CHAPTER 27 Myelodysplastic/myeloproliferative neoplasms
Introduction
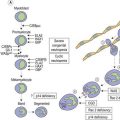
Myelodysplastic/myeloproliferative neoplasms (MDS/MPN) are clonal hematopoietic malignancies with hybrid myelodysplastic and myeloproliferative features. In these disorders, cytopenias and dysplasia may coexist with elevated white blood cell counts, thrombocytosis and/or organomegaly making it difficult to assign individual cases into a myelodysplastic or myeloproliferative category. Even though disorders with mixed myelodysplastic and myeloproliferative features have been recognized for many years, the precise diagnosis and assignment to a biologically defined category has been challenging. Acknowledging the unique characteristics of these diseases, in 2001 the World Health Organization classification of hematopoietic and lymphoid malignancies created a separate group of MDS/MPN.1 Chronic myelomonocytic leukemia (CMML) is perhaps the most well known example of a MDS/MPN. Depending on WBC count, CMML may resemble more closely a myelodysplastic syndrome (MDS) or a myeloproliferative neoplasm (MPN). The disease is clinically heterogeneous and lacks a precise biological connotation. Similar to CMML, other categories of MDS/MPNs are also a genetically heterogeneous group of diseases with variable patient outcomes. They are frequently positioned on a continuum between MDS and MPN, and thus are challenging to diagnose using traditional morphology-based approaches. Integration of morphologic features with laboratory data, clinical presentation, immunophenotype and cytogenetic/molecular tests is required to categorize MDS/MPNs according to current WHO 2008 classification.2 It is critical to diagnose patients at the time of original presentation, i.e. before bone marrow (BM) and peripheral blood (PB) features are modified by therapy or progression of the disease. If a patient has been previously diagnosed elsewhere, a complete review of the original diagnostic material is mandatory. ‘Re-classification’ based on the review of post-treatment and/or follow-up BM sample is strongly discouraged.
Three discrete MDS/MPN entities can be reliably classified using the above described integrated multiparametric approach: chronic myelomonocytic leukemia (CMML), atypical chronic myeloid leukemia (aCML), BCR-ABL1 negative and juvenile myelomonocytic leukemia (JMML). Occasionally, a myeloid neoplasm with myelodysplastic and myeloproliferative features cannot be assigned to any of these ‘classical’ categories. Such cases should be diagnosed as MDS/MPN, unclassifiable.2 This group includes also the provisional entity known as refractory anemia with ring sideroblasts associated with marked thrombocytosis (RARS-T). Additionally, there are rare disorders, which may qualify for inclusion in the MDS/MPN group in the future. One such disease is known as myeloid neoplasm with an isolated isochromosome 17q. This is a rare entity with a rapidly progressive clinical course that may occasionally morphologically resemble CMML.
A detailed discussion of the MDS/MPNs is presented below. The main diagnostic features of these neoplasms are summarized in Table 27.1. Some cytogenetic and genetic features are also addressed in Chapter 21.
Chronic myelomonocytic leukemia
Definition and diagnostic criteria
CMML is a hematopoietic malignancy with hybrid myeloproliferative and myelodysplastic features. The diagnostic criteria include: 1) persistent unexplained monocytosis (>1 × 109/l with >10% monocytes on the differential count); 2) absence of defining genetic features such as BCR-ABL1 fusion or rearrangements of PDGFRA and PDGFRB; 3) less than 20% blasts; and 4) dysplasia in one or more myeloid lineages. In the absence of definitive dysplastic features, the diagnosis of CMML can be rendered if unexplained monocytosis persists longer than 3 months or if cytogenetic/molecular studies demonstrate the presence of acquired clonal cytogenetic or molecular genetic abnormality.2
Epidemiology, clinical and laboratory features
CMML is a rare disease with estimated incidence of 3 cases per 100 000 people.3 The disease is more prevalent in men with a predominance of 1.5–3 : 1 and has a median age of presentation between 65 and 75 years.4,5 The disease is more common in Western countries as compared to Asian population.6 Patients can present with elevated WBC or with leukopenia. Signs and symptoms of BM failure are common and include fatigue, susceptibility to infections and bleeding. Weight loss, night sweats, fever, organomegaly, tissue infiltration (e.g. skin lesions) and serous effusions are also seen. Depending on the WBC (threshold 13 × 109/l), CMML has been previously divided into myeloproliferative or myelodysplastic categories.7 Splenomegaly and increased LDH are more common in the myeloproliferative subtype of CMML.8,9 Other clinical and laboratory features, and prognosis, including overall survival and incidence of transformation into acute leukemia, are similar for both groups.8–10 Whether the myelodysplastic and myeloproliferative CMML should be considered different phases of the disease or represent distinct entities is at present unclear. The separation is not required by the WHO 2008 classification.
Morphology and immunophenotypic features
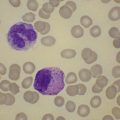
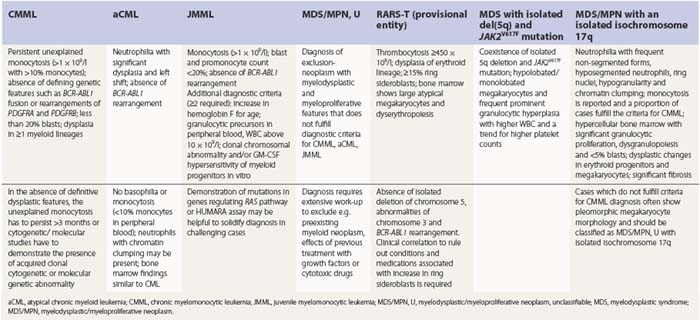
PB shows significant monocytosis (>1 × 109/l with >10% monocytes on the differential count), variable WBC (neutrophilia is seen in about 50% of the cases), anemia and thrombocytopenia. The majority of monocytes are mature, and they frequently display abnormal nuclear and cytoplasmic features. These include abnormal nuclear lobulation or chromatin pattern, prominent granulation or increased basophilia of cytoplasm (Fig. 27.1). Morphological definition of ‘abnormal monocytes’ and promonocytes has been a matter of debate and is thoroughly described in the WHO 2008 classification.2 In most cases, peripheral blood blasts, including monoblasts and promonocytes constitute less than 5% of the differential count. Neutrophilia may be present with dysgranulopoiesis including pseudo-Pelger–Huët cells and hypogranulation. Circulating neutrophilic precursors (promyelocytes and myelocytes) usually constitute less than 10% of white blood cells. In cases with significant eosinophilia (≥1.5 × 109/l), molecular studies excluding the rearrangements in PDGFRA and PDGFRB genes are necessary to further consider the diagnosis of CMML. Basophilia is uncommon. Giant platelets can be seen. Red blood cells are more often normochromic and normocytic but macrocytosis can also be seen.
Even though peripheral blood monocytosis is the single most important diagnostic finding, the final diagnosis of CMML should never be rendered without BM evaluation. The population of promonocytes, which are morphologically more differentiated than blasts in the bone marrow is not infrequently present in PB of patients with acute myelomonocytic and monocytic leukemia. Also, increase in monocytes largely confined to the BM without absolute monocytosis in PB can be seen in rare cases of ‘marrow predominant’ CMML. These diagnoses can be missed if a BM examination is not performed. Conversely, significant monocytosis can also be seen in some cases of BCR-ABL1+ chronic myelogenous leukemia (CML), which should always be excluded (see Chapter 24).
