[level-membership-for-neurosurgery-category]
CHAPTER 327 Neurochemical Pathomechanisms in Traumatic Brain Injury
Traditionally, TBI has been divided into primary and secondary forms of injury. Primary injury is due to the unavoidable direct mechanical forces occurring at the time of the traumatic insult.1 Secondary injury is derived from complications initiated by the primary injury and includes potentially avoidable entities such as hypoxic-ischemic injury, cerebral edema, metabolic dysfunction, alterations in vascular permeability, diminished blood flow, inflammation, diffuse axonal injury, and the consequences of intracranial hypertension. Almost all clinical treatments are aimed at modulating these secondary injury mechanisms.
Both primary and secondary brain injury can be further classified as focal or diffuse. The distinction between focal and diffuse injuries is historically derived from the presence or absence of radiographic mass lesions on computed tomography.2,3 This distinction has now evolved to also consider the distinct pathobiologic mechanisms imparted by the trauma in regions local to and remote from the point of impact. However, any attempt to conclusively classify brain injury remains a difficult task because most TBIs consist of a heterogeneous admixture of focal and diffuse damage. Research efforts and clinical trials have sought to tip the balance of these secondary events toward facilitating neuroprotection rather than autodestruction. Central to research efforts is the neuronal response to brain injury. It is thought that the mortality and morbidity associated with TBI can be greatly reduced by a better understanding of the mechanisms that cause neuronal injury, dysfunction, and death. This information is critical within the clinical realm—not only to minimize neuronal death after TBI but also to potentially facilitate augmentation of neurological reorganization and repair. Accordingly, there is new ongoing research aimed at the use of stem cells for neuroregeneration (see Chapter 328) and for the development of deep brain stimulation techniques that may modulate the sequelae of TBI.4,5
This chapter covers the complex pathophysiology of TBI. It begins by briefly detailing the biomechanical consequences of trauma on the central nervous system (CNS) (also see Chapter 325) and continues with a comprehensive discussion of primary and secondary injury mechanisms.
Primary Injury: Molecular And Microscopic Aspects
Focal versus Diffuse Primary Brain Injury
Diffuse Primary Brain Injury
The best example of a pure diffuse primary brain injury is “mild TBI” or concussion (see Chapter 332). Concussion is a broadly applied term for the clinical manifestations of blunt head trauma that result in rapid-onset, functional disturbance of the CNS (rather than a structural injury) secondary to the inertial forces of TBI. Concussion usually resolves spontaneously and, contrary to popular belief, may or may not be associated with loss of consciousness (LOC). When LOC is present, it is thought that either the magnitude or the biomechanical directionality of the traumatic forces is sufficient to transiently perturb the brainstem reticular activating system and result in LOC.
Focal Primary Brain Injury
Primary focal brain damage is a direct result of the physical forces delivered at the time of injury. These “impact” injuries are manifested clinically as cortical contusions, brain parenchymal lacerations, and vascular lesions with resultant hemorrhage and hematoma formation.6 Focal contusions most commonly result from contact of the brain with more rigid structures such as the skull, dural edges, or physical objects used in assaults. These impact injuries may occur in association with a more significant diffuse injury, such as when the rapidly decelerating cranium strikes a windshield during a motor vehicle collision. The inertial forces of such a diffuse injury are thought to generate widespread damage and are implicated in prolonged unconsciousness. However, focal injuries do not cause LOC but instead may cause permanent discrete neurological deficits because of the immediate effects of the penetrating/focal injury. The forces of impact imparted by primary injuries are largely responsible for rapid necrosis via physical destruction of cellular elements.7 Focal injuries include skull fractures, penetrating TBI, and vascular injuries and are covered additionally in Chapter 324 and Chapter 325.
Relationship between Brain Movement, Mechanical Forces of Injury, and Histologic Effects
The destructive energy imparted by any given trauma is transmitted to the skull—and thereby its contents—in relatively unpredictable patterns. At one end of the spectrum are pure inertial injuries, where rapid deceleration and rotational forces cause devastating diffuse injuries. The cranium may never contact a solid object, yet the brain is irreversibly damaged. Such injuries maximally damage axons and most frequently occur as a result of motor vehicle accidents (although almost always compounded by an additional impact component). At the opposite extreme are unusual impact injuries in which the stationary head (e.g., of a machine operator) is slowly crushed by slow-moving machinery. Such injuries classically produce massive fractures, extra-axial hematomas, and contusions, but these patients do not usually lose consciousness because axonal injury is absent and the reticular activating system/projection fibers are not disturbed. Thibault and Gennarelli used a primate impact acceleration injury model to characterize the relationship between the magnitudes of acceleration/deceleration force, the duration over which it is applied, and the consequences for the intracranial contents.9 A brief, high-intensity deceleration force will tear parasagittal bridging veins and cause an acute subdural hematoma. When the deceleration force is of higher magnitude and longer in duration, as in motor vehicle accidents, diffuse axonal injury may occur. When both the magnitude and the duration of the deceleration force are less, transient unconsciousness (concussion) results but few structural effects are seen when the brain is examined either ultrastructurally or by light microscopy (see Chapter 332).
Brain Movement during Impact
Laboratory models of brain injury have demonstrated that the brain moves substantially within the cranial cavity in response to deceleration forces.10,11 The brain is anchored within the cranial cavity by only the parasagittal bridging veins, parasinusoidal granulations, cranial nerves, and tentorium. Movement of the brain forward toward the anterior cranial-basal structures, particularly the sphenoidal ridges, concentrates force at the bases of the frontal lobes and the tips of the temporal lobes.11 Surface contusions therefore occur very much more frequently at these sites than elsewhere. There is evidence in the human brain that shearing force also concentrates in deep white matter structures such as the corona radiata, thus explaining the frequent finding of parasagittal gliding contusions.12 Finally, it is generally believed that shearing forces transmitted through the brainstem and the reticular activating system are responsible for the immediate LOC.
Damage to Cells/Tissue
Astrocytes
Astrocytes are increasingly becoming understood to have a significant role in the deleterious effects of TBI. Although the surface area and complexity of membranes may be less for astrocytes, there is now clear evidence that astrocytes are excitable, possess ion channels, and may be depolarized (although to a much lesser extent than neurons).13–15 Astrocytic membranes also constitute an important component of the blood-brain barrier (BBB), and there are now extensive data showing that this barrier function is transiently disturbed by mechanical trauma. Furthermore, disturbed ionic and neurotransmitter homeostasis is recognized as one of the most important mechanisms contributing to the secondary brain swelling after TBI, and astrocytes play a role in maintaining this homeostasis. One example is maintenance of potassium homeostasis after TBI. Astrocytes are known to function as potassium uptake buffers in that they have the capacity to rapidly take up potassium from the extracellular space.14,16–18 Kimelberg and Norenberg hypothesized that astrocytes function to conduct potassium away from neurons, particularly in injured brain tissue, and thereby aid in the establishment of ionic homeostasis.14 Thus, there is a net loss of potassium from injured tissue into the microvasculature that begins hours after onset. In addition, stretch-injured astrocytes express a dysfunctional cation current as opposed to an osmoregulatory anion current. This mechanism may contribute to the cytotoxic swelling seen after TBI.19 Such astrocyte swelling is the ultrastructural hallmark of both acute cerebral ischemia and focal cerebral contusion and is almost always seen in animal models of trauma and in humans after trauma.16,20
Axons
About 50 years ago, neuropathologic studies first demonstrated an accumulation of axoplasmic “retraction balls” at sites of axonal discontinuity.12,21 They were chiefly found on large myelinated fibers in patients who were unconscious from the time of injury and subsequently died. These retraction balls were found in high density in white matter tracts in approximately 25% of severely head-injured patients and were thought to occur immediately as a result of tearing.22 Traumatic axonal injury (the experimental correlate to diffuse axonal injury) is now known to be a progressive process involving transient mechanoporation of the axolemma that allows unregulated calcium entry.23 The mechanical insult induces a sequence of events culminating in failure of axoplasmic transport, pooling of intra-axonal contents, and pinching off of the axon from its distal segment (Fig. 327-1). This disconnection occurs within 24 to 72 hours after the traumatic event and is termed delayed or secondary axotomy (because the primary mechanical insult provokes secondary biochemical processes that result in axotomy). This suggests that axons that subsequently show the changes associated with diffuse axonal injury may be functioning, in some capacity, immediately after the injury before eventually degrading. It also suggests that other, less affected axon tracts may not progress to diffuse axonal injury. Thus, diffuse axonal injury is amenable to therapeutic intervention.
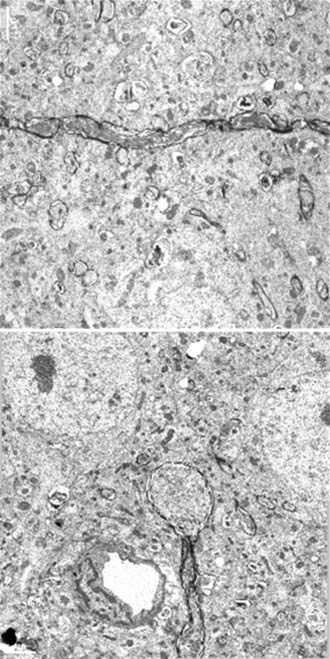
At the molecular and microscopic level, calcium influx initiates activation of calpain24,25 and mitochondrial swelling,26 with release of cytochrome c and activation of caspase27 leading to further axonal injury, apoptosis, and detachment over time. These changes have far-reaching consequences for neuronal function. Interruption of the axon causes proximal wallerian degeneration of the affected neuron. Distally, the axon degenerates, fragments, and disappears, thereby resulting in deafferentation of the affected neuronal fields. The functional consequences of this process may include seizures because of lack of inhibitory effects, spasticity, intellectual decline, and unmodulated behavior patterns. When this process is widespread and wallerian degeneration destroys many neurons, the whole brain becomes atrophic, with ventriculomegaly and, in the worst cases, a persistent vegetative state.22,28
Clinical Implications
The benefits of attenuating secondary axotomy may be enormous. These hopes are bolstered by recent ultrastructural studies revealing that neuron somata show evidence of the potential for reorganization and repair for up to 7 days after traumatically induced axonal injury.29 Cyclosporine is a widely investigated immunosuppressive drug that has been shown to blunt traumatically induced axotomy in experimental models of TBI.30 Its neuroprotective properties are thought to be derived from inhibitory actions on the protein phosphatase calcineurin, as well as from its modulatory effect on mitochondria and the mitochondrial permeability transition pore (see discussion later in this chapter). There has been one clinical trial aimed at taking advantage of these properties via the postinjury administration of cyclosporine in the hope of limiting mitochondrial damage and axonal injury.31 Although the results of this small trial demonstrated that administration of the drug was safe, it did not reveal any benefit in terms of neurological outcome. Future phase III clinical trials are still being considered.
Shear Effect on the Microvasculature
It has been estimated that the magnitude of shear required to damage the pial vasculature may be 5 times greater than that needed to damage axons.9,32 Although this gradation of force suggests that vascular structures should be damaged less frequently than axons and membranes, in reality this may not be the case because traumatic forces are more readily translated to surface vessels than to deeper axons. In the majority of significant head injuries, focal concentrations of force develop at the tips of the frontal and temporal poles and are sufficient to disrupt these pial vessels and cause a focal contusion. Ultrastructural studies in both head-injured humans and appropriate animal models have demonstrated major anatomic changes in the injured microvasculature. Such changes include the following16,17: (1) swelling of perivascular astrocytic end-feet; (2) increased endothelial microvacuolation and micropseudopodial activity; (3) perivascular hemorrhage and transvascular diapedesis of red cells, which may coalesce to form a frank intracerebral hematoma or hemorrhagic contusion; and (4) increased intravascular leukocyte adherence. Frank vascular disruption has been found to be unexpectedly atypical in human pericontusional biopsy material, thus suggesting that small vessels “stretch and leak” much more frequently than they “tear or burst.” These microvascular changes have profound functional consequences, chiefly a reduction in local cerebral blood flow (CBF) and the development of vasogenic and cytotoxic edema with increased intracranial pressure (ICP).20
Ion Channels
Although dendritic spines, synapses, gap junctions, and myelinated axons constitute specialized regions of neurons, ion channels are by far the most frequent structures embedded in neuronal membranes. Using patch clamp techniques and in vitro tissue culture of neurons growing on flexible plastic membranes, studies have shown that ion channel function may be radically altered by mechanical deformation—in this case by delivery of a brief jet air impulse to the flexible membrane.13,33,34 Specific classes of “mechanotransducing” ion channels have been identified by using similar techniques in both neurons and glia.13,33 Some of these ion channels remain perturbed for several hours after mechanical deformation.34 Still other experiments have shown rapid entry of calcium and subsequent neuronal death, along with efflux of lactate and potassium into the culture medium, after mechanical deformation.34 Further implicating channelopathy as a significant factor in TBI are data from in vivo trauma models such as fluid percussion injury and contusion impact models. These models show a massive, approximately threefold to fourfold rapid transient efflux of potassium into the extracellular fluid (ECF) associated with a fall in the sodium content of the ECF.35–38 About a third of the potassium release could be blocked with tetrodotoxin (a selective blocker of voltage-gated sodium channels), thus suggesting that two thirds of the potassium release was occurring through agonist-operated channels. Another investigation similarly revealed that preinjury blockade of voltage-operated ion channels failed to ameliorate the negative neurological and behavioral effects of the trauma and produced only a modest effect on K+ flux in the ECF. This suggests that agonist-operated ion channels are more important in mediating ionic events after TBI.36 Finally, channelopathy has been implicated in the etiology of the calcium influx seen after TBI. Work by Wolf and colleagues suggests that calcium influx occurs, in part, via mechanical alteration of tetrodotoxin-sensitive sodium channels after traumatic axonal injury.39 This implies that excessive calcium influx may not be fully attributable to direct axolemmnal poration but instead is also related to sodium channelopathy. The resulting sodium influx then triggers depolarization-induced calcium influx through voltage-gated calcium channels and reversal of the sodium-calcium exchanger, both acting to increase influx of calcium. Work is ongoing to further explore this phenomenon.
Synapses
Direct investigation of synaptic function is difficult in the acute stages of trauma. Microdialysis studies have investigated the time course of changes in neurotransmitters within the extracellular space after fluid percussion injury, and the results demonstrated brief transient surges in the release of excitatory amino acids (EAAs) and acetylcholine. From experimental models of injury, this posttraumatic excitotoxicity is marked by increases in glutamate, which leads to an increase in extracellular potassium as a result of channel activation. Potassium levels determined by microdialysis techniques were increased in 20% of patients after severe TBI and were also noted to correlate directly with reduced CBF.40 Data from microdialysis studies of patients who have sustained severe head injury and patients with ischemic events superimposed on their primary trauma show that ECF EAAs rise to levels 50 to 60 times higher than normal values when a secondary ischemic event is superimposed on the trauma.35,41 The excitatory neurotransmitters released from damaged cells and neuron processes may be responsible for these increases through a positive feedback loop. EAAs may also come from the intravascular compartment. This conclusion is supported by the finding that serum levels of structural amino acids in these patients were also raised and appeared to fluctuate in parallel with EAAs.42
The behavioral changes that persist up to weeks or months after TBI, even in animals without any evidence of structural damage, have been taken as evidence to support functional changes at the synaptic level or in relation to second messenger systems. Neurochemical studies have shown evidence of synaptic alterations, as well as G protein–coupling variations, within the cell membrane that are manifested as prolonged amplification of protein synthesis in response to activation of muscarinic cholinergic and certain catecholaminergic receptors.43–45 These changes may translate into effects on long-term potentiation in the hippocampus (which have been demonstrated in the absence of structural changes after trauma and may be an important mechanism underlying the traumatic effects on learning and memory). In addition, matrix metalloproteinases (MMPs) are known to modulate molecules forming the extracellular matrix (ECM). MMP proteolysis of ECM molecules may perform a permissive or inductive role in the fiber remodeling or synaptogenesis initiated by deafferentation.46 The significance of this interaction in TBI was explored in animals by intraventricular infusion of the MMP inhibitor FN-439 after unilateral lesions of the entorhinal cortex.46 The lesioned rats receiving the MMP inhibitor failed to develop the capacity for long-term potentiation and showed persistent cellular debris. These results underscore the importance of continuing to improve our understanding of MMPs, the ECM, and other mechanisms involved in remodeling after trauma.
Secondary Injury Processes
The concept of delayed secondary neurological damage after head injury is supported by “lucid interval” statistics. Between 30% and 40% of severely head-injured patients who die will, at some time, have demonstrated a period of lucidity sufficient to obey commands or speak.22,47 This implies that the primary impact events were not sufficiently severe to damage the brain beyond the capacity for function, thus emphasizing the importance of the secondary damage.48 The principal mechanisms to consider are hypoxia-ischemia, edema, excitotoxicity, calcium dysregulation, apoptosis, cytoskeletal proteolysis, metabolic and mitochondrial derangements, oxidative stress, and inflammation.49,50 The deleterious mechanisms at work are diverse and interrelated—often with both sequential and parallel cascades of neuronal reaction and cell death.51
Finally, it is useful to emphasize that many clinical trials, using either physiologic or pharmacologic interventions, view these secondary injury mechanisms as the main therapeutic targets (Table 327-1).52–72 Many of these trials will be discussed briefly throughout this chapter under the most relevant heading. That said, there is no questioning the heterogeneity of TBI and, unfortunately, the lack of favorable results from previous clinical trials. Some of these failures may be due partially to flawed classification systems in terms of optimally reflecting the target population for the drug in question. These classifications have relied on several variables, including clinical severity, pathophysiology, pathoanatomic and prognostic indicators, etiology, and symptomatology. Today, the most commonly used system is the Glasgow Coma Scale, which is based solely on the severity of the neurological injury. As a result, there is growing support to establish a more clinically derived classification system for TBI based chiefly, but not exclusively, on pathoanatomic features. It is hoped that this would improve the application of appropriate treatment strategies targeting the various causes of TBI.73
Hypoxia/Ischemia
A central factor involved in secondary damage after TBI is the onset of hypoxic-ischemic damage. The incidence of ischemic brain damage seen at autopsy in patients who sustained severe TBI is extremely high, with estimates ranging between 60% and 90%.47 During life, most of these patients do not manifest the long periods of low cerebral perfusion pressure (CPP) that are known to be necessary for the generation of ischemic damage. Likewise, in animal models of impact-type head injuries, widespread ischemic damage is not seen other than around the periphery of focal contusions. Thus, there is a fundamental paradox, and the high incidence of ischemic brain damage is not easily explained, although necrosis of neurons, secondary to release of EAAs, may be a factor exacerbating cell death.
The Genesis of Ischemic Brain Damage after Severe Human Traumatic Brain Injury
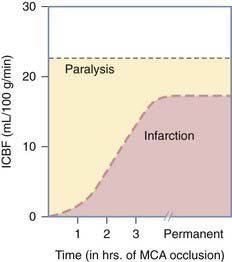
On a global scale, CBF can be decreased by as much as 50% during the first 48 hours after injury and lead to ischemic changes.74 Impaired CBF has well-described cellular consequences, and a time-dependent hierarchy of neuronal events is summarized in Table 327-2.75–77 Both hemorrhage and contusion can lead to local ischemia through compromise of the microcirculation by thrombotic occlusion of blood vessels.1,78–80 Additionally, at the local level, blood flow within focal areas of contusion is dramatically reduced.16 Focal ischemia can also result from the formation of a mass lesion, which will raise ICP and impede blood flow to the damaged region in accordance with the modified Monro-Kellie Doctrine.81 Finally, ischemia secondary to occlusive or hemorrhagic stroke develops as a result of damage to or interruption of the blood supply from a parent vessel to an area of vulnerable parenchyma—such as traumatic carotid dissections.49
TABLE 327-2 Cellular Consequences of Impaired Cerebral Blood Flow
From Jones TH, Morawetz RB, CrowelI RM, et al. Threshold of focal cerebral ischemia in awake monkeys. J Neurosurg. 1981;54:773-782.
| CEREBRAL BLOOD FLOW (mL/100 g/min) | CONSEQUENCES |
|---|---|
| 40-60 | Normal |
| 20-30 | Start of neurological symptoms |
| 16-20 | Isoelectric electroencephalogram, loss of evoked potentials |
| 10-12 | Na+ and K+ pump failure Cytotoxic edema |
| <10 | Complete metabolic failure with gross disturbance of cellular energy homeostasis (infarction) |
 |
|
Infarction versus Selective Neuronal Loss
When flow is profoundly reduced (i.e., <5 to 10 mL/100 g per minute) within the distribution of one cerebral end artery for more than 60 to 90 minutes, infarction ensues (immediate necrosis of all cell types within a zone of the brain). However, when the reduction in flow is to levels of approximately 15 to 18 mL/100 g per minute for a period longer than 30 minutes, selective neuronal loss may occur—especially in the hippocampal neurons (in the molecular layer, CA1 and CA3 sectors), cerebellar granular cells, and cortical neurons (particularly the larger cells in areas such as the cuneate visual cortex).82,83 Within the context of head injury, this type of neuronal loss is especially important in patients with raised ICP, in whom CPP may be marginal (≈30 to 40 mm Hg) for many hours or even days. In such patients, a high frequency of ischemic neuronal loss is seen in the hippocampus.47 This may explain the high frequency of memory disorders and coordination difficulty noted in the majority of severely head-injured survivors. This concept is also in accord with the almost universal finding of marked cerebral atrophy in patients who survive severe head injuries.
Clinical Implications
Clearly, reductions in CBF can have devastating consequences and will directly affect metabolic profiles. Noting that historical strategies for managing severe TBI followed ICP-directed protocols (and therefore would indirectly augment CBF), a phase III clinical trial with CPP-directed therapy was performed.65 To ensure adequate CBF and therefore oxygen delivery, the primary goal in this trial was to maintain higher CPP (versus lowering ICP). The results suggested that CPP-directed therapy improves several physiologic parameters, such as brain perfusion; however, it failed to show any incremental benefit in outcomes when CPP was targeted to levels greater than 70 mm Hg (versus 60 mm Hg). This was predominately due to an increased incidence of acute respiratory distress syndrome, which had a negative impact on mortality measures.65 Still other trials have targeted increased oxygenation by using hemoglobin substitutes, or hyperbaric oxygen, to augment the oxygen-carrying capacity of the microcirculation to damaged tissue.42 One such agent is Oxycyte, a third-generation perfluorocarbon (PFC) that improves the oxygen-carrying capacity of blood. In animals, PFCs have been shown to improve cerebral oxygenation and mitochondrial function after TBI.84 However, increased free radical formation with higher doses was also seen in these same studies. The authors suggested the need for further studies combining PFCs with free radical scavengers. Trials of PFCs and hyperbaric oxygen are ongoing—including the upcoming Brain Tissue Oxygen Monitoring in Traumatic Brain Injury trial, which will aim to implement therapy directed at increasing the partial pressure of oxygen in brain tissue (PbtO2) to further evaluate direct measurements of cerebral oxygenation on outcome.
