Myeloproliferative Neoplasms
After completion of this chapter, the reader will be able to:
1. Define myeloproliferative neoplasms (MPNs).
2. List the most common diseases included in the classification of MPNs and recognize their abbreviations.
3. Define chronic myelogenous (chronic myelocytic) leukemia (CML), with emphasis on the affected cell lines.
4. Discuss the pathogenic mechanism in CML, including the cytogenetic and molecular biology.
5. Describe the peripheral blood and bone marrow findings in CML.
6. List the clinical phases of CML and describe the expected symptoms and laboratory test results in each phase.
7. Discuss the best approach to treatment and mechanisms of drug resistance.
8. Define polycythemia vera (PV), with emphasis on the affected cell lines.
9. Discuss the JAK2 mutation and the proposed pathogenic mechanism in PV.
10. Discuss clinical symptoms commonly observed in patients with PV.
11. Identify major morphologic changes in the bone marrow and peripheral blood in patients with PV.
12. List diagnostic criteria for PV.
13. Discuss the progression of PV and treatment modalities.
14. Define essential thrombocythemia (ET), with emphasis on the affected cell lines.
15. List the diagnostic criteria for ET.
16. Describe the morphologic changes in the peripheral blood in patients with ET.
17. Discuss two complications that may occur in patients with ET.
18. Define primary myelofibrosis (PMF), with emphasis on the affected cell lines and pathogenesis.
19. Describe the key pathologic features of PMF in bone marrow, peripheral blood, and tissues.
20. Describe the course of disease of PMF and current therapy.
21. Briefly describe the other myeloproliferative disorders.
22. Given complete blood count and cytogenetic, molecular, and other laboratory results, recognize the findings consistent with each major MPN.
23. Recommend follow-up testing for suspected MPN and interpret the results of testing.
24. Briefly describe the other MPNs outlined in this chapter.
Case Study
Leukocyte alkaline phosphatase (LAP) score—20 (reference range, 40 to 130)
Lactate dehydrogenase—692 IU (reference range, 140 to 280 IU)
Uric acid—8.1 mg/dL (reference range, 4 to 6 mg/dL)
1. What is the significance of the elevated WBC count and abnormal WBC differential?
2. How does the LAP score aid in the diagnosis?
3. Justify the use of cytogenetic studies in a patient with test results similar to those in this case study.
4. Predict the results of the cytogenetic studies.
5. Describe the molecular mutation resulting from the cytogenetic abnormality.
The myeloproliferative neoplasms (MPNs) are clonal hematopoietic disorders caused by genetic mutations in the hematopoietic stem cells that result in expansion, excessive production, and overaccumulation of erythrocytes, granulocytes, and platelets. Expansion occurs in varying combinations in the bone marrow, peripheral blood, and tissues.1–4 The MPNs have pathogenetic similarities as well as common clinical and laboratory features.5
The World Health Organization (WHO) has classified the MPNs as including four predominant disorders: chronic myelogenous leukemia (CML); polycythemia vera (PV) also known as polycythemia rubra vera; essential (primary) thrombocythemia (ET); and primary myelofibrosis (PMF), also known as agnogenic myelofibrosis with myeloid metaplasia and chronic idiopathic myelofibrosis. Several other less common MPN conditions have been described and are classified as chronic neutrophilic leukemia (CNL); chronic eosinophilic leukemia (CEL), not otherwise specified; mastocytosis; and myeloproliferative disorder, unclassified.6 CML and PV are defined by their overproduction of granulocytes and erythrocytes, respectively.2,7,8 PMF is a combination of overproduction of hematopoietic cells and stimulation of fibroblast production leading to ineffective hematopoiesis with resultant peripheral blood cytopenias.9 ET is characterized by increased megakaryocytopoiesis and peripheral blood thrombocytosis.10
Familial MPNs have been described in families in which two or more members are affected.11
Chronic Myelogenous Leukemia
Chronic myelogenous leukemia (CML) is an MPN arising from a single genetic translocation in a pluripotential hematopoietic stem cell producing a clonal overproduction of the myeloid cell line, which results in a preponderance of immature cells in the neutrophilic line. CML begins with a chronic clinical phase that progresses to an accelerated phase in 3 to 4 years and often terminates as an acute leukemia if left untreated. The clinical features are frequent infection, anemia, bleeding, and splenomegaly, all secondary to massive pathologic accumulations of myeloid progenitor cells in bone marrow, peripheral blood, and extramedullary tissues. Neutrophilia with all maturational stages present, basophilia, eosinophilia, and often thrombocytosis are noted in peripheral blood. The clonal origin of hematopoietic cells in CML has been verified in studies of females heterozygous for glucose-6-phosphate dehydrogenase. Only one isoenzyme is active in affected cells, whereas two isoenzymes are active in nonaffected cells.12
Cytogenetics of the Philadelphia Chromosome
A unique chromosome, the Philadelphia chromosome, is present in proliferating hematopoietic stem cells and their progeny in CML and must be identified to confirm the diagnosis. Although the cause of Philadelphia chromosome formation is unknown, it appears more frequently in populations exposed to ionizing radiation.13,14 In most patients, a cause cannot be identified. Appearance of the Philadelphia chromosome in donor cells after allogeneic bone marrow transplantation indicates the possibility of a transmissible agent.15 The Philadelphia chromosome was first identified as a short chromosome 22 in 1960 by Nowell and Hungerford in Philadelphia.16 In 1973 Rowley, of the University of Illinois at Chicago, discovered that the Philadelphia chromosome is a reciprocal translocation between the long arms of chromosomes 9 and 22 (see Chapter 31).17 This acquired somatic mutation specifically reflects the translocation of an ABL proto-oncogene from band q34 of chromosome 9 to the breakpoint cluster region (BCR) of band q11 of chromosome 22, which results in a unique chimeric gene, BCR/ABL.18 This new gene produces a 210-kD BCR/ABL fusion protein (p210BCR/ABL) that expresses enhanced tyrosine kinase activity compared with its natural enzymatic counterpart.
Molecular Genetics
The t(9;22) translocation that produces the BCR/ABL1 chimeric gene has been observed in four primary molecular forms that produce three versions of the BCR/ABL chimeric protein: p190, p210, and p230 (Figure 34-1). The four genetic variations are based on the area of the BCR gene that houses the breakpoint on chromosome 22, because the breakpoint on chromosome 9 occurs in the same location. The wild-type (normal) ABL1 gene on chromosome 9 is a relatively large gene of approximately 230 kilobases (kb) containing 11 exons. The breakpoint consistently occurs 5′ of the second exon such that exons 2 to 11 are contributed to the BCR/ABL1 fusion gene.
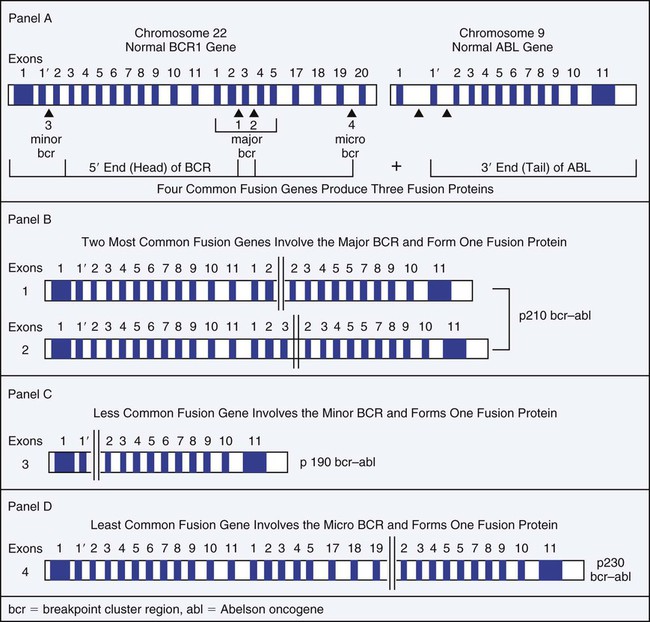
There are four BCR genes in the human genome: BCR1, BCR2, BCR3, and BCR4. It is the BCR1 gene that is involved in the Philadelphia translocation. Wild-type (normal) BCR1 gene is approximately 100 kb with 20 exons. In 1984 Groffen et al identified the BCR on chromosome 22 as a 5-exon region involving exons 12 to 16 that was the area of breakage in the traditional t(9;22) translocation.19 This area was later termed the major BCR. Two other areas of breakage were identified on chromosome 22, one nearer the 5′ (head) of the BCR1 gene, called the minor BCR, and one in the 3′ end (tail) of the BCR1 gene termed the micro BCR.
