28. Neonatal Surgery*
Harold N. Lovvorn III, Joshua Brian Glenn, Annette S. Pacetti and Brian S. Carter
Congenital malformations may be found in up to 3% of all newborns and are an important cause of morbidity, early infant death, and chronic disability. Although overall infant mortality has declined, the mortality attributable to birth defects has increased, and up to 22% of neonatal deaths have been attributed to congenital malformations. 12 In the past decade, improvements in prenatal imaging and perinatology have allowed earlier diagnosis and intervention for surgically correctable malformations. Because of the complexity of accurate prenatal imaging and diagnosis, many anomalous conditions continue to escape early detection and present to the neonatology and surgical teams with advanced developmental consequences. The care of a neonate with a major congenital malformation therefore may be resource intensive and costly. In a study of one regional neonatal intensive care unit (NICU), newborns with major congenital malformations accounted for 27% of NICU referrals, 32% of total NICU days, and 40% of NICU costs. Moreover, surgery was more frequent in newborns with major malformations, and one third required ongoing medical support at the time of discharge. 47 Early diagnosis, comprehensive neonatal care, and a multidisciplinary approach are necessary to ensure an optimal outcome for both parent and child.
This chapter briefly describes the embryology, clinical history, diagnostic evaluation, and therapeutic intervention of common neonatal surgical conditions.
DIAPHRAGMATIC HERNIA
Physiology and Etiology
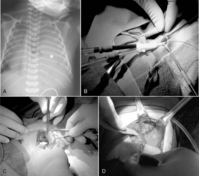
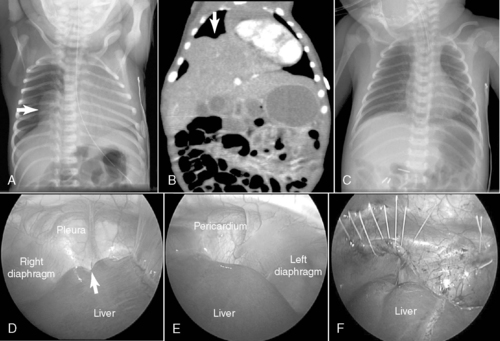
Congenital diaphragmatic hernia (CDH) is a defect in closure of the diaphragm that occurs in 1 in 4000 live births. A posterolateral defect, or Bochdalek diaphragmatic hernia (Figure 28-1), accounts for nearly 95% of all CDHs and may be left-sided (95%) or right-sided (5%). Much less common is the Morgagni diaphragmatic hernia (Figure 28-2), which results from a failure of anteromedial closure and resides in a substernal location. Several theories have been proposed to explain the mechanism for how this maldevelopment of the diaphragm occurs, but currently the most widely accepted theory is failure of the pleuroperitoneal canal to close completely during the 8th week of gestation. If a patent pleuroperitoneal canal persists through the 11th week of gestation, a period when the intestine returns from its normal herniation into the umbilical cord, the stomach, bowel, and spleen may be forced into the chest. Resultant compression of the developing lung leads to a variable extent of pulmonary hypoplasia.
 |
| FIGURE 28-2
(Courtesy Dr. H. Lovvorn, Vanderbilt University Medical Center.)
|
Despite many studies in both animal models and infants over the past four decades, the causes of the pathophysiologic changes that occur in the underdeveloped lung are not well understood. Recognition of the associated abnormal physiologic mechanisms has failed to define these alterations as cause or effect for the malfunctioning lung. In addition to alveolar hypoplasia, a concomitant abnormal development of the pulmonary vasculature is a major contributor to the clinical challenges in managing CDH and its inherent risk for pulmonary hypertension. 67,83 Postnatal blood flow through these hypoplastic lungs is compromised by both a reduced total number of pulmonary arterioles and an increased muscularization of the arteriolar bed. As a result, pulmonary vasculature capacitance is reduced and responsiveness to signals for smooth muscle relaxation may be lost. Persistent pulmonary hypertension after birth maintains patency of and blood flow through natural fetal shunts (the foramen ovale and the ductus arteriosus). Resultant pulmonary hypoperfusion and systemic hypoxemia may be extreme and are often recalcitrant to conventional therapy and irreversible.
Data Collection
HISTORY
Technologic advances in ultrasonography have facilitated earlier and more accurate antenatal diagnosis of diaphragmatic hernias, permitting planning of intervention and counseling of parents, as appropriate. The mother may then be transferred to deliver in a high-risk perinatal center with the appropriate level of neonatal and surgical support on standby, which is clearly less morbid than transferring the ill newborn with CDH. Although in utero surgical techniques to promote lung growth and passive reduction of the herniated abdominal contents seemed promising initially, as of this writing, a moratorium remains in place on this approach in the United States because a demonstrable survival benefit has not been shown, largely because of advances made in postnatal care of CDH. 86 Some fetal surgery centers, particularly European, continue to explore minimally invasive means to promote lung growth in utero and thereby reduce the herniated abdominal contents. The Achilles’ heel of fetal intervention, however, is the risk of preterm labor and the consequent morbidity of prematurity, risks which collectively must be weighed against a term delivery but with profound compromise in lung development.
SIGNS AND SYMPTOMS
Respiratory distress may develop immediately after birth or after an initial period of relative stability. If significant pulmonary hypoplasia is present and fetal circulation persists, the newborn may become rapidly symptomatic, heralded by profound hypoxic respiratory failure and circulatory shock . Because much of the bowel is herniated into the chest, the abdomen appears scaphoid and the anteroposterior diameter of the chest may enlarge as the bowel distends with air. Breath sounds are diminished or absent on the affected side, and the mediastinum may be displaced toward the contralateral side. Associated anomalies of the cardiovascular system may include patent ductus arteriosus, aortic coarctation, and hypoplastic left heart syndrome and require echocardiographic evaluation. Other associated anomalies requiring evaluation include central nervous system (CNS) malformations, genitourinary anomalies, esophageal atresia, omphalocele, cleft palate, and cardiovascular defects. 72
LABORATORY DATA
Mixed respiratory and metabolic acidemia is present on blood gas evaluation. A chest radiograph is obtained and shows bowel herniated into the ipsilateral thoracic space with contralateral displacement of the heart. An echocardiogram is necessary to assess cardiac function, degree of pulmonary hypertension, and presence of significant congenital cardiac anomalies. Nonrotation of the intestine is an understood feature of CDH and does not require specific evaluation as a newborn.
Treatment
PREOPERATIVE CARE (STABILIZATION)
As soon as a diaphragmatic hernia is suspected, an orogastric tube should be placed to prevent further distention of the stomach and bowel and to alleviate compression of the lung. The newborn with CDH commonly requires endotracheal intubation and mechanical ventilation to maintain adequate gas exchange. Because cardiac function may also be compromised, a combination of inotropic and vasopressor medications such as dopamine, dobutamine, and milrinone may be needed. The earlier the infant becomes symptomatic, the more severe the respiratory compromise and the poorer the prognosis may be. Pulmonary hypertension and lung hypoplasia are the major determinants of early outcome.
A significant number of CDH newborns may have such severe pulmonary hypertension refractory to conventional or alternative (e.g., high-frequency oscillation or jet) ventilation that extracorporeal life support (ECLS; venovenous and venoarterial) may be required to establish effective gas exchange and end-organ perfusion. The overriding principle in stabilizing CDH newborns is to reduce barotrauma and oxygen toxicity that result in chronic lung disease. Strategies now emphasize gentle ventilation and permissive hypercapnia, so long as arterial pH does not drift significantly low (<7.25).8
OPERATIVE INTERVENTION
Surgical repair does not alter early outcome. Therefore the baby’s condition should be stabilized and efforts directed toward the management of the associated pulmonary hypoplasia and hypertension. Early repair within the first 72 hours of life is indicated only in infants with little or no pulmonary dysfunction. If severe pulmonary insufficiency is present, medical therapies of conventional or high-frequency mechanical ventilation, inhaled nitric oxide, or ECLS are instituted. If these modalities are successful in stabilizing the baby, surgical repair is generally performed between 4 and 14 days of life.67,87 If ECLS has been needed to stabilize the newborn with CDH, some centers advocate herniorrhaphy while on bypass, whereas other centers recommend repair after decannulation (see Figure 28-1).
Most commonly through a subcostal transabdominal approach, the surgeon reduces the stomach, intestine, and spleen from the chest to the abdominal cavity and repairs the diaphragmatic defect. If the defect is large, a prosthetic patch may be required to complete closure of the hernia. Closure of the abdominal wound may be difficult too because of underdeveloped abdominal wall musculature and loss of abdominal domain. In these circumstances, if abdominal closure is not possible or may result in abdominal hypertension, simple skin closure or prosthetic silo placement may be necessary to cover the abdominal contents, leaving a large ventral hernia for future repair when the abdominal domain is adequate and the baby is more stable. Chest tube placement depends on risk for bleeding, which may be significant if repair is performed on ECLS (because of anticoagulation during extracorporeal membrane oxygenation therapy), or if risk of pneumothorax is anticipated. Minimally invasive techniques to repair CDH may also be used in newborns (see Figure 28-2) but may not be well tolerated in the most fragile neonates. Appropriate patient selection is of prime importance. 28 Thoracoscopic and laparoscopic techniques to repair CDH have been described and may be better suited for infants who present out of the newborn period and without physiologically significant pulmonary hypoplasia or hypertension.
POSTOPERATIVE CARE
The principal postoperative concern remains effective ventilation and oxygenation while imparting the least amount of barotrauma and toxicity. If conventional mechanical ventilation fails, high-frequency oscillation and inhaled nitric oxide are employed. 49,56ECLS is reserved in this setting as a salvage therapy for babies who revert back to fetal circulation and who do not respond to these less invasive modalities.
Complications and Prognosis
The survival rate for newborns with CDH and who require mechanical ventilation in the first 18 to 24 hours of life is approximately 64%. If an infant with a diaphragmatic hernia does not present with respiratory distress in the first 24 hours of life, survival approaches 100%. As improvements in gentle ventilation strategies have emerged, a gradual, albeit small, increase in survival has been realized. 49,56,72,83The primary early pathophysiologic consequence of CDH is pulmonary hypertension. Late complications include chronic lung disease, recurrent diaphragmatic hernia, gastroesophageal reflux, growth restriction, and neurodevelopmental delay.4,25 Basic and clinical research continue in an effort to identify improved therapies for the complex pulmonary dysfunction associated with congenital diaphragmatic hernia.
ESOPHAGEAL ATRESIA AND TRACHEOESOPHAGEAL FISTULA
Physiology and Etiology
Esophageal atresia (EA) occurs between 1 in 3000 and 1 in 4500 live births and represents a spectrum of anomalies that arise early in gestation (3 to 6 weeks) when the trachea normally buds from the primitive foregut. Failure in the normal development of the esophagus and in complete separation of the trachea from the esophagus gives rise to EA and distal tracheoesophageal fistula (TEF) in 85% of cases, isolated EA in 8%, TEF without EA in 5%, and EA with proximal or proximal and distal fistulas in 2%. Etiologies for this collection of defects remain unclear, but it is suspected that genetic alterations in and environmental insults on rapidly proliferating foregut stem cells during this critical period of organogenesis account for such diverse yet predictable esophageal malformations. Aberrations at the cellular level in muscle fibers of the distal esophagus may help explain the nearly universal symptoms of dysmotility and gastroesophageal reflux after operative repair. 46 Because other developing organs are vulnerable to the same insults in this critical period of gestation, associated anomalies are common (50% to 70%), particularly vertebral, anorectal, cardiac, genitourinary, limb, and gastrointestinal (e.g., duodenal atresia). 80 In 11% to 33% of infants with EA and distal TEF, concurrent and severe tracheomalacia is present. Although EA, with or without TEF, has not been associated with a single gene defect, a high incidence has been observed in children with trisomy 21, or Down syndrome. 12,24
Data Collection
HISTORY
Maternal polyhydramnios may suggest esophageal atresia or other conditions in which the fetus does not swallow amniotic fluid normally.
SIGNS AND SYMPTOMS
Babies with esophageal atresia are identified soon after birth because of excessive oropharyngeal secretions and inability to swallow saliva or feedings. Upon feeding, these babies quickly cough and regurgitate undigested formula. When attempting to pass an orogastric tube, obstruction is typically encountered between 8 and 12 cm from the lips, and the diagnosis of EA is established. If a distal TEF is also present, air passes into the stomach and bowel and is present on plain abdominal radiographs. Respiratory distress may arise if oropharyngeal secretions are aspirated or gastric secretions reflux through the TEF and into the lungs, which may incite profound chemical pneumonitis. Symptoms of an “H”-type fistula in the absence of esophageal atresia are less obvious and require a high index of suspicion. Coughing and choking with feedings or recurrent pneumonia over the first months of life suggests the presence of an occult, often “H ”-type, TEF. (See the Critical Findings box below.)
Esophageal Atresia and Tracheoesophageal Fistula
The following are critical assessment findings for esophageal atresia and tracheoesophageal fistula:
• Excessive secretions
• Feeding intolerance
• Inability to pass orogastric tube
• Abdominal distention
• Other findings associated with VACTERL
VACTERL, Vertebrae, Anus, Cardiac system, Trachea, Esophagus, Renal tract, Limbs.
PHYSICAL EXAMINATION
Once esophageal atresia has been established, the infant should be carefully examined to exclude other anomalies of the VACTERL association, a variable sequence of anomalies affecting the Vertebrae, Anus, Cardiac system, Trachea, Esophagus, Renal (urinary) tract, and Limbs. 18,41 Imperforate anus may be obvious or obscure if a perineal fistula allows passage of meconium stool. Echocardiography permits both the identification of significant cardiac anomalies and the presence of a right-sided or left-sided aortic arch, which have important implications in the operative approach.
LABORATORY DATA
After obstruction is met with passing of an orogastric tube, a plain radiograph of the chest and abdomen should be obtained. The orogastric tube is often coiled in the blind proximal esophageal pouch, typically visualized at the second or third thoracic vertebra and above the carina. The stomach and intestines may contain luminal air if a distal TEF is present. If no distal fistula exists, the abdomen on radiograph will appear gasless. In the rare setting of EA, distal TEF, and duodenal atresia, the abdominal gas pattern may show the classic “double bubble” sign, as gas fills the stomach and proximal duodenum only.