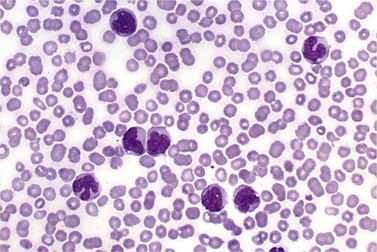
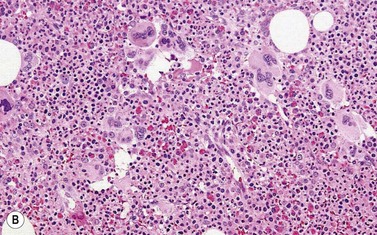
BM is significantly hypercellular in a majority of CMML cases, a finding easily appreciated in a predominantly elderly population, in which this disease occurs (Fig. 27.2A). Rarely, BM may be normo- or hypocellular.11 A prominent myeloid proliferation is usually seen (Fig. 27.2B). This includes both an increased number of neutrophilic forms as well as monocytes, which can be difficult to appreciate in the absence of non-specific esterase cytochemistry. In a significant number of cases, the granulopoiesis may be left shifted and show significant dysplasia including hypogranulation and nuclear abnormalities.7 Due to difficulties in differentiating monocytes from immature granulocytic cells (e.g. myelocytes and metamyelocytes), the quantification of the monocytic component may be challenging, which causes a significant variability between observers. The most significant practical issue is the differentiation of abnormal monocytes from monoblasts and promonocytes. The early stages of monocytic differentiation in the bone marrow of CMML patient are shown in Fig. 27.2C.
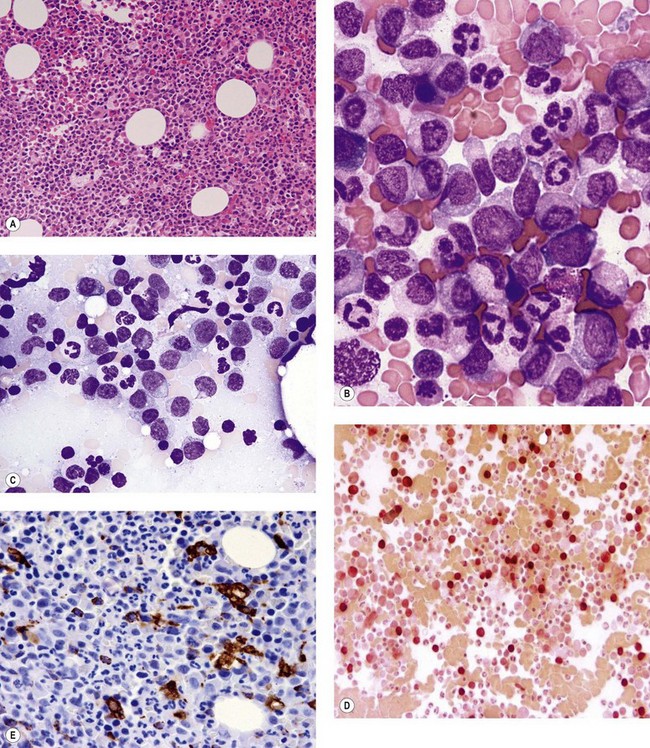
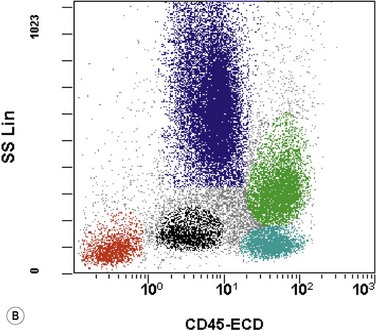
The application of nonspecific esterase and immunophenotyping by flow cytometry or immunohistochemistry are helpful in quantifying the monocytic component. Nonspecific esterase staining (alpha-naphthyl butyrate or alpha-naphthyl acetate esterase) is an effective method of demonstrating monocytic differentiation. The stain is useful to confirm the increased number of esterase positive cells in cases of CMML. Median values of 10–20% esterase positive cells are found in the majority of CMML patients12,13 (Fig. 27.2D). Multiparametric flow cytometry has the added benefits of separating various stages of monocytic differentiation and demonstrating an aberrant antigen expression. The former can be accomplished by profiling the expression of the different epitopes of CD14 antigen in combination with CD64 and CD33, which reveals the presence of promonocytes and more mature monocytic forms.14 Aberrant expression of myeloid antigens, both loss and altered antigen density, and expression of non-myeloid markers, are frequently seen in monocytes and in the granulocytic population. Monocytes show altered density of HLA-DR, CD13 and CD64, and may show co-expression of CD2 and CD56 (Fig. 27.3A). In comparison to reactive monocytes, the aberrant expression of CD56 on CMML monocytes was much higher.15,16 The decrease in right-angle light scatter is not infrequent and reflects the hypogranulation of granulocytic series (Fig. 27.3B). Increase in the number of CD34 and/or CD117 positive blasts may indicate progression of the disease and alert to the impending transformation.
With the exception of demonstrating an increased number of blasts and blast clusters using immunohistochemical stain for CD34, the value of immunohistochemistry in CMML has been limited. Common myeloid/monocyte/macrophage immunohistochemical markers such as lysozyme and CD68 (KP1) applied to BM trephine biopsies (BMTB), are generally not reliable in quantifying monocytic component.12 CD68R and CD163 have been shown to be more restricted to monocytes and macrophages. However, these markers usually stain significantly lower numbers of monocytic cells as compared to naphthyl butyrate esterase12,17 (Fig. 27.2E). Some investigators reported that staining pattern of CD68R (fine vs coarse granules) in conjunction with cell morphology can discriminate between monocyte precursors and mature monocytes.18 The success of this approach may, however, be dependent on the processing of BMTB and staining methodology used, and needs further validation and standardization in a multi-institutional setting. New antibodies applicable to paraffin embedded material have recently been reported to aid in enumeration of the monocytic component in BMTB. A combined use of CD14 and CD16 allowed discriminating between monocyte population and dysplastic granulocytes in this setting.17
Numerous studies confirmed that the number of blasts and promonocytes is the most reliable predictor of patient prognosis and of the risk of transformation to acute leukemia. Recognizing the importance of the increase in blasts, CMML is divided into two groups: CMML-1 with less than 5% blasts in PB and less than 10% in BM; and CMML-2 with 5–19% of blasts in PB and 10–19% in BM. The prognostic value of this classification has been previously confirmed.19
Erythropoiesis is usually normal to decreased and may show dysplastic changes.20 These include megaloblastoid change with asynchrony in maturation of nucleus and cytoplasm, and nuclear abnormalities such as nuclear fragmentation, irregularity of nuclear outline and multinucleation. Increased megakaryopoiesis is not uncommon; however, it is usually less pronounced than that seen in cases of BCR-ABL1+ CML.12 Dysmegakaryopoiesis is seen in the majority of CMML cases.21 Dysplastic megakaryocytes display abnormal nuclear lobation including mononuclear or hypolobated forms, or separated nuclear lobes. Micromegakaryocytes can also be seen. Approximately 20% of cases show significant reticulin fibrosis.13
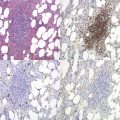
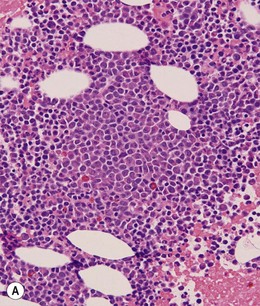
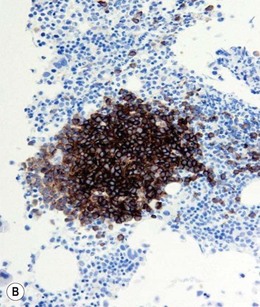
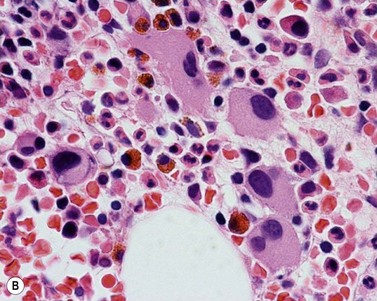
Approximately 20% of CMML cases show nodular proliferations of plasmacytoid dendritic cells12 (Fig. 27.4). The lineage and origin of these cells have long been debated and is reflected by changing nomenclature starting from the designation of ‘T-associated plasma cells’ and ‘plasmacytoid T-cells’ to ‘plasmacytoid monocytes’. Currently, plasmacytoid dendritic cells are believed to be derived from myeloid progenitors. Thus, it is perhaps not surprising that focal proliferations of these cells are seen in association with myeloid neoplasms with monocytic differentiation, most commonly in CMML. These nodules are considered clonal and in select cases have been shown to be clonally related to the underlying myeloid neoplasm.22,23 Plasmacytoid dendritic cells are intermediate in size with round to oval nucleus, with or without indentation, finely dispersed chromatin and moderately abundant cytoplasm with distinct cytoplasmic border. Their identification is facilitated by immunohistochemical stains including CD123, CD68, CD68R, CD4 and CD14. Plasmacytoid dendritic cells are also positive for granzyme B and occasionally for CD56, CD2 and CD5. They are negative for other lymphoid markers, and for CD13, CD11c, myeloperoxidase and CD34.
