[level-membership-for-basic-science-category]
Nasal drug delivery
Gary P. Martin and Alison B. Lansley
Chapter contents
Key points
Introduction
The most common reason for introducing a drug into the nasal cavity is to provide a convenient and accessible route for rapidly and efficiently managing the localized symptoms associated with allergic rhinitis, nasal congestion and nasal infection. Drugs applied topically for such purposes include antihistamines, corticosteroids, sodium cromoglicate, sympathomimetics and antiseptics/antibiotics (Table 38.1). These drugs are administered either in liquid form (from a spray or as drops) or as creams/ointments.
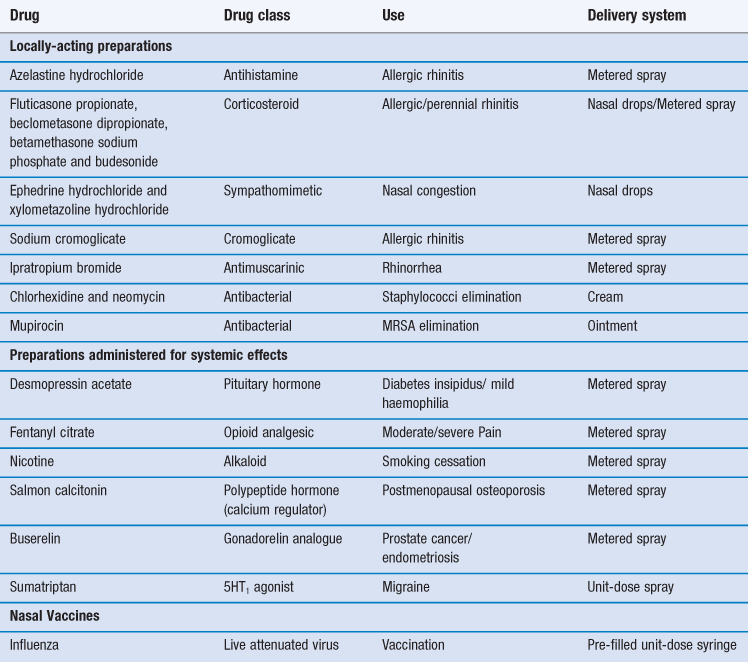
The intranasal route has also been exploited for the delivery of drugs to the systemic circulation (Table 38.1). There are several possible reasons for pharmaceutical companies to consider employing this route for marketed medicines rather than the much more popular (and preferred) oral route. These include:
The nasal cavity has also been utilized, or proposed, as a portal for the delivery of vaccines, particularly for infections associated with the respiratory tract such as influenza and possibly, eventually, tuberculosis. The presentation of an antigen, in combination with an acceptable adjuvant to the nasal-associated lymphoid tissue (NALT) can promote both cellular and humoral responses. However human vaccination need not be restricted to airways infections and systemic immune responses are demonstrable after introduction of appropriate antigens via this route. Indeed, intranasal vaccination has been studied with a view to combating noroviruses, the measles and herpes viruses, diphtheria and tetanus microorganisms. An intranasal vaccine containing live attenuated influenza vaccine has been marketed (Table 38.1).
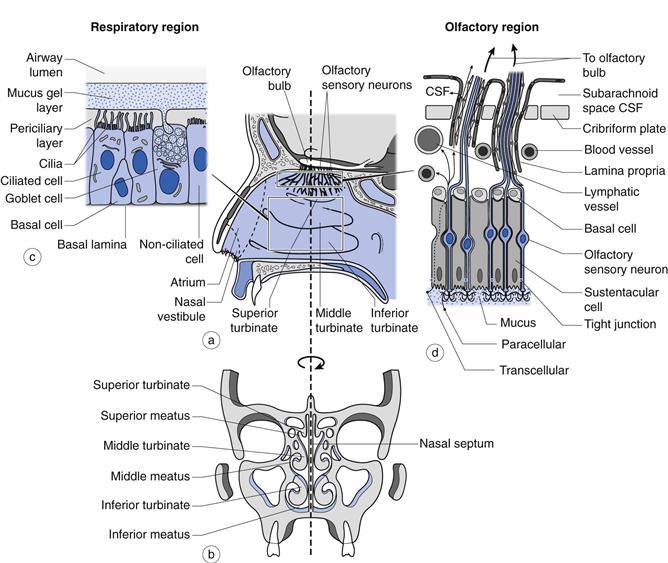
Currently, there is much research interest aimed at establishing whether the olfactory region, positioned in the upper reaches of the nasal cavity (Fig. 38.1), offers a potential means, in humans, of circumventing the obstacles imposed by the blood-brain barrier to the access of many drugs from the bloodstream to the brain. This region contains a direct physiological link between the environment and the central nervous system (CNS). Clearly, if such a route could be established conclusively as being viable for the delivery of therapeutic quantities of drugs in humans, then this would have great potential in treating conditions such as: Alzheimer’s disease, brain tumours, epilepsy, pain and sleep disorders. However, although there are a number of studies that suggest that drugs may be absorbed from the olfactory region, the true significance of many findings are confounded by the use of animal models with the data often being extrapolated uncritically to humans.
So as to appreciate fully the potential of the nasal cavity for drug delivery, it is pertinent to review the relevant anatomy and physiology, consider in more detail the applications of utilising the route, review the physicochemical properties of administered drugs and factors that might affect the choice of formulation and discuss the devices that can be employed to deliver nasal medicines.
Anatomy and physiology
The nasal cavity is 120–140 mm from the nostrils to the nasopharynx (Fig. 38.1) and is divided in two by the nasal septum. The total surface area of both cavities is about 160 cm2 and the total volume is about 15 mL. The first part of the nasal cavity (termed the nasal vestibule) contains the narrowest part of the nasal cavity with a cross-sectional area of 30 mm2 on each side. The lining of the vestibule changes from skin at the entrance, to a stratified squamous epithelium which extends over the anterior third of the entire nasal cavity. The nasal vestibule contains vibrissae (hairs) which filter out inhaled particles with an aerodynamic particle size greater than approximately 10 µm. Progression through the nasal cavity leads to the turbinate region. The turbinates are convoluted projections from the nasal septum which are lined with a pseudostratified columnar epithelium (80–90% of the total surface area of the nasal epithelium in man) composed of mucus-secreting goblet cells, ciliated and non-ciliated cells and basal cells (Fig. 38.1). The apical surfaces of the ciliated and non-ciliated cells are covered with non-motile microvilli, which serve to increase the surface area of the epithelial cells. There are also approximately 100 motile cilia on each ciliated cell which are responsible for mucus transport. Serous and seromucous glands also contribute to nasal secretions. As air moves through the turbinate region via the meatuses (Fig. 38.1), the low rate of airflow in combination with the turbulence created by the shape of the turbinates encourages the air to make contact with the highly vascularized walls, enabling it to be warmed and humidified. Particulates (5–10 µm) within the airstream, such as dust, pollen, microorganisms and pollutants have the potential to deposit on the viscoelastic mucous gel lining the turbinate walls. The cilia, beating within the periciliary fluid, engage with the underside of the mucus and propel the gel and the deposited particles to the nasopharynx, where they are either swallowed or expectorated. This process is termed mucociliary clearance and is able to clear mucus at a rate of about 7 mm min−1. About 20% of the inspired air is directed to the top of the turbinates where the olfactory region is located (Fig. 38.1). This is an area approximately 12.5 cm2 (~ 8% of the total surface area of the nasal epithelium in man) of non-ciliated pseudostratified columnar epithelium traversed by 6–10 million olfactory sensory neurons which pass from the nasal cavity, between the epithelial (sustentacular) cells and through the cribriform plate to the olfactory bulb of the brain.
Drug delivery
Certain constraints are imposed upon formulating preparations for the nasal route and two case studies, one a locally-acting drug (budesonide) and a second systemically-acting peptide drug (calcitonin) are given in Table 38.2.
