Neuroblastoma
Epidemiology
Neuroblastoma is the most common extracranial solid tumor in infants and children, representing 8% to 10% of all childhood tumors. It accounts for approximately 15% of all cancer-related deaths in the pediatric population [1]. The incidence of neuroblastoma is 10.2 cases per million children under 15 years of age [2], and nearly 500 new cases are reported annually. Although 90% of cases are diagnosed before 5 years of age, 30% of those are within the first year. The median age of diagnosis is 22 months [3]. Rarely does it present in adolescence and adulthood, but outcomes are much poorer in this age group. There does not appear to be an increased prevalence among races, but there is a slight predilection for boys (1.2:1.0) [1].
With a family history noted in 1% to 2% of diagnoses, there are reports of autosomal dominant patterns of inheritance [3]. In such pedigrees, patients are frequently diagnosed at an earlier age (median age of 9 months) than those with sporadic disease and are more likely to have multiple associated primary cancers. Neuroblastoma has also been diagnosed in conjunction with other congenital conditions, such as Hirschsprung disease, congenital hypoventilation disorder, and neurofibromatosis type 1 [1]. There was early interest in the co-occurrence of neuroblastoma and neurofibromatosis, as they are both disorders of neural crest cells. However, this may represent coincidence rather than a true association [4].
Outcomes in patients with neuroblastoma have improved steadily over the last 30 years with 5-year survival rates rising from 52% to 74% [2]. The improvement is likely secondary to improved cure rates in the low-risk group, who have survival rates of up to 92%. It is estimated that 50% to 60% of patients in the high-risk group experience relapse [2], and as such, they have only seen a modest decrease in mortality. In a study by the International Neuroblastoma Risk Group, the median time to relapse was 13.2 months, and 73% of those who relapsed were aged 18 months or older. Taken together, their overall survival rates remain quite abysmal (∼20% at 5 years) despite more aggressive therapies [5].
Clinical presentation
Multiple factors play into a patient’s clinical presentation because it depends largely on tumor location, size, degree of invasion, effects from catecholamine secretion, and symptoms caused by paraneoplastic syndromes. Nearly 65% of tumors arise in the abdomen with half of those localized to the medulla of the adrenal gland. However, they can occur in the neck (5%), chest (20%), or pelvis (5%), and 1% of patients have no detectable primary [1,6]. Many patients are asymptomatic, yet some may present with constitutional symptoms (malaise, fevers, and weight loss), an enlarging mass, pain, abdominal distension, lymphadenopathy, or respiratory distress secondary to compression or hepatomegaly. Pelvic masses may cause constipation or difficulty with urination, whereas thoracic involvement can cause dysphagia, dyspnea, or rarely thoracic outlet syndrome [7]. For cervical tumors, a patient may develop Horner syndrome, and in up to 15% of patients, epidural extension may result in neurologic deficits, such as progressive paralysis [1].
At the time of diagnosis, 50% of patients present with localized disease, whereas 35% already have regional lymph node spread [1]. Metastasis can occur by hematogenous or lymphatic route, seeding bone marrow, liver, and bone. Commonly, the orbits are involved, which manifests as periorbital swelling and proptosis (raccoon eyes). When dissemination occurs to the skin, patients develop blue subcutaneous nodules known as blueberry muffin syndrome. Surprisingly, this is associated with a favorable prognosis with likely spontaneous tumor regression.
Because of its neuroendocrine properties, neuroblastoma has the potential to secrete catecholamines, which results in early-onset hypertension and tachycardia. Patients may also experience paraneoplastic syndromes. Examples include intractable diarrhea with electrolyte disturbances caused by release of vasoactive intestinal peptide (VIP), encephalomyelitis, or sensory neuropathy. There have been reports of the development of opsoclonus-myoclonus syndrome (OMS), which occurs when antibodies cross-react with cerebellar tissue [8]. The characteristic symptoms and signs of OMS include rapid, conjugate eye nystagmus with involuntary spasms of the limbs. Interestingly, the patients with intractable diarrhea caused by VIP secretion or OMS generally tend to present with less aggressive neuroblastomas. Nonetheless, symptomatic paraneoplastic syndromes are quite rare and are estimated to occur in less than 0.01% of all cancers [9].
