Chapter 117 Myelomeningocele and Associated Anomalies
History and Epidemiology
When describing NTD at birth, the term incidence is more clinically descriptive than is the term prevalence, because many affected fetuses abort spontaneously. NTDs exhibit wide geographic, ethnic, and gender variation. In the United States, NTD rates have declined from 1.3 per 1000 births in 1970 to 0.6 per 1000 births in 1989.1 Worldwide, numbers range from 1 to 6 per 1000 births.2 Superimposed on a general worldwide decrease in the incidence at birth of myelomeningocele, a number of influences cause both a reduction in prevalence at birth and an increased prevalence in the general population. Reduction of myelomeningocele at birth may result from maternal supplementation with folates and a higher rate of pregnancy termination due to availability of maternal serum α-fetoprotein testing and refined resolution of ultrasound for in utero fetal examination.
Recent improvements in neonatal and postoperative care have resulted in increased survival rates and, therefore, an increased prevalence in the general population. Zachary3 believed that all affected infants should be operated on, even though many survivors inevitably will suffer from multiple handicaps. Worldwide, there are several variables considered and degrees of selection for candidacy for myelomeningocele treatment. The predominant factors are the level of the myelomeningocele and associated paralysis, the severity of associated hydrocephalus, and the presence of gross deformities. The overwhelming majority of untreated infants with myelomeningocele will die early in life.4
The evolution of myelomeningocele treatment over the last few decades also has led to a tremendous change in quality of life in affected patients. McLone5 estimated that more than 75% of surviving infants with myelomeningocele will have normal intelligence; however, the frequency of learning disabilities is likely to be high. More than 80% will be ambulators by school age, while more than 90% will have bladder and bowel control.
Embryogenesis and Pathophysiology
During the last 100 years, various hypotheses have been proposed concerning the embryogenesis of myelomeningocele. Multiple experimental models and autopsy specimens were studied thoroughly. Myelomeningocele was produced in animal models genetically (e.g., curly tail mouse)6 or induced by medical or environmental factors such as vitamin A,7 valproic acid,8 salicylates,9 insulin,10 and hyperglycemic11 and hyperthermic12 conditions.
Four different theories have been described: simple nonclosure, overgrowth and nonclosure, reopening (overdistention), and primary mesodermal insufficiency.7 Since its introduction, the nonclosure theory has gained almost universal acceptance because of its consistency with observations of early human embryos in which NTDs have been studied during or shortly after neural tube closure. Additional support has been provided by animal models of dysraphism, virtually all of which displayed a primary defect of neural tube closure.13
Myelomeningocele includes other consistent anomalies through the CNS axis, including Chiari II malformation and hydrocephalus. After studying the initial developmental defects of the Chiari II malformation using a genetically mutated NTD mouse model, McLone and Knepper14 proposed a unified theory that describes and emphasizes the developmental sequence of associated anomalies. This pathophysiologic sequence starts with cerebrospinal fluid (CSF) leakage from the unclosed spinal defect. As a result, the usual distention and expansion of the developing ventricular system fails. The lack of distention of rhombencephalic vesicles alters the inductive effect of pressure on the surrounding mesenchyme and endochondral bone formation and results in small posterior fossa. Consequently, the development of the cerebellum and brainstem with a small posterior fossa leads to upward herniation and a dysplastic tentorium. Downward herniation results in a large foramen magnum and cerebellar vermis and brainstem displacement into the cervical segments (Chiari II malformation). Hydrocephalus is secondary to maldevelopment of the CSF pathways in the posterior fossa.
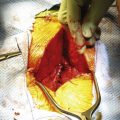
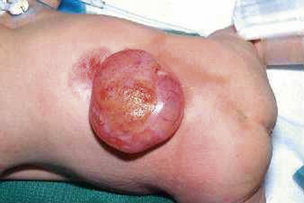
Most myelomeningoceles (85%) occur in the distal thoracic to lumbosacral spine. About 10% are detected in the higher thoracic area, and an additional 5% in the cervical area.15 Typically, a neural placode (plaque), which is unfolded neural tissue, appears at the center with a pia mater on the ventral surface. The ventral and dorsal nerve roots arise from the central surface of the placode, with the dorsal roots originating more laterally. Rostrally, the placode is continuous with the normal spinal cord within the spinal canal. At the periphery of the defect, the placode is circumscribed by arachnoid membrane that fuses with the free margins of the skin, fascia, and dura mater (Fig. 117-1). At involved levels, pedicles of vertebrae are displaced laterally, creating a widened spinal canal diameter. Vertebral bodies may be normal or wedge-shaped in the anteroposterior diameter, resulting in a kyphotic deformity.16
Etiology
The etiology of myelomeningocele is multifactorial and heterogeneous. Experimental teratology has shown that several agents can increase occurrence rates: alcohol, carbamazepine,17 valproic acid,18 salicylates,9 insulin,10 clomiphene, influenza virus,19 and chemotherapeutic agents have all been incriminated. Population studies, however, have provided strikingly little evidence to suggest the role of a single teratogen as the sole cause of a significant number of myelomeningoceles.20 Maternal age and birth order also may contribute to the risk of NTDs. Most studies show an excess of first-born children in the population of NTDs.21 In addition, most mothers of affected children are younger than 20 years of age or older than 35.22
Although rarely clustered in families, a mendelian pattern of inheritance is evident. The recurrence risk for siblings of an affected individual is 2% to 5%, representing a 25- to 50-fold increase in recurrence risk compared with that of the general population.23 A multicenter NTD genetic study reviewed the identification of genes predisposing to NTD through linkage analysis and candidate gene analysis along with characteristics of a large nationally ascertained cohort of families. Results from specific assessments of p53, PAX3, and MTHFR failed to suggest that these genes play a major role in NTD development in these families.24 A frequent association was found between trisomy 13 and 18 and myelomeningocele in the fetus before 24 weeks (as high as 14%) but was rare in full-term infants, suggesting fetal demise.25
Dietary factors related to NTDs were investigated extensively after several studies showed a progressive increase in the prevalence rate of NTDs in lower socioeconomic classes. Zinc deficiency has been considered because of a known increased risk of congenital defects in the offspring of animals fed a zinc-deficient diet,26 but human studies have been inconclusive so far.20
Folic acid antagonists, such as aminopterin, were shown to cause NTDs in animal studies, and since the publication of these studies, the effect of folic acid on prevention of myelomeningocele has been studied. In one study, serum and red blood cell levels of folate were decreased in mothers of children with myelomeningocele compared with controls.27 Folic acid and multivitamin supplementation to high-risk mothers has been demonstrated to reduce the prevalence at birth of myelomeningocele in some populations.27,28 However, adherents to the folic acid prevention philosophy acknowledge that they cannot provide a reasonable mechanism of action.18
Repair of Myelomeningocele
Evaluation of the Newborn
