Chapter 118 Anterior Sacral Meningocele
Sacral meningocele may be congenital or acquired. Most acquired sacral meningoceles are a consequence of dural ectasia in association with the neurofibromatoses, Marfan syndrome, and Ehlers-Danlos syndrome.1–6 These meningoceles usually are single and may expand into the intrasacral, presacral, and parasacral spaces. Traumatic avulsion of sacral nerve roots may produce sacral pseudomeningoceles in conjunction with profound neurologic deficits.7,8 These sacs are more likely to be multiple and located in the lateral presacral space.
The congenital anterior sacral meningocele (ASM) was first described by Bryant in 1837.9 It is rare in comparison to its dorsal counterpart. Matson noted only three examples in his analysis of 1390 cases of spina bifida cystica.10 A congenital ASM characteristically occurs as a cystic presacral mass connected to the caudal thecal sac by a pedicle of variable size. Meningocele volumes as large as 1 to 2 L have been described.11–13 Anorectal, genitourinary, and sacral anomalies also may be present.
Pathogenesis
The processes of spine and spinal cord development span the period from embryogenesis to postnatal development. Similar to more rostral vertebrae, the sacral vertebrae develop from sclerotomes. However, the sacral pattern requires additional centers of ossification. This process occurs slowly and is not completed until the third or fourth decade of life. Developmental sacral osseous anomalies (mesodermal) include sacralization of lumbar and coccygeal segments; lumbarization of the first sacral segment; stenosis or dilatation of the sacral foramina; isolated defects of the dorsal, lateral, and ventral elements; and sacral agenesis.14
The sacrococcygeal neural elements develop after closure of the posterior neuropore. Beneath an intact surface ectoderm, the caudal cell mass enlarges and undergoes canalization. This occurs from the 4th to 6th weeks of life, forming spinal cord segments extending from S2 to S3 to the coccygeal terminus. Extensive cellular degeneration along the distal neural tube produces a fibrous remnant at the caudal-most tip of the developing CNS. This remnant becomes the filum terminale. The ventriculus terminalis lies at the level of the S5 entry zone, marking the point of transition between the conus medullaris and filum terminale.14 Lying at the distal end of the filum terminale, the coccygeal medullary vestige may be the origin of the intrasacral meningocele.15–17
Before the 9th week, spinal cord and vertebral segments are aligned level for level. With the dura mater now forming a complete covering, the spinal cord and dura mater begin to ascend in relation to the growing vertebral column, albeit at different rates. The conus medullaris rises to L3 at birth and to L1-2 by 3 months. The dural sac constricts terminally, rising only to S4 at birth and to S2-3 in the adult. Developmental sacrococcygeal neuroectodermal anomalies include meningocele, myelomeningocele, lipomyelomeningocele, myelocystocele, anomalies of the conus medullaris, tethered filum terminale, intrasacral meningocele, and caudal regression syndromes.14
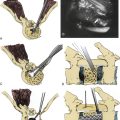
In contrast to dorsal meningoceles that arise from failure of the posterior neuropore to close or dehiscence of a formed neural tube, a congenital ASM arises after failure of one or more sacral sclerotomes to develop. The meningeal sac expands through the sacral defect driven by cerebrospinal fluid (CSF) pulsations. The sacral defect enlarges only slightly, while the developing pelvic viscera offer less resistance to the budding meningocele. The sac enlarges tremendously in the presacral space, remaining attached to the thecal sac by a smaller pedicle. Although spontaneous regression of the meningocele does not occur after birth, progressive enlargement may occur and is associated with the development of symptoms. The large volume attained by some meningoceles causes crowding of the pelvic viscera.11 The ventral sacral defect is usually parasagittal, less commonly midline or lateral. The typical anatomic relationships of an anterior sacral meningocele are depicted in Figure 118-1.
