple endocrine neoplasia type 1 and in a few sporadic families13 |
| Shagreen patch | Up to 48%11 | Occasional |
| Ungual fibromas | Seen in 15% but often not until develop in adulthood11 | Occasionally sporadic or after nail trauma (but typically one lesion)14 |
| Rhabdomyomas | One or more tumors seen in 47%-65% but much more common below 2 years Up to 51% of patients with rhabdomyomas have TSC15-17 |
In 14%-49% of rhabdomyoma patients, there are no other signs of TSC16 |
| Renal angiomyolipoma (AML) | Often multiple AML occur in up to 80% of TSC patients by age 1018 | Sporadic AML occur but are typically solitary |
| Renal cysts | Polycystic kidneys occur in 3%-5% of TSC patients Smaller numbers of renal cysts are present in 15%-20%18 |
There are both dominant and recessive polycystic kidney diseases A few cysts are frequent sporadic findings in adults |
| Cortical dysplasia/tubers | 90%-95% and usually multiple lesions are present (magnetic resonance imaging yields highest detection rate)25 | Sporadic cortical dysplasia (typically 1 lesion) is common among individuals who have epilepsy not due to TSC |
| Subependymal nodules | 83%-93%2,25 | Rare, especially if calcified |
| Subependymal giant cell tumors | Up to 15% (using radiographic criteria)19 | Rare in the absence of TSC |
From Roach, E.S., Sparagana, S.P., 2010. Diagnostic criteria for tuberous sclerosis complex. In: Kwiatkowski, D.J., Whittemore, V.H., Thiele, E.A. (Eds.), Tuberous Sclerosis Complex: Genes, Clinical Features, and Therapeutics. Weinheim, Wiley-VCH Verlag GmbH & Co., pp. 21-25. Used with permission.
Facial angiofibromas (adenoma sebaceum) consist of vascular and connective tissue elements. Although considered specific for TSC, they are found in only three-fourths of affected individuals and often appear several years after other means have established the diagnosis. The lesions typically become apparent during the preschool years as a few small red macules on the malar region; they gradually become papular, larger, and more numerous, sometimes extending down the nasolabial folds or onto the chin (see Fig. 65.2, A). Laser therapy may be a useful therapeutic intervention and particularly helpful in early childhood or just prior to puberty, before a time when growth may be rapid and more aggressive interventions warranted.
The shagreen patch most often is found on the back or flank area; it is an irregularly shaped, slightly raised, or textured skin lesion (see Fig. 65.2, B). Only 20% to 30% of patients with TSC have one patch, which may not be seen in young children. Usually considered specific for TSC, ungual fibromas are nodular or fleshy lesions that arise adjacent to (periungual) or underneath (subungual) the nails (see Fig. 65.2, C). These can occur as a single lesion after trauma in normal individuals. Ungual fibromas occur in only 15% to 20% of patients with TSC, more likely in adolescents or adults.
Neurological Features
Most patients with mental retardation have epilepsy, but many have seizures and normal intelligence. The number of subependymal lesions does not correlate with the clinical severity of TSC, but patients with MRI evidence of numerous cortical lesions tend to have more cognitive impairment and more difficulty with seizure control. The most abnormal regions seen on MRI tend to coincide with focal abnormalities of the electroencephalogram (EEG). The likelihood of intellectual disability in patients with TSC probably is overestimated, and the severity of intellectual dysfunction ranges from borderline to profound mental retardation. However, in addition to intellectual disability, many children with TSC have serious behavioral disorders. Autistic behavior, hyperkinesis, aggressiveness, and frank psychosis sometimes occur, either as isolated problems or in combination with epilepsy or intellectual deficit. The prevalence of autistic spectrum disorders is 25% to 50% and equal between boys and girls. Behavioral problems are frequent and independent of intellectual ability (de Vries, 2010). Mood disorders also are increased.
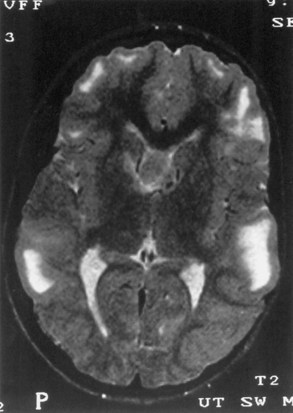
Computed tomography (CT) best demonstrates the calcified subependymal nodules that characterize TSC (Fig. 65.3). CT sometimes shows superficial cerebral lesions, but they are far more obvious with T2-weighted MRI (Fig. 65.4). T2-weighted scans show evidence of abnormal neuronal migration in some patients as high-signal linear lesions running perpendicular to the cortex. SENs along the ventricular surface give the characteristic appearance of candle guttering. More than one-fourth of patients with TSC show cerebellar anomalies.
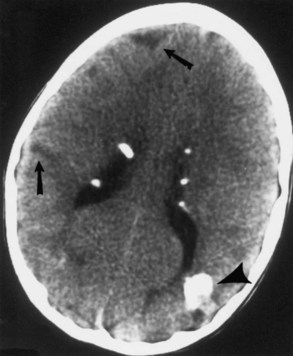
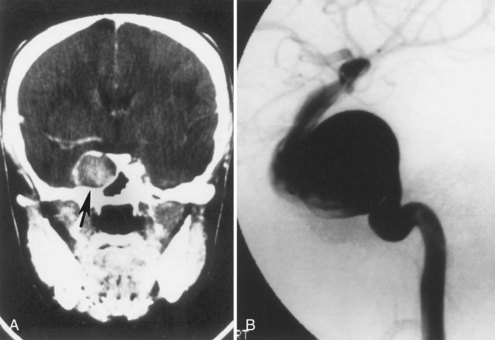
Subependymal giant-cell astrocytomas (SEGAs) develop in 6% to 14% of patients with TSC. Unlike the more common cortical tubers and SENs, giant-cell astrocytomas can enlarge (Fig. 65.5) and cause symptoms of increased intracranial pressure, particularly if extension into the lateral ventricles creates an obstructive hydrocephalus. Clinical features include new focal neurological deficits, increased intracranial pressure, unexplained behavior change, or deterioration of seizure control. Acute or subacute onset of neurological dysfunction may result from sudden obstruction of the ventricular system by an intraventricular SEGA. Rarely, acute deterioration occurs because of hemorrhage into the tumor itself.
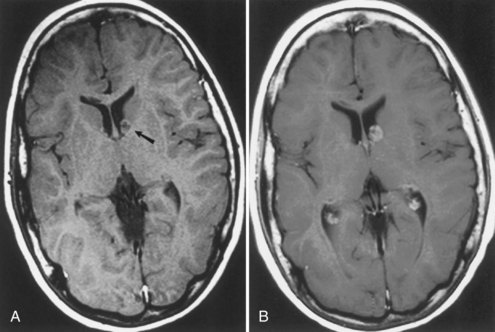
Giant-cell tumors are usually benign but locally invasive, and early surgery can be curative. Identification of an enlarging SEGA before the onset of symptoms of increased intracranial pressure or appearance of new neurological deficits is ideal. Periodic screening to identify SEGA may improve surgical outcome. Recent work suggests that rapamycin inhibits the growth of SEGAs (Franz et al., 2006).
