Neonatal and Pediatric Respiratory Disorders
After reading this chapter you will be able to:
 Discuss the clinical findings, radiographic abnormalities, and treatment of patients with respiratory distress syndrome.
Discuss the clinical findings, radiographic abnormalities, and treatment of patients with respiratory distress syndrome.

 Describe the pathophysiology, presentation, and treatment of meconium aspiration syndrome.
Describe the pathophysiology, presentation, and treatment of meconium aspiration syndrome.

 State the etiology and treatment of apnea of prematurity.
State the etiology and treatment of apnea of prematurity.

 Discuss the pathophysiology, diagnosis, and treatment of congenital diaphragmatic hernia.
Discuss the pathophysiology, diagnosis, and treatment of congenital diaphragmatic hernia.
 Identify the anatomic defects associated with tetralogy of Fallot.
Identify the anatomic defects associated with tetralogy of Fallot.
 Describe the clinical presentation of a ventricular septal defect.
Describe the clinical presentation of a ventricular septal defect.
 Define the epidemiologic factors associated with increased risk of sudden infant death syndrome.
Define the epidemiologic factors associated with increased risk of sudden infant death syndrome.
 Identify the respiratory problems associated with gastroesophageal reflux disease.
Identify the respiratory problems associated with gastroesophageal reflux disease.
 State the clinical findings commonly observed in patients with bronchiolitis.
State the clinical findings commonly observed in patients with bronchiolitis.
 Describe the clinical features and treatment of children with epiglottitis.
Describe the clinical features and treatment of children with epiglottitis.
 Describe the clinical manifestations and treatment of cystic fibrosis.
Describe the clinical manifestations and treatment of cystic fibrosis.
Neonatal Respiratory Disorders
Lung Parenchymal Disease
Respiratory Distress Syndrome
 Rule of Thumb
Rule of ThumbPathophysiology
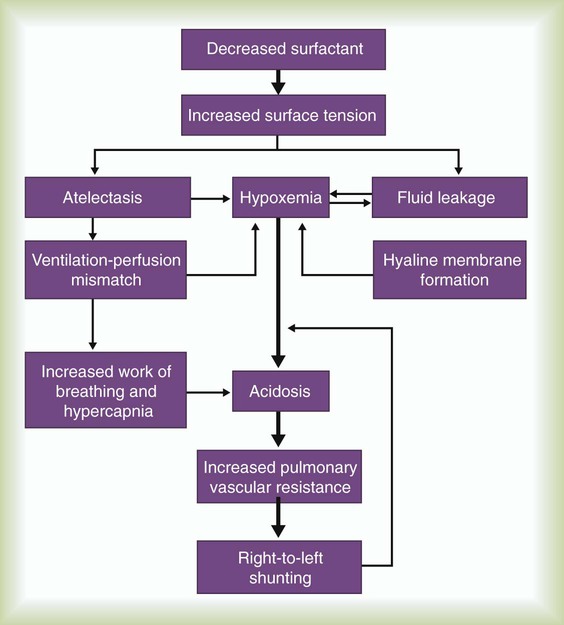
Figure 31-1 outlines the pathophysiologic events associated with RDS. A qualitative decrease in surfactant increases alveolar surface tension forces. This process causes alveoli to become unstable and collapse and leads to atelectasis and increased work of breathing. At the same time, the increased surface tension draws fluid from the pulmonary capillaries into the alveoli. In combination, these factors impair oxygen (O2) exchange and cause severe hypoxemia. The severe hypoxemia and acidosis increase pulmonary vascular resistance (PVR). As pulmonary arterial pressure increases, extrapulmonary right-to-left shunting increases, and hypoxemia worsens. Hypoxia and acidosis also impair further surfactant production. Antenatal steroids have been shown to mature surfactant function.
Clinical Manifestations
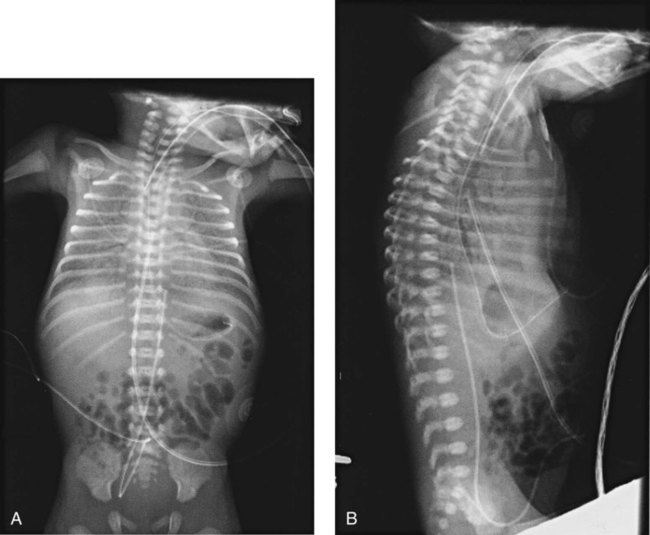
A definitive diagnosis of RDS usually is made with chest radiography (Figure 31-2). Diffuse, hazy, reticulogranular densities with the presence of air bronchograms with low lung volumes are typical of RDS. The reticulogranular pattern is caused by aeration of respiratory bronchioles and collapse of the alveoli. Air bronchograms appear as aerated, dark, major bronchi surrounded by the collapsed or consolidated lung tissue.
Treatment
Continuous positive airway pressure (CPAP) and positive end expiratory pressure (PEEP) are the traditional support modes used to manage RDS. Surfactant replacement therapy and high-frequency ventilation (HFV) have been added to these traditional approaches.1–5 Unless the infant’s condition is severe, a trial of nasal CPAP is indicated (4 to 6 cm H2O).6,7 Because of the hazards of endotracheal tubes, nasal prongs are preferred. If the infant’s clinical condition deteriorates rapidly, a more aggressive approach is required. Endotracheal intubation should be performed under controlled conditions as an elective procedure. Mechanical ventilation with PEEP should be initiated if oxygenation does not improve with CPAP or if the patient is apneic or acidotic. There is significant interest in an approach comprising intubation, delivery of surfactant, extubation, and then nasal CPAP.8 However, more research is needed to understand the risks and benefits of this approach.