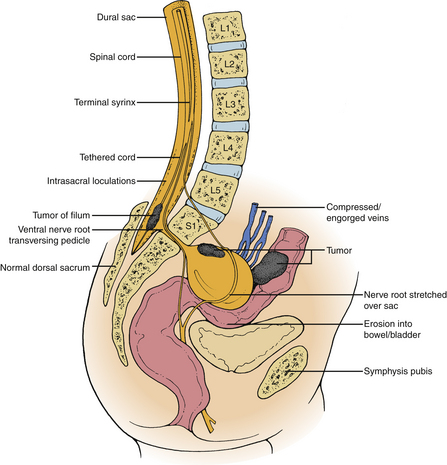
Complex interactions between adjacent germ cell substrata in the embryonic caudal midline give rise to varying cascades of maldevelopment, which include a spectrum of mesectodermal dystrophies. The embryologic event that initiates these patterns of maldevelopment is distinct from the insult that causes failure of the posterior neuropore to close. Although the precise event is not clearly defined, evidence for a vascular etiology exists.14,18,19 In addition to anomalies of the conus medullaris, sacral nerve roots, and sacral dura mater, the clinical manifestations of these patterns of maldevelopment include the development of congenital tumors (e.g., dermoid, epidermoid, hamartoma, lipoma, teratoma, and teratocarcinoma) and anomalies of the colorectal, genitourinary, and reproductive systems.2,14,20–24
Most congenital ASMs appear to occur sporadically, although familial and X-linked dominant inheritance have been reported.20,23,25,26 The seemingly greater incidence in females is a manifestation of the tendency toward symptomatology in the presence of sacral crowding, as well as the greater likelihood that female patients in the second and third decades of life will undergo palpation of the presacral space during routine physical examination. When cases in patients under 20 years of age are considered, the female-to-male ratio is approximately 1:1.12,27
Clinical Presentation
Congenital ASM has a trimodal pattern of presentation.2,12,13,24,28–32 Some ASMs are recognized at birth, when they are discovered in association with anorectal anomalies and sacral defects (Currarino’s triad).33 Most commonly, however, ASMs present during the first and second decades of life with progressive constipation or other symptoms referable to the colorectal, genitourinary, and reproductive systems.2,12,13,24,27,29,30–32,34,35 Less commonly, low back pain, headache, or sacral radiculopathy may be the initial manifestation.12,13,22
A small congenital ASM may remain occult for life, particularly in the male patient.12,13 Occasionally, a meningocele is discovered incidentally during the initiation of routine prenatal care in a new mother. In the rare case of a pregnant woman with no prenatal care, the meningocele may present as dystocia. In this setting, the characteristically benign ASM may threaten serious morbidity and even death.
Colorectal
The most common symptom in patients with congenital ASM is chronic constipation or obstipation, usually manifesting in the first two decades of life. Fecal incontinence and soiling rarely predominate in the clinical picture. Communication between congenital ASM and the rectum may be congenital or acquired and may produce fulminant meningitis. The anal canal may be duplicated or atretic.12,13,20
Genitourinary
Urinary incontinence may be due to tethering of the spinal cord or direct pressure on the bladder, ureters, or S1, S2, and S3 nerve roots, or, rarely, may be the result of congenital deficits in the innervation of the lower urinary tract. Enuresis and recurrent infections affecting either the upper or the lower tract may occur. Coexisting anomalies include horseshoe-shaped kidneys and duplication of the kidneys, renal pelvis, or ureter.12,13
Reproductive
Dysmenorrhea, dystocia, and dyspareunia occur less commonly. Dysmenorrhea may be secondary to compression of retropelvic veins resulting in pelvic venous congestion. Duplication of the uterus and vagina and vaginal atresia may be present.12,13,21
Neurologic
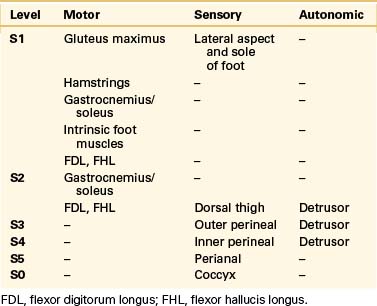
True neurologic symptoms are rare and typically are mild when present because the distal spinal cord and nerve roots develop normally. However, progressive leg or perineal weakness, numbness, and pain may develop as a result of stretching of the sacrococcygeal nerve roots by the meningocele.12,13,27 These patients usually will have some symptoms suggestive of involvement of the pelvic visceral innervation. Chronic pelvic pain may occur due to involvement of the pelvic autonomic plexi. Such symptoms typically are progressive and do not develop until later in life, implying the potential for reversibility after treatment if they are recognized early. Coexisting tethering of the spinal cord actually may lessen the amount of tension exerted on the nerve roots by the enlarging meningocele.2 The presence of severe neurologic dysfunction from birth indicates a more severe myelodysraphic state and is associated with a greater tendency toward anomalous development in adjacent viscera. The patient whose clinical picture is characterized by prominent sacrococcygeal radiculopathy that worsens after a Valsalva maneuver is more likely to harbor an intrasacral meningocele or an ASM based on a large pedicle.2,12,13,15,17 The sacrococcygeal radiculopathies are listed in Table 118-1.