Retinal Features
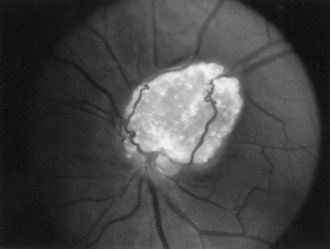
The frequency of retinal hamartomas in TSC varies from almost negligible to 87% of patients, probably reflecting the expertise and technique of the examiner. Pupillary dilatation and indirect ophthalmoscopy are important, particularly in uncooperative children. Findings vary from classic mulberry lesions adjacent to the optic disc (Fig. 65.6) to plaque-like hamartoma or depigmented retinal lesions. Most retinal lesions are clinically insignificant, but some patients have visual impairment caused by large macular lesions, and very few patients have visual loss caused by retinal detachment, vitreous hemorrhage, or hamartoma enlargement. Occasionally, patients have a pigmentary defect of the iris. Funduscopic examination is valuable at the time of diagnosis, to monitor existing abnormalities or evaluate for new symptoms. The histological features of retinal hamartomas, subependymal nodules, and subependymal giant-cell astrocytomas are similar.
Systemic Features
Cardiac
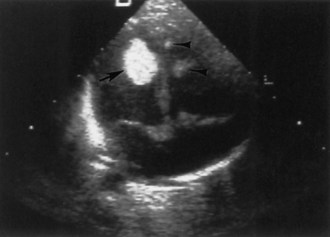
Approximately two-thirds of patients with TSC have a cardiac rhabdomyoma, but few demonstrate clinical symptoms. Cardiac rhabdomyomas are hamartomas, tend to be multiple, and involute with time. These lesions sometimes are evident on prenatal ultrasound testing (Fig. 65.7)—usually after 24 weeks gestational age—and most who develop cardiac dysfunction present soon after birth with heart failure. A few children later develop cardiac arrhythmias or cerebral thromboembolism from the rhabdomyomas. The cause of congestive heart failure is either by obstruction of blood flow by intraluminal tumor or by lack of sufficient normal myocardium to maintain perfusion. Some patients stabilize after medical treatment with digoxin and diuretics and eventually improve; others require surgery. Echocardiography and electrocardiogram establish the diagnosis. Arterial aneurysms can occur. For existing rhabdomyomas, perform surveillance studies every 6 to 12 months until stabilization or involution occurs. The size of these lesions may increase with hormone exposure—a consideration in the neonate, pubertal individual, and child treated with ACTH for infantile spasms.
Renal
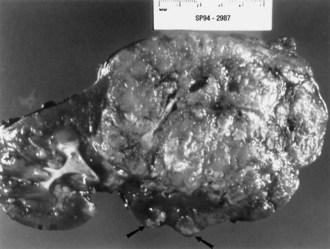
Renal angiomyolipomas occur in up to three-fourths of patients with TSC, usually presenting by 10 years of age. Most of these lesions are histologically benign tumors with varying amounts of vascular tissue, fat, and smooth muscle (Fig. 65.8). Bilateral tumors or multiple tumors in a kidney are common. The prevalence and size of renal tumors increase with age, and tumors larger than 4 cm are much more likely to become symptomatic than smaller tumors. Renal cell carcinoma or other malignancies are less common but affect TSC patient at younger ages than the general population. Coalescing angiomyolipomata can contribute to end-stage renal disease. Endovascular embolization of the larger renal angiomyolipomata prevents hemorrhage and other complications (Ewalt et al., 2005). Rapamycin limits the growth of these tumors, at least transiently.
Neurofibromatosis
A mutation of the NF2 gene on chromosome 22 causes NF2. The NF2 protein product is schwannomin or merlin. The NF2 gene suppresses tumor function. Dysfunction of the NF2 gene accounts for the occurrence of multiple central nervous system (CNS) tumors in patients with NF2. The NF2 gene has several different mutations. The clinical severity may be related to the nature of the NF2 mutation; missense mutations that allow some protein function tend to produce milder clinical forms, whereas frameshift and nonsense mutations that produce stop codons preventing the production of any protein often cause severe disease (Evans, 2004).
If several characteristics are present, the diagnosis of either NF1 or NF2 is obvious, especially when another family member is affected. The diagnosis is difficult when the clinical features are atypical and the family history is negative. Very young children may have fewer apparent lesions, making definitive diagnosis difficult. Diagnostic criteria (Box 65.2) help to resolve some of these questionable cases, but specific gene testing is replacing the use of clinical criteria. Screening for the NF1 gene is technically difficult because the gene is large and several different mutations are causative. Commercially available studies have a 30% false-negative rate. Some suggest the diagnosis of NF2 based on multiple meningiomas or nonvestibular schwannomas even without family history or classic bilateral vestibular schwannomas.
Box 65.2
Diagnostic Criteria for Neurofibromatosis
Neurofibromatosis Type 1 (Any Two or More)
Six or more café-au-lait lesions more than 5 mm in diameter before puberty and more than 15 mm in diameter afterward
Freckling in the axillary or inguinal areas
Two or more neurofibromas or one plexiform neurofibroma
A first-degree relative with neurofibromatosis type 1
A characteristic bony lesion (sphenoid dysplasia, thinning of the cortex of long bones, with or without pseudoarthrosis)
Neurofibromatosis Type 2
Bilateral eighth nerve tumor (shown by magnetic resonance imaging, computed tomography, or histological confirmation)
A first-degree relative with neurofibromatosis type 2 and a unilateral eighth nerve tumor
A first-degree relative with neurofibromatosis type 2 and any two of the following lesions: neurofibroma, meningioma, schwannoma, glioma, or juvenile posterior subcapsular lenticular opacity
Data from Neurofibromatosis. Conference statement, 1988. National Institutes of Health Consensus Development Conference. Arch Neurol 45, 575-578.
Cutaneous Features of Neurofibromatosis Type 1
Cutaneous lesions of NF1 (Fig. 65.9) include café-au-lait spots, subcutaneous neurofibromas, plexiform neurofibromas, and axillary freckling. Café-au-lait spots are flat, light to medium brown areas that vary in shape and size. They typically are present at birth but increase in size and number during the first few years of life. Later in childhood, skin freckling, 1 to 3 mm in diameter, often occurs symmetrically in the axillae (Crowe sign) and other intertriginous regions. Most children with 6 or more café au lait spots as their only diagnostic criterion will go on to meet diagnostic criteria, usually by age 6.
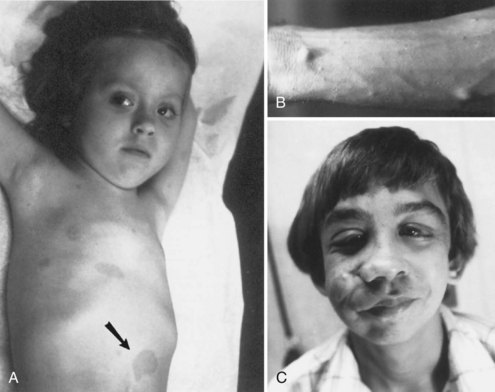
Neurofibromas are benign tumors arising from peripheral nerves (see Fig. 65.9, B). These tumors are composed predominantly of Schwann cells and fibroblasts but contain endothelial, pericytes, and mast cell components. Neurofibromas can develop at any time; their size and number often increase after puberty.