Within the major BCR two specific breakpoints account for the t(9;22) translocation involved in the development of CML. Therefore, two areas of breakage in the major BCR combined with one in the minor BCR and one in the micro BCR produce the four versions of the BCR/ABL1 chimeric gene. Because the two breakpoints in the major BCR differ by only one exon, the chimeric protein product is essentially the same size and is designated as the p210 protein. Breakage in the BCR1 gene in the major BCR contributes exons 1 to 13 or 1 to 14, whereas the ABL1 gene contributes exons 2 to 11. Breakage in the minor BCR contributes only exon one from BCR, which joins with the same exons 2 to 11 of ABL1 to produce a p190 protein. The micro BCR breakpoint contributes exons 2 to 19 from BCR1, which fuse with ABL1 exons 2 to 11, producing the p230 protein. Therefore, the four possible BCR1 breakpoints produce four different chimeric genes, resulting in a total of three different protein products.20
Pathogenetic Mechanism
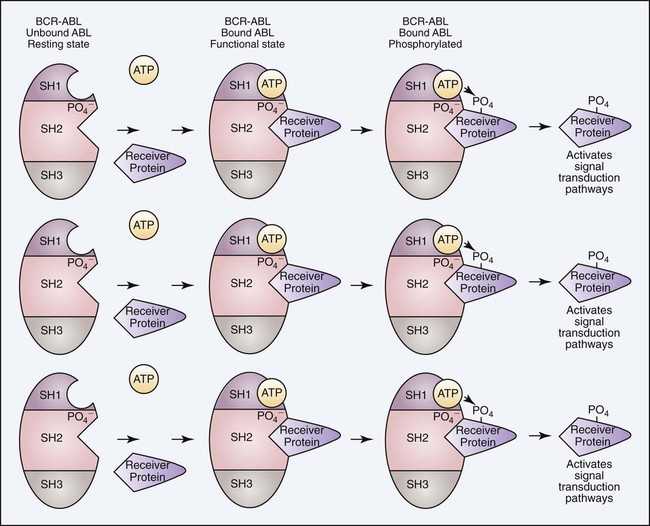
To understand the aberrant function of the BCR/ABL fusion protein, it is first helpful to understand both the normal BCR and ABL proteins. The wild-type ABL protein, when in its usual location on chromosome 9, codes for p125, which exhibits normal tyrosine kinase activity. The BCR1 gene produces p160, expresses serine and threonine kinase activity, and is thought to function in the regulation of cell growth. Protein kinases are enzymes that catalyze the transfer of phosphate groups from adenosine triphosphate (ATP), guanosine triphosphate, and other phosphate donors to receiver proteins. A tyrosine kinase transfers the phosphate group to a tyrosine amino acid on the receiver protein. For the kinase activity of the ABL protein to occur, the ABL protein must first be phosphorylated. This is often accomplished through autophosphorylation. The ABL protein has three primary domains called SR1, SR2, and SR3 that together express and regulate the kinase activity. SR1 is the binding site for ATP; SR2 is the docking point for phosphate receiver proteins; and SR3 is the domain that controls the phosphorylation activity. When ATP binds to the ATP binding site, the phosphate is transferred to the SR2 region of the ABL protein, which initiates a conformational change that alters the tertiary structure of the protein and exposes the active site of the enzyme. When a second ATP binds the ATP binding site and a receiver protein docks in the SR2 domain, the phosphate group is transferred to the receiver protein. In most physiologically normal intracellular pathways, protein phosphorylation activates the receiver proteins (Figure 34-2). This phosphorylation initiates a cascade of phosphorylation events, each activating the next protein until a transcription factor becomes activated. These activation cascades, called signal transduction pathways, are designed to activate genes necessary to control cell proliferation, differentiation, and natural cell death, called apoptosis. There are several signal transduction pathways activated by the ABL tyrosine kinase that function in concert to activate these genes in a precise order and at the required level of activation to control these cellular events.20,21
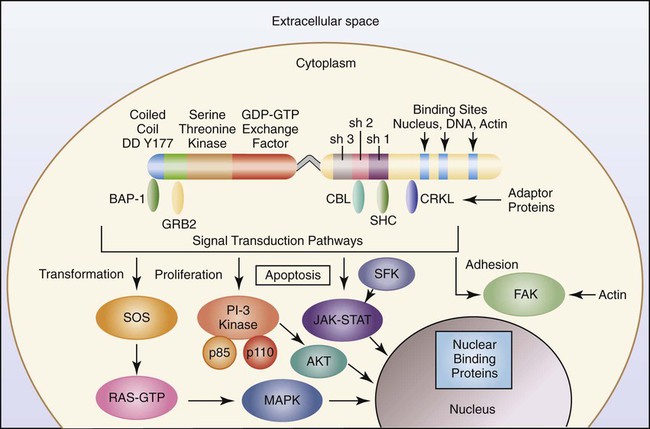
In the case of CML, the BCR/ABL1 translocation occurs next to the SR3 domain of the ABL1 moiety, which is designed to control the rate and timing of phosphorylation. Therefore, the BCR/ABL tyrosine kinase has lost the ability to shut off kinase activity and is said to have constitutive tyrosine kinase activity. The BCR/ABL enzyme continuously adds phosphate groups to tyrosine residues on cytoplasmic proteins, activating several signal transduction pathways. These pathways stimulate gene expression keeping the myeloid cells proliferating, reducing differentiation, reducing adhesion of cells to bone marrow stroma, and virtually eliminating apoptosis. The result is increased clonal proliferation of myeloid cells secondary to a reduction in or loss of sensitivity to protein regulators.22 There is an increase in growth factor–independent cellular proliferation from activation of the RAS gene and a decrease in or resistance to apoptosis. New clones of stem cells vulnerable to additional genetic changes lead to the accelerated and blast phases of CML. In addition, the BCR/ABL protein localizes in the cytoplasm, rather than in the nucleus as does the normal ABL protein. The mutation affects maturation and differentiation of hematopoietic and lymphopoietic cells, whose progeny eventually dominate in the affected individual. Progeny cells that exhibit this chromosome include neutrophils, eosinophils, basophils, monocytes, nucleated erythrocytes, megakaryocytes, and B lymphocytes.7,23
In addition, the loss of genetic segments in the 5′ end of the ABL1 gene results in an altered protein-binding affinity for F-actin, which leads to a reduction in contact binding of hematopoietic CML cells to stromal cells and causing premature release of cells into the circulation.21 Abnormal adhesion between stem cells and stroma may dysregulate hematopoiesis. One action of interferon-α therapy is to reverse the loss of adhesion of CML progenitor cells, which reduces the premature release of these cells into the circulation.24
Peripheral Blood and Bone Marrow
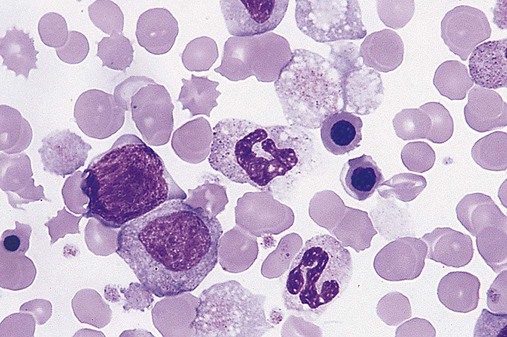
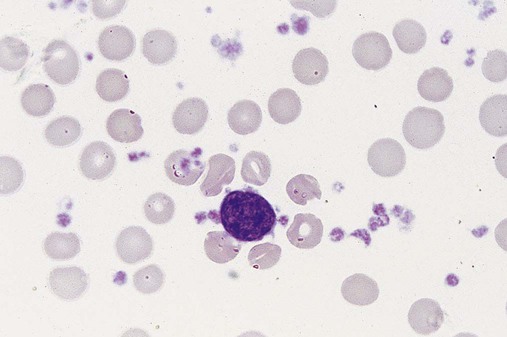
There are dramatic morphologic changes in the peripheral blood and bone marrow that reflect the expansion of the granulocyte pool, particularly in the later maturational stages. Table 34-1 lists the qualitative changes in the peripheral blood, bone marrow, and extramedullary tissues that are commonly observed at the time of diagnosis. A dramatic left shift is noted that extends down to the promyelocyte stage and occasionally even produces a few blasts in the peripheral blood. The platelet count is often elevated, reflecting the myeloproliferative nature of the disease. Extramedullary granulopoiesis may involve sinusoids and medullary cords in the spleen and sinusoids, portal tract zones, and solid areas of the liver.
TABLE 34-1
Common Morphologic Changes in Chronic Myelogenous Leukemia
| Peripheral Blood | |
| Erythrocytes | Normal or decreased |
| Reticulocytes | Normal |
| Nucleated red blood cells | Present |
| Total white blood cells | Increased |
| Lymphocytes | Normal or increased |
| Neutrophils | Increased |
| Basophils | Increased |
| Eosinophils | Increased |
| Myelocytes | Increased |
| Leukocyte alkaline phosphatase | Decreased |
| Platelets | Normal or increased |
| Bone Marrow | |
| Cellularity | Increased |
| Granulopoiesis | Increased |
| Erythropoiesis | Decreased |
| Megakaryopoiesis | Increased or normal |
| Reticulin | Increased |
| Macrophages | |
| Gaucherlike | Sea blue |
| Green-gray crystals | Increased |
| Megakaryocytes | |
| Small | Increased |
| Extramedullary Tissue | |
| Splenomegaly | Present |
| Sinusoidal | Present |
| Medullary | Present |
| Hepatomegaly | Present |
| Sinusoidal | Present |
| Portal tract | Present |
| Local infiltrates | Present |
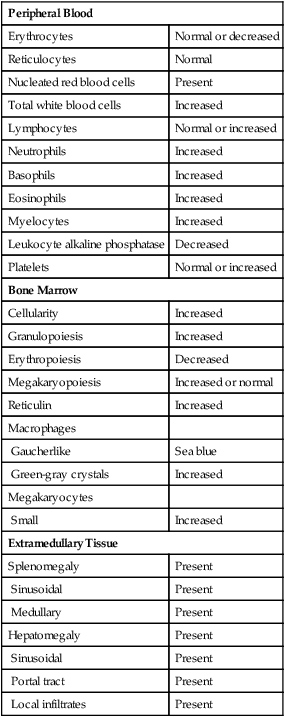
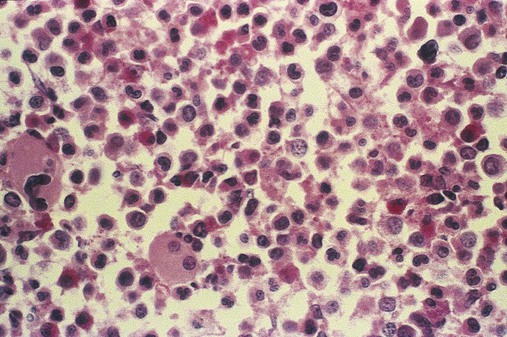
Figure 34-3 illustrates a common pattern in the peripheral blood film of chronic phase CML at the time of diagnosis. Leukocytosis is readily apparent at scanning microscopic powers. Segmented neutrophils, bands, metamyelocytes, and myelocytes predominate, and immature and mature eosinophils and basophils are increased. Myeloblasts and promyelocytes are present at a rate of approximately 1% and 5%, respectively. Lymphocytes and monocytes are present and often show an absolute increase in number but a relative decrease in percentage. Nucleated red blood cells (NRBCs) are rare. Platelets are normal or increased, and some may exhibit abnormal morphology.
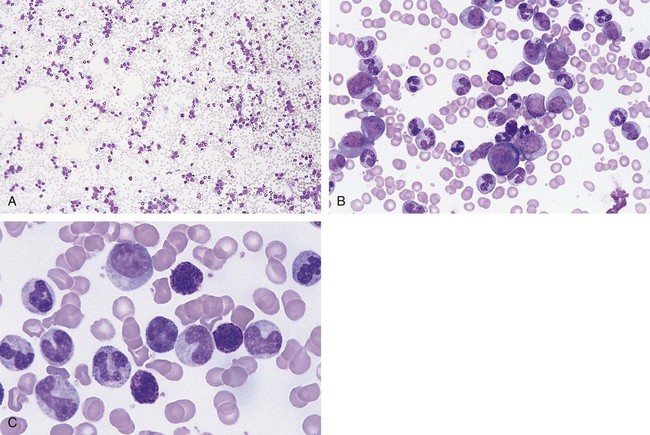
Bone marrow changes are illustrated in Figure 34-4. An intense hypercellularity is present due to granulopoiesis, marked by broad zones of immature granulocytes, usually perivascular or periosteal, differentiating into more centrally placed mature granulocytes. Normoblasts appear reduced in number. Megakaryocytes are normal or increased in number and, when increased, may appear in clusters and exhibit dyspoietic cytologic changes. They often appear small with reduced nuclear size (by approximately 20%) and reduced nuclear lobulations. Reticulin fibers are increased in approximately 20% of patients. Increased megakaryocyte density is associated with an increase in myelofibrosis.25 The presence of pseudo-Gaucher cells (see Chapter 28) usually occurs.