Mild to moderate headache-associated symptoms may occur and are of two forms.2,27,31,36 A high-pressure variant secondary to pressure exerted on the meningocele during pregnancy or after a Valsalva maneuver has been reported occasionally; less commonly, a low-pressure headache may occur on rising to the standing position, caused by the displacement of CSF from the thecal sac into the meningocele. Pressure-related headaches are more likely in the presence of a large communication between the thecal sac and the meningocele.12,13
Congenital ASM may be a cause of meningitis. Bacterial meningitis resulting from erosion of the meningocele into the bowel or bladder lumina or resulting from the presence of a congenital rectothecal or vesiculothecal fistula and aseptic meningitis resulting from leakage of an intraspinal dermoid cyst have been described.2,23,27
Preoperative Evaluation
The most consistent physical finding in congenital anterior sacral meningocele is a soft, cystic, retrorectal mass that appears fixed to the sacrum.12,13 The mass is felt most commonly on rectal examination but may be felt on pelvic, abdominal, gluteal, or inguinal examination as well. A transmitted pulse wave sometimes may be appreciated with a Valsalva maneuver.12,13 Dyck and Wilson37 described a patient who presented with a sliding groin hernia and was found to harbor a congenital ASM. They postulated crowding of the pelvic viscera as a significant contributing factor in the development of the hernia, which contained a portion of the urinary bladder.
Imaging
Preoperative planning for surgery of congenital anterior sacral meningocele begins with the radiologic delineation of the surgical anatomy of the meningocele. Specific considerations are (1) confirmation of the cystic nature of the mass; (2) identification of the pedicle, associated mass lesions, and any other abnormalities of the neural, dural, or vertebral components of the sacrum; (3) determination of the relationship between the meningocele and the sacral nerve roots; and (4) determination of the relationship to the pelvic viscera. Bone window CT and MRI are the primary diagnostic studies performed.8,38–41 Myelography and postmyelographic CT do not offer an equivalent amount of noninvasive surgically useful information.42 Adjuvant evaluation of the pelvic viscera may provide additional useful information in selected cases.
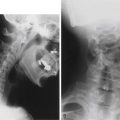
The sacrum is difficult to evaluate with plain radiography owing to its curvilinear shape and overlying soft tissue and bowel gas patterns. The pathognomonic sickle-shaped sacral deformity, or scimitar sign, and a presacral mass may be present (Fig. 118-2). Less obvious findings include widening of isolated sacral foramina, increases in interpedicular distance or flattening of the pedicles, and abnormalities of curvature.12,13 Transforaminal sacral views may better demonstrate these changes. Calcification within an associated presacral mass may be difficult to discern.
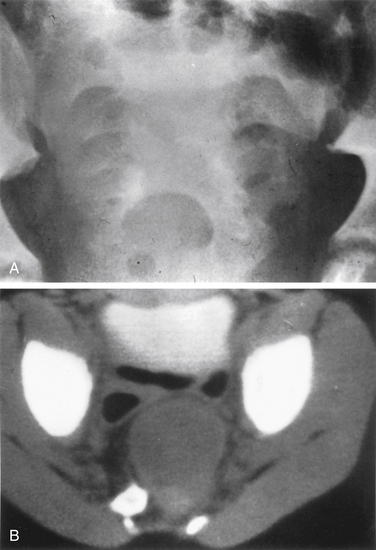
The development of myelography allowed better visualization of the meningocele and its pedicle but contributed little to the diagnosis of associated pelvic visceral anomalies. Only masses within the meningocele could be seen, and in the presence of a very small pedicle, the fistulous communication was not always demonstrated.12,13 Delayed imaging at 24 to 48 hours and the use of large volumes of contrast material increased the chance of identifying the pedicle. Balériaux-Waha et al.42 described the utility of CT in differentiating anterior sacral meningocele from solid masses of the presacral space. CT bone windows are particularly helpful in delineating osseous anatomy. However, as with myelography, CT (even with intrathecal contrast) may fail to demonstrate a small pedicle.