Plexiform neurofibromas often occur on the face and can cause substantial deformity (see Fig. 65.9, C). Patients with plexiform tumors of the head, face, or neck and those who presented before 10 years of age are more likely to do poorly (Needle et al., 1997). Plexiform neurofibromas have a 5% to13% lifetime risk of malignant degeneration into malignant peripheral nerve sheath tumors. Malignant peripheral nerve sheath tumors (MPNST) carry poor 5-year survival rates despite treatment with surgery, chemotherapy, and radiation. PNFs and MPNST are difficult to distinguish radiographically and sometimes even pathologically.
Systemic Features of Neurofibromatosis Type 1
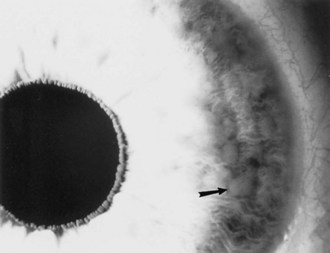
Lisch nodules are pigmented iris hamartomas (Fig. 65.10). They are pathognomonic for NF1. Lisch nodules do not cause symptoms; their significance lies in their implications for the diagnosis of NF1. Lisch nodules are often not apparent during early childhood, so their absence does not exclude the diagnosis of NF1. Rarely, children with NF1 have retinal hamartomas, but these usually remain asymptomatic.
Neurological Features in Neurofibromatosis Type 1
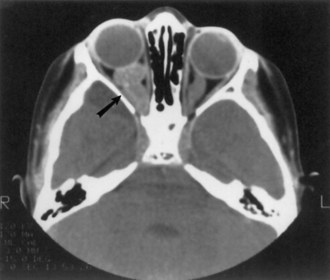
Optic nerve glioma (Fig. 65.11) is the most common CNS tumor caused by NF1. Approximately 15% of patients with NF1 have unilateral or bilateral optic glioma. The growth rate of these tumors varies, but they tend to behave less aggressively in patients with NF1 than those without NF1. When symptomatic, the presenting features are optic atrophy, progressive vision loss, pain, or proptosis. Precocious puberty is a common presenting feature of chiasmatic optic nerve tumors in children with NF1. Management options include observation with serial brain MRI or treatment with radiation, chemotherapy, or excision. Radiation is less favored, especially given possible exacerbation of vasculopathy in this population.
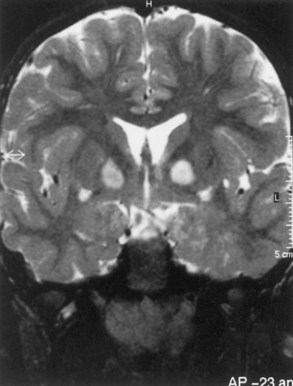
Macrocephaly is seen in half of NF1 patients, typically attributable to megalencephaly related to increases in white-matter volume. Approximately 60% to 78% of patients with NF1 have increased signal lesions within the basal ganglia, thalamus, brainstem, and cerebellum on T2-weighted MRIs (Fig. 65.12). These areas are not routinely visible with CT. The origin and significance of these radiographic lesions are unclear, and they are referred to at times as unidentified bright objects (UBOs). Whether these MRI lesions correlate with the likelihood of cognitive impairment still is debatable; radiographic findings do not correlate with neurological deficits. Patients with NF1 tend to have full-scale intelligence quotient (IQ) scores within the low-normal range and to exhibit behavioral problems. These symptoms may be related to vascular changes in myelin sheath. Deep gray-matter radiological findings tend to decrease with time, while cortical and subcortical findings do not decrease or increase.
Clinical Features of Neurofibromatosis Type 2
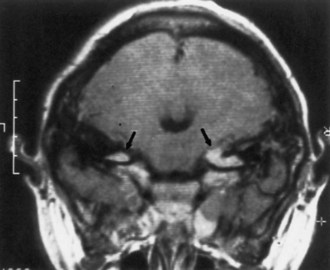
Most patients who meet established diagnostic criteria for NF2 (see Table 65.3) eventually develop bilateral vestibular schwannomas, previously termed acoustic neuromas (Fig. 65.13). Symptoms of NF2 typically develop in adolescence or early adulthood but can begin in childhood. Common complaints with large acoustic tumors include hearing loss, tinnitus, vertigo, facial weakness, poor balance, and headache. Unilateral hearing loss is relatively common in the early stages. Consider screening with annual auditory brainstem responses or brain MRI.
Merlin is a novel regulator of TSC/mTORC1 signaling, such that rapamycin is being evaluated in the management of NF2 tumors (James et al., 2009).
Sturge-Weber Syndrome
The characteristic features of Sturge-Weber syndrome (SWS) are a facial cutaneous angioma (port-wine nevus) and an associated ipsilateral leptomeningeal and brain angioma. In addition to the facial nevus, other findings include mental retardation, seizures, contralateral hemiparesis and hemiatrophy, and homonymous hemianopia (Bodensteiner and Roach, 2010). However, the clinical features are variable, and individuals with cutaneous lesions and seizures but with normal intelligence and no focal neurological deficits are common. The syndrome occurs sporadically and in all races.
Cutaneous Features
The nevus typically involves the forehead and upper eyelid but also may involve both sides of the face and extend onto the trunk and limbs (Fig. 65.14). Nevi that involve only the trunk, or facial nevi that spare the upper face, rarely are associated with intracranial angioma. The facial angioma is usually obvious at birth; it may thicken over time and develop a nodular texture. Reactive hypertrophy of adjacent bone and connective tissue may occur. Some children have the characteristic neurological and radiographic features of SWS yet have no skin lesions. More frequently, the typical cutaneous and ophthalmic findings are present without clinical or radiographic evidence of an intracranial lesion. Only 10% to 20% of children with a port-wine nevus of the forehead have a leptomeningeal angioma. Although the leptomeningeal angioma is typically ipsilateral to a unilateral facial nevus, bilateral brain lesions occur in at least 15% of patients, including some with a unilateral cutaneous nevus.
Diagnostic Studies
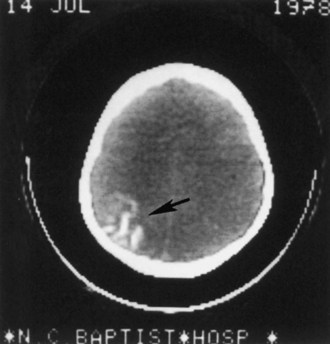
Most children with a facial port-wine nevus do not have an intracranial angioma. Neuroimaging studies help distinguish children with SWS from those with isolated cutaneous lesions. Although gyral calcification is a typical feature of SWS, the tram-track appearance first described on standard radiographs is uncommon and is almost never present in neonates. CT shows intracranial calcification much earlier (Fig. 65.15) than standard skull radiographs Extensive cerebral atrophy is apparent even with CT, but MRI more readily shows subtle atrophy.