Classification and staging
Neuroblastoma belongs to a group collectively known as peripheral neuroblastic tumors, which also includes intermixed ganglioneuroblastoma, ganglioneuroma, and nodular ganglioneuroblastoma. Neuroblastoma can further be divided based on the degree of neuroblastic differentiation (undifferentiated, poorly differentiated, and differentiating) and the mitosis-karyorrhexis index (MKI; low, intermediate, or high) [10]. Histologically, it has limited Schwannian cell production, is stroma-poor, and has abundant neuroblasts [8].
The International Neuroblastoma Pathology Classification has been used to predict prognosis based on the histopathology of the tumor and age of patients. This system takes into account the degree of cell differentiation, MKI, and the presence of Schwann cells. Following these guidelines, the unfavorable group encompasses patients with any tumor more than 60 months; undifferentiated tumors with a high MKI at any age; and undifferentiated or poorly differentiated tumors with intermediate or high MKI in children older than 18 months [11].
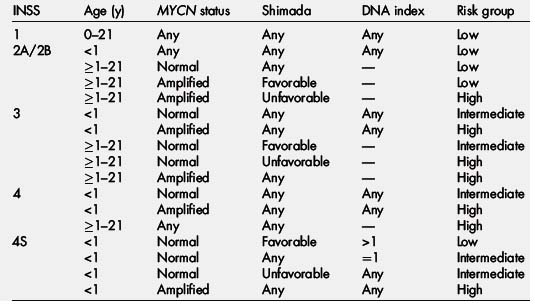
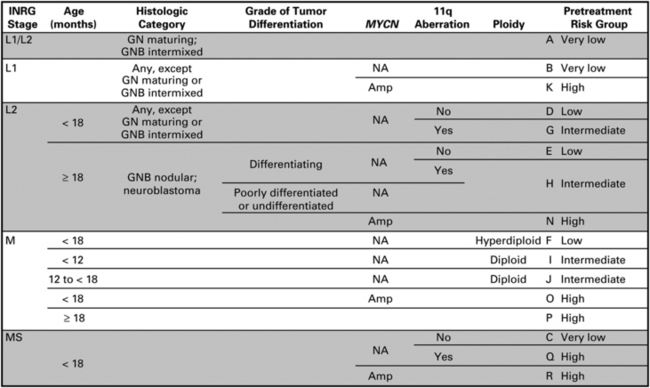
Staging is dictated by the International Neuroblastoma Staging System (INSS) (Box 1). The Children’s Oncology Group currently stratifies patients into low-, intermediate-, or high-risk categories based on patients’ age at diagnosis, INSS stage, tumor histopathology, DNA index, and MYCN amplification status (Table 1) [12]. Because the INSS is based on tumor resection, a new system, called the International Neuroblastoma Risk Group Staging System, was introduced in hopes of better stratifying pretreatment patient risk based on clinical criteria and image-defined risk factors (Fig. 1). This staging system classifies neuroblastoma into L1 (localized disease that does not involve vital structures and is confined to one body compartment); L2 (localized disease with image-defined risk factors); M (distant metastatic disease); and MS (metastatic disease confined to the skin, liver or bone marrow in children younger than18 months) [13]. Based on this, patients can be sorted into pretreatment very low-, low-, intermediate-, and high-risk groups [14]. This classification has implications as to what treatment algorithm is recommended.
Genomics
It has been shown that deletion of the chromosome 1p36 region occurs in 70% of tumors and may correlate with an increased risk of relapse in patients with localized tumors [15,16]. Interestingly, a promising candidate suppressor gene in the 1p36 region is CHD5, which is expressed in the nervous system and controls cellular proliferation, senescence, and apoptosis. Studies have confirmed that CHD5 expression is low in neuroblastoma tumors that have a chromosome 1p deletion, and when these cells were transfected with CHD5, clonogenicity and tumor growth were decreased [17]. Whether this has any prognostic significance is unknown, but it does appear to play a role in neuroblastoma tumorigenesis.