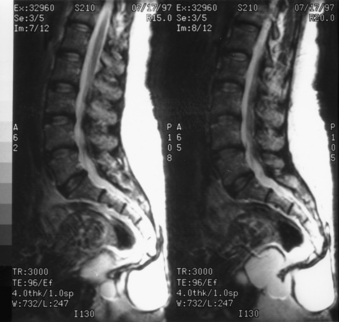
In 1988, Lee et al.39 reported on the use of MRI to demonstrate familial anterior sacral meningoceles in a father and daughter. The authors were able to noninvasively demonstrate a horseshoe-shaped kidney, didelphic uterus, and associated pelvic teratoma, which were not shown by other imaging studies. This is a critical advantage of MRI over other diagnostic imaging studies in the evaluation of congenital ASM, considering the extensive differential diagnoses of a presacral mass (Box 118-1).43–45
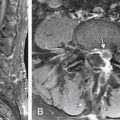
In cases where ASM is present, the MRI appearance of a congenital ASM is characteristic.39–4146 T1-weighted images show a homogeneous low-signal cyst extending from the sacral thecal sac into the presacral space. It usually is possible to identify the communicating pedicle. The thickened filum terminale and any associated tumors may be identified. T2-weighted images show high signals, although occasionally both high and low signals may be present within the meningocele as a consequence of fluid movements during imaging (Fig. 118-3).
Management
Five surgical approaches have been described during the evolution of the surgical treatment of congenital ASM (Fig. 118-4). Transrectal or transvaginal aspiration and the inferior presacral approach (e.g., Kraske approach) are associated with prohibitive risks of morbidity and mortality.2,27,30,31,47 The oblique parasacral approach of Demel48 and Coqui49 was described for the rare gluteal meningocele. The ventral transabdominal-transpelvic approach was recommended by Leibowitz et al.22 to prevent missing an occult tumor. With MRI, this approach is no longer necessary. However, it may still be useful in cases of large abdominopelvic masses or when nerve roots are known to traverse the pedicle of the meningocele. Only the dorsal transsacral approach allows the surgeon to carry out each of the aforementioned goals in a planned, logical progression, while minimizing the risks of injury to the pelvic viscera and presacral neurovascular networks.11,50
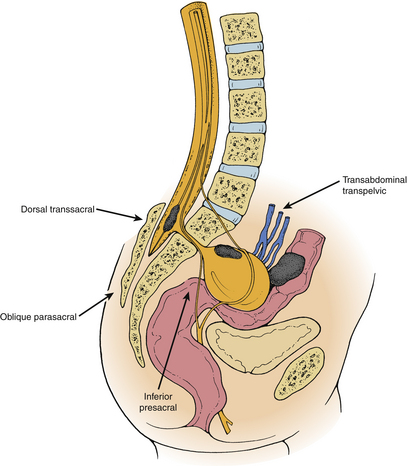
Inferior Presacral Approach (Kraske-Pupovac Approach)
With the inferior presacral approach, an incision is extended from just dorsal to the rectum to the dorsal surface of the coccyx. A plane is developed in the retrorectal space, and the coccyx is excised. The pedicle is identified and ligated, after which the sac may be aspirated via direct puncture. This approach has several disadvantages. First, the pedicle and its relationship with adjacent sacral nerve roots and the filum terminale are not fully visualized. An associated mass lesion within the meningocele or ventral to it can be difficult or impossible to expose via this approach. Inadvertent entry into the bowel or bladder may occur. Finally, because any opening in the dural sac will occur in its most dependent portion, the incidence of a postoperative CSF fistula is increased. Proximity of the incision to the rectum may result in postoperative wound infection, pararectal abscess formation, or meningitis. This proximity is particularly problematic in extremely young children.