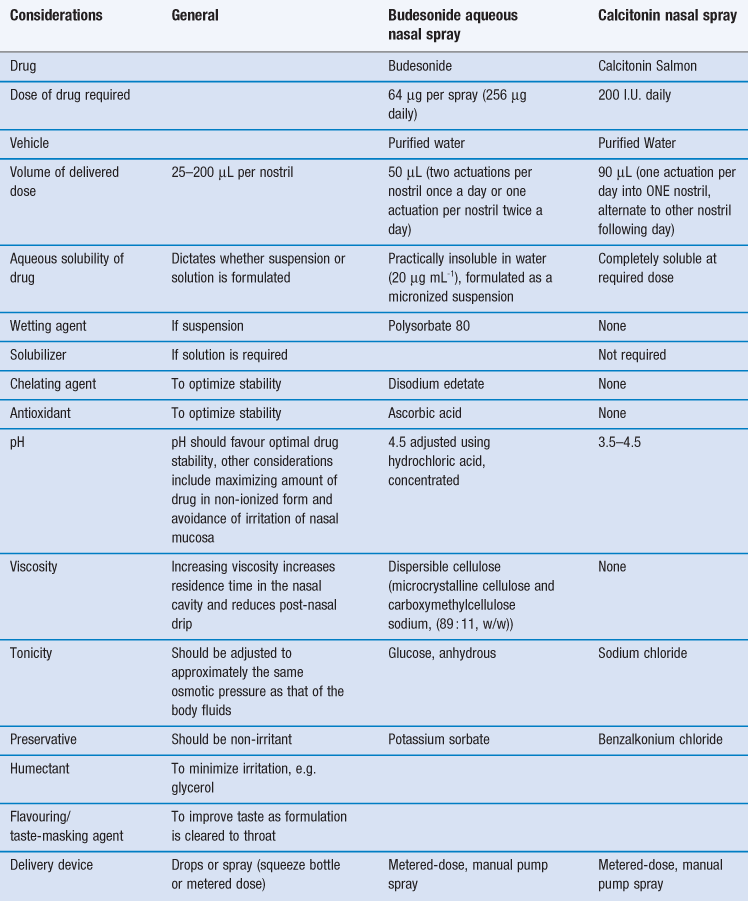
As with the formulation of any medicine, the information garnered from pre-formulation studies (Chapter 23) is an essential prerequisite in the design of an intranasally delivered medicine. The solubility (Chapter 2) of the drug to be administered is a key determinant in the final formulation. The restricted volume that can be applied to the nasal cavity also impacts upon the nature of the resultant formulation. Generally, the premise of presenting the drug in the simplest formulation, containing the fewest excipients possible to ensure a stable medicine with an adequate shelf-life is the course that should be followed in the development process. Currently, delivery devices are usually metered-dose manual pump sprays, since these are cheap, robust and reliable but more sophisticated systems are now under development, as discussed below.
Local delivery
For conditions affecting the nose, it is logical to deliver the drug directly to its site of action. This permits the rapid relief of symptoms with a much lower dose of drug than would be necessary if it were delivered by the oral route, and reduces the chance of systemic side effects. For example, this is particularly pertinent when delivering corticosteroids to reduce local inflammation of the nasal mucosa and sinuses, without causing pituitary-adrenal suppression, or alternatively when using localized antihistamine therapy without inducing drowsiness.
Systemic delivery
The rationale for the use of the nasal cavity for systemic delivery includes its accessibility, avoidance of pre-systemic metabolism and potential to provide a rapid onset of action. Its use for peptides (Tables 38.1 and 38.2) has been successful since, although only a very low percentage of administered drug is absorbed (i.e. low bioavailability), the attained plasma levels are sufficient for therapeutic efficacy. Many of the marketed nasally administered peptides have wide therapeutic windows. Therefore, providing the minimum therapeutic level is exceeded in the bloodstream, a large variability in the final attained plasma level can be tolerated, without systemic toxicity becoming manifest. Any potential localized toxicity can be minimized in chronic administration by alternating nostrils when daily dosing (Table 38.2). Intranasal delivery can also be useful in emergency situations, such as in the treatment of opioid overdose (using naloxone) or in the treatment of intractable childhood seizures (using benzodiazepines). Drug delivery via this route is also well-suited to drugs that, when administered orally, cause emesis e.g. galantamine used to treat dementia.
Anatomical and physiological factors affecting intranasal systemic delivery
For a drug molecule to enter the systemic circulation it must first be absorbed across the nasal epithelium.
This may occur via the mechanisms of passive diffusion via the transcellular or paracellular routes (Chapter 19). The transcellular pathway is the principal route of absorption for lipophilic molecules, while small, hydrophilic molecules diffuse between the epithelial cells (paracellularly) via the tight junctions which are dynamic structures responsible for the integrity of the nasal epithelium. This latter pathway avoids the need for the drug molecules to partition into and out of the lipophilic membrane of the epithelial cells, but imposes a size restriction of between 0.39–0.84 nm. Transcellular absorption can also occur via endocytosis, the route exploited by large hydrophilic molecules (>1 kDa), and via active transport mechanisms where drug molecules with a similar structure to a natural substrate can interact with a carrier protein to cross the epithelial cells.
Since most drug absorption takes place by passive diffusion, the relatively large surface area of the nasal cavity and its rich blood supply (which helps to maintain the concentration gradient across the epithelium) aid this process. Working against these positive attributes of the nasal cavity are the barriers presented by mucus and the epithelium itself and the nasal clearance mechanisms including mucociliary clearance and metabolism. The advantages and disadvantages of the nasal cavity for systemic drug delivery are summarized in Table 38.3.
Table 38.3
Advantages and disadvantages of intranasal drug delivery for systemic activity
| Advantages | Disadvantages |
| Large surface area for absorption (approximately 160 cm2) | Limited to small delivery volumes (25–200 µL) therefore require potent drugs |
| Good blood supply and lymphatic system | Mucociliary clearance, mucus barrier |
| Avoids hepatic first-pass metabolism | Enzymatic activity (pseudo first-pass effect) |
| Epithelium is permeable to small, lipophilic drug molecules; rapid absorption and onset of action | Low epithelial permeability for hydrophilic drugs; require absorption enhancers and large doses |
| Non-invasive, so minimal infection risk during application and low risk of disease transmission (unlike parenteral route) | |
| Easy to self-administer and adjust dose |
Mucociliary clearance
The main drug absorption site is the respiratory epithelium of the nasal turbinates, which is where mucociliary clearance dominates. Drug deposited anterior to this region will remain in the nasal cavity for longer than drug deposited in the turbinates, but absorption from this site is less. Once drug particles (if formulated as a suspension) or molecules (if in solution) find their way on to the mucociliary ‘conveyor belt’ they will be cleared from the nasal cavity and therefore have a limited contact time with the absorption site. For drugs which are in solution and rapidly absorbed (lipophilic, low molecular weight) the limited contact time is likely to be well in excess of that required for complete absorption. However, for drug particles needing time to dissolve prior to absorption, and for polar drug molecules with a low rate of absorption once in solution, the rate of mucociliary clearance is likely to play a significant role in limiting the extent of absorption.
Mucus barrier
The nasal mucosa is protected from the external environment by a layer of mucus. In the nasal cavity this exists as a gel phase which is approximately 1–10 µm thick and found above a watery, sol phase surrounding the cilia (periciliary layer) which is about 7 µm deep (Fig. 38.1). Mucus is secreted continuously by the goblet cells and submucosal glands. Normal mucus is 97% water and 3% solids; with the latter comprising: i) mucins (about 30% of the solid content), ii) non-mucin proteins (e.g. albumin, immunoglobulins, lysozyme and lactoferrin), iii) inorganic salts and iv) lipids. Mucins are extremely large glycoproteins (up to 3 × 106 daltons per monomer) with protein regions rich in serine and threonine which are linked, by their hydroxyl side groups, to sugar chains (O-glycosylation). They are anionic (negatively-charged) because most of their terminal sugars contain carboxyl or sulphate groups. These glycosylated (sugar-rich) regions are separated by regions of non-glycosylated, ‘naked’ protein, rich in cysteine residues, which are believed to form globular domains stabilized by disulfide bonds. These ‘naked’ domains are the most hydrophobic regions of mucins and probably adsorb significant amounts of lipids. They are also the most antigenic sites on mucins. Entanglement of mucin polymers leads to the formation of a mucous gel and the generation of a mesh which is stablized by non-covalent calcium-dependent cross-linking of adjacent polymers. The sugar side chains bind large amounts of water allowing the mucus to act as a lubricant and a reservoir for the periciliary fluid within which the cilia beat. Mucus is a viscoelastic gel with properties of both a deformable solid (elasticity) and a viscous fluid (Chapter 6). Cilia can only transport mucus of the appropriate viscoelasticity and this is controlled by the level of mucus hydration.