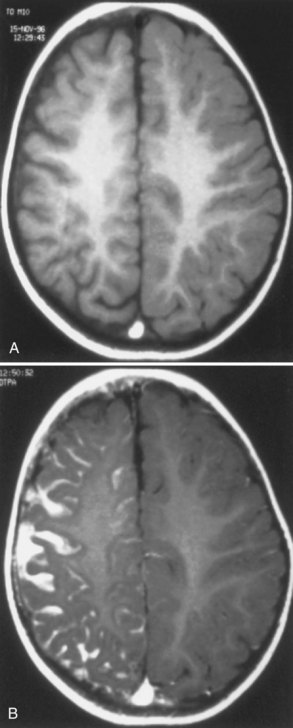
MRI with gadolinium contrast (Fig. 65.16) effectively demonstrates the abnormal intracranial vessels in patients with SWS. Functional imaging with positron emission tomography (PET) demonstrates reduced metabolism of the brain adjacent to the leptomeningeal lesion. However, patients with recent-onset seizures may have increased cerebral metabolism near the lesion. Single-photon emission computed tomography (SPECT) shows reduced perfusion of the affected brain. Both PET and SPECT often reveal vascular changes extending well beyond the area of abnormality depicted by CT (Maria et al., 1998). Although functional imaging is not necessary for all patients, it may help initially to establish a diagnosis and may help localize the lesion before surgery.
Treatment
Generally, the more extensive the intracranial lesion, the more difficult it is to control seizures with medication. Resection of a localized brain vascular lesion or hemispherectomy can often improve seizure control and may promote better intellectual development (Bourgeois et al., 2007). Despite general agreement on the efficacy of surgical resection, debate remains concerning patient selection and the timing of surgery. Almost one patient in five has bilateral cerebral lesions, limiting the surgical options unless one hemisphere is clearly responsible for most of the seizures. Often the patient selected for surgery is one with refractory seizures, clinical dysfunction (e.g., hemiparesis, hemianopia) of the area selected for resection, and failure to respond to an adequate trial of anticonvulsants. Patients with less extensive lesions should have a limited resection rather than a complete hemispherectomy. The limited resection preserves as much normal brain as possible, even at the risk of having to do another operation later. Corpus callosotomy is useful for patients with refractory tonic or atonic seizures and extensive disease. In effect, the surgical considerations in children with SWS are similar to those used with other epileptic patients.
Von Hippel-Lindau Syndrome
Neurological Features
The initial symptoms of VHL usually arise from effects of the vascular anomalies in the CNS. In the CNS, the most common site of hemangioblastomas is the cerebellum in approximately half of patients (Fig. 65.17), followed by spinal and medullary sites. Cerebral hemangioblastomas are present in less than 5% of patients with VHL. The cerebellar hemispheres are affected far more frequently than the cerebellar vermis. Cerebellar hemangioblastomas may begin in the second decade of life.
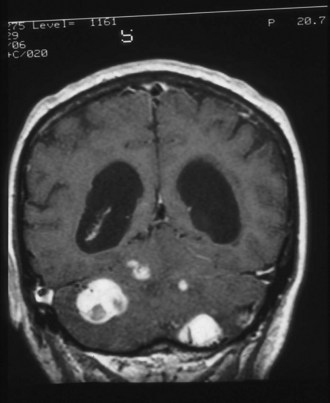
Endolymphatic sac tumors occur in 10% to 15% of these individuals. Sometimes they are bilateral. Presenting symptoms can be abrupt change in hearing accompanying hemorrhage or vertigo and tinnitus (Butman, 2008).
Molecular Genetics
Confirming initial suspicions, the VHL gene is a tumor-suppressor gene located on chromosome 3p25-26. A mutation in the VHL gene also appears in many sporadic clear-cell renal carcinomas that present later in life as compared to those with VHL disease. It demonstrates a role in the function of hypoxia-induced factor, HIF2α. This regulation contributes to increased vascularization and up-regulation of proangiogenic genes and other oxygen-sensitive genes via hypoxia response elements (HRE). These genes include vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), transforming growth factor alpha (TGF-α), glucose transporter-1 (GLUT1), carbonic anhydrase IX, and erythropoietin (EPO), among others. In particular, VEGF is important in angiogenesis; its levels in ocular fluid of patients is significantly higher than in unaffected subjects. Recognition of these affected genes has focused trials of certain therapies (Clarkson and Cookson, 2008).
Hundreds of known mutations exist. Despite complex genotype-phenotype relationships, some clinical correlations are possible. Missense mutations in this gene are associated with pheochromocytoma, whereas nonsense, frameshift, and splice-site mutations as well as deletions predominate in families without pheochromocytomas. Microdeletions and microinsertions, nonsense mutations, or deletions appear in 56% of families with VHL type 1, whereas missense mutations account for 96% of those responsible for VHL type 2 (Chen et al., 1995). Specific mutations in codon 238 account for 43% of the mutations responsible for VHL type 2, and one group of patients (type 2C) appears to be at low risk for any feature of VHL except pheochromocytoma.
Treatment
Careful screening (Box 65.3) is the most important aspect of management of VHL. Screening is mandatory for all first-degree relatives in a family with VHL or pheochromocytoma. Other indications for clinical screening include pancreatic cysts, multiple or bilateral renal cell tumors, retinal hemangiomas, and cerebellar hemangioblastomas. Availability of molecular analysis for the VHL gene reduces the number of asymptomatic relatives requiring surveillance; only relatives who have inherited the VHL mutation need annual screening.
Box 65.3 Cambridge Screening Protocol for von Hippel-Lindau Disease
Affected Patient
Annual physical examination and urine testing
Annual direct and indirect ophthalmoscopy with fluorescein angioscopy or angiography
Cranial magnetic resonance imaging (MRI) or computed tomography (CT) every 3 years to age 50 and every 5 years thereafter
Annual renal ultrasound, with abdominal CT scan every 3 years (more frequently if multiple renal cysts are discovered)
At-Risk Relative
Annual physical examination and urine testing
Annual direct and indirect ophthalmoscopy from age 5 years
Annual fluorescein angioscopy or angiography from age 10 years until age 60
Cranial MRI or CT every 3 years from age 15-40 and every 5 years until age 60
Annual renal ultrasound, with abdominal CT scan every 3 years from age 20-65 years
Hereditary Hemorrhagic Telangiectasia
Treatment
As with most of the neurocutaneous syndromes, treatment of HHT is limited to the management of complications. Because many of the neurological complications of the disease arise secondary to a pulmonary AVF, resection or embolization of the fistula is essential. Treatment of cerebral AVMs is embolization, excision, or radiosurgery. Periodic transfusion and chronic iron administration may be necessary. To reduce risk of brain abscess in cases of undiagnosed pulmonary AVM, antibiotic prophylaxis is a recommendation prior to dental procedures (Faughnan et al., 2011).