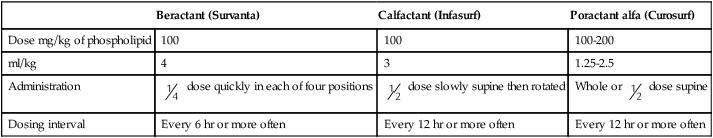
Three surfactant preparations are used in the United States for management of neonatal RDS: beractant (Survanta; Abbott Laboratories, North Chicago, IL), calfactant (Infasurf; ONY, Inc, Amherst, NY), and poractant alfa (Curosurf; Chiesi, Cheadle, United Kingdom).4,5,7–9 Beractant and calfactant are natural bovine surfactant extracts. Poractant alfa is a natural porcine surfactant extract. Each of these three natural surfactants has surfactant proteins B and C as part of the formulation. These surfactant proteins are important for decreasing alveolar surface tension. All of these preparations are liquid suspensions that are instilled directly into the trachea. The current standard of care is to deliver replacement surfactant to all infants with RDS. An additional artificial surfactant, lucinactant, is being actively studied.9 The ability to nebulize with this new surfactant is an exciting possibility. At the present time, no evidence supports the use of a particular brand of surfactant.
Surfactant replacement therapy also is used as both a rescue treatment (of infants who already have RDS) and a prophylactic therapy (in the care of infants delivered prematurely).10–13 Some centers use prophylactic surfactant replacement therapy in the care of all very small infants (<1500 g). Therapies aimed at decreasing pulmonary edema, improving cardiac output, and weaning from O2 and high ventilator pressures are essential in the successful treatment of infants receiving surfactant.
All surfactants are delivered via the endotracheal tube. Animal studies suggest that surfactant is rapidly distributed throughout the lung.14 Each specific surfactant has different dosing volumes and intervals (Table 31-1). The surfactant product insert describes the positioning of the infant for surfactant delivery. Basically, the infant is positioned with different sections of the lung dependent so that the surfactant enters that section of the lung with gravity flow. If the infant is very sick and cannot be repositioned, surfactant can be administered with the infant in a supine position.
TABLE 31-1
| Beractant (Survanta) | Calfactant (Infasurf) | Poractant alfa (Curosurf) | |
| Dose mg/kg of phospholipid | 100 | 100 | 100-200 |
| ml/kg | 4 | 3 | 1.25-2.5 |
| Administration | 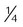 dose quickly in each of four positions dose quickly in each of four positions |
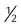 dose slowly supine then rotated dose slowly supine then rotated |
Whole or 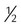 dose supine dose supine |
| Dosing interval | Every 6 hr or more often | Every 12 hr or more often | Every 12 hr or more often |
 Problem
ProblemTransient Tachypnea of the Newborn
Background
Transient tachypnea of the newborn (TTN), often called type II RDS, is probably the most common respiratory disorder of newborns. The cause of TTN is unclear, but it is most likely related to delayed clearance of fetal lung liquid.15–29 During most births, approximately two-thirds of this fluid is expelled by thoracic squeeze in the birth canal; the rest is reabsorbed through the lymphatic vessels during initial breathing. These mechanisms are impaired in infants born by cesarean section or infants with incomplete development of the lymphatic vessels (preterm or small-for-gestational-age infants). The residual lung fluid causes an increase in airway resistance and an overall decrease in lung compliance. Because compliance is low, the infant must generate more negative pleural pressure to breathe. This process can result in hyperinflation of some areas and air trapping in others. Most infants with TTN are born at term without any specific predisposing factors in common. Mothers of neonates who have TTN tend to have longer labor intervals and a higher incidence of failure to progress in labor, which leads to cesarean delivery. In many cases, however, maternal history and labor and delivery are normal.
Meconium Aspiration Syndrome
Pathophysiology
Amniotic fluid consists mainly of fetal lung fluid, fetal urine, and transudate from the uterine wall. Meconium, the contents of the fetal intestine, occasionally is expelled from the fetus into the surrounding amniotic fluid. Meconium consists of mucopolysaccharides, cholesterol, bile acids and salts, intestinal enzymes, and other substances. Meconium normally is not passed until after delivery.30 Infants who have marked perinatal depression or perinatal asphyxia may pass meconium in utero. The pathophysiologic control mechanisms for the passage of meconium in utero are not completely understood. It is widely accepted that infants can have meconium aspiration in utero. Amniotic fluid stained with meconium is found in approximately 12% of all births.30 Meconium-stained amniotic fluid is rare among infants of less than 37 weeks’ gestational age. The clinical syndrome develops in 2 of every 1000 infants. Of infants with inhaled meconium, 95% clear their lungs spontaneously.30Amniotic fluid infusion into the uterus before the delivery of infants with meconium-stained fluid has been shown to improve neonatal outcomes.31,32
For many years, the aspirated meconium itself was considered the primary cause of MAS. More recent evidence suggests that the real causative agent is fetal asphyxia that precedes aspiration.23 Fetal asphyxia causes pulmonary vasospasm and hyperreactivity of the vasculature, which lead to persistent pulmonary hypertension.