Ischemia and Associated Acidosis/Hydrogen
Although hydrogen ions in the extracellular space are powerful cerebral vasodilators, high concentrations of hydrogen ions within cells are harmful because they alter the function of intracellular enzymes.85 For example, low pH causes conformational changes in the N-methyl-D-aspartate (NMDA) ion channel that prevent further ingress of sodium and calcium and egress of potassium during cellular acidosis. The potential benefits of mild acidosis therefore include inactivation of glutamate receptors, decreased free radical generation,86,87 inhibition of phospholipase A2 (which generates free radicals), decreased energy demand because of hyperpolarization, and inhibition of the Na+/H+ exchange transporter, which prevents intracellular entry of Na+ and Ca2+.88
Clinical Implications
The acidosis and elevated lactate levels so often seen accompanying TBI became a therapeutic target in a clinical study in which tris-(hydromethyl)-aminomethane (THAM) was administered to victims of TBI. THAM is an alkalizing agent that can buffer CO2 and acids. Its use in animal models resulted in reduced edema, lower ICP, and more favorable energy kinetics. Unfortunately, this did not translate as well to humans, and no advantage in outcome was observed.61,89
Edema/Increased Intracranial Pressure
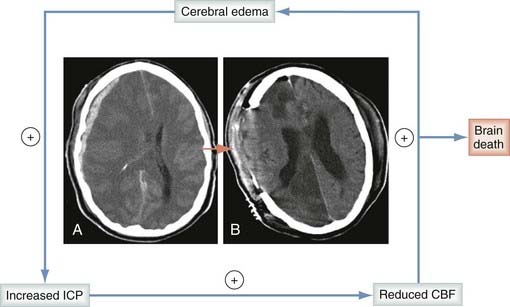
Brain swelling occurs in almost all patients with severe brain injury and in 5% to 10% of those with moderate injuries (also see Chapter 322 and Chapter 324).90,91 The morbidity associated with brain injury was once thought to largely be correlated with the extent of posttraumatic edema. Currently, the significance of edema is acknowledged, but we now appreciate the multifaceted pathobiology constituting TBI.
Without question, posttraumatic edema contributes heavily to intracranial hypertension. The hypertension may then lead to decreased CPP (CPP = MAP − ICP, where MAP is mean arterial pressure) and to ischemia, thereby inciting continued cytotoxic edema and progressive increases in ICP. This vicious cycle is depicted in Figure 327-2. Historically, breakdown of the BBB with protein extravasation (termed vasogenic edema) was believed to be the primary component of the edema seen after TBI.92 This misconception led some practitioners to use corticosteroids for the management of TBI. This has now clearly been shown to be harmful, with the Corticosteroid Randomization After Significant Head Injury trial (>9000 patients enrolled, the largest TBI trial to date) revealing that corticosteroids were associated with worse outcomes when used in patients with severe TBI.93 Although vasogenic edema does contribute to the overall edema seen in TBI, early work by Marmarou and colleagues used mathematical modeling techniques to reveal that the vascular engorgement component of brain swelling after severe brain injury probably represents only about 25% of the overall increase in brain bulk, with the remainder being due predominantly to cytotoxic edema.94 Vasogenic edema probably becomes important around focal contusions on the 2nd through the 10th to 15th days and is most likely associated with transient opening of the BBB to medium-molecular-weight markers (50 to 70 kD).95 In humans studied with both gadolinium-enhanced magnetic resonance imaging (MRI) and pertechnetate-enhanced single-photon emission computed tomography, vasogenic edema is seen only at later time points around contusions and not at all in patients with diffuse nonfocal injuries.96–98 There is now supporting evidence that the majority of early brain edema, both global and focal, is cytotoxic rather than vasogenic. This was further substantiated recently by Marmarou and colleagues, who used diffusion-weighted MRI to evaluate patients with severe TBI. This study revealed that cytotoxic (intracellular) edema is indeed the predominant form of edema present after TBI.99 The mechanisms responsible for generating this cytotoxic edema are a continuing topic of great interest. Identified mechanisms responsible for the generation of cytotoxic edema include hypoxia-ischemia, channelopathy/excitotoxicity, mechanical membrane disruption, and aquaporins.
Hypoxia-Ischemia
Hypoxic-ischemic events are common after TBI and result in failure of ionic pumps, specifically the sodium-potassium pump (see Table 327-2). Pump malfunction causes the cell to lose its innate homeostatic environment via failure of sodium extrusion and potassium uptake. This failure will bring about the accumulation of sodium within the cell and lead to the influx of water because of the altered osmotic gradient.16
Channelopathy/Excitotoxicity
A major source of cytotoxic edema is traumatically induced accumulation of EAA neurotransmitters such as glutamate and glycine.49 These neurotransmitters bind their receptors, activate and open membrane channels, and induce sodium influx. As with ionic pump failure, osmotic pressure dictates that water will follow. In addition to the oncotic effects of sodium entry, sodium influx will also cause membrane depolarization with an influx of chloride—also resulting in swelling.100
Mechanical Membrane Disruption
Trauma induces brief and spontaneously recovering mechanoporation of neuronal and axonal membranes. Membrane disruption causes efflux of potassium from neurons and resultant astrocytic swelling, as confirmed by ultrastructural examination of pericontusional tissue taken as little as 10 minutes after impact.101
Aquaporins
Aquaporins are a relatively new family of at least nine proteins that have been shown to be involved in the formation of cellular water channels under a variety of circumstances.102 At this time, aquaporin expression across injury models has proved inconsistent and is incompletely understood. For example, increased aquaporin expression is present after ischemic injury, whereas it is decreased after a cortical impact model of TBI.103 These conflicting data appear to indicate that aquaporin expression may lead to the accumulation intracellular edema under some circumstances while playing a role in the clearance of edema in others.104 Aquaporin-4 (AQP4) is currently thought to be one of the primary cellular water channels in the brain and is localized to astrocytic foot processes along the basal lamina and brain–cerebrospinal fluid interface. Our understanding of AQP4 has been greatly augmented by the study of AQP4 knockout mice in several models of brain injury.102 In models of cytotoxic edema, AQP4 deletion or alteration has been shown to be protective.104 In contrast, AQP4 deletion in models of vasogenic edema results in decreased clearance of edema and greater progression of disease.104 In addition to AQP4, Tran and associates described a potential role for AQP1 in water homeostasis after experimental TBI.105 AQP1 is another member of the aquaporin family found in the brain and can participate in CO2 transportation across the cellular membrane. Interestingly, AQP1’s promoter contains a glucocorticoid response element. Thus, Tran and colleagues hypothesized that “AQP1 may be involved with edema-related brain injury and might be modulated by external conditions such as the pH and the presence of steroids.” The importance of aquaporins in modulating edema after trauma is thus clear. It further suggests the potential for the development of pharmacologic strategies targeting aquaporin function and expression to dramatically alter our ability to treat cerebral edema, which is currently limited to osmotic agents.
Excitotoxicity
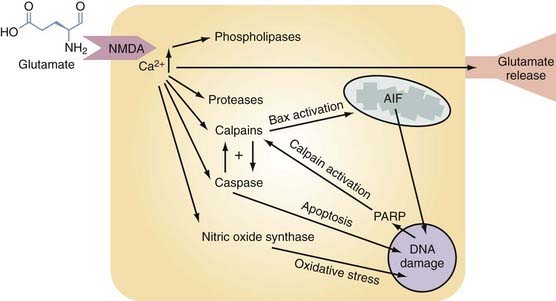
Glutamate excitotoxicity is a self-perpetuating process that triggers numerous injurious intracellular mechanisms. After TBI there is direct release of excessive EAAs, such as glutamate and aspartate, from presynaptic nerve terminals and astrocytes into the extracellular space. This process is depicted in Figure 327-3 and begins when these EAAs bind to the appropriate postsynaptic receptor (NMDA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate [AMPA]). Activation of these ion channels will cause intracellular Ca2+ and Na+ levels to rise, followed passively by movement of Cl− and water. The resultant combination of intracellular volume and Ca2+ overload induces organelle swelling, plasma membrane swelling,106 necrosis,107 and apoptosis and leads to the activation of destructive enzymes108 (phospholipases, calpain, caspase, and nitric oxide synthase [NOS]), as depicted in Figure 327-3. Glutamate-driven excitotoxicity will further depolarize the cell, activate voltage-dependent calcium channels, and thereby propagate a dangerous positive feedback loop.6 The downstream effects of these events are shown in Figure 327-3 and are individually discussed under the appropriate subheading within this chapter.
Clinical Implications
There has been great historical interest in pharmacologic modification of glutamate excitotoxicity. Six phase II-III clinical trials (Eliprodil, Selfotel Int, Selfotel U.S., Cerestat, Saphir/D-CPP-ene, and CP101-606) directed by this strategy have been performed, but only one showed a survival benefit.56 Dexanabinol, a cannabinoid with pluripotent functionality, including noncompetitive NMDA receptor antagonist properties (as well as being a free radical scavenger and inhibitor of tumor necrosis factor-α [TNF-α]), was evaluated in a phase II trial; it was shown to be safe and well tolerated and seemed to decrease ICP. However, the phase III efficacy trial showed no effect on outcome.69
Calcium Dysregulation
As mentioned, TBI always leads to intracellular influx of calcium through numerous routes, including but not limited to (1) opening of voltage-dependent channels induced by mechanical deformation of membrane and ion channels, (2) opening of agonist-dependent channels mediated by neurotransmitter substances released in excess into ECF, and (3) opening of specific calcium channels. It is clear that calcium is strongly implicated in the propagation of several deleterious cascades responsible for the generation of neuronal injury and death after TBI.109,110 It has been linked to many downstream mechanisms of injury, including activation of cysteine proteases and subsequent cytoskeletal proteolysis,111 mitochondrial permeability transition,112 free radical toxicity, and mechanical perturbation of neuronal membranes.113 These mechanisms have been implicated in the initiation of various forms of cell death, including apoptosis and necrosis.114 Regardless of the etiology, unregulated calcium influx can cause calcium to be released from intracellular stores, as well as glutamate-containing exocytotic vesicles, thereby further increasing cytosolic calcium in a vicious cycle of cell destruction. This massive calcium surge can overwhelm cells’ buffering capability, a duty largely handled by mitochondria and ionic pumps under normal circumstances.115 The result is the activation of various enzymes, including the aforementioned cysteine proteases (calpain, caspase, and cathepsin). Induction of these important proteases has been shown in focal116 and diffuse117 models of TBI. Calpain activity has been noted in both necrotic and apoptotic forms of cell death, whereas caspase-3 activity is seen only in apoptosis.118 Finally, mitochondria have a central role in calcium homeostasis, and recent evidence highlights their function in cell death via opening of the mitochondrial permeability transition pore (MPTP), a calcium-dependent process.119
Clinical Implications
The central role of calcium in mediating cell damage and death led to trials of calcium antagonists in head-injured patients. The dihydropyridine calcium antagonist nimodipine has shown little overall benefit in unselected populations with head injury.52,53 Nimodipine has, however, shown a trend for a more favorable outcome in patients with traumatic subarachnoid hemorrhage.120 Poor brain penetration has been implicated as a major factor in the limited effect of nimodipine in TBI.
Cytoskeletal Proteolysis
The cytoskeleton consists of three main protein components: microfilaments, neurofilaments, and microtubules. After TBI, calcium-induced activation of calpain results in proteolysis of the cytoskeleton and may play an integral role in delayed neuronal degeneration—so-called calpain-mediated spectrin proteolysis (CMSP).121 In axonal stretch injury, within minutes there is malalignment and distortion of the cytoskeletal components,122 which leads to loss of microtubules and increased spacing of neurofilaments, especially at the node of Ranvier. In addition to trauma, inhibition of calpains can limit the proteolysis of MAP2 (a type of membrane-associated protein contained in microtubules) after ischemia.123,124 The caspases have also been linked to the breakdown of cytoskeletal proteins such as MAP2, α-spectrin, and neurofilaments.125–127
Clinical Implications
Targeting of CMSP has yet to reach the clinical trial stage. It has been used experimentally with modest success through the use of calpain inhibitors in TBI. In one particular study, a fluid percussion injury model was used to generate TBI in rats.128 The calpain inhibitor MDL-28170 was administered after injury, and axons in the corpus callosum were evaluated with amyloid precursor protein immunohistochemistry. When the drug was given within 30 minutes after injury, there was both structural and functional benefit in terms of axonal injury burden. However, the functional benefits diminished when the drug was given beyond 30 minutes after injury. In short, calpain inhibition has demonstrated potential (i.e., reduction of cytoskeletal breakdown and neuronal degeneration in animal models), yet the results are modest and require further investigation before they can be translated to human trials.
Derangements in Brain Metabolism after Traumatic Brain Injury
Because the brain is dependent on aerobic metabolism for delivery of substrate (oxygen and glucose) and because of the frequent impairment of oxygenation and perfusion that occurs after severe head injury, metabolic derangement is an extremely common and important consequence of TBI. The metabolic changes may be global or focal, with evidence demonstrating metabolic derangement after TBI coming from several methodologies, including (1) a 2-deoxyglucose technique to measure glucose metabolism, (2) positron emission tomography (PET) studies using fluorodeoxyglucose in humans, (3) measurement of the jugular/arterial differences in oxygen and lactate to yield global measures of oxygen consumption and lactate production in humans (AVDO2 and CMRO2), (4) measurements of whole-brain metabolic indices with magnetic resonance spectroscopy, and (5) measurements of focal ECF indices using microdialysis and PbtO2 monitors. The data from these studies allow certain generalized conclusions. TBI induces massive ion fluxes across neuronal membranes, widespread loss of resting membrane potential, and release of neurotransmitters into the extracellular space. Within minutes of these events, the brain attempts to restore ionic homeostasis by ionic pumping and reuptake of neurotransmitters. These processes are intensely energy dependent and result in an abrupt increase in glucose utilization. Studies based on the fluid percussion model in rats have shown that this increase in glucose metabolism, to facilitate the generation of adenosine triphosphate (ATP), is brief and maximally localized to parts of the brain that are maximally deformed by the shearing forces.128 When focal lesions such as subdural hematoma, focal infarction, or cerebral contusion are present, glucose use increases for a period in the penumbral border zone.129,130 Furthermore, in humans, PET studies have shown that these increases in glucose are maximal in the penumbral zone of contusions and in the hemisphere underlying hematomas when this part of the brain is viable.131 This increase may persist for 2 to 4 hours in the rat and 5 to 7 days in humans.129,131 In both animal models and humans, glucose use is depressed when measured days after diffuse injury and remains so for weeks after impact, which is consistent with the reduced metabolic needs of the comatose brain.132
Clinical Implications
Hypothermia was an initially promising intervention intended to improve outcomes after TBI that is best discussed as a targeted therapy for the metabolic derangements seen after TBI. Historically, clinical interest in induced hypothermia is derived from experimental models of head injury in which hypothermia has been shown to (1) reduce energy requirements of the brain, (2) stabilize cell membranes,133 (3) improve posttraumatic CBF-metabolic uncoupling, (4) attenuate axonal injury, (5) reduce inflammation, and (6) reduce ICP. There have been at least 12 clinical trials performed, with approximately 6 ongoing trials, and thus far no outcome benefit has been revealed. As of this writing, it is unclear why no benefit has been observed. Hypothermia does have many associated potential systemic complications, and these may be negating the beneficial effects. Therefore, prophylactic hypothermia has currently lost momentum as a standalone therapy for TBI. However, ongoing research is being performed to see whether it might expand the window of opportunity to introduce other therapeutic strategies, both pharmacologic and physiologic. In addition, work is ongoing to more carefully evaluate the role of therapeutic hypothermia, in particular for intracranial hypertension, in the management of TBI.
Mitochondrial Permeability Transition
Clearly, mitochondrial integrity is pivotal in the maintenance of cellular metabolic homeostasis. The occurrence of a process involving the increased permeability of mitochondrial membranes en route to cellular death has long been speculated. Kroemer and coauthors proposed the term mitochondrial permeability transition (MPT) to describe a calcium-induced process of increased mitochondrial membrane permeability.134 Since that time there has been a tremendous explosion in research on MPT and cell death. MPT has been described as increased permeability of the inner membrane of mitochondria via the opening of channels called MPTPs. Opening of MPTPs is a devastating event with resultant loss of transmembrane potential, mitochondrial swelling, and eventual rupture of the outer mitochondrial membrane. This loss of mitochondrial function produces profound deficiencies in neuronal metabolism and ionic equilibrium. To clarify terminology, mitochondrial permeability transition is the term used to identify the entire process, whereas mitochondrial permeability transition pore is the term used to describe the actual channel (therefore, opening of the MPTP allows the process of MPT to occur).
Clinical Implications
Cyclophilin D (CyD) is a member of the cyclophilin family involved in protein folding that is normally found in the mitochondrial matrix but migrates to the inner membrane during MPT. In experiments using the drug cyclosporine to block CyD, MPT activity was significantly attenuated, thereby establishing CyD as a critical player in MPT. As discussed previously, cyclosporine is a Food and Drug Administration–approved drug that has been shown to confer neuroprotection after TBI in multiple animal models and is being evaluated for potential use in neurocritical care.135 Interestingly, a recent in vitro study using isolated mitochondria revealed that the presence of phosphate was necessary for the inhibition of MPTP opening by cyclosporine administration or CyD knockout.136 Investigations are ongoing to determine the relationship, if any, of the neuroprotective effect of cyclosporine specifically on the blunting of MPT. Studies have investigated the role of MPT in apoptosis. Although cyclosporine-dependent MPT has been tied to the release of cytochrome c137 and thus to apoptosis, other studies have failed to show inhibition of apoptosis by cyclosporine-mediated blockage of MPT.138 Furthermore, one study indicated that CyD-dependent MPT regulates necrotic cell death but not apoptotic cell death.139 Finally, overexpression of CyD has been shown to be somewhat protective of apoptotic death. Overall, it is clear that MPT is an important mediator of necrotic cell death and can be implicated in apoptosis. However, its exact role in apoptosis remains unclear and warrants further investigation.
DNA Damage
Figure 327-3 illustrates several of the pathways that lead to DNA damage, including caspase-independent apoptosis (via apoptosis-inducing factor [AIF]), caspase-dependent apoptosis, and oxidative stress (through the generation of nitric oxide [NO]). DNA damage will then activate poly(adenosine diphosphate ribose) polymerase (PARP), which will induce the release of AIF through sequential activation of calpain and then Bax. It should be pointed out that DNA damage is usually associated with apoptosis, but this is not always the case because apoptosis can occur without DNA fragmentation.140 Furthermore, the presence of DNA fragmentation should not be used to exclusively indicate apoptosis.
Neuronal injury mechanisms involving DNA damage are also often mentioned in concert with induction of the tumor suppressor gene p53, which can serve as a transcription factor for a variety of genes with many actions (e.g., growth arrest, promotion of apoptosis). Increased expression of p53 with an associated increase in DNA fragmentation and neuronal apoptosis has been shown after focal141 and diffuse TBI.142 Increased endonuclease activity resulting in DNA fragmentation has been demonstrated in animal models of head injury.143 These delayed apoptotic mechanisms may be more amenable to pharmacologic blockage because of their long “window of opportunity” in contrast to other neuroprotective mechanisms, and at least two current neuroprotective trials involving TBI (Solvay SL334 and Neuren NZ1366) are in part targeting this mechanism.
Free Radical Formation
Free radicals are highly reactive ionic molecules bearing an unpaired electron in their outer electron shell. This unpaired electron confers high chemical reactivity. Free radicals are the normal by-product of oxidative metabolism within mitochondria, and they fulfill important physiologic roles such as signaling and polymorphonuclear leukocyte–mediated destruction of bacteria.144,145 Reactive oxygen species (ROSs) are inherently injurious, however, and contribute to many diseases affecting the CNS, including Parkinson’s disease, Alzheimer’s disease, multiple sclerosis, and amyotrophic lateral sclerosis.146 ROSs are also important contributors to secondary injury and are produced early after neurotrauma.147,148 Indeed, many secondary injury processes lead to free radical production, and in turn, free radicals feedback positively to increase the activity of many of these harmful processes.
Excess levels of free radicals after TBI can damage all components of the cell, including proteins, carbohydrates, nucleic acids, and lipids, inhibit their function, and eventually lead to cell death.149 The brain is thought to be particularly vulnerable to ROSs because it contains high concentrations of polyunsaturated fatty acids, which are readily damaged by oxidative stress.150 In addition, the brain has lower levels of antioxidants than other organs do.151 Within the brain, neurons are especially vulnerable because of lower levels of glutathione than in other CNS cell types.152
A number of different free radical species are produced after trauma, as depicted in Figure 327-4A. Superoxide (O2−) is produced when the mitochondrial electron transport chain is disrupted, as well as by reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (particularly in inflammatory cells), xanthine oxidase, and cytochrome P450–dependent oxygenases.151 NO, another important species, is produced from arginine by a family of NOSs. Both O2− and NO are relatively inert, but their interaction generates peroxynitrite (ONOO−), which is a potent oxidant. Most free radicals act locally, but superoxides and hydrogen peroxide readily cross the cell membrane and can cause remote damage.153
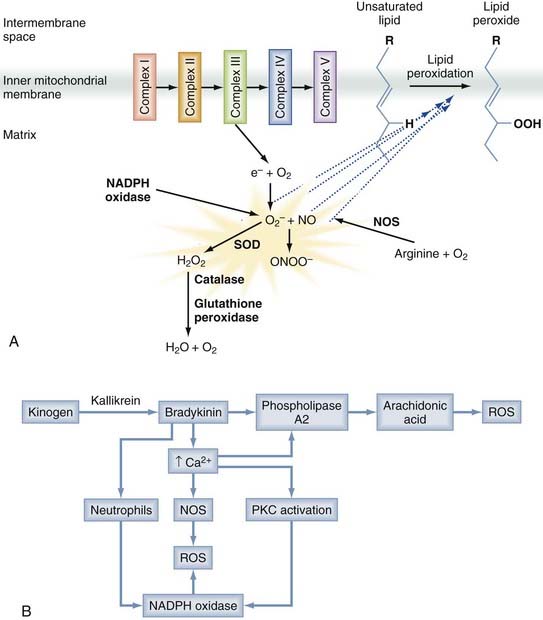
Mitochondrial dysfunction is a predominant contributor to free radical generation, but free radicals are produced in a number of additional ways, as depicted in Figure 327-4B. ROSs are also generated as a by-product of inflammation and elevated intracellular calcium. Bradykinin, formed early after nerve injury in a process catalyzed by kallikrein, activates phospholipase A2, which cleaves membrane phospholipids to produce arachidonic acid. The subsequent metabolism of arachidonic acid can generate free radical species. Moreover, the elevated calcium levels prominent after injury activate NOS, as well as NADPH oxidase.148 Inflammatory cells such as microglia and macrophages contain such enzymes, and they generate and release ROSs after trauma, thereby contributing to the oxidative damage. Free radicals are also generated in presence of free ferrous iron released by erythrocyte breakdown after trauma. This free iron acts as a catalyst in the Haber-Weiss reaction, which generates hydroxyl radicals from hydrogen peroxide and superoxide.