Gain of chromosome 17q occurs in approximately 80% of neuroblastomas, making it the most common genetic aberration. Although whole-chromosome 17 gain is associated with a good prognosis [16], unbalanced translocations of chromosome 17q with either 1p or 11q have been reported to be an independent poor prognostic factor. Likewise, deletions of chromosome 11q have been identified in 15% to 22% of neuroblastomas and are also associated with unfavorable patient outcomes and a lessened time of progression-free survival [8]. Chromosome 11q and 3p deletions are often found together and occur in high-stage tumors without MYCN amplification that is typical of aggressive neuroblastoma [16]. With 3p deletions frequently noted in patients who are older at the time of diagnosis (median age of 60.5 months), some speculate that 3p loss is an event that occurs later in oncogenesis [18].
In instances of familial neuroblastoma, recent evidence points to mutations of the anaplastic lymphoma kinase (ALK) oncogene as the putative cause. ALK is a tyrosine kinase receptor whose expression is important in neural differentiation, proliferation, and survival. Its malignant transformation is thought to arise from missense mutations that are linked to chromosomal region 2p23–24 and result in constitutive kinase activity. However, ALK can also be mutated at specific hotspots, such as R1275Q and F1174L, in up to 12% of sporadic cases, with the latter mutation being the most common [19]. Currently, efforts are being made to determine the pathways by which ALK exerts its effects and whether inhibitors of ALK would be feasible therapeutically.
Lastly, the MYCN oncogene, which is amplified at chromosome 2p24 in 25% of cases, is used as a biomarker for disease stratification. MYCN amplification is defined as greater than or equal to 10 gene copies per nuclei because tumors with fewer than this do not behave as aggressively [20]. MYCN amplification is found in 30% to 40% of stage 3 and 4 neuroblastomas, but only in 5% of localized or stage 4S neuroblastomas. It encodes for multiple transcription factors that, when overexpressed, lead to deregulated growth and proliferation [4]. Several MYCN downstream targets have been identified, including p53, Aurora A kinase, and MDM2 [21]. However, the exact mechanisms remain unknown.
Diagnosis
Based on clinical presentation, the index of suspicion must be high. Initial diagnostic testing should include basic blood work (complete blood count, serum electrolytes, liver function tests) and a chest radiograph, which may reveal calcifications or a posterior mediastinal mass. Additional diagnostic findings will include increased levels of urine or serum catecholamines or catecholamine metabolites (dopamine, vanillylmandelic acid, and homovanillic acid). Patients may have elevated levels of nonspecific biomarkers, such as lactate dehydrogenase (>1500 U/mL), ferritin (>142 ng/mL), and neuron-specific enolase (>100 ng/mL), which may be associated with advanced stage or relapse [15].
For diagnostic imaging, a computed tomography scan of the neck, chest, or abdomen is the gold standard, as it can simultaneously localize the tumor and determine the degree of involvement (see Fig. 1). Ultrasound may be used initially to characterize the mass. A magnetic resonance imaging may be useful if there is concern for spinal extension, and imaging of the brain is only necessary in the setting of neurologic symptoms. Although not routinely used, a 123/131iodine-meta-iodobenzylguanidine (MIBG) scan is valuable in both the detection of primary tumor and metastases because metaiodobenzylguanidine, a norepinephrine analog, is selectively concentrated in sympathetic tissue. It has also proven to be practical in surveillance of treatment response and recurrence. The role of fluorodeoxyglucose positive emission tomography (FDG-PET) remains controversial, but a small retrospective study has suggested that although MIBG is generally more sensitive for the detection of lesions, FDG-PET may be better at localizing soft-tissue metastases [22].
Treatment
The mainstay of treatment consists of chemotherapy, surgical resection, or radiotherapy. However, many aggressive neuroblastomas have developed resistance to chemotherapeutic agents, making the likelihood of relapse quite high. Treatment can be hindered by aggressive neuroblastomas that exhibit drug resistance, and this may be related to the selection of clones that express multidrug resistant-associated protein (MRP1) [23]. The treatment algorithm is dependent on the patients’ stage and risk stratification. The goal of induction chemotherapy is to achieve remission by reducing the tumor burden, which then allows for a more complete resection when indicated. Induction chemotherapy consists of some combination of cyclophosphamide, doxorubicin, cisplatin, melphalan, carboplatin, etoposide, topotecan, ifosfamide, and vincristine. After induction, treatment is consolidated with one or more courses of high-dose chemotherapy to induce bone marrow ablation, which necessitates autologous hematopoietic stem cell support. Rescue is not without complication as it can lead to growth failure, endocrinopathy, and the occurrence of secondary malignancies [24].