Dorsal Transsacral Approach (Adson Approach)
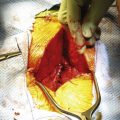
The dorsal transsacral approach allows inspection of intrasacral anatomy, direct visualization of the sacral nerve roots, aspiration and collapse of the meningocele, detethering of the spinal cord, ligation of the pedicle, excision of associated tumors, and access to the sacral nerve roots for intraoperative stimulation. Rarely, a large abdominopelvic mass cannot be completely excised via this exposure. If sacral nerve roots traverse the pedicle, creation of a dural sleeve around the exiting roots may also be difficult by a dorsal exposure.23 Because the meningocele is not excised, dissection in the presacral space is minimized. There have been no reported surgical deaths with this approach since 1965. A staged dorsal transsacral and transabdominal-transpelvic approach may be required in some cases.
Surgical Technique
Care must be taken to avoid using inappropriate surgical approaches, particularly ventral approaches, that may not provide the most direct exposure of the pathology and that, in addition, may be associated with significant morbidity. For the vast majority of cases, the dorsal transsacral approach of Adson, or a variant thereof, is recommended.50 With this approach, the patient is positioned prone on two longitudinal chest rolls, and all pressure points are carefully padded. The bowel is prepared preoperatively. Prophylactic antibiotics are administered and continued for 48 hours postoperatively. Intraoperative monitoring, if employed, is planned in advance and is now checked. A Foley catheter, an arterial line, and two large-bore peripheral intravenous lines are placed. The skin is cleansed with 0.25% triclosan (Septisol) and alcohol and prepared with povidone-iodine (Betadine).
If entry into the meningocele is desired for approach to an associated mass lesion or for plication of a dural sleeve around nerve roots traversing the pedicle, an opening should be made in the ventral surface of the dural sac adjacent to the pedicle. A fibrous plane usually is found between the ventral surface of the thecal sac and the dorsal surface of the meningocele, although venous bleeding from this space occasionally may be problematic. The sac is then opened, and the associated mass lesion is excised. The thecal sac is irrigated with warm antibiotic-containing solution to remove any blood that may have run down into the exposure. A watertight closure of the dural openings with #4-0 to #6-0 monofilament or braided nylon is confirmed by a Valsalva maneuver. After closure of the dorsal thecal sac, the paraspinous tissues are reapproximated. With meticulous hemostasis, a drain is not placed. Skin closure should be cosmetic. In the infant or young child, #5-0 to #4-0 Vicryl sutures are placed in inverted fashion at the subdermal plane. The skin edges may then be held in approximation using Steri-Strips (3M Nexcare, St. Paul, MN) or #5-0 monofilament nylon in simple interrupted fashion. In the older child or adult, the subdermal closure is done with #3-0 Vicryl in a similar fashion, and the final layer is closed with #4-0 monofilament nylon in a simple interrupted manner.
Complications
Some of the operative and postoperative complications associated with the dorsal transsacral approach of Adson and the transabdominal-transpelvic approach are listed in Box 118-2. The following discussions address some of these procedures’ common complications and their prevention and management.
BOX 118-2 Perioperative Complications
Operative Complications
Pelvic Visceral Injury
Injury to the pelvic viscera is minimized by use of the dorsal transsacral approach and by limiting dissection in the presacral space. In addition to direct injury to the ureters, rectum, and bladder and their neurovascular supply, delayed hematoma formation secondary to inadequate hemostasis of retropelvic veins and venous thrombosis with central extension are potential serious risks of transabdominal-transpelvic approaches. Retropelvic hemorrhage may be life threatening.
Barberá J., Broseta J., Argüelles F., et al. Traumatic lumbosacral meningocele. J Neurosurg. 1977;46:536.
Bryant T. Case of deficiency of the anterior part of the sacrum with a thecal sac in the pelvis, similar to the tumor of spina bifida. Lancet. 1837;1:358.
Coller F.A., Jackson R.G. Anterior sacral meningocele. Surg Gynecol Obstet. 1943;76:703.