The presence of mucus at the epithelial surface of the nasal cavity provides an additional potential diffusion barrier to drug delivery. The ability of a molecule to diffuse through the gel is a function of the size of the drug molecule, the effective mesh size of the mucous gel formed by the mucin molecules and any interactions between the drug and the components of the mucous gel. The permeability of small, uncharged molecules appears to be less affected by a mucous barrier than the permeability of larger, cationic molecules. However, several large molecular weight, globular proteins (e.g. bovine serum albumin) and even 500 nm PEG nanoparticles have been observed to readily diffuse through mucus (cervical) at a rate comparable to their diffusion through water. Mucus seems to present a barrier to the permeability of small, relatively hydrophobic molecules like testosterone and this is believed to result from their interaction with the lipid component of the mucous gel or the hydrophobic (non-glycosylated) region of the mucin molecules. It is thought that such small molecules are only able to form low-affinity, monovalent bonds with the mucins which persist for just a short time. A number of studies indicate that positively-charged (cationic), low molecular weight drugs, such as amikacin, gentamicin, tobramycin and some β-lactam antibiotics bind electrostatically to negatively-charged components in mucus. It is believed that such molecules bind tightly and polyvalently to the negatively-charged sugar residues of the mucins. Large positively-charged nanoparticles, such as those coated with chitosan bind especially tightly to mucous gels by a similar mechanism.
Enzymatic activity
A broad range of enzymes are present in the nasal cavity, including those involved with Phase 1 metabolism (e.g. monooxygenase, carboxyl esterases, epoxide hydrolases and cytochrome P450 isoenzymes) and also conjugative Phase II metabolism (e.g. UDP-glucuronyltransferase and glutathionetransferase). In addition, proteolytic enzymes (proteases and aminopeptidases) provide a potential barrier to the absorption of certain peptides. Drugs may be metabolized in the lumen of the nasal cavity or as they pass across the nasal epithelium. However, the metabolic activity of the nasal cavity is less than that of the gastrointestinal tract (on a nmol/mg protein basis) and, in addition, there are a number of factors that will affect the relevance of metabolism to drug absorption. These include, the amount of drug applied to the contained nasal surface area, the chemical nature of the drug, the rate of removal of drug from the cavity and its rate of absorption across the mucosa.
Epithelial barrier – efflux transporters
The absorption of certain drugs across the nasal epithelium can be limited by the presence of efflux transporters. One such transporter, belonging to the superfamily of adenosine triphosphate (ATP)-binding cassette (ABC) transporters has been found in the nasal respiratory mucosa and is termed P-glycoprotein 1 (P-gp), multi-drug resistance protein 1 (MDR1) or ABCB1. This transporter is also expressed by cells within the intestine and poses a similar barrier to drugs that are orally administered (Chapter 19). P-gp is a 170 kDa glycosylated transmembrane protein found in the apical membranes of the cells. It is able to bind a wide variety of hydrophobic and amphiphilic substrates, including certain peptides, to its binding site located cytoplasmically at the inner leaflet of the apical cell membrane and actively pump them from the cell, back into the nasal cavity. Hence, drugs that are substrates for P-gp will be less well absorbed across the nasal epithelium than their physicochemical properties (molecular size, lipophilicity, degree of ionization) might predict. Active transport is concentration-dependent, saturable and can be competitively inhibited by other substrates for the binding site. Thus, co-administration of an inhibitor of P-gp, such as rifampicin or verapamil can enhance drug absorption. P-gp is also found in the olfactory epithelium, at a higher concentration than is found in the respiratory epithelium, where it reduces drug absorption into the brain
Physicochemical properties of drugs affecting intranasal systemic delivery
In general, for a drug to be absorbed it must be in solution (molecularly dispersed). Since the volume of liquid that can be administered intranasally is relatively low (25–200 µL) drugs with low aqueous solubility and/or those requiring a high dose can be problematic. Such issues can be overcome by formulating the drug as a suspension or powder (generally in the micrometre size range), in which case the drug will be required to dissolve in the fluid of the nasal cavity prior to absorption. There is some evidence that nanoparticles (which are an order of magnitude smaller) can be transported from the nasal cavity into the systemic circulation without dissolving, and it is possible that uptake may involve the NALT.
Solubility
Strategies to increase the solubility of a drug can involve modifying the molecular form and include the use of prodrugs and choice of salt form, or the use of appropriate excipients, such as co-solvents, when the drug is formulated (considered below).
Prodrugs are often developed to increase the lipophilicity of a drug molecule and hence its absorption across a biological membrane. However, in the case of nasal delivery the principle has been explored to increase the aqueous solubility of the parent drug to enable a clinically relevant dose of drug to be dissolved in less than 150 µL of solution and has been successful for several drugs. For instance, the solubility of L-dopa (aqueous solubility = 1.65 mg mL−1) is increased 400 fold by producing it as a butyl ester prodrug, enabling an effective dose of 10 mg to be delivered in 125 µL. The prodrug is rapidly converted to the active parent drug once it enters the bloodstream.
The appropriate choice of salt form of an ionizable drug can be used to increase its aqueous solubility. This is an empirical process since it is hard to predict reliably the effect of a particular counter ion on the solubility of the resulting salt. Nevertheless, examples exist where this approach has been successful. For instance, the solubilities of galantamine hydrobromide and morphine sulphate have been increased sufficiently by exchanging the bromide or sulphate ions for gluconate to make nasal delivery feasible for these compounds. However, a change in salt form can result in irritancy to the nasal mucosa and this has to be considered when choosing an appropriate counter-ion.
Lipophilicity/hydrophilicity and molecular size
Once in solution, lipophilic drugs such as propranolol, progesterone and fentanyl are rapidly absorbed from the nasal cavity by the transcellular route and have a nasal bioavailability similar to that obtained after intravenous administration (almost 100%). The absorption of hydrophilic (polar) drugs occurs via the paracellular route (between the epithelial cells via the tight junctions) and the rate and extent of absorption is inversely proportional to the molecular weight of the drug. Since the paracellular route provides a much smaller area for absorption than the transcellular route (the paracellular route comprises about 0.01% of the transcellular route in the gastrointestinal tract), the absorption of hydrophilic compounds is much slower than that of lipophilic drugs. For both lipophilic and hydrophilic molecules, absorption is relatively efficient for drugs with a molecular weight below 1 kDa but then declines. Nevertheless, calcitonin (salmon) is successfully used to reduce the risk of vertebral fractures in postmenopausal osteoporosis (Table 38.2) despite being a hydrophilic peptide with a molecular weight of 3432 Da and having a nasal bioavailability that is just 3% of its bioavailability when delivered intramuscularly. When considering dose reproducibility from the nasal cavity, dosing is relatively consistent for low molecular weight drugs when compared to the oral or parenteral routes, whereas for compounds with a high molecular weight, such as peptides and proteins, relatively high variability is exhibited compared to injections.
Degree of ionization
For drugs that are weak acids or bases, the pH of the nasal cavity will affect the degree of ionization of the drug. The pH at the surface of the nasal mucosa has been reported to be 7.4 while the pH of the mucus is in the range 5.5–6.5. In addition, the pH of the formulation itself can alter the local pH, particularly if buffered vehicles are employed. Studies have indicated that the non-ionized form of a drug, which has a higher partition oil/water partition coefficient than its ionized counterpart, is better absorbed than the ionized form (pH partition hypothesis) (Chapter 20). The ionized form of the drug also shows some permeability, the degree of which may be dependent upon the nature of the counter-ion.
Formulation factors affecting intranasal systemic delivery
The same general formulation considerations apply to drugs formulated for systemic action as for local action, as indicated by the case examples shown in Table 38.2. However, additional strategies can be employed to increase absorption across the nasal epithelium. In essence, the bioavailability of nasally administered drugs can be limited by:
• rapid and extensive enzymatic degradation of the drug in the nasal cavity
• poor permeability of the drug across the respiratory epithelium.
Approaches that have been used to overcome these limitations are summarized in Table 38.4 and include the use of prodrugs (see above), enzymatic inhibitors, mucoadhesive formulations and permeation enhancers which affect the epithelial barrier.