Prognosis
The survival of patients with CMML is extremely variable, ranging from 1 to 100 months.2,4,12,24 Considerable efforts have been made in the past to design a predictor of clinical outcome. All scoring systems shown to be useful in predicting the clinical outcome (such as modified Bournemouth score, Dusseldorf score, MD Anderson prognostic score and Spanish CMML score) used a combination of BM blast count with PB counts to predict survival.5,24 In multivariate analysis, PB and BM blast percentage proved to be the strongest predictor of survival and of transformation into acute myeloid leukemia (AML).4,13,19,25,26 PB and BM blast counts are the keystones to distinguish between CMML-1 and CMML-2. These two subcategories were shown to be highly predictive of overall survival and correlated well with transformation into AML. Specifically, at 5-year follow-up, 63% of patients with a BM blast count of 10% or greater developed AML as compared to 18% of patients with blast count below 10%.19 Previously discussed myelodysplastic and myeloproliferative subtypes of CMML were not significant for predicting patient outcome.19
As our knowledge on the pathogenesis of myeloid neoplasms expands, new prognostic factors emerge. TET2 gene mutations have been recently reported to occur in 50% of CMML patients (see Chapter 21) and have been associated with poor survival in the CMML-1 subgroup.27 RUNX1 mutations occur in up to 40% of CMML cases and abnormalities of C-terminal portion of this gene are associated with progression to AML.28
Special considerations
Chronic myelomonocytic leukemia developing in a course of myelodysplastic syndrome
The evolution of previously diagnosed MDS to CMML is well documented.29–32 The pathogenesis of this phenomenon is not well understood with only rare studies testing select genes implicated in myeloid neoplasms.32 The interval between the initial MDS diagnosis and the evolution to CMML ranged between 2 and 59 months.
Approximately 20% of MDS patients show relative peripheral blood monocytosis (more than 10% monocytes but with absolute monocyte count less than 1 × 109/l) at the time of their initial diagnosis. Over time, a significant proportion of these patients may develop CMML. Conversely, up to 24% of patients with CMML have a documented preceding MDS phase.13,32 Both groups, whether originally presenting with or without relative monocytosis, initially show less than 1 × 109/l monocytes in PB. Rigolin et al. reported the distinct clinicopathological features of MDS cases with relative monocytosis (>10% monocytes).29 In comparison to MDS without monocytosis, these patients showed higher marrow cellularity, higher incidence of chromosomal abnormalities, and a more aggressive disease with high incidence of progression to CMML and transformation into acute myelomonocytic or monocytic leukemia.29 When CMML evolving from the MDS phase was compared to myelodysplastic type of de novo CMML, the former showed a superior survival.31,32
Therapy-related CMML
The majority of CMML cases represent de novo disease arising in patients without a pre-existing hematologic or non-hematopoietic neoplasm. Occasionally, CMML may develop after treatment with chemotherapeutic agents and/or radiotherapy for malignancies or non-neoplastic diseases.33–35 Even though the numbers of reported cases are low, therapy-related CMML seems to show morphologic and immunophenotypic features similar to those of de novo CMML. However, a proportion of these cases show the rearrangement of MLL gene, a finding which is usually seen only in acute leukemia. Regardless of morphologic features, current WHO classification recommends to classify all therapy-related cases under the umbrella of therapy-related myeloid neoplasms. Therefore, all therapy-related CMML cases should be included in this category. Thus, the detailed knowledge of patient’s previous medical history is critical while diagnosing CMML.
Atypical chronic myeloid leukemia, BCR-ABL1 negative
Epidemiology, clinical and laboratory features
The aCML is a rare myeloid neoplasm with an incidence of 1–2 cases for every 100 cases of Philadelphia-positive CML.36 Patients present in the 7th or 8th decade of life with leukocytosis, splenomegaly, frequent anemia and variable platelet counts. Both genders are equally affected. The JAK2V617F mutation is absent.37
Morphology and immunophenotypic features
By definition, the WBC is elevated in the range of 18–300 × 109/l (median 24–36 × 109/l)36–39 and is frequently accompanied by anemia and thrombocytopenia. The majority of cases show more than 10% of circulating immature granulocytic cells, which is roughly similar to what is seen in CML. Blast count is low, usually less than 5%. Prominent dysgranulopoiesis differentiates aCML from CML (Fig. 27.5A). CML usually does not show dysgranulopoieis at the time of the initial diagnosis but may develop severe dysplastic changes upon progression of the disease.
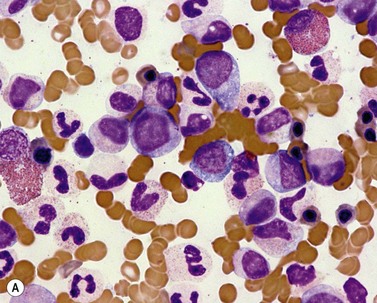
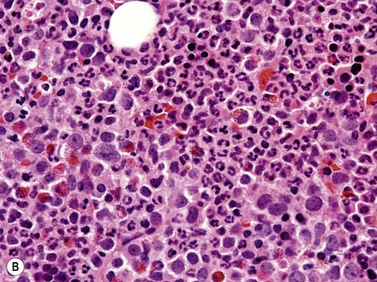
Fig. 27.5 (A, B) aCML. (A) Bone marrow aspirate smear of aCML showing dysplastic granulocytes. (B) Bone marrow biopsy of aCML showing a hypercellular marrow.