Other Laboratory Findings
Hyperuricemia and uricosuria from increased cell turnover may be associated with secondary gout, urinary uric acid stones, and uric acid nephropathy.26 Approximately 15% of patients exhibit total white blood cell (WBC) counts greater than 300 × 109/L.27 Symptoms in these patients are secondary to vascular stasis and possible intravascular consumption of oxygen by the leukocytes. Symptoms are reversible with the lowering of the total WBC count.28
Progression
In the pre-imatinib era, most patients’ disease would eventually transform into acute leukemia.29 Before blastic transformation, some patients proceed through an intermediate metamorphosis or accelerated phase. Disease progression is accompanied by an increase in the frequency and number of clinical symptoms, adverse changes in laboratory values, and poorer response to therapy than in the chronic phase. Additional chromosome abnormalities may appear, associated with enhanced dyshematopoietic cell maturation patterns and increases in morphologic and functional abnormalities in blood cells. There is often an increasing degree of anemia and, in the peripheral blood, fewer mature leukocytes, more basophils, and fewer platelets, with a greater proportion of abnormal platelets, micromegakaryocytes, and megakaryocytic fragments. The circulating blast count increases to 10% to 19%. This total blast percentage, or a combination of 20% blasts and promyelocytes, has been proposed as a diagnostic criterion for the accelerated phase.30
Blast crisis involves the peripheral blood, bone marrow, and extramedullary tissues. By definition, blasts constitute more than 20% of total bone marrow cellularity; the peripheral blood exhibits increased blasts.29 Blast crisis leukemia usually is AML or ALL, but origins from other hematopoietic clonal cells are possible. Extramedullary growth may occur as lymphocytic or myelogenous cell proliferations; the latter are often referred to as granulocytic sarcoma. Extramedullary sarcoma is observed at many sites or locations in the body and may precede a marrow blast crisis. The clinical symptoms of blast crisis mimic those of acute leukemia, including severe anemia, leukopenia of all WBCs except blasts, and thrombocytopenia. Chromosome abnormalities such as additional Philadelphia chromosome(s), isochromosome 17, trisomy 8, loss of Y chromosome, and trisomy 19 accumulate with disease progression.31,32 These generally occurred in approximately 75% of patients in the pre-imatinib era.
Related Diseases
Several diseases exist that are clinically similar to CML but do not exhibit the Philadelphia chromosome and express only a few pseudo-Gaucher cells. Chronic neutrophilic leukemia is another MPN that manifests with peripheral blood, bone marrow, and extramedullary infiltrative patterns similar to those of CML, except that only neutrophilic granulocytes are present and fewer than 10% of peripheral blood neutrophils are immature.33 Similarly, chronic monocytic leukemia involves a comparable expansion of monocytes, including functional monocytes.34
Juvenile myelomonocytic leukemia and adult chronic myelomonocytic leukemia are classified by the WHO as myelodysplastic/myeloproliferative diseases because of the overlap in clinical, laboratory, or morphologic findings. Juvenile myelomonocytic leukemia is observed in children younger than 4 years of age and is accompanied by an expansion in the number of monocytes and granulocytes, including immature granulocytes, and manifestations of dyserythropoiesis.35
The peripheral blood of adults with chronic myelomonocytic leukemia may have characteristics similar to those seen in the refractory anemias, such as oval macrocytes and reticulocytopenia. The peripheral WBC concentration may reach 100 × 109/L. According to WHO criteria, absolute monocytosis (more than 1 × 109 monocytes/L) must be present to make the diagnosis. Clinical features include prominent splenomegaly, symptoms of anemia, fever, bleeding, and infection. Before presence of the Philadelphia chromosome was established as a requirement for the diagnosis of CML, some cases that were classified as Philadelphia chromosome–negative CML likely represented misdiagnoses of chronic myelomonocytic leukemia.36 Chronic myelomonocytic leukemia is discussed further with myelodysplastic syndromes in Chapter 35.
A puzzling group of patients exhibit Philadelphia chromosome–positive acute leukemia. Studies reveal that 2% of patients with AML exhibit Philadelphia chromosome in a significant proportion of blasts. Further, 5% of patients with childhood-onset ALL and 20% of those with adult-onset ALL test positive for Philadelphia chromosome.37–40 The proper alignment of these cases within the spectrum of CML is speculative. It is understood that some of these cases likely represent undiagnosed CML that rapidly progressed to an acute leukemia prior to diagnosis. However, because rapidly dividing malignant cells are more prone to genetic mutation, the presence of the Philadelphia chromosome in acute leukemias may reflect a late-stage mutation that contributed little to acute leukemia leukemogenesis.
Treatment
Early treatment approaches for CML were unable to produce remission, so the goal of therapy became the reduction of tumor burden. The first forms of therapy for CML included alkylating agents such as nitrogen mustard,41 introduced in the late 1940s, and busulfan,42 which came into use in the early 1950s. Later, busulfan in combination with 6-thioguanine was used to achieve the goal of tumor burden reduction. Other drugs like hydroxyurea and 6-mercaptopurine were introduced later and found to improve patient survival. The discovery of interferon-α in 1983 dramatically improved outcomes of patients with CML by inducing the suppression of the Philadelphia chromosome, reducing the rate of cellular progression to blast cells, and increasing the frequency of long-term patient survival.43
Interferon-α stimulates a cell-mediated antitumor host response that reduces myeloid cell numbers, induces cytogenetic remissions, and increases survival.44 It improves the frequency and duration of hematologic remission and reduces the frequency of detection of the Philadelphia chromosome. In some patients, a complete cytogenetic remission is achieved for a time.
In 1997 it was discovered that cytarabine given with interferon-α improved the frequency of hematologic remissions but did not eliminate the BCR/ABL gene, which was still detected by molecular and fluorescent methods.45 Also, in some patients the side effects of therapy became severe, drug resistance appeared, and relapse rates were not improved compared with other chemotherapies.
Bone marrow and stem cell transplantation with either autologous or allogeneic hematopoietic stem cells have been reported as curative, especially in patients younger than age 55. Relapses occur, but long-term, disease-free survival is possible. Optimal survival occurs when the patient is treated during the chronic phase within 1 year of diagnosis and is younger than age 50. Treatment requires ablative chemotherapy followed by transplantation of mobilized normal progenitor cells that exhibit CD34+ surface markers. Allogeneic bone marrow transplants are more successful in patients up to age 55 when donors are matched for HLA antigens A, B, and DR. Donor-matched lymphocyte infusions after allogeneic transplantation of marrow from a sibling donor may assist in producing complete remissions.46
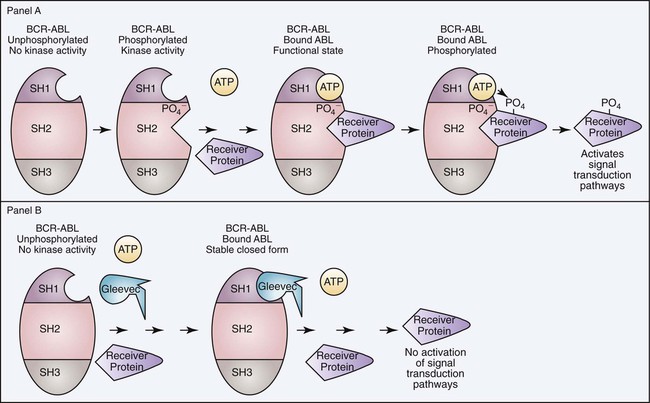
Modern therapies involve the use of synthetic proteins that bind the abnormal BCR/ABL protein, blocking the constitutive tyrosine kinase activity and reducing signal transduction activation. Imatinib mesylate is a synthetic tyrosine kinase inhibitor designed to selectively bind the ATP binding site and thus inhibit the tyrosine kinase activity of the BCR/ABL fusion protein. When imatinib binds the ATP binding site, ATP is unable to bind to provide the phosphate group necessary for kinase activity. Imatinib binds the BCR/ABL protein in the inactive conformation, which precedes the autophosphorylation necessary to generate the kinase active site (Figure 34-5).47
Goals of therapy include complete hematologic remission and cytogenetic remission induced in part by the reactivation of apoptotic pathways.48 The effectiveness of imatinib therapy and stem cell transplantation is best monitored using quantitative real-time reverse transcriptase polymerase chain reaction (RT-PCR). These monitoring tools are used to determine complete cytogenetic and molecular remission. The most sensitive measure of the effectiveness of imatinib therapy is the number of log reductions of BCR/ABL transcripts using real-time RT-PCR.49 Expected therapeutic response to imatinib is complete hematologic remission in 3 to 6 months, complete cytogenetic response in 6 months to 1 year, and a 2 to 3 log reduction in BCR/ABL transcripts. When real-time RT-PCR is used, the greatest log reduction possible is a more than 4 log reduction, which represents the maximum sensitivity of the assay. However, discontinuation of imatinib therapy in patients who achieve a more than 4 log reduction will result in relapse.
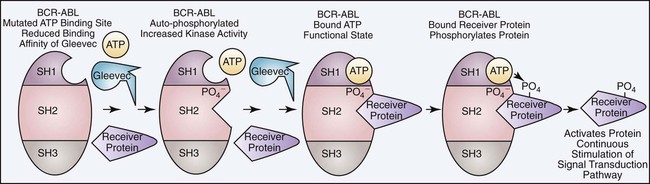
Although imatinib has proven to be a successful form of therapy, a major limitation is the development of imatinib resistance resulting in relapse. Approximately 4% of patients with newly diagnosed CML will become imatinib resistant. There are two major categories of imatinib resistance, primary and secondary. Primary resistance is defined as the inability to reach the remission milestones. This form of resistance is rare and probably results from the presence of mutations other than the BCR/ABL mutation at the time of diagnosis. Secondary resistance involves the loss of a previous response and occurs at a rate of 16% at 42 months. The majority of cases of imatinib resistance result from two primary causes: acquisition of additional BCR/ABL mutations and expression of point mutations in the ATP binding site. Additional BCR/ABL mutations can occur through the usual translocation of the remaining unaffected chromosomes 9 and 22, which converts the hematopoietic stem cell from heterozygous to homozygous for the BCR/ABL mutation. A double dose of BCR/ABL can also be acquired from gene duplication during mitosis and accounts for 10% of secondary mutations. An additional BCR/ABL mutation will double the tyrosine kinase activity, making the imatinib dosage inadequate. In these cases higher dosages of imatinib will restore remission in most patients (Figure 34-6). The majority of patients who do not respond to higher dosages of imatinib express point mutations in the ATP binding site. Over 50 mutations have been identified in the ATP binding site, and these account for the remaining 50% to 90% of secondary mutations. Mutations in the ATP binding site reduce the binding affinity of imatinib, producing some level of resistance (Figure 34-7). Second- and third-generation tyrosine kinase inhibitors like dasatinib (Sprycel), nilotinib (Dasigna), and bosutinib (still in clinical trials at the time of publication) overcome the ATP binding site mutations except the T315I mutation. However, drugs designed to bind the A loop (receiver protein binding site) will inhibit tyrosine kinase activity and overcome the T315I mutation.50,51 Currently, studies are under way to evaluate modification of the dosage of imatinib used, to identify and develop other tyrosine kinase inhibitors, and to discover new classes of inhibitors that may be more effective than currently known tyrosine kinase inhibitors.