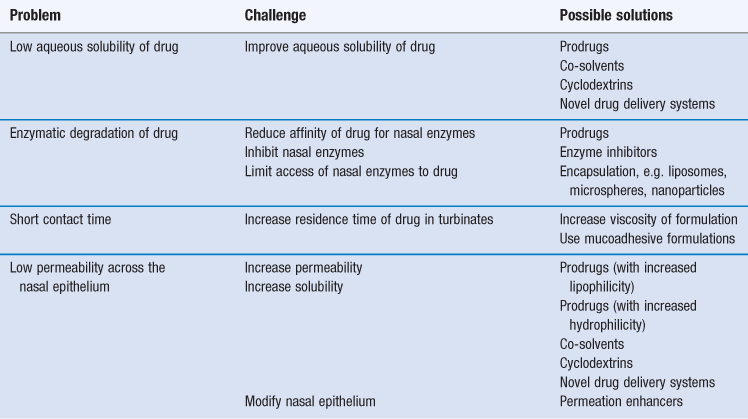
Increasing aqueous solubility
As discussed above, for a drug to be absorbed it should normally be in solution. Drug solubility can be increased by using a mixed solvent system or a co-solvent in the formulation. Solvents used with water for nasal delivery include glycerol, ethanol, propylene glycol and polyethylene glycol (PEG). It is important that any co-solvents do not irritate the nasal mucosa and it is likely that ethanol, used at high concentrations, would not be well-tolerated. However, PEG 300 has been used successfully to increase the solubility of buprenorphine hydrochloride and melatonin, and has enabled clinically relevant doses to be administered with low nasal irritation being observed in humans.
Cyclodextrins (Chapter 24) are cyclic compounds composed of α-D-glucopyranose units. They tend to be water-soluble due to their hydrophilic/polar outer surface, but have a hydrophobic/less polar centre. They are able to increase the aqueous solubility of lipophilic compounds by forming dynamic inclusion complexes where the lipophilic part of the drug molecule is incorporated into the lipophilic central cavity of the cyclodextrin ring. An intranasal formulation containing 17-β-estradiol solubilized in dimethyl-β-cyclodextrin (seven glucopyranose units) was available for the treatment of menopausal symptoms, until it was withdrawn in 2006. The formulation was well-tolerated and as effective as transdermal and oral formulations of estradiol. The dimethyl-β-cyclodextrin was reported to increase absorption of the drug by both enhancing its solubility and increasing the permeability of the nasal epithelium.
pH of the formulation
Many drugs are weak acids or bases and their degree of absorption will depend on their pKa and the pH of the absorption site. The pH of a formulation is generally dictated by the stability of the drug but, within these constraints, a pH favouring more unionized molecules would be expected to enhance absorption. It is important to recognize that the formulation should be non-irritant to the nasal mucosa and formulating at a pH close to that of the nasal cavity (5.0–6.5) may also be desirable although, unexpectedly, it has been shown that pH values ranging from 3–10 can be tolerated by the nasal mucosa (Table 38.2).
Use of enzyme inhibitors
Should peptides be administered via the nasal cavity, they are clearly potentially prone to degradation by the enzymes of the nasal mucus and epithelium. Proteolytic enzyme inhibitors could prevent the hydrolysis of peptide and protein drugs in the nasal cavity improving their stability at the absorption site. As examples, the aminopeptidase and trypsin inhibitor, camostat mesilate, improved the nasal absorption of the peptide vasopressin and its analogue, desmopressin, and the absorption of calcitonin can also be enhanced by the use of trypsin inhibitors. However, proteolytic enzyme inhibitors do not improve the ability of peptide and protein drugs to cross the epithelium of the nasal cavity and therefore do not dramatically improve nasal bioavailability, as clearance mechanisms continue to operate to remove the drug from the absorption site.
Increasing nasal residence time
Unless a drug molecule possesses the ideal characteristics for rapid absorption, the percentage of the administered dose entering the systemic circulation is likely to be affected by the residence time of the nasal formulation in the turbinates. One way of increasing the time that the formulation is in contact with the absorptive mucosa is by the use of mucoadhesive polymers, such as cellulose derivatives, polyacrylates, starch and chitosan. Most of these polymers are ‘Generally Regarded As Safe’ (i.e. given GRAS status as categorized by the FDA) and if included as pharmaceutical excipients within the vehicle, have been shown to increase the absorption of hydrophilic macromolecules. The polymers themselves are not absorbed and therefore would not be expected to cause any systemic toxicity.
Adhesion of a polymeric material can occur to both the nasal epithelial surface (bioadhesion) and nasal mucus (mucoadhesion). Mucoadhesive formulations can be administered to the nasal cavity in the form of solid powders or particulates, gels or liquids. For good mucoadhesion, the formulation should spread well on the nasal mucosa (solid formulations should flow well and be readily wettable) after which the hydration of the polymer and the intimate contact it has with the nasal mucosa is very important. Mucoadhesives can increase absorption by three mechanisms:
With time, the continuous production of mucus will cause further hydration of the polymer (beyond the optimum required for mucoadhesion), the strength of mucoadhesion will diminish and normal mucociliary clearance will resume, so clearing the polymer from the nasal cavity.
Examples of polymers and drugs that have been used in studies of nasal mucoadhesion are given in Table 38.5. When the polymers are formulated in solution, the viscosity of the preparation will be greater than that of a simple solution. Whilst an increased formulation viscosity leads to a prolonged residence time, it does not always result in increased absorption. This might be due to the decreased rate of diffusion of the drug molecules through a solution of higher viscosity. However, the viscosity of a solution for nasal delivery has to be limited to about 500 mPa s since, although more viscous solutions show better mucoadhesion they are too viscous to instil easily and accurately into the nasal cavity. To overcome this problem a type of gel has been developed (in-situ gel) that is liquid prior to administration (allowing convenient and accurate dosing) but forms a gel once in contact with the nasal mucosa. The temperature or pH of the mucus promotes the transition from liquid to gel. In studies, this approach has been used successfully to increase the nasal absorption of metoclopramide, sumatripan and insulin. A marketed product (PecFent™) containing the analgesic fentanyl and low methoxyl (LM) pectins is administered to the nasal cavity as a solution, but interacts with calcium ions in nasal secretions to form a muco/bioadhesive gel. Fentanyl is a low molecular weight, lipophilic molecule that readily crosses the nasal epithelium and is useful for the treatment of break-through pain with a more rapid onset of action and better bioavailability from the nasal cavity than oral transmucosal fentanyl (buccal or sublingual). Nevertheless, it has a relatively short duration of action. The LM pectin in the formulation causes a slight delay in the onset of action compared with a nasal formulation without LM pectin (longer tmax and lower Cmax) (Chapter 21) but prolongs the residence time of the fentanyl in the nasal cavity, extending its duration of action until the product is cleared.
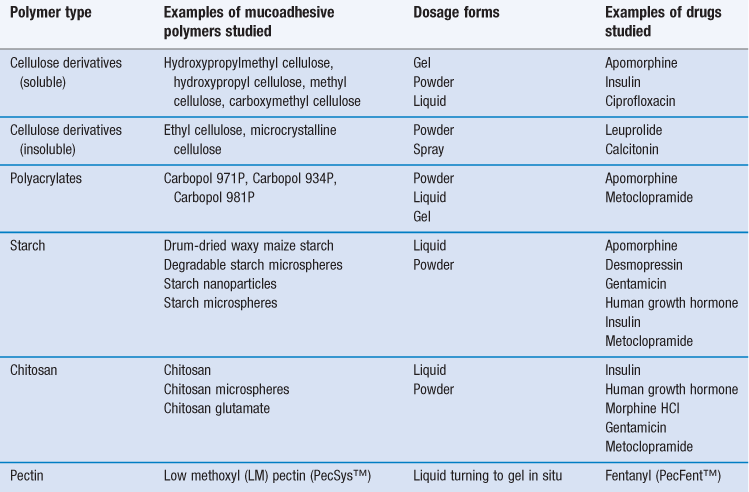
Polymers can also be formulated as dry powders; these are not mucoadhesive when dry, which allows them to be easily administered by metered-dose insufflations, but they become mucoadhesive once in contact with the nasal mucosa by absorbing water from the nasal mucus. Powders have certain advantages over liquid formulations and these include:
• a larger amount of drug can be delivered
• no need for preservatives since they do not support microbial growth
• no requirement for storage at reduced temperatures due to improved stability.
These advantages make the study of dry powder formulations popular for the administration of small hydrophobic drugs, peptides and vaccines. Disadvantages of powder administration include the possible irritation to the nasal mucosa and a possible gritty texture. The aerodynamic size of the particles (see Chapter 37) will affect the deposition site in the nasal cavity, and manufacturing particles of the correct aerodynamic particle size to deposit in the respiratory region of the nasal cavity, where maximum absorption takes place, can be expensive.