Treatment
PREOPERATIVE CARE
Once the diagnosis of EA is established, an orogastric tube is placed in the upper esophageal pouch and set to low continuous suction to prevent aspiration of oral secretions. While awaiting operation, the neonate should be kept in a supine position with the head of the bed elevated about 30 degrees and antacid and antireflux medication should be considered to minimize gastroesophageal reflux and the consequent risk of acid-induced pneumonitis. Operative repair, in general, is not an emergency procedure. Patients first should be evaluated thoroughly for other associated anomalies by physical examination, echocardiography to delineate the anatomy of the heart and great vessels, abdominal ultrasound of the kidneys and genitourinary tract, and plain radiographs of the spine and limbs. Newborns with cyanotic congenital heart disease may require a palliative cardiac procedure before reconstruction of the esophagus.
If babies with EA and distal TEF are born prematurely and have respiratory distress syndrome (i.e., “stiff,” noncompliant lungs), a significant portion of mechanical tidal volumes, delivered under positive pressure, may be shunted preferentially through the fistula and into the stomach. As a result, effective ventilation is lost and critical gastric distention ensues, further restricting diaphragmatic excursion. Emergent ligation of the TEF with gastrostomy tube insertion is necessary in such instances to restore effective ventilation.
OPERATIVE INTERVENTION
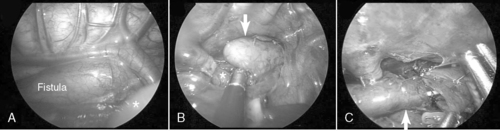
The type of esophageal malformation dictates the surgical approach. Surgical repair of EA with or without TEF is generally not an emergency but should be carried out as soon as the patient is stable. If the infant is in otherwise good health and the gap between esophageal elements is not too large, primary anastomosis is indicated through a right thoracotomy and retropleural approach. To reduce the pain and potential morbidities associated with a thoracotomy, some surgeons recommend repair thoracoscopically (Figure 28-3).
 |
| FIGURE 28-3
(Courtesy Dr. H. Lovvorn, Vanderbilt University Medical Center.)
|
Unfortunately, in isolated EA, the gap distance is generally too long to allow early primary repair. If the gap length is considered too great, with or without TEF, or if the child is too ill, a delayed or staged repair is planned. An early gastrostomy is placed for decompression and feeding, and the TEF, if present, is divided to prevent reflux into the tracheobronchial tree. After a variable period of time to allow resolution of pneumonitis or maximum growth of the distal esophagus, a second operation completes the repair. To promote lengthening and growth of the distal esophagus in cases of isolated EA, bougienage or balloon dilation may be performed via a mature gastrostomy tract.
POSTOPERATIVE CARE
Postoperative care includes appropriate pain control and pulmonary care, total parenteral nutrition (TPN), and a brief course of systemic antibiotics. Tracheal and esophageal suction catheters should not come in contact with the newly repaired esophagus and trachea, because suture line disruption may cause a leak or recurrent fistula, both potentially catastrophic complications. Antibiotic therapy is continued for 48 to 72 hours. A chest tube and/or retropleural drain is placed to control an anastomotic leak should it occur. Before initiating oral feedings, an esophagram is usually obtained within 7 to 10 days of repair to verify complete anastomotic healing and absence of leak. All EA babies have some degree of esophageal dysmotility and gastroesophageal reflux (GER) after repair. Elevating the head of the bed 30 to 45 degrees, administering histamine H2 antagonists or proton pump inhibitors, and slow feeding may help control reflux symptoms.
If delayed or staged repair is planned, secretions must be controlled. Suction catheters placed in the upper pouch are maintained to reduce the risk of aspiration. In rare cases, reconstruction using the native esophagus is not possible. In these circumstances, esophageal replacement using gastric or colon transposition is necessary. If the infant is not a candidate for early operation because of a lethal chromosomal defect or severe congenital heart disease, cervical esophagostomy and gastrostomy are performed to palliate the infant and esophageal replacement is performed later, as indicated.
Complications and Prognosis
Postoperative complications include anastomotic leak and/or stricture and esophageal dysmotility. Anastomotic leaks may occur in up to 20% of patients and generally are treated conservatively with chest tube drainage, parenteral nutrition, antibiotics, and tincture of time. The vast majority of leaks close without operative intervention but tend to heal with some degree of stricture, commonly amenable to dilation. Strictures often are associated with or exacerbated by GER and also may be treated successfully by esophageal dilation. If GER is complicated by stricture and is refractory to maximal medical therapy, a fundoplication procedure rarely may be necessary. Some degree of esophageal dysmotility usually exists because of poor peristalsis in the distal esophagus. The child may adapt to a poorly functioning esophagus by altering his or her feeding habits. However, in infancy, gastrostomy feeding may be necessary to prevent vomiting and aspiration.
With modern neonatal care and surgical techniques, long-term survival after repair of EA and TEF is excellent. 21Prognosis depends largely on two factors: (1) the presence and type of cardiac anomalies, and (2) the presence of prematurity and respiratory distress syndrome. A useful system to predict survival is the Spitz classification, which stratifies infant survival by birth weight and major cardiac anomaly74:
I: Birth weight equal to or greater than 1500 g, no major congenital heart disease (CHD), survival is greater than 97%
II: Birth weight less than 1500 g or major CHD, survival is 59%
III: Birth weight less than 1500 g and major CHD, survival is 22%
CONGENITAL CHEST MASSES
Physiology and Etiology
The most common congenital chest masses requiring surgical intervention in the newborn period are congenital pulmonary airway malformations (CPAMs, also known as cystic adenomatoid malformations), pulmonary sequestrations (both intralobar and extralobar types), bronchogenic cysts, and congenital lobar emphysema. Each of these malformations may exist alone or in combination with other anomalies. 7,44
CPAM lesions are believed to arise from focal interruption in coordinated pulmonary progenitor cell growth, resulting in abnormal development of pulmonary tissues and structural distortion. Histologically, CPAM is associated with increased cell proliferation and decreased apoptosis when compared with normal lung tissue. The CPAM lesion receives its blood supply from the pulmonary system but does not communicate with normally formed bronchial structures.
Anomalous development of the foregut is the accepted underlying etiology of both the bronchogenic cyst and pulmonary sequestration. Bronchogenic cysts are lined by ciliated columnar and/or cuboidal epithelium. The surrounding tissues resemble those of the normal bronchus and are generally, although not exclusively, located within the mediastinum along the tracheobronchial tree. Extralobar sequestrations are masses of primitive pulmonary parenchyma with no bronchial connection and are supplied by the systemic, not pulmonary, vasculature. Congenital lobar emphysema presents in the newborn period as a fluid-filled over-distended lobe that, under positive-pressure ventilation, may trap air and generate tension physiology. In many cases, though not all, congenital lobar emphysema is associated with the absence or hypoplasia of cartilaginous rings of the major and segmental bronchi. These structurally underdeveloped bronchi are prone to collapse on expiration, thereby trapping air.
Data Collection
HISTORY/SIGNS AND SYMPTOMS
Although rare, congenital lung malformations may lead to considerable morbidity, such as infection, hemorrhage, respiratory failure, and pulmonary hypoplasia, and may even prove lethal. Some lesions may escape prenatal detection and so appear later in development. Failure to recognize a malformation may lead to inappropriate intervention. For example, placement of a chest tube to manage suspected tension pneumothorax in a baby with congenital lobar emphysema may lead to lung injury and loss of tidal volume through the thoracostomy tube instead of into the remaining healthy lung.
Congenital Pulmonary Airway Malformation
CPAMs are usually detected prenatally and are nicely characterized on ultrasound but, if not, may be further delineated by fetal magnetic resonance imaging (MRI), as indicated. 42,81 In utero, these lesions may cause a variety of problems, from pulmonary hypoplasia (both ipsilateral and contralateral) to non-immune hydrops fetalis with congestive heart failure.
Polyhydramnios may also be present if the lesion compresses the esophagus and compromises fetal swallowing of amniotic fluid. Fetal intervention may be indicated if the gestation has not yet reached 34 weeks, in which case premature delivery might be planned. Large, fluid-filled cystic lesions may be amenable to thoracoamniotic shunt placement while in utero to relieve compression of intrathoracic structures and to restore hemodynamic status. Solid CPAM lesions arising early in gestation and causing similar complications have been resected in fetuses with promising results. If these lesions do not manifest with in utero pathophysiology (yet are of sufficient size), the neonate may develop respiratory distress shortly after birth. This process is responsible for the cystic appearance on radiographs. Babies may have mediastinal shift and a large air space, easily confused with a pneumothorax or diaphragmatic hernia. Sonography may be helpful to delineate a solid or cystic mass and should establish the diagnosis. CPAM may result in recurrent infections because mucociliary clearance is poor. Rarely, malignancy may arise in a CPAM in the form of pulmonary blastoma, rhabdomyosarcoma, or bronchoalveolar carcinoma. 42
Pulmonary Sequestration
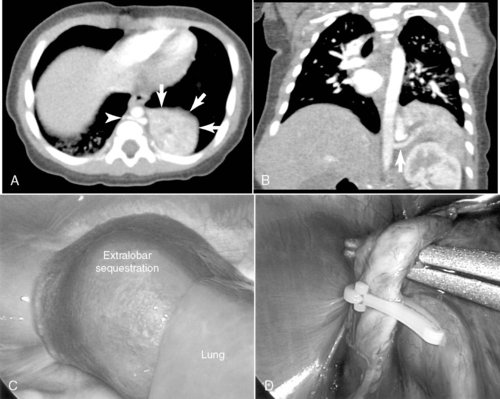
Pulmonary sequestration accounts for fewer than 10% of all congenital lung malformations and mostly occurs in the lower lobes.7,44 A sequestration represents a mass of disorganized bronchopulmonary tissue without a normal bronchial communication and may have either a pulmonary or systemic vascular supply. The abnormal sequestered lung may be intralobar or extralobar and is classified according to pleural coverage, either within the pleural investment of the whole lung itself (intralobar) or outside of this normal pleural lining (extralobar). Sequestrations rarely may have some sort of communication with the foregut. Infants with an intralobar sequestration not detected prenatally may present outside of the newborn period and often with recurrent respiratory problems, such as chronic cough, or with recurrent pneumonias, either in the lesion or in the surrounding normal but compressed lung tissue. Plain radiographs may show simple consolidation. Anomalies associated with extralobar sequestration include diaphragmatic hernia and eventration and may share a similar dysregulated embryologic event, as approximately 95% of extralobar lesions are left-sided. Extralobar lesions may reside either above or below the left diaphragm. Older children may have exercise intolerance if a large systemic arteriovenous shunt exists. Systemic arterial flow through the lesion may produce a murmur and may lead to congestive cardiac failure. Squamous cell carcinoma, adenocarcinoma, and rhabdomyosarcoma may rarely arise in the sequestration.
Bronchogenic Cyst
Bronchogenic cysts may be considered a foregut duplication and arise from an abnormal budding of the ventral foregut. Approximately 85% are mediastinal and 15% are intrapulmonary. Bronchogenic cysts may be filled with air or fluid and may show air-fluid levels on plain radiographs. 77 As a result, bronchogenic cysts may become infected or simply grow over time and so may behave as a space-occupying and compressive lesion. Many cysts are asymptomatic or have vague symptoms and are discovered on routine chest radiographs. Infection, hemorrhage, and, in rare cases, late malignancy may occur. Associated respiratory symptoms include stridor or wheezing. Chronic air trapping may lead to emphysema, atelectasis, or both. Dysphagia, chest pain, and epigastric discomfort may also occur.
Congenital Lobar Emphysema
Although generally not discovered in utero, congenital lobar emphysema typically manifests in neonates as hyperinflation of one or more lung lobes. Causes include intrinsic absence or abnormality (bronchomalacia) of cartilaginous rings or external compression of a segmental bronchus by a large pulmonary artery. 44,65 Hyperinflation of a pulmonary lobe develops after birth as inspired air enters the affected lobe but cannot exit, because the positive pressure of expiration collapses the poorly structured airway. Congenital lobar emphysema most commonly involves the upper lobes. The left upper lobe is involved in roughly 41% of patients, the right middle lobe in 34%, and the right upper lobe in 21%. Involvement of the lower lobes is rare, occurring in fewer than 5% of patients. Neonates may present with mild to moderate respiratory distress. Mediastinal shift may develop with progressive air trapping, and decreased breath sounds are noted on the involved side. Infants who have a milder form of lobar emphysema will present with nonspecific findings, including cough, wheezing, respiratory distress, and cyanosis. Older children may present with recurrent chest infections. On plain radiographs obtained in neonates, the affected lobe may be hyperlucent or slightly opacified if alveoli remain fluid-filled. Associated cardiac anomalies may occur in as many as 10% of patients.
LABORATORY DATA
Routine chest radiograph is the initial evaluation tool in distinguishing congenital chest masses and is the principle study to establish the diagnosis of diaphragmatic hernia and congenital lobar emphysema in newborns. Sonography and/or computed tomography (CT) scan of the chest are useful means to evaluate CPAM, sequestrations and bronchogenic cysts, and lobar emphysema in older infants and children. The differential diagnosis of a hyperlucent hemithorax with mediastinal shift on chest x-ray in the newborn includes tension pneumothorax, cystic CPAM, diaphragmatic hernia with air-filled stomach or intestine in the chest, and congenital lobar emphysema.
Treatment
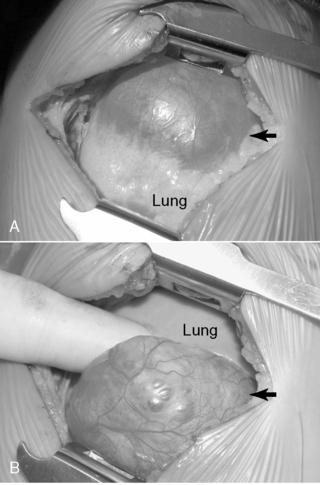
Surgical resection of these congenital chest masses is curative. Some small, asymptomatic lesions of sequestration or CPAM may be followed expectantly, as reports of spontaneous regression may be found in the literature. However, most lesions may be removed with little morbidity in an effort to minimize long-term complications of the various lesions. Operative approach to these lesions may be either via thoracotomy or thoracoscopy, depending on the suitability of the baby and the skill of the surgeon (Figures 28-4 and 28-5).
 |
| FIGURE 28-4
(Courtesy Dr. H. Lovvorn, Vanderbilt University Medical Center.)
|
 |
| FIGURE 28-5
(Courtesy Dr. H. Lovvorn, Vanderbilt University Medical Center.)
|
Special consideration to resection of the pulmonary lobe involved with congenital lobar emphysema should be given (Figure 28-6). Extreme caution must be followed upon induction of general anesthesia and endotracheal intubation with positive-pressure ventilation. Because of the collapsible airway and the propensity for air-trapping in congenital lobar emphysema, rapid development of tension physiology may ensue, compromising the well-being of the baby and necessitating emergent decompressive thoracotomy. Such pathophysiology is possible in any neonate with congenital lobar emphysema and requiring positive-pressure ventilation.