Hypomelanosis of Ito
Cutaneous Features
The skin findings are distinctive and in fact are the only constant feature of HI. Hypopigmented whorls, streaks, and patches are present at birth and tend to follow Blaschko lines (Fig. 65.18), pathways demarcating embryonic skin development. In HI, the hypopigmented skin lesions are usually multiple, involve several body segments, and may be unilateral or bilateral. They may be observable at birth but commonly develop in infancy, depending on the degree of skin pigmentation. Woods lamp examination may enable detection of hypopigmented lesions. The degree or distribution of skin depigmentation does not appear to correlate with either the severity of neurological symptoms or associated organ pathology. The hypopigmented lesions follow Blaschko lines in two-thirds of patients and are patchy in others. Other skin findings in patients with HI include café-au-lait spots, cutis marmorata, aplasia cutis, nevus of Ota, trichorrhexis, focal hypertrichosis, and nail dystrophy. Electron microscopy of the hypopigmented lesions consistently shows a marked reduction of melanocytes (Cavallari et al., 1996). In the proximity of preserved melanocytes, the basal keratinocytes contain a nearly normal content of melanosomes. Depigmented areas contain an increased number of Langerhans cells.
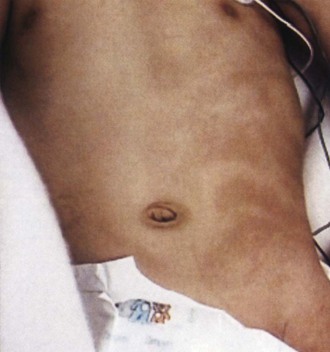
Neurological Features
The frequency of neurological abnormalities in patients with typical skin lesions ranges from 50% to 80% (Nehal et al., 1996). Seizures and mental retardation are the most common neurological abnormalities. Approximately half of patients with HI have seizures, usually with onset in the first year of life. Focal seizures are most common.
Incontinentia Pigmenti
Genetics
Transmission is X-linked dominant, and nuclear factor kappa B (NF-κB) essential modulator (NEMO) is the causative gene. Deletion in exons 4 to 10 is the responsible mutation in 80% to 90% of patients (Nelson, 2006). This protein is involved in prevention of apoptosis. The mutation is expected to be lethal in males; however, males with somatic mosaicism or an additional X chromosome, as in Klinefelter syndrome, have survived. These individuals demonstrate immunodeficiency and ectodermal dysplasia rather than the characteristic skin findings described.
Ataxia-Telangiectasia
Neurological Features
Ataxia, the first manifestation of AT, appears when the child learns to walk in the second year of life. Truncal ataxia predominates early in the course of the disorder, affecting sitting, balance, and gait. Muscle strength is normal, and attainment of early gross motor milestones is usually on time. The ataxia is slowly progressive, and children typically require a wheelchair by the age of 12 years. As the child matures, limb ataxia, intention tremor, and segmental myoclonus become apparent. Choreoathetosis may be difficult to distinguish from dysmetria and intention tremor, but it may dominate the clinical picture in older children. At times, the choreoathetosis may resemble segmental myoclonus of the limbs or trunk. Progressive dystonia of the fingers may appear in the second and third decades of life. Axial muscles are affected, and a stooped posture gradually develops. Progressive dysarthria is present. (See also Chapter 72.)
Laboratory Diagnosis
Useful laboratory tests in the diagnosis of AT include serum α-fetoprotein, immunoglobulins, and cellular radiosensitivity tests. Nearly all patients with AT have an elevated α-fetoprotein level, which is utilized as a screening diagnostic test. Approximately 80% have decreased serum immunoglobulin (Ig)A, IgE, or IgG, especially the IgG2 subclass. Characteristic cellular features are reduced lifespan in culture, cytoskeletal abnormalities, chromosomal instability, hypersensitivity to ionizing radiation and radiomimetic agents, defective radiation-induced checkpoints at the G1, S, and G2 phases of the cell cycle, and defects in signal transduction pathways (Rotman and Shiloh, 1997).
Epidermal Nevus Syndrome
The term epidermal nevus syndrome (ENS) encompasses several disorders that have in common an epidermal nevus and neurological manifestations such as seizures or hemimegalencephaly. The syndrome name represents the predominant cell type of the nevus; for example, nevus verrucosus (keratinocytes), nevus comedonicus (hair follicles), and nevus sebaceous (sebaceous glands). Several subtypes of ENS exist and should be differentiated from one another: nevus sebaceous syndrome (Schimmelpenning-Feuerstein-Mims syndrome), Proteus syndrome, CHILD (congenital hemidysplasia with ichthyosiform nevus and limb defects) syndrome, Becker nevus associated with extracutaneous involvement (pigmented hairy epidermal nevus syndrome), nevus comedonicus syndrome, and phakomatosis pigmentokeratotica (Happle, 1995). Terms such as Schimmelpenning syndrome, organoid nevus syndrome, and Jadassohn nevus phakomatosis describe combinations of neurological findings and sebaceous nevi. It is probably best to consider ENS a heterogeneous group of disorders characterized by epidermal and adnexal hamartomas and other organ system involvement.
Neurocutaneous Melanosis
Although the precise pathogenesis is not well understood, a disorder involving neural crest cell differentiation and melanocyte embryogenesis is suspected. The prominent involvement of the leptomeninges and skin over the spine supports the suggestion that the primary defect is abnormal migration of nevus cell precursors, although the embryological origin of nevus cells has not been determined. Alternatively, melanin-producing cells may be produced in excessive numbers. It has also been speculated that nevi located over the spine result from an error early in nevus cell migration or differentiation, whereas nevi are restricted to the extremities if the error occurs later in development (Pavlidou et al., 2008).
Cutaneous Features
The characteristic lesions are dark to light brown hairy nevi present at birth (Fig. 65.19). Multiple small nevi (satellite nevi) usually are present around one giant nevus that most commonly appears on the lower trunk and perineal area (swimming trunk nevus). A giant nevus is absent in 34% of patients with NCM. Approximately one-third of patients have a large nevus over the upper back (cape nevus). The giant nevi may fade over time, but satellite nevi continue to appear during the first few years of life.
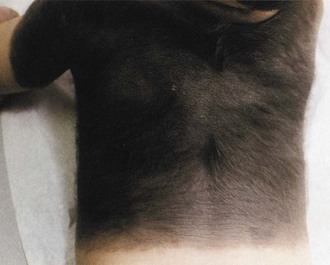
Fig. 65.19 Large, dark, hairy nevus covering most of the back of an infant with neurocutaneous melanosis.
Biopsy of a congenital nevus reveals extension of the nevus cells into the deep dermis or even the subcutis between collagen bundles and around nerves, hair follicles, and blood vessels. Nevus cells tend to form cords or nests. Sheets of nevomelanocytes in the dermis may display a few mitoses and large atypical cells positive for S100 and HMB45 antibodies and formaldehyde-induced green-specific fluorescence. The occurrence of atypical mitoses in the dermis may constitute an early stage of malignant melanoma (Sasaki et al., 1996). The greatest risks in NCM are the high incidence of malignant transformation of melanotic cells and spinal and intracranial pathology.
