[level-membership-for-neurology-category]
Chapter 68 Movement Disorders
Introduction
Movement disorders are syndromes characterized by impaired voluntary movement, the presence of involuntary movements, or both. There may be impaired targeting and velocity of intended movements, abnormal involuntary movements, abnormal postures, or excessive normal-appearing movements at inappropriate or unintended times. Movement disorders in children include athetosis, chorea, dystonia, myoclonus, parkinsonism, stereotypies, tics, and tremor [Sanger, 2003a]. Movement disorders may be accompanied by weakness, spasticity, hypotonia, ataxia, apraxia [Koski et al., 2002], and other motor deficits, although many authors do not include these accompanying deficits.
Movement disorder terminology has been well defined for adults, but less so for children. Therefore, it is likely that movement disorders are under-reported in children, and that there is inconsistent terminology. Recently, there have been attempts to provide specific definitions of childhood motor disorders [Sanger et al., 2003, 2006, 2010]. According to these definitions, childhood disorders can be divided into three major categories: hypertonic disorders, hyperkinetic disorders, and negative signs. Hypertonic disorders include spasticity, dystonia, and rigidity. Hyperkinetic disorders include chorea, dystonia, athetosis, myoclonus, tremor, stereotypies, and tics. Negative signs include weakness, reduced selective motor control, ataxia, apraxia, and developmental dyspraxia, although we will not discuss these here. Consensus definitions for these terms have been established, although the list is not intended to be exhaustive and disorders of gait, balance, speech, and eye movement are not included. Several important terms, including hypotonia and bradykinesia, have not yet been defined for children. The prevalence in children of different types of disorders is not known, although there have been studies investigating symptoms in certain populations, including children with cerebral palsy.
Characteristic Features of Pediatric Movement Disorders
Movement disorders in children differ from those in adults in several aspects. Perhaps the most important is that movement disorders in childhood are primarily symptoms of other diseases, rather than diseases in themselves [Sanger, 2003a, b]. In adults, dystonia and parkinsonism are usually due to primary dystonia or idiopathic Parkinson’s disease, respectively. However, dystonia or parkinsonism in children are more likely to be features of an underlying static or progressive neurological disorder. Diagnosis in children is complicated by the fact that many symptoms have more than one cause, and any particular underlying pathophysiology may lead to a complex combination of symptoms. The diagnostic work-up in children is guided by symptoms, but the existence of a large class of diseases that can lead to the same set of symptoms often necessitates a broad etiologic work-up. There may be both specific etiologic treatments, as well as symptomatic treatments, both of which may be beneficial in an individual child. In particular, many of the causes of childhood movement disorders do not yet have any specific treatment, yet symptomatic treatment for the resulting movement disorder can be extremely helpful and lead to improvement in quality of life.
Diagnosis of Movement Disorders
Classification of a movement disorder based upon the spatial and temporal pattern is essential for diagnosis. It is also important to define the context in which the movements occur. While it is often helpful to list the characteristics of the movements (Table 68-1), the diagnosis relies on pattern recognition, and the clinician must see the movements. If the movements are not apparent during the neurological examination, repeating the examination at another time or obtaining video recordings of the movements is important to making an accurate diagnosis. The widespread availability of video cameras has substantially improved diagnosis of movement disorders.
Table 68-1 Phenomenological Classification of Movement Disorders
| Movement Disorder | Brief Description |
|---|---|
| Athetosis | Slow, continuous, writhing movements of distal body parts, especially the fingers and hands |
| Chorea/ballism | Chaotic, random, repetitive, brief, purposeless movements. Rapid, but not as rapid as myoclonus. When very large in amplitude and affecting proximal joints, choreic limb movements are often called ballism |
| Dystonia | Repetitive, sustained, abnormal postures and/or movements. Abnormal postures typically have a twisting quality |
| Myoclonus | Sudden, brief, shocklike movements that may be repetitive or rhythmic |
| Parkinsonism | Hypokinetic syndrome characterized by a combination of rest tremor, slow movement (bradykinesia), rigidity, and postural instability |
| Stereotypy | Patterned, episodic, repetitive, purposeless, rhythmic movements |
| Tics | Stereotyped intermittent, sudden, discrete, repetitive, nonrhythmic movements, most frequently involving head and upper body |
| Tremor | Rhythmic oscillation about a central point or position, involving one or more body parts |
When approaching a patient with a movement disorder, it is helpful to address some key questions:
The Role of the Basal Ganglia in Movement Disorders
The basal ganglia are subcortical structures comprising several interconnected nuclei in the forebrain, midbrain, and diencephalon (Figure 68-1). They include the striatum (caudate, putamen, nucleus accumbens), the subthalamic nucleus (STN), the globus pallidus (internal segment [GPi]); external segment [GPe]; and ventral pallidum), and the substantia nigra (pars compacta [SNpc] and pars reticulata [SNpr]). The striatum and STN are the primary input structures of the basal ganglia, receiving excitatory input from cerebral cortex. The GPi and SNpr are the primary output nuclei, sending inhibitory output to thalamus and brainstem targets. Acting through the thalamus, the basal ganglia output influences frontal lobe cortical neurons. By virtue of the inhibitory output from the basal ganglia, conditions associated with destruction of the output nuclei may be associated with unwanted and nonspecific overactivity of thalamocortical and brainstem targets.