For high-risk patients, radiotherapy is needed for local and metastatic control. It is indicated when there is minimal residual disease post-induction chemotherapy and resection. Current radiation protocols use a dose of 2100 cGy, and based on the practices at the Memorial Sloan-Kettering Cancer Center, the combination of chemotherapy, surgery, and radiation therapy has resulted in a local relapse rate of less than 10% [20]. Radiation is contraindicated for intraspinal tumors because it can lead to vertebral damage, growth arrest, and scoliosis. Yet, it may be necessary for palliation in the setting of pain or acute neurologic symptoms caused by cord compression [8].
Presently, patients receive 6 courses of 13-cis-retinoic acid (CRA) to eradicate residual disease that may still be present despite meeting imaging criteria for complete remission. This treatment is based on the finding that high-dose therapy with 13-cis-retinoic acid given after chemoradiation significantly improved event-free survival in high-risk neuroblastoma. Side effects, such as skin dryness and cheilitis, are the dose-limiting factor and, consequently, CRA therapy consists of 2-week courses alternating with 2 weeks for mucocutaneous recovery [25].
Trials involving myeloablative chemotherapy and 131I-MIBG have been underway in an effort to minimize adverse side effects by making therapies more targeted. Previous studies have shown that 131I-MIBG exhibits activity against refractory neuroblastoma with response rates ranging from 10% to 50% [26]. In a phase I trial of 131I-MIBG therapy for relapsed neuroblastoma, myelosuppression was the most significant toxicity at doses greater than 15 mCi/kg, as nearly half of the patients enrolled required hematopoietic cell transfusion. Despite this, the response rate (36%), event-free survival (18% at 1 year), and overall survival (49% at 1 year; 29% at 2 years) were found to be significantly higher in patients older than 12 years and who had fewer than 3 prior treatment regimens [27]. Subsequently, a phase I dose-escalation study of 131I-MIBG with myeloablative chemotherapy and stem cell rescue showed a significant response rate of 25% in patients with primary refractory disease. Given these findings, 131I-MIBG may prove to be useful in conjunction with other treatment modalities.
The total length of therapy averages nearly 1 year, and most treatment failures are caused by minimal residual disease that was not eradicated following high-dose chemotherapy. Although the aim of further treatment is remission, prolonged disease stabilization is usually the reality because most patients who relapse eventually die of disease progression. Even patients who achieve a cure with initial therapy remain at risk for developing long-term complications related to treatment, including hearing loss, infertility, and second malignancies [28].
Current research milestones and proposed novel therapies
Retinoic acid
Retinoids are vitamin A derivatives that can cause arrest of cell growth and induce differentiation of human neuroblastoma cells. Although 13-cis-retinoic acid is being used clinically for this purpose, fenretinide (4-HPR) is another retinoid derivative currently in phase I trials which has been reported to inhibit growth in vitro in a dose-dependent manner. At the higher end of the dose spectrum, it also seems to be active against retinoic acid-resistant neuroblastoma cell lines that harbor mutations in the retinoic acid receptor (RAR). Unlike all-trans and 13-cis retinoic acids, 4-HPR does not induce differentiation, but is actually cytotoxic and pro-apoptotic and may do so in a receptor-independent manner [25,29]. So far, it appears that 4-HPR is minimally toxic with no evidence of myelosuppression; the most common side effect reported is decreased night vision.