All these processes are linked and self-perpetuating. For instance, bradykinin increases Ca2+ levels, which increases free radical production directly and indirectly, in addition to triggering and perpetuating many other secondary injury processes. In addition, transcription of the nuclear factor NFκB is activated by ROSs and in turn upregulates the inflammatory cascade. Furthermore, ONOO− can inactivate oxidative respiration and generate further ROSs. Lipid peroxidation (Fig. 327-5A) has been associated with disruption of the BBB, thereby leading to vasogenic edema and the potential for further brain damage through elevation of ICP. Also problematic is that pyruvate dehydrogenase is highly vulnerable to oxidation and inactivation. Because this enzyme is the sole bridge between anaerobic and aerobic metabolism, damage to this enzyme significantly limits the brain’s ability to generate ATP.154
A number of endogenous enzymes normally protect against oxidative damage. Superoxide dismutases convert superoxide to hydrogen peroxide. Glutathione peroxidase and catalase can reduce hydrogen peroxide to water and molecular oxygen with the use of glutathione. As might be expected, elevated intracellular calcium can inactivate these enzymes.144
Clinical Implications
Medical science has long endeavored to augment endogenous defenses against free radicals, which are overwhelmed and insufficient after neurotrauma. Corticosteroids and the related lazaroid compounds inhibit the phospholipase A2 and cyclooxygenase pathways but have lacked efficacy or have been harmful in human clinical trials.155 Another strategy also shown to be ineffective in human clinical trials was the administration of polyethylene glycol–conjugated superoxide dismutase. It has been postulated that failure of these agents could relate to inefficient penetration into brain tissue. A number of new agents are being explored and are showing some promise. A vitamin E analogue, MDL74, was effective in a rat model of TBI. Erythropoietin and propofol have both been shown to inhibit lipid peroxidation, and both are being evaluated in human clinical trials (NCT00313716 and NCT00336882, respectively). Erythropoietin is a naturally occurring cytokine (and endocrine/paracrine marrow proliferation–inducing agent) being explored for its potential neuroprotective qualities. It is unclear how erythropoietin provides its benefits, but recognized mechanisms include anti-inflammatory, antiapoptotic, angiogenic (therefore combating ischemia), and neurotrophic qualities, with receptors being demonstrated on astrocytes and microglia. Edaravone is a novel antioxidant that has been in development in Japan since the 1980s. Edaravone showed significant improvement in human stroke and has been approved as a neuroprotectant in Japan since 2001.156 Edaravone scavenges all radicals except superoxide and thus inhibits lipid peroxidation.157 Edaravone therefore has promise for application in TBI. Finally, the newer free radical scavenger Tempol may show better brain penetration and efficacy in animal models.
Other Metabolic Derangements
Coupled Lactate Metabolism
Usually, aerobic glycolysis is the only form of metabolism used in the unstressed brain. Until recently, it has been dogma that neurons and glia use glucose exclusively as their sole energy source. An increasing body of evidence now suggests that astrocytes and glia may have the ability to use “coupled lactate metabolism” to meet their energy needs. Thus, neurons and astrocytes are functionally coupled in their metabolic activity such that when astrocytes transport glucose across the BBB, they can anaerobically metabolize it to lactate. Lactate, released into the extracellular space, is taken up by neurons and metabolized aerobically, by mitochondrial oxidative phosphorylation, to generate energy (36 ATP molecules). As neuronal activity is increased, potassium and glutamate are released into the extracellular space and taken up by the astrocytes in an energy-dependent fashion, thereby resulting in more astrocytic glycolysis. Under extreme conditions, such as TBI, aerobic metabolism may be overwhelmed either by a reduction in oxygen delivery or by increased release of excitatory neurotransmitters with resultant increased lactate accumulation. Evidence suggests that ionic pumping and glutamate surges in astrocytes both preferentially activate anaerobic glycolysis, thus producing lactate, especially in astrocytes.158 It has been proposed that in this model of coupled energy metabolism, the cerebral metabolic rate of glucose may be reflective of astrocytic metabolic function, whereas CMRO2 may be indicative of neuronal function.159–161 Recently, it has been shown in animal models that lactate uptake is significantly increased at the injury site, where metabolism is probably the greatest.162 Lactate infusion initiated 30 minutes after a fluid percussion injury resulted in the animals demonstrating significantly reduced cognitive deficits when compared with their saline infusion counterparts.163 Finally, there is growing experimental evidence that the brain can use alternative energy substrates after injury. Specifically, a shift of brain metabolism toward ketone metabolism was studied in rats after TBI induced by controlled cortical impact.164 The ability to use ketones for metabolism was found to occur in an age-dependent manner. In this investigation, rats of varying age were given a ketogenic diet after TBI. Younger animals demonstrated a reduction in cortical contusion volume. The data suggest that although alternative metabolic substrates may be used after TBI and can offer some degree of neuroprotection, this protection may be limited to younger patients. This complex relationship continues to be investigated in experimental models of TBI.
Cell Death
Nomenclature of Cell Death
Over time it has become difficult to draw definitive conclusions on the neuronal reactions to trauma because of inconsistencies in the use of cell death terminology. Therefore, an understanding of cell death mechanisms, particularly necrosis and apoptosis, is central to a discussion on the neuronal response to TBI. One of the prevailing messages from the Nomenclature Committee on Cell Death (NCCD)142 is that more than one methodology should be used before associating a specific type of cell death with a particular event. It is also clear (at least with currently available information) that despite implementing more rigorous nomenclature standards, different cell death pathways may not be completely distinguishable from one another. Many times apoptosis and necrosis mechanisms can cooperate to execute cell death via the enzymatic process involved in the generation of that morphology. The NCCD recognizes four distinct modalities of cell death: (1) apoptosis, (2) autophagy, (3) necrosis, and (4) cornification (this will not be discussed because it occurs in the epidermis). There are also atypical forms of cell death relevant to TBI mentioned by the NCCD. These processes combine features of the recognized modalities listed but do not warrant a distinct classification of cell death, and there are situations in which mixed features of the three principal modalities are demonstrated, including the commonly mentioned TBI pathways such as excitotoxicity and wallerian degeneration. Some have suggested that a continuum exists between apoptosis and necrosis and that the eventual fate of a cell is a result of the dynamic, regional, and evolving microenvironment induced by TBI. Necrosis may be more likely with severe injuries,165 whereas apoptosis plays a greater role in milder injury after TBI.166 Some evidence suggests that low intracellular calcium levels lead to apoptosis whereas high intracellular calcium leads to necrosis.108,114 Previous studies have also described neurons undergoing necrotic cell death as being shrunken with Nissl staining, undergoing vacuolization, and exhibiting eosinophilic material.167 This observation has been supported by the finding of isolated necrotic neurons in the neocortex, cerebellum, and the C1 and C3 pyramidal layers of the dentate hilus of the hippocampus in many animal and human studies after TBI.113,168 A key determinant may be whether the cell has an energy supply (i.e., ATP). Apoptosis is an energy-dependent process. Because one of the prominent features of TBI is mitochondrial damage, ATP concentrations may become exhausted and cell death mechanisms may shift from apoptosis to necrosis.169–171 Neuronal loss initiated via apoptosis may convert to necrosis if the local microenvironment sustains mitochondrial damage that results in the depletion of intracellular ATP concentrations. It is generally accepted that contusions, coupled with the inevitable perilesional ischemia, cause primarily localized necrosis with only scattered apoptotic cell death.172,173 This conclusion is supported by morphologic observations with electron microscopy in humans and animals that demonstrated neuronal changes most consistent with necrosis (swelling of mitochondria and cell, pyknotic nuclei, and vacuolated cytoplasm) in the early posttraumatic period, particularly after focal TBI.174,175 These mechanisms seem to persist with time, at least partially, in the subacute and chronic stages of injury, as noted by an increase in cortical lesion size with time.167,176
Autophagic cell death is characterized by “the sequestration of cytoplasmic material with autophagosomes.”140 Morphologic features include (1) the absence of chromatin condensation (as opposed to apoptosis), (2) the presence of vacuolization of the cytoplasm, and (3) no association with phagocytes (also different from apoptosis).
Necrosis is morphologically characterized by “increased cell volume (oncosis), swelling of organelles, and plasma membrane rupture with loss of intracellular contents.”140 Evidence is accruing that necrosis is not simply an uncontrolled passive process but is regulated through a cascade of events similar to other forms of cell death. Potential biochemical reactions indicative of necrotic cell death in TBI include activation of calpains and cathepsins.
The morphologic features of apoptosis are “rounding-up of the cell, retraction of pseudopodes, reduction of cellular volume (pyknosis), chromatin condensation, nuclear fragmentation (karyorrhexis), classically little or no ultrastructural modifications of cytoplasmic organelles, plasma membrane blebbing, and engulfment by resident phagocytes.”140 To be classified as apoptosis, several of these features must be present with concomitant demonstration of related enzymatic or biochemical processes, such as activation of Bcl-2 family proteins, DNA fragmentation, and mitochondrial transmembrane dissipation.140 Apoptotic cell death also has a prominent role in diffuse TBI.177 Apoptotic cells have been seen in the cortex, diencephalon, and hippocampus, as demonstrated by suggestive biochemical assays and further supported via TUNEL staining (terminal deoxynucleotidyl transferase dUTP-biotin nick end labeling).166,178,179 Previous investigations have suggested that some cells display characteristics of both apoptosis and necrosis and continue to refer to this as a hybrid form of cell death termed necroptosis or aponecrosis.7,180 The significance of differentiating different forms of cell death revolves around two main issues. Specific drugs have been shown to block the apoptotic mechanisms (ZVAD, minocycline, and cyclosporine among others). Second, because apoptotic cell death is slow in evolving, drug treatment may more easily be initiated before the apoptosis process has become inevitable. At least two new drugs, Solvay SL334 and Neuren NZ, address the apoptotic mechanisms in part and are beginning evaluation in clinical trials.
Calpains have been shown to be involved in both apoptosis and necrosis. As detailed previously, severe TBI causes an excess of calcium influx that leads to the activation of calpains, which then induce cytoskeletal damage and membrane permeability through cleavage of target substrates.7,118 Calpains and caspases will cleave spectrin at specific points and produce characteristic cleavage products based on their weight. These specific cleavage products are referred to as spectrin breakdown products (SBDPs) and include SBDP-120 (from cleavage of caspase), SBDP-145 (cleavage of calpain), and SBDP-150 (more indicative of general protease activity).118 SBDPs have frequently been shown to be present after TBI in humans and animals.181 In fact, the presence of SBDPs is now routinely used to verify the occurrence of CMSP. Furthermore, there are ongoing trials, the “Banyan Biomarker” clinical studies, to determine whether SBDPs could be of clinical use as a biomarker for phases of TBI.182,183
Caspases are another member of the cysteine protease family activated by TBI. Caspases have an integral role in neuronal death across many modes of CNS insult, including ischemia, free radical production, radiation, ischemia, and TBI.7 Caspases are a large family of 14 proteins and have been divided into two functional categories—activator and executioner caspases.184 CNS trauma causes autocleavage and activation of capsase-8 or caspase-9 (or both).185,186 This initiator caspase subsequently activates the executioner caspase-3, which acts downstream to promote apoptosis via multiple mechanisms, including release of cytochrome c, cleavage of antiapoptotic regulators, cleavage and activation of DNA fragmentation endonucleases, and cleavage of the cytoskeletal elements actin and spectrin in the BBB.186 Increased caspase-3 activity has been shown to be involved after focal and diffuse TBI in animal and human brain injury.178,182 Therapeutic strategies using varying techniques to suppress caspase activity (i.e., inhibitors, transgenics) have exerted neuronal protection in models of focal TBI187 and diffuse TBI.188 Generally speaking, caspases act downstream from the actions of calpains and are more closely associated with apoptosis.118 Apoptosis can occur via caspase-dependent and caspase-independent pathways. Moreover, there are two caspase-dependent pathways leading to activation of caspase-3 and apoptosis—the intrinsic and extrinsic pathways.189 The caspase-dependent pathways, both intrinsic and extrinsic, are depicted in Figure 327-3 and are described in the following sections.
Intrinsic Pathway of Caspase-Dependent Apoptosis
The intrinsic pathway is initiated by a pathologic intracellular process (such as DNA damage or compromised integrity of the mitochondrial membrane) brought about by an apoptotic trigger (e.g., mechanical forces, protease activity, an increase in proapoptotic proteins such as Bax or Bak) (see Fig. 327-5). This increase in outer membrane permeability will permit the release of several proteins from the intermembrane space into the cytoplasm. Many of these released proteins have pre-apoptotic function, including cytochrome c, Smac/Diablo, HtrA2 (Omi), AIF, and DNaseG.190 Cytochrome c then induces apoptosis via the following cascade of events. In the presence of ATP, cytochrome c will bind and activate apoptosis protease-activating protein-1 (Apaf-1). Together, this complex (cytochrome c/Apaf-1) will activate caspase-9; caspase-9 then cleaves the proenzyme form of caspase-3, which leads to its activation and subsequent apoptosis. For apoptosis to proceed, inhibitor of apoptosis proteins must be inactivated. This is achieved by the release of Smac/Diablo from the mitochondria.
Extrinsic Pathway of Caspase-Dependent Apoptosis
In the extrinsic pathway, activated death ligands (Fas/CD95, TNF-α1) bind to their corresponding receptor and bring about oligomerization of apoptotic death receptors with subsequent initiation of an ordered sequence of events (see Fig. 327-5).186 The death receptor then forms a complex with adapter proteins, such as fas-associated death domain (FADD), to induce the autolytic activation of caspase-8. Caspase-8 will then ultimately activate caspase-3. This process is not thought to require gene induction or protein synthesis, both of which are necessary for the intrinsic process.191
Unique Neuronal And Glial Responses To Traumatic Brain Injury
“Nonlethal” Neuronal Reactive Change to Diffuse Traumatic Brain Injury
Neuronal cell death after diffuse TBI was described earlier, but neuronal injury exclusive of cell death has received little attention. Much of the neuronal injury sustained after TBI is caused by damage initiated via enzymatic processes; however, recent investigations have demonstrated that the mechanical forces inherent in TBI may serve as a pathologic mechanism itself. It had previously been believed that primary axotomy always elicited some neuronal necrosis,192 but it is now known that evidence exists for cellular reorganization and repair as denoted by reestablishment of protein synthesis, lengthening of the axotomized segment, and a reduction in axonal swelling after TBI.29,69 It is important to point out, as described in previous investigations,108 that this study also showed scattered necrotic and apoptotic neurons after diffuse TBI in somata not associated with perisomatic secondary axotomy.
Cell Membrane Poration after Traumatic Brain Injury
Recent studies have shown four distinct types of reversible membrane pathology: (1) those that reseal early after early perturbation, (2) those that exhibit delayed resealing after early perturbation, (3) those that have enduring permeability after early perturbation, and (4) those with delayed permeability. Liu and Schnellmann demonstrated that there is not a causal relationship between CMSP and membrane permeability.193 Thus, the forces of trauma, when not directly causing axotomy, may alter permeability of the neuronal plasmalemma and the axolemmal membranes. The magnitude or duration of the forces may determine the fate of the damaged soma or axon (or both). A temporal progression of membrane poration also appears to occur, with a redistribution of poration subtypes taking place between 4 and 8 hours. As time evolved, more neurons revealed delayed poration as opposed to resealed neurons, thus suggesting ongoing secondary mechanisms that continue to result in changes in permeability. Finally, the long-term fate of neurons undergoing perisomatic axotomy in the thalamus was explored via central fluid percussion injury, a diffuse model of TBI. The cells were observed for up to 28 days and were shown to atrophy rather than degenerate as determined by microscopic evaluation and measurements of nuclear cytoplasmic volumes.194
Neuroinflammation
Multiple studies have shown that neuroinflammatory events play important dual and opposing roles: on one hand, brain damage mediated by the release of neurotoxic substances and, on the other hand, initiation of demolition and repair of the injured tissue. Polymorphonuclear leukocytes begin to accumulate in damaged brain tissue within 24 hours after acute injury.195,196 Thirty-six to 48 hours after injury, macrophages are seen197 and secrete many factors, including cytokines.198 Cytokines are vasoactive substances that have been shown to increase vascular permeability and induce edema formation,199,200 and they have direct cytotoxic effects on glial and neuronal cells.201 Multiple cytokines appear to trigger these processes; for example, TNF-α is upregulated in animal models of TBI 1 to 4 hours after injury. This rapid response is thought to be due to the synthesis of TNF-α by primary cells of the CNS (glia and neurons) rather than by systemic inflammatory cells.201,202 TNF-α plays an important role in regulation of cell growth, immunomodulation, inflammation, and autoimmune processes.203 Furthermore, TNF-α and interleukin-1β (IL-1β) may exert deleterious effects on the CNS by facilitating secondary damage, such as vasogenic edema and delayed neuronal death, and thereby contribute to the pathophysiology of acute TBI.204,205 Conversely, it has also been reported that these inflammatory cytokines initiate wound-healing processes such as fibrosis and neovascularization in the CNS197 and that TNF-α may play a neuroprotective role after ischemic brain injury in the immune response against excitotoxic, metabolic, and oxidative insults.206 Toulmond and Rothwell have shown that treatment with an IL-1β receptor antagonist decreases experimentally induced TBI cell death and functional deficits.207
Early perivascular aggregation of polymorphonuclear inflammatory cells that correlates temporally and anatomically with early opening of the BBB has been demonstrated experimentally.193,208 Holmin and colleagues have shown a varied inflammatory response in human cerebral contusions.209 In patients who underwent resection of their contusions less than 24 hours after injury, the inflammation was mainly intravascular and dominated by polymorphonuclear cells, whereas in patients undergoing surgery 3 to 5 days after trauma, the inflammation was parenchymal and consisted of monocytes/macrophages, reacting microglia, polymorphonuclear cells, and CD4- and CD8-positive T lymphocytes. These findings correlate with a rat model of closed head injury in which biphasic development of edema was detectable, with the delayed phase reaching a maximum on day 6 after trauma that was correlated with an inflammatory infiltrate consisting of monocytes and lymphocytes.209,210 An increase in the inflammatory cytokines IL-1β, IL-6, and TNF-α was detected in the cerebrospinal fluid of patients with severe brain injuries.211 More recently, it has been shown in experimental TBI that TNF-α and NFκB (p50 and p65) may play an important role in the injury-induced immune response.212 Furthermore, improved TBI outcome has been linked to peak IL-6 levels after severe TBI.213
In addition to cytokines, animal studies first linked the complement cascade to secondary injury mechanisms after TBI. Working on this premise, a study was undertaken to analyze human brain tissue resected during the surgical management of TBI because of intractable increased ICP.214 Analysis of this resected tissue revealed increased immunoreactivity for the complement components C1q, C3b, and C3d and the C5b-9 membrane attack complex in the penumbra of the contusion (Fig. 327-6). There was also an indication of local synthesis of complement. These results suggest traumatically induced complement activation, which leads to the development of secondary brain damage through excessive activation of inflammatory cascades.215,216
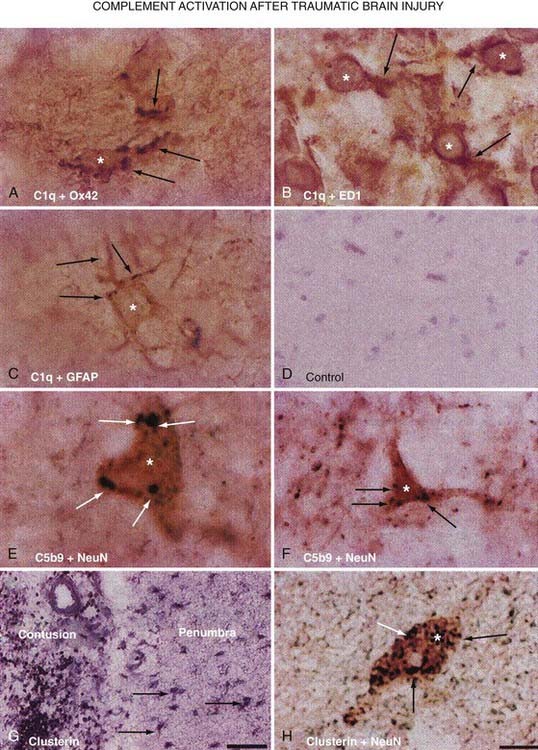
Gahm and colleagues described the temporal profiles of expression of the three isoforms of NOS in a rat model of focal brain contusion.217 They demonstrated that the invading inflammatory cells (polymorphonuclear cells and macrophages) express neuronal NOS and inducible NOS (iNOS), thus identifying an important therapeutic target for amelioration of NO-mediated injury in trauma studies. A postmortem study of human brains from patients who had died after TBI showed upregulation of iNOS more than 48 hours after TBI and absent 8 days after injury.218 Their findings confirmed prolonged iNOS induction.