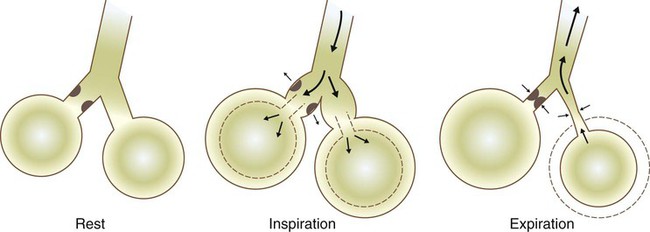
MAS involves three primary problems: pulmonary obstruction, lung tissue damage, and pulmonary hypertension.33 Obstruction occurs because of plugging of the airways with particulate meconium. This obstruction often is of the ball-valve type, which allows gas entry but prevents gas exit. Ball-valve obstruction causes air trapping and can lead to volutrauma (Figure 31-3). The lung tissue injury caused by MAS is chemical pneumonitis. Additionally, there are various chemical effects, inflammatory responses, cytokine and chemokine activations, complement activation, and phospholipase A2 activation.33–37 Persistent pulmonary hypertension with intracardiac and extracardiac right-to-left shunting frequently complicates MAS.30
Clinical Manifestations
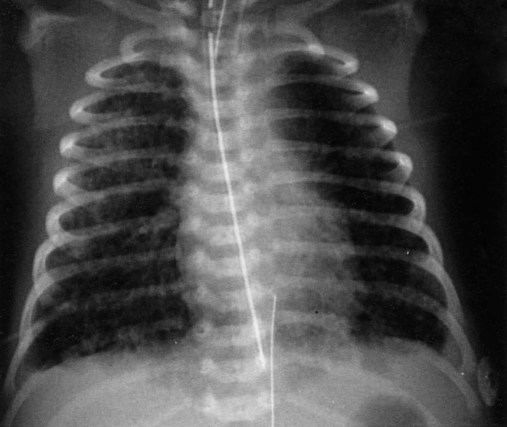
Before birth, thick meconium, fetal tachycardia, and absent fetal cardiac accelerations during labor are evidence that the fetus is at high risk of MAS.38 After delivery, if the infant has a low umbilical artery pH, an Apgar score less than 5, and meconium aspirated from the trachea, intensive care and close observation for MAS are warranted. Infants with MAS typically have gasping respirations, tachypnea, grunting, and retractions. The chest radiograph usually shows irregular pulmonary densities, which represent areas of atelectasis, and hyperlucent areas, which represent hyperinflation secondary to air trapping (Figure 31-4). Arterial blood gases typically show hypoxemia with mixed respiratory and metabolic acidosis. In the most severe cases, there is right-to-left shunting and persistent pulmonary hypertension.30
Treatment
It is no longer recommended that vigorous infants with meconium-stained fluid be intubated and suctioned.31,39–41 However, it is important that an endotracheal tube be inserted immediately in severely depressed infants with thick meconium, and suction should be applied directly to the endotracheal tube.40 The endotracheal tube is removed and inspected for meconium. If meconium is present, the procedure is repeated with a new endotracheal tube until no further meconium is aspirated or until two to four aspirations have been performed. The endotracheal tube should be left in place, and mechanical ventilation should be started. For prevention of hypoxemia, a flow of warmed 100% O2 should be blown across the infant’s face during the aspiration efforts. No evidence suggests an improved outcome because of endotracheal suctioning in the care of infants who have meconium and are vigorous and would not otherwise require intubation.42,43 There is evidence that tracheal lavage with dilute surfactant improves the clinical course and outcome of infants with MAS.44–46
If the infant’s condition worsens, CPAP or mechanical ventilation may be indicated. CPAP is indicated if the primary problem is hypoxemia. By distending the small airways, CPAP can sometimes overcome the ball-valve obstruction and improve both oxygenation and ventilation. If respiratory acidosis is severe or clinical assessment indicates excessive work in breathing, mechanical ventilation should be started. Figure 31-3 shows the ball-valve effect. At rest, the airway lumen is partially obstructed. With inspiration, negative intrathoracic pressure opens the airway and relieves the obstruction. Gas enters and expands the alveoli. With expiration, intrathoracic pressure changes to a positive force, which narrows the airway and causes total occlusion. Gas cannot be expelled and is trapped within the alveoli. It is difficult to provide ventilation to infants with severe MAS. These infants often retain CO2 and need increased ventilator support. Because of high airways resistance, the lungs have a long time constant. High ventilator rates and pressures increase the risk of air trapping and volutrauma.
Evidence suggests that both HFV and synchronous intermittent mechanical ventilation decrease the risk of air leak.47 Various studies have shown improvement in MAS with the use of HFV and surfactant.48 Nitric oxide has become a major adjunct in the management of persistent pulmonary hypertension.49 Corticosteroids have not yet been shown to improve outcomes for infants with MAS.50 High mean airway pressures may worsen pulmonary hypertension and aggravate right-to-left cardiac shunting.38
Mini Clini
Meconium Aspiration Syndrome
 Problem
ProblemThe RT should have the standard resuscitation equipment available. Current recommendations for resuscitating a newborn with meconium staining do not include immediate intubation and tracheal suctioning for a vigorous infant. If the infant is depressed and not breathing, the infant should be resuscitated similar to any other depressed and apneic infant, which includes intubation. However, there is no evidence for whether the depressed infant would benefit from intubation and tracheal suctioning.43
Bronchopulmonary Dysplasia
Background
Infants, especially preterm infants, with severe respiratory failure in the first few weeks of life may develop a chronic pulmonary condition called bronchopulmonary dysplasia (BPD). BPD is a complex disease that is poorly defined.51–54 Historical definitions have included radiographic patterns and the requirement for supplemental O2 at fixed time points in the infant’s life. Immaturity, genetics, malnutrition, O2 toxicity, and mechanical ventilation all have been implicated in the origin of BPD.51,55–62
Pathophysiology
The development of BPD is complex and involves many pathways. The initiating factors are related to atelectrauma (lung collapse) and volutrauma (large tidal volume [VT]). Factors such as hyperoxia and hypoxia, mechanical forces, vascular maldevelopment, inflammation, nutrition, and genetics contribute to the abnormal development of the lung and lead to BPD.56,59,63–67 Atelectrauma is a term coined to describe loss of alveolar volume that is both a consequence and a cause of lung injury. Volutrauma is the term used to describe local overinflation (and stretch) of airways and alveoli. Atelectrauma leads to derecuitment (e.g., areas of alveolar collapse) of the lung. Volutrauma leads to damage to airways, pulmonary capillary endothelium, alveolar and airway epithelium, and basement membranes. The combination of atelectrauma and volutrauma synergistically increases lung injury.
The response of the lungs to the combination of trauma and O2 toxicity is the production and release of soluble mediators. These mediators probably are released from granulocytes residing in the lung. The release of these mediators can injure the alveolar-capillary barrier and induce an inflammatory response.62 A “new” BPD is being described that shows decreased alveolarization rather than the prominent airway damage of the “old” BPD. This change in the pathologic characteristics of BPD is thought to be related to improvements in ventilator management, the use of surfactant, and processes that interrupt alveolar development (e.g., postnatal steroid therapy).54,68–70 Some authors speculate that the “new” BPD and “old” BPD are the same disease. The difference is that clinicians are better at performing mechanical ventilation in infants and do less damage to the airway compared with 20 years ago.71
Clinical Manifestations
BPD has various clinical manifestations. Some very immature infants may start with little or no O2 requirement and little or no mechanical ventilation requirement. Progressive respiratory distress develops at approximately 2 to 3 weeks of life, and then the infant needs O2 and mechanical ventilation. Other immature infants may begin with pneumonia or sepsis and need very high levels of O2 and mechanical ventilation. In either of these scenarios, progressive vascular leakage and areas of atelectasis and emphysema develop in the lungs, and progressive pulmonary damage occurs. The chest radiograph in severe disease shows areas of atelectasis, emphysema, and fibrosis diffusely intermixed throughout the lung (Figure 31-5).67,72 Arterial blood gas measurements reveal varying degrees of hypoxemia and hypercapnia secondary to airway obstruction, air trapping, pulmonary fibrosis, and atelectasis. There is a marked increase in airway resistance with an overall decrease in lung compliance.