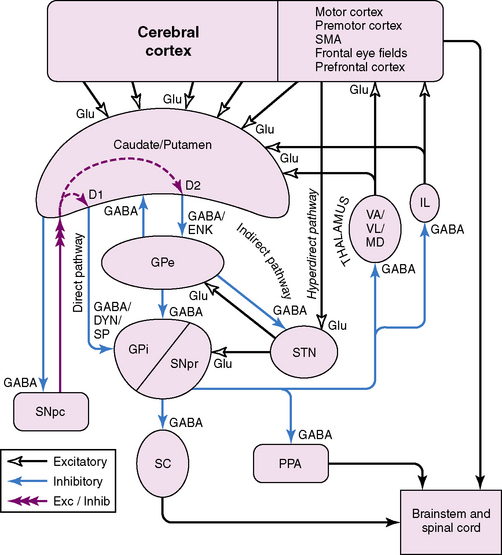
Medium spiny striatal neurons contain the inhibitory neurotransmitter GABA and are inhibitory to their targets. In addition, they have peptide neurotransmitters that are co-localized with GABA. Based on the type of neurotransmitters and the predominant type of dopamine receptor they contain, the medium spiny neurons can be divided into two populations. One population contains GABA, dynorphin, and substance P, and primarily expresses D1 dopamine receptors. These neurons project to the basal ganglia output nuclei, GPi, and SNpr, and form the “direct pathway” [Alexander and Crutcher, 1990; Albin et al., 1989; DeLong, 1990]. The second population contains GABA and enkephalin, and primarily expresses D2 dopamine receptors. These neurons project to GPe and form the first limb of the “indirect pathway” [Alexander and Crutcher, 1990; Albin et al., 1989; DeLong, 1990].
The STN receives an excitatory glutamatergic input from many areas of frontal lobes with particularly large inputs from motor areas of cortex. The STN also receives an inhibitory GABA input from GPe. The output from the STN is glutamatergic and excitatory to the basal ganglia output nuclei, GPi and SNpr. This projection forms the second limb of the “indirect pathway” [Alexander and Crutcher, 1990; Albin et al., 1989; DeLong, 1990].
According to the most common model of basal ganglia function, there are several distinct routes through which information flows from the cerebral cortex to the basal ganglia output nuclei. The two most direct are the disynaptic, inhibitory, “direct pathway” from cortex to striatum to GPi and SNpr, and the disynaptic, excitatory, “hyperdirect pathway” [Nambu et al., 2000] from cortex to STN to GPi and SNpr. The “direct pathway” is inhibitory to GPi and SNpr; the “hyperdirect pathway” is excitatory to GPi and SNpr. The “hyperdirect” pathway is the fastest route through the basal ganglia. There are several indirect routes, but the most important is the “indirect pathway” from cortex to striatum to GPe to STN to GPi and SNpr. Organization of these pathways confers a pattern of fast, powerful, and relatively broad excitation via the hyperdirect pathway, and more focused inhibition via the direct pathway (Figure 68-2) [Nambu et al., 2000; Parent and Hazrati, 1993; Mink, 1996]. The indirect pathway confers additional focus.
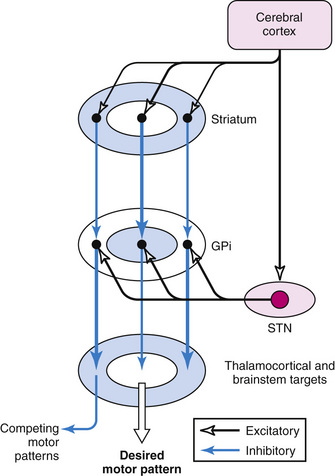
Output from basal ganglia to thalamocortical circuits appears to be segregated anatomically and possibly functionally. Thalamic targets of GPi and SNpr project, in turn, to frontal lobe, with the strongest output going to motor areas. The basal ganglia motor circuit has a somatotopic organization, with separate representation of different body parts maintained throughout the basal ganglia. There also appears to be relative segregation of motor from nonmotor basal ganglia circuits [Hoover and Strick, 1993].
A popular model of basal ganglia dysfunction in movement disorders was developed in the late 1980s (Figure 68-3) [Albin et al., 1989, 1995; DeLong, 1990]. In simple terms, the model proposes that hypokinetic disorders (e.g., parkinsonism) can be distinguished from hyperkinetic movement disorders (e.g., chorea, dystonia, tics), based on the magnitude and pattern of the basal ganglia output neurons in GPi and SNpr [Wichmann and DeLong, 1996]. Because basal ganglia output neurons are inhibitory to thalamus and the PPA, their function is analogous to a braking mechanism, such that increased activity inhibits and decreased activity facilitates motor pattern generators in cerebral cortex and brainstem [Mink, 1996]. As described above, the anatomic organization of the basal ganglia confers a pattern of focused facilitation and surround inhibition of motor mechanisms in thalamocortical and brainstem circuits (see Figure 68-2). The normal function of this organization is selectively to facilitate desired movements and to inhibit potentially competing movements [Mink, 1996, 2003].

Basal Ganglia Pathophysiology in Movement Disorders
Lesions in the striatum produce variable results that depend in part on the location of the lesion, in part on the lesion mechanism, and in part on what is measured. Lesions in the caudate nucleus most commonly cause behavioral disturbance, such as abulia, but may also cause chorea or dystonia. Lesions in the putamen are likely to produce movement deficits and typically cause dystonia (most common) or parkinsonism [Bhatia and Marsden, 1994].