Prenatal diagnosis and counseling have a significant effect on the preparation and decision-making process in cases of myelomeningocele.29 Prenatal diagnosis of myelomeningocele involves the combined use of maternal serum α-fetoprotein screening and fetal ultrasonography.30 Advances in ultrasound imaging have led to an increased rate of identification of myelomeningocele before birth. On detection, obstetricians usually refer the mother and family to a multidisciplinary team that includes a pediatric neurosurgeon, neonatologist, social worker, and spina bifida team coordinator. This referral facilitates postnatal treatment, including surgery, to be planned in a timely fashion, and informs and educates the family about the nature of myelomeningocele and its associated anomalies.15
Early closure of the spinal defect remains an important part of the modern management of children born with myelomeningocele. The rationale for early closure includes the prevention of ascending infection; preservation of motor, sensory, and intellectual function; establishment of a suitable environment for continued development of neural tissue; and cosmetic reconstruction.31 Most pediatric neurosurgeons agree that closure of the open myelomeningocele within the first 24 to 48 hours after birth decreases morbidity and mortality rates. Initiating surgery after 72 hours carries a significant risk of meningitis and ventriculitis,32 a decrease of motor function, and an increase in neurologic deficits.15
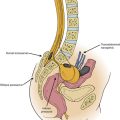
More than 90% of neonates with myelomeningocele have some form of neurogenic bladder. Dribbling of urine as the neonate cries or moves is a strong indicator of future urinary incontinence, whereas periodic micturition with a good stream suggests a possibility of partial incontinence.33 These infants also should be carefully inspected for characteristic external features that point to severe chromosomal abnormalities. Examination of the placode includes noting its shape and circumference, skin integrity, and extent of the cutaneous and epithelialized layers. The spinal column is examined for early congenital scoliosis, kyphosis, and palpable prominent laminae at the lateral margin of the lesion.
Surgical Repair
The dura mater is located just beneath the skin edge laterally. Its lateral attachment is incised and the epidural space identified (often marked by the presence of epidural fat). The dura mater should be freed rostrally and caudally to the apices of the defect on each side so that the free edges come together in the midline dorsally. The dura mater is closed in a watertight fashion. The closure should be patulous to prevent spinal cord ischemia and tethering. Occasionally, a dural graft may be required. Intradural hemostasis should be meticulous to minimize late fibrosis and scarring. With all these surgical procedures, latex reactions should be considered.34
Reapproximation of the paraspinous muscles over the repair may require some lateral dissection for mobilization. In the presence of a kyphos, the paraspinous muscles can act as spine flexors because of their lateral position, which, owing to the deformity, is ventral to the neural axis of the spine. The paraspinous muscles may then be repositioned in the normal anatomic location: dorsal and paramedian. The dorsal fascia can now be reapproximated. A continuous suture with intermittent, interrupted sutures can be used for this layer to further reinforce the watertight dural closure. It is important to note that adequate mobilization of the paraspinous muscles is crucial to prevent constriction of the underlying neural elements.15
Closure of the skin in the midline can be facilitated by a generous blunt subcutaneous dissection for mobilization. The proper plane for this blunt dissection is just superficial to the dorsal fascia. Continuous monofilament suture is used for skin closure, which further reinforces the layer by watertight closure. Occasionally, the defect is so large that skin closure cannot be accomplished without undue tension. Consultation with a plastic surgeon may be obtained if adequate skin closure is difficult or even impossible. Several surgical techniques have been used in this situation, including local rotated skin flaps, tissue expansion,35 relaxation of the lateral incisions with a skin graft,36 and a Limberg-latissimus dorsi myocutaneous flap.37
A recent development in myelomeningocele closure involves the in utero closure of myelomeningocele defects. The rationale for fetal repair of myelomeningoceles and initial clinical outcomes have been discussed over the last few years. The development of techniques to close open neural tube malformations prior to birth has generated great interest and hope for fetal interventions and their outcomes. In most recent series of patients, intrauterine myelomeningocele repair appeared to decrease the incidence of hindbrain herniation and shunt-dependent hydrocephalus in infants with myelomeningocele, but increased the incidence of premature delivery. Long-term improvement of neurologic outcome and prevention of hindbrain herniation and hydrocephalus has yet to be proven.38–41
Associated Anomalies
A multitude of associated anomalies are common in infants with myelomeningocele. CNS anomalies are more common than other systemic anomalies. Microgyria, polygyria, enlargement of the massa intermedia, agenesis or dysgenesis of the corpus callosum, and cerebellar dysgenesis, along with Chiari II malformation, are common findings that can be encountered on MRI. The midbrain, especially the tectal area, can be beaked, and the aqueduct of Sylvius can be anomalous. The pons and medulla oblongata are bowed dorsally and often extend into the rostral cervical spinal canal along with the cerebellar vermis and tonsils.15
In a series of autopsies on infants with the myelomeningocele and Chiari malformations, cerebral and cerebellar cortical malformations were found in 92% and 72% of cases, respectively. Heterotopias (i.e., displaced, well-formed gyri and folia), heterotaxias (i.e., disordered combinations of mature neurons and germinal cells), immature germinal cell collections, microgyria, and polymicrogyria were observed. Ventricular system anomalies were present in 92% of cases in the same series, including atresia, stenosis, and forking of the cerebral aqueduct. Atresia of the third ventricle and stenosis of the fourth ventricle occurred rarely. Other uncommon findings included septum pellucidum cysts and agenesis of the olfactory tracts and bulbs.42
Systemic anomalies also occur and should be considered independently when assessing infants with myelomeningocele and Chiari malformations for surgical intervention. Associated systemic anomalies appear in the gastrointestinal, pulmonary, and cardiovascular systems and in the craniofacial structures.43 The most common anomalies in the genitourinary system include hydroureter and hydronephrosis, which usually occur after long-standing neurogenic bladder. Gastrointestinal anomalies include inguinal hernia, Meckel diverticulum, malrotation, omphalocele, and imperforate anus. Cardiovascular anomalies include ventricular or atrial septal defects, patent ductus arteriosus, and coarctation of the aorta.44
Hydrocephalus
CSF shunting for hydrocephalus is required in approximately 80% of infants born with myelomeningocele.45 Most clinical signs develop in the first 3 weeks of life, but clinical signs occasionally develop at an older age.45 It is possible that the rapid onset of hydrocephalus after birth is due to the myelomeningocele acting as a compensatory reservoir that is terminated by defect closure.16
Both advanced gestational age and severity of posterior fossa deformity have been shown independently to correlate with the size of ventricles in infants born with myelomeningocele.46 The sites of obstruction of CSF pathways are variable and include the aqueduct, outlet of the fourth ventricle, arachnoid space at the level of the foramen magnum, and tentorium. Wills et al.47 showed that hydrocephalus itself has a very minor effect on intellectual development; however, prior shunt infections and ventriculitis in the neonatal period are major factors limiting intelligence.