Treatment
Multiple pharmacologic treatments have been advocated for infants with BPD.73 Diuretics are given as needed to decrease pulmonary edema; antibiotics are given to manage existing pulmonary infection.74 Chest physical therapy may help mobilize secretions and prevent further atelectasis. Bronchodilator therapy may be useful in decreasing airway resistance.75 Steroid therapy with dexamethasone can produce substantial short-term improvement in lung function, often allowing rapid weaning from ventilatory support. However, steroid therapy has little effect on long-term outcome such as mortality and duration of O2 therapy.76,77 Steroid therapy also has been implicated in decreased alveolarization and increased developmental delay.78 Although steroids are still given in clinical practice, they should be used cautiously and only after the risks have been thoroughly explained to the parents.
Control of Breathing
Apnea of Prematurity
Background
Apnea of prematurity is a common, controllable disorder among premature infants. It usually resolves over time.79–84 Premature infants frequently have periodic respiration, which comprises sequential short apneic episodes of 5 to 10 seconds followed by 10 to 15 seconds of rapid respiration. Apneic spells are abnormal if (1) they last longer than 15 seconds; or (2) they are associated with cyanosis, pallor, hypotonia, or bradycardia.
If no effort to breathe occurs during a spell, the apnea is called central apnea. If breathing efforts occur, but obstruction prevents airflow, the apnea is termed obstructive. Mixed apnea is a combination of the central and obstructive types that starts as obstructive apnea and then develops into central apnea.79–83,85
Etiology
Premature infants have immature control of respiratory drive in response to oxygen and carbon dioxide. In mature animals, an increase in alveolar PaCO2 elicits an increase in VT and respiratory rate. A decrease in FiO2 below room air also triggers an increase in VT. Conversely, in premature animals, an increase in PaCO2 temporarily increases VT but does not increase respiratory rate. A decrease in FiO2 below room air decreases VT and respiratory rate. This effect can lead to apnea in a premature infant. Several factors in addition to prematurity can cause apnea in infants. Table 31-2 summarizes the potential causes, associated signs, and diagnostic indicators.86
TABLE 31-2
Evaluation of an Infant With Apnea
| Possible Cause | Associated Signs | Investigation |
| Infection | Lethargy, respiratory distress, temperature instability | Complete blood count, sepsis evaluation |
| Metabolic disorder | Poor feeding, lethargy, jitteriness | Glucose, calcium, electrolyte levels |
| Impaired oxygenation | Respiratory distress, tachypnea, cyanosis | O2 monitoring, arterial blood gases, chest radiograph |
| Maternal drugs | Maternal history, hypotonia, central nervous system depression | Magnesium level, urine drug screen |
| Intracranial lesion | Abnormal neurologic findings, seizures | Cranial ultrasonography |
| Environmental | Lethargy | Monitor temperature (infant and environment) |
| Gastroesophageal reflux | Feeding difficulty | Specific observation, radiographic barium swallow examination |
From Stark AR: Disorders of respiratory control in infants. Respir Care 36:673, 1991.
Treatment
Infants with apnea need continuous monitoring of heart and respiratory rates. Continuous noninvasive monitoring of oxygenation by transcutaneous electrode or pulse oximetry is recommended. Most apneic incidents can be quickly terminated with gentle mechanical stimulation, such as picking the infant up, flicking the sole of the foot, or rubbing the skin.79,81,82,85,87 If the cause of apnea is not prematurity, treatment must be directed at resolving the underlying condition. Table 31-3 outlines current treatment strategies for infants with apnea.86 Apnea secondary to prematurity responds well to methylxanthines, especially theophylline and caffeine.82,85,87 These agents stimulate the central nervous system and increase the infant’s responsiveness to CO2. For infants with apnea that is refractory to treatment with theophylline, doxapram can be used.88–90 However, doxapram is delivered by continuous infusion and has multiple toxicities.
TABLE 31-3
Treatment Strategies for Infants With Apnea
| Treatment | Rationale |
| Manage underlying cause if identified | Removes precipitating factor |
| Tactile stimulation | Increases respiratory drive by sensory stimulation |
| CPAP | Reduces mixed and obstructive apnea by splinting the upper airway |
| Theophylline or caffeine | Increases respiratory center output and CO2 response, enhances diaphragm strength, adenosine antagonist |
| Doxapram | Stimulates respiratory center and peripheral chemoreceptors |
| Transfusion | Decreases hypoxic depression by increasing O2-carrying capacity |
| Mechanical ventilation | Provides support when respiratory effort is inadequate |
From Stark AR: Disorders of respiratory control in infants. Respir Care 36:673, 1991.
CPAP can also be used to manage infant apnea.91 Although the mechanism of action is not established, CPAP probably increases FRC and improves arterial partial pressure of oxygen (PaO2) and PaCO2. CPAP may also stimulate vagal receptors in the lung, increasing the output of the brainstem respiratory centers. Severe or recurrent apnea that is unresponsive to these interventions may necessitate mechanical ventilatory support.
Apnea monitoring can allow infants who are otherwise ready for discharge but still having occasional episodes of apnea to go home.79,81,85,92–95 However, the presence of a home apnea monitor is a significant inconvenience to the family. Home monitors lack the sophisticated filtering systems of hospital monitors, and they have very frequent false alarms.
Pulmonary Vascular Disease
Persistent Pulmonary Hypertension of the Newborn
Background
Persistent pulmonary hypertension of the newborn (PPHN) is a complex syndrome with many causes.96 The common denominator in PPHN is a return to fetal circulatory pathways, usually because of elevated PVR. This condition results in further right-to-left shunting, severe hypoxemia, and metabolic and respiratory acidosis.
Pathophysiology
There are three fundamental types of PPHN: vascular spasm, increased muscle wall thickness, and decreased cross-sectional area of pulmonary vessels.97 Vascular spasm is an acute event that can be triggered by many different conditions, including hypoxemia, hypoglycemia, hypotension, and pain. Increased muscle wall thickness is a chronic condition that develops in utero in response to several different etiologic factors, including chronic fetal hypoxia, increased pulmonary blood flow (e.g., intrauterine closure of the ductus arteriosus), and pulmonary venous obstruction (e.g., total anomalous pulmonary venous return with obstructed below-diaphragm return). Decreased cross-sectional area is related to hypoplasia of the lungs and occurs with congenital diaphragmatic hernia, Potter sequence (absent kidneys), and oligohydramnios syndromes (decreased amniotic fluid).