The development of a care plan for treating a patient with newly diagnosed CML requires not only the formulation of alternative approaches to achieve complete cellular remission, but also the establishment of parameters to follow that confirm long-term success of therapy. Previous chemotherapies have provided cellular remission but usually do not suppress or delay clinical transitions to accelerated or blast phases. Bone marrow transplantation for patients who qualify is likely the preferred choice, but the long-term success (cure) rate remains at 50% to 70%. For a patient to qualify for transplantation, the patient must be younger than 50 years of age, must be in the first year of the disease, and must have CML that is still in the chronic phase, and a histocompatible donor must be available. Today, imatinib is considered first-line therapy for all patients with newly diagnosed CML. For the small subset of patients who qualify for hematopoietic stem cell transplantation, imatinib is used to induce hematologic remission prior to transplantation. For all other CML patients, imatinib is used as first-line therapy unless remission is not achieved (primary resistance) or until relapse occurs following remission (secondary resistance). Once the cause of relapse has been determined by cytogenetic and molecular testing, either a higher dosage of imatinib can be given (for an additional BCR/ABL mutation) or a second- or third-generation tyrosine kinase inhibitor (dasatinib, nilotinib, bosatinib) can be prescribed unless the mutation is the T315I mutation in the ATP binding site. If the T315I mutation is detected, the patient can be given an A-loop inhibitor (ONO12380) or other drugs like MK 0457 or BIRB-796 that inhibit the T315I mutation.51
Polycythemia Vera
Polycythemia vera (PV) is a neoplastic clonal myeloproliferative disorder that commonly manifests with panmyelosis in the bone marrow and increases in erythrocytes, granulocytes, and platelets in the peripheral blood.2 Splenomegaly is common. The disease arises in a hematopoietic stem cell. The hypothesis of a clonal origin for PV is supported by studies of X-linked restriction fragment length deoxyribonucleic acid (DNA) polymorphisms that demonstrate monoclonal X chromosome inactivation in all blood cells.52
Pathogenic Mechanism
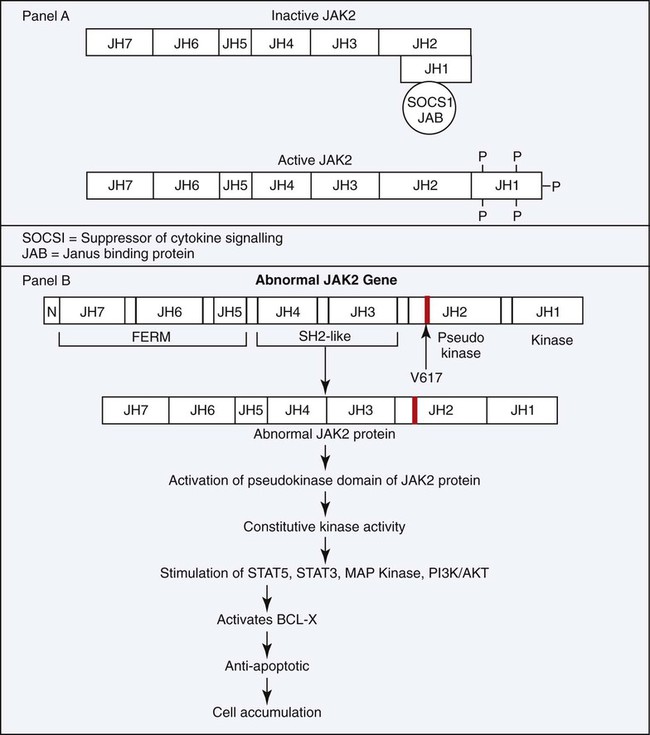
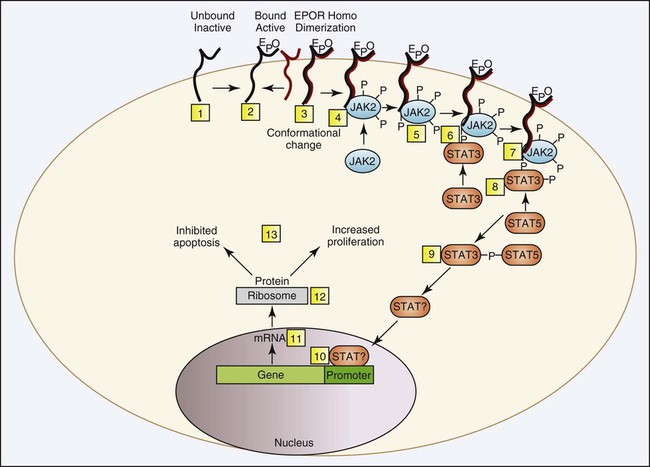
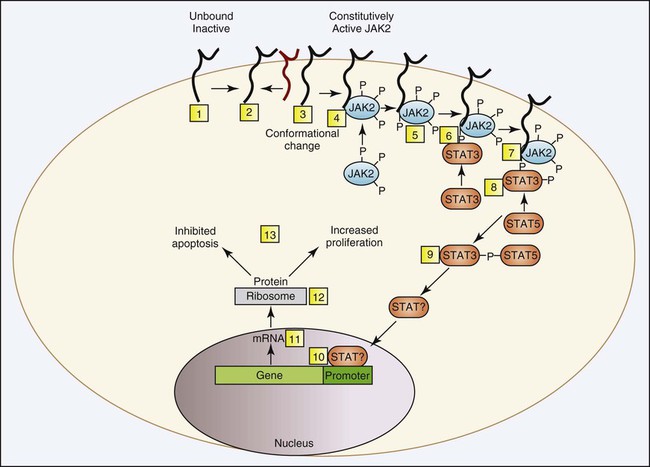
In PV, neoplastic clonal stem cells are hypersensitive to, or function independently of, erythropoietin for cell growth. Trace levels of erythropoietin in serum stimulate the growth of erythroid progenitor cells in in vitro colony-forming growth systems. There is preservation of hypersensitive and normosensitive erythroid colony-forming units, however, which indicates some level of normal hematopoiesis.53 Adverse clinical progression seems to correlate with the propagation of the erythropoietin-sensitive colony-forming units.54 Understanding of the pathologic mechanism explaining this phenomenon in PV was significantly advanced in 2005 with the discovery of a consistent mutation in the JAK2 gene. The specific JAK2 mutation, JAK2 V617F, is detected in 90% to 97% of patients with PV. Shortly after the JAK2 V617F mutation was reported, several groups corroborated the finding using other approaches and showed that the mutation is acquired, clonal, present in the hematopoietic stem cell, constitutively active, and capable of activating the erythropoietic signal transduction pathway in the absence of erythropoietin. The point mutation changes the amino acid at position 617 from valine to phenylalanine, which unlocks the inhibition of the tyrosine kinase and converts it to the active conformation. More specifically, the phenylalanine mutation in the kinase domain is unable to bind the corresponding amino acid in the pseudokinase domain, as can the wild-type counterpart valine, which prevents the protein from folding and remaining inactive (Figure 34-8). Normally, erythropoietin is released from the kidney into the blood in response to hypoxia and binds to erythropoietin receptors on the surface of erythroid precursor cells. The resulting conformational change in the erythropoietin receptor causes two erythropoietin receptors to dimerize. This produces a docking point for inactive JAK2, which becomes activated through phosphorylation. Phosphorylated JAK2 undergoes a conformational change, and the valine releases from the pseudokinase domain, which converts it to an active tyrosine kinase. JAK2 phosphorylates several cytoplasmic proteins, but the signal transducer and activator of transcription (STAT) proteins are the main target. A cascade of phosphorylations produce activated transcription factors that activate a host of genes designed to drive and control cell proliferation and differentiation while also initiating apoptosis (Figure 34-9). Constitutive tyrosine kinase activity of the JAK2 protein causes continuous activation of several signal transduction pathways that are normally activated following erythropoietin stimulation via the erythropoietic receptor. Active JAK2 will phosphorylate STAT proteins in the absence of erythropoietin or will overphosphorylate in its presence (Figure 34-10). Hematopoietic stem cells that bear the JAK2 mutation are resistant to erythropoietin-deprivation apoptosis by upregulation of BCL-X, an antiapoptotic protein. PV progenitor cells do not divide more rapidly but accumulate because they do not die normally.55–57
Diagnosis
Based on the WHO standards, the diagnosis of PV requires that two major criteria and one minor criterion be met or that the first major criterion listed and two minor criteria be met. The two major criteria are an elevated hemoglobin (Hb) level (greater than 18.5 g/dL men and greater than 16.5 g/dL in women) and the identification of the JAK2 V617F mutation, the JAK2 exon 12 mutation, or a similar JAK2 mutation. The three minor criteria are panmyelosis in the bone marrow, low serum erythropoietin levels, and autonomous, in vitro erythroid colony formation.6 Additional diagnostic features of PV include an increased RBC mass of 36 mL/kg or greater in males and 32 mL/kg or greater in females, an arterial oxygen saturation of 92% (normal) or greater, and splenomegaly. Other features of PV are thrombocytosis of greater than 400 × 109 platelets/L; leukocytosis of greater than 12 × 109 cells/L without fever or infection; and increases in leukocyte alkaline phosphatase (LAP), serum vitamin B12, or unbound vitamin B12 binding capacity.58,59 Recent research indicates that the JAK2 V617F mutation can be expected in more than 90% to 95% of cases.6 The WHO criteria for the diagnosis of PV are summarized in Box 34-1.