Polymers can also be formulated as microparticles/microspheres and nanoparticles (Chapter 45). These systems can protect the drug from enzymatic degradation, improve contact with the absorptive epithelium and enhance uptake. Nanoparticulate systems are taken up by the NALT, suggesting potential application for the delivery of vaccines.
Enhancing the permeability of nasal epithelium
It is possible to increase the absorption of both small and large hydrophilic drug molecules by administering them with permeation enhancers which modify the structure of the nasal epithelium. However, it is important that any alteration to the barrier function of the epithelium is short-term and reversible, since the epithelium constitutes one of the body’s primary defence mechanisms against insult from the external environment. A range of nasal products is on the market, none of which contains a permeation enhancer. This is because the drug molecules are either both small and lipophilic, and have adequate absorption without the need for a permeation enhancer, e.g. sumatriptan, fentanyl and nicotine, or because the nasal bioavailability, although low, is still sufficient for the drug to exert a therapeutic effect, as is the case for the peptides calcitonin, desmopressin, buserelin and naferelin. For this latter group, it is also likely that the permeation enhancers available at the time of marketing were too toxic for use. Thus, there is a need for safe and efficient permeation enhancers to enable less potent biological drugs to be delivered intranasally and to improve the bioavailability of those currently on the market.
The requirements of an ideal permeation enhancer include the following:
• rapidly-acting with a transient and reversible effect on the nasal epithelium
• non-toxic, non-irritant and non-allergenic
• does not permit entry of dangerous environmental material
• compatible with drugs and other excipients in the formulation
• safe for chronic use (depending on the condition to be treated).
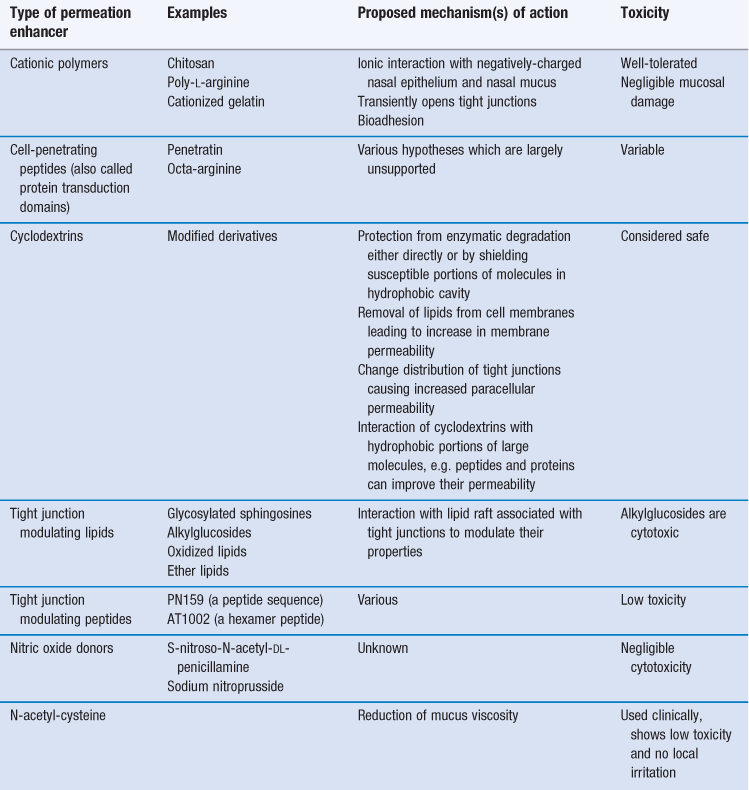
Examples of permeation enhancers that have been studied to increase the absorption from the nasal cavity include surfactants (such as bile salts and their derivatives) and certain phospholipids. These have proved effective in promoting absorption by a variety of different mechanisms, including: solubilization of the drug, inhibition of enzymatic activity, extraction of lipid or protein from the cell membrane, alteration of the mucus layer and alteration of tight junctions. However, many of these enhancers cause severe irritation and damage to the nasal mucosa at the concentrations required to promote nasal absorption. Some of the materials able to enhance permeability while possessing a better toxicity profile are detailed in Table 38.6.
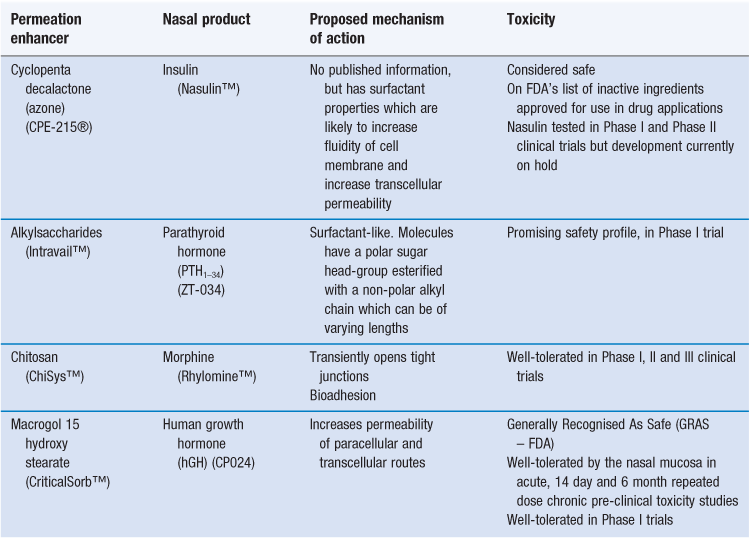
A number of permeation enhancers are currently being developed commercially for clinical use (Table 38.7). These have achieved a better balance between efficacy and safety/toxicity and have been assessed in humans in clinical trials. The permeation enhancers are generally being developed for use with peptide and protein drugs, but none of these products has yet reached the market. The product most advanced in its development is that containing the small hydrophilic molecule morphine in a formulation with chitosan (Rhylomine™), which is currently in Phase III trials.
Patient factors affecting intranasal systemic delivery
Patient compliance.
If a patient does not use a medication appropriately, then it cannot be expected to be effective. Thus, good patient compliance is paramount for successful treatment. For systemic treatment, the nasal route is usually chosen when the oral route is not available. Thus, use of the nasal route is generally compared with parenteral delivery or with other transmucosal routes. The nasal route is accessible to the patient using simple dosage forms (sprays and drops) permitting self-medication over extended periods of time. Unlike parenteral delivery, it is non-invasive and therefore has a reduced risk of introducing infection upon application and a low risk of disease transmission. In addition, provided that the formulation does not cause irritation, it should be comfortable to use. In studies comparing intranasal delivery with the parenteral route, fewer patients preferred the parenteral route. When compared with another transmucosal route, e.g. rectal, the use of intranasal midazolam for the treatment of childhood seizures was found to be safe and effective, easier for care-givers to administer and more dignified for the patient than rectally administered medication which is commonly used to control break-through seizures in the home.
Disease.
A number of nasal pathologies might be expected to affect the absorption of drugs into the systemic circulation, either by altering nasal mucociliary clearance, e.g. the common cold or by affecting the permeability of the nasal epithelium, e.g. allergic rhinitis. However, there is little information in the literature to substantiate this. This is possibly because for those drugs that are rapidly absorbed, the effect is negligible and for those that are poorly absorbed, provided they have a sufficiently wide therapeutic window, the variability is acceptable. Clearly, for a poorly absorbed drug with a narrow therapeutic window the potential unpredictability in absorption caused by such pathologies would be undesirable.
Nasal vaccines
Mucosal tissues are attractive sites for vaccination due to their accessibility, immunological competence and because local immune responses can be elicited which can protect against infection at the point of virus entry. Intranasal vaccination targets the nasal-associated lymphoid tissue (NALT) which is situated beneath the nasal epithelium and consists of groups of dendritic cells, T-cells and B-cells.