A unilateral STN lesion is the classic cause of hemiballism, but smaller-amplitude chorea can also result from STN lesions [Carpenter and Carpenter, 1951]. When bilateral, STN lesions cause bilateral chorea or ballism. Most commonly, the chorea or hemiballism resulting from STN lesions involves the lower extremities more than the upper extremities, and can persist for days to months. The intensity of hemiballism usually diminishes over time. Globus pallidus lesions usually involve GPe and GPi, GPe and putamen, or GPi and SNpr. Such lesions can cause dystonia, parkinsonism, or both [Mink, 1996; Bhatia and Marsden, 1994; Heidenreich et al., 1988]. Chorea rarely accompanies globus pallidus lesions. Focal lesions of SNpr can cause involuntary eye movements [Hikosaka and Wurtz, 1985]. Lesions of SNpc dopamine neurons cause depletion of dopamine in the striatum, resulting in parkinsonism, dystonia, or both [Calne et al., 1997].
Focal basal ganglia lesions are uncommon in children. Movement disorders in children are more likely to result from global dysfunction of basal ganglia circuits. The model shown in Figure 68-3 has been used to explain the basis for hypokinetic and hyperkinetic movement disorders, but does not distinguish among the different hyperkinetic movement disorders. However, based on what is known about these disorders and the underlying neural circuitry, specific models have been proposed [Mink, 2003]. Diseases affecting the basal ganglia cause movement disorders that can be understood as failure to facilitate desired movements (e.g., Parkinson’s disease), failure to inhibit unwanted movements (e.g., chorea, dystonia, tics), or both [Albin et al., 1989; DeLong, 1990; Mink, 1996, 2003]. The involuntary movements of chorea, dystonia, and tics differ in important spatial and temporal characteristics that reflect important pathophysiologic differences. More extensive discussion of the neural basis of these disorder is contained in Mink [2003].
Etiology of Movement Disorders in Children
The causes of pediatric movement disorders are extensive. The most common cause of secondary disorders is likely to be cerebral palsy, with a prevalence of 2 per 1000. However, cerebral palsy itself represents a constellation of injuries and symptoms, and there is a wide range of types of injury, localization, and combinations of symptoms [SCPE, 2000]. Cerebral palsy can be associated with almost all forms of childhood movement disorders, and despite the lack of an on-going destructive process, the clinical picture may change during development. The diagnosis and management of cerebral palsy is complex and is discussed in Chapter 69.
Specific types of movement disorder may represent dysfunction of particular localized regions of the central nervous system [Sanger, 2003b]. Ataxia most likely occurs due to injury to the cerebellum or its afferent and efferent pathways [Taroni and DiDonato, 2004]. Bradykinesia most likely occurs with injury to the substantia nigra or striatum, leading to presynaptic or postsynaptic failure of dopaminergic transmission [Albin et al., 1989]. Chorea typically occurs with injury to the subthalamic nucleus, but it also can occur with widespread cerebral injury (e.g., following encephalitis). Dystonia most likely involves injury to the basal ganglia, but cortical or cerebellar abnormalities cannot be excluded as contributors [Hallett, 1998; Sanger, 2003b]. Myoclonus most likely involves cortical, brainstem, or spinal injury to gray matter [Caviness and Brown, 2004]. Localization of tremor depends upon the type, with some forms involving cerebellar or brainstem circuits [Uddin and Rodnitzky, 2003; Jankovic et al., 2004]. Tic disorders probably involve an abnormality of the basal ganglia, but cortical mechanisms may also contribute [Mink, 2001a].
Classification of Childhood Movement Disorders
The first step toward diagnosing and treating a movement disorder is to define the disorder (see Table 68-1). Individual names of movement disorders can refer to neurological signs or to neurological syndromes or diseases, which can cause some confusion. In this section, we will limit the definitions to the neurological signs to which they refer. In subsequent sections, we will present a more complete discussion of the syndromes and diseases in which the different signs are seen, either alone or in combination with other signs.
The most common movement disorders in the pediatric population are tic disorders. The prevalence of tic disorders in schoolchildren may be as high as 25 percent [Snider et al., 2002]. Most tic disorders are not severe, and the level of morbidity is low. Nevertheless, because of the lack of association with other neurological symptoms, it is not unreasonable to consider tic disorders as a primary disorder in idiopathic cases. There are rare exceptions in which tics are due to secondary disorders, such as brain injury or neuroacanthocytosis.
Dystonia is a relatively prevalent disorder in children. This most likely is due to its occurrence in cerebral palsy. Dystonia as the primary symptom in cerebral palsy is less common, but dystonia as a complicating factor, particularly in the upper extremities of children with cerebral palsy otherwise characterized by weakness or spasticity, is probably more common. The incidence and classification of cerebral palsy are discussed in Chapter 69.
Choreoathetosis is a term that has been applied to a specific subset of children with a dyskinetic form of cerebral palsy. This term is not generally used in adults. It is not known whether or not choreoathetosis represents a form of dystonia, chorea, or an entirely different disorder [Turny et al., 2004], but a recent consensus definition [Sanger et al., 2010] suggests that it is most appropriately considered to be a combination of chorea (random-appearing, more rapid movements) and athetosis (slower, flowing movements without intervening periods of rest). Certainly, choreoathetotic cerebral palsy (also commonly termed dyskinetic cerebral palsy) is an easily recognized syndrome, and since many of these children also possess some degree of dystonia as well as chorea, choreoathetosis may simply be the expression of a combination of other movement disorders [Morris et al., 2002a]. Spasticity and weakness are very common in childhood motor disorders, again due to the higher prevalence of cerebral palsy.