Dysgranulopoiesis seen in aCML includes the presence of neutrophils with abnormally lobated nuclei such as pseudo-Pelger–Huët cells, abnormally condensed chromatin and hypogranulation. In a proportion of cases, the dysplastic neutrophils have abnormally hyperlobulated nuclei and abnormal chromatin clumping (syndrome of abnormal chromatin clumping). Due to leukocytosis, the absolute monocyte count can be higher than 1 × 109/l. However, monocytes never exceed 10% of the differential count. Significant basophilia, as seen in CML cases, is not present and basophils rarely exceed 2% of the differential count. Anemia may be macrocytic with macroovalocytes as evidence of dysplastic erythroid maturation. Platelets are frequently decreased. However, cases with thrombocytosis have been reported (range 9–2675 × 109/l). In the original study by the French-American-British (FAB) Cooperative Leukemia Group, the parameters most strongly associated with the diagnosis of aCML were: peripheral blood leukocytosis with immature myeloid forms, dysgranulopoiesis, and the absence of basophilia and monocytosis.40
In the majority of cases, BM is hypercellular with a significantly increased granulopoiesis (Fig. 27.5B). Dysgranulopoiesis is seen in all cases. By definition, blast count is below 20% and, in most instances, blasts represent less than 5% of marrow elements. The M : E ratio is commonly increased with erythroid series frequently accounting for less than 10% of BM elements.40 Dyserythropoiesis is common. The number of megakaryocytes is variable and not uncommonly decreased. Dysmegakaryopoiesis is usually seen. Hypolobated and/or monolobated forms and micromegakaryocytes are most common. As in other subtypes of MDS/MPN, reticulin fibers may be increased.
Prognosis
aCML is an aggressive disease with a median survival of 14–37 months depending on the therapy received.36,38,39,41 The majority of patients succumb to BM failure and approximately 20–40% of cases transform to AML. The rate and time to transformation is variable, dependent on the initial treatment (conservative vs intensive induction-like therapy). In younger patients, the outcome is improved by hematopoietic stem cell transplantation.42
Variables associated with shorter survival times include older age, female gender, leukocyte count of more than 50 × 109/l, and circulating immature precursors. The percentage of BM blasts (>5%) and marked dyserythropoiesis were the most significant factors associated with leukemic transformation.36
Special considerations
Syndrome of abnormal chromatin clumping
The syndrome of abnormal chromatin clumping, which has long been recognized as representing a hematologic neoplasm with both myelodysplastic and myeloproliferative features is, in most cases, simply a morphologic variant of aCML.43–47 The majority of patients from the original series with abnormal chromatin clumping developed leukocytosis, anemia and thrombocytopenia. Rare patients showed neutropenia at the onset of the disease. The defining feature of this disease is a characteristic neutrophil morphology – excessive ‘clumping’ of chromatin into large well separated blocks of heterochromatin and euchromatin. This feature is commonly accompanied by a loss of segmentation and is best appreciated in mature neutrophils (Fig. 27.6).
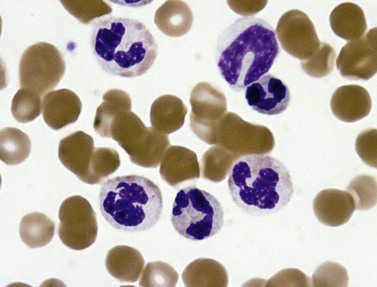
Abnormal chromatin clumping is not limited to the syndrome described above. Rare cases of BCR-ABL1+ CML have been reported to show an identical morphology.48 In addition, reversible chromatin clumping is seen in neutrophils of patients treated with mycophenolate mofetil, in HIV infection, and in select patients treated for lymphoproliferative disorders.49,50 Thus, as in all hematologic malignancies, the final diagnosis has to be made in correlation with laboratory data and in the context of clinical presentation.
Atypical chronic myeloid leukemia with t(8;9)(p22;p24)/PCM1-JAK2
A number of cases classified as aCML and carrying fusion of PCM1-JAK2 genes have been described.51,52 Detailed description of the morphologic features of PB and BM is not available in all cases. However, in many instances, significant eosinophilia of a degree comparable to that seen in chronic eosinophilic leukemia has been reported. Other cases showed more complex morphologic characteristics and multilineage involvement. PCM1-JAK2 abnormality can also present as de novo AML or acute lymphoblastic leukemia. Thus, in cases with t(8;9)(p22;p24), a detailed review of cellular morphology and correlation with laboratory values is necessary for a definitive classification.
Juvenile myelomonocytic leukemia (JMML)
Epidemiology, clinical and laboratory features
In the past, JMML has been referred to as juvenile chronic myeloid leukemia, chronic myelomonocytic leukemia and infantile monosomy 7 syndrome. The incidence of this disease is approximately 1 case per million children younger than 14 years.53–55 It is most common in children below 3 years of age; however, early adolescent patients have also been reported. Males are affected more frequently than females. There is an association with neurofibromatosis type 1 and to lesser extent with Noonan syndrome, related to the activation of RAS pathway through mutations in tumor suppressor gene NF1, and PTPN11, KRAS or SOS1 genes. Presenting features include leukocytosis with monocytosis, anemia and thrombocytopenia leading to pallor, malaise, infections and bleeding. Similar to adult CMML, a maculopapular rash also occurs. Organ infiltration by leukemic cells leads to hepatosplenomegaly and lymphadenopathy. Leukemic infiltrates in the lungs and gastrointestinal tract clinically manifest as respiratory failure and diarrhea. In cases of neurofibromatosis type 1 or Noonan syndrome, additional features related to primary disorder, such as café au lait spots, may also be present.
Two additional laboratory features are keystones of JMML diagnosis. At least half of the patients show elevation of the HbF level corrected for age. This feature is seen predominantly in patients without monosomy 7.56 In vitro hypersensitivity of myeloid progenitors to GM-CSF is another important diagnostic parameter.57 In contrast to other neoplasms with myeloproliferative features, this hypersensitivity is limited to GM-CSF. The response of JMML cells to interleukin(IL)-3 or G-CSF is normal. In vitro hypersensitivity to GM-CSF is a labor intensive and challenging assay and may, in the future, be replaced by flow cytometry testing hyperphosphorylation of signal transducer and activator of transcription (STAT)-5 . In this assay, BM cells are stimulated by a low-dose of GM-CSF with subsequent measurement of STAT3 phosphorylation. JMML marrows show a significantly higher percentage of phospho-STAT5 positive cells upon stimulation than that observed in normal BM and in select other pediatric myeloid disorders. This test can be easily performed in clinical flow cytometry laboratory.58
Morphology and immunophenotypic features
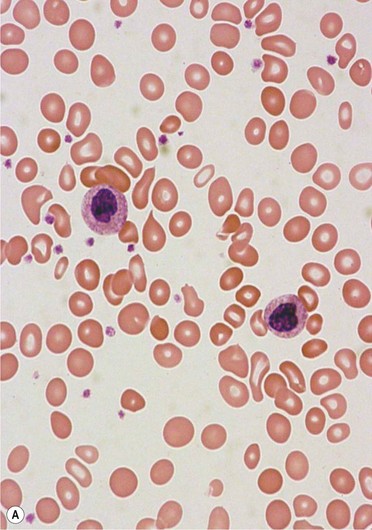
PB shows leukocytosis with reported medians in the range of 30 × 109/l. Approximately 10% of patients have WBC exceeding 100 × 109/l. Absolute monocytosis is the defining feature of this disease and is even evident in rare patients who present with WBC within normal limits (Fig. 27.7A). The degree of monocytosis is variable with the majority of patients showing more than 5 × 109/l monocytes. The reported range for an absolute monocyte count is between 1.1 and 60.8 × 109/l.56 Both mature monocytes and their precursors can be seen in the PB. However, the combined blast and promonocyte count is below 5% in the majority of patients. Immature granulocytes and erythroid precursors can also be seen in PB. A proportion of cases show macrocytic red blood cells; however, normocytic or microcytic anemia are not infrequent. Thrombocytopenia is common with close to 20% of patients showing platelet count below 20 × 109/l.56
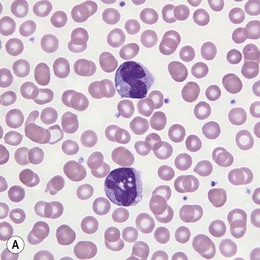
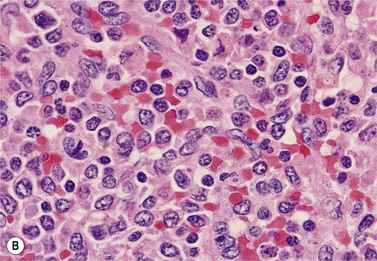
Fig. 27.7 (A, B) JMML. (A) Monocytes in peripheral blood. (B) Extramedullary manifestation (spleen) in JMML.