Currarino G., Coln D., Votteler T. Triad of anorectal, sacral, and presacral anomalies. Am J Radiol. 1981;137:395.
Pang D. Sacral agenesis and caudal spinal cord malformations. Neurosurgery. 1993;32:755.
Stern W.E. Dural ectasia and the Marfan syndrome. J Neurosurg. 1988;69:221.
Wilkins R.H., Odom G.L. Anterior and lateral spinal meningocele. In: Vinken P.J., Bruyn G.W., editors. Handbook of clinical neurology. New York: Elsevier Biomedical; 1978:193.
1. Klenerman L., Merrick M.V. Anterior sacral meningocele occurring in a family. J Bone Joint Surg [Br]. 1973;55:331.
2. Oren M., Laber B., Lee S.H., et al. Anterior sacral meningocele: report of five cases and review of the literature. Dis Colon Rectum. 1977;20:492.
3. Raftopoulous C., Pierard G.E., Retif C., et al. Endoscopic cure of a giant sacral meningocele associated with Marfan’s syndrome: case report. Neurosurgery. 1992;30:765.
4. Smith M.D. Large sacral dural defect in Marfan syndrome. J Bone Joint Surg [Am]. 1993;75:1067.
5. Stern W.E. Dural ectasia and the Marfan syndrome. J Neurosurg. 1988;69:221.
6. Strand R.D., Eisenberg H.M. Anterior sacral meningocele in association with Marfan’s syndrome. Radiology. 1971;99:653.
7. Barberá J., Broseta J., Argüelles F., et al. Traumatic lumbosacral meningocele. J Neurosurg. 1977;46:536.
8. McLennan J.E., McLaughin W.T., Skillicorn S.A. Traumatic lumbar nerve root meningocele: case report. J Neurosurg. 1973;39:528.
9. Bryant T. Case of deficiency of the anterior part of the sacrum with a thecal sac in the pelvis, similar to the tumor of spina bifida. Lancet. 1837;1:358.
10. Matson D.D. Neurosurgery of infancy and childhood, ed 2. Springfield, IL, 1969: Charles C Thomas; 1969.
11. Smith H.P., Davis C.H. Anterior sacral meningocele: two case reports and discussion of surgical approach. Neurosurgery. 1980;7:61.
12. Villarejo F., Scavone C., Blazquez M.G., et al. Anterior sacral meningocele: review of the literature. Surg Neurol. 1983;19:57.
13. Wilkins R.H., Odom G.L. Anterior and lateral spinal meningocele. In: Vinken P.J., Bruyn G.W., editors. Handbook of clinical neurology. New York: Elsevier Biomedical; 1978:193.
14. Pang D. Sacral agenesis and caudal spinal cord malformations. Neurosurgery. 1993;32:755.
15. Doty J.R., Thomason J., Sinods G., et al. Occult intrasacral meningocele: clinical and radiographic diagnosis. Neurosurgery. 1989;24:616.
16. Enderle C. Meningocoele intrasacrale occulto (rivelato con la mielografia). Riv Neurol. 1932;5:418.
17. Lamas E., Lobato R.D., Amor T. Occult intrasacral meningocele. Surg Neurol. 1977;8:181.
18. Bavinck J.N., Weaver D.D. Subclavian artery supply disruption sequence: hypothesis of a vascular etiology for Poland, Klippel-Feil, and Mobius anomalies. Am J Med Genet. 1986;23:903.
19. Brill C.B., Peyster R.G., Keller M.S., et al. Isolation of the right subclavian artery with subclavian steal in a child with Klippel-Feil anomaly: an example of the subclavian artery supply disruption sequence. Am J Med Genet. 1987;26:933.
20. Aaronson I. Anterior sacral meningocele, anal canal duplication cyst, and covered anus occurring in one family. J Pediatr Surg. 1970;5:559.
21. Henley R.B., Lawrence L.B. Pelvic meningocele: a case report. J Neurosurg. 1965;23:206.
22. Leibowitz E., Barton W., Sadighi P., et al. Anterior sacral meningocele contiguous with a pelvic hamartoma: case report. J Neurosurg. 1984;61:188.