Chorea
Chorea describes an apparently random, nonrhythmic, purposeless set of movements of either distal or proximal muscles that appears to flow from one muscle or muscle group to another without any pattern. The causes of chorea and chorealike movements in childhood are summarized in Box 68-1. Chorea occurs at rest and with action, and gives the child a “fidgety” appearance and the inability to remain still. It is associated with motor impersistence (for example, the inability to maintain the tongue extended). Chorea may worsen or improve with voluntary movement, but even very severe chorea may not prevent accurate voluntary movement for some children, suggesting that compensatory mechanisms exist. Many individuals with chorea will incorporate the involuntary movements into a voluntary movement in order to mask the movements. However, the involuntary movements can lead to significant disability, and some children will injure themselves or others due to rapid, ballistic, flinging movements of the arms or legs. Tone is normal or reduced in pure chorea but, in children, chorea may occur in the presence of hypertonic disorders, including dystonia. As with most movement disorders, chorea disappears in sleep but it may be at its worst when the child is drowsy. The term “choreiform” is often used to describe the minimal twitching or “piano-playing” movements seen in many normal young children when arms are extended during the neurological exam. The movements of chorea are briefer than the sustained muscle contractions seen in dystonia and are longer in duration than the “shocklike” movements of myoclonus [Marsden et al., 1983].
Box 68-1 Causes of Chorea in Childhood










































Drugs/Toxins


































Huntington’s Disease
Huntington’s disease (HD) is transmitted as an autosomal-dominant trait, caused by a trinucleotide repeat expansion of the IT-15 gene on chromosome 4 [Li et al., 1993]. This disorder is characterized by a combination of dystonia, chorea, myoclonus, behavioral abnormalities, ataxia, and, ultimately, dementia. When HD begins in childhood, the typical presentation of the movement disorder is dystonia and rigidity, and not chorea. Seizures are not infrequently the first manifestation of HD in childhood. This has been referred to as the Westphal variant of HD [Quinn and Schrag, 1998; Topper et al., 1998]. The age of onset is earlier for children with a higher number of repeats [Langbehn et al., 2004]. There tends to be amplification of the number of repeats, particularly when it is transmitted from father to child, and therefore in most children with juvenile HD disease is inherited from the father and involves repeat lengths that are significantly higher than those seen in adults.
Diagnosis is based upon identification of the trinucleotide repeat sequence. Although at least 38 repeats are usually required for the occurrence of symptoms in adults, a larger number of repeats would be expected when symptoms present in childhood [Quinn and Schrag, 1998; Langbehn et al., 2004]. Magnetic resonance imaging (MRI) shows atrophy of the heads of the caudate nucleus and, in later stages, generalized cerebral and cerebellar atrophy.
In teenagers, the initial presentation of HD may be with psychiatric illness and, in particular, major depression [Tost et al., 2004]. Dystonia or chorea usually supervenes later. The pathology includes basal ganglia, cerebellar, and cortical degeneration [Vonsattel et al., 1985; de la Monte et al., 1988; Myers et al., 1988]. The origin of the chorea has been hypothesized to be a primary loss of medium spiny cells in the striatum that bear D2-like receptors [Deng et al., 2004]. In children, there may be a relatively symmetric loss of D1-bearing and D2-bearing medium spiny neurons, which could account for the primarily dystonic rather than choreic presentation [Augood et al., 1997; Albin et al., 1990].
Treatment of Huntington’s chorea in adults is typically based upon modification of the chorea or myoclonus. Since the chorea has been presumed to be due to loss of medium spiny neurons in the indirect pathway, neuroleptic medications have been most frequently used in adults [Bonelli and Hofmann, 2004; Bonelli et al., 2004]. Neuroleptics would be expected to compensate partially for early loss of indirect pathway neurons by blocking the inhibitory effects of dopamine and disinhibiting the remaining indirect pathway neurons. There is much less experience in children, for whom dystonia and rigidity may be greater contributors to disability. Other medications that have been used successfully in HD include tetrabenazine [Asher and Aminoff, 1981; Ondo et al., 2002], clonazepam [Thompson et al., 1994], and valproic acid [Grove et al., 2000]. There is currently no treatment that modifies the progression of the disease.
Ataxia-Telangiectasia
Despite the name of this disorder, ataxia is not always the presenting finding, and ataxia may be a much less prominent early symptom than upper-extremity chorea. However, the chorea is usually less disabling than the ataxia. Other symptoms associated with ataxia-telangiectasia include dystonia, which is often proximal [Bodensteiner et al., 1980]. Ataxia-telangiectasia (Louis–Bar syndrome) is most commonly due to mutations in the ATM (ataxia-telangiectasia mutated) gene on 11q22–23 [Chun and Gatti, 2004; Coutinho et al., 2004; Gatti et al., 1988]. Mutation in this gene leads to decreased inhibition of cell cycling in the context of injury to nuclear DNA, and this leads to an accumulation of mutations at known “hot spots,” including translocations between regions on chromosome 7 and 14 known to be involved with immune function [Farina et al., 1994]. Therefore, one of the cardinal features of this disorder is a history of frequent sinopulmonary infections [Centerwall and Miller, 1958], which requires very close attention to pulmonary function and aggressive treatment of pulmonary infections at all ages. In later stages, hematologic malignancy may occur [Stankovic et al., 1998]. Children with ataxia-telangiectasia may be particularly vulnerable to injury from ionizing radiation and certain chemotherapy agents, and this must be taken into consideration in planning any treatment for malignancy.