The leukemic infiltrates of extramedullary organs such as lymph nodes, spleen, liver, lungs and skin are composed of a myelomonocytic population similar to that seen in the BM (Fig. 27.7B).
Prognosis
JMML is an aggressive disease with a 5-year survival of 40%.53 Prognostic factors associated with shorter survival include platelet count below 40 × 109/l, an increased HbF level, and age above 2 years. Combined HbF, platelet count and cytogenetics (FPC) score has previously been shown to be highly predictive of survival in JMML.53,59
Rare cases of spontaneous improvement of the disease were reported in the association with Noonan syndrome and in de novo JMML with glycine to serine substitution of codon 12 of RAS genes.60–62 The latter observation has been subsequently disputed.63 Thus, further large-scale studies are required to determine the association of genotype with clinical features and to warrant the withholding of currently available and potentially curative treatment regimens.
Special considerations
Differential diagnosis between JMML and viral infection
The most significant differential diagnosis, especially in young children, is viral infection. Similar to JMML, infections can be accompanied by a leukemoid reaction and monocytosis. Infections with EBV, CMV and HHV6, and other microorganisms such as Histoplasma, Toxoplasma and mycobacteria can mimic BM findings and clinical presentation of JMML.64–67 BM morphology is not useful in distinguishing between JMML and an infectious process. The demonstration of clonality, either with cytogenetic or molecular analysis, is most helpful to resolve this differential diagnosis. Even though cytogenetic abnormalities are seen only in a third of patients with JMML, 75% of children harbor mutations in genes regulating RAS pathway (NRAS, KRAS, PTPN11 and NF1). Recently described mutations in CBL gene bring the number of JMML cases with identifiable genetic lesion to over 90%. In female patients with normal cytogenetics and no mutations of genes involved in the regulation of RAS activation, human androgen receptor gene analysis (HUMARA assay) may be helpful in establishing a definitive diagnosis.61
JMML and transient polyclonal myeloproliferation of Noonan syndrome
Patients with Noonan syndrome are at increased risk of developing JMML due to mutations in genes regulating the RAS pathway. They can also exhibit a constitutional non-clonal myelomonocytic proliferation, which can fulfill current diagnostic criteria for JMML and present with similar clinical features.60,61 These patients can develop a clonal monocyte population and/or cytogenetic abnormality leading to progression of the disease or they can undergo spontaneous remission despite persistent hypersensitivity to GM-CSF.61 It is critical to distinguish between the transient polyclonal myeloproliferation of Noonan syndrome and true JMML to avoid unnecessary therapy.
Myelodysplastic/myeloproliferative neoplasm, unclassifiable
Morphology and immunophenotypic features
PB can show leukocytosis and/or thrombocytosis with variable degrees of dysplastic features such as pelgeroid and hypogranular forms, and giant and hypogranular platelets. Macrocytic anemia or dimorphic red blood cells may be seen. Increase in blasts cells above 10% may indicate the progression of the disease.2
There are no specific cytochemical features. Immunophenotype has not been well studied.
Provisional entity in myelodysplastic/myeloproliferative neoplasm, unclassifiable: refractory anemia with ring sideroblasts associated with marked thrombocytosis
Epidemiology, clinical and laboratory features
RARS-T is rare and accounted for only 0.7% of FAB-defined MDS cases (data from the MDS registry, Dusseldorf, Germany).68,69 It predominantly affects older adults with a reported median age of presentation ranging from 59 to 73 years. Men and women are equally affected. Anemia and thrombocytosis can be significant; however, the majority of patients present with a median hemoglobin level of 10.1 g/dl and moderate thrombocytosis below 1000 × 109/l.70 In a recent study, Raya et al. compared RARS-T cases with marked thrombocytosis to those with only borderline platelet elevation.71 The former group showed more prominent myeloproliferative features including a higher WBC, higher incidence of splenomegaly and more frequent JAK2V617F mutations. However, rare cases of RARS-T with the JAK2V617F mutation and platelet counts within the normal range have also been reported.
Morphology and immunophenotypic features
Most cases show hypercellular BM (Fig. 27.8A). The most significant BM findings differentiating RARS-T from other myeloid neoplasms presenting with thrombocytosis and increased numbers of ring sideroblasts, are megakaryocyte morphology accompanied by dyserythropoiesis (Fig. 27.8B). Large pleomorphic megakaryocytes similar to those seen in BCR-ABL1 negative MPN are frequently seen. Although small hypolobated and monolobated forms can also be present, true micromegakaryocytes (diameter of less than 15 microns) are not common. Dyserythropoiesis is a constant finding and includes maturation asynchrony, nuclear budding and megaloblastoid chromatin. Ring sideroblasts are defined as erythroid cells with at least five siderotic granules surrounding at least one third of the nuclear circumference72 (Fig. 27.8C). In RARS-T, by definition, ring sideroblasts constitute at least 15% of erythroid precursors. It is important to remember that ring sideroblasts per se are not a feature of dysplasia. They can also be seen in a variety of reactive states and appear transiently in association with exposure to medications and toxins. The review of clinical history to exclude reactive causes of ring sideroblasts is an integral part of BM evaluation. BM blasts constitute less than 5% of the differential count. Abnormal localization of immature precursors has not been reported in this entity. Mast cell hyperplasia can be seen in some cases. The majority of cases show at least a mild increase in reticulin fibers. Significant reticulin fibrosis can also be observed.