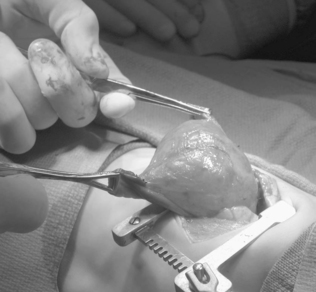 |
| FIGURE 28-6
(Courtesy Dr. H. Lovvorn, Vanderbilt University Medical Center.)
|
MALROTATION AND VOLVULUS
Physiology and Etiology
In the 4th week of gestation, the midgut exists as a straight tube deriving its blood supply from the superior mesenteric artery (SMA). The proximal limb of primitive intestine, representing the future duodenum, jejunum, and proximal ileum, lies in the midline and anterior to the SMA. The distal limb, destined to become the terminal ileum, ascending colon, and transverse colon, lies posterior to the SMA. During the 6th week of gestation, these segments of bowel, known collectively as the midgut, can lengthen rapidly by herniating through the incompletely closed abdominal wall and into the umbilical stalk. While lengthening outside of the coelomic cavity, the midgut undergoes a 270° counterclockwise rotation around the SMA axis. On return to the abdominal cavity, the duodenojejunal junction comes to rest in the left upper quadrant and becomes fixed in this location by the ligament of Treitz. At the end of the 11th week of gestation, midgut rotation is completed and the cecum now resides anterior and to the right of the SMA and is fixed in the right lower quadrant. Because of the counterclockwise nature of this intestinal rotation, the ascending colon and transverse colon lie to the right of the SMA. The hindgut (splenic flexure of the colon to the rectum) then fixes in the left hemi-abdomen and derives its blood supply largely from the inferior mesenteric artery (IMA).
Failure of this rotation and fixation results in the clinical condition termed malrotation, which covers a wide spectrum of rotational anomalies. Complete nonrotation is characterized by the entire small bowel existing on the right side of the abdomen and the colon principally to the left. Partial malrotation involves the improper fixation of a single segment. Complete malrotation is thought to occur from a lax umbilical ring allowing the gut to return en masse to the abdomen. Because proper rotation does not occur, the root of the mesentery is not anchored in the left upper quadrant and the superior mesenteric artery and vein loosely suspend the entire bowel without fixation. This unfixed, narrow mesenteric pedicle predisposes the midgut and its tenuous blood supply to twisting or volvulus. If volvulus occurs, the blood supply to the midgut may be compromised, leading rapidly to ischemia and bowel infarction. The majority of patients with midgut malrotation are diagnosed in the first month of life but may be seen with decreasing frequency in the older child or, rarely, the adult. 50,88
By definition, malrotation also exists in several anomalies, including gastroschisis, omphalocele, and congenital diaphragmatic hernia, because the midgut is trapped and cannot rotate and fix properly in these conditions.
Data Collection
HISTORY
Malrotation may manifest in the newborn simply as a proximal mechanical bowel obstruction caused by abnormal attachments, or Ladd’s bands, between the cecum and porta hepatis. These babies typically show some degree of feeding intolerance early on with or without bilious emesis. A more worrisome presentation of malrotation may arise should the bowel volvulize around its unfixed, narrow vascular pedicle. These babies present with an acute, high-grade proximal bowel obstruction. In a neonate who develops midgut volvulus, the first few days of life usually are unremarkable but then the baby develops acute feeding intolerance and bilious emesis in the absence of abdominal distention. If diagnosis is delayed, intestinal ischemia sets in and the symptoms may progress rapidly to an acute abdomen and profound shock as a result of gangrenous bowel. Abdominal wall erythema and distention are usually present in advanced stages of intestinal ischemia and are ominous findings.50,78
SIGNS AND SYMPTOMS
The symptoms of nonvolvulized malrotation mimic those of duodenal stenosis or atresia, proximal jejunal atresia, or other conditions resulting in proximal intestinal obstruction. These babies develop feeding intolerance followed by bilious emesis and typically have a scaphoid abdomen on examination. Midgut volvulus presents with a more sudden onset of signs, suggesting acute proximal intestinal obstruction. If diagnosis is delayed, signs of intestinal ischemia become evident and include abdominal distention, lethargy, hypovolemic shock, and anuria. The presence of bloody emesis or stools suggests intestinal ischemia with mucosal injury or necrosis. In this setting, rapid diagnosis and prompt surgical intervention are essential to avoid extensive bowel loss or death. (See the Critical Findings box above.)
LABORATORY DATA
Plain abdominal radiographs may show a dilated stomach and proximal duodenum or, rarely, pneumoperitoneum in the presence of advanced intestinal necrosis. However , the definitive study is an upper gastrointestinal (UGI) series, which shows both abnormal rotation of the duodenum (malrotation) and partial obstruction (from Ladd’s bands) or complete obstruction with a bird’s beak suggesting midgut volvulus. A contrast enema may show an abnormal location of the cecum but is not diagnostic alone of malrotation and provides no information about the presence or absence of midgut volvulus. 75 Laboratory data are generally unremarkable unless bowel ischemia is present, as suggested by leukocytosis, anemia, and metabolic acidosis.
Treatment
PREOPERATIVE CARE
Although distinguishing between symptomatic malrotation with obstruction (Ladd’s bands) and volvulus in its early stages may be difficult, these conditions should be managed similarly. Gastric decompression, fluid resuscitation, correction of electrolyte and acid-base abnormalities, and parenteral antibiotics are instituted in the preoperative period. Emergent abdominal exploration should be considered in any infant with suspected or confirmed volvulus, because the bowel will be irreparably damaged in as little as 4 hours. In this setting, prompt surgical intervention with continued intraoperative resuscitation is indicated to maximize the chances for bowel salvage and survival.
OPERATIVE INTERVENTION
Operative correction of malrotation without midgut volvulus includes division of Ladd’s bands (to relieve duodenal obstruction), correction of the malrotation (by placement of the small bowel in the right of the abdominal cavity and the colon on the left), broadening the base of the mesentery by dividing its peritoneum, and appendectomy (the appendix and cecum will reside in the left upper quadrant). This procedure is best performed via laparotomy, and a laparoscopic approach has limited utility in the newborn. 9 The long-term results of the laparoscopic approach remain to be elucidated. 50 Cases of nonrotation in the older child may be amenable to laparoscopic techniques.
If volvulus is present, the bowel is detorsed and allowed to reperfuse (Figure 28-7). Necrotic segments of bowel are resected and stomas created as indicated. In select instances, substantial resection may result in short bowel syndrome. In these cases, marginal intestine may be left in place rather than removed and a planned reoperation performed within 24 to 36 hours to reevaluate the need for additional bowel resection. The objective of a second-look laparotomy is to allow continued resuscitation and marginally viable intestine the necessary time to recover. Bowel that is not salvageable will become more obviously nonviable but should not have perforated in this short period. This approach is designed to minimize the amount of total intestine resected.
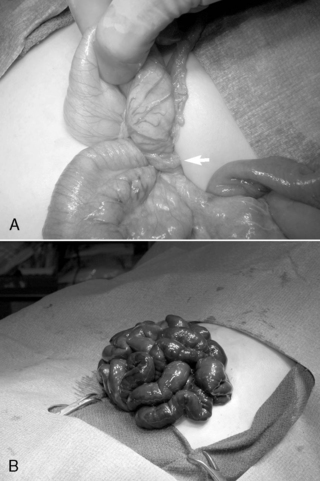 |
| FIGURE 28-7
(Courtesy Dr. H. Lovvorn, Vanderbilt University Medical Center.)
|
Complications and Prognosis
Proximal obstruction related to Ladd’s bands is corrected by the Ladd’s procedure, and recurrent obstruction is rare. The risk for subsequent volvulus is greatly reduced with Ladd’s procedure but is not entirely eliminated. Adhesive small bowel obstruction may occur later in life at the same rare incidence as after any other laparotomy.
The immediate postoperative care consists of nasogastric decompression and intravenous (IV) fluid therapy until the return of gastrointestinal function (4 to 6 days). 23 Conversely, the outcome after malrotation with midgut volvulus is predicated on the degree of intestinal resection. 48,50 Midgut volvulus is a leading cause of short bowel syndrome in infants and may render the infant dependent on TPN if extensive intestinal necrosis has occurred.
INTESTINAL ATRESIA
Physiology and Etiology
Any segment of the bowel may be narrowed (stenosis) or become discontinuous (atresia). Duodenal atresia is the most commonly involved bowel segment, followed by ileum, jejunum, colon, and stomach. 17
Duodenal atresia results from failure of vacuolization (5th to 6th week of gestation) and recanalization (8th to 10th week of gestation). A vascular accident or segmental volvulus occurring later in utero is thought to give rise to jejunal, ileal, or colonic atresia. 47,66 Because duodenal atresia results from an early in utero event, a high incidence (30%) of anomalies may be associated and include trisomy 21, congenital heart disease, and VACTERL association. 24,27,61 Conversely, intestinal atresia occurs later in gestation and so is rarely associated with significant anomalies. Atresias are classified as membranes, fibrous cords, gap defect including mesentery, and “apple-peel” atresia. 84
Data Collection
HISTORY AND PHYSICAL EXAMINATION
Commonly, a history of maternal polyhydramnios may be provided and the affected neonate may appear small for gestational age (SGA). The more proximal the site of intestinal atresia, the more likely the history of maternal polyhydramnios. Newborns with a proximal atresia (duodenum or jejunum) present with early feeding intolerance and emesis and a scaphoid abdomen. Bilious emesis is present when the obstruction is distal to the ampulla of Vater, as is the case in approximately 85% of duodenal atresias. However, in 15% of cases, duodenal atresia occurs proximal to the ampulla of Vater and therefore the baby does not show bile-stained emesis or gastric aspirates.
The more distal the site of atresia and obstruction, the more likely that the infant will manifest significant abdominal distention. Babies with a distal intestinal atresia (ileum or colon) show typical features of a distal intestinal obstruction and develop abdominal distention often with visible intestinal loops. 66 If the atresia occurs early in gestation, the infant fails to pass meconium and only mucus is passed after birth. (See the Critical Findings box above.)
Intestinal Atresia
The following are critical assessment findings for intestinal atresia:
• Maternal polyhydramnios
• Emesis (nonbilious vs. bilious depending on location of atresia)
• Abdominal radiograph suggestive of intestinal obstruction
• Abdominal radiograph with “double bubble” suggestive of duodenal atresia
LABORATORY DATA
Initial evaluation of suspected duodenal atresia begins with a plain abdominal radiograph (flat and left lateral decubitus views), which classically shows a dilated, air-filled stomach and proximal duodenum in a pattern called the “double bubble” sign. A contrast study is not indicated unless air is present in the distal bowel, in which case malrotation with midgut volvulus cannot be excluded.
Abdominal radiographs that show multiple distended loops of bowel suggest a distal intestinal obstruction. In the setting of atresia of the small intestine or colon, abdominal radiographic images demonstrate dilation of intestinal segments proximal to the site of obstruction with absence of air in the distal bowel. For intestinal atresia distal to the duodenum, a contrast enema is generally performed and typically shows a microcolon or unused colon and no reflux of the contrast agent into the proximal bowel is observed.
Treatment
PREOPERATIVE CARE
Preoperative care includes orogastric tube decompression to reduce the risk of vomiting and aspiration, fluid resuscitation, and correction of electrolyte abnormalities. Preoperative antibiotics will be initiated.
OPERATIVE INTERVENTION
All forms of intestinal atresia require surgical correction to restore gastrointestinal tract continuity. Duodenal atresia is repaired through a diamond-shaped end-to-end anastomosis of the proximal and distal duodenum, and care must be exercised to prevent injury to the bile and pancreatic ducts. Repair of duodenal atresia may be performed either through a standard transverse right upper quadrant incision or laparoscopically (Figure 28-8). Other intestinal atresias are generally repaired by a standard end-to-end anastomosis. The size disparity between the dilated proximal loop and the decompressed distal loop may require that the proximal bowel be tapered or partially resected or the distal bowel may be cut obliquely to allow anastomosis (Figure 28-9). If these methods are not possible because of size discrepancy, then the segments just proximal and distal to the atresia may be brought out as stomas and intestinal anastomosis delayed to allow reduction in size of the dilated proximal segment and growth of the distal segment. When the caliber of the bowel becomes more comparable in size, anastomosis is performed.