So far, the intranasal route has been successfully used (Table 38.1) for a commercial influenza (live-attenuated) vaccine (FluMist™). Conventional needle-based intramuscular vaccinations are able to induce the production of serum antibodies (IgG) which, by transudation to the lungs, protect the lower respiratory tract against influenza infection and the more severe complications of influenza. However, vaccines given intranasally can induce local IgA responses in the upper respiratory tract (as well as systemic IgG responses) which can neutralize the target virus immediately after it is inspired and protect against early disease symptoms. Secretory IgA is also more cross-reactive than IgG and can provide protection against different strains of the virus. Nevertheless, since the nasal mucosa is exposed to a multitude of antigens present in the environment, tolerance mechanisms limit the resultant immune reaction. Consequently, these mechanisms have to be overcome for successful vaccination and, unless the vaccine contains live attenuated viruses, it is essential to incorporate an effective mucosal adjuvant within the final formulation. The benefits of nasal vaccination when compared to needle-based delivery systems include a reduced risk of needle-stick injuries and risk of infection from the re-use of needles, increased patient compliance among patients with needle phobia, a decreased need for vaccines to be administered by trained healthcare professionals and possibly a decreased need for cold chain storage and distribution, if vaccines can be formulated as dry powders. In addition, use of the route provides a ready means of vaccinating large population groups. Vaccine formulation and delivery is described in greater detail in Chapter 46.
CNS delivery
The blood-brain barrier (BBB) restricts the entry of potentially harmful substances into the brain but also limits the access of potentially useful drugs. It exists at the level of the cerebral microvasculature. In contrast to the leaky barrier presented by the endothelial cells of the capillaries in the peripheral circulation, the endothelial cells in the brain exhibit low rates of pinocytosis and are joined by tight junctions which limit the paracellular diffusion of hydrophilic solutes from the blood into the brain. In addition, the BBB expresses a high number of efflux transporters, such as P-glycoprotein (P-gp), which further reduce access to the brain for those molecules that might be predicted to be well-absorbed from their size and lipophilicity.
Drugs delivered intranasally that enter the systemic circulation would have to cross the BBB to enter the CNS. However, it has been proposed that there is a route from the olfactory region of the nasal cavity (Fig. 38.1) to the brain that avoids the BBB and which can be exploited to deliver drugs directly to the brain. This is currently an area of great research interest and studies have shown that both low molecular weight drugs and high molecular weight peptides and proteins appear to be able to access the brain following intranasal delivery. It is possible that a drug will gain access to the brain following its absorption into the systemic circulation and not via a direct route, and it is important to eliminate or account for this possibility in the design of studies seeking to establish or quantify direct nose-to-brain transport. It should also be noted that many of the studies of direct nose-to-brain transport have been undertaken in rats which differ in their nasal anatomy compared to humans; the nasal passages of the rat have a higher surface area-to-volume ratio (SA/V) than those of man (51.5 and 6.4 respectively), mucociliary clearance in the rat is in the anterior direction whereas mucus is moved posteriorly in man and a significantly higher percentage of the nasal epithelium is concerned with olfaction in the rat (50%) compared to man (8%). In addition, the experimental conditions employed in some studies could not be considered applicable to man; formulations containing penetration enhancers at concentrations damaging to the nasal mucosa have been used and some formulations were applied to the nasal cavity in a manner that would be inappropriate in man (too large a volume or application at too high a pressure over an extended time which might be expected to damage the epithelium). Nevertheless, studies in rats are important and provide useful information on the pathways and mechanisms of drug absorption both into the systemic circulation and CNS.
The olfactory mucosa is composed of the olfactory epithelium and its underlying lamina propria. The routes by which the molecules cross the olfactory epithelium have yet to be fully elucidated, but a number of possible pathways have been suggested (Fig. 38.1). An intracellular, axonal pathway has been proposed where substances are taken into the olfactory sensory neurons (OSNs) via adsorptive, receptor-mediated or non-specific fluid phase endocytosis and transported within the cell along the axon to the olfactory bulb. Another pathway involves substances crossing the other cells of the olfactory epithelium (e.g. sustentacular (supporting) cells) via transcellular or paracellular passive diffusion to reach the lamina propria. Tight junctions exist between cells in the olfactory epithelium but the regular turnover of cells in the epithelium may lead to loosening of the tight junctions, helping the paracellular transport of larger molecular weight substances.
Once at the lamina propria, entry into the CNS is believed to occur, via diffusion or convection, extracellularly along the perineural space (which is the space surrounding the olfactory nerve bundles), through the cribriform plate and into the cerebrospinal fluid (CSF) or olfactory bulb. However, a proportion of molecules is also likely to enter the blood vessels of the systemic circulation or lymphatic vessels and will therefore be prevented from entering the CNS by this direct route. Of the two routes into the CNS, it has been suggested that intracellular transport along the axon of the OSN would be too slow to account for the experimental results observed and that the other extracellular pathway is the most likely.
Immunohistochemical studies have found the efflux transporter P-gp localized to the endothelial cells lining the olfactory bulb and the olfactory epithelium. P-gp is able to reduce entry into the CNS of those drugs which are substrates for the transporter. Since drugs need to be inside the epithelial cell to interact with the binding site of P-gp, this will mainly affect those drugs crossing the supporting cells of the olfactory epithelium via the transcellular route.
Interestingly, the trigeminal nerve which innervates the respiratory epithelium of the nasal cavity also feeds into various areas of the brain and could potentially be exploited for nose to brain drug delivery. However, so far, this route has not been implicated as providing a pathway for CNS drug delivery.
It should be noted that in the many studies of drug transport (low molecular weight drugs and peptides and proteins) from the nose to the brain, the amount of drug reaching the CNS is small compared to the amount administered to the nasal cavity, generally less than 1%. One major problem is the inaccessibility of the olfactory region of the nasal cavity coupled with the poor permeability of certain types of molecule (including peptides and proteins) across the olfactory epithelium. There is a need for a formulation containing an acceptable nasal permeation enhancer and a bioadhesive material which can be delivered from a nasal device that is able to target the formulation to the olfactory region.
Nasal delivery systems
Nasally administered medicines can be formulated as ointments or creams but most usually as a liquid (solution, gel or suspension) or as a powdered solid (Tables 38.1 and 38.2). The formulation issues with each of these dosage forms have been considered in previous chapters. With multi-dose liquid dosage forms, the possibility of ‘suck-back’ exists which is when a portion of the administered dose is sucked back into the remaining liquid in the delivery device. As a consequence, multi-dose liquid dosage forms can require the inclusion of antimicrobial preservatives to prevent the growth of contaminating microorganisms. There is evidence that some of these preservatives can cause irritation to the nasal mucosa and/or damage the cilia and therefore compromise mucociliary clearance, especially if used over a long period. Strategies to minimize or obviate such effects include the use of alternate nostrils, if chronic daily dosage is required, and the use of pressurized containers or unit-dose delivery systems (Table 38.8) which do not require the inclusion of a preservative. There is a move towards delivery systems that deliver an accurate metered dose and away from dosage forms such as nasal drops, which require considerable skill, dexterity and even flexibility (in terms of mobility) to apply uniformly across the mucosa. Smaller doses (< 100 µL) tend to persist longer than larger doses which may drip from the nostril after delivery.
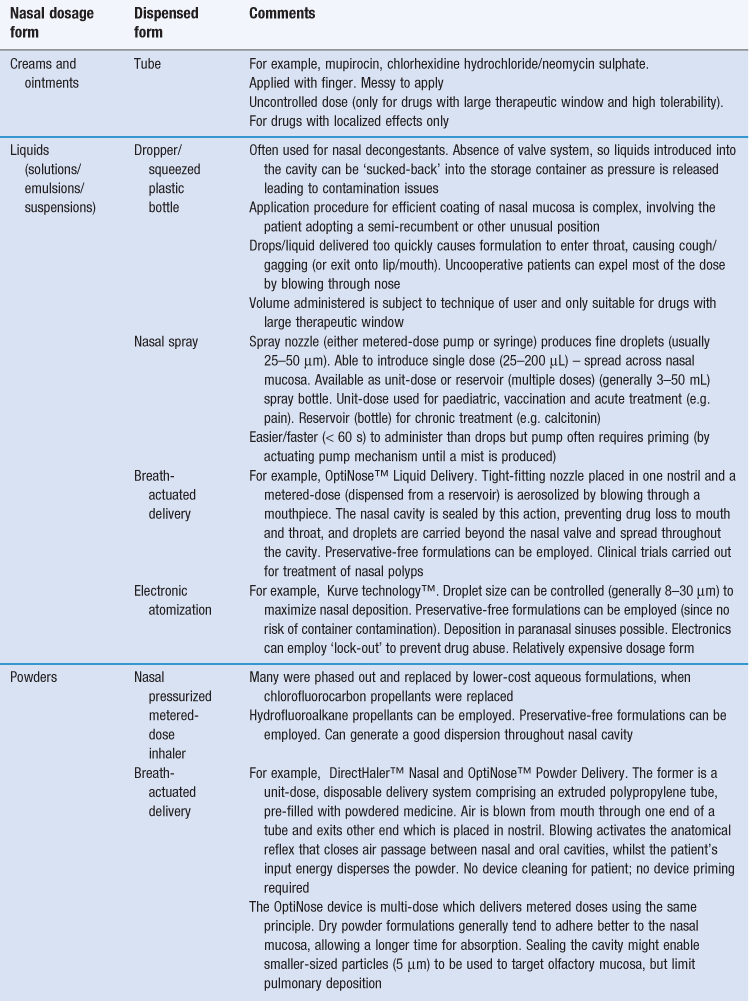
Powdered solids tend to remain in the nasal cavity for longer periods than liquids, since a preliminary hydration step generally occurs before mucociliary clearance reaches maximal efficiency. This can prolong the window over which systemic drug absorption can occur or the duration of action of a locally-acting drug. Creams and ointments can also be utilized to prolong retention in the cavity. Moreover, the use of solids or creams limits the rapid introduction of fluid to the throat and mouth, which can often initiate an unwelcome taste and possibly induce coughing and gagging. Control of particle size is important since particles with sizes less than 10 µm can move beyond the nasal turbinates towards the lung, whereas particles larger than 50 µm can be cleared more rapidly by mucociliary clearance and nose blowing.