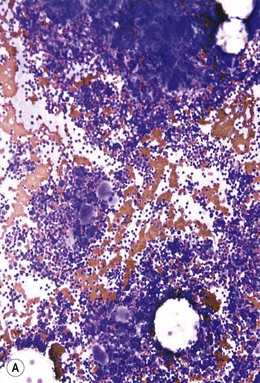
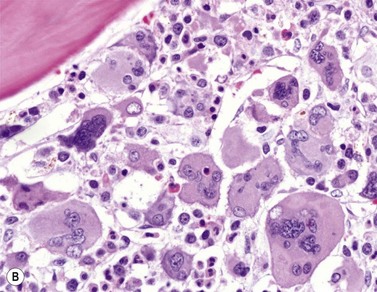
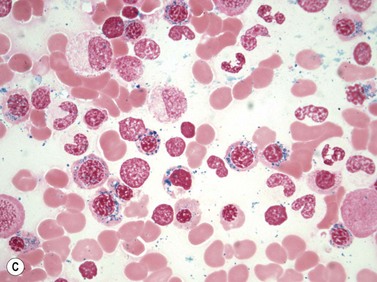
Prognosis
Prognosis of patients with RARS-T is better than that of other subtypes of MDS/MPNs. Reported median survivals are variable (range between 71 and 101 months).73,74 However, most reports agree that survival in RARS-T is inferior to that seen in essential thrombocythemia (ET) and comparable to that of RA-RS category of MDS. The initial report on the JAK2V617F mutation suggested that cases with mutated JAK2 have a survival advantage.75 This finding has not been confirmed in more recent reports and requires further study.71 Transformation to acute leukemia has been reported.76
Special considerations
Differentiation of RARS-T from myeloproliferative neoplasms with thrombocytosis and ring sideroblasts
The presence of ring sideroblasts in MPN has been linked to mutations in genes associated with hemochromatosis.77 Ring sideroblasts can be seen in approximately 5% of BCR-ABL1 negative MPN including ET and primary myelofibrosis (PMF). These disorders can overlap with RARS-T in regards to the presence of thrombocytosis, degree of megakaryocytic proliferation seen in the bone marrow, and the presence of the JAK2V617F mutation. ET presents with normocellular or slightly hypercellular BM and, in most cases, lacks erythroid or granulocytic hyperplasia. Rare cases can show a borderline increase in the erythroid series; however, dyserythropoiesis is not present. On the contrary, the diagnosis of RARS-T requires the presence of dyserythropoiesis. Similarly, in early prefibrotic stage, PMF presents with thrombocytosis and hypercellular marrow with megakaryocytic hyperplasia. In the majority of these cases, the reduction in the erythroid series, more pronounced dysmegakaryopoiesis and megakaryocyte clustering are helpful in establishing the diagnosis of PMF.
RARS and RARS-T, distinct entities or clinical continuum
Considering overlapping clinicopathological features of this disease, the designation of RARS-T as a distinct entity has been previously disputed. The overlap with myeloproliferative neoplasms has been discussed above. Cases overlapping with MDS of RA-RS category have also been reported. Perhaps the most compelling evidence for the existence of the biological and clinical continuum between these two entities is the development of elevated platelet counts in classical cases of RARS.70 In a few reported cases of RARS, the onset of thrombocytosis coincided with the appearance of the JAK2V617F mutation in a subset of granulocytes. Of note, JAK2 mutation has been reported in 60% to over 90% of RARS-T cases, and is more frequent in cases with marked thrombocytosis.71 The latter cases show also higher WBC and more frequent splenomegaly. Thus, the acquisition of the JAK2V617F mutation may indeed confer a myeloproliferative phenotype to what would be otherwise a case of RARS. The more definitive studies to elucidate pathogenesis of RARS-T are needed to clarify its position in the taxonomy of MPN. Until these data are available, the set of features most useful in separating RARS-T from RARS is megakaryocyte morphology and the presence of the JAK2V617F mutation.
Miscellaneous myeloid neoplasms with myelodysplastic and myeloproliferative features
Myelodysplastic syndrome with isolated del(5q) and JAK2V617F mutation
A small subset of MDS with isolated 5q deletion has been recently shown to harbor JAK2V617F mutations.78 In addition to typical features of MDS with isolated del(5q) such as hypolobated/monolobated megakaryocytes, these cases more frequently demonstrated a prominent granulocytic hyperplasia with a higher WBC (5.21 vs. 4.45 × 109/l in cases with wild type JAK2 gene) and a trend for higher platelet counts (Fig. 27.9). A proportion of these cases are responsive to lenalidomide treatment.79 Until further clinicopathological data are available, the WHO classification recommends that these cases be classified as MDS with isolated del(5q) and the JAK2V617F mutation.
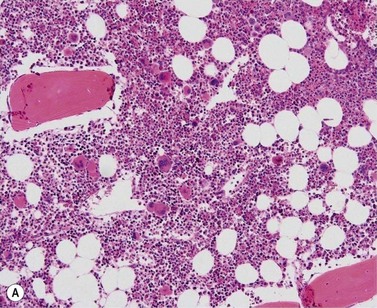
Myelodysplastic/myeloproliferative neoplasm with isolated isochromosome 17q
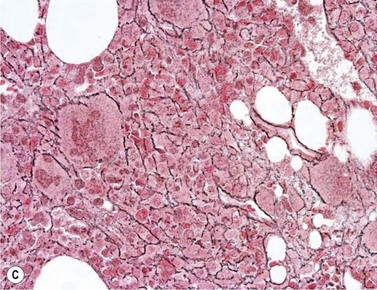
MDS/MPN with isochromosome 17q is not uncommon in myeloid neoplasms both within a complex karyotype and as secondary abnormality in patients with CML in accelerated or blast phase.80 It can also present as a sole abnormality in cases with mixed myelodysplastic and myeloproliferative features.81–83 The patient age varies with a median of 60 years. There is a male predominance. The majority of patients present with anemia, leukocytosis due to neutrophilia and organomegaly. Monocytosis is commonly reported and a proportion of cases fulfill the criteria for CMML-1.83 Dysgranulopoiesis including frequent non-segmented forms, hyposegmented neutrophils (pseudo-Pelger–Huët cells), ring nuclei, hypogranularity and chromatin clumping is prominent (Fig. 27.10A). Blasts represent less than 5% of the differential count in the majority of cases. Platelet counts are variable and abnormal hypogranular and/or giant platelets are often seen. The BM is markedly hypercellular with significant granulocytic proliferation and dysgranulopoiesis (Fig. 27.10B). Blasts constitute less than 5% of the cellularity. Dysplastic changes are also seen in erythroid progenitors and megakaryocytes (Fig. 27.10B). Dysplastic small megakaryocytes and large multinucleated ones have both been reported. Significant fibrosis is frequent (Fig. 27.10C). The median survival was 2.5 years and 64% of patients progressed to AML.83
1 Jaffe E, Harris N, Stein H, editors. WHO Classification of Tumours Pathology and Genetics of Tumours of Hematopoietic and Lymphoid Tissues. Lyon: IARC Press, 2001.
2 Swerdlow S, Campo E, Harris N, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press, 2008.
3 Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol. 1982;51:189-199.
4 Fenaux P, Beuscart R, Lai JL, et al. Prognostic factors in adult chronic myelomonocytic leukemia: an analysis of 107 cases. J Clin Oncol. 1988;6:1417-1424.
5 Onida F, Kantarjian HM, Smith TL, et al. Prognostic factors and scoring systems in chronic myelomonocytic leukemia: a retrospective analysis of 213 patients. Blood. 2002;99:840-849.
6 Bowen DT. Chronic myelomonocytic leukemia: lost in classification? Hematol Oncol. 2005;23:26-33.
7 Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. 1976;33:451-458.
8 Germing U, Gattermann N, Minning H, et al. Problems in the classification of CMML – dysplastic versus proliferative type. Leuk Res. 1998;22:871-878.
9 Voglová J, Chrobák L, Neuwirtová R, et al. Myelodysplastic and myeloproliferative type of chronic myelomonocytic leukemia – distinct subgroups or two stages of the same disease? Leuk Res. 2001;25:493-499. 2001
10 Nösslinger T, Reisner R, Grüner H, et al. Dysplastic versus proliferative CMML – a retrospective analysis of 91 patients from a single institution. Leuk Res. 2001;25:741-747.
11 Storniolo AM, Moloney WC, Rosenthal DS, et al. Chronic myelomonocytic leukemia. Leukemia. 2002;4:766-770.
12 Orazi A, Chiu R, O’Malley DP, et al. Chronic myelomonocytic leukemia: the role of bone marrow biopsy immunohistology. Mod Pathol. 2006;19:1536-1545.