The release of arachidonic acid with its subsequent metabolism to prostaglandins and leukotrienes is regarded as a key early response linked to neuronal signal transduction. Upregulation of phospholipase A2 after TBI results in the accumulation of bioactive metabolites such as arachidonic acid, oxygenated derivatives of arachidonic acid, and platelet-activating factor. Exogenous prostaglandin E2 (a product of cyclooxygenation) was found to significantly inhibit microglial activation and TNF-α in experimental TBI and may play an important role in modulating the immune response occurring at the injured site.219 Cyclooxygenase, an obligate rate-limiting enzyme for the conversion of arachidonic acid to prostaglandins, is known to be induced after TBI220 and may serve as a potential pharmacologic target in the future.221
Other Molecules And Pathways Important After Traumatic Brain Injury
Second-Messenger Systems and Neurotransmitters
Second messengers are large molecules, usually situated within the neuronal membrane or adjacent to its inner surface within the cytoplasm, that have the capability of modulating or amplifying external signals brought to the cell via neurotransmitters and mediators, such as glutamate, adenosine, steroids, and acetylcholine. A number of studies have shown that second-messenger systems, probably because of their large molecular size and the complexity of their stearic interactions, are vulnerable to the shear forces of neurotrauma. In some circumstances, second-messenger systems may be amplified (up to 200-fold or more) by neurotrauma, whereas other types of second messengers are downregulated or deactivated.43,222 It is thought that such systems may play an important role in complex neurological processes, such as encoding of memory, and thus these changes in second-messenger systems could constitute a mechanism for the behavioral and memory changes that occur in both animals and humans after neurotrauma.223 In the central fluid percussion model, few or no anatomic changes are seen in the presence of these long-lasting neurobehavioral deficits.44,45
Acetylcholine production has been reported to be upregulated in brain tissue and cerebrospinal fluid after TBI in humans224 and experimental models.225,226 Although postmortem studies of injured patient brains have revealed a marked reduction in choline acetyltransferase activity in the temporal cortex, preservation of muscarinic receptor binding sites supports the view that the reduced choline acetyltransferase may be associated with cognitive impairment in survivors of head injuries.227 It has also been suggested that cholinergic mechanisms may be responsible for the cognitive dysfunction in experimental models of TBI.228,229 Finally, it has been proposed that changes in catecholamine and monoamine neurotransmitters after head injury may be indicators of the severity of brain damage.230,231 Changes in tissue concentrations of dopamine, noradrenaline, and adrenaline have been found in experimental models of TBI.232
Transcriptional Response/Stress Proteins/Immediate Early Genes
Immediate Early Genes
Immediate early genes (IEGs) are increased transiently after the insult and represent an important interface between environmental signals and the cellular genome and thus are an important signal transduction event. Many IEGs act as “cellular switches” that program transcription patterns important for neuronal survival, intercellular organization, and synaptic reorganization. IEGs are regulators of RNA expression for “late response genes” such as cyclins and growth factors. DNA-binding proteins are usually generated in the form of transcription factors. In other words, these IEGs link short-term extracellular insults to long-term phenotypic changes via alterations in gene expression. Binding of transcription factors to the DNA promotor site allows the generation of late secondary genes.233 Studies in both animal models234 and humans235 have demonstrated that fos proteins are expressed in neuronal and glial cells within 5 minutes after TBI.236 The glucose transporter gene is also upregulated in neurons that do not usually progress to death after a stressful event. An investigation of IEGs after contusional head injury found increased expression of c-fos and c-jun in more than 50% of patients.237 Those with poorer outcomes had higher levels of gene expression. It was thus concluded that IEGs may be involved in the pathogenic mechanisms of contusional head injury.
Stress Response Proteins
The most important group, heat shock proteins, is involved in the folding and intracellular transport of damaged proteins. Heat shock protein 70 (HSP 70) is upregulated after TBI and has been speculated to be part of a cellular response that involves repair of damaged proteins and is thought to occur in stressed neurons destined to survive an insult.238–240 Upregulation of HSP 72 may be increased during neuronal injury and appears to be involved in cellular repair mechanisms and cytoprotection.241 HSP 72 upregulation may also serve as a marker during neuronal injury.242 It is not yet clear whether their appearance is beneficial or an index of an irreversible injury.243
Bcl-2 Family of Proteins
As is becoming apparent, the neuronal response to TBI, particularly as it pertains to cell death, does not follow a rigid sequence of events. In vivo investigations continue to reveal a dynamic interrelationship among many mediators, including the B-cell lymphoma (Bcl-2) proto-oncogene family. This family has been projected to participate in both necrosis and apoptosis across several models of CNS injury, including human and experimental TBI.177,244,245 This large family of proteins has been divided into prodeath (e.g., Bax, Bad) and prosurvival (e.g., Bcl-2, Bcl-xL) groups based on their enzymatic activity.246 When considering apoptosis, it is helpful to categorize factors that initiate, modulate, or execute apoptotic cell death. The Bcl-2 family is closely involved in modulation of the intrinsic and extrinsic pathways of caspase-dependent apoptosis and serves to either promote or inhibit apoptotic cell death,247 as previously depicted in Figure 327-5. It is clear that a complex and dynamic relationship exists among the Bcl-2 family proteins after TBI. A precise understanding of the family’s protein expression patterns has not been achieved to date. For example, studies have shown both increased178 and decreased248 expression of prosurvival members after experimental diffuse TBI. An example of how this family influences cell death is detailed as follows: after an appropriate apoptotic trigger, such as p53 expression after TBI, expression of Bax is upregulated and it is subsequently translocated from the cytoplasm to the mitochondria, where it induces the release of cytochrome c.246 This was also shown to occur in a focal model of brain injury.141 As reviewed elsewhere, cytochrome c then binds Apaf-1 and activates the caspases involved in apoptosis. Cernak and coworkers demonstrated the presence of this process in diffuse TBI.178 A final example of Bcl-2 family involvement in supporting apoptotic death after trauma is the proapoptotic member Bid. In an investigation of focal TBI via a focal cortical impact model of injury, increased Bid cleavage was detected, with possible subsequent activation of the initiator enzymes caspase-8 and caspase-9 to support apoptosis.249
In contrast to Bax, the prosurvival members of the family can directly and indirectly affect proapoptotic signaling pathways. Yang and colleagues demonstrated that overexpression of Bcl-2 in the mitochondrial outer membrane facilitated cell survival by preventing the release of cytochrome c and thereby halting activation of caspase-3.250 Using a transgenic strain of mouse overexpressing Bcl-2, Nakamura and colleagues confirmed the neuronal protective ability conferred by Bcl-2 in a focal model of TBI.251 This same feature has been shown in animal models and human head injury.108,187,245 Furthermore, Bcl-2 and alike family members can indirectly alter a critical pore-forming function of these complexes and block the release of cytochrome c.252 In this case, Bcl-2 forms a heterodimer with Bax, and this complex interferes with Bax-generated release of cytochrome c. Alternatively, a decrease in Bcl-2 expression has been linked to a propensity for cell death.166
In addition to involvement of Bcl-2 family proteins in caspase-mediated apoptosis, Tsujimoto and Shimizu have also described a relationship between MPTP regulation and the Bcl-2 family of proteins.253 Bcl-2 family proteins have been localized to the mitochondrial outer membrane and can regulate MPTP and outer membrane permeability.254–256 In experimental paradigms, two prosurvival members of this family, Bcl-2 and Bcl-xL, have been shown to directly inhibit activity of the voltage-dependent anion channel component of the MPTP, thereby blocking MPT.257,258
Mitogen-Activated Protein Kinases
Mitogen-activated protein kinases (MAPKs) are another regulatory family of proteins involved in translating extracellular stress signals into intracellular signaling pathways. They have been studied in a number of injury paradigms and have a relatively complex regulatory pattern that has varied in accordance with the specific experimental insult implemented.259 Because they induce intracellular signaling in response to extracellular cues, there has been a great deal of interest in exploring their therapeutic modulation as a means of modifying apoptosis after an assortment of CNS insults. As with Bcl-2 family proteins, they are in dynamic balance with one another. Depending on the signal, MAPK members can either promote or resist cell death when activated through phosphorylation. Neurons with an apoptotic phenotype have been linked to the activation and increase in c-Jun N-terminal kinase (JNK) or p38 MAPK in models of epilepsy260 and ischemia.261 Typically balancing the actions of JNK and p38 MAPK are extracellular signal–regulated kinases (ERK1/2) and Akt kinase.259 Pharmacologic inhibition of an ERK activator increased cell survival, whereas suppression of p38 kinase and JNK did not confer protection. More current studies supporting these findings have revealed phosphorylation and activation of ERK1/2 after diffuse and focal TBI.262–264 The precise mechanisms that determine the actions of MAPK family members, whether it be prosurvival or proapoptotic for a given insult, remain unclear.
Lysosomal Pathway
Limited membrane disruption, as can result from mechanical microporation of neuronal membranes, yields the release of cathepsin, whereas generalized membrane rupture typically leads to necrosis. Cathepsins are also part of the cysteine protease family and are primarily located in lysosomes.265 Furthermore, in predominantly ischemic models of injury, Yamashima and associates demonstrated calcium-induced calpain-mediated activation of the lysosomal hydrolytic pathways.181,265,266 Autophagy is a cell death mechanism that is dependent on the actions of lysosomes and their hydrolytic enzymes for the sequestration, clearance, and bulk degradation of cytoplasmic material.125 Evidence for the presence of autophagic cell death in TBI comes through the identification of biochemical and morphologic (presence of autophagosomes) markers of autophagy.267
Modifiers Of Response To Traumatic Brain Injury
Age
Age is generally recognized as the strongest predictor of outcome after TBI, even after adjustment for potential confounders of this effect.268,269 Although an age threshold has been suggested,270 current evidence suggests a continuous relationship between increasing age and worsening outcome after TBI.267 It is believed that this may reflect a decreased capacity for brain repair, as well as increased susceptibility to the complications of TBI.
TBI is the most common cause of death and disability in the pediatric population, and in this group, age 4 years or younger is associated with poorer outcome despite the plasticity inherent in the immature brain.271,272 The pediatric brain is more susceptible to diffuse swelling, although the reason for this remains to be characterized.273 It may also be more susceptible to apoptotic cell loss.273 It is uncertain, however, whether these factors account for this poor prognosis.
Genomics and Apolipoprotein E
As we learn more about the human genome, it is increasingly being recognized that inheritance of specific alleles can alter the incidence and course of many illnesses. Alleles that may alter the course of TBI are increasingly being described, although this work remains in its infancy. The most recognized association between a genetic polymorphism and outcome involves the apolipoprotein E (apo E) gene. Apo E is produced by glial cells and is the major lipid transport lipoprotein in cerebrospinal fluid. It is also responsible for maintenance of the structural integrity of microtubules within the axon or neuron. Apo E is a 299–amino acid protein encoded by a single gene and has three allelic variants that yield three isoforms (ε2, ε3, and ε4) with frequencies of 7%, 78%, and 15%, respectively, in white individuals.259 Even a single copy of the ε4 allele has been associated with poorer outcome after TBI in multiple studies,274–279 including a recent meta-analysis.280 It has now been demonstrated that the apo E ε4 isoform is associated with an increased risk for Alzheimer’s disease, and it seems to play a role in the deposition of β-amyloid, especially after TBI. Mayeux and colleagues in 1995 showed that a history of a previous head injury and apo E ε4 interact synergistically to cause a 10-fold increase in the risk for Alzheimer’s disease versus a 2-fold increase in risk with apo E ε4 alone.281 Additionally, apo E ε4 is associated with larger intracerebral hematomas and greater ischemia after TBI.277
Other genetic variations are being described that appear to influence outcome after TBI, although these associations are as yet supported by a smaller body of research. Polymorphisms of IL-1, angiotensin-converting enzyme, p53, the dopamine D2 receptor, and catechol-O-methyltransferase have recently been associated with poorer outcomes.274,282 Confirmation of these findings and an understanding of their molecular basis may improve prognostication after TBI and could suggest novel treatment strategies.
Our increased understanding of genetics is rapidly bringing us into an era of molecular medicine. DNA microarray technology is currently being used by neuroscientists to evaluate changes in expression of very large numbers of genes after TBI. A recent microarray analysis performed in vitro looked at changes in gene expression in response to modeled TBI.283 Mild injury resulted in the differential expression of 999 genes, whereas 587 were differentially expressed with more severe injury. In both injury severities, the majority of the alterations in expression consisted of downregulation. With milder injury, the differentially expressed genes were found to be important for structural damage to the cellular architecture, cell signaling, and regulation of transcription. With more severe injury, genes involved in inflammation, necrosis, and apoptosis were upregulated.283 A second analysis examined gene expression in the rat brain 1 and 4 days after injury. At the early time point, changes in expression were predominantly noted in genes involved in cellular repair and metabolism. Fewer genes were differentially expressed at the 4-day time point, and a large proportion of these genes are involved in the immune response.284 Better understanding of these extremely complex data will also be an important step in our understanding of TBI.
The Role of Gender
Important sex differences exist in many aspects of TBI. Women suffer TBI less often than men do and are typically older at the time of injury.285,286 Gender differences in the physiologic response to TBI are increasingly being described. These sex-related differences may involve differential presence of the Y chromosome or the obviously different hormonal milieu.287,288 Anatomic and physiologic sex differences are known to exist in the uninjured as well as the injured brain. In the absence of injury, females have higher hemispheric blood flow and a larger volume of cortex than males do.289,290 Moreover, ovarian steroids are known to have complex and important effects on gene transcription and neuronal excitability.291 Male and female brains may thus experience the same insults in a different fashion as a result of these baseline differences. After injury, the female brain is less prone to edema.292 In addition, female neurons are less vulnerable to insult in vitro.276
An increasing body of literature suggests that there are important sex differences in the molecular consequences of TBI as well. These have largely been studied in the context of the effects of estrogen and progesterone. Interestingly, both estrogen and progesterone have been ascribed neuroprotective properties. Estrogen has been shown to increase CBF, and it has antioxidant effects.293 Estrogen also affords protection from ischemia,294 and this may, in part, stem from direct antagonism of NMDA receptors.295 Progesterone has receptors throughout the CNS and may reduce edema,291 lipid peroxidation,296,297 excitotoxicity,298 and neuronal loss299 and increase the seizure threshold. Indeed, progesterone administration has been studied in a recent phase II human clinical trial (ProTECT trial) in which it was demonstrated that males and females randomized to progesterone therapy had a significantly lower mortality rate than did those randomized to placebo. Both estrogen and progesterone may have harmful effects, however. Progesterone may exacerbate tissue swelling, and estrogen decreases the seizure threshold.300
It is unclear, however, whether females fare better, worse, or the same as males after TBI. Some studies report that females fare worse than males do,301–305 whereas others report that they fare better306 or that there is no difference.307 This relationship is made increasingly complex by the fact that sex is confounded by age and injury mechanism: females with TBI are typically older and more likely to be injured in a fall than men.288,290,301,302,308
Adams JK, Doyle D, Ford L. Diffuse axonal injury in head injury: definition, diagnosis, and grading. Histopathology. 1989;15:49-59.
Edwards P, Arango M, Balica L, et al. Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury—outcomes at 6 months. for the CRASH trial collaborators. Lancet. 2005;365;:1957-1959.
Farin A, Deutsch R, Biegon A, et al. Sex-related differences in patients with severe head injury: greater susceptibility to brain swelling in female patients 50 years of age and younger. J Neurosurg. 2003;98:32-36.
Gennarelli TA. Mechanisms of brain injury. J Emerg Med. 1993;11(suppl 1):5-11.
Gennarelli TA, Graham DI. Neuropathology of the head injuries. Semin Clin Neuropsychiatry. 1998;3:160-175.
Hukkelhoven CW, Steyerberg EW, Habbema JD, et al. Predicting outcome after traumatic brain injury: development and validation of a prognostic score based on admission characteristics. J Neurotrauma. 2005;22:1025-1039.
Katayama Y, Becker DP, Tamura T, et al. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J Neurosurg. 1990;73:889-900.
Maas AI, Murray G, Henney H3rd, et al. for the Pharmos TBI investigators. Efficacy and safety of dexanabinol in severe traumatic brain injury: results of a phase III randomised, placebo-controlled, clinical trial. Lancet Neurol. 2006;5:38-45.
Marmarou A. Traumatic brain edema: an overview. Acta Neurochir (Wien). 1994;60(suppl):421-424.
Marshall LF, Maas AI, Marshall SB, et al. A multicenter trial on the efficacy of using tirilazad mesylate in cases of head injury. J Neurosurg. 1998;89:519-525.
Mazzeo AT, Brophy GM, Gilman CB, et al. Safety and tolerability of cyclosporin a in severe traumatic brain injury patients: results from a prospective randomized trial. J Neurotrauma. 2009;26:2195-2206.
Morganti-Kossmann MC, Rancan M, Otto VI, et al. Role of cerebral inflammation after traumatic brain injury: a revisited concept. Shock. 2001;16:165-177.
Morris GF, Bullock R, Marshall SB, et al. Failure of the competitive N-methyl-D-aspartate antagonist Selfotel (CGS 19755) in the treatment of severe head injury: results of two phase III clinical trials. The Selfotel Investigators. J Neurosurg. 1999;91:737-743.
Pettus EH, Christman CW, Gieble ML, et al. Traumatically induced altered membrane permeability: its relationship to traumatically induced reactive axonal change. J Neurotrauma. 1994;11:507-522.
Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathol. 2004;14:215-222.
Reilly P, Bullock R. Head Injury: Pathophysiology and Management. London: Hodder Arnold; 2005.
Robertson CS, Valadka AB, Hannay HJ, et al. Prevention of secondary ischemic insults after severe head injury. Crit Care Med. 1999;27:2086-2095.
Rockswold GL, Ford SE, Anderson DC, et al. Results of a prospective randomized trial for treatment of severely brain-injured patients with hyperbaric oxygen. J Neurosurg. 1992;76:929-934.
Saatman KE, Duhaime AC, Bullock R, et al. for the Workshop Scientific Team and Advisory Panel Members. Classification of traumatic brain injury for targeted therapies. J Neurotrauma. 2008;25:719-738.
Singleton RH, Zhu J, Stone JR, et al. Traumatically induced axotomy adjacent to the soma does not result in acute neuronal death. J Neurosci. 2002;22:791-802.
Strich SJ. Diffuse degeneration of the cerebral white matter in severe dementia following head injury. J Neurol Neurosurg Psychiatry. 1956;19:163-185.
Temkin NR, Anderson GD, Winn HR, et al. Magnesium sulfate for neuroprotection after traumatic brain injury: a randomised controlled trial. Lancet Neurol. 2007;6:29-38.
Wolf JA, Stys PK, Lusardi T, Meaney D, et al. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci. 2001;21:1923-1930.
Wright DW, Kellermann AL, Hertzberg VS, et al. ProTECT: a randomized clinical trial of progesterone for acute traumatic brain injury. Ann Emerg Med. 2007;49:391-402.
Yakovlev AG, Faden AI. Mechanisms of neural cell death: implications for development of neuroprotective treatment strategies. NeuroRx. 2004;1:5-16.
Zhou W, Xu D, Peng X, et al. Meta-analysis of APOE4 allele and outcome after traumatic brain injury. J Neurotrauma. 2008;25:279-290.
1 Reilly P, Bullock R. Head injury: Pathophysiology and Management. London: Hodder Arnold; 2005.
2 Gennarelli TA, Thibault LE. Biomechanics of acute subdural hematoma. J Trauma. 1982;22:680-686.
3 Zimmerman RA, Bilaniuk LT, Gennarelli T, et al. Cranial computed tomography in diagnosis and management of acute head trauma. AJR Am J Roentgenol. 1978;131:27-34.
4 Hentall ID, Pinzon A, Noga BR. Spatial and temporal patterns of serotonin release in the rat’s lumbar spinal cord following electrical stimulation of the nucleus raphe magnus. Neuroscience. 2006;142:893-903.
5 Schiff ND, Giacino JT, Kalmar K, et al. Behavioural improvements with thalamic stimulation after severe traumatic brain injury. Nature. 2007;448:600-603.
6 Gennarelli TA. Mechanisms of brain injury. J Emerg Med. 1993;11(suppl 1):5-11.
7 Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathol. 2004;14:215-222.
8 Gennarelli TA, Graham DI. Neuropathology of the head injuries. Semin Clin Neuropsychiatry. 1998;3:160-175.
9 Gennarelli TA, Thibault LE, Adams JH. Diffuse axonal injury and traumatic coma in the primate. In: Dacey RG, editor. Trauma of the Central Nervous System. New York: Raven Press; 1985:169-193.
10 Holbourne AHS. Mechanics of head injury. Lancet. 1943;2:438-441.
11 Gurdjian ES, Lissner HR, Hodgson VR. Mechanisms of head injury. Clin Neurosurg. 1966;12:112-128.
12 Strich SJ. Diffuse degeneration of the cerebral white matter in severe dementia following head injury. J Neurol Neurosurg Psychiatry. 1956;19:163-185.
13 Bowman CL, Ding JP, Sachs F, et al. Mechanotransducing ion channels in astrocytes. Brain Res. 1992;584:272-286.
14 Kimelberg HK, Norenberg MD. Astrocytes. Sci Am. 1989;260:66-76.
15 Orkand RK, Nicholls JG, Kuffler SW. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J Neurophysiol. 1966;29:778-806.
16 Bullock R, Maxwell WL, Graham DI, et al. Glial swelling following human cerebral contusion: an ultrastructural study. J Neurol Neurosurg Psychiatry. 1991;54:427-434.
17 Bullock R, Landolt H, Maxwell WL, et al. Massive astrocytic swelling in response to extracellular glutamate—a possible mechanism for post traumatic brain swelling? Acta Neurochir (Wien). 1994;60:465-467.
18 Newman EA. High potassium conductance in astrocyte end feet. Science. 1986;223:453-454.
19 Di X, Goforth PB, Bullock R, et al. Mechanical injury alters volume of activated ion channels in cortical astrocytes. Acta Neurochir Suppl. 2000;76:379-383.
20 Schroeder ML, Muizelaar JP, Kuta AJ. Documented reversal of global ischemia immediately after removal of an acute subdural hematoma. J Neurosurg. 1994;80:324-327.
21 Peerless SJ, Rewcastle NM. Shear injuries of the brain. Can Med Assoc J. 1967;96:557-582.
22 Adams JK, Doyle D, Ford l. Diffuse axonal injury in head injury: definition, diagnosis, and grading. Histopathology. 1989;15:49-59.
23 Pettus EH, Christman CW, Gieble ML, et al. Traumatically induced altered membrane permeability: its relationship to traumatically induced reactive axonal change. J Neurotrauma. 1994;11:507-522.
24 Buki A, Siman R, Trojanowski JQ, et al. The role of calpain-mediated spectrin proteolysis in traumatically induced axonal injury. J Neuropathol Exp Neurol. 1999;58:365-375.
25 Shields DC, Schaecher KE, Hogan EL, et al. Calpain activity and expression increased in activated glial and inflammatory cells in penumbra of spinal cord injury lesion. J Neurosci Res. 2000;61:146-150.
26 Okonkwo DO, Povlishock JT. An intrathecal bolus of cyclosporin A before injury preserves mitochondrial integrity and attenuates axonal disruption in traumatic brain injury. J Cerebral Blood Flow Metab. 1999;19:443-451.
27 Buki A, Okonkwo DO, Wang KKW, et al. Cytochrome c release and caspase activation in traumatic axonal injury. J Neurosci. 2000;20:2825-2834.
28 McLellan DR, Adams JH, Graham DI, et al. The structural basis of the vegetative state and prolonged coma after non-missile head injury. In: Papo I, Cohadon F, Massarotti M, editors. Le Coma Traumatique. Padua, Italy: Liviana Editrice; 1986:165-185.
29 Singleton RH, Zhu J, Stone JR, et al. Traumatically induced axotomy adjacent to the soma does not result in acute neuronal death. J Neurosci. 2002;22:791-802.
30 Singleton RH, Stone JR, Okonkwo DO, et al. The immunophilan ligand FK506 attenuates axonal injury in an impact-acceleration model of traumatic brain injury. J Neurotrauma. 2001;18:607-613.