Peripheral Blood and Bone Marrow
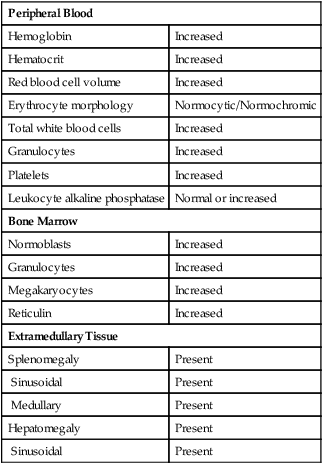
Common peripheral blood, bone marrow, and tissue findings in the early or proliferative phase of PV are listed in Table 34-2. Figures 34-11 and 34-12 show common morphologic patterns in peripheral blood and bone marrow morphologic and cellular changes. Not only are quantitative changes seen, but bone marrow normoblasts may collect in large clusters, megakaryocytes are enlarged and exhibit lobulated nuclei, and bone marrow sinuses are enlarged without fibrosis. Pseudo-Gaucher cells are rare.26 Approximately 80% of patients manifest bone marrow panmyelosis, and 100% of bone marrow volume may exhibit hematopoietic cellularity. Although the bone marrow pattern may mimic that in other MPNs, the peripheral blood cells appear normal, with normocytic, normochromic erythrocytes; mature granulocytes; and normal-sized, granulated platelets. The other 20% of patients exhibit lesser degrees of cellularity in the bone marrow and peripheral blood. Splenomegaly, hepatomegaly, generalized vascular engorgement, and circulatory disturbances increase the risk of hemorrhage, tissue infarction, and thrombosis.
TABLE 34-2
Common Morphologic Changes in Polycythemia Vera
| Peripheral Blood | |
| Hemoglobin | Increased |
| Hematocrit | Increased |
| Red blood cell volume | Increased |
| Erythrocyte morphology | Normocytic/Normochromic |
| Total white blood cells | Increased |
| Granulocytes | Increased |
| Platelets | Increased |
| Leukocyte alkaline phosphatase | Normal or increased |
| Bone Marrow | |
| Normoblasts | Increased |
| Granulocytes | Increased |
| Megakaryocytes | Increased |
| Reticulin | Increased |
| Extramedullary Tissue | |
| Splenomegaly | Present |
| Sinusoidal | Present |
| Medullary | Present |
| Hepatomegaly | Present |
| Sinusoidal | Present |
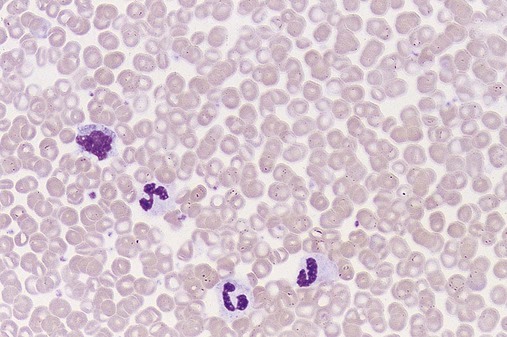
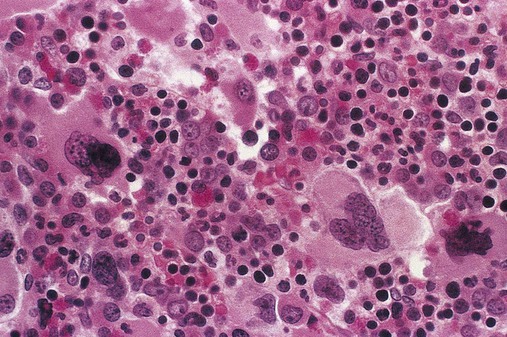
Clinical Presentation
PV initially manifests in a proliferative phase independent of normal regulatory mechanisms. PV is always associated with increased RBC mass. This is the stable phase of PV, which progresses to a spent phase in a few patients. In the spent phase, patients experience progressive splenomegaly (palpable spleen) or hypersplenism (large spleen with bone marrow hyperplasia and peripheral blood cytopenias) and pancytopenia. They may also exhibit the triad of bone marrow fibrosis, splenomegaly, and anemia with teardrop-shaped poikilocytes. The latter pattern is called postpolycythemic myeloid metaplasia, and its morphologic features are similar to those of PMF. Peripheral WBC and RBC counts vary, and nucleated erythrocytes, immature granulocytes, and large platelets are present. Usually, splenomegaly is secondary to extramedullary hematopoiesis.60 Myelofibrosis occurs within the bone marrow and may come to occupy a significant proportion of bone marrow volume, with subsequent ineffective hematopoiesis.61
Treatment and Prognosis
The disease progresses to acute leukemia in 15% of patients. The use of myelosuppressive therapy such as phosphorus P 32 (32P) or alkylating agents seems to increase the risk.59 Only 1% to 2% of patients treated with phlebotomy alone experience leukemic transformation. However, the risk of thrombosis and bleeding is increased in patients treated with phlebotomy alone, so the use of alkylating myelosuppressive agents may be required to control these complications. Some patients may manifest a temporary disease pattern similar to myelodysplasia, and the cell morphology in transformation to acute leukemia may be difficult to classify. Patients with both early and advanced PV may show clinical, peripheral blood, bone marrow, and extramedullary features that mimic those of other MPNs.
Essential Thrombocythemia
Essential thrombocythemia (ET) is a clonal MPN with increased megakaryopoiesis and thrombocytosis, usually with a count greater than 600 × 109/L and sometimes with a count greater than 1000 × 109/L.62 However, WHO criteria require a sustained thrombocytosis with a platelet count of 450 × 109/L or greater. Over the years, ET has been known as primary thrombocytosis, idiopathic thrombocytosis, and hemorrhagic thrombocythaemia.63
Incidence
In the absence of a well-defined diagnostic algorithm, determining the true incidence of ET has been difficult. When the diagnostic system developed by the Polycythemia Vera Study Group (PVSG) is applied, however, the incidence is estimated to be between 0.6 and 2.5 cases per 100,000 persons per year. The majority of cases occur in individuals between the ages of 50 and 60 years, but a second peak occurs primarily in women in the childbearing years, approximately 30 years of age.63
Clinical Presentation
In more than one half of the patients diagnosed with ET, the disorder is discovered in the laboratory by virtue of an unexpectedly elevated platelet count on a routine complete blood count; the remaining patients see a physician due to vascular occlusion or hemorrhage. Vascular occlusions are often the result of microvascular thromboses in the digits or thromboses in major arteries and veins that occur in a variety of organ systems, including splenic or hepatic veins as in Budd-Chiari syndrome. Bleeding occurs most frequently from mucous membranes in the gastrointestinal and upper respiratory tracts. Splenomegaly is observed at presentation in 50% of patients when the PVSG diagnostic criteria are used but at a much lower frequency when the WHO standards are applied. This difference is largely due to the elimination of the diagnosis of ET in patients who meet the criteria for PMF in the prefibrotic stage.63
Diagnosis
ET must be differentiated from secondary or reactive thrombocytoses and from other MPNs. Thrombocytosis may be secondary to chronic active blood loss, hemolytic anemia, chronic inflammation or infection, or nonhematogenous neoplasia. The diagnostic criteria for ET first proposed by the PVSG were intended to distinguish ET from other MPNs. These features included a platelet count of greater than 600 × 109/L, a hemoglobin of less than 13 g/dL or a normal erythrocyte mass, and stainable iron in the bone marrow or a failure of iron therapy. Philadelphia chromosome negativity, absence of marrow collagen fibrosis (less than one third of a biopsy specimen is fibrous), no splenomegaly, absence of leukoerythroblastic reaction, and no known cause of reactive thrombocytosis all support the diagnosis.64
The newest WHO group now requires the documentation of four major criteria to establish a diagnosis of ET. First, the WHO group lowered the platelet count threshold to 450 × 109/L or greater to capture patients who would eventually meet diagnostic criteria but who were experiencing hemorrhage or thrombosis with platelet counts of between 450 × 109/L and 600 × 109/L.63 Because a lower platelet threshold could lead to false-positive ET diagnoses, all the WHO criteria must be met to eliminate such patients. Second, the bone marrow must show significant megakaryopoiesis characterized by large, mature-looking megakaryocytes with no substantial increase in erythropoiesis or granulopoiesis or left shift in the neutrophil line. Third, the condition cannot meet the criteria of any other MPN, myelodysplasia, or other myeloid neoplasm. Fourth, patients must demonstrate either the JAK2 V617F or other clonal mutation or, in the absence of a clonal marker, the absence of reactive thrombocytosis. Careful analysis of the bone marrow biopsy specimen is useful in distinguishing ET from myelodysplastic syndromes (MDSs) associated with the del(5q) mutation, refractory anemia with ringed sideroblasts with thrombocytosis, and the prefibrotic phase of PMF. Likewise, the identification of the JAK2 V617F mutation excludes cases of reactive thrombocytosis.63
The JAK2 V617F mutation is found in 50% to 60% of ET patients and supports the diagnosis of ET.65,66 WHO diagnostic criteria were modified to include a minimum platelet count of 450 × 109/L when the JAK2 mutation is present.67 JAK2 mutations have not been identified in the germline of any patient with MPN disorder, which supports the view that the mutation is acquired. MPL W515K/L, a mutation in the thrombopoietin receptor (MPL), has been reported in 1% of ET cases and is also used to exclude a diagnosis of reactive thrombocytosis. Other genetic mutations are uncommon but have been reported to be found in 5% to 10% of cases when the diagnostic criteria proposed by the PVSG are applied. The most commonly reported additional mutations are +8, 9q, and (del)20q.63
Peripheral Blood and Bone Marrow
Figure 34-13 shows a peripheral blood film that exhibits early-phase thrombocytosis with variation in platelet diameter and shape, including giantism, agranularity, and pseudopods. Commonly, platelets are present in clusters and tend to accumulate near the thin edge of the blood film. Segmented neutrophils may be increased; basophils are not. Erythrocytes are normocytic and normochromic, unless iron deficiency is present secondary to excessive bleeding.
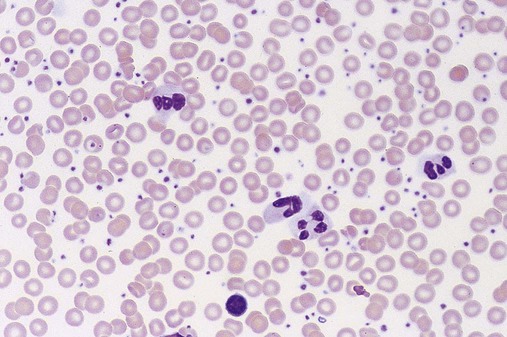
Early-phase bone marrow shows marked megakaryocytic hypercellularity, clustering of megakaryocytes, and increased megakaryocyte diameter with nuclear hyperlobulation and density (Figure 34-14). Special studies reveal increased numbers of smaller and less mature megakaryocytes.68 Increased granulopoiesis and erythropoiesis may contribute to bone marrow hypercellularity, and in a few patients, reticulin fibers may be increased. The major peripheral blood, bone marrow, and extramedullary findings are listed in Table 34-3.