Treatment
Initial therapy for PPHN is removal of the underlying cause, such as administration of O2 for hypoxemia, surfactant for RDS, glucose for hypoglycemia, and inotropic agents for low cardiac output and systemic hypotension. If correction of the underlying problem does not correct hypoxemia, the infant needs intubation and mechanical ventilation. Because pain and anxiety may contribute to PPHN, the infant may need sedation and, frequently, paralysis. If these measures do not improve oxygenation, the next step is HFV. This mode of ventilation allows a higher FRC without a large VT. Inhaled nitric oxide is considered the next intervention.98–100 If all of these modalities fail to improve oxygenation, the infant may be a candidate for extracorporeal membrane oxygenation (ECMO).99,101–103 Even with all of these therapeutic modalities, PPHN remains a complex disease with high morbidity.
Congenital Abnormalities Affecting Respiration
Airway Diseases
Airway disruptions usually are related to tracheoesophageal fistula (TEF) in a newborn. This malformation usually is associated with esophageal atresia. There are five types of TEF: esophageal atresia with a proximal fistula, esophageal atresia with a distal fistula, esophageal atresia with both a proximal and a distal fistula, esophageal atresia without either fistula, and an intact esophagus with a so-called H fistula.104 The most common of these malformations is esophageal atresia with a distal fistula, which accounts for 85% to 90% of all TEFs. The least common is the H fistula. All of these malformations manifest as difficulty swallowing, bubbling and frothing at the mouth, and choking, in particular, during attempts at feeding. These anomalies can occur in isolation or as part of an association of defects. The most common is the VATER or VACTERL association of vertebral anomalies, imperforate anus, tracheoesophageal fistula, and renal or radial anomalies. In VACTERL, cardiac anomalies are added, and renal and limb anomalies replace renal or radial anomalies in the acronym. These associated anomalies must be sought in any infant with TEF. TEF is managed with surgical ligation of the fistula and reconnection of the interrupted esophagus.105 Most infants with TEF have a good outcome; however, some infants have severe malformations that can cause chronic problems. Infants with TEF usually need only supportive respiratory care. They usually do not have lung disease. However, some infants need HFV because the air leak through the fistula can become larger than the airflow to the alveoli.
Lung Malformations
There is a broad spectrum of rare lung malformations that occur in the newborn period.106–108 These lesions are thought to be part of a continual spectrum of diseases that originate as defects in lung segmentation. The most common is congenital pulmonary adenomatoid malformation (CPAM); this was previously known as cystic adenomatoid malformation of the lung. CPAM is classified into five types on the basis of the type and size of the cyst.109,110 The disease may affect entire lobes of the lung. The affected parts of the lung do not exchange gas and can become infected. The usual treatment is surgical removal of the affected lobe. There is also the potential for malignant transformation.
Congenital Diaphragmatic Hernia
Congenital diaphragmatic hernia is a severe disease that usually manifests in newborns as severe respiratory distress. The pathophysiologic mechanism is a complex combination of lung hypoplasia, including decreased alveolar count and decreased pulmonary vasculature; pulmonary hypertension; and unusual anatomy of the inferior vena cava.111 This disorder varies between asymptomatic (rare) and severe life-threatening disease (frequent). There are two types of hernia: Bochdalek hernia (lateral and posterior defect, usually on the left) and Morgagni hernia (medial and anterior, may be on either side). Hernias that occur in the right hemidiaphragm may be less severe because the liver can block the defect and decrease the volume of abdominal contents that can enter the thorax.112
The general treatment of infants with congenital diaphragmatic hernia involves neonatologists and pediatric surgeons. Initial treatment is insertion of an endotracheal tube, paralysis, and mechanical ventilation. A large sump tube is placed in the stomach and connected to continuous suction. These therapies allow adequate ventilation and oxygenation and prevent gas insufflation of the intestine. Most centers delay surgical repair for several days to allow the natural decrease in PVR. On day 7 to 10 of life, a surgeon closes the defect. This scenario occurs only for infants with easily correctable pulmonary hypertension. Infants with severe pulmonary hypertension may need HFV and ECMO. At some centers, the diaphragm is repaired during ECMO. Most centers try to wean the infant from ECMO and then perform the repair. Despite all these advanced therapies, the mortality for this disease is high.113 Survival depends on many complex variables (e.g., liver herniation into the thorax, fetal head-to-lung ratio, initial PaO2 and PaCO2).113,114
Abdominal Wall Abnormalities
Because all newborns are primarily abdominal breathers, the abdominal wall is an intrinsic part of the respiratory system. Large defects in the abdominal wall can cause severe respiratory compromise.115 One of the most common of these defects is omphalocele. An omphalocele is an abdominal wall defect that involves the insertion of the umbilical cord. The umbilical cord goes into the omphalocele. The bowel of an infant with an omphalocele is usually covered by a membrane that looks like the surface of the umbilical cord. Occasionally, the omphalocele membrane ruptures and exposes the bowel of the infant. Omphaloceles must be distinguished from gastroschisis. Gastroschisis is an abdominal wall defect that is completely separate from the insertion of the umbilical cord. The bowel of an infant with a gastroschisis is not covered by a membrane. Usually only large omphaloceles cause respiratory distress. When they are greater than 10 cm in diameter, these defects can cause severe respiratory distress and frequently necessitate prolonged mechanical ventilation.
 Problem
ProblemNeuromuscular Control
Many diseases of poor neuromuscular control affect newborns,116–119 including spinal muscular atrophy, congenital myasthenia gravis, and myotonic dystrophy. These diseases frequently necessitate respiratory support in the newborn and pediatric periods. The morbidity and mortality of these diseases are extremely variable. New technologies may allow noninvasive respiratory support of some patients.120 Some diseases can be quite severe in the newborn period and be relieved with age. It is important to make an accurate diagnosis to be able to estimate prognosis and to provide genetic counseling. Many of these diseases are inherited with known inheritance patterns.
Congenital Heart Disease
A full discussion of congenital heart disease is beyond the scope of this chapter. However, basic knowledge of the common defects is essential to good practice in pediatric and neonatal respiratory care. Congenital heart diseases usually are divided into two large categories: cyanotic and acyanotic heart disease. Cyanotic heart diseases are diseases in which blood shunts from right to left, bypassing the lungs, and is deoxygenated. Acyanotic heart diseases are diseases in which blood shunts from left to right causing congestive heart failure. Figure 31-6 compares normal cardiac anatomy with the features of the five most common congenital defects.