31 Mazzeo AT, Brophy GM, Gilman CB, et al. Safety and tolerability of cyclosporin A in severe traumatic brain injury patients: results from a prospective randomized trial. J Neurotrauma. 2009;26:2195-2206.
32 Hayashi K, Handa H, Nagasawa S, et al. Stiffness and elastic behavior of human intracranial and extracranial arteries. J Biomech. 1980;13:175-184.
33 Sachs F. Mechanical transduction by membrane ion channels: a mini review. Mol Cell Biochem. 1991;104:57-60.
34 Tavalin SJ, Ellis E, Satin SJ. Mechanical perturbation of cultured cortical neurons reveals a stretch-induced delayed depolarization. J Neurophysiol. 1995;74:2767-2773.
35 Bullock R, Zauner A, Tsuji O, et al. Patterns of excitatory amino acids release and ionic flux after severe human head trauma. In: Proceedings of the 9th International Symposium on ICP and Brain Damage. Tokyo: Springer-Verlag; 1995:64-71.
36 Di X, Lyeth B, Hamm R, et al. Voltage dependent Na/K ion channel blockade fails to ameliorate behavioral deficits after traumatic brain injury in the rat. J Neurotrauma. 1996;13:497-504.
37 Katayama Y, Becker DP, Tamura T, et al. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J Neurosurg. 1990;73:889-900.
38 Nilsson P, Hillered L, Olsson Y, et al. Regional changes in interstitial potassium and calcium levels following cortical compression, contusion, trauma in rat. J Cerebral Blood Flow Metab. 1993;13:83-192.
39 Wolf JA, Stys PK, Lusardi T, et al. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci. 2001;21:1923-1930.
40 Reinert M, Khaldi A, Zauner A, et al. High level of extracellular potassium and its correlates after severe head injury: relationship to high intracranial pressure. J Neurosurg. 2001;94:1025-1027.
41 Zauner A, Bullock R, Kuta J, et al. Glutamate release and cerebral blood flow after severe head injury. Acta Neurochir (Wien). 1996;67:40-44.
42 Di X, Harpold T, Watson JC, et al. Excitotoxic damage in neurotrauma: fact or fiction? Restor Neurol Neurosci. 1996;9:231-241.
43 Delahunty TK, Jiang JY, Black RT, et al. Differential modulation of carbachol and trans-ACPB–stimulated phosphoinositide turnover following traumatic brain injury. Neurochem Res. 1995;20:405-411.
44 Miyazaki S, Katayama Y, Lyeth BG, et al. Enduring suppression of hippocampal long-term potentiation following traumatic brain injury in the rat. Brain Res. 1992;585:335-339.
45 Prasad RM, Dhillon HS, Carbary S, et al. Enhanced phosphodiestetic breakdown of phosphatidylinositol bisphosphate after experimental brain injury. J Neurochem. 1994;63:773-776.
46 Reeves D, Ursell T, Sens P, et al. Matrix metalloproteinase inhibition alters functional and structural correlates of deafferentation-induced sprouting in the dentate gyrus. J Neurosci. 2003;23:10182-10189.
47 Graham DI. The pathology of brain ischaemia and possibilities for therapeutic intervention. Br J Anaesth. 1985;57:3-25.
48 Reilly PL, Adams RH, Graham DI, et al. Patients with head injury who talk and die. Lancet. 1975;2:375-377.
49 Gaetz M. The neurophysiology of brain injury. Clin Neurophysiol. 2004;115:4-18.
50 Povlishock JT, Katz DI. Update of neuropathology and neurological recovery after traumatic brain injury. J Head Trauma Rehabil. 2005;20:76-94.
51 Yakovlev AG, Faden AI. Mechanisms of neural cell death: implications for development of neuroprotective treatment strategies. NeuroRx. 2004;1:5-16.
52 Bailey I, Bell A, Gray J, et al. A randomized trial of the effect of nimodipine on outcome of head injury. Acta Neurochir (Wien). 1991;110:97-105.
53 A multicenter trial of the efficacy of nimodipine on outcome after severe head injury. The European Study Group on Nimodipine in Severe Head Injury. J Neurosurgery. 1994;80:797-804.
54 Harders A, Kakarieka A, Braakman R. Traumatic subarachnoid hemorrhage and its treatment with nimodipine. German tSAH Study Group. J Neurosurgery. 1996;85:82-89.
55 Morris GF, Bullock R, Marshall SB, et al. Failure of the competitive N-methyl-D-aspartate antagonist selfotel (CGS 19755) in the treatment of severe head injury: results of two phase III clinical trials. The Selfotel Investigators. J Neurosurg. 1999;91:737-743.
56 Yurkewicz L, Weaver J, Bullock MR, et al. The effect of the selective NMDA receptor antagonist traxoprodil in the treatment of traumatic brain injury. J Neurotrauma. 2005;22:1428-1443.
57 Grumme T, Baethmann A, Kolodziejczyk D, et al. Treatment of patients with severe head injury by triamcinolone: a prospective, controlled multicenter clinical trial of 396 cases. Res Exp Med (Berl). 1995;195:217-229.
58 Gaab MR, Trost HA, Alcantara A, et al. “Ultrahigh” dexamethasone in acute brain injury. Results from a prospective randomized double-blind multicenter trial (GUDHIS). German Ultrahigh Dexamethasone Head Injury Study Group. Zentralbl Neurochir. 1994;55:135-143.
59 Edwards P, Arango M, Balica L, et al. for the CRASH trial collaborators. Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury—outcomes at 6 months. Lancet. 2005;365:1957-1959.
60 Hatton J, Rosbolt B, Empey P, et al. Dosing and safety of cyclosporine in patients with severe brain injury. J Neurosurg. 2008;109:699-707.
61 Wolf AL, Levi L, Marmarou A, et al. Effect of THAM upon outcome in severe head injury: a randomized prospective clinical trial. J Neurosurg. 1993;78:54-59.
62 Marmarou A, Nichols J, Burgess J, et al. Effects of the bradykinin antagonist Bradycor (deltibant, CP-1027) in severe traumatic brain injury: results of a multi-center, randomized, placebo-controlled trial. American Brain Injury Consortium Study Group. J Neurotrauma. 1999;16:431-444.
63 Young B, Runge JW, Waxman KS, et al. Effects of pegorgotein on neurologic outcome of patients with severe head injury. A multicenter, randomized controlled trial. JAMA. 1996;276:538-543.
64 Marshall LF, Maas AI, Marshall SB, et al. A multicenter trial on the efficacy of using tirilazad mesylate in cases of head injury. J Neurosurg. 1998;89:519-525.
65 Robertson CS, Valadka AB, Hannay HJ, et al. Prevention of secondary ischemic insults after severe head injury. Crit Care Med. 1999;27:2086-2095.
66 Rockswold GL, Ford SE, Anderson DC, et al. Results of a prospective randomized trial for treatment of severely brain-injured patients with hyperbaric oxygen. J Neurosurg. 1992;76:929-934.
67 Rockswold SB, Rockswold GL, Zaun DA, et al. A prospective, randomized clinical trial to compare the effect of hyperbaric to normobaric hyperoxia on cerebral metabolism, intracranial pressure, and oxygen toxicity in severe traumatic brain injury. J Neurosurg. 2010;112:1080-1094.
68 Wright DW, Kellermann AL, Hertzberg VS, et al. ProTECT: a randomized clinical trial of progesterone for acute traumatic brain injury. Ann Emerg Med. 2007;49:4391-4402.
69 Maas AI, Murray G, Henney H3rd, et al. for the Pharmos TBI investigators. Efficacy and safety of dexanabinol in severe traumatic brain injury: results of a phase III randomised, placebo-controlled, clinical trial. Lancet Neurol. 2006;5:38-45.
70 Temkin NR, Anderson GD, Winn HR. Magnesium sulfate for neuroprotection after traumatic brain injury: a randomised controlled trial. Lancet Neurol. 2007;6:29-38.
71 Hatton J, Rapp RP, Kudsk KA, et al. Intravenous insulin-like growth factor-I (IGF-I) in moderate-to-severe head injury: a phase II safety and efficacy trial. Neurosurg Focus. 1997;2(5):ECP1.
72 Hatton J, Kryscio R, Ryan M, et al. Systemic metabolic effects of combined insulin-like growth factor-I and growth hormone therapy in patients who have sustained acute traumatic brain injury. J Neurosurg. 2006;105:843-852.
73 Saatman KE, Duhaime AC, Bullock R, et al. for the Workshop Scientific Team and Advisory Panel Members. Classification of traumatic brain injury for targeted therapies. J Neurotrauma. 2008;25:719-738.
74 Bouma GJ, Muizelaar JP, Stringer WA, et al. Ultra-early evaluation of regional cerebral blood flow in severely head-injured patients using xenon-enhanced computerized tomography. J Neurosurg. 1992;77:360-368.
75 Astrup J, Siesjö PK, Symon L. Thresholds in cerebral ischemia: the ischemic penumbra. Stroke. 1981;12:723-725.
76 Branston NK, Symon L, Crockard HA, et al. Relationships between the cortical evoked potential and local cortical blood flow following acute middle cerebral artery occlusion in the baboon. Exp Neurol. 1974;45:195-208.
77 Jones TH, Morawetz RB, CrowelI RM, et al. Threshold of focal cerebral ischemia in awake monkeys. J Neurosurg. 1981;54:773-782.
78 Lafuente JV, Cervós-Navarro J. Craniocerebral trauma induces hemorheological disturbances. J Neurotrauma. 1999;16:425-430.
79 Sayeed I, Wali B, Stein DG. Progesterone inhibits ischemic brain injury in a rat model of permanent middle cerebral artery occlusion. Restor Neurol Neurosci. 2007;25:151-159.
80 Smith WS, Sung G, Saver J, et al. Mechanical thrombectomy for acute ischemic stroke: final results of the Multi MERCI trial. Stroke. 2008;39:1205-1212.
81 Greenberg MS, Arredondo N. Handbook of Neurosurgery. Tampa, FL: Greenberg Graphics, 1997.
82 DeGirolami U, Crowell RM, Marcoux SW. Selective necrosis and total necrosis in focal cerebral ischemia. Neuropathologic observations on experimental middle cerebral artery occlusion in the macaque monkey. Neuropathol Exp Neurol. 1984;43:57-64.
83 Pulsinelli WA, Brierley JB, Plum F. Temporal profiles of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:499-504.
84 Daugherty WP, Levasseur JE, Sun D, et al. Perfluorocarbon emulsion improves cerebral oxygenation and mitochondrial function after fluid percussion brain injury in rats. Neurosurgery. 2004;54:1223-1230.
85 Siesjö BK. Pathophysiology and treatment of focal cerebral ischemia. Part II: mechanisms of damage and treatment. J Neurosurg. 1992;77:337-354.
86 Kraig RP, Petito CK, Plum F, et al. Hydrogen ions kill brain at concentrations reached in ischemia. J Cereb Blood Flow Metab. 1987;7:379-386.
87 Tonnessen TI. Biological basis for PCO2 as a detector of ischaemia. Acta Anaesthesiol Scand. 1997;41:659-669.
88 Kaku DA, Giffard RG, Choi DW. Neuroprotective effects of glutamate antagonists and extracellular acidity. Science. 1993;260:1516-1518.
89 Marmarou A, Holdaway R, Ward JD, et al. Traumatic brain tissue acidosis: experimental and clinical studies. Acta Neurochir Suppl (Wien). 1993;57:160-164.
90 Miller JD, Butterworth JF, Gudeman SK, et al. Further experience in the management of severe head injury. J Neurosurg. 1981;54:289-299.
91 Marmarou A. Traumatic brain edema: an overview. Acta Neurochir Suppl (Wien). 1994;60:421-424.
92 Kuroiwa T, Cahn R, Juhler M, et al. Role of extracellular proteins in the dynamics of vasogenic brain edema. Acta Neuropathol. 1985;66:3-11.
93 Roberts, Yates D, Sandercock P, et al. Effect of Intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRC CRASH trial): randomized placebo-controlled trial. Lancet. 2004;364:1321-1328.
94 Marmarou A, Fatouros P, Bandoh K, et al. The contribution of brain edema to brain swelling, ICP and craniospinal dynamic. In: Avezaat CJJ, van Eindhoven JHM, Maas AIR, et al, editors. Tans Intracranial Pressure VIII. Berlin: Springer-Verlag; 1993:525-528.
95 Ito J, Marmarou A, Barzo P, et al. Characterization of edema by diffusion weighted imaging in experimental traumatic brain injury. J Neurosurg. 1996;84:97-103.
96 Bullock R, Statham P, Patterson J, et al. The time course of vasogenic edema after focal human head injury—evidence from SPECT mapping of blood brain barrier deficit. Acta Neurochir Suppl (Wien). 1990;51:286-288.
97 Lang DA, Hadley DM, Teasdale GT, et al. Gadolinium DTPA enhanced magnetic resonance imaging in acute head injury. Acta Neurochir (Wien). 1991;109:5-11.
98 Todd NV, Graham DI. Blood brain barrier damage in traumatic brain contusion. Acta Neurochir Suppl (Wien). 1990;51:296-299.
99 Marmarou A, Signoretti S, Fatouros PP, et al. Predominance of cellular edema in traumatic brain swelling in patients with severe head injuries. J Neurosurg. 2006;104:720-730.
100 Marmarou A. A review of progress in understanding the pathophysiology and treatment of brain edema. Neurosurg Focus. 2007;22(5):E1.
101 Schröder ML, Muizelaar JP, Bullock MR, et al. Focal ischemia due to traumatic contusions documented by stable xenon-CT and ultrastructural studies. J Neurosurg. 1995;82:966-971.
102 Manley GT, Fujimura M, Ma T, et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159-163.
103 Ke C, Poon WS, Ng HK, et al. Heterogeneous responses of aquaporin-4 in oedema formation in a replicated severe traumatic brain injury model in rats. Neurosci Lett. 2001;301:21-24.
104 Bloch O, Manley GT. The role of aquaporin-4 in cerebral water transport and edema. Neurosurg Focus. 2007;22(5):E3.
105 Tran ND, Kim S, Vincent HK, et al. Aquaporin-1–mediated cerebral edema following traumatic brain injury: effects of acidosis and corticosteroid administration. J Neurosurg. 2010;112:1095-1104.
106 Choi DW. Calcium: still center-stage in hypoxic-ischemic neuronal death. Trends Neurol Sci. 1995;18:58-60.
107 Wyllie A, Kerr J, Currie A. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251-306.
108 Zipfel GJ, Babcock DJ, Lee JM, et al. Neuronal apoptosis after CNS injury: the roles of glutamate and calcium. J Neurotrauma. 2000;17:857-869.
109 Fineman I, Hovda DA, Smith M, et al. Concussive brain injury is associated with a prolonged accumulation of calcium: a 45Ca autoradiographic study. Brain Res. 1993;624:94-102.
110 Shapira Y, Yadid G, Cotev S, et al. Accumulation of calcium in the brain following head trauma. Neurol Res. 1989;11:169-172.
111 Young W. Secondary CNS injury. J Neurotrauma. 1988;5:219-221.
112 Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria. III. Transitional Ca2+ release. Arch Biochem Biophys. 1979;195:468-477.
113 Farkas O, Lifshitz J, Povlishock JT. Mechanoporation induced by diffuse traumatic brain injury: an irreversible or reversible response to injury? J Neurosci. 2006;26:3130-3140.
114 Gwag BJ, Canzoniero LM, Sensi SL, et al. Calcium ionophores can induce either apoptosis or necrosis in cultured cortical neurons. Neuroscience. 1999;90:1339-1348.
115 Suarez JI. Critical Care Neurology and Neurosurgery. Totowa, NJ: Humana Press; 2004.
116 Kampfl A, Posmantur R, Nixon R, et al. Mu-Calpain activation and calpain-mediated cytoskeletal proteolysis following traumatic brain injury. J Neurochem. 1996;67:1575-1583.
117 Saatman KE, Bozyczko-Coyne D, Marcy V, et al. Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J Neuropathol Exp Neurol. 1996;55:850-860.
118 Wang KK. Calpain and caspase: can you tell the difference? Trends Neurosci. 2000;23:59.
119 Tsujimoto Y, Nakagawa T, Shimizu S. Mitochondrial membrane permeability transition and cell death. Biochim Biophys Acta. 2006;1757:1297-1300.
120 Harders A, Kakarieka A, Braakman R, et al. Traumatic subarachnoid hemorrhage and its treatment with nimodipine. J Neurosurg. 1996;85:82-89.
121 Kampfl A, Postmantur RM, Zhao X, et al. Mechanisms of calpain proteolysis following traumatic brain injury: implications for pathology and therapy: a review and update. J Neurotrauma. 1997;14:121-134.
122 Maxwell WL, Povlishock JT, Graham DL. A mechanistic analysis of nondisruptive axonal injury: a review. J Neurotrauma. 1997;14:419-440.
123 Inuzuka T, Tamura A, Sato S, et al. Suppressive effect of E-64c on ischaemic degradation of cerebral proteins following occlusion of middle cerebral artery in rat. Brain Res. 1990;526:177-179.
124 Inuzuka T, Tamura A, Sato S, et al. Changes in the concentration of cerebral proteins following occlusion of the middle cerebral artery in rats. Stroke. 1990;21:917-922.
125 Harris AS, Croall DE, Morrow JS. The calmodulin-binding site in alpha-fodrin is near the calcium-dependent protease-1 cleavage site. J Biol Chem. 1988;263:15734-15761.
126 Johnson GV, Litersky JM, Jope RS. Degradation of microtubule-associated protein 2 and brain spectrin by calpain: a comparative study. J Neurochem. 1991;56:1630-1638.
127 Kamakura K, Ishiura S, Suzuki K, et al. Calcium-activated neutral protease in the peripheral nerve, which requires µM order Ca2+, and its effects on the neurofilament triplet. J Neurosci Res. 1985;13:391-403.
128 Kawamata T, Katayama Y, Hovda DA, et al. Lactate accumulation following concussive brain injury: the role of ionic fluxes induced by excitatory amino acid. Brain Res. 1995;674:196-204.
129 Kuroda Y, Bullock R. Failure of cerebral blood flow–metabolism coupling after acute subdural hematoma in the rat. Neurosurgery. 1992;31:1062-1071.
130 Sutton RL, Hovda DA, Adelson PD, et al. Metabolic changes following cortical contusion: relationships to edema and morphological changes. Acta Neurochir Suppl. 1994;60:446-448.
131 Bergsneider M, Hovda DA, Shalmon E. Cerebral hyperglycolysis following severe traumatic brain injury in humans: a positron emission tomography study. J Neurosurg. 1997;86:241-251.
132 Yoshino A, Hovda DA, Kawamata T, et al. Dynamic changes in local cerebral glucose utilization following cerebral concussion in rats: evidence of a hyper- and subsequent hypo-metabolic state. Brain Res. 1991;561:106-119.
133 Ginsberg MD, Sternau LL, Globus MY, et al. Therapeutic modulation of brain temperature: relevance to ischemic brain injury. Cerebrovasc Brain Metab Rev. 1992;4:189-225.
134 Kroemer G, Petit P, Zamzami N, et al. The biochemistry of programmed cell death. FASEB J. 1995;9:1277-1287.
135 Clausen T, Bullock R. Medical treatment and neuroprotection in traumatic brain injury. Curr Pharm Des. 2001;7:1517-1532.
136 Basso E, Petronilli V, Forte MA, et al. Phosphate is essential for inhibition of the mitochondrial permeability transition pore by cyclosporin A and by cyclophilin D ablation. J Biol Chem. 2008;283:26307-26311.
137 Scorrano L, Ashiya M, Buttle K, et al. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell. 2002;2:55-67.
138 Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112:481-490.
139 Nakagawa T, Shimizu S, Watanabe T, et al. Cyclophilin D–dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652-658.
140 Kroemer G, Galluzzi L, Vandenabeele P, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3-11.
141 Kaya SS, Mahmood A, Li Y, et al. Apoptosis and expression of p53 response proteins and cyclin D1 after cortical impact in rat brain. Brain Res. 1999;818:23-33.
142 Napieralski JA, Raghupathi R, McIntosh TK. The tumor-suppressor gene, p53, is induced in injured brain regions following experimental traumatic brain injury. Brain Res Mol Brain Res. 1999;71:78-86.
143 Zhang C, Raghupathi R, Saatman KE, et al. Regional and temporal alterations in DNA fragmentation factor (DFF)-like proteins following experimental brain trauma in the rat. J Neurochem. 1999;73:1650-1659.
144 Kontos HA. Oxygen radicals in cerebral vascular injury. Circ Res. 1985;57:508-516.
145 Siesjö BK. Pathophysiology and treatment of focal cerebral ischemia. Part I: Pathophysiology. J Neurosurg. 1992;77:169-184.
146 Adibhatla RM, Hatcher JF. Phospholipase A(2), reactive oxygen species, and lipid peroxidation in CNS pathologies. BMB Rep. 2008;41:560-567.
147 Kontos HA, Povlishock JT. Oxygen radicals in brain injury. Cent Nerv Syst Trauma. 1986;3:257-263.
148 Pun PB, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res. 2009;43:348-364.
149 Hall ED, Braughler JM. Free radicals in CNS injury. Res Publ Assoc Res Nerv Ment Dis. 1993;71:81-105.
150 Hall ED, Braughler JM. Central nervous system trauma and stroke—pathophysiological and pharmacological evidence for involvement of oxygen radicals and lipid peroxidation. Free Radic Biomed. 1989;6:303-313.
151 Adibhatla RM, Hatcher JF. Altered lipid metabolism in brain injury and disorders. Subcell Biochem. 2008;49:241-268.
152 Dringen R, Gutterer JM, Hirrlinger J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur J Biochem. 2000;267:4912-4916.
153 Yoshida H, Yanai H, Namiki Y, et al. Neuroprotective effects of edaravone: a novel free radical scavenger in cerebrovascular injury. CNS Drug Rev. 2006;12:9-20.
154 Fiskum G, Rosenthal RE, Vereczki V, et al. Protection against ischemic brain injury by inhibition of mitochondrial oxidative stress. J Bioenerg Biomembr. 2004;36:347-352.
155 Rigg JL, Zafonte RD. Corticosteroids in TBI: is the story closed? J Head Trauma Rehabil. 2006;21:285-288.
156 The Edaravone Acute Brain Infarction Study Group. Effect of a novel free radical scavenger, edaravone (MCI-186), on acute brain infarction. Cerebrovasc Dis. 2003;15:222-229.
157 Watanabe T, Yuki S, Egawa M, et al. Protective effects of MCI-186 on cerebral ischemia: possible involvement of free radical scavenging and antioxidant actions. J Pharmacol Exp Ther. 1994;268:1597-1604.