TABLE 34-3
Common Morphologic Changes in Essential Thrombocythemia
| Peripheral Blood | |
| Hemoglobin | Slightly decreased |
| Hematocrit | Slightly decreased |
| Red blood cell volume | Normal |
| Total white blood cells | Normal or slightly increased |
| Neutrophils | Normal or slightly increased |
| Platelets | Increased |
| Platelet function | Decreased |
| Bone Marrow | |
| Normoblasts | Normal or increased |
| Granulocytes | Normal or slightly increased |
| Megakaryocytes | |
| Clusters | Present |
| Large | Present |
| Hyperlobulated | Present |
| Dense nuclei | Present |
| Variability in size | Increased |
| Reticulin | Normal or slightly increased |
| Extramedullary Tissue | |
| Splenomegaly | Present |
| Sinusoidal | Present |
| Medullary | Present |
| Megakaryocytic proliferation | Present |
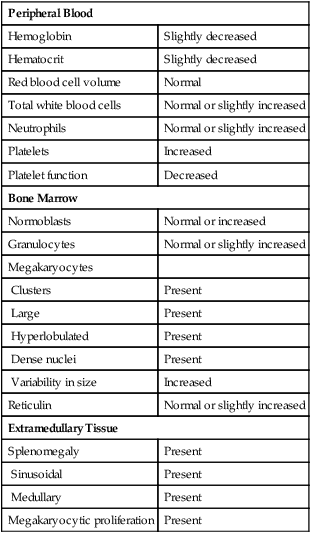
A diagnosis of ET is questionable in patients with a platelet count of more than 450 × 109/L if certain features are observed on the bone marrow biopsy specimen. For example, increased erythropoiesis or granulopoiesis in the bone marrow is a questionable finding for ET and suggests an alternative diagnosis of PV or PMF, respectively, especially if bizarre or significantly atypical megakaryocytes are also observed. Dyserythropoiesis and/or dysgranulopoiesis suggests a myelodysplastic disorder and should prompt an investigation for (del)5q, (inv)3, and/or t(3 ; 3).63
Treatment and Prognosis
Patients with ET experience relatively long survival provided they remain free of serious thromboembolic or hemorrhagic complications. Clinical symptoms associated with thromboembolic vasoocclusive events include the syndrome of erythromelalgia (throbbing and burning pain in the hands and feet, accompanied by mottled redness of areas), transient ischemic attacks, seizures, and cerebral or myocardial infarction. Other symptoms include headache, dizziness, visual disturbances, and dysesthesias (decreased sensations). Hemorrhagic complications include bleeding from oral and nasal mucous membranes or gastrointestinal mucosa and the appearance of cutaneous ecchymoses (see Chapter 43).
Treatment involves prevention or early alleviation of hemorrhagic or vasoocclusive complications that occur as the platelet count increases. The production of platelets must be reduced by suppressing marrow megakaryocyte production with 32P or one of several alkylating agents. As observed in PV, ET patients so treated may incur an increased risk for disease transformation to acute leukemia or myelofibrosis. However, malignant transformation occurs at a frequency of less than 5%.63 Hydroxyurea therapy may achieve a desired reduction of peripheral platelets without the risk of complications experienced with myelosuppressive agents. This may relate to the youth of ET patients, in whom the risk of leukemic transformation seems relatively low. More recently, cytoreduction has been achieved with interferon-α.
Studies commonly show a median survival of longer than 10 years, including cases in which the process arises in younger patients. However, some patients may develop post-ET myelofibrosis, which reduces survival. Patients whose cells manifest chromosome abnormalities may have a poorer prognosis.37
Primary Myelofibrosis
PMF clonality was manifest in studies in which cytogenetic abnormalities were detected in normoblasts, neutrophils, macrophages, basophils, and megakaryocytes. Female patients heterozygous for glucose-6-phosphate dehydrogenase isoenzymes have PMF cells of a single enzyme isotype, whereas tissue cells, including marrow fibroblasts, contain both enzyme isotypes.4
Myelofibrosis
The myelofibrosis in this disease comprises three of the five types of collagen: I, III, and IV. Increases in type III collagen are detected by silver impregnation techniques, increases in type I by staining with trichrome, and increases in type IV by the presence of osteosclerosis, which may be diagnosed from increased radiographic bone density.69 In approximately 30% of patients, biopsy specimens show no fibrosis.37 Increases in these collagens are not a part of the clonal proliferative process but are considered secondary to an increased release of fibroblastic growth factors, such as platelet-derived growth factor, transforming growth factor α from megakaryocyte α-granules, tumor necrosis factor-α, and interleukin-1α, and interleukin-1β. Marrow fibrosis causes expansion of marrow sinuses and vascular volume with an increased rate of blood flow. Bone marrow fibrosis is not the sole criterion for the diagnosis of PMF, because increases in marrow fibrosis may reflect a reparative response to injury from benzene or ionizing radiation, may be a consequence of immunologically mediated injury, or may represent a reactive response to other hematologic conditions.
Type IV collagen and laminin normally are discontinuous in sinusoidal membranes but appear as stromal sheets in association with neovascularization and endothelial cell proliferation in regions of fibrosis. In addition, deposition of type VII collagen is observed, and this may form a linkage between type I fibers, type III fibers, and type I plus type III fibers.70
Hematopoiesis and Extramedullary Hematopoiesis
Extramedullary hematopoiesis, clinically recognized as hepatomegaly or splenomegaly, seems to originate from release of clonal stem cells into the circulation.71 The cells accumulate in the spleen, liver, or other organs, including adrenals, kidneys, lymph nodes, bowel, breasts, lungs, mediastinum, mesentery, skin, synovium, thymus, and lower urinary tract. The cause of extramedullary hematopoiesis is unknown. In experimental animal models, chemicals, hormones, viruses, radiation, and immunologic factors have been implicated. The disease is associated with an increase in circulating hematopoietic cells, but fibroblasts are a secondary abnormality and not clonal.72 B and T cells may be involved.73 There is an increase in circulating unilineage and multilineage hematopoietic progenitor cells,74 and the number of CD34+ cells may be 300 times normal.75 The increase in circulating CD34+ cells separates PMF from other MPNs and predicts the degree of splenic involvement and risk of conversion to acute leukemia.
Body cavity effusions containing hematopoietic cells may arise from extramedullary hematopoiesis in the cranium, the intraspinal epidural space, or the serosal surfaces of pleura, pericardium, and peritoneum. Portal hypertension, with its attendant consequences of ascites, esophageal and gastric varices, gastrointestinal hemorrhage, and hepatic encephalopathy, arises from the combination of a massive increase in splenoportal blood flow and a decrease in hepatic vascular compliance secondary to fibrosis around the sinusoids and hematopoietic cells within the sinusoids.76
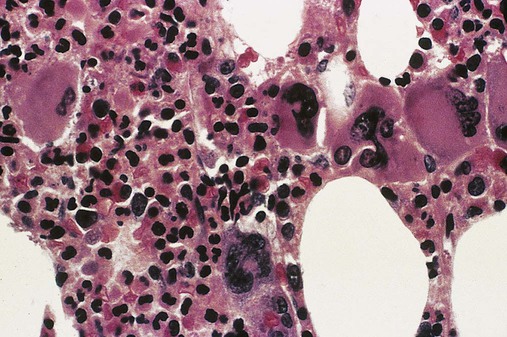
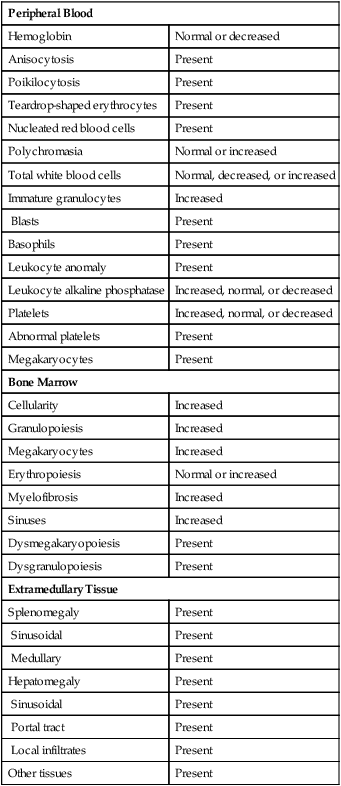
Peripheral Blood and Bone Marrow
PMF presents with a broad range of changes in laboratory test values and peripheral blood film results, but examination of the bone marrow biopsy specimen provides most of the information for diagnosis. Changes commonly observed in peripheral blood and bone marrow examinations are summarized in Table 34-4.
TABLE 34-4
Common Morphologic Changes in Primary Myelofibrosis
| Peripheral Blood | |
| Hemoglobin | Normal or decreased |
| Anisocytosis | Present |
| Poikilocytosis | Present |
| Teardrop-shaped erythrocytes | Present |
| Nucleated red blood cells | Present |
| Polychromasia | Normal or increased |
| Total white blood cells | Normal, decreased, or increased |
| Immature granulocytes | Increased |
| Blasts | Present |
| Basophils | Present |
| Leukocyte anomaly | Present |
| Leukocyte alkaline phosphatase | Increased, normal, or decreased |
| Platelets | Increased, normal, or decreased |
| Abnormal platelets | Present |
| Megakaryocytes | Present |
| Bone Marrow | |
| Cellularity | Increased |
| Granulopoiesis | Increased |
| Megakaryocytes | Increased |
| Erythropoiesis | Normal or increased |
| Myelofibrosis | Increased |
| Sinuses | Increased |
| Dysmegakaryopoiesis | Present |
| Dysgranulopoiesis | Present |
| Extramedullary Tissue | |
| Splenomegaly | Present |
| Sinusoidal | Present |
| Medullary | Present |
| Hepatomegaly | Present |
| Sinusoidal | Present |
| Portal tract | Present |
| Local infiltrates | Present |
| Other tissues | Present |
Abnormalities in erythrocytes noted on peripheral blood films include the presence of dacryocytes, other bizarre shapes, nucleated RBCs, and polychromatophilia. Granulocytes are increased, normal, or decreased in number and may include immature granulocytes, blasts, and cells with nuclear or cytoplasmic anomalies. Platelets may be normal, increased, or decreased in number with a mixture of normal and abnormal morphologic features (Figure 34-15). Micromegakaryocytes may be observed (Figure 34-16).