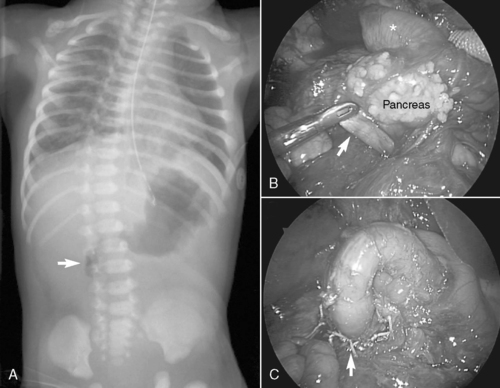 |
| FIGURE 28-8
(Courtesy Dr. H. Lovvorn, Vanderbilt University Medical Center.)
|
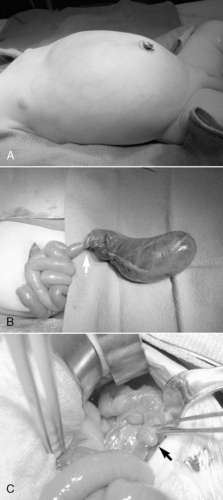 |
| FIGURE 28-9
(Courtesy Dr. H. Lovvorn, Vanderbilt University Medical Center.)
|
POSTOPERATIVE CARE
Postoperatively, an orogastric tube remains in place to decompress the stomach until bowel function begins. Stomas must be protected from desiccation by covering with petrolatum gauze or a stoma appliance. Proximal ostomies have high output because of a lack of absorptive capacity and therefore require replacement of both fluid and electrolyte losses. The proximal output may be re-fed into a distal mucous fistula if present, but this technique may be challenging because of problems intubating the distal stoma and securing the feeding catheter to permit infusion. All infants endure a significant period of bowel dysfunction after surgery, and therefore temporary central venous access should be established to permit TPN support. 61,66,68 Furthermore, infants with intestinal atresia should undergo screening for cystic fibrosis (CF), which may contribute to both the development of the anomaly and ongoing bowel dysfunction. 11
Complications and Prognosis
The overall prognosis for these patients is excellent unless severe associated anomalies are present. Prolonged bowel dysfunction is the primary complication after surgical correction of intestinal atresia. In select cases, tapering of dilated bowel segments is attempted to enhance the recovery of bowel function. 61,73 Some infants fail to recover sufficient bowel function and require long-term parenteral nutritional support, either because of dysmotility or inadequate bowel length resulting from long segment atresia. Fortunately, the majority of patients have no long-term problems after postoperative recovery and return of bowel function.
NECROTIZING ENTEROCOLITIS
Physiology and Etiology
Necrotizing enterocolitis (NEC) is an inflammatory condition of the bowel of uncertain cause and occurs at a rate of 1 in 1000 live births and in 5% of infants born weighing less than 1500 grams (very low birth weight [VLBW]). NEC is fatal in 17% of all cases, in 20% of VLBW infants who develop the disease, and in as many as 40% to 50% of infants with a birth weight less than 1000 grams. NEC is primarily a disease of premature infants, although approximately 5% of cases occur in term babies. Despite intensive study, major advances in newborn intensive care, and improved survival of the VLBW preterm infant in the past two decades, the incidence of and mortality associated with NEC have changed little. In fact, the increase in survival of VLBW preterm infants has increased the size of the population at risk.
Perinatal stressors, an immature intestinal barrier, intestinal ischemia, bacterial colonization of the gut, and nutritional substrate in the gut lumen have all been implicated as contributing factors in the development of NEC. Other conditions that result in mucosal injury also have been linked to NEC: hypoxia; polycythemia; hyperosmolar feedings; gastrointestinal infection (bacterial or viral); and severe cardiopulmonary disease. Current research using animal models implicates up-regulation of multiple inflammatory mediators in the injured intestinal epithelia affected by NEC, and the presence of certain anti-inflammatory mediators may be cytoprotective. 14,29,37,63
Necrotizing Enterocolitis
The following are critical assessment findings for necrotizing enterocolitis:
• Feeding intolerance
• Abdominal distention
• Bloody or Hemoccult-positive stools
• Thrombocytopenia, leukocytosis, leukopenia, and metabolic acidosis
• Pneumatosis intestinalis on abdominal radiograph
• Free air on abdominal radiograph in the presence of perforated viscus
Inflammation and ischemia initially involve the intestinal mucosa, but as the disease progresses, the muscular and subserosal layers of the bowel become involved. The intestinal wall becomes hemorrhagic and attenuated, with evidence of intramural gas (pneumatosis). Histologically, the intestine shows features of acute and chronic inflammation with areas of coagulative necrosis. The ileocecal region is most commonly involved (50%), followed by disease limited to the colon (25%), or both large and small intestines (25%). Up to 15% of infants will develop pan-intestinal necrosis, or NEC-totalis, which is a non-survivable insult. Further, NEC is a significant risk factor for developing short bowel syndrome chronically if less than 40 cm of small bowel remains in the absence of the ileocecal valve or if, in its presence, less than 20 cm of bowel remains. 2,85
Data Collection
HISTORY/SIGNS AND SYMPTOMS
The onset of NEC is heralded by the development of feeding intolerance, abdominal distention, and bloody stools in a premature infant receiving enteral feedings. A history of perinatal hypoxia, respiratory distress, congenital heart disease, or indomethacin administration for patent ductus arteriosus (PDA) closure often is elicited. As the disease progresses, the infant develops signs and symptoms of septic shock (lethargy, respiratory distress, temperature instability, hypotension, apnea, and oliguria). Examination reveals a distended and tender abdomen that may demonstrate erythema, induration, and pitting edema in severe cases. (See the Critical Findings box above.)
LABORATORY DATA
CBC and serum electrolyte evaluations typically reveal thrombocytopenia, leukocytosis or leukopenia, and metabolic acidosis. Blood gas analysis reveals that metabolic acidemia and lactate levels are elevated . C-reactive protein (CRP) levels are increasingly obtained and appear to be a good marker of onset, persistence, and resolution of NEC. 62Stool tests may be positive for occult blood and reducing substances in more than 50% of cases. The diagnostic test of choice is the flat plate (prone) plain abdominal radiograph and a left lateral decubitus abdominal radiograph. Plain radiographs are carefully reviewed for the characteristic finding of pneumatosis intestinalis, or intramural bowel gas. The radiograph should be assessed for free air (pneumoperitoneum), which would suggest intestinal perforation. Other findings may include dilated bowel, portal venous gas, ascites, or a fixed bowel loop that does not change on repeated studies. Recently, ultrasound has been introduced in the diagnostic and prognostic evaluation of NEC. 71
Treatment
PREOPERATIVE CARE
The only absolute and immediate indication for surgical intervention is intestinal perforation, which may be detected radiographically by the finding of pneumoperitoneum. In a clinically stable infant without findings of perforation, medical management consisting of bowel rest, gastric decompression, fluid resuscitation, broad-spectrum antibiotic therapy, and TPN is indicated. The infant is monitored carefully (serial abdominal examinations and radiographs every 8 hours) for signs of intestinal gangrene. More than half of infants respond to medical management, but up to 30% of infants treated medically may develop an intestinal stricture requiring surgical management.
A subset of infants continue to deteriorate despite maximum medical therapy, suggesting intestinal gangrene without intestinal perforation. Presence of intestinal gangrene should be considered in infants with persistent thrombocytopenia, leukopenia, hyperkalemia, refractory shock and acidemia, erythema of the abdominal wall, or a fixed, dilated intestinal loop on plain abdominal radiograph. 82
OPERATIVE INTERVENTION
The principle of surgical management is to resect all necrotic bowel while preserving as much of the intestinal length as possible. In cases of extensive bowel involvement, only necrotic segments of intestine are removed and reoperation at 12 to 24 hours may be planned (a second look). After bowel resection, proximal and distal ostomies are created and the abdomen is thoroughly irrigated to reduce bacterial and fecal contamination. In severely premature infants weighing less than 1000 grams, primary peritoneal drainage (PPD) to decompress the abdomen after intestinal perforation may be performed in the NICU as an alternative to laparotomy. PPD and laparotomy may have comparable survival rates, but it is interesting to note that infants treated with PPD have substantial improvement in residual bowel length, which may be in part because up to one third of these patients do not require further operative therapy. 19 Nevertheless, laparotomy provides more definitive therapy and further aids in establishing the extent of diseased bowel, which, if NEC-totalis is discovered, may reduce futile care. In most cases, PPD should be considered a temporizing means, or “bridge to laparotomy,” to allow complete resuscitation before definitive surgery (Figure 28-10). 10
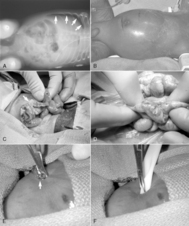 |
| FIGURE 28-10
(Courtesy Dr. H. Lovvorn, Vanderbilt University Medical Center.)
|
POSTOPERATIVE CARE
After laparotomy, supportive care (fluids, TPN, antibiotics) and bowel rest are continued for 10 to 14 days. 26 At 2 weeks postoperatively, low-osmolar elemental feedings may be started and advanced as tolerated. A stoma closure procedure is planned for 6 to 8 weeks after the initial surgery. All infants should undergo a preoperative contrast enema before ostomy closure to make certain the intestine and colon distal to the ostomy have not strictured.
Complications and Prognosis
Stomal prolapse or retraction, wound infection, intraabdominal abscess, and intestinal obstruction are early complications. Recurrent NEC is uncommon but may occur in approximately 5% of infants treated medically or surgically. The most significant late complication is that of inadequate intestinal length (short bowel syndrome) and the need for long-term parenteral nutrition. Preservation of the ileocecal valve is critically important to slowing intestinal transit and thereby limiting sequelae of short bowel syndrome. Babies at highest risk for short bowel syndrome have the ileocecal valve but less than 20 cm of small intestine or have no ileocecal valve and less than 40 cm of small bowel. Survival in infants weighing more than 1000 g has improved from 50% to 80% over the past two decades. Severely premature infants weighing less than 1000 g still have a mortality rate in excess of 50%. 34,45
MECONIUM ILEUS
Physiology and Etiology
Meconium ileus is an intestinal obstruction caused by hyperviscous secretions from the intestinal glands coupled with a lack of pancreatic enzymes necessary to help digest intestinal secretions and contents. The result is tenacious, viscous meconium that creates a sticky plug obstructing the lumen of bowel. The obstruction generally occurs within the terminal ileum, mimicking ileal atresia. More than 90% of infants with meconium ileus have cystic fibrosis (CF), a three-base-pair deletion on chromosome 7. This autosomal recessive gene defect results in alteration of the chloride channel transporter and therefore fluid flow across the apical surface of epithelial cells. Meconium ileus occurs in 10% to 25% of patients with CF; 20% of mothers have polyhydramnios. A family history of CF is present in 10% to 30% of cases.
This ileus associated with CF and retained meconium is in contrast to the meconium plug syndrome (MPS), which manifests as a failure to pass stool with obstruction in the colon and is not specific to CF. MPS is generally recognized as immaturity of the ganglion cells and generally is benign. 40 Rectal dilation and/or contrast enema usually results in passage of the meconium plug(s), and recurrence is uncommon; however, significant obstruction can develop with rare incidences of perforation. MPS, again in contrast to meconium ileus, is associated with Hirschsprung’s disease in 15% of cases.
Data Collection
SIGNS AND SYMPTOMS
Meconium ileus is classified as simple (obstruction and obturation) or complicated (volvulus, intestinal atresia, perforation, meconium peritonitis). Uncomplicated meconium ileus presents as distal ileal obstruction caused by inspissated meconium (pellets) and proximal intestinal dilation. The onset of symptoms associated with simple meconium ileus begins 24 to 48 hours after birth. Bilious emesis, progressive abdominal distention, and the failure to pass meconium suggest intestinal obstruction. The differential diagnosis in addition to meconium ileus includes ileal atresia, meconium plug, and Hirschsprung’s disease. Physical examination shows a patent anus that may express a small amount of gray meconium. Examination of the abdomen reveals moderate distention with a characteristic doughlike sensation on palpation because of the thickened meconium contained in the dilated bowel. Complicated meconium ileus often manifests more abruptly and progresses more quickly. Symptoms include abdominal distention within 24 hours of birth, respiratory distress (especially if a postnatal perforation has occurred), and an edematous, erythematous abdomen. 64 (See the Critical Findings box below).
LABORATORY DATA
Abdominal radiograph demonstrates a “soap bubble” appearance of the bowel caused by trapped gas within the meconium and also shows large, dilated (with air) loops of bowel with few air-fluid levels because of the viscous nature of the meconium. A contrast enema shows a microcolon and pellets of inspissated meconium at the site of distal obstruction. If in utero perforation has occurred, microcalcifications may also be present on plain abdominal radiographs.
Treatment
PREOPERATIVE CARE
If meconium ileus is suspected, orogastric decompression, IV hydration, and electrolyte replacement are instituted. Once the infant is appropriately hydrated, a diluted Gastrografin or Cysto-CONRAY enema should be attempted. 55Note: The infant should be adequately hydrated before the enema because of the hyperosmolarity of the Gastrografin or Cysto-CONRAY. Intra-colonic instillation of these water-soluble agents draws fluid into the bowel lumen, diluting the viscous meconium and facilitating passage. If the Gastrografin enema results in incomplete evacuation, it may be repeated over the next several days. However, if the Gastrografin enema fails to result in passage of meconium, complicated meconium ileus or ileal atresia may be present and operative intervention is indicated. 39,55
OPERATIVE INTERVENTION
The goal of operative treatment is to relieve intestinal obstruction. For uncomplicated meconium ileus, several operative approaches are described to eliminate the obstructing inspissated meconium. Techniques include the following:
• Enterotomy with extraction of the tenacious meconium and irrigation of the bowel with saline solution or 2% N-acetylcysteine (Mucomyst)
• Resection of the affected segment with anastomosis
• Formation of chimney ostomies just proximal to the obstruction (Bishop-Koop procedure: bowel is divided to create an ostomy of the distal ileum for continued irrigations, and an internal anastomosis of the proximal to distal ileum is fashioned to maintain intestinal continuity)
More commonly now, a T-tube enterostomy may be created, in which a soft, small-caliber tube is securely placed within the bowel lumen and delivered through a separate wound in the abdominal wall (Figure 28-11). This tube, like the chimney ostomy, allows continued postoperative irrigations with normal saline or diluted Mucomyst to complete or maintain passage of intestinal contents. If complicated meconium ileus is identified, the obstructed segment is resected and ostomies are performed to permit postoperative irrigation. Ostomy closure usually is performed 4 to 6 weeks later.
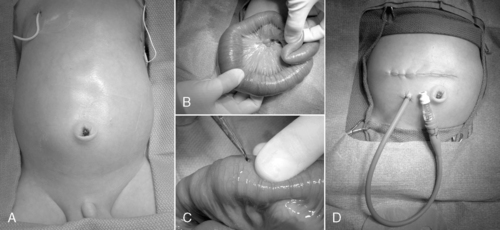 |
| FIGURE 28-11
(Courtesy Dr. H. Lovvorn, Vanderbilt University Medical Center.)
|
POSTOPERATIVE CARE
Postoperatively, nasogastric tube decompression, nutritional support with TPN, and irrigation of the rectum or ostomies with saline solution or N-acetylcysteine (Mucomyst) are instituted. Once gastrointestinal function returns, feedings using protein hydrolysate or elemental formula and pancreatic enzyme supplements are started. The diagnosis of cystic fibrosis is confirmed with genetic testing and sweat chloride testing.