158 Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucoutilization. Proc Natl Acad Sci U S A. 1994;91:10625-10629.
159 Zauner A, Bullock R. Brain oxygenation and energy metabolism: Part 1: biological function and pathophysiology. Neurosurgery. 2002;51:219-230.
160 Golding EM, Robertson CS, Bryan RM. The consequences of traumatic brain injury on cerebral blood flow and autoregulation. Clin Exp Hypertens. 1999;21:299-332.
161 Magistretti PJ, Pellerin L. Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philos Trans R Soc Lond B Biol Sci. 1999;354:1155-1163.
162 Chen T, Qian YZ, Rice A, et al. Brain lactate uptake increases at the site of impact after traumatic brain injury. Brain Res. 2002;861:281-287.
163 Rice AC, Zsoldos R, Chen T, et al. Lactate administration attenuates cognitive deficits following traumatic brain injury. Brain Res. 2002;928:156-159.
164 Prins ML, Fujima LS, Hovda DA. Age-dependent reduction of cortical contusion volume by ketones after traumatic brain injury. J Neurosci Res. 2005;82:413-420.
165 Conti AC, Raghupathi R, Trojanowski JQ, et al. Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post-traumatic period. J Neurosci. 1998;18:5663-5672.
166 Raghupathi R, Conti AC, Graham DI, et al. Mild traumatic brain injury induces apoptotic cell death in the cortex that is preceded by decreases in cellular bcl-2 immunoreactivity. Neuroscience. 2002;110:605-616.
167 Hicks R, Soares H, Smith D, et al. Temporal and spatial characterization of neuronal injury following lateral fluid-percussion brain injury in the rat. Acta Neuropathol. 1996;91:236-246.
168 Singleton RH, Povlishock JT. Identification and characterization of heterogeneous neuronal injury and death in regions of diffuse brain injury: evidence for multiple independent injury phenotypes. J Neurosci. 2004;24:3543-3553.
169 Eguchi Y, Shimizu S, Tsujimoto Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997;57:1835-1840.
170 Tsujimoto Y. Apoptosis and necrosis: intracellular ATP level as a determinant for cell death modes. Cell Death Differ. 1997;4:429-434.
171 Green DR, Reed JC. Mitochondia and apoptosis. Science. 1998;281:1309-1312.
172 Fox GB, Fan L, Levasseur RA, et al. Sustained sensory/motor and cognitive deficits with neuronal apoptosis following controlled cortical impact brain injury in the mouse. J Neurotrauma. 1998;15:599-614.
173 Newcomb JK, Zhao X, Pike BR, et al. Temporal profile of apoptotic-like changes in neurons and astrocytes following controlled cortical impact injury in the rat. Exp Neurol. 1999;158:76-88.
174 Dietrich WD, Alonso O, Halley M. Early microvascular and neuronal consequences of traumatic brain injury: a light and electron microscopic study in rats. J Neurotrauma. 1994;11:289-301.
175 Sutton RL, Lescaudron L, Stein DG. Unilateral cortical contusion injury in the rat: vascular disruption and temporal development of cortical necrosis. J Neurotrauma. 1993;10:135-149.
176 Colicos MA, Dixon CE, Dash PK. Delayed, selective neuronal death following experimental cortical impact injury in rats: possible role in memory deficits. Brain Res. 1996;739:111-119.
177 Rink A, Fung KM, Trojanowski JQ, et al. Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am J Pathol. 1995;147:1575-1583.
178 Cernak I, Chapman SM, Hamlin GP, et al. Temporal characterisation of pro- and anti-apoptotic mechanisms following diffuse traumatic brain injury in rats. J Clin Neurosci. 2002;9:-72.
179 Cernak I, Vink R, Zapple DN, et al. The pathobiology of moderate diffuse traumatic brain injury as identified using a new experimental model of injury in rats. Neurobiol Dis. 2004;17:29-43.
180 Formigli L, Papucci L, Tani A, et al. Aponecrosis: morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J Cell Physiol. 2000;182:41-49.
181 Farkas O, Povlishock JT. Cellular and subcellular change evoked by diffuse traumatic brain injury: a complex web of change extending far beyond focal damage. Prog Brain Res. 2007;161:43-59.
182 Pineda JA, Wang KK, Hayes RL. Biomarkers of proteolytic damage following traumatic brain injury. Brain Pathol. 2004;14:202-209.
183 Pineda JA, Lewis SB, Valadka AB, et al. Clinical significance of alphaII-spectrin breakdown products in cerebrospinal fluid after severe traumatic brain injury. J Neurotrauma. 2007;24:354-366.
184 Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312-1316.
185 Beer R, Franz G, Krajewski S, et al. Temporal and spatial profile of caspase 8 expression and proteolysis after experimental traumatic brain injury. J Neurochem. 2001;78:862-873.
186 Yakovlev AG, Ota K, Wang G, et al. Differential expression of apoptotic protease-activating factor-1 and caspase-3 genes and susceptibility to apoptosis during brain development and after traumatic brain injury. J Neurosci. 2001;21:7439-7446.
187 Clark RS, Kochanek PM, Watkins SC, et al. Caspase-3 mediated neuronal death after traumatic brain injury in rats. J Neurochem. 2000;74:740-753.
188 Yakovlev AG, Knoblach SM, Fan L, et al. Activation of cpp32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J Neurosci. 1997;17:7415-7424.
189 Harwood SM, Yaqoob MM, Allen DA. Caspase and calpain function in cell death: bridging the gap between apoptosis and necrosis. Ann Clin Biochem. 2005;42:415-431.
190 Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922-2933.
191 Cory S, Adams JM. The bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647-656.
192 Barron KD, Dentinger MP. Cytologic observations on axotomized feline Betz cells. 1. Qualitative electron microscopic findings. J Neuropathol Exp Neurol. 1979;38:128-151.
193 Liu X, Schnellmann RG. Calpain mediates progressive plasma membrane permeability and proteolysis of cytoskeleton-associated paxillin, talin, and vinculin during renal cell death. J Pharmacol Exp Ther. 2003;304:63-70.
194 Lifshitz J, Kelley BJ, Povlishock JT. Perisomatic thalamic axotomy after diffuse traumatic brain injury is associated with atrophy rather than cell death. J Neuropathol Exp Neurol. 2007;66:218-229.
195 Soares HD, Hicks RR, Smith DH, et al. Inflammatory leukocytic recruitment and diffuse neuronal degeneration are separate pathological processes resulting from traumatic brain injury. J Neurosci. 1995;15:8223-8233.
196 Biagas KV, Uhl MV, Schiding JK, et al. Assessment of post-traumatic polymorphonuclear leucocyte accumulation in rat brain using tissue myeloperoxidase assay and vinblastine treatment. J Neurotrauma. 1992;9:363-371.
197 Guilian D, Chen J, Ingeman JE, et al. The role of mononuclear phagocytes in wound healing after traumatic injury to mammalian brain. J Neurosci. 1989;9:4416-4429.
198 Morganti-Kossmann MC, Kossmann T, Wahl SM. Cytokines and neuropathology. Trends Pharmacol Sci. 1992;13:286-291.
199 Kim KS, Wass CA, Cross AS, et al. Modulation of blood-brain barrier permeability by tumor necrosis factor and antibody to tumor necrosis factor in the rat. Lymphokine Cytokine Res. 1992;11:293-298.
200 Megyeri P, Abraham CS, Temesvari P, et al. Recombinant human tumor necrosis factor α constricts pial arterioles and increases blood-brain barrier permeability in new-born piglets. Neurosci Lett. 1992;148:137-140.
201 Talley AK, Dewhurst S, Perry SW, et al. Tumor necrosis factor α–induced apoptosis in human neuronal cells: protection by the antioxidant N-acetylcysteine and the genes bcl-2 and crmA. Mol Cell Biol. 1995;15:2359-2366.
202 Knoblach SM, Fan L, Faden AI. Early neuronal expression of tumor necrosis factor-α after experimental brain injury contributes to neurological impairment. J Neuroimmunol. 1999;95:115-125.
203 Merril JE, Benveniste EN. Cytokines in inflammatory brain lesions. Helpful and harmful. Trends Neurosci. 1996;19:331-338.
204 Gordon CR, Merchant RS, Marmarou A, et al. Effect of murine recombinant interleukin-1 on brain edema in the rat. Acta Neurochir Suppl. 1990;51:268-270.
205 Selmaj KW, Raine CS. Tumor necrosis factor mediates myelin and oligodendrocyte damage in vitro. Ann Neurol. 1988;23:339-346.
206 Bruce AJ, Boling W, Kindy MS, et al. Altered neuronal and microglial responses to excitoxic and ischaemic brain injury in mice lacking TNF receptors. Nat Med. 1996;2:788-794.
207 Toulmond S, Rothwell NJ. Interleukin-1 receptor antagonist inhibits neuronal damage caused by fluid percussion injury in the rat. Brain Res. 1995;671:261-266.
208 Schoettle RJ, Kocanek PM, Magargee MJ, et al. Early polymorphonuclear leukocyte accumulation correlates with the development of post-traumatic cerebral edema in rats. J Neurotrauma. 1990;7:207-217.
209 Holmin S, Soderland J, Biberfeld P, et al. Intracerebral inflammation after human brain contusion. Neurosurgery. 1998;42:291-299.
210 Holmin S, Mathiesen T. Dexamethasone and colchicines reduce inflammation and delayed edema following experimental brain contusion. Acta Neurochir (Wien). 1996;138:418-424.
211 Ross SA, Halliday MI, Campbell GC, et al. The presence of tumor necrosis factor in CSF and plasma after severe head injury. Br J Neurosurg. 1994;8:419-425.
212 Nomoto Y, Yamamoto M, Fukushima T, et al. Expression of nuclear factor κB and tumor necrosis factor α in the mouse brain after experimental thermal ablation injury. Neurosurgery. 2001;48:158-166.
213 Singhal A, Baker AJ, Hare GM, et al. Association between cerebrospinal fluid interleukin-6 concentrations and outcome after severe human traumatic brain injury. J Neurotrauma. 2002;19:929-937.
214 Bellander BM, Singhrao SK, Ohlsson M, et al. Complement activation in the human brain after traumatic head injury. J Neurotrauma. 2001;18:1295-1311.
215 Morganti-Kossmann MC, Rancan M, Otto VI, et al. Role of cerebral inflammation after traumatic brain injury: a revisited concept. Shock. 2001;16:165-177.
216 Stahel PF, Morganti-Kossmann MC, Kossmann T. The role of the complement system in traumatic brain injury. Brain Res Brain Res Rev. 1998;27:243-256.
217 Gahm C, Holmin S, Mathiesen T. Temporal profiles and cellular sources of three nitric oxide synthetase isoforms in the brain after experimental contusion. Neurosurgery. 2000;46:169-177.
218 Orihara Y, Ikematsu K, Tsuda R, et al. Induction of nitric oxide synthase by traumatic brain injury. Forensic Sci Int. 2001;123:142-149.
219 Zhang J, Rivest S. Anti-inflammatory effects of prostaglandins E2 in the central nervous system in response to brain injury and circulating lipopolysaccharide. J Neurochem. 2001;76:855-864.
220 Schwab JM, Seid K, Schluesner HJ. Traumatic brain injury induces prolonged accumulation of cyclooxygenase-1 expressing microglia/brain macrophages in rats. J Neurotrauma. 2001;18:881-890.
221 Hurley SD, Olschowka JA, O’Banion MK. Cyclooxygenase inhibition as a strategy to ameliorate brain injury. J Neurotrauma. 2002;19:1-15.
222 Kuroda Y, Dewar D, Bullock R. Early changes in second messenger but not receptor binding sites after acute subdural hematoma—an in vitro autoradiographic study. J Neurotrauma. 1993;10:47-55.
223 Bortolotto ZA, Bashir ZL, Davies CK, et al. A molecular switch activated by metabotropic glutamate receptors regulates induction of long-term potentiation. Nature. 1994;368:740-743.
224 Tower D, McEachern D. Acetylcholine and neuronal activity in craniocerebral trauma. J Clin Invest. 1948;27:558-559.
225 Ruge D. The use of cholinergic blocking agents in the treatment of craniocerebral injuries. J Neurosurg. 1954;11:77-83.
226 Bornstein M. Presence and action of acetylcholine in experimental brain trauma. J Neurophysiol. 1946;9:349-366.
227 Dewar D, Graham DI. Depletion of choline acetyltransferase but preservation of M1 and M2 muscarinic receptor binding sites in temporal cortex following head injury. A preliminary human post-mortem study. J Neurotrauma. 1996;13:181-187.
228 Jenkins LW, Monzynski K, Lyeth BG, et al. Increased vulnerability of the mildly traumatized brain to cerebral ischemia: the use of controlled secondary ischemia as a research tool to identify common or different mechanisms contributing to mechanical and ischemic brain injury. Brain Res. 1989;477:212-224.
229 Lyeth BG, Dixon CE, Hamm RJ, et al. Effects of anticholinergic treatment on transient behavioral suppression and physiological responses following concussive brain injury to the rat. Brain Res. 1994;488:88-97.
230 Hamill RW, Woolf PD, McDonald JB, et al. Catecholamines predict outcome in traumatic brain injury. Ann Neurol. 1987;21:438-443.
231 Woolf PD, Hamill RW, Lee LA, et al. The predictive value of catecholamines in assessing outcome in traumatic brain injury. J Neurosurg. 1987;66:875-882.
232 McIntosh TK, Yu T, Gennarelli TA. Alterations in regional brain catecholamine concentrations after experimental brain injury in rat. J Neurochem. 1994;63:1426-1433.
233 Curran T, Morgan J. Fos: An immediate-early transcription factor in neurons. J Neurobiol. 1994;26:403-412.
234 Dragunow M, Robertson H. Brain injury induces c-fos protein(s) in nerve and glial-like cells in adult mammalian brain. Brain Res. 1988;455:295-299.
235 Dutcher SA, Underwood BD, Walker PD, et al. Patterns of immediate early gene mRNA expression following rodent and human traumatic brain injury. Neurol Res. 1999;21:234-242.
236 Yang K, Mu XS, Xue JJ, et al. Increased expression of c-fos mRNA and AP-1 transcription factors after cortical impact injury in rats. Brain Res. 1994;664:141-147.
237 Whitfield PC, Pickard JD. Expression of the immediate early genes c-Fos and c-Jun after head injury in man. Neurol Res. 2000;22:138-144.
238 Brown I, Rush S, Ivy G. Induction of a heat shock protein gene at the site of tissue injury in the rat brain. Neuron. 1989;2:1559-1564.
239 Tanno H, Nockels R, Pitts L, et al. Immunolocalization of heat shock proteins after fluid percussive brain injury and relationship to breakdown of the blood-brain barrier. J Cereb Blood Flow Metab. 1993;13:116-124.
240 Dutcher SA, Underwood BD, Walker PD, et al. Patterns of heat-shock protein 70 biosynthesis following human traumatic brain injury. J Neurotrauma. 1998;15:411-420.
241 Amin V, Cummings V, Latchman D. Over-expression of heat shock protein 70 protects neuronal cells against both thermal and ischaemic stress but with different efficiencies. Neurosci Lett. 1996;206:45-48.
242 Gonzalez M, Shiraiishi K, Hisanaga K, et al. Heat shock proteins as markers of neural injury. Mol Biol Res. 1989;6:93-100.
243 Latchman DS. Cell stress genes and neuronal protection. Neuropathol Appl Neurobiol. 1995;21:475-477.
244 Clark RS, Chen J, Watkins SC, et al. Apoptosis-suppressor gene bcl-2 expression after traumatic brain injury in rats. J Neurosci. 1997;17:9172-9182.
245 Clark RS, Kochanek PM, Chen M, et al. Increases in bcl-2 and cleavage of caspase-1 and caspase-3 in human brain after head injury. FASEB J. 1999;13:813-821.
246 Graham SH, Chen J, Clark RS. Bcl-2 family gene products in cerebral ischemia and traumatic brain injury. J Neurotrauma. 2000;17:831-841.
247 Reed JC. Bcl-2 family proteins. Oncogene. 1998;17:3225-3236.
248 Felderhoff-Mueser U, Sifringer M, Pesditschek S, et al. Pathways leading to apoptotic neurodegeneration following trauma to the developing rat brain. Neurobiol Dis. 2002;11:231-245.
249 Franz G, Beer R, Intemann D, et al. Temporal and spatial profile of bid cleavage after experimental traumatic brain injury. J Cereb Blood Flow Metab. 2002;22:951-958.
250 Yang J, Liu X, Bhalla K, et al. Prevention of apoptosis by bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129-1132.
251 Nakamura M, Raghupathi R, Merry DE, et al. Overexpression of bcl-2 is neuroprotective after experimental brain injury in transgenic mice. J Comp Neurol. 1999;412:681-692.
252 Rossé T, Olivier R, Monney L, et al. Bcl-2 prolongs cell survival after bax-induced release of cytochrome c. Nature. 1998;391:496-499.
253 Tsujimoto Y, Shimizu S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis. 2007;12:835-840.
254 Harris MH, Thompson CB. The role of the bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death Differ. 2000;7:1182-1191.
255 Nguyen M, Millar DG, Yong VW, et al. Targeting of bcl-2 to the mitochondrial outer membrane by a cooh-terminal signal anchor sequence. J Biol Chem. 1993;268:25265-25268.
256 Tanaka S, Saito K, Reed JC. Structure-function analysis of the bcl-2 oncoprotein. Addition of a heterologous transmembrane domain to portions of the bcl-2 beta protein restores function as a regulator of cell survival. J Biol Chem. 1993;268:10920-10926.
257 Brenner C, Cadiou H, Vieira HL, et al. Bcl-2 and bax regulate the channel activity of the mitochondrial adenine nucleotide translocator. Oncogene. 2000;19:329-336.
258 Shimizu S, Eguchi Y, Kamiike W, et al. Bcl-2 prevents apoptotic mitochondrial dysfunction by regulating proton flux. Proc Natl Acad Sci U S A. 1998;95:1455-1459.
259 Xia Z, Dickens M, Raingeaud J, et al. Opposing effects of ERK and jnk-p38 MAP kinases on apoptosis. Science. 1995;270:1326-1331.
260 Mielke K, Brecht S, Dorst A, et al. Activity and expression of JNK1, p38 and ERK kinases, c-jun N-terminal phosphorylation, and c-jun promoter binding in the adult rat brain following kainate-induced seizures. Neuroscience. 1999;91:471-483.
261 Ozawa H, Shioda S, Dohi K, et al. Delayed neuronal cell death in the rat hippocampus is mediated by the mitogen-activated protein kinase signal transduction pathway. Neurosci Lett. 1999;262:57-60.
262 Dash PK, Mach SA, Moore AN. The role of extracellular signal–regulated kinase in cognitive and motor deficits following experimental traumatic brain injury. Neuroscience. 2002;114:755-767.
263 Otani N, Nawashiro H, Fukui S, et al. Temporal and spatial profile of phosphorylated mitogen-activated protein kinase pathways after lateral fluid percussion injury in the cortex of the rat brain. J Neurotrauma. 2002;19:1587-1596.
264 Otani N, Nawashiro H, Fukui S, et al. Differential activation of mitogen-activated protein kinase pathways after traumatic brain injury in the rat hippocampus. J Cereb Blood Flow Metab. 2002;22:327-334.
265 Yamashima T. Implication of cysteine proteases calpain, cathepsin and caspase in ischemic neuronal death of primates. Prog Neurobiol. 2000;62:273-295.
266 Yamashima T, Tonchev AB, Tsukada T, et al. Sustained calpain activation associated with lysosomal rupture executes necrosis of the postischemic CA1 neurons in primates. Hippocampus. 2003;13:791-800.
267 Liu CL, Chen S, Dietrich D, et al. Changes in autophagy after traumatic brain injury. J Cereb Blood Flow Metab. 2008;28:674-683.
268 Hukkelhoven CW, Steyerberg EW, Habbema JD, et al. Predicting outcome after traumatic brain injury: development and validation of a prognostic score based on admission characteristics. J Neurotrauma. 2005;22:1025-1039.
269 Hukkelhoven CW, Steyerberg EW, Rampen AJ, et al. Patient age and outcome following severe traumatic brain injury: an analysis of 5600 patients. J Neurosurgery. 2003;99:666-673.
270 Sun D, Colello RJ, Daugherty WP, et al. Cell proliferation and neuronal differentiation in the dentate gyrus in juvenile and adult rats following traumatic brain injury. J Neurotrauma. 2005;22:95-105.
271 Babikian T, Asarnow R. Neurocognitive outcomes and recovery after pediatric TBI: meta-analytic review of the literature. Neuropsychology. 2009;23:283-296.
272 Levin BE, Dunn-Meynell A. Adult rat barrel cortex plasticity occurs at 1 week but not at 1 day after vibrissectomy as demonstrated by the 2-deoxyglucose method. Exp Neurol. 1991;113:237-248.
273 Kochanek PM. Pediatric traumatic brain injury: quo vadis? Dev Neurosci. 2006;28:244-255.
274 Wilson M, Montgomery H. Impact of genetic factors on outcome from brain injury. Br J Anaesth. 2007;99:43-48.
275 Crawford FC, Vanderploeg RD, Freeman MJ, et al. APOE genotype influences acquisition and recall following traumatic brain injury. Neurology. 2002;58:1115-1118.
276 Friedman G, Froom P, Sazbon L, et al. Apolipoprotein E-epsilon4 genotype predicts a poor outcome in survivors of traumatic brain injury. Neurology. 1999;52:244-248.
277 Liaquat I, Dunn LT, Nicoll JA, et al. Effect of apolipoprotein E genotype on hematoma volume after trauma. J Neurosurg. 2002;96:90-96.
278 Lichtman SW, Seliger G, Tycko B, et al. Apolipoprotein E and functional recovery from brain injury following postacute rehabilitation. Neurology. 2000;55:1526-1531.
279 Teasdale GM, Nicoll JAR, Murray G, et al. Association of apolipoprotein E polymorphism with outcome after head injury. Lancet. 1997;350:1069-1071.
280 Zhou W, Xu D, Peng X, et al. Meta-analysis of APOE4 allele and outcome after traumatic brain injury. J Neurotrauma. 2008;25:279-290.
281 Mayeux R, Ottman R, Maestre G, et al. Synergistic effects of traumatic head injury and apolipoprotein ε4 in patients with Alzheimer’s disease. Neurology. 1995;45:555-557.
282 Jordan BD, Relkin NR, Ravdin LD, et al. Apolipoprotein E epsilon-4 associated with chronic traumatic brain injury in boxing. JAMA. 1997;278:136-140.
283 Di Pietro V, Amin D, Pernagallo S, et al. Transcriptomics of traumatic brain injury: gene expression and molecular pathways of different grades of insult in a rat organotypic hippocampal culture model. J Neurotrauma. 2010;27:349-359.