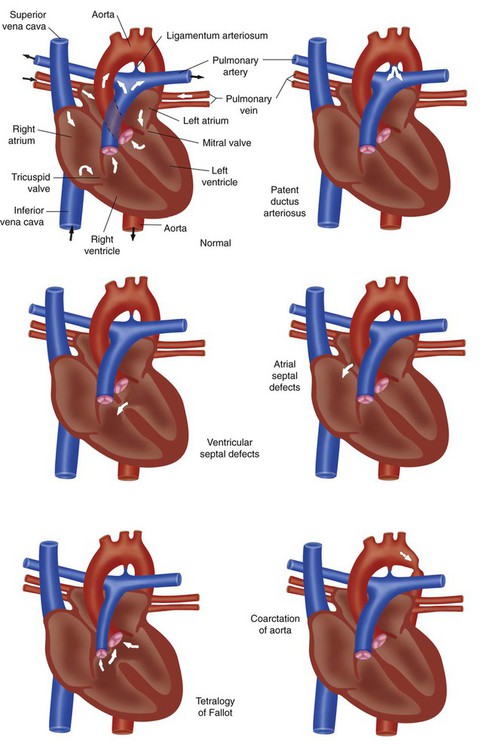
Cyanotic Heart Diseases
Tetralogy of Fallot
Tetralogy of Fallot is a defect that includes (1) obstruction of right ventricular outflow (pulmonary stenosis), (2) ventricular septal defect (a hole between the right and left ventricles), (3) dextroposition of the aorta, and (4) right ventricular hypertrophy. Tetralogy of Fallot varies between mild disease, which is initially diagnosed in early childhood, and severe disease, which is diagnosed in the newborn period.121–123 The mild form of the disease manifests as a heart murmur, intermittent severe cyanotic spells, a history of the infant squatting or entering a knee-chest position, or a combination of these features. The severe form of the disease manifests as a heart murmur and severe continuous cyanosis. Most types of tetralogy of Fallot can be managed surgically. All infants with tetralogy of Fallot should be evaluated for deletions on chromosome 22 (22q11).124 The type and timing of the surgery depend on the anatomy of the defects. Children with this defect are at increased risk of sudden death from arrhythmia later in life.
Transposition of the Great Arteries
Transposition of the great arteries is the heart disease that most frequently causes severe cyanosis.125–128 It usually manifests as moderate to severe cyanosis immediately after birth. A murmur may be present. Infants with this abnormality frequently need emergency atrial septostomy (cutting a hole in the wall between the two atria). This procedure historically has been performed in heart catheterization laboratories. Many pediatric cardiologists who perform invasive procedures have begun performing this procedure with ultrasound guidance in the neonatal intensive care unit. The condition of infants who need atrial septostomy usually stabilizes. The goal is to allow PVR to decrease and then to perform the arterial switch operation in week 2 or 3 of life.
 Rule of Thumb
Rule of ThumbAcyanotic Heart Diseases
 Problem
ProblemPatent Ductus Arteriosus
In a fetus, most of the pulmonary blood flow is shunted through the ductus arteriosus to the aorta. Closure of the ductus normally occurs 5 to 7 days after birth of a term infant. Patent ductus arteriosus usually is a disease of immature, preterm infants. Factors altering pressure gradients or affecting smooth muscle contraction can cause the ductus not to close or to reopen after it has closed. Depending on the pressure gradients established, shunting through an open ductus may be either right to left (pulmonary pressure greater than aortic) or left to right (aortic pressure greater than pulmonary). Treatment is either pharmacologic (indomethacin) or surgical (ligation). In recent years, the best timing of treatment and the treatment mechanism have become quite controversial.129
Left Ventricular Outflow Obstructions
Hypoplastic left heart syndrome (Figure 31-7), interrupted aortic arch, and coarctation of the aorta have in common obstruction of left ventricular outflow.130–132 They all manifest in the newborn period with symptoms of acute heart failure. Systemic blood flow depends on patency of the ductus arteriosus. When the ductus spontaneously closes (usually at 5 to 7 days of age), severe congestive heart failure develops. The symptoms range from moderate respiratory distress to complete cardiovascular collapse. Initial treatment is intravenous administration of prostaglandin E1. Most infants with these defects need support with mechanical ventilation. These infants do not have lung disease. The pressures and rates used should be set appropriately.
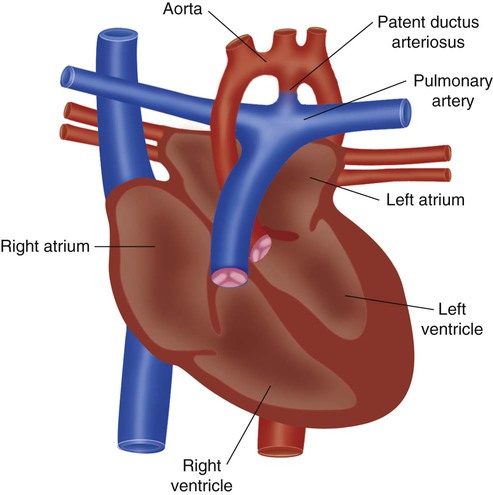
There are standard surgical repairs for both interrupted aortic arch and coarctation of the aorta. Hypoplastic left heart syndrome has several accepted treatments, including a palliative surgical procedure (Norwood) and transplantation.130–132 Neither the Norwood procedure nor transplantation is ideal, and each option has significant associated problems. The decision must be made in consultation with the family.
Neonatal Resuscitation
Resuscitation of a newborn is a subset of resuscitation techniques. Most infant resuscitations occur in the delivery room. Although these resuscitations can range from minimal intervention to full resuscitation, more than 90% of them can be successfully dealt with by stimulation, ensuring the presence of an airway, and providing breathing support.133–135 RTs are a vital part of any resuscitation team. Their expertise in establishing and supporting an airway and initiating respiratory support is essential. It is beyond the scope of this chapter to delineate the guidelines of neonatal resuscitation. The reader should refer to the neonatal resuscitation guidelines published by the American Academy of Pediatrics (AAP).133–139
Pediatric Respiratory Disorders
Sudden Infant Death Syndrome
Sudden infant death syndrome (SIDS) is the leading cause of death (40%) among infants younger than 1 year in the United States. Approximately 7000 infants die of SIDS each year in the United States.83,92,140 A presumptive diagnosis is based on the conditions of death in which a previously healthy infant dies unexpectedly, usually during sleep. Autopsy shows that many infants who die of SIDS have evidence of repeated episodes of hypoxemia or ischemia. Factors associated with increased frequency of SIDS are presented in Box 31-1. If the infant is found and resuscitation is successful, the diagnosis would be apparent life-threatening event.