23. Quigley M.R., Schinco F., Brown J.T. Anterior sacral meningocele with an unusual presentation: case report. J Neurosurg. 1984;61:790.
24. Silvis R.S., Riddle L.R., Clark G.G. Anterior sacral meningocele. Am Surg. 1956;22:554.
25. Cohn J., Bay-Nielsen E. Hereditary defect of the sacrum and coccyx with anterior sacral meningocele. Acta Pediatr Scand. 1969;58:268.
26. Solopaev A.A., Mylkinokov P.I., Gerber I.V.M., et al. Anterior sacral meningocele (in Russian). Zh Vopr Neiro. 1977;5:18.
27. Amacher A.I., Drake C.G., McLachlin A.D. Anterior sacral meningocele. Surg Gynecol Obstet. 1968;126:986.
28. Anderson F.M., Burke B.L. Anterior sacral meningocele: a presentation of three cases. JAMA. 1977;237:39.
29. Coller F.A., Jackson R.G. Anterior sacral meningocele. Surg Gynecol Obstet. 1943;76:703.
30. Eder D. Anterior sacral meningocele: survey of literature and report of case. Bull Los Angeles Neurol Soc. 1949;14:104.
31. Haddad F.S. Anterior sacral meningocele: report of two cases and review of the literature. Can J Surg. 1958;1:230.
32. Thierry A., Archimbaud J.P., Ficher G., et al. La meningocele sacree anterieure: revue de la literature et presentation d’un cas. Neurochirurgie. 1969;15:389.
33. Currarino G., Coln D., Votteler T. Triad of anorectal, sacral, and presacral anomalies. Am J Radiol. 1981;137:395.
34. Ivamoto H.S., Wallman L.J. Anterior sacral meningocele. Arch Neurol. 1974;31:345.
35. Jabre A., Ball J.B., Tew J.M. Anterior sacral meningocele: current diagnosis. Surg Neurol. 1985;23:9.
36. Brown M.H., Powell L.D. Anterior sacral meningocele. J Neurosurg. 1945;2:535.
37. Dyck P., Wilson C.B. Anterior sacral meningocele. J Neurosurg. 1980;53:548.
38. Davis S.W., Levy L.M., LeBihan D., et al. Sacral meningeal cysts: evaluation with MR imaging. Radiology. 1993;187:445.
39. Lee K.S., Gower D.J., McWhorter J.M., et al. The role of MR imaging in the diagnosis and treatment of anterior sacral meningocele. J Neurosurg. 1988;69:628.
40. Martin B., de Latour F.B. MR imaging of anterior sacral meningocele. J Comput Tomogr. 1988;12:166.
41. Wetzel L.H., Levie E. MR imaging of sacral and presacral lesions. Am J Radiol. 1990;154:771.
42. Baleriaux-Waha D., Osteaux M., Terwinghe G., et al. The management of anterior sacral meningocele with computed tomography. Neuroradiology. 1977;14:42.
43. Rengachary S.S. Masses of the sacrum. In: Wilkins R.H., Rengachary S.S., editors. Neurosurgery. New York: McGraw-Hill; 1985:1079.
44. Tarlov I.M. Perineurial cysts of the spinal nerve roots. Arch Neurol Psychiatry. 1938;40:1067.
45. Turner M.L., Mulhern C.B., Dalinka M.K. Lesions of the sacrum: differential diagnosis and radiological evaluation. JAMA. 1981;245:275.
46. Enzmann D.R., DeLaPaz R.L., Rubin J.B. Magnetic resonance of the spine. St Louis: Mosby; 1990.
47. Pupovac C.D. Zur Kenntnis der pathologischen Anatomie und Genese der Hydromeningocele sacralis anterior. Arb Geb Klin Chir. 1903;1:533.
48. Demel R. Meningocele sacralis anterior. Deutsch Z Chir. 1928;209:90.
49. Coqui. Beitrag zur Kasuistik, Diagnose und Therapie der Meningocele sacralis anterior. Z Geburtshilfe Gynakol. 1916;78:609.
50. Adson A.W. Spina bifida cystica of the pelvis: diagnosis and surgical treatment. Minn Med. 1938;21:468.