An interesting advance in nasal delivery devices which shows useful potential in delivering nasal formulations involves breath-actuated bi-directional delivery (Table 38.8). The device is constructed such that the aerosolization of the powder is initiated by the patient themselves exhaling through the mouth against a resistance. This action closes the soft palate and separates the nasal cavity from the oral cavity. A communication pathway remains between the two nostrils, located behind the nasal septum. The expired air (and aerosolized powder) blown into one nostril is turned through 180°, passes through the pathway and leaves via the second nostril, ensuring that powder deposits throughout the cavity.
Summary
The potential of delivering drugs and vaccines to the body via the nasal cavity is far from being fully realized. There are active programmes within a number of pharmaceutical and biotechnological companies seeking to utilize the route. The development of more nasal vaccines is almost certain. However, the use of the nose for systemic delivery is contemplated only if the oral route, for whatever reason, is not viable. It is limited in terms of the dose that can be delivered, either as a result of solubility of the drug (given the volume of liquid that can be delivered comfortably) or in terms of the quantity of powder or semi-solid that can be tolerated per dose. Once drug is delivered, then it must be absorbed rapidly otherwise the normal clearance mechanisms will remove it from the absorbing epithelium and result in reduced bioavailability. Strategies are therefore being employed to either extend the absorption window (through the use of mucoadhesive excipients) or increase permeability (by a number of means, including the use of permeation enhancers). As with pulmonary delivery however, the primary packaging is an important component of the final medicine, since this also comprises the delivery device. There is much scope to develop novel devices with improved dose dispensing and superior targeting (perhaps to the olfactory region) or more efficient coating of mucosa. Drug development programmes need to consider both the device and the formulation as a whole, taking into account the therapeutic purpose of the medicine. Finally, as with pulmonary dosage forms, patient counselling will be especially important in advising patients of their medicines to gain maximum effectiveness, since whatever formulation is marketed, it will not be as easy to administer as the swallowing of a solid dosage form.
Bibliography
1. Amorij J-P, Hinrichs WLJ, Frijlink HW, Wilschut JC, Huckriede A. Needle-free influenza vaccination. Lancet Infectious Diseases. 2010;10:699–711.
2. Cone RA. Barrier properties of mucus. Advanced Drug Delivery Reviews. 2009;61:75–85.
3. Costantino HR, Illum L, Brandt G, Johnson PH, Quay SC. Intranasal delivery: physicochemical and therapeutic aspects. International Journal of Pharmaceutics. 2007;337:1–24.
4. Duan X, Mao S. New strategies to improve the intranasal absorption of insulin. Drug Discovery Today. 2010;15:416–427.
5. Fahy JV, Dickey BF. Airway mucus function and dysfunction Medical progress. The New England Journal of Medicine. 2010;363:2233–2247.
6. Illum L. Nasal drug delivery – recent developments and future prospects. Journal of Controlled Release. 2012;161:254–263.
7. Jiang L, Gao L, Wang X, Tang L, Ma J. The application of mucoadhesive polymers in nasal drug delivery. Drug Development and Industrial Pharmacy. 2010;36:323–336.
8. Khanvilkar K, Donovan MD, Flanagan DR. Drug transfer through mucus. Advanced Drug Delivery Reviews. 2001;48:173–193.
9. Lochhead JJ, Thorne RG. Intranasal delivery of biologics to the central nervous system. Advanced Drug Delivery Reviews. 2012;64:614–628.
10. Malerba F, Paoletti F, Capsoni S, Canttaneo A. Intranasal delivery of therapeutic proteins for neurological diseases. Expert Opinion on Drug Delivery. 2011;8:1277–1296.
11. Merkus FWHM, van den Berg MP. Can nasal drug delivery bypass the blood-brain barrier? Drugs in Research and Development. 2007;8:133–144.
12. Pires A, Fortuna A, Alves G, Falcão A. Intranasal Drug Delivery: How, why and what for? Journal of Pharmacy and Pharmaceutical Science. 2009;12:288–311.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Nasal drug delivery
Gary P. Martin and Alison B. Lansley
Chapter contents
Key points
Introduction
The most common reason for introducing a drug into the nasal cavity is to provide a convenient and accessible route for rapidly and efficiently managing the localized symptoms associated with allergic rhinitis, nasal congestion and nasal infection. Drugs applied topically for such purposes include antihistamines, corticosteroids, sodium cromoglicate, sympathomimetics and antiseptics/antibiotics (Table 38.1). These drugs are administered either in liquid form (from a spray or as drops) or as creams/ointments.
The intranasal route has also been exploited for the delivery of drugs to the systemic circulation (Table 38.1). There are several possible reasons for pharmaceutical companies to consider employing this route for marketed medicines rather than the much more popular (and preferred) oral route. These include:
The nasal cavity has also been utilized, or proposed, as a portal for the delivery of vaccines, particularly for infections associated with the respiratory tract such as influenza and possibly, eventually, tuberculosis. The presentation of an antigen, in combination with an acceptable adjuvant to the nasal-associated lymphoid tissue (NALT) can promote both cellular and humoral responses. However human vaccination need not be restricted to airways infections and systemic immune responses are demonstrable after introduction of appropriate antigens via this route. Indeed, intranasal vaccination has been studied with a view to combating noroviruses, the measles and herpes viruses, diphtheria and tetanus microorganisms. An intranasal vaccine containing live attenuated influenza vaccine has been marketed (Table 38.1).
Currently, there is much research interest aimed at establishing whether the olfactory region, positioned in the upper reaches of the nasal cavity (Fig. 38.1), offers a potential means, in humans, of circumventing the obstacles imposed by the blood-brain barrier to the access of many drugs from the bloodstream to the brain. This region contains a direct physiological link between the environment and the central nervous system (CNS). Clearly, if such a route could be established conclusively as being viable for the delivery of therapeutic quantities of drugs in humans, then this would have great potential in treating conditions such as: Alzheimer’s disease, brain tumours, epilepsy, pain and sleep disorders. However, although there are a number of studies that suggest that drugs may be absorbed from the olfactory region, the true significance of many findings are confounded by the use of animal models with the data often being extrapolated uncritically to humans.
So as to appreciate fully the potential of the nasal cavity for drug delivery, it is pertinent to review the relevant anatomy and physiology, consider in more detail the applications of utilising the route, review the physicochemical properties of administered drugs and factors that might affect the choice of formulation and discuss the devices that can be employed to deliver nasal medicines.