Head circumference is below the 50th percentile in most infants born with myelomeningocele. However, serial measurements of head circumference and ultrasound examination of the head should be obtained after closure of the myelomeningocele defect to identify infants who will subsequently need shunting. A mean increase of 2 mm per day was found to have a predictive value. Infants born with overt hydrocephalus signs such as large head circumference, bulging fontanelle, split cranial sutures, and dilated scalp veins generally require early CSF diversion to relieve the intracranial hypertension and prevent myelomeningocele wound dehiscence. Signs of increased intracranial pressure in the newborn, such as bradycardia, poor sucking, apnea, and decreased spontaneous activity, indicate the need for shunting. The function of the shunt should always be considered and checked before any surgical treatment of Chiari II malformation or syringomyelia after symptoms deteriorate.5 A common practice is to wait 5 days or more after myelomeningocele closure before shunting. Most infants will tolerate this waiting period.16 In one series, shunt infection and malfunction rates were comparable between infants undergoing a simultaneous myelomeningocele closure and shunting procedures and those infants with the delayed-onset hydrocephalus and shunting.
Chiari Malformation and Syringomyelia
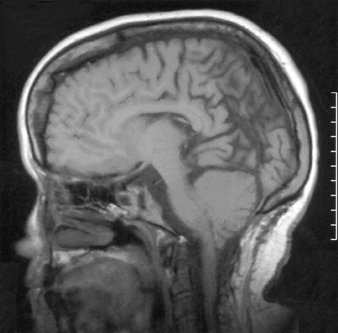
Virtually all children with myelomeningocele have a Chiari II malformation.48 The hallmark of the Chiari II malformation is the caudal displacement of the cerebellar vermis, medulla, and lower brainstem below the level of the foramen magnum into or below the craniocervical junction. The caudal descent of the lower brainstem into the cervical canal may create a kink or spur of medulla behind the cervical spinal cord in up to 70% of patients with Chiari II malformation.49 The cerebellum of these patients is smaller than normal; however, it resides in a proportionately smaller posterior fossa. The tentorium has a low insertion and often is hypoplastic (Fig. 117-2).
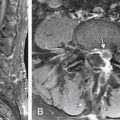
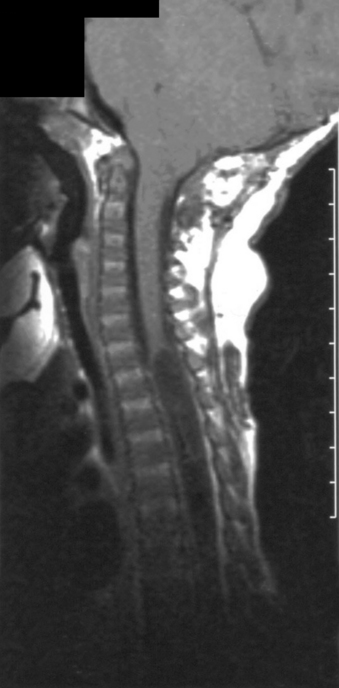
The prevalence of hydrosyringomyelia in patients with Chiari II malformation has been variably reported as 44% to 88%.50,51 The true frequency in recent series may be underestimated secondary to collapse of the syrinx after shunting and/or Chiari decompression52 (Fig. 117-3).
Between 20% and 33% of patients with Chiari II malformation become symptomatic from hindbrain herniation.52 Of these symptomatic children, nearly one third do not survive beyond infancy, making the symptomatic hindbrain herniation the leading cause of mortality in treated myelomeningocele in the first 2 years of life.5 Between 1983 and 1992, four different retrospective studies indicated that there are basically two types of presentation that are clearly age dependent and may reflect different pathophysiologies.48,52–54 Neonatal patients present with symptoms referable to lower cranial nerve and brainstem dysfunction, and older children present with spasticity, cerebellar signs, and weakness.52 Infantile symptoms include swallowing difficulty, stridor, aspiration, apnea, bradycardia, arm weakness, and opisthotonic posturing.