Complications and Prognosis
One-year survival for infants with simple or complicated meconium ileus is favorable (>90%), but long-term survival is limited primarily because of the pulmonary complications of cystic fibrosis. 53,54Late gastrointestinal complications of CF include distal intestinal obstruction syndrome (meconium ileus equivalent), appendicitis, intussusception, rectal prolapse, intestinal stricture, pancreatitis, and cholestatic liver disease. 53
HIRSCHSPRUNG’S DISEASE
Physiology and Etiology
Hirschsprung’s disease is a congenital intestinal disorder caused by a lack of ganglion cells in the bowel wall that prevents effective peristalsis. During development, neural crest cells (the progenitor or stem cells of the enteric nervous system) migrate along the intestinal tube to populate the entire gut in a craniocaudal fashion, with the distal colon, rectum, and sphincter being the last to be colonized. These progenitor cells divide, differentiate, and proliferate to form the enteric nervous system, of which the ganglion cells are a critical component. Arrest of migration, proliferation, and differentiation results in the aganglionosis found in Hirschsprung’s disease, a well-recognized cause of distal intestinal obstruction in the newborn. At the site of arrest, a transition from normal to abnormal innervation is present and all intestine distal to this site will be aganglionic and therefore dysfunctional. The result is a functional obstruction that mimics mechanical intestinal obstruction. In brief, the pathophysiologic consequence of the absence of ganglion cells is loss of the ability of the involved rectum and colon to relax, and therefore the fecal stream cannot be passed effectively through this region and beyond. Rectosigmoid aganglionosis is most common (85%), with the remainder of patients developing variable lengths of more proximal colonic and, rarely, small intestine disease. Total colonic aganglionosis occurs in roughly 10% of cases.
Hirschsprung’s disease occurs in 1 in 5000 live births, with a 4:1 male-to-female predominance. The majority of cases are sporadic (80% to 90%), but familial occurrences are well recognized and multiple genetic alterations have been identified in affected pedigrees. Associated anomalies are rare in sporadic cases but may be seen in as many as 25% of the familial cases. Babies born with Down syndrome also carry a higher incidence of Hirschsprung’s disease (2%) than the population at large, and between 5% and 10% of Hirschsprung’s patients will have Down syndrome.
Approximately 15% of Hirschsprung’s neonates will present with meconium plug syndrome (MPS), an obstruction of the colon by inspissated meconium. However, in general, MPS is associated with immature ganglion development and is not indicative of the more serious Hirschsprung’s disease.
Data Collection
SIGNS AND SYMPTOMS
Ninety-eight percent of normal infants pass meconium in the first 24 to 48 hours of life. Failure to pass meconium early, feeding intolerance, and abdominal distention suggest a diagnosis of Hirschsprung’s disease. In some babies with a short segment of aganglionic bowel, spontaneous evacuation of stool may be noted and the infant may appear otherwise healthy. If vomiting, abdominal distention, and constipation (or paradoxic diarrhea resulting from watery stool escaping around the obstipated stool) continue, further investigation is indicated. Hirschsprung’s disease may rarely escape detection during the newborn period, and in older children, a history of refractory and chronic obstipation may be the only symptom. Approximately 5% to 10% of affected infants will present with a picture of enterocolitis (toxic megacolon), characterized by fever, vomiting, abdominal distention/tenderness, foul-smelling diarrhea, and septic shock. The infant may rapidly deteriorate, with a 50% risk of death if the colon is not rapidly decompressed, either by transanal soft rubber tube irrigations or emergency colostomy. Fortunately, in most cases of Hirschsprung’s disease, the infant is only mildly ill, allowing time for definitive diagnostic studies before surgical correction is undertaken. 15 (See the Critical Findings box on p. 833.)
Hirschsprung’s Disease
The following are critical assessment findings for Hirschsprung’s disease:
• Failure to pass meconium within 48 hours of birth
• Feeding intolerance
• Abdominal distention
• Enterocolitis (fever, abdominal distention, foul-smelling diarrhea, sepsis)
• Transition zone on barium enema
• Absent ganglion cells and nerve hypertrophy on rectal biopsy
LABORATORY DATA
The diagnostic evaluation begins with a contrast enema. Surgical dogma states that the first enema should be a contrast enema. This study typically shows a contracted or spastic rectosigmoid colon, with contrast material entering the proximal dilated bowel. The area between the contracted and dilated bowel is called the transition zone. If the contrast enema is equivocal, an abdominal x-ray film should be obtained on the next day to evaluate extent of retained contrast material. Significant contrast material retained within the distal colon and rectum suggests the presence of Hirschsprung’s disease. Definitive diagnosis is made by performing a bedside suction rectal biopsy, a well-tolerated procedure in neonates that does not require any analgesics or sedatives. On histology, the biopsy shows an absence of ganglion cells and a presence of hypertrophic nerve trunks within the submucosal and intermyenteric plexus. Conversely, if ganglion cells are observed on histologic examination, a diagnosis of Hirschsprung’s disease is excluded.
Treatment
PREOPERATIVE CARE
In infants with Hirschsprung’s-associated enterocolitis, orogastric tube decompression, IV fluid resuscitation, broad-spectrum antibiotics, and correction of acid-base deficits and electrolyte abnormalities are promptly initiated. Infants who are less ill with symptoms of obstruction are placed on nothing-by-mouth (NPO) status, and IV antibiotic therapy and orogastric decompression are instituted, permitting time to complete the diagnostic evaluation. Once the diagnostic evaluation is completed, transanal rectal irrigations, not enemas, are performed twice daily until surgery. Babies should have return to normal bowel function with these irrigations and may be fed enterally (so long as rectal irrigations continue) until definitive surgery.
OPERATIVE INTERVENTION
In the presence of profound enterocolitis, an emergency colostomy may be indicated. If necessary, a colostomy is performed at a site of normal bowel (ganglion cells present), as confirmed by a frozen section histologic examination. In select cases, the neonate will be too ill so operative time is minimized by the creation of a right-sided colostomy, because most affected babies have more distal colonic involvement. If the neonate presents with milder symptoms, one of several surgical options may be selected: (1) primary laparoscopically assisted endorectal pull-through (Figure 28-12), (2) primary transanal pull-through, or (3) staged reconstruction (temporary colostomy, followed by a pull-through in 3 to 6 months). 70 In all cases, multiple intestinal seromuscular biopsies are performed at the time of operation until the normally innervated bowel is identified. A coloanal anastomosis or colostomy is performed at the site of bowel that was proven normal by histologic examination. If the anastomosis or colostomy site is absent of ganglion cells, the neonate will remain symptomatic—the bowel will not function normally because of aganglionosis.
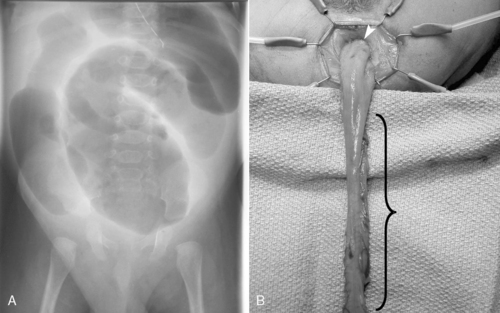 |
| FIGURE 28-12
(Courtesy Dr. H. Lovvorn, Vanderbilt University Medical Center.)
|
Several factors influence the decision to perform a primary pull-through procedure in neonates. 70 The neonate should be of sufficient gestational age and size (>2 kg), have rectosigmoid disease as demonstrated by contrast enema, not have significant proximal bowel distention, and not have evidence of advanced enterocolitis. Neonates who do not meet these criteria should be treated in a staged manner, with an immediate colostomy and a pull-through procedure delayed until later in infancy. 30,76
POSTOPERATIVE CARE
The recovery period is generally straightforward, and supportive care is provided until the return of bowel function. For babies treated initially with colostomy, postoperative care includes teaching the parents about stoma maintenance and hygiene (Box 28-1). The pull-through procedure entails resecting abnormal aganglionic bowel and bringing ganglionic bowel to the anus. Several variations of the pull-through operation have been described; each has unique advantages and disadvantages, but in general, the results are similar regarding long-term stooling patterns. For neonates having a primary laparoscopically assisted endorectal pull-through, first stool is usually passed within 24 hours of the procedure, at which time breast milk or Pedialyte may be introduced. Feeds may be advanced to goal (according to tolerance) over the next 24 to 48 hours.
BOX 28-1
Supplies for Pouch Changes
| Dry washcloths | Pouch |
| Warm, wet washcloths or adhesive remover | Stomahesive paste |
| Mild soap | Clamp or rubber band |
| Pattern or guide | Scissors |
| Barrier wipes/Stomahesive powder (if skin is reddened or irritated) | Optional: gauze pads, dry paper towel, cotton balls, or toilet paper |
Directions
1. Gather supplies. (It is helpful to keep all supplies together in one place.)
2. Wash hands.
3. Trace pattern on the paper side of the pouch (wafer). The opening should fit closely around the stoma but not tightly or on top of the stoma.
4. Cut an opening in the wafer; use scissors to cut along the lines you traced on the wafer paper. (Cut several wafer patterns and pouches at once to save time when changing pouches.)
5. Gently remove the old wafer/pouch using adhesive remover or warm, wet washcloths.
6. Clean skin with a warm, soapy washcloth. Rinse well with warm water. (Do not use soaps that contain lotions or oils.)
7. If skin is reddened or irritated, apply a light dusting of Stomahesive powder and seal with barrier wipes. (The barrier wipes contain a skin sealer that protects the skin from stool and from tearing when the pouch is removed.)
8. Apply a small, thin line of Stomahesive paste around the stoma or around the opening cut in the wafer. (Stomahesive paste acts as caulking or filler, smoothing creases, dips, or scars, and helps prevent liquid from seeping under the wafer.)
9. Apply the wafer to the skin. Press around the stoma to ensure a good seal.
10. Hold your hand over the pouch for 1 minute to warm the wafer so it will stick to the skin.
11. If stool is liquid, you can place a gauze pad, for example, in the pouch to absorb liquid.
12. Place clamp or rubber band at the end of the pouch.
Helpful Hints
1. Always change a leaking pouch immediately. Do not use tape to seal a leak. As a rule, empty the pouch when it is ⅓ to ½ full. A full pouch can be heavy enough to fall off.
2. When emptying the pouch, turn the bottom of the pouch inside out before emptying; this makes it easier to clean the bottom of the pouch before applying rubber band or clamp.
3. You can squirt water into a pouch to clean it after emptying, but it is not necessary. You can also insert a ½-capful of mouthwash or an ostomy deodorizer into the pouch after emptying to control odors.
4. Never leave home without extra supplies (including a wafer/pouch that is already cut and ready for application) and clothing. Because checked baggage can be lost or stolen, always keep an extra supply in your “carry-on” luggage. Do not leave supplies in the car when the weather is either hot or freezing unless they are in a well-insulated bag, because extreme temperature will ruin pouches.
5. Use one-piece outfits to keep the infant’s hands away from the pouch. Use empty paper towel or toilet paper rolls to help keep the infant’s legs out of the way when emptying or changing the ostomy bag. Slit the roll up the side and wrap it around the infant’s legs.
Special thanks to Elliott Douglass, RN, BSN, CWOCN, Wound, Ostomy, and Continence Nurse, Monroe Carell, Jr. Children’s Hospital at Vanderbilt.
Complications and Prognosis
Early complications of the pull-through operation include inadequate blood supply to the coloanal anastomosis, anastomotic stricture, anastomotic dehiscence, and cuff abscess. Later complications include Hirschsprung’s-associated enterocolitis, perianal excoriation, and recurrent constipation.51 The infant usually thrives postoperatively and grows normally. It is not uncommon for the infant to have frequent stools during the immediate postoperative period, which gradually normalize to a lesser frequency. However, some children, despite a technically satisfactory operation, will experience recurrent constipation requiring some form of bowel management program with or without placement of a colostomy tube for antegrade enemas. 89
ANORECTAL MALFORMATIONS
Physiology and Etiology
Anorectal malformations (ARMs) encompass a broad spectrum of hindgut anomalies, from isolated imperforate anus in boys and girls, which may include fistulous communications between the urogenital tract and rectum, to complicated persistent cloaca in females. Although the development of the cloaca and its subsequent septation into urogenital and anorectal tracts is not well understood, each organ system is recognizable as a separate entity by the 7th week of gestation. Therefore persistent cloaca in females arises from an arrest in development of the gut and its complete separation from urogenital tract between the 4th and 6th week of gestation. Cloacal exstrophy arises if disruption of the cloacal membrane occurs before the urorectal septum has separated the urinary bladder from the hindgut. Disruption of the cloacal membrane after septation results in exstrophy of the bladder only. Any insult occurring at this critical period of organogenesis places a number of organ systems at risk and accounts for the fact that 60% of infants with a persistent cloaca will have concomitant anomalies. 32
Imperforate anus is the most common ARM, occurring in 1 in 5000 live births, and predominantly affects males (58%) more than females (42%). Imperforate anus is characterized as low, intermediate, or high. Note: The higher the defect, the more likely the presence of other associated malformations. A high imperforate anus is defined as the end of the rectum terminating above the levator ani muscles. Conversely, in low imperforate anus, the rectum descends below the levator complex. A fistulous connection to the perineum or urogenital tract is almost always present. In high lesions, the rectal fistula enters the membranous urethra in the male or, rarely, the vagina in the female. In low lesions, the rectal fistula empties on the perineum of both boys and girls or the posterior fourchette of the introitus, the most common site in girls. 59 Congenital VACTERL anomalies and trisomy 21 are common and require evaluation. Moreover, a high incidence of spinal dysraphism is observed with anorectal malformation; imaging of the spine is indicated. 79
Data Collection
SIGNS AND SYMPTOMS
Most anorectal malformations are apparent on physical examination but may be missed if a careful inspection of the buttocks and anus is not performed. Once the diagnosis is made, a fistula should be sought. In low lesions, a thin membrane may be over the anal orifice or a fistula may be along the perineum and scrotal raphe of boys. In girls, the fistula most commonly terminates in the vestibule or fourchette of the introitus. If meconium passes in the urine or from the vagina, a high lesion is present. If the condition remains unrecognized, the infant develops signs and symptoms of distal intestinal obstruction. Down syndrome babies with an ARM usually (95%) have a high type variant of rectal atresia without genitourinary tract communication. (See the Critical Findings box below.)