284 von Gertten C, Flores Morales A, Holmin S, et al. Genomic responses in rat cerebral cortex after traumatic brain injury. BMC Neurosci. 2005;6:69.
285 Bell KR, Pepping M. Women and traumatic brain injury. Phys Med Rehabil Clin N Am. 2001;12:169-182.
286 Bruns JJr, Hauser WA. The epidemiology of traumatic brain injury: a review. Epilepsia. 2003;44(suppl 10):2-10.
287 Sperry JL, Nathens AB, Frankel HL, et al. for the Inflammation and the Host Response to Injury Investigators. Characterization of the gender dimorphism after injury and hemorrhagic shock: are hormonal differences responsible? Crit Care Med. 2008;36:1838-1845.
288 Stein DG. Sex differences in brain damage and recovery of function: experimental and clinical findings. Prog Brain Res. 2007;161:339-351.
289 Gur RE, Gur RC. Gender differences in regional cerebral blood flow. Schizophr Bull. 1990;16:247-254.
290 Ng SC, Poon WS, Chan MT. Cerebral hemisphere asymmetry in cerebrovascular regulation in ventilated traumatic brain injury. Acta Neurochir Suppl. 2006;96:21-23.
291 Vagnerova K, Koerner IP, Hurn PD. Gender and the injured brain. Anesth Analg. 2008;107:201-214.
292 Roof RL, Duvdevani R, Stein DG. Gender influences outcome of brain injury: progesterone plays a protective role. Brain Res. 1993;607:333-336.
293 Liu R, Wen Y, Perez E, et al. 17-beta-Estradiol attenuates blood-brain barrier disruption induced by cerebral ischemia-reperfusion injury in female rats. Brain Res. 2005;1060:55-61.
294 Roof RL, Hall ED. Gender differences in acute CNS trauma and stroke: neuroprotective effects of estrogen and progesterone. J Neurotrauma. 2000;17:367-388.
295 Weaver CEJr, Marek P, Park-Chung M, et al. Neuroprotective activity of a new class of steroidal inhibitors of the N-methyl-D-aspartate receptor. Proc Natl Acad Sci U S A. 1997;94:10450-10454.
296 Roof RL, Hoffman SW, Stein DG. Progesterone protects against lipid peroxidation following traumatic brain injury in rats. Mol Chem Neuropathol. 1997;31:1-11.
297 Bayir H, Marion DW, Puccio AM, et al. Marked gender effect on lipid peroxidation after severe traumatic brain injury in adult patients. J Neurotrauma. 2004;21:1-8.
298 Smith SS. Progesterone administration attenuates excitatory amino acid responses of cerebellar Purkinje cells. Neuroscience. 1991;42:309-320.
299 Roof RL, Duvdevani R, Braswell L, et al. Progesterone facilitates cognitive recovery and reduces secondary neuronal loss caused by cortical contusion injury in male rats. Exp Neurol. 1994;129:64-69.
300 Farin A, Deutsch R, Biegon A, et al. Sex-related differences in patients with severe head injury: greater susceptibility to brain swelling in female patients 50 years of age and younger. J Neurosurg. 2003;98:32-36.
301 Farace E, Alves WM. Do women fare worse? A metaanalysis of gender differences in outcome after traumatic brain injury. Neurosurg Focus. 2000;8(1):e6.
302 Kraus JF, Peek-Asa C, McArthur D. The independent effect of gender on outcomes following traumatic brain injury: a preliminary investigation. Neurosurg Focus. 2000;8(1):e5.
303 Klauber MR, Barrett-Connor E, Marshall LF, et al. The epidemiology of head injury: a prospective study of an entire community—San Diego County, California, 1978. Am J Epidemiol. 1981;113:500-509.
304 Kraus JF, Nourjah P. The epidemiology of mild, uncomplicated brain injury. J Trauma. 1988;28:1637-1643.
305 Jensen OK, Thulstrup AM. Gender differences of post-traumatic headache and other post-commotio symptoms. A follow-up study after a period of 9-12 months. Ugeskr Laeger. 2001;163:5029-5033.
306 Groswasser Z, Cohen M, Keren O. Female TBI patients recover better than males. Brain Inj. 1998;12:805-808.
307 Coimbra R, Hoyt DB, Potenza BM, et al. Does sexual dimorphism influence outcome of traumatic brain injury patients? The answer is no!. J Trauma. 2003;54:689-700.
308 Slewa-Younan S, van den Berg S, Baguley IJ, et al. Towards an understanding of sex differences in functional outcome following moderate to severe traumatic brain injury: a systematic review. J Neurol Neurosurg Psychiatry. 2008;79:1197-1201.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 327 Neurochemical Pathomechanisms in Traumatic Brain Injury
Traditionally, TBI has been divided into primary and secondary forms of injury. Primary injury is due to the unavoidable direct mechanical forces occurring at the time of the traumatic insult.1 Secondary injury is derived from complications initiated by the primary injury and includes potentially avoidable entities such as hypoxic-ischemic injury, cerebral edema, metabolic dysfunction, alterations in vascular permeability, diminished blood flow, inflammation, diffuse axonal injury, and the consequences of intracranial hypertension. Almost all clinical treatments are aimed at modulating these secondary injury mechanisms.
Both primary and secondary brain injury can be further classified as focal or diffuse. The distinction between focal and diffuse injuries is historically derived from the presence or absence of radiographic mass lesions on computed tomography.2,3 This distinction has now evolved to also consider the distinct pathobiologic mechanisms imparted by the trauma in regions local to and remote from the point of impact. However, any attempt to conclusively classify brain injury remains a difficult task because most TBIs consist of a heterogeneous admixture of focal and diffuse damage. Research efforts and clinical trials have sought to tip the balance of these secondary events toward facilitating neuroprotection rather than autodestruction. Central to research efforts is the neuronal response to brain injury. It is thought that the mortality and morbidity associated with TBI can be greatly reduced by a better understanding of the mechanisms that cause neuronal injury, dysfunction, and death. This information is critical within the clinical realm—not only to minimize neuronal death after TBI but also to potentially facilitate augmentation of neurological reorganization and repair. Accordingly, there is new ongoing research aimed at the use of stem cells for neuroregeneration (see Chapter 328) and for the development of deep brain stimulation techniques that may modulate the sequelae of TBI.4,5
This chapter covers the complex pathophysiology of TBI. It begins by briefly detailing the biomechanical consequences of trauma on the central nervous system (CNS) (also see Chapter 325) and continues with a comprehensive discussion of primary and secondary injury mechanisms.
Primary Injury: Molecular And Microscopic Aspects
Focal versus Diffuse Primary Brain Injury
Diffuse Primary Brain Injury
The best example of a pure diffuse primary brain injury is “mild TBI” or concussion (see Chapter 332). Concussion is a broadly applied term for the clinical manifestations of blunt head trauma that result in rapid-onset, functional disturbance of the CNS (rather than a structural injury) secondary to the inertial forces of TBI. Concussion usually resolves spontaneously and, contrary to popular belief, may or may not be associated with loss of consciousness (LOC). When LOC is present, it is thought that either the magnitude or the biomechanical directionality of the traumatic forces is sufficient to transiently perturb the brainstem reticular activating system and result in LOC.
Focal Primary Brain Injury
Primary focal brain damage is a direct result of the physical forces delivered at the time of injury. These “impact” injuries are manifested clinically as cortical contusions, brain parenchymal lacerations, and vascular lesions with resultant hemorrhage and hematoma formation.6 Focal contusions most commonly result from contact of the brain with more rigid structures such as the skull, dural edges, or physical objects used in assaults. These impact injuries may occur in association with a more significant diffuse injury, such as when the rapidly decelerating cranium strikes a windshield during a motor vehicle collision. The inertial forces of such a diffuse injury are thought to generate widespread damage and are implicated in prolonged unconsciousness. However, focal injuries do not cause LOC but instead may cause permanent discrete neurological deficits because of the immediate effects of the penetrating/focal injury. The forces of impact imparted by primary injuries are largely responsible for rapid necrosis via physical destruction of cellular elements.7 Focal injuries include skull fractures, penetrating TBI, and vascular injuries and are covered additionally in Chapter 324 and Chapter 325.
Relationship between Brain Movement, Mechanical Forces of Injury, and Histologic Effects
The destructive energy imparted by any given trauma is transmitted to the skull—and thereby its contents—in relatively unpredictable patterns. At one end of the spectrum are pure inertial injuries, where rapid deceleration and rotational forces cause devastating diffuse injuries. The cranium may never contact a solid object, yet the brain is irreversibly damaged. Such injuries maximally damage axons and most frequently occur as a result of motor vehicle accidents (although almost always compounded by an additional impact component). At the opposite extreme are unusual impact injuries in which the stationary head (e.g., of a machine operator) is slowly crushed by slow-moving machinery. Such injuries classically produce massive fractures, extra-axial hematomas, and contusions, but these patients do not usually lose consciousness because axonal injury is absent and the reticular activating system/projection fibers are not disturbed. Thibault and Gennarelli used a primate impact acceleration injury model to characterize the relationship between the magnitudes of acceleration/deceleration force, the duration over which it is applied, and the consequences for the intracranial contents.9 A brief, high-intensity deceleration force will tear parasagittal bridging veins and cause an acute subdural hematoma. When the deceleration force is of higher magnitude and longer in duration, as in motor vehicle accidents, diffuse axonal injury may occur. When both the magnitude and the duration of the deceleration force are less, transient unconsciousness (concussion) results but few structural effects are seen when the brain is examined either ultrastructurally or by light microscopy (see Chapter 332).
Brain Movement during Impact
Laboratory models of brain injury have demonstrated that the brain moves substantially within the cranial cavity in response to deceleration forces.10,11 The brain is anchored within the cranial cavity by only the parasagittal bridging veins, parasinusoidal granulations, cranial nerves, and tentorium. Movement of the brain forward toward the anterior cranial-basal structures, particularly the sphenoidal ridges, concentrates force at the bases of the frontal lobes and the tips of the temporal lobes.11 Surface contusions therefore occur very much more frequently at these sites than elsewhere. There is evidence in the human brain that shearing force also concentrates in deep white matter structures such as the corona radiata, thus explaining the frequent finding of parasagittal gliding contusions.12 Finally, it is generally believed that shearing forces transmitted through the brainstem and the reticular activating system are responsible for the immediate LOC.
Damage to Cells/Tissue
Astrocytes
Astrocytes are increasingly becoming understood to have a significant role in the deleterious effects of TBI. Although the surface area and complexity of membranes may be less for astrocytes, there is now clear evidence that astrocytes are excitable, possess ion channels, and may be depolarized (although to a much lesser extent than neurons).13–15 Astrocytic membranes also constitute an important component of the blood-brain barrier (BBB), and there are now extensive data showing that this barrier function is transiently disturbed by mechanical trauma. Furthermore, disturbed ionic and neurotransmitter homeostasis is recognized as one of the most important mechanisms contributing to the secondary brain swelling after TBI, and astrocytes play a role in maintaining this homeostasis. One example is maintenance of potassium homeostasis after TBI. Astrocytes are known to function as potassium uptake buffers in that they have the capacity to rapidly take up potassium from the extracellular space.14,16–18 Kimelberg and Norenberg hypothesized that astrocytes function to conduct potassium away from neurons, particularly in injured brain tissue, and thereby aid in the establishment of ionic homeostasis.14 Thus, there is a net loss of potassium from injured tissue into the microvasculature that begins hours after onset. In addition, stretch-injured astrocytes express a dysfunctional cation current as opposed to an osmoregulatory anion current. This mechanism may contribute to the cytotoxic swelling seen after TBI.19 Such astrocyte swelling is the ultrastructural hallmark of both acute cerebral ischemia and focal cerebral contusion and is almost always seen in animal models of trauma and in humans after trauma.16,20
Axons
About 50 years ago, neuropathologic studies first demonstrated an accumulation of axoplasmic “retraction balls” at sites of axonal discontinuity.12,21 They were chiefly found on large myelinated fibers in patients who were unconscious from the time of injury and subsequently died. These retraction balls were found in high density in white matter tracts in approximately 25% of severely head-injured patients and were thought to occur immediately as a result of tearing.22 Traumatic axonal injury (the experimental correlate to diffuse axonal injury) is now known to be a progressive process involving transient mechanoporation of the axolemma that allows unregulated calcium entry.23 The mechanical insult induces a sequence of events culminating in failure of axoplasmic transport, pooling of intra-axonal contents, and pinching off of the axon from its distal segment (Fig. 327-1). This disconnection occurs within 24 to 72 hours after the traumatic event and is termed delayed or secondary axotomy (because the primary mechanical insult provokes secondary biochemical processes that result in axotomy). This suggests that axons that subsequently show the changes associated with diffuse axonal injury may be functioning, in some capacity, immediately after the injury before eventually degrading. It also suggests that other, less affected axon tracts may not progress to diffuse axonal injury. Thus, diffuse axonal injury is amenable to therapeutic intervention.
At the molecular and microscopic level, calcium influx initiates activation of calpain24,25 and mitochondrial swelling,26 with release of cytochrome c and activation of caspase27 leading to further axonal injury, apoptosis, and detachment over time. These changes have far-reaching consequences for neuronal function. Interruption of the axon causes proximal wallerian degeneration of the affected neuron. Distally, the axon degenerates, fragments, and disappears, thereby resulting in deafferentation of the affected neuronal fields. The functional consequences of this process may include seizures because of lack of inhibitory effects, spasticity, intellectual decline, and unmodulated behavior patterns. When this process is widespread and wallerian degeneration destroys many neurons, the whole brain becomes atrophic, with ventriculomegaly and, in the worst cases, a persistent vegetative state.22,28
Clinical Implications
The benefits of attenuating secondary axotomy may be enormous. These hopes are bolstered by recent ultrastructural studies revealing that neuron somata show evidence of the potential for reorganization and repair for up to 7 days after traumatically induced axonal injury.29 Cyclosporine is a widely investigated immunosuppressive drug that has been shown to blunt traumatically induced axotomy in experimental models of TBI.30 Its neuroprotective properties are thought to be derived from inhibitory actions on the protein phosphatase calcineurin, as well as from its modulatory effect on mitochondria and the mitochondrial permeability transition pore (see discussion later in this chapter). There has been one clinical trial aimed at taking advantage of these properties via the postinjury administration of cyclosporine in the hope of limiting mitochondrial damage and axonal injury.31 Although the results of this small trial demonstrated that administration of the drug was safe, it did not reveal any benefit in terms of neurological outcome. Future phase III clinical trials are still being considered.
Shear Effect on the Microvasculature
It has been estimated that the magnitude of shear required to damage the pial vasculature may be 5 times greater than that needed to damage axons.9,32 Although this gradation of force suggests that vascular structures should be damaged less frequently than axons and membranes, in reality this may not be the case because traumatic forces are more readily translated to surface vessels than to deeper axons. In the majority of significant head injuries, focal concentrations of force develop at the tips of the frontal and temporal poles and are sufficient to disrupt these pial vessels and cause a focal contusion. Ultrastructural studies in both head-injured humans and appropriate animal models have demonstrated major anatomic changes in the injured microvasculature. Such changes include the following16,17: (1) swelling of perivascular astrocytic end-feet; (2) increased endothelial microvacuolation and micropseudopodial activity; (3) perivascular hemorrhage and transvascular diapedesis of red cells, which may coalesce to form a frank intracerebral hematoma or hemorrhagic contusion; and (4) increased intravascular leukocyte adherence. Frank vascular disruption has been found to be unexpectedly atypical in human pericontusional biopsy material, thus suggesting that small vessels “stretch and leak” much more frequently than they “tear or burst.” These microvascular changes have profound functional consequences, chiefly a reduction in local cerebral blood flow (CBF) and the development of vasogenic and cytotoxic edema with increased intracranial pressure (ICP).20
Ion Channels
Although dendritic spines, synapses, gap junctions, and myelinated axons constitute specialized regions of neurons, ion channels are by far the most frequent structures embedded in neuronal membranes. Using patch clamp techniques and in vitro tissue culture of neurons growing on flexible plastic membranes, studies have shown that ion channel function may be radically altered by mechanical deformation—in this case by delivery of a brief jet air impulse to the flexible membrane.13,33,34 Specific classes of “mechanotransducing” ion channels have been identified by using similar techniques in both neurons and glia.13,33 Some of these ion channels remain perturbed for several hours after mechanical deformation.34 Still other experiments have shown rapid entry of calcium and subsequent neuronal death, along with efflux of lactate and potassium into the culture medium, after mechanical deformation.34 Further implicating channelopathy as a significant factor in TBI are data from in vivo trauma models such as fluid percussion injury and contusion impact models. These models show a massive, approximately threefold to fourfold rapid transient efflux of potassium into the extracellular fluid (ECF) associated with a fall in the sodium content of the ECF.35–38 About a third of the potassium release could be blocked with tetrodotoxin (a selective blocker of voltage-gated sodium channels), thus suggesting that two thirds of the potassium release was occurring through agonist-operated channels. Another investigation similarly revealed that preinjury blockade of voltage-operated ion channels failed to ameliorate the negative neurological and behavioral effects of the trauma and produced only a modest effect on K+ flux in the ECF. This suggests that agonist-operated ion channels are more important in mediating ionic events after TBI.36 Finally, channelopathy has been implicated in the etiology of the calcium influx seen after TBI. Work by Wolf and colleagues suggests that calcium influx occurs, in part, via mechanical alteration of tetrodotoxin-sensitive sodium channels after traumatic axonal injury.39 This implies that excessive calcium influx may not be fully attributable to direct axolemmnal poration but instead is also related to sodium channelopathy. The resulting sodium influx then triggers depolarization-induced calcium influx through voltage-gated calcium channels and reversal of the sodium-calcium exchanger, both acting to increase influx of calcium. Work is ongoing to further explore this phenomenon.
Synapses
Direct investigation of synaptic function is difficult in the acute stages of trauma. Microdialysis studies have investigated the time course of changes in neurotransmitters within the extracellular space after fluid percussion injury, and the results demonstrated brief transient surges in the release of excitatory amino acids (EAAs) and acetylcholine. From experimental models of injury, this posttraumatic excitotoxicity is marked by increases in glutamate, which leads to an increase in extracellular potassium as a result of channel activation. Potassium levels determined by microdialysis techniques were increased in 20% of patients after severe TBI and were also noted to correlate directly with reduced CBF.40 Data from microdialysis studies of patients who have sustained severe head injury and patients with ischemic events superimposed on their primary trauma show that ECF EAAs rise to levels 50 to 60 times higher than normal values when a secondary ischemic event is superimposed on the trauma.35,41 The excitatory neurotransmitters released from damaged cells and neuron processes may be responsible for these increases through a positive feedback loop. EAAs may also come from the intravascular compartment. This conclusion is supported by the finding that serum levels of structural amino acids in these patients were also raised and appeared to fluctuate in parallel with EAAs.42
The behavioral changes that persist up to weeks or months after TBI, even in animals without any evidence of structural damage, have been taken as evidence to support functional changes at the synaptic level or in relation to second messenger systems. Neurochemical studies have shown evidence of synaptic alterations, as well as G protein–coupling variations, within the cell membrane that are manifested as prolonged amplification of protein synthesis in response to activation of muscarinic cholinergic and certain catecholaminergic receptors.43–45 These changes may translate into effects on long-term potentiation in the hippocampus (which have been demonstrated in the absence of structural changes after trauma and may be an important mechanism underlying the traumatic effects on learning and memory). In addition, matrix metalloproteinases (MMPs) are known to modulate molecules forming the extracellular matrix (ECM). MMP proteolysis of ECM molecules may perform a permissive or inductive role in the fiber remodeling or synaptogenesis initiated by deafferentation.46 The significance of this interaction in TBI was explored in animals by intraventricular infusion of the MMP inhibitor FN-439 after unilateral lesions of the entorhinal cortex.46 The lesioned rats receiving the MMP inhibitor failed to develop the capacity for long-term potentiation and showed persistent cellular debris. These results underscore the importance of continuing to improve our understanding of MMPs, the ECM, and other mechanisms involved in remodeling after trauma.
Secondary Injury Processes
The concept of delayed secondary neurological damage after head injury is supported by “lucid interval” statistics. Between 30% and 40% of severely head-injured patients who die will, at some time, have demonstrated a period of lucidity sufficient to obey commands or speak.22,47 This implies that the primary impact events were not sufficiently severe to damage the brain beyond the capacity for function, thus emphasizing the importance of the secondary damage.48 The principal mechanisms to consider are hypoxia-ischemia, edema, excitotoxicity, calcium dysregulation, apoptosis, cytoskeletal proteolysis, metabolic and mitochondrial derangements, oxidative stress, and inflammation.49,50 The deleterious mechanisms at work are diverse and interrelated—often with both sequential and parallel cascades of neuronal reaction and cell death.51
Finally, it is useful to emphasize that many clinical trials, using either physiologic or pharmacologic interventions, view these secondary injury mechanisms as the main therapeutic targets (Table 327-1).52–72 Many of these trials will be discussed briefly throughout this chapter under the most relevant heading. That said, there is no questioning the heterogeneity of TBI and, unfortunately, the lack of favorable results from previous clinical trials. Some of these failures may be due partially to flawed classification systems in terms of optimally reflecting the target population for the drug in question. These classifications have relied on several variables, including clinical severity, pathophysiology, pathoanatomic and prognostic indicators, etiology, and symptomatology. Today, the most commonly used system is the Glasgow Coma Scale, which is based solely on the severity of the neurological injury. As a result, there is growing support to establish a more clinically derived classification system for TBI based chiefly, but not exclusively, on pathoanatomic features. It is hoped that this would improve the application of appropriate treatment strategies targeting the various causes of TBI.73
Hypoxia/Ischemia
A central factor involved in secondary damage after TBI is the onset of hypoxic-ischemic damage. The incidence of ischemic brain damage seen at autopsy in patients who sustained severe TBI is extremely high, with estimates ranging between 60% and 90%.47 During life, most of these patients do not manifest the long periods of low cerebral perfusion pressure (CPP) that are known to be necessary for the generation of ischemic damage. Likewise, in animal models of impact-type head injuries, widespread ischemic damage is not seen other than around the periphery of focal contusions. Thus, there is a fundamental paradox, and the high incidence of ischemic brain damage is not easily explained, although necrosis of neurons, secondary to release of EAAs, may be a factor exacerbating cell death.