Anatomy and physiology
The nasal cavity is 120–140 mm from the nostrils to the nasopharynx (Fig. 38.1) and is divided in two by the nasal septum. The total surface area of both cavities is about 160 cm2 and the total volume is about 15 mL. The first part of the nasal cavity (termed the nasal vestibule) contains the narrowest part of the nasal cavity with a cross-sectional area of 30 mm2 on each side. The lining of the vestibule changes from skin at the entrance, to a stratified squamous epithelium which extends over the anterior third of the entire nasal cavity. The nasal vestibule contains vibrissae (hairs) which filter out inhaled particles with an aerodynamic particle size greater than approximately 10 µm. Progression through the nasal cavity leads to the turbinate region. The turbinates are convoluted projections from the nasal septum which are lined with a pseudostratified columnar epithelium (80–90% of the total surface area of the nasal epithelium in man) composed of mucus-secreting goblet cells, ciliated and non-ciliated cells and basal cells (Fig. 38.1). The apical surfaces of the ciliated and non-ciliated cells are covered with non-motile microvilli, which serve to increase the surface area of the epithelial cells. There are also approximately 100 motile cilia on each ciliated cell which are responsible for mucus transport. Serous and seromucous glands also contribute to nasal secretions. As air moves through the turbinate region via the meatuses (Fig. 38.1), the low rate of airflow in combination with the turbulence created by the shape of the turbinates encourages the air to make contact with the highly vascularized walls, enabling it to be warmed and humidified. Particulates (5–10 µm) within the airstream, such as dust, pollen, microorganisms and pollutants have the potential to deposit on the viscoelastic mucous gel lining the turbinate walls. The cilia, beating within the periciliary fluid, engage with the underside of the mucus and propel the gel and the deposited particles to the nasopharynx, where they are either swallowed or expectorated. This process is termed mucociliary clearance and is able to clear mucus at a rate of about 7 mm min−1. About 20% of the inspired air is directed to the top of the turbinates where the olfactory region is located (Fig. 38.1). This is an area approximately 12.5 cm2 (~ 8% of the total surface area of the nasal epithelium in man) of non-ciliated pseudostratified columnar epithelium traversed by 6–10 million olfactory sensory neurons which pass from the nasal cavity, between the epithelial (sustentacular) cells and through the cribriform plate to the olfactory bulb of the brain.
Drug delivery
Certain constraints are imposed upon formulating preparations for the nasal route and two case studies, one a locally-acting drug (budesonide) and a second systemically-acting peptide drug (calcitonin) are given in Table 38.2.
As with the formulation of any medicine, the information garnered from pre-formulation studies (Chapter 23) is an essential prerequisite in the design of an intranasally delivered medicine. The solubility (Chapter 2) of the drug to be administered is a key determinant in the final formulation. The restricted volume that can be applied to the nasal cavity also impacts upon the nature of the resultant formulation. Generally, the premise of presenting the drug in the simplest formulation, containing the fewest excipients possible to ensure a stable medicine with an adequate shelf-life is the course that should be followed in the development process. Currently, delivery devices are usually metered-dose manual pump sprays, since these are cheap, robust and reliable but more sophisticated systems are now under development, as discussed below.
Local delivery
For conditions affecting the nose, it is logical to deliver the drug directly to its site of action. This permits the rapid relief of symptoms with a much lower dose of drug than would be necessary if it were delivered by the oral route, and reduces the chance of systemic side effects. For example, this is particularly pertinent when delivering corticosteroids to reduce local inflammation of the nasal mucosa and sinuses, without causing pituitary-adrenal suppression, or alternatively when using localized antihistamine therapy without inducing drowsiness.
Systemic delivery
The rationale for the use of the nasal cavity for systemic delivery includes its accessibility, avoidance of pre-systemic metabolism and potential to provide a rapid onset of action. Its use for peptides (Tables 38.1 and 38.2) has been successful since, although only a very low percentage of administered drug is absorbed (i.e. low bioavailability), the attained plasma levels are sufficient for therapeutic efficacy. Many of the marketed nasally administered peptides have wide therapeutic windows. Therefore, providing the minimum therapeutic level is exceeded in the bloodstream, a large variability in the final attained plasma level can be tolerated, without systemic toxicity becoming manifest. Any potential localized toxicity can be minimized in chronic administration by alternating nostrils when daily dosing (Table 38.2). Intranasal delivery can also be useful in emergency situations, such as in the treatment of opioid overdose (using naloxone) or in the treatment of intractable childhood seizures (using benzodiazepines). Drug delivery via this route is also well-suited to drugs that, when administered orally, cause emesis e.g. galantamine used to treat dementia.
Anatomical and physiological factors affecting intranasal systemic delivery
For a drug molecule to enter the systemic circulation it must first be absorbed across the nasal epithelium.
This may occur via the mechanisms of passive diffusion via the transcellular or paracellular routes (Chapter 19). The transcellular pathway is the principal route of absorption for lipophilic molecules, while small, hydrophilic molecules diffuse between the epithelial cells (paracellularly) via the tight junctions which are dynamic structures responsible for the integrity of the nasal epithelium. This latter pathway avoids the need for the drug molecules to partition into and out of the lipophilic membrane of the epithelial cells, but imposes a size restriction of between 0.39–0.84 nm. Transcellular absorption can also occur via endocytosis, the route exploited by large hydrophilic molecules (>1 kDa), and via active transport mechanisms where drug molecules with a similar structure to a natural substrate can interact with a carrier protein to cross the epithelial cells.
Since most drug absorption takes place by passive diffusion, the relatively large surface area of the nasal cavity and its rich blood supply (which helps to maintain the concentration gradient across the epithelium) aid this process. Working against these positive attributes of the nasal cavity are the barriers presented by mucus and the epithelium itself and the nasal clearance mechanisms including mucociliary clearance and metabolism. The advantages and disadvantages of the nasal cavity for systemic drug delivery are summarized in Table 38.3.
Table 38.3
Advantages and disadvantages of intranasal drug delivery for systemic activity
| Advantages | Disadvantages |
| Large surface area for absorption (approximately 160 cm2) | Limited to small delivery volumes (25–200 µL) therefore require potent drugs |
| Good blood supply and lymphatic system | Mucociliary clearance, mucus barrier |
| Avoids hepatic first-pass metabolism | Enzymatic activity (pseudo first-pass effect) |
| Epithelium is permeable to small, lipophilic drug molecules; rapid absorption and onset of action | Low epithelial permeability for hydrophilic drugs; require absorption enhancers and large doses |
| Non-invasive, so minimal infection risk during application and low risk of disease transmission (unlike parenteral route) | |
| Easy to self-administer and adjust dose |
Mucociliary clearance
The main drug absorption site is the respiratory epithelium of the nasal turbinates, which is where mucociliary clearance dominates. Drug deposited anterior to this region will remain in the nasal cavity for longer than drug deposited in the turbinates, but absorption from this site is less. Once drug particles (if formulated as a suspension) or molecules (if in solution) find their way on to the mucociliary ‘conveyor belt’ they will be cleared from the nasal cavity and therefore have a limited contact time with the absorption site. For drugs which are in solution and rapidly absorbed (lipophilic, low molecular weight) the limited contact time is likely to be well in excess of that required for complete absorption. However, for drug particles needing time to dissolve prior to absorption, and for polar drug molecules with a low rate of absorption once in solution, the rate of mucociliary clearance is likely to play a significant role in limiting the extent of absorption.
Mucus barrier
The nasal mucosa is protected from the external environment by a layer of mucus. In the nasal cavity this exists as a gel phase which is approximately 1–10 µm thick and found above a watery, sol phase surrounding the cilia (periciliary layer) which is about 7 µm deep (Fig. 38.1). Mucus is secreted continuously by the goblet cells and submucosal glands. Normal mucus is 97% water and 3% solids; with the latter comprising: i) mucins (about 30% of the solid content), ii) non-mucin proteins (e.g. albumin, immunoglobulins, lysozyme and lactoferrin), iii) inorganic salts and iv) lipids. Mucins are extremely large glycoproteins (up to 3 × 106 daltons per monomer) with protein regions rich in serine and threonine which are linked, by their hydroxyl side groups, to sugar chains (O-glycosylation). They are anionic (negatively-charged) because most of their terminal sugars contain carboxyl or sulphate groups. These glycosylated (sugar-rich) regions are separated by regions of non-glycosylated, ‘naked’ protein, rich in cysteine residues, which are believed to form globular domains stabilized by disulfide bonds. These ‘naked’ domains are the most hydrophobic regions of mucins and probably adsorb significant amounts of lipids. They are also the most antigenic sites on mucins. Entanglement of mucin polymers leads to the formation of a mucous gel and the generation of a mesh which is stablized by non-covalent calcium-dependent cross-linking of adjacent polymers. The sugar side chains bind large amounts of water allowing the mucus to act as a lubricant and a reservoir for the periciliary fluid within which the cilia beat. Mucus is a viscoelastic gel with properties of both a deformable solid (elasticity) and a viscous fluid (Chapter 6). Cilia can only transport mucus of the appropriate viscoelasticity and this is controlled by the level of mucus hydration.
[/not-level-membership-for-basic-science-category]