Neurological Features
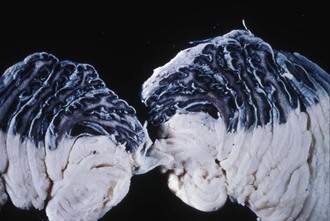
Neurological symptoms may result from leptomeningeal melanosis, intracranial melanoma, or intracerebral or subarachnoid hemorrhage. Malformations of the vertebral column, spine, and brain also may impair neurological function. The median age of neurological complications is 2 years, but infants may be affected (DeDavid et al., 1996). Leptomeningeal melanosis is probably the most common cause of neurological symptoms, especially in children. This tends to occur at the base of the brain along the interpeduncular fossa, ventral brainstem, upper cervical cord, and ventral surface of the lumbosacral cord. Marked leptomeningeal melanosis (Fig. 65.20) is present in the vast majority of patients with NCM and associated with interruption of cerebrospinal fluid (CSF) flow. This leads to hydrocephalus and increased intracranial pressure with typical symptoms of irritability, vomiting, seizures, and papilledema. In infants, symptoms may include rapidly increasing head circumference or tense anterior fontanel. Cranial nerve deficits such as limitation of upgaze and lateral gaze are common. Myelopathy occurs when leptomeningeal proliferation affects the spinal cord or spinal nerves.
Neuroimaging
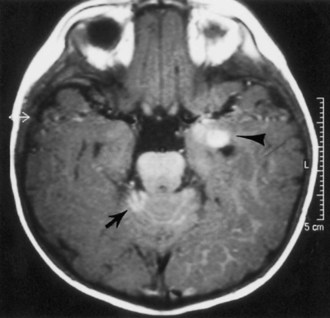
Approximately half of neurologically asymptomatic children with NCM have abnormal cranial neuroimaging study results. Cranial MRI demonstrates lesions with T1 shortening in the cerebellum, in the anterior temporal lobe (especially the amygdala), and along the basilar meninges (Fig. 65.21). Some of these lesions also show T2 shortening. The pons, medulla, thalami, and base of the frontal lobe often are affected. Gadolinium-enhanced MRIs rarely may show enhancement of the pia-arachnoid. In one study, all five children with neurological symptoms of increased intracranial pressure showed leptomeningeal thickening and enhancement. Conversely, asymptomatic children never showed leptomeningeal thickening. Inferior vermian hypoplasia and Dandy-Walker malformation have been reported. Spinal MRI study results are usually normal.
Ehlers-Danlos Syndrome
At least 10 subtypes of Ehlers-Danlos syndrome (EDS) exist, defined by the clinical features, inheritance pattern, and even specific molecular defects. Together these syndromes are characterized by fragile or hyperelastic skin (Fig. 65.22), hyperextensible joints, vascular lesions, easy bruising, poor wound healing, and excessive scarring. Some patients develop peripheral neuropathy caused by lax ligaments, and others may present with neonatal hypotonia or weakness. Neuromuscular kyphoscoliosis can develop. However, vascular lesions such as aneurysm (Fig. 65.23) and arterial dissection are the most serious threat to the nervous system. (See also Chapter 51A.)
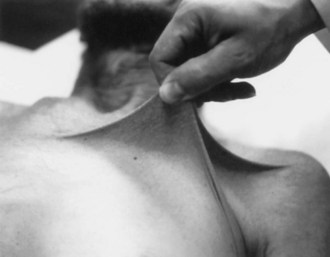
More than 80% of EDS patients have type I, II, or III, and the other subtypes are individually uncommon. Type IV most often leads to neurovascular complications, and its prevalence is 1 in 50,000 to 500,000. Often, a delay in the diagnosis of type IV EDS occurs because of a decreased incidence of hyperelastic skin or hyperextensible joints compared to other types. Transmission of all familial type IV EDS cases with a documented abnormality of type III collagen is autosomal dominant. Various defects of the COL3A1 gene (which codes for the α1 chain of type III collagen) on chromosome 2 have been identified (Schwartze et al., 1997). Mutations of this gene are rare in patients with aneurysm without EDS type IV (Hamano et al., 1998).
Cerebrotendinous Xanthomatosis
Cerebrotendinous xanthomatosis (CTX) is an autosomal recessive disorder of bile acid synthesis characterized by tendon xanthomas, cataracts, and progressive neurological deterioration (Box 65.4). The underlying defect consists of the enzyme, sterol 27-hydroxylase, whose gene (CYP27A1) is located on chromosome 2q. The enzyme deficiency leads to deposits of cholesterol and cholestanol, a metabolic derivative of cholesterol, in virtually every tissue, particularly the Achilles tendons, brain, and lungs. Bile acid production decreases markedly, which leads to reduced chenodeoxycholic acid (CDCA) concentration in bile. Excretion of bile acid precursors increases in bile and urine. Serum cholesterol levels are typically not elevated in CTX syndrome.
Neurological Features
Personality changes and decline in school performance may be the earliest neurological manifestations of this syndrome. Progressive loss of cognitive function typically begins in childhood, but some patients remain intellectually normal for many years. EEG shows nonspecific characteristics of metabolic encephalopathy such as slowing. Seizures may occur. Ataxia with gait disturbance, dysmetria, nystagmus, and dysarthria are common. Psychosis with auditory hallucinations, paranoid ideation, and catatonia occur rarely, but examination for cataracts and tendon xanthomas should be included in the evaluation of patients with new-onset psychosis. Parkinsonism may be the only neurological symptom. Cranial MRI typically shows involvement of the dentate nuclei (Gallus et al., 2006). Other findings include cerebral and cerebellar atrophy and diffusely abnormal white matter, presumably reflecting sterol infiltration with demyelination. Focal lesions of the cerebral white matter and globus pallidus are sometimes demonstrable on MRI.
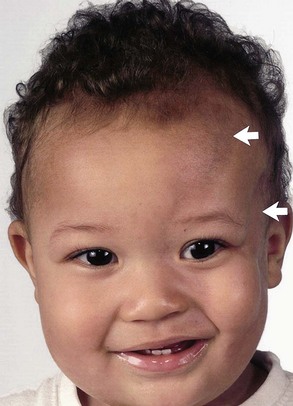
Progressive Facial Hemiatrophy
Progressive facial hemiatrophy (Parry-Romberg syndrome) occurs sporadically. The relationship of this disorder to coup de sabre, morphea, and linear scleroderma still is debated (Peterson et al., 1996). Traditionally, progressive facial hemiatrophy involves the upper cranium, whereas coup de sabre tends to affect the lower face as well. Scleroderma and morphea affect other parts of the body. However, understanding of pathogenesis is poor, and they may prove to have a similar origin. An arbitrary distinction based on the anatomical distribution does have at least one practical use: as a rule, only patients whose upper face and head are affected are likely to develop cerebral complications.
Clinical Features
Progressive facial hemiatrophy is characterized by unilateral atrophy of the skin, subcutaneous tissue, and adjacent bone (Fig. 65.24). The atrophic area is characteristically oblong or linear and sometimes begins as a raised erythematous lesion. The lesion sometimes begins after trauma to the area. The atrophy eventually stabilizes, leaving facial disfigurement.
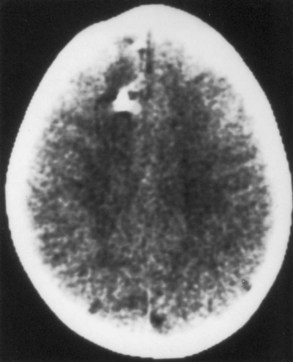
Epilepsy is probably the most common neurological problem associated with progressive facial hemiatrophy. Some patients develop a usually mild hemiparesis. Less common neurological features include cognitive impairment, cranial neuropathy, or even brainstem signs. Cerebral calcifications and white-matter lesions are common neuroimaging findings (Fig. 65.25).