The cause of the cerebellar degeneration is not known [Farina et al., 1994], but the pathology shows a characteristic loss of Purkinje cells, such that, by late stages in the disease, Purkinje cells are often completely absent. The cause of the chorea is not known, and basal ganglia pathology is minimal. Eye movement disorders are well described, and include an inability to suppress, as well as an inability to initiate, saccades [Lewis et al., 1999; Bundey, 1994]. The inability to initiate saccades leads to a characteristic finding of oculomotor apraxia, in which a child will use thrusts of the head in order to position the eyes on the target. Diagnosis is based upon laboratory testing, including decreased immunoglobulin (Ig) G2 and IgA, abnormalities in T-cell subsets, elevated α-fetoprotein, lymphocyte radiosensitivity, and decreased radiation-induced mitotic suppression [Stray-Pedersen et al., 2004]. Treatment of this disorder is primarily supportive, although there are isolated reports of dystonic symptoms responding to trihexyphenidyl or l-DOPA. The chorea generally is mild and does not require specific treatment. There is no known treatment for the ataxia.
Ataxia-oculomotor apraxia is an autosomal-recessive disorder with similar motor symptoms, caused in some cases by mutations in the aprataxin gene AOA-1 [Tranchant et al., 2003; Shimazaki et al., 2002; Moreira et al., 2001]. This disorder commonly presents with chorea, ataxia, and oculomotor apraxia [Sano et al., 2004]. It is most prevalent in Japan and Europe. AOA-1 does not seem to be involved with cell cycle regulation, and therefore the DNA breakage, immune abnormalities, and malignancies seen in ataxia-telangiectasia do not occur. These patients have hypoalbuminemia [Shimazaki et al., 2002]. AOA-2 is a similar autosomal-recessive disorder due to mutations in Senataxin (SETX). Like ATM, Senataxin is involved with DNA repair, although mutations are not associated with increased radiosensitivity [Suraweera et al., 2009]. Clinical features include ataxia, oculomotor apraxia, peripheral neuropathy, and, more rarely, chorea or dystonia [Criscuolo et al., 2006; Le Ber et al., 2004]. The long-term prognosis of AOA-1 and AOA-2 is considerably better than for ataxia-telangiectasia.
Other Genetic Choreas
Other genetic causes of chorea include familial benign hereditary chorea [Kleiner-Fisman et al., 2003; Breedveld et al., 2002a], which is an autosomal-dominant disorder that, in some cases, is due to mutations in the TITF-1 gene on chromosome 14 [Breedveld et al., 2002b]. Affected family members have normal or near-normal intelligence, and chorea is often the only complaint, although mild truncal dystonia and gait ataxia have been reported [Fernandez et al., 2001]. The severity of chorea can vary between different affected family members. It may be accompanied by hypothyroidism, pulmonary disease, or both in some family members with TITF-1 mutations. Symptomatic treatment may yield some mild benefit.
Neuroacanthocytosis is also a potential cause of chorea [Marson et al., 2003], although classification as a primary or secondary chorea is not universally agreed upon.
Sydenham’s Chorea
Of the secondary causes of childhood chorea, Sydenham’s chorea is the most common [Jordan and Singer, 2003; Jummani and Okun, 2001; Garvey and Swedo, 1997]. It has onset weeks or months after an acute infection with group A beta-hemolytic streptococcus (GABHS) and is one of the cardinal symptoms of rheumatic fever. Symptoms may persist for weeks or months, but chorea almost always resolves spontaneously within 6 months [Jordan and Singer, 2003]. There have been rare reported cases of continued or recurring symptoms [Faustino et al., 2003]. The chorea typically involves the distal musculature, initially of one hand and then of both, with a “piano-playing” pattern, but other forms of chorea, including ballism, have been observed. Large, generalized body movements in some patients previously inspired the term “St. Vitus’ dance.”
Anti-basal ganglia antibodies (ABGA) are found in some children [Church et al., 2002] and it has been hypothesized that production of these antibodies is triggered due to molecular mimicry by streptococcal antigens [Loiselle and Singer, 2001; Kirvan et al., 2003; Singer et al., 2003; Goldenberg et al., 1992]. ABGA can be detected in cerebrospinal fluid [Singer et al., 2003], and anti-streptolysin (ASLO) antibodies can be detected in serum. However, the high prevalence of positive ASLO titers in the general population means that both acute and convalescent ASLO titers must be measured in order to probe for an acute infection. The clinical situation often provides a strong indication for the diagnosis, but if other neurological symptoms are present or there is doubt about etiology, a more complete work-up may be needed, including tests for thyroid function, toxins, metabolic disorders, or encephalitis.
Sydenham’s chorea usually does not require treatment, although the acute streptococcal infection should be treated. All children diagnosed with Sydenham’s chorea, even in cases of isolated chorea, should be treated with penicillin, both acutely for treatment and long-term for prophylaxis, according to American Academy of Pediatrics guidelines [American Academy of Pediatrics, 2006].
Penicillin or an acceptable alternative is effective as secondary prevention of recurrent chorea, but more importantly reduces the likelihood that future GABHS infections will cause carditis and permanent valvular damage. Current recommendations in the United States are for prophylactic treatment until age 21 years, regardless of the age of chorea onset. Valproic acid, carbamazepine, or neuroleptics may have some symptomatic benefit in more severe or persistent cases [Pena et al., 2002; Genel et al., 2002; Kulkarni and Anees, 1996]. Treatment with immune suppressant medication, including corticosteroids or intravenous immunoglobulin preparations, has been studied, but the natural history, with spontaneous resolution of symptoms, makes interpretation of efficacy in open clinical trials difficult [Garvey and Swedo, 1997; Green, 1978; Cardoso et al., 2003]. One randomized, blinded, placebo-controlled study showed that a 4-week, 2-mg/kg daily oral dose of prednisone, followed by a taper, reduced duration of chorea and accelerated the reduction in symptoms. Weight gain was substantial by the end of 2 months, and long-term outcome including recurrence rates was not different between groups [Paz et al., 2006]. There are sometimes associated obsessive-compulsive or behavioral symptoms [Moore, 1996; Wilcox and Nasrallah, 1988], and these symptoms are often managed with selective serotonin reuptake inhibitors.