The Genesis of Ischemic Brain Damage after Severe Human Traumatic Brain Injury
On a global scale, CBF can be decreased by as much as 50% during the first 48 hours after injury and lead to ischemic changes.74 Impaired CBF has well-described cellular consequences, and a time-dependent hierarchy of neuronal events is summarized in Table 327-2.75–77 Both hemorrhage and contusion can lead to local ischemia through compromise of the microcirculation by thrombotic occlusion of blood vessels.1,78–80 Additionally, at the local level, blood flow within focal areas of contusion is dramatically reduced.16 Focal ischemia can also result from the formation of a mass lesion, which will raise ICP and impede blood flow to the damaged region in accordance with the modified Monro-Kellie Doctrine.81 Finally, ischemia secondary to occlusive or hemorrhagic stroke develops as a result of damage to or interruption of the blood supply from a parent vessel to an area of vulnerable parenchyma—such as traumatic carotid dissections.49
TABLE 327-2 Cellular Consequences of Impaired Cerebral Blood Flow
From Jones TH, Morawetz RB, CrowelI RM, et al. Threshold of focal cerebral ischemia in awake monkeys. J Neurosurg. 1981;54:773-782.
| CEREBRAL BLOOD FLOW (mL/100 g/min) | CONSEQUENCES |
|---|---|
| 40-60 | Normal |
| 20-30 | Start of neurological symptoms |
| 16-20 | Isoelectric electroencephalogram, loss of evoked potentials |
| 10-12 | Na+ and K+ pump failure Cytotoxic edema |
| <10 | Complete metabolic failure with gross disturbance of cellular energy homeostasis (infarction) |
|
|
|
Infarction versus Selective Neuronal Loss
When flow is profoundly reduced (i.e., <5 to 10 mL/100 g per minute) within the distribution of one cerebral end artery for more than 60 to 90 minutes, infarction ensues (immediate necrosis of all cell types within a zone of the brain). However, when the reduction in flow is to levels of approximately 15 to 18 mL/100 g per minute for a period longer than 30 minutes, selective neuronal loss may occur—especially in the hippocampal neurons (in the molecular layer, CA1 and CA3 sectors), cerebellar granular cells, and cortical neurons (particularly the larger cells in areas such as the cuneate visual cortex).82,83 Within the context of head injury, this type of neuronal loss is especially important in patients with raised ICP, in whom CPP may be marginal (≈30 to 40 mm Hg) for many hours or even days. In such patients, a high frequency of ischemic neuronal loss is seen in the hippocampus.47 This may explain the high frequency of memory disorders and coordination difficulty noted in the majority of severely head-injured survivors. This concept is also in accord with the almost universal finding of marked cerebral atrophy in patients who survive severe head injuries.
Clinical Implications
Clearly, reductions in CBF can have devastating consequences and will directly affect metabolic profiles. Noting that historical strategies for managing severe TBI followed ICP-directed protocols (and therefore would indirectly augment CBF), a phase III clinical trial with CPP-directed therapy was performed.65 To ensure adequate CBF and therefore oxygen delivery, the primary goal in this trial was to maintain higher CPP (versus lowering ICP). The results suggested that CPP-directed therapy improves several physiologic parameters, such as brain perfusion; however, it failed to show any incremental benefit in outcomes when CPP was targeted to levels greater than 70 mm Hg (versus 60 mm Hg). This was predominately due to an increased incidence of acute respiratory distress syndrome, which had a negative impact on mortality measures.65 Still other trials have targeted increased oxygenation by using hemoglobin substitutes, or hyperbaric oxygen, to augment the oxygen-carrying capacity of the microcirculation to damaged tissue.42 One such agent is Oxycyte, a third-generation perfluorocarbon (PFC) that improves the oxygen-carrying capacity of blood. In animals, PFCs have been shown to improve cerebral oxygenation and mitochondrial function after TBI.84 However, increased free radical formation with higher doses was also seen in these same studies. The authors suggested the need for further studies combining PFCs with free radical scavengers. Trials of PFCs and hyperbaric oxygen are ongoing—including the upcoming Brain Tissue Oxygen Monitoring in Traumatic Brain Injury trial, which will aim to implement therapy directed at increasing the partial pressure of oxygen in brain tissue (PbtO2) to further evaluate direct measurements of cerebral oxygenation on outcome.
Ischemia and Associated Acidosis/Hydrogen
Although hydrogen ions in the extracellular space are powerful cerebral vasodilators, high concentrations of hydrogen ions within cells are harmful because they alter the function of intracellular enzymes.85 For example, low pH causes conformational changes in the N-methyl-D-aspartate (NMDA) ion channel that prevent further ingress of sodium and calcium and egress of potassium during cellular acidosis. The potential benefits of mild acidosis therefore include inactivation of glutamate receptors, decreased free radical generation,86,87 inhibition of phospholipase A2 (which generates free radicals), decreased energy demand because of hyperpolarization, and inhibition of the Na+/H+ exchange transporter, which prevents intracellular entry of Na+ and Ca2+.88
Clinical Implications
The acidosis and elevated lactate levels so often seen accompanying TBI became a therapeutic target in a clinical study in which tris-(hydromethyl)-aminomethane (THAM) was administered to victims of TBI. THAM is an alkalizing agent that can buffer CO2 and acids. Its use in animal models resulted in reduced edema, lower ICP, and more favorable energy kinetics. Unfortunately, this did not translate as well to humans, and no advantage in outcome was observed.61,89
Edema/Increased Intracranial Pressure
Brain swelling occurs in almost all patients with severe brain injury and in 5% to 10% of those with moderate injuries (also see Chapter 322 and Chapter 324).90,91 The morbidity associated with brain injury was once thought to largely be correlated with the extent of posttraumatic edema. Currently, the significance of edema is acknowledged, but we now appreciate the multifaceted pathobiology constituting TBI.
Without question, posttraumatic edema contributes heavily to intracranial hypertension. The hypertension may then lead to decreased CPP (CPP = MAP − ICP, where MAP is mean arterial pressure) and to ischemia, thereby inciting continued cytotoxic edema and progressive increases in ICP. This vicious cycle is depicted in Figure 327-2. Historically, breakdown of the BBB with protein extravasation (termed vasogenic edema) was believed to be the primary component of the edema seen after TBI.92 This misconception led some practitioners to use corticosteroids for the management of TBI. This has now clearly been shown to be harmful, with the Corticosteroid Randomization After Significant Head Injury trial (>9000 patients enrolled, the largest TBI trial to date) revealing that corticosteroids were associated with worse outcomes when used in patients with severe TBI.93 Although vasogenic edema does contribute to the overall edema seen in TBI, early work by Marmarou and colleagues used mathematical modeling techniques to reveal that the vascular engorgement component of brain swelling after severe brain injury probably represents only about 25% of the overall increase in brain bulk, with the remainder being due predominantly to cytotoxic edema.94 Vasogenic edema probably becomes important around focal contusions on the 2nd through the 10th to 15th days and is most likely associated with transient opening of the BBB to medium-molecular-weight markers (50 to 70 kD).95 In humans studied with both gadolinium-enhanced magnetic resonance imaging (MRI) and pertechnetate-enhanced single-photon emission computed tomography, vasogenic edema is seen only at later time points around contusions and not at all in patients with diffuse nonfocal injuries.96–98 There is now supporting evidence that the majority of early brain edema, both global and focal, is cytotoxic rather than vasogenic. This was further substantiated recently by Marmarou and colleagues, who used diffusion-weighted MRI to evaluate patients with severe TBI. This study revealed that cytotoxic (intracellular) edema is indeed the predominant form of edema present after TBI.99 The mechanisms responsible for generating this cytotoxic edema are a continuing topic of great interest. Identified mechanisms responsible for the generation of cytotoxic edema include hypoxia-ischemia, channelopathy/excitotoxicity, mechanical membrane disruption, and aquaporins.
Hypoxia-Ischemia
Hypoxic-ischemic events are common after TBI and result in failure of ionic pumps, specifically the sodium-potassium pump (see Table 327-2). Pump malfunction causes the cell to lose its innate homeostatic environment via failure of sodium extrusion and potassium uptake. This failure will bring about the accumulation of sodium within the cell and lead to the influx of water because of the altered osmotic gradient.16
Channelopathy/Excitotoxicity
A major source of cytotoxic edema is traumatically induced accumulation of EAA neurotransmitters such as glutamate and glycine.49 These neurotransmitters bind their receptors, activate and open membrane channels, and induce sodium influx. As with ionic pump failure, osmotic pressure dictates that water will follow. In addition to the oncotic effects of sodium entry, sodium influx will also cause membrane depolarization with an influx of chloride—also resulting in swelling.100
Mechanical Membrane Disruption
Trauma induces brief and spontaneously recovering mechanoporation of neuronal and axonal membranes. Membrane disruption causes efflux of potassium from neurons and resultant astrocytic swelling, as confirmed by ultrastructural examination of pericontusional tissue taken as little as 10 minutes after impact.101
Aquaporins
Aquaporins are a relatively new family of at least nine proteins that have been shown to be involved in the formation of cellular water channels under a variety of circumstances.102 At this time, aquaporin expression across injury models has proved inconsistent and is incompletely understood. For example, increased aquaporin expression is present after ischemic injury, whereas it is decreased after a cortical impact model of TBI.103 These conflicting data appear to indicate that aquaporin expression may lead to the accumulation intracellular edema under some circumstances while playing a role in the clearance of edema in others.104 Aquaporin-4 (AQP4) is currently thought to be one of the primary cellular water channels in the brain and is localized to astrocytic foot processes along the basal lamina and brain–cerebrospinal fluid interface. Our understanding of AQP4 has been greatly augmented by the study of AQP4 knockout mice in several models of brain injury.102 In models of cytotoxic edema, AQP4 deletion or alteration has been shown to be protective.104 In contrast, AQP4 deletion in models of vasogenic edema results in decreased clearance of edema and greater progression of disease.104 In addition to AQP4, Tran and associates described a potential role for AQP1 in water homeostasis after experimental TBI.105 AQP1 is another member of the aquaporin family found in the brain and can participate in CO2 transportation across the cellular membrane. Interestingly, AQP1’s promoter contains a glucocorticoid response element. Thus, Tran and colleagues hypothesized that “AQP1 may be involved with edema-related brain injury and might be modulated by external conditions such as the pH and the presence of steroids.” The importance of aquaporins in modulating edema after trauma is thus clear. It further suggests the potential for the development of pharmacologic strategies targeting aquaporin function and expression to dramatically alter our ability to treat cerebral edema, which is currently limited to osmotic agents.
Excitotoxicity
Glutamate excitotoxicity is a self-perpetuating process that triggers numerous injurious intracellular mechanisms. After TBI there is direct release of excessive EAAs, such as glutamate and aspartate, from presynaptic nerve terminals and astrocytes into the extracellular space. This process is depicted in Figure 327-3 and begins when these EAAs bind to the appropriate postsynaptic receptor (NMDA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate [AMPA]). Activation of these ion channels will cause intracellular Ca2+ and Na+ levels to rise, followed passively by movement of Cl− and water. The resultant combination of intracellular volume and Ca2+ overload induces organelle swelling, plasma membrane swelling,106 necrosis,107 and apoptosis and leads to the activation of destructive enzymes108 (phospholipases, calpain, caspase, and nitric oxide synthase [NOS]), as depicted in Figure 327-3. Glutamate-driven excitotoxicity will further depolarize the cell, activate voltage-dependent calcium channels, and thereby propagate a dangerous positive feedback loop.6 The downstream effects of these events are shown in Figure 327-3 and are individually discussed under the appropriate subheading within this chapter.
Clinical Implications
There has been great historical interest in pharmacologic modification of glutamate excitotoxicity. Six phase II-III clinical trials (Eliprodil, Selfotel Int, Selfotel U.S., Cerestat, Saphir/D-CPP-ene, and CP101-606) directed by this strategy have been performed, but only one showed a survival benefit.56 Dexanabinol, a cannabinoid with pluripotent functionality, including noncompetitive NMDA receptor antagonist properties (as well as being a free radical scavenger and inhibitor of tumor necrosis factor-α [TNF-α]), was evaluated in a phase II trial; it was shown to be safe and well tolerated and seemed to decrease ICP. However, the phase III efficacy trial showed no effect on outcome.69
Calcium Dysregulation
As mentioned, TBI always leads to intracellular influx of calcium through numerous routes, including but not limited to (1) opening of voltage-dependent channels induced by mechanical deformation of membrane and ion channels, (2) opening of agonist-dependent channels mediated by neurotransmitter substances released in excess into ECF, and (3) opening of specific calcium channels. It is clear that calcium is strongly implicated in the propagation of several deleterious cascades responsible for the generation of neuronal injury and death after TBI.109,110 It has been linked to many downstream mechanisms of injury, including activation of cysteine proteases and subsequent cytoskeletal proteolysis,111 mitochondrial permeability transition,112 free radical toxicity, and mechanical perturbation of neuronal membranes.113 These mechanisms have been implicated in the initiation of various forms of cell death, including apoptosis and necrosis.114 Regardless of the etiology, unregulated calcium influx can cause calcium to be released from intracellular stores, as well as glutamate-containing exocytotic vesicles, thereby further increasing cytosolic calcium in a vicious cycle of cell destruction. This massive calcium surge can overwhelm cells’ buffering capability, a duty largely handled by mitochondria and ionic pumps under normal circumstances.115 The result is the activation of various enzymes, including the aforementioned cysteine proteases (calpain, caspase, and cathepsin). Induction of these important proteases has been shown in focal116 and diffuse117 models of TBI. Calpain activity has been noted in both necrotic and apoptotic forms of cell death, whereas caspase-3 activity is seen only in apoptosis.118 Finally, mitochondria have a central role in calcium homeostasis, and recent evidence highlights their function in cell death via opening of the mitochondrial permeability transition pore (MPTP), a calcium-dependent process.119
Clinical Implications
The central role of calcium in mediating cell damage and death led to trials of calcium antagonists in head-injured patients. The dihydropyridine calcium antagonist nimodipine has shown little overall benefit in unselected populations with head injury.52,53 Nimodipine has, however, shown a trend for a more favorable outcome in patients with traumatic subarachnoid hemorrhage.120 Poor brain penetration has been implicated as a major factor in the limited effect of nimodipine in TBI.
Cytoskeletal Proteolysis
The cytoskeleton consists of three main protein components: microfilaments, neurofilaments, and microtubules. After TBI, calcium-induced activation of calpain results in proteolysis of the cytoskeleton and may play an integral role in delayed neuronal degeneration—so-called calpain-mediated spectrin proteolysis (CMSP).121 In axonal stretch injury, within minutes there is malalignment and distortion of the cytoskeletal components,122 which leads to loss of microtubules and increased spacing of neurofilaments, especially at the node of Ranvier. In addition to trauma, inhibition of calpains can limit the proteolysis of MAP2 (a type of membrane-associated protein contained in microtubules) after ischemia.123,124 The caspases have also been linked to the breakdown of cytoskeletal proteins such as MAP2, α-spectrin, and neurofilaments.125–127
Clinical Implications
Targeting of CMSP has yet to reach the clinical trial stage. It has been used experimentally with modest success through the use of calpain inhibitors in TBI. In one particular study, a fluid percussion injury model was used to generate TBI in rats.128 The calpain inhibitor MDL-28170 was administered after injury, and axons in the corpus callosum were evaluated with amyloid precursor protein immunohistochemistry. When the drug was given within 30 minutes after injury, there was both structural and functional benefit in terms of axonal injury burden. However, the functional benefits diminished when the drug was given beyond 30 minutes after injury. In short, calpain inhibition has demonstrated potential (i.e., reduction of cytoskeletal breakdown and neuronal degeneration in animal models), yet the results are modest and require further investigation before they can be translated to human trials.
Derangements in Brain Metabolism after Traumatic Brain Injury
Because the brain is dependent on aerobic metabolism for delivery of substrate (oxygen and glucose) and because of the frequent impairment of oxygenation and perfusion that occurs after severe head injury, metabolic derangement is an extremely common and important consequence of TBI. The metabolic changes may be global or focal, with evidence demonstrating metabolic derangement after TBI coming from several methodologies, including (1) a 2-deoxyglucose technique to measure glucose metabolism, (2) positron emission tomography (PET) studies using fluorodeoxyglucose in humans, (3) measurement of the jugular/arterial differences in oxygen and lactate to yield global measures of oxygen consumption and lactate production in humans (AVDO2 and CMRO2), (4) measurements of whole-brain metabolic indices with magnetic resonance spectroscopy, and (5) measurements of focal ECF indices using microdialysis and PbtO2 monitors. The data from these studies allow certain generalized conclusions. TBI induces massive ion fluxes across neuronal membranes, widespread loss of resting membrane potential, and release of neurotransmitters into the extracellular space. Within minutes of these events, the brain attempts to restore ionic homeostasis by ionic pumping and reuptake of neurotransmitters. These processes are intensely energy dependent and result in an abrupt increase in glucose utilization. Studies based on the fluid percussion model in rats have shown that this increase in glucose metabolism, to facilitate the generation of adenosine triphosphate (ATP), is brief and maximally localized to parts of the brain that are maximally deformed by the shearing forces.128 When focal lesions such as subdural hematoma, focal infarction, or cerebral contusion are present, glucose use increases for a period in the penumbral border zone.129,130 Furthermore, in humans, PET studies have shown that these increases in glucose are maximal in the penumbral zone of contusions and in the hemisphere underlying hematomas when this part of the brain is viable.131 This increase may persist for 2 to 4 hours in the rat and 5 to 7 days in humans.129,131 In both animal models and humans, glucose use is depressed when measured days after diffuse injury and remains so for weeks after impact, which is consistent with the reduced metabolic needs of the comatose brain.132
Clinical Implications
Hypothermia was an initially promising intervention intended to improve outcomes after TBI that is best discussed as a targeted therapy for the metabolic derangements seen after TBI. Historically, clinical interest in induced hypothermia is derived from experimental models of head injury in which hypothermia has been shown to (1) reduce energy requirements of the brain, (2) stabilize cell membranes,133 (3) improve posttraumatic CBF-metabolic uncoupling, (4) attenuate axonal injury, (5) reduce inflammation, and (6) reduce ICP. There have been at least 12 clinical trials performed, with approximately 6 ongoing trials, and thus far no outcome benefit has been revealed. As of this writing, it is unclear why no benefit has been observed. Hypothermia does have many associated potential systemic complications, and these may be negating the beneficial effects. Therefore, prophylactic hypothermia has currently lost momentum as a standalone therapy for TBI. However, ongoing research is being performed to see whether it might expand the window of opportunity to introduce other therapeutic strategies, both pharmacologic and physiologic. In addition, work is ongoing to more carefully evaluate the role of therapeutic hypothermia, in particular for intracranial hypertension, in the management of TBI.
Mitochondrial Permeability Transition
Clearly, mitochondrial integrity is pivotal in the maintenance of cellular metabolic homeostasis. The occurrence of a process involving the increased permeability of mitochondrial membranes en route to cellular death has long been speculated. Kroemer and coauthors proposed the term mitochondrial permeability transition (MPT) to describe a calcium-induced process of increased mitochondrial membrane permeability.134 Since that time there has been a tremendous explosion in research on MPT and cell death. MPT has been described as increased permeability of the inner membrane of mitochondria via the opening of channels called MPTPs. Opening of MPTPs is a devastating event with resultant loss of transmembrane potential, mitochondrial swelling, and eventual rupture of the outer mitochondrial membrane. This loss of mitochondrial function produces profound deficiencies in neuronal metabolism and ionic equilibrium. To clarify terminology, mitochondrial permeability transition is the term used to identify the entire process, whereas mitochondrial permeability transition pore is the term used to describe the actual channel (therefore, opening of the MPTP allows the process of MPT to occur).
Clinical Implications
Cyclophilin D (CyD) is a member of the cyclophilin family involved in protein folding that is normally found in the mitochondrial matrix but migrates to the inner membrane during MPT. In experiments using the drug cyclosporine to block CyD, MPT activity was significantly attenuated, thereby establishing CyD as a critical player in MPT. As discussed previously, cyclosporine is a Food and Drug Administration–approved drug that has been shown to confer neuroprotection after TBI in multiple animal models and is being evaluated for potential use in neurocritical care.135 Interestingly, a recent in vitro study using isolated mitochondria revealed that the presence of phosphate was necessary for the inhibition of MPTP opening by cyclosporine administration or CyD knockout.136 Investigations are ongoing to determine the relationship, if any, of the neuroprotective effect of cyclosporine specifically on the blunting of MPT. Studies have investigated the role of MPT in apoptosis. Although cyclosporine-dependent MPT has been tied to the release of cytochrome c137 and thus to apoptosis, other studies have failed to show inhibition of apoptosis by cyclosporine-mediated blockage of MPT.138 Furthermore, one study indicated that CyD-dependent MPT regulates necrotic cell death but not apoptotic cell death.139 Finally, overexpression of CyD has been shown to be somewhat protective of apoptotic death. Overall, it is clear that MPT is an important mediator of necrotic cell death and can be implicated in apoptosis. However, its exact role in apoptosis remains unclear and warrants further investigation.
DNA Damage
Figure 327-3 illustrates several of the pathways that lead to DNA damage, including caspase-independent apoptosis (via apoptosis-inducing factor [AIF]), caspase-dependent apoptosis, and oxidative stress (through the generation of nitric oxide [NO]). DNA damage will then activate poly(adenosine diphosphate ribose) polymerase (PARP), which will induce the release of AIF through sequential activation of calpain and then Bax. It should be pointed out that DNA damage is usually associated with apoptosis, but this is not always the case because apoptosis can occur without DNA fragmentation.140 Furthermore, the presence of DNA fragmentation should not be used to exclusively indicate apoptosis.
Neuronal injury mechanisms involving DNA damage are also often mentioned in concert with induction of the tumor suppressor gene p53, which can serve as a transcription factor for a variety of genes with many actions (e.g., growth arrest, promotion of apoptosis). Increased expression of p53 with an associated increase in DNA fragmentation and neuronal apoptosis has been shown after focal141 and diffuse TBI.142 Increased endonuclease activity resulting in DNA fragmentation has been demonstrated in animal models of head injury.143 These delayed apoptotic mechanisms may be more amenable to pharmacologic blockage because of their long “window of opportunity” in contrast to other neuroprotective mechanisms, and at least two current neuroprotective trials involving TBI (Solvay SL334 and Neuren NZ1366) are in part targeting this mechanism.
Free Radical Formation
Free radicals are highly reactive ionic molecules bearing an unpaired electron in their outer electron shell. This unpaired electron confers high chemical reactivity. Free radicals are the normal by-product of oxidative metabolism within mitochondria, and they fulfill important physiologic roles such as signaling and polymorphonuclear leukocyte–mediated destruction of bacteria.144,145 Reactive oxygen species (ROSs) are inherently injurious, however, and contribute to many diseases affecting the CNS, including Parkinson’s disease, Alzheimer’s disease, multiple sclerosis, and amyotrophic lateral sclerosis.146 ROSs are also important contributors to secondary injury and are produced early after neurotrauma.147,148 Indeed, many secondary injury processes lead to free radical production, and in turn, free radicals feedback positively to increase the activity of many of these harmful processes.
[/not-level-membership-for-neurosurgery-category]