Kinky Hair Syndrome (Menkes Disease)
Kinky hair syndrome, also known as Menkes disease or trichopoliodystrophy, is an X-linked recessive disorder of connective tissue and neuronal metabolism caused by inborn disorders of copper metabolism, namely impaired cellular export of copper leading to accumulation in all tissues except the liver and brain. The estimated frequency is 1 in 40,000 to 1 in 298,000 live births. In the classic form of kinky hair syndrome, the neurological symptoms begin in the first year of life, and the course is rapidly progressive, with death by the third year of life in more involved cases. Cause of death commonly relates to infection, cerebrovascular complications, or the neurodegenerative process. Documented cases of late-onset cases and apparently asymptomatic individuals are in the literature. The basis for diagnosis in a few affected females was a low copper content in liver and high copper content in an intestinal biopsy sample. Clinical features in these girls were similar to but milder than those seen in typical neonatal-onset cases. Genetic analysis revealed inactivation of the normal X chromosome. (See also Chapter 62.)
Cutaneous Features
Connective tissue abnormalities are a major feature of kinky hair syndrome and include loose skin, hyperextensible joints, bladder diverticula, and skeletal anomalies. The enzyme, lysyl oxidase, has copper-dependent steps that are impaired in the establishment of elastin and collagen cross-linking. Keratin cross-linking and melanin production also depend on copper, and copper deficiency leads to characteristic cutaneous features. The hair is light colored and brittle and on microscopic examination (Fig. 65.26) appears as pili torti (twisted hair) and trichorrhexis nodosa (complete or incomplete fractures of the hair shafts at regular intervals). Trichorrhexis nodosa is not pathognomonic of kinky hair syndrome; it also occurs in biotinidase deficiency and argininosuccinic aciduria. Skin may be diffusely hypopigmented or normal.

Other Clinical Features
Kinky hair syndrome covers a clinical continuum from nearly normal to the severe classic infantile-onset form (Box 65.5). Newborns may be more prone to cephalohematomas or spontaneous bone fractures and develop temperature instability, diarrhea, and failure to thrive in early infancy. Sympathetic adrenergic dysfunction, including hypotension, hypothermia, anorexia, and somnolence, is attributable to the impairment of dopamine β-hydroxylase that requires copper for the synthesis of norepinephrine and other neurotransmitters.
Neurological Features
Intracranial and extracranial blood vessels may be tortuous, kinked, and dilated (Kim and Suh, 1997); the cause may be defective or deficient elastin fibers in the walls of these blood vessels. Neuropathological studies show diffuse neuronal loss and gliosis that is particularly prominent in the cerebrum and cerebellum. Microscopic findings include abnormal dendritic arborization in the cerebellar cortex.
Genetic Studies
Infantile-onset kinky hair syndrome results from extensive mutations of ATP7A (e.g., large deletions, frame-shift mutations), whereas occipital horn syndrome is associated with promoter and splicing efficiency mutations, possibly leading to reduced levels of an otherwise normal protein product. Mutations of the ATP7A gene in the patients with the occipital horn syndrome have been base pair substitutions affecting normal messenger ribonucleic acid (mRNA) splicing (Tumer and Horn, 1997). Copper deficiency impairs the function of multiple enzymes that require copper as a cofactor: tyrosinase, cytochrome C oxidase, dopamine β-hydroxylase, and Cu/Zn superoxide dismutase, among others.
Xeroderma Pigmentosum
Complementation Groups
Complementation analysis has been important in understanding the genetic basis of XP. If two particular cell types with different metabolic abnormalities are fused, the cell produced may function normally. These two cell types have different complementation groups and presumably have a different genetic basis. In XP, eight complementation groups have been identified (XP-A through XP-G and a variant group). Although some general genotype-phenotype correlations among these complementation groups exist, considerable clinical overlap exists between them (Copeland et al., 1997). Complementation groups XP-A, XP-C, and XP-D are the most common. Despite the few individuals with XP-B available, cloning of the responsible gene (XPBC) was successful. The XPBC gene is located on chromosome 2q21 and encodes a protein that is a component of the basal transcription factor, TFIIH/BTF2. This protein helps regulate both DNA transcription initiation and nucleotide excision repair.
Cutaneous and Ocular Features
Cutaneous and ocular features of XP result primarily from ultraviolet light exposure (Box 65.6). The onset of cutaneous symptoms in XP is usually early; the median age of onset is 1 to 2 years, typically freckling or erythema and bullae formation after sun exposure. Nearly half of patients develop malignant skin lesions, with the median age of first skin neoplasm being only 8 years. The estimated incidence of basal cell carcinoma or squamous cell carcinoma is 4800 times greater than that observed for the general U.S. population. Light-skinned infants develop erythema and bullae after even brief sun exposure. Sun exposure also induces prominent macule formation (freckling or solar lentigenes), which over time enlarge and coalesce. Telangiectasias and epidermal and dermal atrophy develop in later years, and the skin becomes dry. Actinic keratosis, angiomas, keratoacanthomas, and fibromas also occur. Ocular tissues are particularly susceptible to ultraviolet damage. Keratitis and conjunctivitis with photophobia are common in patients with XP. Atrophy of the eyelids leads to loss of lashes and ectropion or entropion. Neoplasms of the eyelid, conjunctiva, and cornea include squamous cell carcinoma, epithelioma and basal cell carcinoma, and melanoma. The tip of the tongue, gingiva, and palate also are susceptible to sun exposure.
Box 65.6 Cutaneous and Ocular Features of Xeroderma Pigmentosum
Most of what is known about the neurological features in XP comes from studies of Japanese patients with XP-A. Research indicates that the severity of neurological symptoms correlates with particular mutations within the XPAC gene, and presumably this is true in other types of XP (Maeda et al., 1995). The principal neurological symptoms in XP-A are progressive dementia, sensorineural hearing loss, tremor, choreoathetosis, and ataxia. Progressive dementia begins in patients with XP-A during the preschool years, and IQ scores after 10 years of age are invariably less than 50. Sensorineural hearing loss has a later onset, but most patients older than 10 years have hearing impairment. Cerebellar signs develop at approximately the same time as the hearing loss. Microcephaly is present in about half of patients.
Treatment
Cancer surveillance and avoidance of precipitating factors is the most important aspect of health screening of individuals with XP. Optimism exists for genetic therapy to reduce cancer risk and perhaps improve neurological outcomes. In vitro studies offer hope that eventually, recombinant retroviruses can transfer and stably express the human DNA repair genes in XP cells to correct defective DNA repair (Zeng et al., 1997). Using the recombinant retroviral vector, LXSN, successful transfer of human XP-A, XP-B, and XP-C complementary DNAs (cDNAs) into primary and immortalized fibroblasts obtained from patients with XP-A, XP-B, and XP-C occurred. After transduction, monitoring of the complete correction of DNA repair deficiency and functional expression of the transgenes included ultraviolet survival, unscheduled DNA synthesis, and recovery of RNA synthesis. In a similar study, cloning of XP-F cDNA into a mammalian expression vector plasmid and introduced into group F XP (XP-F) cells. The XP-FR2 cells expressed a high level of XP-F protein and ERCC1 protein. They showed ultraviolet resistance comparable to that in normal human cells and had normal levels of ultraviolet-induced unscheduled DNA synthesis and normal capability to remove DNA adducts. This demonstrates that the nucleotide excision repair defect in XP-F cells is fully corrected by overexpression of XP-F cDNA alone.