mTOR inhibitors
One promising target is the mammalian target of rapamycin (mTOR), which is a serine/threonine protein kinase that regulates cell growth, proliferation, motility, metabolism, and survival. It can exert its effects through several different signaling pathways, notably the phosphatidylinositol-3 kinase (PI3K)-AKT pathway. Rapamycin, a bacterial byproduct, is the classically cited mTOR inhibitor, and it does so by complexing with the intracellular receptor FKBP12 to bind directly to the FKBP12-rapamycin binding (FRB) domain on mTOR [30]. This process leads to activation of downstream effectors, such as cyclin D1, p21, and HIF1α/β. Treatment with mTOR inhibitors leads to downregulation of MYCN and vascular endothelial growth factor (VEGF). Temsirolimus, a rapamycin analog, is being investigated in phase II trials in patients with relapsed neuroblastoma. One study showed that treatment with an insulin-like growth factor (IGF)-1 receptor inhibitor and temsirolimus in vitro was more effective at inducing cell death than either agent alone [31]. Known side effects include skin rashes, stomatitis, and hyperglycemia.
Aurora A kinase
Aurora A kinase is a serine/threonine kinase important in regulating progression through the cell cycle, particularly during the G2 to M phase transition. Aurora A kinase has an important role in centrosome maturation, spindle assembly, meiotic maturation, and spindle orientation. Selective inhibition of Aurora A kinase results in inhibition of autophosphorylation and p53 phosphorylation, monopolar spindles, and G2-M arrest [32]. MLN8237, a reversible Aurora A kinase inhibitor, is being investigated in phase I clinical trials by the COG for patients who have experienced relapse. In vitro and in vivo studies with MLN8237 have shown that in addition to inducing apoptosis, it upregulates p53 and the tumor suppressor genes p21 and p27 [33]. Dose-escalation and combination therapy studies are currently underway.
TrkB inhibitors
The Trk family is a group of tropomyosin kinases that are known to modulate neuronal differentiation and survival. There are 3 isoforms (TrkA, TrkB, and TrkC) that each has an affinity for a specific ligand (NGF, BDNF, and NT-3, respectively). Ligand binding causes receptor homodimerization, which leads to autophosphorylation, docking of downstream effectors, and activation of PI3K/AKT, phospholipid C (PLC) γ1, and Ras/mitogen-activated protein kinase (Ras-MAPK) signaling [34]. Their ligands can similarly bind p75, a nonselective and low-affinity death receptor, to induce apoptosis via the c-Jun amino terminal kinase (JNK) pathway [35]. Although TrkA expression is associated with favorable prognosis, TrkB has been found to correlate with MYCN amplification and clinically aggressive neuroblastoma. In studies, neuroblastoma tumor cells treated with brain-derived nerve factor (BDNF) appear to be less sensitive to cytotoxic drugs, survive in a less than optimal microenvironment, are more invasive, and produce increased levels of VEGF. Furthermore, inhibitors were found to slow growth and induce apoptosis. Thus, the Trk inhibitor CEP-701 is presently being used as an oral phase I agent with the premise that it will be most effective in conjunction with conventional chemotherapeutic agents.
Immunotherapy
The ganglioside GD2 is expressed in the surface of tumors of neuroectodermal origin, including neuroblastoma. Anti-GD2 monoclonal antibodies have been developed because they are specific, high affinity, and fairly nontoxic. They kill tumor cells through both complement and cell-mediated lysis. The antibody 3F8 is a murine IgG3 monoclonal antibody that has been shown to activate complement on malignant neuroblastomas with ensuing lysis. Moreover, it is thought that granulocyte-macrophage colony-stimulating factor (GM-CSF) can potentially amplify 3F8 antitumor activity by increasing granulocytes [36]. However, its clinical use has been complicated by the formation of human antimouse antibodies. To circumvent this, chimeric mouse-human antibodies, such as Ch14.18 (a monoclonal antibody against the tumor-associated GD2), have been generated. Although they were thought to be less immunogenic, 28% of patients reportedly still developed antibodies in a phase I trial [37]. In other studies, beta-glucan has been found to enhance anti-GD2 antibody antitumor effects through increasing iC3b-mediated cytotoxicity. An ongoing phase I trial is comparing the efficacy of 3F8 when given with oral beta-glucan. Other trials are focused on using Ch14.18 in conjunction with other agents, such as interleukin-2 and GM-CSF, after high-dose chemotherapy with stem-cell rescue. A recent study demonstrated that immunotherapy with Ch14.18, GM-CSF, and interleukin-2 was found to be associated with significantly improved overall outcome when compared with standard therapy in high-risk group patients with neuroblastoma [38].