Imperforate Anus
The following are critical assessment findings for imperforate anus:
• Absence of anus or presence of anteriorly displaced perineal fistula
• Signs and symptoms of obstruction if diagnosis not made
• Perineal ultrasound study will identify the level of defect in the absence of a perineal fistula
LABORATORY DATA
A plain abdominal radiograph may show features of distal intestinal obstruction without rectal gas, but it will not reliably show the distal termination of the rectum. Perineal ultrasonography may be used to establish the termination of the rectum and its distance from the skin—data that may help operative planning. In boys with imperforate anus without a perineal fistula, a contrast study of the urethra should delineate a rectourethral fistula, if present. In girls without a perineal or vestibular fistula, contrast genitogram may help define the anatomic relationships of a persistent cloaca. A perineal fistula visible on physical examination does not usually warrant imaging of ARM. However, because of the possibility for VACTERL association, echocardiography and abdominal sonography of the genitourinary tract are indicated, as is a plain radiograph of the spine and limbs.
Treatment
PREOPERATIVE CARE
The infant should be kept on NPO status while evaluation of ARM is underway, and an orogastric tube should be placed to exclude esophageal atresia and to decompress the stomach. If a fistula is present on the perineum or at the fourchette, an early anoplasty may be performed, assuming the baby has no associated cardiac anomaly and is otherwise deemed to be a suitable candidate for a general anesthetic. If early repair is contraindicated, then the fistula tract may be dilated twice daily to promote elimination of fecal contents until the baby is a more suitable candidate for surgery. If no fistula is visualized and a high lesion or cloaca is present, a divided colostomy is performed and staged reconstruction is planned for later in infancy.
OPERATIVE INTERVENTION
For low imperforate anus, early reconstruction is performed either in the newborn period or in the first months of life if the infant can produce stools adequately through the fistulous tract with dilations. After colostomy for high imperforate anus, a formal repair is undertaken when the child is 3 to 6 months of age. Approached through a posterior sagittal incision (buttock), the fistula is separated from the urethra in boys or vagina in girls and the rectum is mobilized to lie within the center of the sphincter mechanism. The levator muscles and sphincter are closed anteriorly and posteriorly around the rectum, which is then anastomosed to the perineal skin in a procedure known as a posterior sagittal anorectoplasty ( PSARP, or Peña procedure). 60 Alternatively, to minimize wound complications and postoperative pain, a laparoscopic-assisted anorectoplasty may be performed (Figure 28-13). A Foley catheter should always be placed in boys at time of the definitive repair to assist in separation of the rectum from the urethra and to facilitate bladder drainage in the postoperative period; this catheter will be in place typically for 7 to 14 days after PSARP.
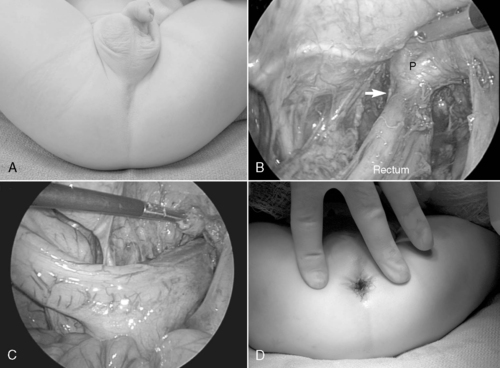 |
| FIGURE 28-13
(Courtesy Dr. H. Lovvorn, Vanderbilt University Medical Center.)
|
POSTOPERATIVE CARE
After anorectoplasty, simple skin care is all that is necessary, and a program of anal dilation is instituted 10 to 14 days postoperatively and will continue for 4 to 6 months. After a colostomy, stoma care and teaching are begun with the parents. Colostomy closure will be performed within 6 to 8 weeks of PSARP, assuming the neoanus is of adequate size and not strictured.
Complications and Prognosis
Mechanical complications of the stoma (prolapse, stenosis, and skin breakdown) may arise but generally do not require revision and should be temporized until stomal closure. 58Urinary tract infection (UTI) or hyperchloremic metabolic acidosis may result from the fistulous connection of the rectum to the urinary tract. Antibiotic prophylaxis is instituted, and select infants may require bicarbonate supplement until the fistula is divided.
Constipation is the primary long-term problem after correction of low imperforate anus. Stricture of the anoplasty should always be considered and may be treated with anal dilation or, rarely, revision anoplasty.
High imperforate anus is most often complicated by incontinence and frequent soiling. Long-term results are influenced by the degree of sphincter muscle development and innervation. Approximately 25% of infants have good continence, 50% have fair continence, and 25% have poor results. Bowel programs and strategies have been developed to permit some degree of social continence so that permanent colostomy can be avoided.
OMPHALOCELE AND GASTROSCHISIS
Physiology and Etiology
Omphalocele and gastroschisis are distinct defects of the abdominal wall at or near the umbilicus. 6,43Omphalocele is characterized by the persistent herniation of the abdominal viscera through the umbilical ring, and the herniated contents are covered by the normal components of the umbilical cord: the peritoneum, Wharton’s jelly, and amnion. Omphalocele, also known as exomphalos, is a defect in abdominal wall development that may result from failure of embryonic enfolding as early as the 4th to 7th week of gestation or from failure of closure of the exocoelomic space, which is usually completed by the 12th week of gestation. As many as 50% of neonates presenting with omphalocele will have an underlying chromosomal abnormality, most often trisomies 12 and 18 but also trisomy 21. Congenital heart lesions are seen in as many as 50% of affected infants. Congenital syndromes involving an omphalocele are potentially lethal, usually as a result of the associated abnormalities. Cloacal exstrophy, occurring in 1 in 200,000 pregnancies, and the constellation of defects known as pentalogy of Cantrell likely represent the earliest of embryonic failures in the development of this spectrum of anomalies including omphalocele.
Gastroschisis is a full-thickness defect of the abdominal wall that occurs most commonly to the right of the umbilicus and exposes the extruded bowel to the amniotic fluid. It has been hypothesized that this defect results from a weakening of the anterior abdominal wall caused by a vascular accident involving the right omphalomesenteric artery, which takes over perfusion of the anterior abdominal wall during the 7th week of gestation. Gastroschisis occurs 3 to 4 times more frequently than omphalocele and is rising in developed countries for unknown reasons. This increasing incidence seems to be occurring in younger mothers, most frequently in those younger than 20 years, although no clear epidemiology has been correlated with this finding. Gastroschisis typically is not associated with major congenital anomalies or syndromes, although 20% or more of affected infants have a concomitant intestinal atresia.33 This atresia is likely secondary to either the initial vascular accident thought to initiate the defect or compromise of the affected bowel segment arising from a constricting fascial defect.
Nonrotation of the intestine, by definition, is uniformly present in all incidences of these two conditions.
Data Collection
HISTORY
Abdominal wall defects are readily diagnosed by antenatal ultrasonography, which is helpful in planning future delivery and therapy. 13 Spontaneous or induced vaginal delivery should be considered in most cases of gastroschisis, because risk of bowel injury during delivery is minimal. 1,20,36 Babies with an omphalocele may also be delivered vaginally, but liver herniation or associated anomalies may dictate cesarean section.
In cases of gastroschisis, severe serositis resulting from exposure of the bowel to amniotic fluid makes closure more difficult and delays the return of bowel function. Early delivery may be recommended for certain fetuses who have gastroschisis if sonographic evidence reveals progressive bowel distention and thickening, suggesting intestinal obstruction or severe serositis. In cases of omphalocele, prenatal sonography should thoroughly evaluate the fetus for other potential anomalies and may be supplemented by fetal MRI.
SIGNS AND SYMPTOMS
Both anomalies present as a mass of abdominal contents extruding through an anterior abdominal wall defect. Eviscerated bowel without a peritoneal covering characterizes gastroschisis, whereas an omphalocele is defined by a peritoneal covering of herniated bowel and often a segment of liver. Gastroschisis defects are most commonly to the right of the midline and are found adjacent to the umbilical stalk, whereas omphalocele occurs through a central defect at the base of the umbilical cord.
In contradistinction to gastroschisis, omphalocele carries a high incidence of significant associated anomalies, with cardiac and urinary tract malformations being most prevalent. Omphalocele also is a feature of several recognizable syndromes, including Beckwith-Wiedemann, prune belly, cloacal exstrophy, and pentalogy of Cantrell. Chromosomal defects are also identified with greater frequency in cases of omphalocele than gastroschisis and include trisomies 13, 18, and 21. Most babies with an omphalocele will deliver at term. Gastroschisis, in contrast, is associated with few anomalies outside of the gastrointestinal tract. Malrotation is understood to exist with gastroschisis, and so the most common associated anomaly is intestinal stenosis or atresia (10% to 15%), a rare finding in omphalocele. Babies with gastroschisis are more commonly preterm and SGA. (See the Critical Findings box above.)
Omphalocele and Gastroschisis
The following are critical assessment findings for omphalocele and gastroschisis:
• Typically detected on antenatal ultrasound examination
• Eviscerated bowel without peritoneal covering in gastroschisis
• Eviscerated bowel with peritoneal covering in omphalocele
• Diagnosis of omphalocele necessitates search for other associated anomalies
LABORATORY DATA
In a neonate with omphalocele, a careful search for associated anomalies is performed before closure is attempted. Echocardiography and an x-ray examination of the chest and spine are performed to rule out cardiac, chest wall, diaphragmatic, and spinal anomalies. Abdominal sonography is obtained to evaluate integrity of the urinary tract. Newborns with gastroschisis do not require routine imaging of the heart and kidneys unless otherwise indicated.
Treatment
PREOPERATIVE CARE
Initial management of gastroschisis includes preservation of body heat and fluid, orogastric decompression, protection of the intestine, and prophylaxis against infection. Covering the exposed viscera minimizes heat and fluid loss. Wrapping the intestines in warm, saline-soaked gauze covered with a plastic wrap or placing the infant’s torso into an impermeable, clear plastic bowel bag is generally employed. Both methods decrease evaporative losses; the bowel bag further allows continuous visual monitoring of the bowel. With either method, it is imperative that the bowel be positioned to prevent constriction of the blood supply at the fascial level.
Intravenous fluids and broad-spectrum antibiotics should be instituted early. To prevent bowel distention, an orogastric tube is placed to low continuous suction. In omphalocele, because the abdominal contents are covered naturally, less evaporative fluid and heat loss is encountered.
OPERATIVE INTERVENTION
Rarely, an infant with omphalocele may be too ill to undergo operation, may have other lethal malformations, or may have a “giant omphalocele,” defined as the sac containing a significant portion of the liver (Figure 28-14). Under these circumstances, the newborn is given palliative care with a daily application of a desiccant or silver sulfadine to the abdominal sac. The result is eschar formation and subsequent epithelialization in 10 to 20 weeks. A large ventral hernia will remain, and if the patient survives, repair may be performed later in infancy. For cases of omphalocele without liver herniation or significant associated anomalies, the sac may be removed and primary fascial closure accomplished shortly after birth; alternatively, at a minimum, simple skin closure may be performed, with ventral hernia repair scheduled for later in infancy once the abdominal domain has been restored. Some cases of omphalocele will require biosynthetic fascial substitutes to accomplish visceral coverage.
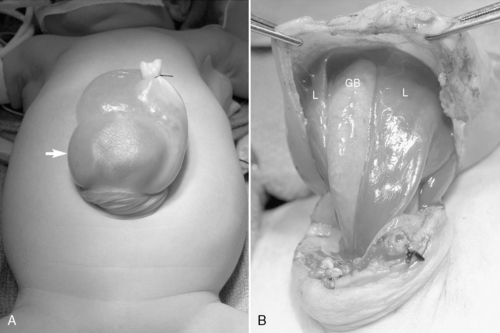 |
| FIGURE 28-14
(Courtesy Dr. H. Lovvorn, Vanderbilt University Medical Center.)
|
For babies born with gastroschisis, primary surgical repair entails reduction of the herniated abdominal contents into the peritoneal cavity without increasing abdominal pressure to a point that ventilation, venous return, and intestinal blood supply are compromised. If the amount of eviscerated abdominal contents is small or moderate, primary repair is simple and safe. Before closure of gastroschisis, the surgeon searches carefully for associated atresia but may or may not attempt to restore intestinal continuity during the initial operation. If atresia is identified but inflammation and matting of the intestine will not permit safe anastomosis, the atretic bowel may be placed within a silo or returned to the abdomen primarily; re-exploration may be planned for 4 to 6 weeks later to allow the thickened, edematous bowel wall to normalize, facilitating anastomosis or enterostomy. If atresia is found but the bowel is not inflamed or matted, intestinal continuity may be restored at the initial operation or an ostomy created, with plans for delayed reconstitution 4 to 6 weeks later.
In select cases, the fascial defect may have to be enlarged to allow replacement of the herniated organs into the abdomen because of the small size of the fascial ring, loss of abdominal domain, and the amount of intestinal herniation. The inability to perform primary closure necessitates placement of a Silastic silo or patch to permit staged reduction (Figure 28-15). Over the ensuing 2 to 7 days, the herniated viscera are gradually returned to the abdomen on a daily basis by gravity (baby remains supine) and by applying gentle and constant pressure to the silo at the bedside (umbilical tape is tied sequentially along the silo until the intestine has reached the fascial level). Once all of the silo contents have been successfully reduced, the infant is returned to the operating room for removal of the silo and closure of the fascia. 69 While a silo is in place, most neonates will remain on antibiotics.
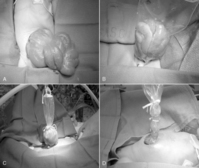 |
| FIGURE 28-15
(Courtesy Dr. H. Lovvorn, Vanderbilt University Medical Center.)
|
Complications and Prognosis
Bowel injury, respiratory compromise, and diminished venous return caused by abdominal hypertension may complicate recovery after primary fascial closure of abdominal wall defects. In this setting, the infant is returned to the operating room for placement of a silo. Rarely, a silo may become infected or separate from the fascia, complicating closure by this method.