The prognosis for the movement disorder is excellent, and complete resolution occurs in most cases. Recurrence is rare and is sometimes associated with recurrence of other symptoms of rheumatic fever [Korn-Lubetzki et al., 2004]. There appears to be a higher risk of chorea gravidarum in women with a previous history of Sydenham’s chorea [Korn-Lubetzki et al., 2004].
The rapid onset of Sydenham’s chorea has raised the question of whether an autoimmune mechanism could be responsible for other acute-onset movement disorders. This has led to the hypothesis of pediatric autoimmune neuropsychiatric disorders associated with Streptococcus (PANDAS) [March, 2004; Trifiletti and Packard, 1999]. PANDAS is characterized by an abrupt or explosive onset of tics, obsessive-compulsive behavior, and a movement disorder (usually chorea) that is temporally associated with streptococcal infection [Snider and Swedo, 2004; Chmelik et al., 2004; Pavone et al., 2004; Swedo, 2002]. Symptoms may persist, with a relapsing and remitting course. The existence of this disorder has been debated [Singer and Loiselle, 2003] [Swedo et al., 2004; Kurlan and Kaplan, 2004; Kurlan, 2004], and ABGA have not been shown to be elevated [Singer et al., 2004]. It has not been possible to transmit the disorder passively to animals by inoculation with antibodies from affected humans [Loiselle et al., 2004]. Nevertheless, there have been an increasing number of case reports of explosive onset of neurobehavioral disorders, including tic disorders, chorea, and obsessive-compulsive disorder following streptococcal infections, and therefore the hypothesis merits further investigation. It is not clear that there is any specific treatment, although some authors advocate immune modulation, long-term antibiotics, or tonsillectomy [Green, 1978], [Araujo et al., 2002; Heubi and Shott, 2003; van Toorn et al., 2004; Leonard and Swedo, 2001; Gebremariam, 1999].
Medication-Induced Chorea
Chorea is frequently associated with administration of medications, and any child with chorea needs to be carefully examined for the possibility of iatrogenic causes. In particular, treatment of other movement disorders with anticholinergic medication, such as trihexyphenidyl, can precipitate chorea, even at relatively low doses [Nomoto et al., 1987]. Antiepileptic medications, including carbamazepine and phenytoin, have been associated with precipitation of chorea, and in children with diffuse brain injury, sedating medications can sometimes precipitate chorea.
Ballism
Ballism is a high-amplitude, flinging movement, usually due to involuntary movements of proximal joints. Ballism is part of the spectrum of chorea and involves similar pathophysiological mechanisms [Albin et al., 1989; DeLong, 1990]. When ballism involves one side of the body, it is called hemiballism. Hemiballism is the classical manifestation of vascular events affecting the subthalamic nucleus, but can be associated with lesions in other parts of the basal ganglia.
Treatment of Chorea
Chorea is often difficult to treat. Secondary chorea may respond to a combination of clonazepam and valproic acid. Sedation may be helpful for short-term management. In some cases, tetrabenazine [Asher and Aminoff, 1981; Jankovic and Orman, 1988; Chatterjee and Frucht, 2003], reserpine, or neuroleptics may be helpful, but the use of neuroleptics requires caution, as chorea may mask the onset of tardive dyskinesia or tardive dystonia. If dystonia is present with chorea, then neuroleptics should be used only with great caution due to their ability to precipitate acute or tardive dystonia and the potential for worsening dystonia due to dopaminergic failure. Trihexyphenidyl, l-DOPA, carbamazepine, and phenytoin can worsen chorea, and should be avoided in most cases. Chorea that is due to global cerebral injury, such as following encephalitis, may worsen when the child is sleepy, and therefore sedating medications should be avoided if possible, although clonazepam is sometimes helpful. Some forms of chorea are time-limited and may only require brief treatment. It is possible that there may be a role for deep brain stimulation in some patients with chorea [Thompson et al., 2000]. Before any treatment for chorea, it is important to evaluate the child’s current medications to rule out an iatrogenic cause.
Dystonia
Dystonia is probably the next most common movement disorder in children after tics. This is because dystonia can occur as an associated feature in many static and degenerative childhood disorders. In children, dystonia is defined as “a movement disorder in which involuntary sustained or intermittent muscle contractions cause twisting and repetitive movements, abnormal postures, or both” [Sanger et al., 2003; Fahn and Williams, 1988]. Although the term seems to imply an abnormality of tone, dystonia is not primarily a disorder of tone, but rather a disorder of posture and/or movement. When individuals with dystonia are at rest, tone is often diminished, although, in severe dystonia, involuntary contractions persist during attempted rest and tone may be increased. Dystonia can manifest as either hypertonic dystonia with increased stiffness, hyperkinetic dystonia with increased movements, or a combination of the two. Dystonia can also be reflected in very slow twisting or writhing movements or very quick ballistic movements, and can have rhythmicity, appearing like tremor, in which case it would be referred to as dystonic tremor.