13 Tefferi A, Hoagland HC, Therneau TM, et al. Chronic myelomonocytic leukemia: natural history and prognostic determinants. Mayo Clin Proc. 1989;64:1246-1254.
14 Yang DT, Greenwood JH, Hartung L, et al. Flow cytometric analysis of different CD14 epitopes can help identify immature monocytic populations. Am J Clin Pathol. 2005;124:930-936.
15 Xu Y, McKenna R, Karandikar N, et al. Flow cytometric analysis of monocytes as a tool for distinguishing chronic myelomonocytic leukemia from reactive monocytosis. Am J Clin Pathol. 2005;124:799-806.
16 Lacronique-Gazaille C, Chaury M, Le Guyader A, et al. A simple method for detection of major phenotypic abnormalities in myelodysplastic syndromes: expression of CD56 in CMML. Haematologica. 2007;92:859-860.
17 Qubaja M, Marmey B, Le Tourneau A, et al. The detection of CD14 and CD16 in paraffin-embedded bone marrow biopsies is useful for the diagnosis of chronic myelomonocytic leukemia. Virchows Arch. 2009;454:411-419.
18 Ngo N, Lampert I, Naresh K. Bone marrow trephine morphology and immunohistochemical findings in chronic myelomonocytic leukaemia. Br J Haematol. 2008;141:771-781.
19 Germing U, Strupp C, Knipp S, et al. Chronic myelomonocytic leukemia in the light of the WHO proposals. Haematologica. 2007;92:974-977.
20 Van der Weide M, Sizoo W, Nauta JJ, et al. Myelodysplastic syndromes: analysis of clinical and prognostic features in 96 patients. Eur J Haematol. 1988;41:115-122.
21 Martiat P, Michaux JL, Rodhain J. Philadelphia-negative (Ph−) chronic myeloid leukemia (CML): comparison with Ph+ CML and chronic myelomonocytic leukemia. Groupe Français de Cytogénétique Hématologique. Blood. 1, 1991. 205–111
22 Chen YC, Chou JM, Ketterling RP, et al. Histologic and immunohistochemical study of bone marrow monocytic nodules in 21 cases with myelodysplasia. Am J Clin Pathol. 2003;120:874-881.
23 Vermi W, Facchetti F, Rosati S, et al. Nodal and extranodal tumor-forming accumulation of plasmacytoid monocytes/interferon-producing cells associated with myeloid disorders. Am J Surg Pathol. 2004;28:585-595.
24 Germing U, Kündgen A, Gattermann N. Risk assessment in chronic myelomonocytic leukemia (CMML). Leuk Lymphoma. 2004;45:1311-1318.
25 Storniolo AM, Moloney WC, Rosenthal DS, et al. Chronic myelomonocytic leukemia. Leukemia. 1990;4:766-770.
26 Germing U, Strupp C, Aivado M, et al. New prognostic parameters for chronic myelomonocytic leukemia. Blood. 2002;100:731-732.
27 Kosmider O, Gelsi-Boyer V, Ciudad M, et al. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica. 2009;94:1676-1681.
28 Kuo MC, Liang DC, Huang CF, et al. RUNX1 mutations are frequent in chronic myelomonocytic leukemia and mutations at the C-terminal region might predict acute myeloid leukemia transformation. Leukemia. 2009;23:1426-1431.
29 Rigolin GM, Cuneo A, Roberti MG, et al. Myelodysplastic syndromes with monocytic component: hematologic and cytogenetic characterization. Haematologica. 1997;82:25-30.
30 Rosati S, Mick R, Xu F, et al. Refractory cytopenia with multilineage dysplasia: further characterization of an ‘unclassifiable’ myelodysplastic syndrome. Leukemia. 1996;10:20-26.
31 Breccia M, Cannella L, Frustaci A, et al. Chronic myelomonocytic leukemia with antecedent refractory anemia with excess blasts: further evidence for the arbitrary nature of current classification systems. Leuk Lymphoma. 2008;49:1292-1296. 2008
32 Wang SA, Galili N, Cerny J, et al. Chronic myelomonocytic leukemia evolving from preexisting myelodysplasia shares many features with de novo disease. Am J Clin Pathol. 2006;126:789-797.
33 George R, Pearson AD, Evans J, et al. Secondary chronic myelomonocytic leukemia with t(9;11) in a child. Cancer Gen Cytogenet. 1994;75:64-66.
34 Noriko S, Yasushi I, Yoshiko O, et al. Novel MLL-CBP fusion transcript in therapy-related chronic myelomonocytic leukemia with a t(11;16) (q23;p13) chromosome translocation. Genes Chromosome and Cancer. 1997;20:60-63.
35 Czader M, Orazi A. Therapy-related myeloid neoplasms. Am J Clin Pathol. 2009;132:410-425.
36 Breccia M, Biondo F, Latagliata R, et al. Identification of risk factors in atypical chronic myeloid leukemia. Haematologica. 2006;91:1566-1568.
37 Fend F, Horn T, Koch I, et al. Atypical chronic myeloid leukemia as defined in the WHO classification is a JAK2 V617F negative neoplasm. Leukemia Res. 2008;32:1931-1935.
38 Hernández JM, del Cañizo MC, Cuneo A, et al. Clinical, hematological and cytogenetic characteristics of atypical chronic myeloid leukemia. Ann Oncol. 2000;11:441-444.
39 Kurzrock R, Bueso-Ramos CE, Kantarjian H, et al. BCR rearrangement-negative chronic myelogenous leukemia revisited. J Clin Oncol. 2001;19:2915-2926.
40 Bennett JM, Catovsky D, Daniel MT, et al. The chronic myeloid leukaemias: guidelines for distinguishing chronic granulocytic, atypical chronic myeloid, and chronic myelomonocytic leukaemia. Proposals by the French-American-British Cooperative Leukaemia Group. Br J Haematol. 1994;87:746-754.
41 Costello R, Sainty D, Lafage-Pochitaloff M, et al. Clinical and biological aspects of Philadelphia-negative/BCR-negative chronic myeloid leukemia. Leuk Lymphoma. 1997;25:225-232.
42 Koldehoff M, Beelen DW, Trenschel R, et al. Outcome of hematopoietic stem cell transplantation in patients with atypical chronic myeloid leukemia. Bone Marrow Transplant. 2004;34:1047-1050.
43 Morel P, Bryon P, Guyon J, et al. Hémopathie maligne de type aplastique avec anomalies nucléaires majeures des granulocytes. Semaine des Hôpitaux. 1968;49:3026-3028.
44 Gustke SS, Becker GA, Garancis JC, et al. Chromatin clumping in mature leukocytes: a hitherto unrecognized abnormality. Blood. 1970;35:637-658.
45 Felman P, Bryon PA, Gentilhomme O, et al. The syndrome of abnormal chromatin clumping in leucocytes: a myelodysplastic disorder with proliferative features? Br J Haematol. 1988;70:49-54.
46 Brizard A, Huret JL, Lamotte F, et al. Three cases of myelodysplastic-myeloproliferative disorder with abnormal chromatin clumping in granulocytes. Br J Haematol. 1989;72:294-295.
47 Jaen A, Irriguible D, Milla F, et al. Abnormal chromatin clumping in leucocytes: a clue to a new subtype of myelodysplastic syndrome. Eur J Haematol. 1990;45:209-214.
48 Adhya A, Ahluwalia J, Varma N, et al. Abnormal chromatin clumping in leucocytes of Ph positive chronic myeloid leukemia cases – extending the morphological spectrum. Indian J Pathol Microbiol. 2008;51:548-550.