Angiogenesis inhibitors
VEGF has been established as an important growth factor that promotes tumor angiogenesis by inducing endothelial cell proliferation. It has been found to play a significant role in tumorigenesis because in hypoxic environments, such as those in rapidly growing tumors, hypoxia-inducing factor (HIF) stimulates VEGF production. This process results in downstream activation of several signaling pathways, including PI3K/AKT (increased vascular permeability) and MAPK/ERK (endothelial cell proliferation). In neuroblastoma murine models, VEGF inhibitors were found to decrease cell proliferation [39]. Bevacizumab, a human-derived VEGF monoclonal antibody, is approved by the Food and Drug Administration for the treatment of colorectal, breast, and lung cancer, and it is now being used in phase II studies in neuroblastoma, as it has not previously been used in children.
MDM2 and ODC1 as a MYCN targets
Mutations of the gene p53 are rare in neuroblastoma and several studies have shown that it appears to be functional because it is capable of activating downstream apoptotic effectors in response to DNA damage [40]. MDM2 is a negative regulator of p53, being a key player in its ubiquitination. It has been suggested that deregulated MDM2 expression may explain aberrant p53 function. Notably, MDM2 is transactivated by MYCN, and downregulation of MYCN leads to decreased MDM2 expression, p53 stabilization, and cell death. Therefore, dysregulation of MDM2 is thought to decrease p53 activity, and in vitro studies with the MDM2 inhibitor nutlin showed that it could stabilize p53 and induce G1 cell cycle arrest and apoptosis [41,42]. Further investigations are needed, but this class of agents may represent an untapped resource.
Recently, ODC1, an oncogene that encodes for an enzyme needed in polyamine biosynthesis, was tagged as a potential therapeutic candidate. Polyamines enhance transcription, translation, and replication and are, therefore, essential for cell survival as depletion leads to growth arrest and apoptosis. MYCN functions as a transcription factor that dimerizes with another factor known as MAX to form a complex that can then activate ODC1 transcription [43]. MYCN-amplified tumors have elevated levels of ODC1 and exhibit deregulated polyamine synthesis. In mouse models, studies revealed that treatment with cyclophosphamide and α-difluoromethylornithine (DFMO), an ODC inhibitor, led to an increased overall survival (80% vs 20% at 3 months) than with cyclophosphamide alone [44]. It is now being piloted in phase I clinical trials with and without etoposide administration.
GRP receptor
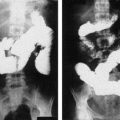
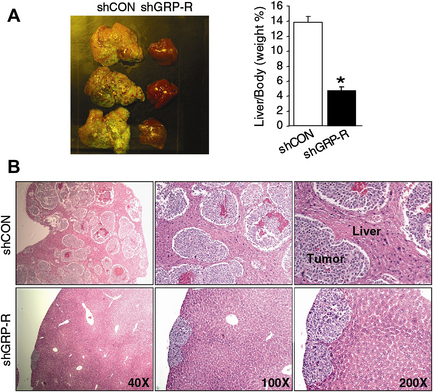
Gastrin-releasing peptide (GRP) receptors, a member of G-protein coupled receptor family, are abundantly expressed in undifferentiated neuroblastomas. As in other well-characterized cell surface tyrosine kinase receptor’s function in cancers (eg, epidermal growth factor receptor), GRP receptor may also be an important therapeutic cell membrane target in neuroblastomas. GRP ligand is produced and secreted by neuroblastomas to stimulate its tumor growth, and hence, it functions as an autocrine growth factor [45]. GRP binds to GRP receptors to activate the PI3K/AKT pathway in promoting tumorigenesis in neuroblastomas [46]. Targeted silencing of GPR receptors in neuroblastoma cells showed significantly attenuated xenograft growth as well as liver metastases when compared with controls with constitutive expression of GRP receptors [47]. Knockdown of GRP receptors demonstrated inhibition of tumor metastasis to liver in vivo (Fig. 2) [47].
Acknowledgments
References
This work was supported by grant R01 DK61470 from the National Institute of Health and Rally Foundation for Cancer Research.