Recovery of bowel function is uniformly delayed, especially in those with gastroschisis caused by exposure of the bowel to amniotic fluid and in those with intestinal problems (atresias, stenosis, perforation, necrosis, or volvulus).5 A central venous catheter should be placed at the time of initial surgery for long-term TPN support. Intestinal stricture, incisional hernia, and adhesive bowel obstruction are possible short-term and long-term complications. The principle long-term morbidity associated with gastroschisis is short bowel syndrome. Morbidity for omphalocele is principally secondary to any associated anomalies and the challenges of abdominal closure. Otherwise, prognosis for both types of abdominal wall defects should be good.
NEONATAL TUMORS
Neonatal tumors are discovered in every 12,500 to 25,000 live births and account for 2% of all childhood malignancies. 52 The majority of affected neonates present with a mass at birth (or by day 28 of life), which may or may not have been identified on prenatal screening. The two most frequently encountered neonatal tumors are teratoma (principally sacrococcygeal [Figure 28-16] but also cervical [Figure 28-17]) and neuroblastoma. Soft tissue sarcomas, infantile myofibromatosis, renal tumors (benign mesoblastic nephroma and malignant Wilms’ tumor), hepatoblastoma, and central nervous system tumors follow in frequency. Malignant tumors rarely arise in the newborn; however, some benign tumors encountered at birth may acquire malignant features later in infancy.
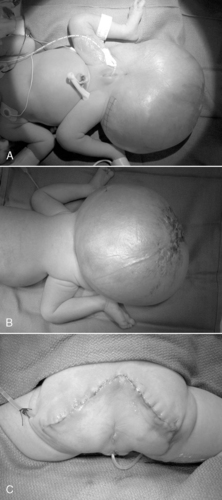 |
| FIGURE 28-16
(Courtesy Dr. H. Lovvorn, Vanderbilt University Medical Center.)
|
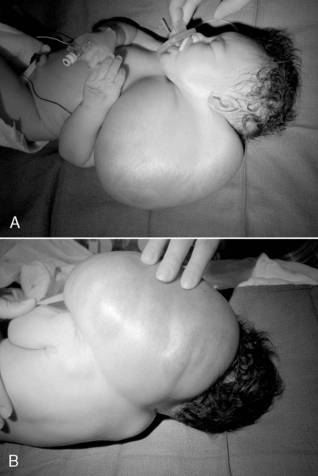 |
| FIGURE 28-17
(Courtesy Dr. H. Lovvorn, Vanderbilt University Medical Center.)
|
Nevertheless, although a neonatal tumor may be histologically benign, these tumors may be life threatening because of size, location, arteriovenous shunts, or rupture with hemorrhage. Some tumors show invasive or infiltrative characteristics, yet these may not have metastatic potential. Furthermore, screening programs have identified potentially malignant tumors earlier in development, such as for neuroblastoma in Japan, but have as yet failed to improve overall survival. Some neonatal tumors even show the potential for spontaneous regression. Taken together, neonatal tumors represent a protean mix of diseases that have a low malignant potential yet the biologic behavior may not be entirely predictable.
The etiology of solid malignancies in infants and children is an area of great interest. Many of these “congenital” tumors are classified as embryonal tumors because of retained features of embryonic development within the organ in which each tumor arises. Carcinomas, typical of adulthood, are virtually nonexistent in neonates. Under the microscope, many of these tumors show a recapitulation of cell types found in early embryonic development of the particular organ, but terminal differentiation of the progenitor cells has not been completed and so no functional tissue architecture is appreciated. Associated anomalies may be found in 15% of neonates with congenital tumors, and genetic defects are also relatively common.
Routine prenatal ultrasonography has contributed to an increasing diagnosis of fetal and neonatal tumors. It is interesting to note that several fetal tumors, particularly neuroblastoma, have a unique property to undergo spontaneous regression and involution by 12 months of age. As a result, the natural history of any given adrenal tumor and optimal management remain unclear. The Children’s Oncology Group has an ongoing study to determine the natural history of adrenal masses in newborns or infants, presumed to be neuroblastoma. Patients are being enrolled into an “expectant management” arm, which should shed light on the natural history of these lesions.
Commonly, fetuses harboring an embryonal or germ cell tumor will be identified on antenatal screening. Some tumors—in particular, sacrococcygeal teratomas—may be so large as to cause dystocia or may present a significant risk for rupture during delivery, and either scenario could be disastrous. Further, a cervical teratoma or cystic hygroma may cause airway obstruction at birth. Fetuses with these occasionally huge cervical tumors should be delivered by cesarean section, bronchoscoped, and intubated before clamping of the umbilical cord, referred to as an EXIT procedure ( EX-utero Intra-partum Therapy). The tumor then may be resected electively once the extent of the disease and any associated anomalies have been fully evaluated. 35
Most neonatal tumors are benign or of low malignant potential and tend to behave more favorably than the same type of tumor in older children. Neuroblastoma, for example, generally presents as stage I disease in 90% of infants younger than 1 year and is amenable to observation, if small, or complete resection. Hepatoblastomas presenting in the newborn period also tend to behave more favorably than when presenting later in infancy. Despite a collective rarity, presentation of malignant tumors in the newborn period does occur, and much work remains to identify determinants of tumorigenesis and pathogenesis.
MINIMALLY INVASIVE SURGERY
Laparoscopic or minimally invasive techniques are beginning to supplant many surgical procedures previously performed by laparotomy or thoracotomy. Laparoscopic procedures have been associated with decreased postoperative pain, earlier return to gastrointestinal function, shorter hospital stays, fewer wound complications, and improved cosmetic results, when compared with the corresponding open procedures. 31
Laparoscopic techniques have been applied to many of the neonatal procedures described in this chapter. Indeed, laparoscopic Nissen fundoplication for gastroesophageal reflux disease (GERD) and laparoscopic-assisted pull-through procedures for Hirschsprung’s disease are being performed routinely in the neonate and show shorter time to postoperative feeding, decreased hospital stay, and superior cosmesis. For example, thoracoscopic repair of esophageal atresia with tracheoesophageal fistula is beginning to be advocated as an equally effective approach, and supporters claim improved pain management and musculoskeletal benefits. Laparoscopic repair of duodenal atresia is also being performed with good results. Both thoracoscopic and laparoscopic techniques are being used increasingly to repair Bochdalek and Morgagni diaphragmatic hernias with good outcomes in neonates who do not require ECLS. Laparoscopic-assisted endorectal pull-through for Hirschsprung’s disease and for anorectoplasty to correct ARM is becoming more and more a part of the neonatal surgeon’s armamentarium. 70 However, pathophysiologic effects of pneumoperitoneum or iatrogenic pneumothorax are only recently being analyzed. Adverse consequences include the following38:
• Reduced intraoperative arterial saturation
• Increased carbon dioxide (CO 2) retention (CO 2 is used for abdominal insufflation, and diaphragmatic excursion may be compromised because of increased abdominal pressure)
• Oliguria or even anuria for up to 4 hours postoperatively
• Hypothermia
• Need for extended postoperative intubation if procedures exceed 100 minutes, which they often may
As experience increases and technical refinements are seen with improved optical systems and smaller instruments, a broader application of laparoscopy inevitably will occur in the neonatal patient population.
PARENT TEACHING
Preoperative
The advent of ever-increasing mechanisms of prenatal diagnosis allows parents to begin adjusting to the presence of certain malformations—some minor and others potentially lethal—before birth. 3 Research shows that parents experience various stages of grief, including shock, denial, anger, and sadness, when faced with such news. Although the intensity of the negative emotions associated with the initial diagnosis may lessen by the time of delivery, resolution of these emotions should not be expected. Expressions of fear, anxiety, and guilt are likely to remain for some time. 57
Because parents’ fears and fantasies about their infant’s surgical diagnosis are frequently worse than reality, they should see their infant as soon as possible after birth and before surgery. Pictures of the infant should be taken before surgery and should include views with and without the defect if possible. Whenever time and patient condition allow, parents should be encouraged to hold their infant and pictures should be taken of the family with their newborn. In the event of a neonate’s death, these pictures may be very valuable to the family. In the event neonatal transport from the birth hospital to a referral center is required, the transport team should do everything possible to ensure that the mother, who may be in the early recovery phase from her delivery, sees her infant before departure. 16
The planned operative procedure and its expected results, as well as risks and alternatives, are discussed with the parents, ensuring that all questions and concerns are addressed. An informed consent for surgery is signed and witnessed. The bedside nurse should be present when the physician meets with the family to discuss the operation, because the nurse is often the most consistent individual hearing explanations from the neonatologist, surgeon, and anesthesiologist and must answer questions, interpret information, and reassure an anxious family when these teams leave. The nurse has a further important role in reassuring parents about postoperative analgesia and sedation for there infant. Preoperative teaching can involve educating parents in recognizing pain cues in their baby, and they should be encouraged to discuss their concerns if they perceive their baby is experiencing discomfort. Care providers’ sensitivity to the neonate’s pain and advocating for pain relief are comforting for parents (see Chapter 12).
Parents often fear what the infant will look like on return from surgery. Providing written material with simple drawings of the defects and operative procedure may help prevent postoperative surprises. Seeing another patient who has had a similar procedure and has the postoperative equipment that has been described (e.g., colostomy, orogastric tube, chest tube) may be helpful to the family as well. 57 The nursing staff must be careful, however, to protect the privacy of other patients and should get parental consent before visiting another infant for this purpose.
Intraoperative
Accompanying the infant to the preoperative area and seeing the infant as soon as possible after surgery are comforting to the parents. Progress reports of the surgery, if possible, are helpful for the anxious family members. After the operation, the pediatric surgeon should immediately see the parents to explain the procedure and any unexpected problems that occurred during the operation. It should be clear to both the family and the surgeon where this important communication will take place, and privacy should be protected during the interchange.
Postoperative
After surgery, the nurse should help parents focus on their infant rather than the surrounding intensive care environment. Although the nursing staff should identify the monitors, equipment, dressings, and foreign bodies on the baby and in the room and should explain the purpose of each to the family, encouraging parents to comfort their baby will be of greatest benefit to their postoperative child (and to them). Early involvement in caregiving helps the parents feel as though they are essential to their child’s recovery. Even in the immediate postoperative period, a parent can quietly sit and hold the infant’s hand or take an axillary temperature. After most surgical procedures, mothers should be encouraged to express breast milk and may benefit from a lactation nurse consultation. Feeding issues may often remain a potential barrier to maternal confidence and maternal-infant interaction and warrant education and assistance during the postoperative period long before NICU discharge. 22
If an infant is to be discharged home with an ostomy, parents begin to participate in stoma care as early as possible. Consultation with an enterostomal therapy nurse is of further benefit. Parents begin by learning to cleanse the skin around the ostomy or to prepare the appliance and peristomal salves. Gradually, parents will learn to increase their responsibilities of caregiving as their infant improves. Frequent practice improves proficiency and empowers parents for taking their infant home. Delay until a few days before discharge does not give parents adequate time for practice and familiarity for home care and does not serve the infant or family well. The same is true for infants who are discharged with other types of complicated care, such as home TPN or feedings through a gastrostomy tube and/or via a continuous feeding pump. Parent teaching includes the possibility of late postoperative complications and recognition of problems that may develop and a plan of action for dealing with them.
The importance of follow-up care is emphasized to the parents. It may be helpful for parents to talk with a “graduate” parent who had an infant with similar problems. Contact information about available resources, such as visiting nurses, graduate parents, and parent support groups is provided to parents before discharge. A concise history of hospitalization and the discharge plan are made available to all post-hospitalization health care providers, and a copy is provided to parents at the time of discharge. Encouraging parents to keep a copy of this discharge summary in their diaper bag increases the chances they will have it available should they be required to take their medically complex and vulnerable infant emergently to the hospital.
REFERENCES
1. Abdel-Latif, N.S.; Bolisetty, S.; Abeywardana, S.; et al., Mode of delivery and neonatal survival of infants with gastroschisis in Australia and New Zealand, J Pediatr Surg 43 (2008) 1685.
2. Adzick, NS.; Nance, M.L., Pediatric surgery, N Engl J Med 342 (2000) 1651.
3. Aite, L.; Zaccara, A.; Nahom, A.; et al., Mother’s adaptation to antenatal diagnosis of surgically correctable anomalies, Early Hum Dev 82 (2006) 649.
4. American Academy of Pediatrics, Section on Surgery and the Committee on Fetus and Newborn, Postdischarge follow-up of infants with congenital diaphragmatic hernia, Pediatrics 121 (2008) 627.
5. Arnold, M.A.; Chang, D.C.; Nabaweesi, R.; et al., Risk stratification of 4344 patients with gastroschisis into simple and complex categories, J Pediatr Surg 42 (2007) 1520.
6. Aspelund, G.; Langer, J.C., Abdominal wall defects, Curr Pediatr 16 (2006) 192.
7. Azizkhan, R.G.; Crombleholme, T.M., Congenital cystic lung disease: contemporary antenatal and postnatal management, Pediatr Surg Int 24 (2008) 643.
8. Bagolan, P.; Casaccia, G.; Crescenzi, F.; et al., Impact of a current treatment protocol on outcome of high-risk congenital diaphragmatic hernia, J Pediatr Surg 39 (2004) 313.
9. Bax, N.M.; van der Zee, D.C., Laparoscopic treatment of intestinal malrotation in children, Surg Endosc 12 (1998) 1314.
10. Blakely, M.L.; Gupta, H.; Lally, K.P., Surgical management of necrotizing enterocolitis and isolated intestinal perforation in premature neonates, Semin Perinatol 32 (2008) 122.
11. Casaccia, G.; Trucchi, A.; Nahom, A.; et al., The impact of cystic fibrosis on neonatal intestinal obstruction: the need for prenatal/neonatal screening, Pediatr Surg Int 19 (2003) 75.
12. Centers for Disease Control and Prevention, Trends in infant mortality attributable to birth defects: United States, 1980-1995, MMWR Morb Mortal Wkly Rep 47 (1998) 773.
13. Chen, C.P.; Liu, F.F.; Jan, S.W.; et al., Prenatal diagnosis and perinatal aspects of abdominal wall defects, Am J Perinatol 13 (1996) 355.