The surgical treatment of Chiari II malformation usually consists of decompressive laminectomy at the C1 level and lower as needed, excision of the constrictive dural band at the C1 level, and a cautious suboccipital craniectomy due to the low-lying transverse sinus. Fourth ventricular exploration may be performed to ensure that a CSF outlet and dural graft may be used in closure. The timing of surgery is controversial.53 The surgical treatment of older children can be rewarding, with almost all patients showing stabilization or return to normal function.52 Irreversible compressive or ischemic brainstem injury has been proposed as the cause of the high mortality rate and as an argument for relatively emergent surgical intervention. Two of the retrospective studies have noted no obvious evidence of brainstem dysfunction at birth.54 In autopsy specimens from infants with Chiari II malformation who had died, ischemic medullary hemorrhages confirmed that vascular compromise is a contributing cause.55 It is likely that in the one third of myelomeningocele patients who become symptomatic before 3 years of age, there is a greater degree of brainstem dysmorphism, creating a brainstem at risk that would have little tolerance for compression or ischemia. In these patients, rapid deterioration would be expected, and the results of prompt surgical intervention on the initial symptoms may be favorable.52
The indications for surgery are clearer in patients with syringomyelia. Between 60% and 73% improved after treatment for a symptomatic syrinx, and none worsened after surgery in two series.50,56 Typically, patients have an initial stable neurologic examination and then present with progressive spasticity and scoliosis. An expanding cervical hydrosyringomyelic cavity classically results in dissociative sensory loss of the upper extremities caused by interruption in the crossing spinothalamic tracts. Weakness, hyporeflexia, and atrophy of the upper musculature may result from involvement of the anterior horn cells. Motor and sensory findings can be asymmetrical, and a worsening scoliosis may be the sole manifestation of an expanding cavity of the thoracic spinal cord.53,57 Also, an expanding cervical cavity can result in a worsening lower cranial nerve dysfunction.52 Different treatment strategies have been followed. In the presence of ventriculomegaly, ventricular shunting reduces the syrinx size when there is a communication between the syrinx and the fourth ventricle (in about 10% of cases).57,58 When a shunt is already in place, its function should be evaluated and corrected. A decompressive laminectomy and opening of the foramen of Magendie to restore normal CSF hydrodynamics is indicated when a functional shunt is confirmed.57 Plugging of the obex, fourth ventricle-to-subarachnoid shunts, syringosubarachnoid shunts, and syringopleural shunts have all been used, but their use is controversial.57–60 However, syrinx shunts have been used successfully when decompression alone fails to result in clinical improvement and as a primary treatment of thoracic lesions.59
Tethered Spinal Cord
After primary repair of myelomeningocele, late progressive neurologic deterioration commonly occurs due to a tethered spinal cord at the site of previous closure secondary to scarring and adherence of the neural placode to the dorsal dura mater61 (Fig. 117-4). Tethered cord is the result of fixation of the spinal cord, which limits motion in the caudal-rostral direction. Yamada et al.62 correlated the pattern of gradually developing tethered cord symptoms with impairment of oxidative metabolism and mitochondrial anoxia as a result of excessive steady traction of the spinal cord in experimental animal models.
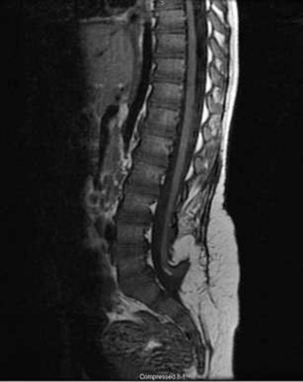
FIGURE 117-4 Sagittal T1-weighted MRI showing tethering of the spinal cord 10 years after myelomeningocele closure.
In some series, neurologic deterioration due to tethered cord occurred in 27% of children.5 The average onset of symptoms occurs at 10 years of age and correlates with a period of accelerating growth. Presenting symptoms include gait imbalance, lower back pain, weakness in the lower extremities, bladder or bowel incontinence, scoliosis, and orthopaedic deformities. MRI of the lumbar spine is the gold standard of anatomic diagnosis. However, because all spinal cords in patients with myelomeningocele appear elongated and tethered, the decision to operate depends on the demonstration of clinical progression. The decision to treat may result from deterioration seen on a urodynamic study or ascending sensory or motor neurologic loss of function.
Surgical release involves laminectomy at the vertebral level just rostral to the defect and lysis of adhesions. Additional pathology such as hydromyelia, thickened filum terminale, dermal sinus tracts, and diastematomyelia can present in 30% of patients.61 The stretched spinal cord tissue should appear relaxed at the completion of the procedure. Duraplasty may be considered to prevent postoperative dural adhesions in the repair of the tethered spinal cord.63
The timing of the surgical release of the tethered spinal cord is controversial. However, early diagnosis and early treatment are essential to provide patients with the best chance of recovery. In one report, 82% of patients who were followed closely and treated early had improvement, whereas only 54% of those not followed closely had improvement.61 Another report showed that delayed untethering, following the onset of a neurologic deficit, may reverse some lost motor function but is unlikely to restore bladder and bowel function.64
Scoliosis
Deformity of the spine is common in patients with myelomeningocele. The most commonly encountered spinal deformities are scoliosis and kyphosis. Scoliotic deformities typically develop over years as a result of imbalance of the spinal musculature and abnormal posture.65 The frequency of spinal deformities is related to the neurologic level, with nearly 100% of children with thoracic level myelomeningocele requiring corrective surgery.66 Other important factors include age, the presence of syringomyelia,67 and tethering of the spinal cord.68
Scoliosis presents in two forms in these patients: congenital and developmental.69 Congenital scoliosis arises from vertebral anomalies, particularly hemivertebrae and segmentation defects. Developmental scoliosis, the more common type, has a delayed onset and a progressive course. It produces considerable functional decline at advanced stages and generally is believed to result from muscular imbalance, which leads to progressive deformity during growth. Scoliotic curves usually develop between 5 and 10 years of age, but most deterioration occurs between 10 and 15 years of age.69
The higher the level of paralysis, the greater the incidence of spinal deformity.70 Scoliosis develops in up to 100% of patients with thoracic lesions, but in fewer than 10% of patients with sacral lesions. In a large series of 465 patients, Carstens et al.70 showed that the patient’s neurologic level of the lesion is the most important factor determining the development of scoliosis in myelomeningocele. A 3.5-degree deterioration of the angle occurred every year in patients with levels of paralysis between T3 and T12, and a 2.5-degree deterioration in patients with levels of paralysis between L1 and L3.