49 Banerjee R, Halil O, Bain BJ, et al. Neutrophil dysplasia caused by mycophenolate mofetil. Transplantation. 2000;70:1608-1610.
50 Daliphard S, Accard F, Delattre C, et al. Reversible abnormal chromatin clumping in granulocytes from six transplant patients treated with mycophenolate mofetil: a rare adverse effect mimicking abnormal chromatin clumping syndrome. Br J Haematol. 2002;116:725-728.
51 Reiter A, Walz C, Watmore A, et al. The t(8;9)(p22;p24) is a recurrent abnormality in chronic and acute leukemia that fuses PCM1 to JAK2. Cancer Res. 2005;65:2662-2667.
52 Bousquet M, Quelen C, De Mas V, et al. The t(8;9)(p22;p24) translocation in atypical chronic myeloid leukaemia yields a new PCM1-JAK2 fusion gene. Oncogene. 2005;24:7248-7252.
53 Passmore SJ, Chessells JM, Kempski H, et al. Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia in the UK: a population-based study of incidence and survival. Br J Haematol. 2003;121:758-767.
54 Hasle H, Niemeyer CM, Chessells JM, et al. A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia. 2003;17:277-282.
55 Chan RJ, Cooper T, Kratz CP, et al. Juvenile myelomonocytic leukemia: a report from the 2nd International JMML Symposium. Leuk Res. 2009;33:355-362.
56 Niemeyer CM, Arico M, Basso G, et al. Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS). Blood. 89, 1997. 3534–3343
57 Emanuel PD, Bates LJ, Castleberry RP, et al. Selective hypersensitivity to granulocyte-macrophage colony-stimulating factor by juvenile chronic myeloid leukemia hematopoietic progenitors. Blood. 1991;77:925-929.
58 Kotecha N, Flores N, Irish J, et al. Single cell profiling identifies aberrant STAT5 activation in myeloid malignancies with specific clinical and biologic correlates. Cancer Cell. 2008;14:335-343.
59 Passmore SJ, Hann IM, Stiller CA, et al. Pediatric myelodysplasia: a study of 68 children and a new prognostic scoring system. Blood. 1995;1:1742-1750.
60 Bader-Meunier B, Tchernia G, Miélot F, et al. Occurrence of myeloproliferative disorder in patients with Noonan syndrome. J Pediatr. 1997;130:885-889.
61 Lavin VA, Hamid R, Patterson J, et al. Use of human androgen receptor gene analysis to aid the diagnosis of JMML in female Noonan syndrome patients. Pediatr Blood Cancer. 2008;51:298-302.
62 Matsuda K, Shimada A, Yoshida N, et al. Spontaneous improvement of hematologic abnormalities in patients having juvenile myelomonocytic leukemia with specific RAS mutations. Blood. 2007;109:5477-5480.
63 Flotho C, Kratz C, Bergstrasser E, et al. Genotype-phenotype correlation in cases of juvenile myelomonocytic leukemia with clonal RAS mutations. Blood. 2008;111:966-967.
64 Herrod H, Dow L, Persistent SullivanJ. Epstein–Barr virus infection mimicking juvenile chronic myelogenous leukemia: immunologic and hematologic studies. Blood. 1983;61:1098-1104.
65 Kirby M, Weitzman S, Freedman MH. Juvenile chronic myelogenous leukemia: differentiation from infantile cytomegalovirus infection. Am J Pediatr Hematal Oncol. 1990;12:292-296.
66 Pinkel D. Differentiating juvenile myelomonocytic leukemia from infectious disease. Blood. 1998;91:365-367.
67 Lorenzana A, Lyons H, Sawaf H, et al. Human herpesvirus 6 infection mimicking juvenile myelomonocytic leukemia in an infant. J Pediatr Hematol. 2002;24:136-141.
68 Gattermann N, Billiet J, Kronenwett R, et al. High frequency of the JAK2 V617F mutation in patients with thrombocytosis (platelet count>600 × 109/L) and ringed sideroblasts more than 15% considered as MDS/MPD, unclassifiable. Blood. 2007;1:1334-1335.
69 Orazi A, Germing U. The myelodysplastic/myeloproliferative neoplasms: myeloproliferative diseases with dysplastic features. Leukemia. 2008;22:1308-1319.
70 Malcovati L, Della Porta MG, Pietra D, et al. Molecular and clinical features of refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Blood. 2009;22:3538-3545.
71 Raya JM, Arenillas L, Domingo A, et al. Refractory anemia with ringed sideroblasts associated with thrombocytosis: comparative analysis of marked with non-marked thrombocytosis, and relationship with JAK2 V617F mutational status. Int J Hematol. 2008;88:387-395.
72 Mufti GJ, Bennett JM, Goasguen J, et al. Diagnosis and classification of myelodysplastic syndrome: International Working Group on Morphology of Myelodysplastic Syndrome (IWGM-MDS) consensus proposals for the definition and enumeration of myeloblasts and ring sideroblasts. Haematologica. 2008;93:1712-1717.
73 Shaw GR. Ringed sideroblasts with thrombocytosis: an uncommon mixed myelodysplastic/myeloproliferative disease of older adults. Br J Haematol. 2005;131:180-184.
74 Wang SA, Hasserjian RP, Loew JM, et al. Refractory anemia with ringed sideroblasts associated with marked thrombocytosis harbors JAK2 mutation and shows overlapping myeloproliferative and myelodysplastic features. Leukemia. 2006;20:1641-1644.
75 Schmitt-Graeff A, Thiele J, Zuk I, et al. Essential thrombocythemia with ringed sideroblasts: a heterogeneous spectrum of diseases, but not a distinct entity. Haematologica. 2002;87:392-399.
76 Pich A, Riera L, Sismondi F, et al. JAK2V617F activating mutation is associated with the myeloproliferative type of chronic myelomonocytic leukaemia. J Clin Pathol. 2009;62:798-801.
77 Szpurka H, Tiu R, Murugesan G, et al. Refractory anemia with ringed sideroblasts associated with marked thrombocytosis (RARS-T), another myeloproliferative condition characterized by JAK2 V617F mutation. Blood. 2006;1:2173-2181.
78 Ingram W, Lea NC, Cervera J, et al. The JAK2 V617F mutation identifies a subgroup of MDS patients with isolated deletion 5q and a proliferative bone marrow. Leukemia. 2006;20:1319-1321.
79 Melchert M, Kale V, List A. The role of lenalidomide in the treatment of patients with chromosome5q deletion and other myelodysplastic syndromes. Curr Opin Hematol. 2007;14:123-129.
80 Hernandez-Boluda JC, Cervantes F, Costa D, et al. Blast crisis of Philadelphia-positive chronic myeloid leukemia with isochromosome 17q: report of 12 cases and review of the literature. Leuk Lymphoma. 2000;38:83-90.
81 Weh HJ, Kuse R, Hossfeld DK. Acute nonlymphocytic leukemia (ANLL) with isochromosome i(17q) as the sole chromosomal anomaly: a distinct entity? Eur J Haematol. 1990;44:312-314.
82 Sole F, Torrabadella M, Granada I, et al. Isochromosome 17q as a sole anomaly: a distinct myelodysplastic syndrome entity? Leuk Res. 1993;17:717-720.
83 McClure RF, Dewald GW, Hoyer JD, et al. Isolated isochromosome 17q: a distinct type of mixed myeloproliferative disorder/myelodysplastic syndrome with aggressive clinical course. Br J Haematol. 1999;106:445-454.