14. Chokshi, N.K.; Guner, Y.; Henri, R.F., The role of IL-10 in experimental necrotizing enterocolitis, J Am Coll Surg 207 (2008) S14; (abstract).
15. Coran, A.G.; Teitelbaum, D.H., Recent advances in the management of Hirschsprung’s disease, Am J Surg 180 (2000) 382.
16. Cornette, L., Transporting the sick neonate, Curr Paediatr 14 (2004) 20.
17. Dalla Vecchia, L.K.; Grosfeld, J.L.; West, K.W.; et al., Intestinal atresia and stenosis: a 25-year experience with 277 cases, Arch Surg 133 (1998) 490.
18. de Jong, E.M.; Felix, J.F.; Deurloo, J.A.; et al., Non-VACTERL-type anomalies are frequent in patients with esophageal atresia/tracheo-esophageal fistula and full or partial VACTERL association, Birth Defects Res A Clin Mol Teratol 82 (2008) 92.
19. Dimmitt, R.A.; Meier, A.H.; Skarsgard, E.D.; et al., Salvage laparotomy for failure of peritoneal drainage in necrotizing enterocolitis in infants with extremely low birth weight, J Pediatr Surg 35 (2000) 856.
20. Drewitt, M.; Michailidis, G.D.; Burge, D., The perinatal management of gastroschisis, Early Hum Dev 82 (2006) 305.
21. Engum, S.A.; Grosfeld, J.L.; West, K.W.; et al., Analysis of morbidity and mortality in 227 cases of esophageal atresia or tracheoesophageal fistula over two decades, Arch Surg 130 (1995) 502.
22. Faugli, A.; Emblem, R.; Veenstra, M.; et al., Does esophageal atresia influence the mother-infant interaction?J Pediatr Surg 43 (2008) 1796.
23. Feitz, R.; Vos, A., Malrotation: the postoperative period, J Pediatr Surg 32 (1997) 1322.
24. Freeman, S.B.; Torfs, C.P.; Romitti, P.A.; et al., Congenital gastrointestinal defects in Down syndrome: a report from the Atlanta and National Down Syndrome Projects, Clin Genet 75 (2) ( 2009) 180; Epub ahead of print: November 17, 2008.
25. Friedman, S.; Chen, C.; Chapman, J.S.; et al., Neurodevelopmental outcomes of congenital diaphragmatic hernia survivors followed in a multidisciplinary clinic at ages 1 and 3, J Pediatr Surg 43 (2008) 1035.
26. Gibbins, S.; Maddalena, P.; Golec, L., Evidenced-based care for the infant with necrotizing enterocolitis, Newborn Infant Nurs Rev 8 (2008) 144.
27. Grosfeld, J.L.; Rescorla, F.J., Duodenal atresia and stenosis: reassessment of treatment and outcome based on antenatal diagnosis, pathologic variance, and long-term follow-up, World J Surg 17 (1993) 301.
28. Guner, Y.S.; Chokshi, N.; Aranda, A.; et al., Thoracoscopic repair of neonatal diaphragmatic hernia, J Laparoendosc Adv Surg Tech 18 (2008) 875; Bottom of Form.
29. Guner, Y.S.; Chokshi, N.; Petrosyan, M.; et al., Necrotizing enterocolitis—bench to bedside: novel and emerging strategies, Semin Pediatr Surg 17 (2008) 255.
30. Haricharan, R.N.; Georgeson, K.E., Hirschsprung disease, Semin Pediatr Surg 17 (2008) 266.
31. Harres, A.E., Minimally invasive neonatal surgery, J Perinat Neonatal Nurs 21 (2007) 39.
32. Hendren, W.H., Cloaca, the most severe degree of imperforate anus: experience with 195 cases, Ann Surg 228 (1998) 331.
33. Henrich, K.; Huemmer, H.P.; Reingruber, B.; et al., Gastroschisis and omphalocele: treatments and long-term outcomes, Pediatr Surg Int 24 (2008) 167.
34. Henry, M.C.W.; Moss, R.L., Neonatal necrotizing enterocolitis, Semin Pediatr Surg 17 (2008) 98.
35. Hirose, S.; Farmer, D.L.; Lee, H.; et al., The ex utero intrapartum treatment procedure: looking back at the EXIT, J Pediatr Surg 39 (2004) 375.
36. How, H.Y.; Harris, B.J.; Pietrantoni, M.; et al., Is vaginal delivery preferable to elective cesarean delivery in fetuses with a known ventral wall defect?Am J Obstet Gynecol 182 (2000) 1527.
37. Hunter, J.H.; Williams, M.; Petrosyan, M.; et al., Lactobacillus species abrogates pathogen induced experimental necrotizing enterocolitis by attenuating inducible nitric oxide synthase production, J Am Coll Surg 207 (2008) S54; (abstract).
38. Kalfa, N.; Ecochard, A.; Patte, C.; et al., Tolerance of laparoscopy and thoracoscopy in neonates, Pediatrics 115 (2005) 785.
39. Kao, S.C.; Franken Jr, E.A., Nonoperative treatment of simple meconium ileus: a survey of the Society for Pediatric Radiology, Pediatr Radiol 25 (1995) 97.
40. Keckler, S.J.; St. Peter, S.D.; Spilde, T.L.; et al., Current significance of meconium plug syndrome, J Pediatr Surg 43 (2008) 896.
41. Keckler, S.J.; St. Peter, S.D.; Valusek, P.A.; et al., VACTERL anomalies in patients with esophageal atresia: an updated delineation of the spectrum and review of the literature, Pediatr Surg Int 23 (2007) 309.
42. Lakhoo, K., Management of congenital cystic adenomatous malformations of the lung, Arch Dis Child Fetal Neonatal Ed 94 (2009) F73.
43. Langer, J.C., Gastroschisis and omphalocele, Semin Pediatr Surg 5 (1996) 124.
44. Langston, C., New concepts in the pathology of congenital lung malformations, Sem Pediatr Surg 12 (2003) 17.
45. Lemons, J.A.; Bauer, C.R.; Oh, W.; et al., Very low birth weight outcomes of the National Institute of Child Health and Human Development Neonatal Research Network, January 1995 through December 1996, Pediatrics 107 (2001) e1.
46. Li, K.; Zheng, S.; Xiao, X.; et al., The structural characteristics and expression of neuropeptides in the esophagus of patients with congenital esophageal atresia and tracheoesophageal fistula, J Pediatr Surg 42 (2007) 1433.
47. Lindower, J.; Atherton, H.; Kotagal, U., Outcomes and resource utilization for newborns with major congenital malformations: the initial NICU admission, J Perinatol 19 (1999) 212.
48. Messineo, A.; MacMillan, J.H.; Palder, S.B.; et al., Clinical factors affecting mortality in children with malrotation of the intestine, J Pediatr Surg 27 (1992) 1343.
49. Migliazza, L.; Bellan, C.; Alberti, D.; et al., Retrospective study of 111 cases of congenital diaphragmatic hernia treated with early high-frequency oscillatory ventilation and presurgical stabilization, J Pediatr Surg 42 (2007) 1526.
50. Millar, A.J.W.; Rode, H.; Cywes, S., Malrotation and volvulus in infancy and childhood, Semin Pediatr Surg 12 (2003) 229.
51. Mills, J.L.A.; Konkin, D.E.; Milner, R.; et al., Long-term bowel function and quality of life in children with Hirschsprung’s disease, J Pediatr Surg 43 (2008) 899.
52. Moore, S.W.; Satge, D.; Sasco, A.J.; et al., The epidemiology of neonatal tumors, Pediatr Surg Int 19 (2003) 509.
53. Munck, A.; Gérardin, M.; Alberti, C.; et al., Clinical outcome of cystic fibrosis presenting with or without meconium ileus: a matched cohort study, J Pediatr Surg 41 (2006) 1556.
54. Mushtaq, I.; Wright, V.M.; Drake, D.P.; et al., Meconium ileus secondary to cystic fibrosis: the East London experience, Pediatr Surg Int 13 (1998) 365.
55. Navarro, O.M.; Daneman, A.; Miller, S.F., Contrast enema depiction of small-bowel volvulus in complicated neonatal bowel obstruction, Pediatr Radiol 34 (2004) 1020.
56. Ng, G.Y.T.; Derry, C.; Marston, L.; et al., Reduction in ventilator-induced lung injury improves outcome in congenital diaphragmatic hernia?Pediatr Surg Int 24 (2008) 145.
57. Nisell, M.; Ojmyr-Joelsson, M.; Frenckner, B.; et al., How a family is affected when a child is born with anorectal malformation: interviews with three patients and their parents, J Pediatr Nurs 18 (2003) 423.
58. Patwardhan, N.; Kiely, E.M.; Drake, D.P.; et al., Colostomy for anorectal anomalies: high incidence of complications, J Pediatr Surg 36 (2001) 795.
59. Peña, A., Anorectal malformations, Semin Pediatr Surg 4 (1995) 35.
60. Peña, A.; Hong, A., Advances in the management of anorectal malformations, Am J Surg 180 (2000) 370.
61. Piper, H.G.; Alesbury, J.; Waterford, S.D.; et al., Intestinal atresias: factors affecting clinical outcomes, J Pediatr Surg 43 (2008) 1244.
62. Pourcyrous, M.; Korones, S.B.; Yang, W.; et al., C-reactive protein in the diagnosis, management, and prognosis of neonatal necrotizing enterocolitis, Pediatrics 116 (2005) 1064.
63. Radulescu, A.; Yu, X.; Chen, Y.; et al., HB-EGF knockout mice have increased susceptibility to necrotizing enterocolitis, J Am Coll Surg 207 (2008) S54; (abstract).
64. Rescorla, F.J.; Grosfeld, J.L., Contemporary management of meconium ileus, World J Surg 17 (1993) 318.
65. Riedlinger, W.F.J.; Vargas, S.O.; Jennings, R.W.; et al., Bronchial atresia is common to extralobar sequestration, intralobar sequestration, congenital cystic adenomatoid malformation, and lobar emphysema, Pediatr Dev Path 9 (2006) 361.
66. Robb, A.; Lander, A., Duodenal and small intestinal atresias and stenosis, Surgery (Oxford) 25 (2007) 287.
67. Roessingh, A.S.; de Buys, A.T.; Dinh-Xuan, Congenital diaphragmatic hernia: current status and review of the literature, Eur J Pediatr 168 (2009) 393; Epub ahead of print: December 23, 2008.
68. Sato, S.; Nishijima, E.; Muraji, T.; et al., Jejunoileal atresia: a 27-year experience, J Pediatr Surg 33 (1998) 1633.
69. Sauter, E.R.; Falterman, K.W.; Arensman, R.M., Is primary repair of gastroschisis and omphalocele always the best operation?Am Surg 57 (1991) 142.
70. Shinall Jr, M.C.; Koehler, E.; Shyr, Y.; et al., Comparing cost and complications of primary and staged surgical repair of neonatally diagnosed Hirschsprung’s disease, J Pediatr Surg 43 (2008) 2220.
71. Silva, C.T.; Daneman, A.; Navarro, O.M.; et al., Correlation of sonographic findings and outcome in necrotizing enterocolitis, Pediatr Radiol 37 (2007) 274.
72. Skari, H.; Bjornland, K.; Haugen, G.; et al., Congenital diaphragmatic hernia: a meta-analysis of mortality factors, J Pediatr Surg 35 (2000) 1187.
73. Spigland, N.; Yazbeck, S., Complications associated with surgical treatment of congenital intrinsic duodenal obstruction, J Pediatr Surg 25 (1990) 1127.
74. Spitz, L.; Kiely, E.M.; Morecroft, J.A.; et al., Oesophageal atresia: at-risk groups for the 1990s, J Pediatr Surg 29 (1994) 723.
75. Strouse, P.J., Disorders of intestinal rotation and fixation (malrotation), Pediatr Radiol 34 (2004) 837.
76. Teitelbaum, D.H.; Cilley, R.E.; Sherman, N.J.; et al., A decade of experience with the primary pull-through for Hirschsprung disease in the newborn period: a multicenter analysis of outcomes, Ann Surg 232 (2000) 372.
77. Tireli, G.A.; Özbey, H.; Temiz, A.; et al., Bronchogenic cysts: a rare congenital cystic malformation of the lung, Surg Today 34 (2004) 573.
78. Torres, A.M.; Ziegler, M.M., Malrotation of the intestine, World J Surg 17 (1993) 326.
79. Torres, R.; Levitt, M.A.; Tovilla, J.M.; et al., Anorectal malformations and Down’s syndrome, J Pediatr Surg 33 (1998) 194.
80. Tovar, J.A., The neural crest in pediatric surgery, J Pediatr Surg 42 (2007) 915.
81. Tran, H.; Fink, M.A.; Cramer, J.; et al., Congenital cystic adenomatoid malformation: monitoring the antenatal and short-term neonatal outcome, Aust N Z J Obstet Gynaecol 48 (2008) 462.
82. Ververidis, M.; Kiely, E.M.; Spitz, L.; et al., The clinical significance of thrombocytopenia in neonates with necrotizing enterocolitis, J Pediatr Surg 36 (2001) 799.
83. Waag, K-L.; Loff, S.; Zahn, K.; et al., Congenital diaphragmatic hernia: a modern day approach, Semin Pediatr Surg 17 (2008) 244.
84. Waldhausen, J.H.; Sawin, R.S., Improved long-term outcome for patients with jejunoileal apple peel atresia, J Pediatr Surg 32 (1997) 1307.
85. Walesa, P.W.; de Silva, N.; Kim, J.H.; et al., Neonatal short bowel syndrome: a cohort study, J Pediatr Surg 40 (2005) 755.
86. Walsh, D.S.; Adzick, N.S., Fetal surgical intervention, Am J Perinatol 17 (2000) 277.
87. Weber, T.R.; Kountzman, B.; Dillon, P.A.; et al., Improved survival in congenital diaphragmatic hernia with evolving therapeutic strategies, Arch Surg 133 (1998) 498.
88. Williams, H., Green for danger: intestinal malrotation and volvulus, Arch Dis Child Educ Pract Ed 92 (2007) ep87.
89. Yanchar, N.L.; Soucy, P., Long-term outcome after Hirschsprung’s disease: patients’ perspectives, J Pediatr Surg 34 (1999) 1152.