Immune Response
Humoral immune responses are altered in approximately 50% of patients and include the appearance of autoantibodies to erythrocyte antigens, nuclear proteins, gamma globulins, phospholipids, and organ-specific antigens.77 Circulating immune complexes, increased proportions of marrow-reactive lymphocytes, and the development of amyloidosis are evidence for active immune processes. Collagen disorders coexist with PMF, which suggests that immunologic processes may stimulate marrow fibroblast activity.
Treatment and Prognosis
Average survival from the time of diagnosis is about 5 years, but patients have lived 15 years. During this time, increasing numbers and pleomorphy of megakaryocytes lead to progressive marrow failure. Marrow blasts may increase.37 Adverse prognostic indicators include more severe anemia and thrombocytopenia, greater hepatomegaly, unexplained fever, and hemolysis. Mortality is associated with infection, hemorrhage, postsplenectomy complications, and transformation to acute leukemia.
Chemotherapy is partially successful in reducing the number of CD34+ cells and immature hematopoietic cells, marrow fibrosis, and splenomegaly.78 Single-agent chemotherapy is most helpful in the early clinical phases of the disease, and agents such as busulfan, 6-thioguanine, and chlorambucil, alone or in combination with other chemotherapy, are useful. Other therapies include interferon-α, hydroxyurea, and combinations of the previously mentioned drugs.79 The most successful treatment to date for patients younger than age 60 is allogeneic stem cell transplantation. Five-year survival approaches 50% in patients undergoing transplantation, but 1-year mortality is 27%, and graft-versus-host disease occurs in 33%.80
Interconnection Between Essential Thrombocythemia, Polycythemia Vera, and Primary Myelofibrosis
Currently four hypotheses exist to account for this apparent discordance. One prevailing thought suggests that the resulting phenotype is dependent on the stage of differentiation of the hematopoietic stem cell. For example, if the hematopoietic stem cell has developed a predilection toward platelet development at the time of the JAK2 mutation, ET will develop. Reports have described differences in differentiation programs81,82 and in JAK2 mutations among ET, PV, and PMF.83 A second hypothesis proposes that the genetic background of the patient predisposes the patient to a particular phenotype. Mutations in the erythropoietin receptor, thrombopoietin receptor (MPL), and granulocyte colony-stimulating factor receptor have all been implicated.84 The third hypothesis suggests that the phenotype depends of the level of JAK2 tyrosine kinase activity, called the dosage effect. Patients diagnosed with PV showed greater tyrosine kinase activity than patients presenting with an ET phenotype. Experiments in which erythroid progenitors were collected from these patients and tested in colony-forming assays showed that nearly all the cell cultures developing a PV phenotype were homozygous for the JAK2 mutation, whereas the ET phenotype was observed in the vast majority of cell cultures expressing a heterozygous genotype.85,86 This phenomenon was corroborated in experiments with transgenic mice.87,88 The last hypothesis proposes that a pre-JAK2 mutation produces a premalignant clone89–91 predisposing the hematopoietic stem cell to a particular phenotype and that the JAK2 mutation drives the malignant transformation. Groups have reported mutations coexisting with the JAK2 V617F mutation, like BCR/ABL,92,93 MPL mutations,94 and another version of the JAK2 mutation.95,96 Familial MPN provides the strongest support for a pre-JAK2 mutation.97,98
The most appealing model includes all of the hypotheses previously presented and suggests that ET, PV, and PMF may represent a continuum of diseases. It seems reasonable to assume that a pre-JAK2 mutation occurs in the hematopoietic stem cell most if not all of the time to create a hyperproliferative clone that predisposes to additional mutations like the JAK2 mutation. The pre-JAK2 mutation can be familial, congenital, or somatic. Because MPL is expressed in high levels on megakaryocyte precursors, one JAK2 V617F mutation (heterozygous) is sufficient to induce MPL signaling and thus stimulate megakaryocyte production. This could lead to the ET phenotype. In contrast, the erythropoietin receptor is expressed in low density on the surface of erythroid precursors, which requires the higher amount of JAK2 V617F tyrosine kinase that is produced by two JAK2 mutations (homozygous). This could lead to the PV phenotype. Because MPL stimulation begins with the first JAK2 mutation and continues with the second JAK2 mutation, the MPL receptor undergoes continuous stimulation. It has been shown that excessive thrombopoietin stimulation leads to myelofibrosis, which may result in a progression to PMF.99
Other Myeloproliferative Neoplasms
Chronic Neutrophilic Leukemia
Chronic neutrophilic leukemia (CNL) is a clonal disorder in which a hyperproliferation of neutrophilic cells in the bone marrow produces sustained neutrophilia in the peripheral blood and hepatosplenomegaly. CNL must be differentiated from CML, based on the absence of the Philadelphia chromosome and the BCR/ABL fusion gene, as well as from both a reactive neutrophilic process and other MPNs.100
Clinical Presentation
Hepatosplenomegaly is the most common finding, but 25% to 30% of patients report bleeding from mucocutaneous sites like the gastrointestinal tract. Other symptoms include gout from WBC turnover and pruritus that may be associated with neutrophil infiltration of tissues and organs.100
Peripheral Blood and Bone Marrow
Patients have a WBC count of more than 25 × 109/L with a slight left shift. Neutrophils dominate but the increase in bands, metamyelocytes, myelocytes, and promyelocytes in combination usually comprise fewer than 5% of WBCs but can be as many as 10%. Neutrophils do not appear dysplastic, but they often contain toxic granules. RBC and platelet morphology are normal in the peripheral blood.100
The bone marrow reflects the peripheral blood in that it is hypercellular with predominately a proliferation of neutrophils, including myelocytes, metamyelocytes, bands, and segmented neutrophils. The myeloid-to-erythroid ratio is at least 20 : 1 or greater. RBCs and platelets are normal in number, and no cell line exhibits significant dysplastic morphology.100
Diagnosis
The WBC count must be greater than 25 × 109/L with greater than 90% mature neutrophils, fewer than 10% immature neutrophilic cells, and fewer than 1% blasts in the peripheral blood. The bone marrow shows an increase in normal-appearing neutrophilic cells with fewer than 5% myeloblasts. Megakaryocytes are normal or slightly left-shifted. Splenomegaly must be present and is often accompanied by hepatomegaly. Reactive neutrophilia must be excluded by eliminating infection, inflammation, and tumors as a cause of the neutrophilia. A diagnosis of CNL can still be made in the presence of a reactive process if clonality of the myeloid line can be documented by karyotyping or molecular analysis. There must be no evidence of the Philadelphia chromosome, BCR/ABL mutation, or rearrangements of the PDGFRA, PDGFRB, or FGRF1 genes. Lastly, there can be no evidence of PV, PMF, ET, MDS, or MDS/MPN disorders.100
Genetics
Approximately 90% of CNL patients have a normal karyotype, but chromosomal abnormalities are observed, particularly as the disease progresses, including +8, +9, +21, del(20q), del(11q), and del(12p). The Philadelphia chromosome or a BCR/ABL mutation cannot be expressed in CNL; otherwise a diagnosis of CML is required. JAK2 mutations have been observed but rarely.100
Chronic Eosinophilic Leukemia, Not Otherwise Specified
Chronic eosinophilic leukemia (CEL) is a clonal proliferation of eosinophils from eosinophil precursors that dominate in the bone marrow and peripheral blood. Eosinophils are found in other peripheral tissues, including heart, lungs, central nervous system, gastrointestinal tract, and skin. Hepatosplenomegaly is observed in approximately 30% to 50% of patients. Infiltrating eosinophils degranulate to release cytokines, enzymes, and other granular proteins that damage the surrounding tissue, which results in organ dysfunction.101
Clinical Presentation
Although some patients may be asymptomatic when found to have eosinophilia, most have signs and symptoms of fever, fatigue, cough, angioedema, muscle pain, and pruritus. A more severe sequela of CEL involves the heart. Fibrosis can form in the heart (endomyocardial fibrosis), which can evolve into cardiomegaly. Within the heart scar tissue may form in the mitral and tricuspid valves, affecting valve function and predisposing to thrombi formation. Other serious complications include peripheral neuropathy, central nervous system dysfunction, pulmonary symptoms from eosinophilic infiltrates, and rheumatologic problems.101
Peripheral Blood and Bone Marrow
Peripheral eosinophilia must be observed, with the majority of eosinophils appearing normal. Some evidence of eosinophil abnormality is found, however, and includes the presence of eosinophilic myelocytes and metamyelocytes, hypogranulation, and vacuolization. Neutrophilia is a common finding; other features such as mild monocytosis, basophilia, and the presence of blasts are less common. The bone marrow is hypercellular owing to eosinophilic proliferation and can demonstrate Charcot-Leyden crystals. Myeloblast numbers are elevated but below the 20% threshold necessary to classify the disorder as an acute leukemia. Erythrocytes and megakaryocytes are normal in number but sometimes demonstrate dysplastic morphologic features. Bone marrow fibrosis occurs due to the release of eosinophilic basic protein and eosinophilic cationic proteins from the eosinophil granules. Bone marrow fibrosis contributes to the premature release of eosinophils into the circulation, and they deposit in a variety of tissues.101
Diagnosis
The diagnosis of CEL requires eosinophilia with a count of more than 1.5 × 109 cells/L and the presence of malignant features, and the elimination of reactive eosinophilia and other malignancies that have concomitant eosinophilia. Reactive conditions like parasitic infections, allergies, Loeffler syndrome (pulmonary disease), cyclical eosinophilia, angiolymphoid hyperplasia of the skin, collagen vascular disorders, and Kimura disease must be excluded in the differential diagnosis. Likewise, other malignancies that can produce a concomitant eosinophilia include T-cell lymphoma, Hodgkin lymphoma, systemic mastocytosis, chronic myelomonocytic leukemia, atypical CML, and ALL. These disorders lead to the release of a variety of interleukins that can drive a secondary eosinophil reaction. No single genetic abnormality is specific for CEL, but CEL can be ruled out by the presence of several karyotypic abnormalities, such as PDGFRA, PDGFRB, FGFR1, and BCR/ABL. Common myeloid mutations like +8, i(17q), and JAK2 can support a diagnosis of CEL.101
Mastocytosis
Mastocytosis is a broad term referring to a clonal neoplastic proliferation of mast cells, which accumulate in one or more organ systems, but it can present differently and manifest in a range of severities. The WHO group has classified mastocytosis into seven subcategories: cutaneous mastocytosis, indolent systemic mastocytosis, systemic mastocytosis with associated clonal hematologic non–mast-cell-lineage disease, aggressive systemic mastocytosis, mast cell leukemia, mast cell sarcoma, and extracutaneous mastocytoma.102
Incidence