Etiology
The cause of SIDS is unknown. Apnea of prematurity is not a predisposing factor, and there is no evidence that immaturity of the respiratory centers is a cause. Although infants in families in which two or more SIDS deaths have occurred are at slightly higher risk, there is no evidence of a genetic link. The best knowledge of SIDS comes from population or epidemiologic studies and is summarized in Box 31-2. An infant who dies of SIDS typically is a preterm African-American boy born to a poor mother younger than 20 years who received inadequate prenatal care. Infants 1 to 3 months old are most susceptible, and death is most likely to occur at night during the winter. The risk of SIDS also is high among infants who previously experienced an apparent life-threatening event. Such an event occurs when an infant becomes apneic, cyanotic, or limp enough to frighten the parent or caregiver. The prone sleeping position has been strongly associated with increased risk of SIDS. It is difficult to differentiate death of SIDS from death of intentional suffocation. The possibility of intentional suffocation must be investigated but with great sensitivity.141
Prevention
Because of the unknown causation and unexpected occurrence, there is no therapy for SIDS. Prevention is the goal. Successful prevention requires that infants at high risk be identified through a history of risk factors and documented monitoring or event recording. After identification that an infant is at risk, the family is trained in apnea monitoring and cardiopulmonary resuscitation (CPR). The AAP recommends placing infants in either the supine or the side-lying position for the first 6 months of life and reducing soft objects in the infant’s sleeping environment.92,140–142 To define the need and appropriate approach for home monitoring of infants, the AAP has developed a policy statement on infantile apnea and home monitoring.92 The AAP recommendations for the need for and use of home monitoring are summarized in Box 31-3.
Gastroesophageal Reflux Disease
Gastroesophageal reflux disease (GERD) is the regurgitation of stomach contents into the esophagus and is common in childhood. Some causes of GERD are not pathologic. There is general agreement that there are important interactions between GERD and various disorders of the respiratory system.143 Respiratory problems caused by gastroesophageal reflux include reactive airways disease, aspiration pneumonia, laryngospasm, stridor, chronic cough, choking spells, and apnea.143–147 GERD should be considered when an infant has faced a sudden life-threatening event and when an older child has unexplained chronic head and neck problems. GERD can be diagnosed with esophageal pH testing, upper gastrointestinal contrast studies, and gastric scintiscan. When GERD has been diagnosed, medical therapy can begin.148–150 Occasional cases that do not respond to medical management may require surgical intervention.
Bronchiolitis
Bronchiolitis is an acute infection of the lower respiratory tract, usually caused by respiratory syncytial virus (RSV). Nearly 1 in 10 infants younger than 2 years acquires a bronchiolitis infection. The outcome is generally good, although approximately 1% of infants hospitalized for bronchiolitis die of respiratory failure. Infants most prone to respiratory failure as a consequence of bronchiolitis are very young and immunodeficient and have comorbidity, such as congenital heart disease, BPD, CF, or childhood asthma.151–155
Clinical Manifestations
Prophylaxis
In recent years, passive immunization for RSV has become available.156–158 Initially, passive immunization was recommended only for preterm infants with BPD. However, passive immunization is now recommended for infants younger than 2 years who require medical therapy for chronic lung disease, infants born at less than 32 weeks’ gestational age, and infants with congenital heart disease who have cardiovascular compromise (Box 31-4).156–160
Treatment
Treatment of a patient with bronchiolitis varies with the severity of the infection and the clinical signs and symptoms. Many patients can be treated at home with humidification and oral decongestants. Patients with more severe symptoms (apnea) and comorbidity usually are hospitalized, and treatment is directed at relieving the airway obstruction and associated hypoxemia. Hospitalized children frequently are treated with systemic hydration and O2 hood, croup tent, or nasal cannula and assisted with airway clearance.154,155,161–163 Antibiotics may be administered to control secondary bacterial infections. If bronchiolitis progresses to acute respiratory failure, mechanical ventilation is required. Because of the obstructive nature of this disorder, low respiratory rates and long expiratory times may be needed to prevent air trapping. Heliox has been used for severe airways disease requiring mechanical ventilation.164 Vigorous bronchial hygiene, occasionally including tracheobronchial aspiration, usually is needed to maintain a patent airway.
Croup
Clinical Manifestations
Symptoms become evident after 2 or 3 days of nasal congestion, fever, and coughing. A child typically has slow, progressive inspiratory and expiratory stridor and a barking cough. As the disease progresses, dyspnea, cyanosis, exhaustion, and agitation occur. A radiograph of the upper airway is helpful in confirming the diagnosis and ruling out epiglottitis but is usually not needed in most cases of croup.84 Classic croup is seen on an anteroposterior radiograph as characteristic subglottic narrowing of the trachea, called the steeple sign (Figure 31-8).
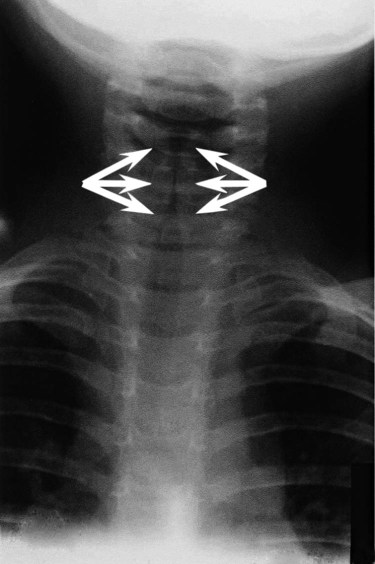
Treatment
The evaluation and treatment of a child with croup must focus on the degree of respiratory distress and associated clinical findings. If stridor is mild or occurs only on exertion and cyanosis is not present, hospitalization is generally not required, and the child is treated at home. If there is stridor at rest (accompanied by harsh breath sounds, suprasternal retractions, and cyanosis with breathing of room air), hospitalization is indicated. The traditional treatment of a child with mild to moderate croup has involved cool mist therapy with or without supplemental O2. However, there is no evidence that this practice is beneficial.165 Corticosteroids and epinephrine have been shown to have the greatest benefit for decreasing the length and severity of respiratory symptoms associated with viral croup.166,167 The addition of budesonide has been shown to reduce the severity of symptoms in mild to moderate cases of croup.166,167 Progressive worsening of the clinical signs despite treatment indicates the need for intubation and mechanical ventilation. Heliox has been used for infants and children with severe disease. However, there is insufficient evidence to show whether this is beneficial.168
Epiglottitis
Epiglottitis is an acute and often life-threatening infection of the upper airway that causes severe obstruction secondary to supraglottic swelling. Evidence suggests that the incidence of epiglottitis is decreasing among children and increasing in adults,169 probably because of the use of vaccines. The most common cause is H. influenzae type B infection. Other organisms that are increasingly found to be causes of acute epiglottitis include group A S. pneumoniae, S. aureus, Klebsiella pneumoniae, Haemophilus parainfluenzae, and beta-hemolytic streptococci (groups A, B, C, and F).170
Clinical Manifestations
A child with epiglottitis usually has a high fever, sore throat, stridor, and labored breathing.170–172 The patient does not have a croupy bark but instead has a muffled voice. Older children may report a sore throat and difficulty swallowing. Difficulty swallowing may cause drooling. Lateral radiographs of the neck (Figure 31-9) show the epiglottis is markedly thickened and flattened (thumb sign) and the aryepiglottic folds are swollen; the vallecula may not be visualized. Visual examination of the upper airway is dangerous in these children and always should be performed in a controlled setting by personnel expert in emergency intubation. Inadvertent traction of the tongue can cause further and immediate swelling of the epiglottis and abrupt and total upper airway obstruction. Children with suspected epiglottitis should be accompanied by personnel expert in emergency intubation during any transport for diagnostic procedures.