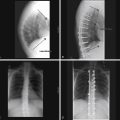
The natural history of untreated scoliosis in myelomeningocele is that of deformity progression, with a rate of deterioration significantly higher than in idiopathic scoliosis (Fig. 117-5). The aims of treatment are preservation of respiratory function, maintenance of sitting stability, achievement of maximal trunk length, and stabilization of the trunk.69 Corrective bracing is used as the initial management while the child matures. Children with myelomeningocele should be examined yearly for the presence or progression of scoliosis. Once spinal curvature surpasses 30 degrees, bracing usually fails and fusion surgery is required to control progression.65 However, spinal orthoses in children with myelomeningocele may lead to deformity of the ribs, decreased pulmonary function, ischemia of the skin, and pressure ulcerations.71 Progressive scoliosis is often the result of other correctable neurologic pathologies such as syringomyelia, hydromyelia, tethered spinal cord, or hydrocephalus shunt malfunction.66 Stability or improvement of the scoliosis can be anticipated following untethering of the spinal cord.68 Tomlinson et al.72 found that syringomyelia is associated with a high incidence of developmental scoliosis and that decompression of the syrinx either leads to improvement in, or stabilization of, the majority of scoliotic curves or postpones the need for fusion surgery. If a neurosurgical cause is discovered and treated, spontaneous improvement in the scoliosis may be seen if the curve is small in magnitude. Large curves do not resolve despite neurosurgical intervention.73
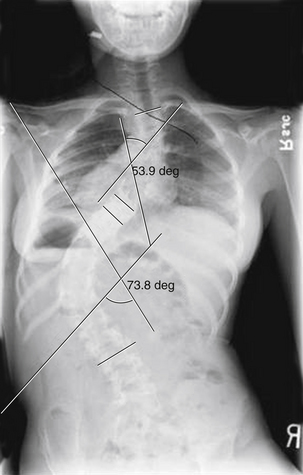
The definitive treatment of scoliosis in the myelomeningocele patient is spinal fusion. The entire curve must be included in the fusion. Distally, the fusion should extend to include the lumbosacral joint if there is pelvic obliquity and the patient does not walk.66 Combined ventral and dorsal spinal fusions have been recommended because dorsal fusion alone can result in a high rate of pseudarthroses due to incomplete solid fusion at the defective laminar defects. Most arthroses occur at the lumbosacral junction. However, bilateral transpedicular fixation of the L5 and S1 levels can be performed as a prophylactic procedure.74 The role of ventral fusion is controversial, and it may not be necessary for routine curves.75 The most common complications are infection, loss of correction, and pseudarthrosis.76
Kyphosis
A kyphotic deformity of the spine, in association with a myelomeningocele, is present in approximately 10% of patients.77 Progression of the kyphotic deformity can be seen in the sagittal plane of affected children.78 Unrelenting progression of 8 degrees per year is seen in most of them.79 Congenital kyphoses progress not only because of the deficiencies of the dorsal elements of the spine but also because of the effects of gravity during sitting and standing, as well as the subluxation of the extensor spinal muscles laterally and the unopposed psoas muscles anteriorly.80 A kyphotic deformity can interfere with sitting and prevents supination. Common associated symptoms include skin ulceration over the gibbus and crowding of the thoracic and abdominal contents. Kyphosis usually is associated with pulmonary and digestive compromise, owing to the crowding of the lungs by the intra-abdominal contents, which are forced upward by the flexion of the trunk.66,81 Neurologic deterioration usually is not a concern, because there is no useful neurologic function distal to the kyphosis in most children so affected.
The goal of a kyphectomy procedure is restoration of complete sagittal alignment, balance, and stability, while allowing for growth.82 The preferred age for surgery appears to be 2 to 5 years, when the anteroposterior vertebral diameter reaches a 25-mm minimum. McLone5 recommended that kyphectomy be performed prior to 2 years of age for children born with a thoracolumbar kyphosis and motor function at or below the L4 level. Corrective surgery also may be performed at the time of myelomeningocele closure, with resection of misaligned segments. In severe cases, this may be needed to obtain adequate closure.
Optimal surgical results require resection of the vertebral bodies at the apex of the kyphos to facilitate complete correction, without which recurrence is likely.83 Published reports of long-term follow-up of resection of the rostral portion of the kyphotic deformity demonstrate that most patients improved, but a residual deformity persisted and that some degree of loss of correction of sagittal balance was observed over time.82,84 A number of methods of internal fixation have been described. Aggressive deformity correction procedures that achieve satisfactory results have been described. However, this was associated with significant blood loss and a significant recurrence at 1 year of age, when limited internal fixation is used.81
In general, a correction of 45 degrees per vertebral level can be expected, and the lumbar lordosis can be restored, along with thoracic kyphosis and comprehensive sagittal balance. Postoperative wound breakdown, a common complication, can be managed by a postoperative full-thickness flap advancement or preoperative placement of tissue expanders.79,81
Other Spinal Anomalies
Other vertebral anomalies include an absence of the spinous processes and laminae, a reduction in the anteroposterior dimension of the vertebral body, an increase of the interpedicular distance, a decrease in the height of the pedicle, lateral extension of the large transverse processes, hemivertebrae, partial or complete vertebral fusion, and fusion of transverse processes.15 Spinal cord anomalies other than open myelomeningocele include hydromyelia, diplomyelia, diastematomyelia, and defective myelinization; one or more of these were found in 88% of autopsy cases having spinal cord anomalies.42
Postoperative and Chronic Care
About 15% of patients with myelomeningocele are born with clinical signs of hydrocephalus, but 80% or more develop hydrocephalus in early infancy. In most instances, the clinical features and diagnosis become obvious within a week or two.85 Performing a CSF diversion procedure (e.g., ventriculoperitoneal shunt) will relieve the ventricular pressure and tension on the myelomeningocele closure site to permit healing without CSF leakage. Third ventriculostomy may be considered for treatment of aqueductal stenosis in patients with myelomeningocele; reported success rates range between 65% and 80%. Third ventriculostomy also is a safe and effective means of treating hydrocephalus in the older myelomeningocele population even after years of shunting and offers the possibility of a long-term, shunt-independent life for selected patients.86,87
A thorough urologic evaluation should determine the degree of sphincter impairment, bladder dysfunction, and infection. In the United States, clean intermittent catheterization is used by 90% of children with myelomeningocele, 75% of whom perform self-catheterization.5 Bowel management programs are easily achieved by supplementing proper nutrition, using laxatives, and taking advantage of the normal physiologic response of the gastrointestinal tract.15
Over the last 30 years, continual progress has been made in the outcomes of children born with myelomeningocele. Survival rates dramatically improved once it became possible to control hydrocephalus. Survival rates range between 80% and 85% at 15-year follow-up in one treated group of patients.5 Hindbrain dysfunction caused by Chiari II malformation remains the principal cause of mortality. With an ongoing coordinated care plan, the affected patient’s level of function can be expected to be maintained.5
Adinolfi M., Beck S., Embury S., et al. Levels of alpha-fetoprotein in amniotic fluids of mice (curlytail) with neural tube defects. J Med Genet. 1976;13:511-513.