Other Neurological Conditions with Cutaneous Manifestations
Many other conditions with cutaneous stigmata also manifest neurological symptoms primarily or secondarily. Fabry disease is an X-linked lysosomal disorder with prominent cutaneous angiokeratoma corporis diffusum, sensory neuropathy, and risk of stroke (see Chapters 51A and 76). A connective tissue disorder known as pseudoxanthoma elasticum demonstrates characteristic skin plaques, and vascular involvement leads to cerebrovascular compromise (see Chapter 51A). Wyburn-Mason syndrome (or Bonnet-Dechaume-Blanc syndrome) is a rare sporadic neurocutaneous syndrome characterized by retinal, facial, and intracranial AVMs.
Au K.S., Williams A.T., Roach E.S., et al. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med. 2007;9:88-100.
Bodensteiner J., Roach E.S. Sturge-Weber Syndrome, second ed. Mt. Freedom, NJ: Sturge-Weber Foundation; 2010.
Bourgeois M., Crimmins D., de Oliviera R.S., et al. Surgical treatment of epilepsy in Sturge-Weber syndrome in children. J Neurosurg. 2007;106:20-28.
Butman J.A., Linehan W.M., Lonser R.R. Neurologic manifestations of von Hippel-Lindau disease. JAMA. 2008;300:1334-1342.
Cavallari V., Ussia A.F., Siragusa M., et al. Hypomelanosis of Ito: electron microscopical observations on two new cases. J Dermatol Sci. 1996;13:87-92.
Chen F., Kishida T., Yao M., et al. Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: correlations with phenotype. Hum Mutat. 1995;5:66-75.
Clarkson P.E., Cookson M.S. The von Hippel Lindau gene: turning discovery into therapy. Cancer. 2008;113:1768-1778.
Copeland N.E., Hanke C.W., Michalak J.A. The molecular basis of xeroderma pigmentosum. Dermatol Surg. 1997;23:447-455.
DeDavid M., Orlow S.J., Provost N., et al. Neurocutaneous melanosis: clinical features of large congenital melanocytic nevi in patients with manifest central nervous system melanosis. J Am Acad Dermatol. 1996;35:529-538.
de Vries P.J. Neurodevelopmental, psychiatric and cognitive aspects of tuberous sclerosis complex. In: Kwiatkowski D.J., Whittemore V.H., Thiele E.A. Tuberous Sclerosis Complex: Genes, Clinical Feature, and Therapeutics. Weinheim: Wiley-VCH Verlag GmbH & Co.; 2010:229-267.
Evans D.G. Neurofibromatosis type 2. In: Roach E.S., Miller V.S. Neurocutaneous Syndromes. London: Cambridge University Press; 2004:50-59.
Ewalt D.H., Diamond N., Rees C., et al. Long-term outcome of transcatheter embolization of renal angiomyolipomas due to tuberous sclerosis complex. J Urol. 2005;174:1764-1766.
Faughnan M.E., Palda V.A., Garcia-Tsao G., et al. International guidelines for the diagnosis and management of hereditary hemorrhagic telangiectasia. J Med Genet. 2011;48:73-87.
Franz D.N., Leonard J., Tudor C., et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59:490-498.
Gallus G.N., Dotti M.T., Federico A. Clinical and molecular diagnosis of cerebrotendinous xanthomatosis with a review of the mutations in the CYP27A1 gene. Neurol Sci. 2006;27:143-149.
Hamano K., Kuga T., Takahashi M., et al. The lack of type III collagen in patients with aneurysms and an aortic dissection. J Vasc Surg. 1998;28:1104-1106.
Happle R. Epidermal nevus syndrome. Semin Dermatol. 1995;14:111-121.
James M.F., Han S., Polizzano C., et al. NF2/Merlin is a novel negative regulator of mTOR complex 1, and activation of mTORC1 is associated with meningioma and schwannoma growth. Mol Cell Biol. 2009;29:4250-4261.
Kim O.H., Suh J.H. Intracranial and extracranial MR angiography in Menkes disease. Pediatr Radiol. 1997;27:782-784.
Maeda T., Sato K., Minami H., et al. Chronological difference in walking impairment among Japanese group A xeroderma pigmentosum (XP-A) patients with various combinations of mutation sites. Clin Genet. 1995;48:225-231.
Maria B.L., Neufeld J.A., Rosainz L.C., et al. High prevalence of bihemispheric structural and functional defects in Sturge-Weber syndrome. J Child Neurol. 1998;13:595-605.
Needle M.N., Cnaan A., Dattilo J., et al. Prognostic signs in the surgical management of plexiform neurofibroma: the Children’s Hospital of Philadelphia experience, 1974-1994. J Pediatr. 1997;131:678-682.
Nehal K.S., PeBenito R., Orlow S.J. Analysis of 54 cases of hypopigmentation and hyperpigmentation along the lines of Blaschko. Arch Dermatol. 1996;132:1167-1170.
Nelson D.L. NEMO, NFκB signaling and incontinentia pigmenti. Curr Opin Genet Dev. 2006;16:282-288.
Pavlidou E., Ahgel C., Papavasillou A., et al. Neurocutaneous melanosis: report of 3 cases and up-to-date review. J Child Neurol. 2008;12:1382-1391.
Peterson L.S., Nelson A.M., Su W.P. Classification of morphea (localized scleroderma). Mayo Clin Proc. 1996;70:1068-1076.
Roach E.S., Sparagana S.P. Diagnosis of tuberous sclerosis complex. J Child Neurol. 2004;19:463-469.
Rotman G., Shiloh Y. The ATM gene and protein: possible roles in genome surveillance, checkpoint controls and cellular defense against oxidative stress. Cancer Surv. 1997;29:285-304.
Sancak O., Nellist M., Goedbloed M., et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype-phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. Eur J Hum Genet. 2005;13:731-741.
Sasaki Y., Kobayashi S., Shimizu H., et al. Multiple nodular lesions seen in a patient with neurocutaneous melanosis. J Dermatol. 1996;23:828-831.
Schwartze U., Goldstein J.A., Byers P.H. Splicing defects in the COL3A1 gene: marked preference for 5’ (donor) splice-site mutations in patients with exon-skipping mutations and Ehlers- Danlos syndrome type IV. Am J Hum Genet. 1997;61:1276-1286.
Tumer Z., Horn N. Menkes disease: recent advances and new aspects. J Med Genet. 1997;34:265-274.
Zeng L., Quilliet X., Chevallier-Lagente O., et al. Retrovirus-mediated gene transfer corrects DNA repair defect of xeroderma pigmentosum cells of complementation groups A, B and C. Gene Ther. 1997;4:1077-1084.