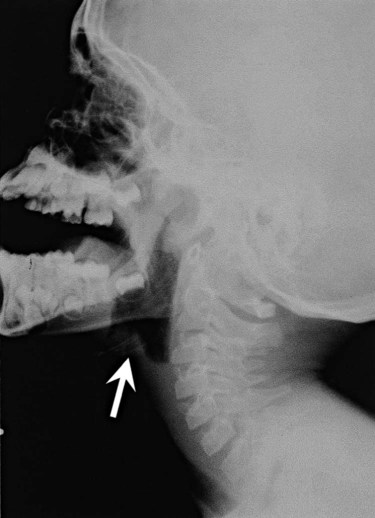
 Problem
ProblemTreatment
Children with epiglottitis need elective intubation under general anesthesia in the operating room. Tracheostomy may be needed if the patient’s condition warrants it; however, this procedure is rarely used. There should be no attempt to lie the child down or attempts to intubate until the child is sedated. Premature attempts at intubation can precipitate acute airway obstruction and respiratory arrest. After an airway is secured, a sample for bacterial culture should be obtained, and antibiotic therapy should be started. Corticosteroids may decrease the swelling.170 Children with an endotracheal tube should be sedated and restrained to prevent inadvertent extubation. Extubation should not be attempted until an upper airway leak is readily detected.
Cystic Fibrosis
Cystic fibrosis (CF) is the most common lethal genetic disease among whites in the United States. It is inherited as an autosomal recessive trait that affects approximately 30,000 persons in the United States.173 The disease is caused by a genetic mutation of the gene coding for a large protein that controls the movement of chloride ions through the cell membrane.174 This protein is called the cystic fibrosis transmembrane conductance regulator (CFTR). Movement of chloride ions is vital to the proper production and regulation of secretions. CFTR can be mutated in more than 1000 different ways to cause CF.175 Some CF mutations cause more severe CFTR dysfunction than others. The variety of mutations that can be inherited explains some of the variability in the severity of clinical CF. In CF, abnormalities of chloride movement through the surface of exocrine glands cause most of the clinical manifestations.
 Rule of Thumb
Rule of ThumbClinical Manifestations
Patients with CF experience abnormalities in nearly all organs with exocrine function. The most severely affected organs are the sweat glands, pancreas, and lungs. Normal sweat is produced as a saline solution that has much of the salt removed on its way to the surface of the skin. Sweat glands of patients with CF are unable to remove salt from sweat properly.176 As a result, the skin of patients with CF develops a salty taste, and they are prone to dehydration during hot weather.177 The sweat chloride test used for diagnosis of the disease is based on the high salt concentration in the sweat of patients with CF.178 Most patients with CF also have exocrine pancreatic insufficiency, which usually starts in infancy. Exocrine pancreatic insufficiency dramatically reduces the number of digestive enzymes, and patients lacking digestive enzymes do not break down large proteins, carbohydrates, and fats for absorption, causing malnutrition and diarrhea. Digestion of fats is particularly compromised in patients with CF. These patients often have deficiencies of the fat-soluble vitamins A, D, E, and K and have large amounts of undigested fat in the stool (steatorrhea).
Diagnosis
The diagnosis of CF is suspected when a patient has clinical manifestations of the disease or has close family members with CF. Findings of recurrent pulmonary infections or failure to grow as rapidly as expected are clinical clues that often prompt physicians to initiate testing for CF. The diagnosis usually is confirmed with the sweat chloride test, which is done with electrical stimulation of the skin to produce sweat (iontophoresis). In a child, a sweat chloride level greater than 60 mEq/L confirms the diagnosis of CF.179
Treatment
The deficiency of pancreatic enzymes that occurs in patients with CF is managed with pancreatic enzyme supplementation. Several important steps are taken to help patients with CF maintain lung function. Regular chest physical therapy improves lung function and clearance of secretions.180 When patients with CF become teenagers or young adults, they may have no partner to assist with chest physiotherapy. For these patients, strenuous exercise, external pneumatic vests,181 PEEP devices, or autogenic drainage may substitute for regular chest physiotherapy. Another mucus clearance adjunct is inhaled recombinant deoxyribonuclease (DNase or DNAase). The routine daily use of inhaled DNase reduces the frequency of respiratory infections.182,183 The daily use of nebulized 7% saline both preserves lung function and decreases the likelihood of bronchiectatic flare-ups.184
Antibiotics directed against the usual infectious organisms are required with each bronchiectatic exacerbation. As CF progresses, the bacteria causing exacerbations can become progressively more resistant to antibiotic treatments. Intravenous antibiotics are often required if a bronchiectatic exacerbation is caused by resistant bacteria. A nebulized form of the antibiotic tobramycin is used to prevent infection. When inhaled tobramycin is used twice daily every other month, there is a marked reduction in the number of bronchiectatic exacerbations.185 The regular use of azithromycin helps preserve lung function and decreases the frequency of pulmonary flare-ups.186
High doses of the antiinflammatory drug ibuprofen reduce the rate of lung function loss in patients younger than 13 years old.187 Many patients with CF have asthma symptoms. These patients benefit from bronchodilators.
Lung transplantation is commonly performed in patients with advanced CF lung disease. Double-lung transplantation is the most commonly used form of lung transplantation in the treatment of patients with CF. New therapies for CF focus on improving the function of specific CFTR mutations.188