Dias M.S., Walker M.L. The embryogenesis of complex dysraphic malformations: a disorder of gastrulation? Pediatr Neurosurg. 1992;18:229-253.
Laurence K.M., James N., Miller M.H., et al. Double-blind randomised controlled trial of folate treatment before conception to prevent recurrence of neural-tube defects. Br Med J (Clin Res Ed). 1981;282:1509-1511.
McLone D.G. Treatment of myelomeningocele: arguments against selection. Clin Neurosurg. 1986;33:359-370.
McLone D.G. Continuing concepts in the management of spina bifida. Pediatr Neurosurg. 1992;18:254-256.
McLone D.G., Knepper P.A. The cause of Chiari II malformation: a unified theory. Pediatr Neurosci. 1989;15:1-12.
Park T.S., Cail W.S., Maggio W.M., Mitchell D.C. Progressive spasticity and scoliosis in children with myelomeningocele: radiological investigation and surgical treatment. J Neurosurg. 1985;62:367-375.
Pollack I.F., Pang D., Albright A.L., Krieger D. Outcome following hindbrain decompression of symptomatic Chiari malformations in children previously treated with myelomeningocele closure and shunts. J Neurosurg. 1992;77:881-888.
Urui S., Oi S. Experimental study of the embryogenesis of open spinal dysraphism. Neurosurg Clin North Am. 1995;6:195-202.
1. Yen I.H., Khoury M.J., Erickson J.D., et al. The changing epidemiology of neural tube defects: United States, 1968-1989. Am J Dis Child. 1992;146:857-861.
2. Myrianthopoulos N.C., Melnick M. Studies in neural tube defects. I. Epidemiologic and etiologic aspects. Am J Med Genet. 1987;26:783-796.
3. Zachary R.B. Moral and social problems in intensive treatment of myelomeningocele. Moral, philosophical, religious and social bases of intensive treatment of severe forms of myelomeningocele. Ann Chir Infant. 1969;10:7-14.
4. Lorber J. Results of treatment of myelomeningocele: an analysis of 524 unselected cases, with special reference to possible selection for treatment. Dev Med Child Neurol. 1971;13:279-303.
5. McLone D.G. Continuing concepts in the management of spina bifida. Pediatr Neurosurg. 1992;18:254-256.
6. Adinolfi M., Beck S., Embury S., et al. Levels of alpha-fetoprotein in amniotic fluids of mice (curlytail) with neural tube defects. J Med Genet. 1976;13:511-513.
7. Urui S., Oi S. Experimental study of the embryogenesis of open spinal dysraphism. Neurosurg Clin North Am. 1995;6:195-202.
8. Naruse I., Collins M.D., Scott W.J.Jr. Strain differences in the teratogenicity induced by sodium valproate in cultured mouse embryos. Teratology. 1988;38:87-96.
9. Warkany J., O’Toole B.A. Experimental spina bifida and associated malformations. Childs Brain. 1981;8:18-30.
10. Cole W.A., Trasler D.G. Gene-teratogen interaction in insulin-induced mouse exencephaly. Teratology. 1980;22:125-139.
11. Reece E.A., Pinter E., Leranth C.Z., et al. Ultrastructural analysis of malformations of the embryonic neural axis induced by in vitro hyperglycemic conditions. Teratology. 1985;32:363-373.
12. Finnell R.H., Moon S.P., Abbott L.C., et al. Strain differences in heat-induced neural tube defects in mice. Teratology. 1986;33:247-252.
13. Dias M.S., Walker M.L. The embryogenesis of complex dysraphic malformations: a disorder of gastrulation? Pediatr Neurosurg. 1992;18:229-253.
14. McLone D.G., Knepper P.A. The cause of Chiari II malformation: a unified theory. Pediatr Neurosci. 1989;15:1-12.
15. Hahn Y.S. Open myelomeningocele. Neurosurg Clin North Am. 1995;6:231-241.
16. Pang D. Surgical complications of open spinal dysraphism. Neurosurg Clin North Am. 1995;6:243-257.
17. Rosa F.W. Spina bifida in infants of women treated with carbamazepine during pregnancy. N Engl J Med. 1991;324:674-677.
18. Shurtleff D.B., Lemire R.J. Epidemiology, etiologic factors, and prenatal diagnosis of open spinal dysraphism. Neurosurg Clin North Am. 1995;6:183-193.
19. Elwood J.H. Epidemics of anencephalus and spina bifida in Ireland since 1900. Int J Epidemiol. 1973;2:171-175.
20. Campbell L.R., Dayton D.H., Sohal G.S. Neural tube defects: a review of human and animal studies on the etiology of neural tube defects. Teratology. 1986;34:171-187.