Dystonia is commonly triggered or exacerbated by voluntary movement, and it may fluctuate in presence and severity over minutes, weeks, or months. Dystonia can be movement-specific, so that a muscle may exhibit involuntary contraction only during certain voluntary movements and not others. For example, walking forward may elicit severe lower-extremity and truncal twisting, yet walking backward, running, or swimming may be completely unaffected. Stress or excitement exacerbates most forms of dystonia. Dystonic contractions resolve during sleep. Individuals with dystonia sometimes discover that touching one part of the body may relieve the dystonic spasms; this phenomenon is called a sensory trick or geste antagoniste. Dystonia may be generalized or focal, involving just a single body part. Childhood-onset dystonia is more likely to be generalized than adult dystonia [Bressman et al., 1994]. Task-specific dystonia, in which symptoms are seen only during performance of a specific skilled action (e.g., writer’s cramp), does not occur commonly in children, although focal dystonia that affects one part of the body during many actions certainly is seen.
Causes of dystonia and dystonia-like movements in childhood are summarized in Box 68-2.
Box 68-2 Causes of Dystonia in Childhood















































Drugs/Toxins






































Classification
Dystonia is classified by etiology as either primary or secondary, and by location as focal, segmental, hemidystonia, multifocal, or generalized dystonia [Fahn et al., 1998]. Focal dystonia involves a single part of the body, such as a limb, the neck, or the face. Examples include blepharospasm (forceful eye closure), oromandibular dystonia (involving the jaw and/or mouth musculature), spasmodic dysphonia (vocal cord involvement), torticollis (twisting or tilting of the neck), or focal hand or foot dystonia. Segmental dystonia involves the muscles of two contiguous body parts. For example, cranial segmental dystonia involves multiple muscles of the face and neck, axial dystonia involves the neck and trunk, brachial dystonia involves the arm and trunk, and crural dystonia involves one or both legs and the trunk. Hemidystonia involves all or most muscles on one side of the body. Multifocal dystonia involves two or more noncontiguous body regions. Generalized dystonia involves multiple limbs on both sides of the body, including at least one leg.
Dystonia is considered primary when it is the predominant or only clinical feature. At least 17 different inherited phenotypes designated with the “DYT” genetic locus specification have been defined so far [Bressman, 2004; Misbahuddin and Warner, 2001; Nishiyama et al., 2000]. Most of these are quite rare. Only seven of the genes have been identified. Many of these disorders do not present in childhood. Many of these disorders represent “dystonia plus” syndromes that include dystonia as a primary feature but which also have other associated movement disorders. Examples of dystonia-plus syndromes include dopa-responsive dystonia (DRD), myoclonus dystonia, and rapid-onset dystonia-parkinsonism (RDP).
Diagnosis
Dystonia must be distinguished from other disorders, including spasticity, rigidity, tics, tremor, and ataxia, although in some cases this is difficult. Spasticity is associated with increased reflexes and frequently shows a spastic catch, whose position depends on the velocity of movement [Sanger et al., 2003; Jobin and Levin, 2000]. Rigidity does not tend to have a particular fixed posture; it has a “lead-pipe” quality and resists attempts at passive movement of the arm or leg, although the limb can be placed into an arbitrary posture by the examiner [Sanger et al., 2003]. Tics can be differentiated by their episodic and repeatable nature, and normal posture and function between tics [Sanger et al., 2010]. In addition, tics rarely have the same degree of motor disability, as is seen in dystonia. Dystonic tremor tends to be less rhythmic than regular tremor, and often exhibits a “null point,” in which there exists a particular joint angle at which the tremor decreases or is absent [Deuschl, 2003]. To one side of that joint angle, movement in one direction will cause the tremor to worsen, but often beating in a particular direction that is different from the direction that the tremor will beat in if the joint is moved to the opposite direction. Ataxia does not show characteristic postures, does not have overflow of muscle activity, and quality of movement may improve at slow speeds [Sanger et al., 2006; Paulson and Ammache, 2001].
Dystonic Storm
A particularly dramatic presentation of dystonia that is seen primarily in children with pre-existing motor abnormalities is dystonic storm [Opal et al., 2002; Dalvi et al., 1998]. Usually, this is seen in the context of an acute illness in a child with a previous history of dystonia, cerebral palsy, or other movement disorder. The child will become ill with an otherwise unremarkable disease, such as a respiratory illness, but then will rapidly develop severe and intractable hyperkinetic dystonia or chorea with ballistic or flinging movements of the arms, legs, or head. This can also be associated with severe limb extension or opisthotonic posturing, with dorsiflexion at the neck or trunk. In many cases, these symptoms will not have been seen previously. In some cases, this clinical picture is associated with encephalitis or other causes of acute striatal injury. Treatment is extremely difficult, and may require general anesthesia to abate the movement. Multiple medications have been tried, including benzodiazepines, l-DOPA, anticholinergic medications, tetrabenazine, reserpine, dantrolene, and baclofen. The combination of trihexyphenidyl and tetrabenazine is sometimes effective, but any causative or triggering disease must be treated aggressively, and hyperthermia, rhabdomyolysis, and myoglobinuria must be watched for and controlled. It is possible that intrathecal baclofen [Dalvi et al., 1998] or deep brain stimulation [Angelini et al., 2000] could be helpful in severe cases when medications are ineffective [Kyriagis et al., 2004]. Dystonic storm can continue for months and can be life-threatening, usually requiring intensive care monitoring. It can recur in the context of a subsequent illness.