Mastocytosis can occur at any age, with cutaneous mastocytosis seen most often in children. Babies can be born with cutaneous mastocytosis, and half of affected children develop the disease before 6 months of age. In contrast, systemic mastocytosis generally occurs after the second decade of life. Approximately 80% of patients with mastocytosis show skin involvement regardless of the type of mastocytosis diagnosed. Cutaneous mastocytosis occurs in the skin; systemic mastocytosis usually involves the bone marrow and other organ systems like the spleen, lymph nodes, liver, and gastrointestinal tract; and mast cell leukemia is characterized by mast cells in the peripheral blood.102
Clinical Presentation
Patients present with urticarial lesions (wheel and flare) that may become activated when stroked upon physical examination. Skin lesions also tend to have melanin pigmentation. Four categories of symptom severity have been described in mastocytosis: (1) constitutional systems like fatigue and weight loss; (2) skin manifestations; (3) mediator-related systemic events such as abdominal pain, gastrointestinal distress, headache, and respiratory symptoms; and (4) musculoskeletal complaints like bone pain, arthralgias, and myalgias. Hematologic findings include anemia, leukocytosis, eosinophilia, neutropenia, and thrombocytopenia. In patients with systemic mastocytosis with associated clonal hematologic non–mast-cell-lineage disease the most common associated hematologic finding is chronic myelomonocytic leukemia, but any myeloid or lymphoid malignancy can occur, although myeloid versions predominate.102
Diagnosis
The typical skin lesion is the first diagnostic clue to mastocytosis. Cutaneous mastocytosis occurs in three forms: urticaria pigmentosa, diffuse cutaneous mastocytosis, and mastocytosis of the skin, all of which occur predominantly in children. In urticaria pigmentosa mast cells are confined to the skin and form aggregates in the dermis, whereas in diffuse cutaneous mastocytosis, mast cells are found in more than one cutaneous location. In systemic mastocytosis, mast cells are observed in one area of the bone marrow with fibrosis and other areas of the bone marrow are hypercellular with panmyelosis, and/or mast cells are identified in other extracutaneous sites. In addition to the major criteria just described, at least one of four minor criteria must be met. These include the following: (1) More than 25% of mast cells must be immature or have atypical morphology like a spindle shape. (2) Mast cells must express a KIT mutation at codon 816. (3) Mast cells must express normal markers and CD2 and/or CD25. (4) Total serum tryptase must be above 20 mg/mL. The key diagnostic features that separate the six types of systemic mastocytosis are as follows: (1) Indolent systemic mastocytosis is characterized by a low mast cell burden. (2) Systemic mastocytosis with associated clonal hematologic non–mast-cell-lineage disease presents with one of the following WHO classifications of neoplasms: myelodysplastic syndrome, myeloproliferative neoplasm, AML, lymphoma, or other hematopoietic neoplasm. (3) Aggressive systemic mastocytosis usually does not manifest with skin lesions or mast cells in circulation but does have one or more of the following: mast cells in bone marrow, dysplastic hematopoietic changes, or hepatosplenomegaly. (4) Mast cell leukemia is characterized by more than 20% atypical mast cells in the bone marrow and more than 10% in the peripheral blood. (5) Mast cell sarcoma presents as a single unifocal mast cell tumor with a high-grade pathology. (6) Extracutaneous mastocytoma also exhibits a unifocal mast cell tumor, but it is of low-grade pathology.102
Genetics
The most common genetic mutation in patients with mastocytosis involves codon 816 in the KIT gene and occurs in about 95% of adults and 33% of children with systemic mastocytosis. This mutation replaces aspartic acid with valine, which alters the tyrosine kinase receptor activity so as to cause constitutive kinase activity in the absence of ligand. Usually the mutation is somatic, but a few cases of familial KIT mutations have been reported. Additional mutations can push proliferation of hematopoietic clones, causing systemic mastocytosis with associated clonal hematologic non–mast cell lineage disease. These include mutations of RUNX1/RUNX1T in AML, JAK1 in MPN, and FIP1L1/PDGFRA in myeloid neoplasms with eosinophilia.102
Prognosis
Cutaneous mastocytosis in children has a favorable prognosis and may regress spontaneously around puberty. Milder versions like cutaneous mastocytosis and indolent cutaneous mastocytosis follow a benign course and are associated with a normal life span. Hematologic involvement usually evolves into the corresponding hematologic disease. Patients with aggressive systemic mastocytosis, mast cell leukemia, and mast cell sarcoma are often treated with cytoreductive chemotherapy but may survive only a few months after diagnosis. Signs and symptoms that predict a poorer prognosis include elevated lactate dehydrogenase and alkaline phosphatase, anemia, thrombocythemia, abnormal peripheral blood morphology, bone marrow hypercellularity, and hepatosplenomegaly.102
Myeloproliferative Neoplasm, Unclassifiable
The category myeloproliferative neoplasm, unclassifiable (MPN-U) is designed to capture disorders that clearly express myeloproliferative features but either fail to meet the criteria of a specific condition or have features that overlap two or more specific conditions. Most patients with MPN-U fall into one of three groups: (1) patients with an early stage of PV, ET, or PMF in which the criteria that define the disorders are not yet fully developed; (2) patients presenting with features indicative of advanced disease resulting from clonal evolution that masks the potential underlying condition; and (3) patients who have clear evidence of an MPN but who have a concomitant condition like a second neoplasm or an inflammatory condition that alters the MPN features. MPN-U may account for as many as 10% to 15% of MPN disorders, but caution should be exercised so that morphologic changes caused by the patient’s cytotoxic drug therapy or growth factor therapy, or poor collection of samples are not confused with the features of MPN-U. In patients with MPN-U in the early stages of development or with a concomitant disorder like inflammation, the MPN-U may be reclassified to a specific category of MPN once the disease begins to express typical features or the secondary conditions subsides. Likewise, in patients with an advanced MPN, the disorder may be reclassified as an acute leukemia once the blast criterion of more than 20% blasts in the bone marrow is met.103
Summary
• MPNs are clonal hematopoietic stem cell disorders that result in excessive production and overaccumulation of erythrocytes, granulocytes, and platelets in some combination in bone marrow, peripheral blood, and body tissues.
• Within the classification of MPN the four major conditions are CML, PV, ET, and PMF.
• In CML, there are large numbers of myeloid precursors in the bone marrow, peripheral blood, and extramedullary tissues.
• The peripheral blood exhibits leukocytosis with increased myeloid series, particularly the later maturation stages, often with increases in eosinophils and basophils.
• The LAP score is dramatically decreased in CML.
• The Philadelphia chromosome, t(9;22), either at the chromosomal or molecular level, must be present in all cases of CML.
• In CML, the bone marrow exhibits intense hypercellularity with a predominance of myeloid precursors. Megakaryocyte numbers are normal to increased.
• Patients with CML progress from a chronic stable phase through an accelerated phase into transformation to acute leukemia.
• Bone marrow transplantation has been successful in CML, and imatinib mesylate, a tyrosine kinase inhibitor, produces remission in most cases.
• Approximately 4% of CML patients given imatinib as first-line therapy develop imatinib resistance.
• Dosage escalation or administration of second-generation tyrosine kinase inhibitors restores remission in most patients with imatinib resistance.
• PV manifests with panmyelosis in the bone marrow with increases in erythrocytes, granulocytes, and platelets.
• The clinical diagnosis of PV requires a hemoglobin of greater than 18.5 g/dL in men and greater than 16.5 g/dL in women or other evidence of increased RBC volume and the presence of JAK2 V617F or another mutation in the JAK2 gene. If only one of these major criteria is met, two of the minor criteria must be satisfied.
• The JAK2 V617F mutation is found in 90% to 95% of PV patients and contributes to the pathogenesis of the disease.
• ET involves an increase in megakaryocytes with a sustained platelet count greater than 450 × 109/L.
• Other diagnostic criteria include normal RBC mass, stainable iron in the bone marrow, absence of the Philadelphia chromosome, lack of marrow collagen fibrosis, absence of splenomegaly or leukoerythroblastic reaction, and absence of any known cause of reactive thrombocytosis.
• In the early phases of ET, peripheral blood shows increased numbers of platelets with abnormalities in size and shape. Bone marrow megakaryocytes are increased in number and in size.
• Complications of ET include thromboembolism and hemorrhage.
• The JAK2 V617F mutation is observed in 50% to 60% of patients with ET and PMF and contributes to the pathogenesis of the disorders.
• PMF manifests with ineffective hematopoiesis, sparse areas of marrow hypercellularity (especially with increased megakaryocytes), bone marrow fibrosis, splenomegaly, and hepatomegaly.
• The peripheral blood in PMF exhibits immature granulocytes and nucleated RBCs; teardrop-shaped cells are a common finding.
• Platelets may be normal, increased, or decreased in number with abnormal morphology. Micromegakaryocytes may be present.
• Immune responses are altered in about 50% of patients.
• Other MPNs include CNL, CEL not otherwise specified, mastocytosis, and unclassifiable MPN.
Review Questions
1. A peripheral blood film that shows increased neutrophils, basophils, eosinophils, and platelets is highly suggestive of:
2. Which of the following chromosome abnormalities is associated with CML?
3. A patient has a WBC count of 30 × 109/L and the following WBC differential:
Which of the following test results would be helpful in determining whether the patient has CML?
a. Nitroblue tetrazolium reduction product increased
4. A patient in whom CML has previously been diagnosed has circulating blasts and promyelocytes that total 30% of leukocytes. The disease is considered to be in what phase?
5. The most common mutation found in patients with primary PV is:
6. The peripheral blood in PV typically manifests:
7. A patient has a platelet count of 700 × 109/L with abnormalities in the size, shape, and granularity of platelets; a WBC count of 12 × 109/L; hemoglobin of 11 g/dL. The Philadelphia chromosome is not present. The most likely diagnosis is:
8. Complications of ET include all of the following except:
9. Which of the following patterns is characteristic of the peripheral blood in patients with PMF?
a. Teardrop-shaped erythrocytes, nucleated RBCs, immature granulocytes
c. Hypochromic erythrocytes, immature granulocytes, and normal platelets
d. Spherocytes, immature granulocytes, and increased numbers of platelets
10. The myelofibrosis associated with PMF is a result of:
a. Apoptosis resistance in the fibroblasts of the bone marrow
b. Impaired production of normal collagenase by the mutated cells
c. Enhanced activity of fibroblasts owing to increased stimulatory cytokines
d. Increased numbers of fibroblasts owing to cytokine stimulation of the pluripotential stem cells