21. Carter C.O., Evans K.A., Campbell S. Letter: neural tube malformations in offspring of spina-bifida patients. Lancet. 1975;1:685.
22. Carter C.O., Evans K. Children of adult survivors with spina bifida cystica. Lancet. 1973;2:924-926.
23. Elwood M. Folic acid prevents neural tube defects. Med J Aust. 1991;155:579-581.
24. Melvin E.C., George T.M., Worley G., et al. Genetic studies in neural tube defects: NTD Collaborative Group. Pediatr Neurosurg. 2000;32:1-9.
25. Luthy D.A., Wardinsky T., Shurtleff D.B., et al. Cesarean section before the onset of labor and subsequent motor function in infants with meningomyelocele diagnosed antenatally. N Engl J Med. 1991;324:662-666.
26. Warkany J., Petering H.G. Congenital malformations of the central nervous system in rats produced by maternal zinc deficiency. Teratology. 1972;5:319-334.
27. Smithells R.W., Nevin N.C., Seller M.J., et al. Further experience of vitamin supplementation for prevention of neural tube defect recurrences. Lancet. 1983;1:1027-1031.
28. Laurence K.M., James N., Miller M.H., et al. Double-blind randomised controlled trial of folate treatment before conception to prevent recurrence of neural-tube defects. Br Med J (Clin Res Ed). 1981;282:1509-1511.
29. Charney E.B., Weller S.C., Sutton L.N., et al. Management of the newborn with myelomeningocele: time for a decision-making process. Pediatrics. 1985;75:58-64.
30. Babcook C.J. Ultrasound evaluation of prenatal and neonatal spina bifida. Neurosurg Clin North Am. 1995;6:203-218.
31. McLone D.G. Technique for closure of myelomeningocele. Childs Brain. 1980;6:65-73.
32. McLone D.G., Dias M.S. Complications of myelomeningocele closure. Pediatr Neurosurg. 1991;17:267-273.
33. Stark G. Prediction of urinary continence in myelomeningocele. Dev Med Child Neurol. 1971;13:388-389.
34. Banta J.V., Bonanni C., Prebluda J. Latex anaphylaxis during spinal surgery in children with myelomeningocele. Dev Med Child Neurol. 1993;35:543-548.
35. Teichgraeber J.F., Riley W.B., Parks D.H. Primary skin closure in large myelomeningoceles. Pediatr Neurosci. 1989;15:18-22.
36. Munro I.R., Neu B.R., Humphreys R.P., Lauritzen C.G. Limberg-latissimus dorsi myocutaneous flap for closure of myelomeningocele. Childs Brain. 1983;10:381-386.
37. McDevitt N.B., Gillespie R.P., Woosley R.E., et al. Closure of thoracic and lumbar dysgraphic defects using bilateral latissimus dorsi myocutaneous flap transfer with extended gluteal fasciocutaneous flaps. Childs Brain. 1982;9:394-399.
38. Bruner J.P., Tulipan N., Paschall R.L., et al. Fetal surgery for myelomeningocele and the incidence of shunt-dependent hydrocephalus. JAMA. 1999;282:1819-1825.
39. Cochrane D.D., Irwin B., Chambers K. Clinical outcomes that fetal surgery for myelomeningocele needs to achieve. Eur J Pediatr Surg. 2001;11(suppl 1):S18-S20.
40. Sutton L.N., Adzick N.S., Bilaniuk L.T., et al. Improvement in hindbrain herniation demonstrated by serial fetal magnetic resonance imaging following fetal surgery for myelomeningocele. JAMA. 1999;282:1826-1831.
41. Walsh D.S., Adzick N.S., Sutton L.N., Johnson M.P. The rationale for in utero repair of myelomeningocele. Fetal Diagn Ther. 2001;16:312-322.
42. Gilbert J.N., Jones K.L., Rorke L.B., et al. Central nervous system anomalies associated with meningomyelocele, hydrocephalus, and the Arnold-Chiari malformation: reappraisal of theories regarding the pathogenesis of posterior neural tube closure defects. Neurosurgery. 1986;18:559-564.
43. McLone D.G. Treatment of myelomeningocele: arguments against selection. Clin Neurosurg. 1986;33:359-370.
44. Brown S.F. Congenital malformations associated with myelomeningocele. J Iowa Med Soc. 1975;65:101-104.
45. Stein S.C., Schut L. Hydrocephalus in myelomeningocele. Childs Brain. 1979;5:413-419.
46. Babcook C.J., Drake C.M., Goldstein R.B. Spinal level of fetal myelomeningocele: does it influence ventricular size? AJR Am J Roentgenol. 1997;169:207-210.
47. Wills K.E., Holmbeck G.N., Dillon K., McLone D.G. Intelligence and achievement in children with myelomeningocele. J Pediatr Psychol. 1990;15:161-176.
48. Vandertop W.P., Asai A., Hoffman H.J., et al. Surgical decompression for symptomatic Chiari II malformation in neonates with myelomeningocele. J Neurosurg. 1992;77:541-544.
49. el Gammal T., Mark E.K., Brooks B.S. MR imaging of Chiari II malformation. AJR Am J Roentgenol. 1988;150:163-170.
50. Hoffman H.J., Neill J., Crone K.R., et al. Hydrosyringomyelia and its management in childhood. Neurosurgery. 1987;21:347-351.
51. Samuelsson L., Bergstrom K., Thuomas K.A., et al. MR imaging of syringohydromyelia and Chiari malformations in myelomeningocele patients with scoliosis. AJNR Am J Neuroradiol. 1987;8:539-546.
52. Rauzzino M., Oakes W.J. Chiari II malformation and syringomyelia. Neurosurg Clin North Am. 1995;6:293-309.
53. Park T.S., Hoffman H.J., Hendrick E.B., Humphreys R.P. Experience with surgical decompression of the Arnold-Chiari malformation in young infants with myelomeningocele. Neurosurgery. 1983;13:147-152.
54. Pollack I.F., Pang D., Albright A.L., Krieger D. Outcome following hindbrain decompression of symptomatic Chiari malformations in children previously treated with myelomeningocele closure and shunts. J Neurosurg. 1992;77:881-888.
55. Morley A.R. Laryngeal stridor, Arnold-Chiari malformation, and medullary haemorrhages. Dev Med Child Neurol. 1969;11:471-474.
56. Park T.S., Cail W.S., Maggio W.M., Mitchell D.C. Progressive spasticity and scoliosis in children with myelomeningocele: radiological investigation and surgical treatment. J Neurosurg. 1985;62:367-375.
57. Wisoff J.H., Epstein F. Management of hydromyelia. Neurosurgery. 25, 1989. 562–571
58. Vaquero J., Martinez R., Arias A. Syringomyelia-Chiari complex: magnetic resonance imaging and clinical evaluation of surgical treatment. J Neurosurg. 1990;73:64-68.
59. Milhorat T.H., Johnson W.D., Miller J.I., et al. Surgical treatment of syringomyelia based on magnetic resonance imaging criteria. Neurosurgery. 1992;31:231-244.
60. Tognetti F., Calbucci F. Syringomyelia: syringo-subarachnoid shunt versus posterior fossa decompression. Acta Neurochir (Wien). 1993;123:196-197.
61. Herman J.M., McLone D.G., Storrs B.B., Dauser R.C. Analysis of 153 patients with myelomeningocele or spinal lipoma reoperated upon for a tethered cord: presentation, management, and outcome. Pediatr Neurosurg. 1993;19:243-249.
62. Yamada S., Iacono R.P., Andrade T., et al. Pathophysiology of tethered cord syndrome. Neurosurg Clin North Am. 1995;6:311-323.
63. Aliredjo R.P., de Vries J., Menovsky T., et al. The use of Gore-Tex membrane for adhesion prevention in tethered spinal cord surgery: technical case reports. Neurosurgery. 1999;44:674-677.
64. McLone D.G., La Marca F. The tethered spinal cord: diagnosis, significance, and management. Semin Pediatr Neurol. 1997;4:192-208.
65. Hall J.E., Poitras B. The management of kyphosis in patients with myelomeningocele. Clin Orthop Relat Res. 1977;128:33-40.
66. Karol L.A. Orthopedic management in myelomeningocele. Neurosurg Clin North Am. 1995;6:259-268.
67. Hall P.V., Kalsbeck J.E., Wellman H.N. Clinical radiosotope investigations in hydrosyringomyelia and myelodysplasia. J Neurosurg. 1976;45:188-194.
68. McLone D.G., Herman J.M., Gabrieli A.P., Dias L. Tethered cord as a cause of scoliosis in children with a myelomeningocele. Pediatr Neurosurg. 1990;16:8-13.
69. Eysel P., Hopf C., Schwarz M., Voth D. Development of scoliosis in myelomeningocele: differences in the history caused by idiopathic pattern. Neurosurg Rev. 1993;16:301-306.
70. Carstens C., Vetter J., Niethard F.U. Development of paralytic scoliosis in myelomeningocele. Z Ortho Ihre Grenzgeb. 1990;128:174-182.
71. Winter R.B., Carlson J.M. Modern orthotics for spinal deformities. Clin Orthop Relat Res. 1977;126:74-86.
72. Tomlinson R.J.Jr., Wolfe M.W., Nadall J.M., et al. Syringomyelia and developmental scoliosis. J Pediatr Orthop. 1994;14:580-585.
73. Hall P.V., Campbell R.L., Kalsbeck J.E. Meningomyelocele and progressive hydromyelia: progressive paresis in myelodysplasia. J Neurosurg. 1975;43:457-463.
74. Geiger F., Parsch D., Carstens C. Complications of scoliosis surgery in children with myelomeningocele. Eur Spine J. 1999;8:22-26.
75. Ward W.T., Wenger D.R., Roach J.W. Surgical correction of myelomeningocele scoliosis: a critical appraisal of various spinal instrumentation systems. J Pediatr Orthop. 1989;9:262-268.
76. Osebold W.R., Mayfield J.K., Winter R.B., Moe J.H. Surgical treatment of paralytic scoliosis associated with myelomeningocele. J Bone Joint Surg [Am]. 1982;64:841-856.
77. Hoppenfeld S. Congenital kyphosis in myelomeningocele. J Bone Joint Surg [Br]. 1967;49:276-280.
78. Mintz L.J., Sarwark J.F., Dias L.S., et al. The natural history of congenital kyphosis in myelomeningocele: a review of 51 children. Spine (Phila Pa 1976). 1991;16:S348-S350.
79. Sarwark J.F. Kyphosis deformity in myelomeningocele. Orthop Clin North Am. 1999;30:451-455. i-ix
80. Banta J.V. The evolution of surgical treatment of spinal deformity in myelomeningocele. Z Kinderchir. 1987;42(suppl 1):10-12.
81. Lindseth R.E., Stelzer L.Jr. Vertebral excision for kyphosis in children with myelomeningocele. J Bone Joint Surg [Am]. 1979;61:699-704.
82. Lowe G.P., Menelaus M.B. The surgical management of kyphosis in older children with myelomeningocele. J Bone Joint Surg [Br]. 1978;60:40-45.
83. Torode I., Godette G. Surgical correction of congenital kyphosis in myelomeningocele. J Pediatr Orthop. 1995;15:202-205.
84. Leatherman K.D., Dickson R.A. Congenital kyphosis in myelomeningocele: vertebral body resection and posterior spine fusion. Spine (Phila Pa 1976). 1978;3:222-226.
85. Stark G.D. Neonatal assessment of the child with a myelomeningocele. Arch Dis Child. 1971;46:539-548.
86. Elbabaa S.K., Steinmetz M., Ross J., et al. Endoscopic third ventriculostomy for obstructive hydrocephalus in the pediatric population: evaluation of outcome. Eur J Pediatr Surg. 2001;11(suppl 1):S52-S54.
87. Teo C., Jones R. Management of hydrocephalus by endoscopic third ventriculostomy in patients with myelomeningocele. Pediatr Neurosurg. 1996;25:57-63.