Associated Movement Disorders
Athetosis literally means “not fixed,” which refers to the continuous writhing movements that characterize this disorder [Morris et al., 2002b]. Athetotic movements are often caused or worsened by attempts at voluntary movement. The use of term athetosis has diminished substantially in recent years. It has been argued that, in most cases, athetosis is actually a form of dystonia [Turny et al., 2004], although a recent consensus suggests that it can be defined as a separate entity [Sanger et al., 2006]. Currently, the term is used most commonly to describe a form of cerebral palsy (athetoid), but it is also employed in conjunction with the term chorea (choreoathetosis) to describe the apparent random and hyperkinetic quality of the movements [Morris et al., 2002a]. Pure athetosis may be rare in children.
Dystonia in children is frequently associated with bradykinesia [Nygaard and Duvoisin, 1986]. This may be due to the fact that both dystonia and bradykinesia can be caused by failure of dopamine transmission [Segawa et al., 1999; Riederer and Foley, 2002]. Bradykinesia may be a relatively common finding in children with hypertonic disorders, but it is likely to be under-reported since the slow movement may be attributed to the hypertonia rather than to the underlying bradykinesia.
Primary Dystonias
DYT-1 Dystonia
DYT-1 dystonia is known as Oppenheim’s dystonia, after Hermann Oppenheim, who first described it in 1911. It has also been referred to as dystonia musculorum deformans, which was Oppenheim’s original term. This is probably the most common genetically determined cause of dystonia in children. In some populations, this is the most common explanation for dystonia with onset in the foot before the age of 20 years [Klein et al., 1999; O’Riordan et al., 2004]. The median age at onset is 10 years, with a range of 4 years to adulthood. Usually, onset is in a limb, either upper or lower. The general features include a gradually progressive dystonia that eventually involves multiple limbs and progresses to generalized dystonia, often affecting both distal and proximal musculature [Bressman et al., 2002]. However, there is great variation in the progression and severity. When DYT-1 dystonia presents in adulthood, it is usually mild.
DYT-1 dystonia is autosomal-dominant, but it has approximately 30 percent penetrance [Saunders-Pullman et al., 2004a]. Recent evidence suggests that nonmanifesting carriers do have abnormalities of brain networks on FDG positron emission tomography (PET), but the reason that they do not express symptoms of the disease is currently not known [Eidelberg et al., 1998]. The gene is on chromosome 9Q34 and its product is torsin-A [Bressman et al., 1994; Ozelius et al., 1999]. Torsin-A is an adenosine triphosphate (ATP)-binding protein with homology to heat shock and chaperone proteins [Ozelius et al., 1997]. The homology to heat shock proteins has been proposed as a possible explanation for the onset of symptoms following injury, surgery, or other situations where such proteins would be expected to be expressed. The common deletion is a three basepair GAG deletion, resulting in loss of a single glutamate from a highly conserved region of the protein [Ozelius et al., 1999]. This mutation seems to have arisen independently from founders in different populations. The prevalence of manifesting DYT-1 dystonia is higher in Ashkenazi Jews, for whom it is approximately 1 in 5000. The population of non-Ashkenazi Jews has a rate of 1 in 15,000 worldwide, but there may be other groups for whom higher prevalence occurs.
DYT-1 dystonia usually does not respond to dopaminergic medication. The mainstay of treatment is anticholinergic medication, but often very high doses are required [Bressman and Greene, 2000; Bressman, 2000]. Other medications, such as benzodiazepines, baclofen, carbamazepine, neuroleptics, or tetrabenazine, have been tried with varied success, and these will be summarized below. In multiple case reports, pallidotomy or deep brain stimulation seems to be particularly helpful in DYT-1 patients with dystonia [Eltahawy et al., 2004; Coubes et al., 2000; Coubes et al., 2004; Cif et al., 2003]. Untreated, DYT-1 dystonia will typically progress gradually over many years, although it may reach a plateau in adulthood. The prognosis and life span depend on the severity of the disorder. There do not appear to be associated abnormalities in other organ systems, and mortality is usually due to pulmonary complications of the motor dysfunction.
Dopa-Responsive Dystonia
DRD is labeled DYT-5 dystonia [Bandmann et al., 1998a] and is also known as Segawa’s disease after its first description in 1976 [Segawa et al., 1999, 2003; Bandmann and Wood, 2002]. The presentation is very similar to DYT-1 dystonia in many cases, although it can have onset at an earlier age. The average age of onset is 6 years, with a range of onset from 1 to 12 years. It frequently starts in a limb in children. In adolescents and adults, it is often accompanied by parkinsonism and may even present in adulthood as parkinsonism rather than limb dystonia [Uncini et al., 2004; Nygaard et al., 1990]. The combination of dystonia and parkinsonism is due to decreased but not absent dopamine production. Diurnal variation is present in 77 percent of children [Nygaard et al., 1988]. Such children appear to be significantly improved upon awakening in the morning or after a nap, with a gradual worsening of symptoms the longer they remain awake and active. DRD can mimic many of the features of cerebral palsy [Jan, 2004], and there have been reported cases of associated hyperreflexia [Bandmann et al., 1998b]. Untreated, DRD is a progressive disorder and this helps to distinguish it from static disorders, such as cerebral palsy. Untreated cases can develop secondary orthopedic deformities, including joint contractures, muscle and tendon shortening, and scoliosis.
DRD is due to mutations in either the GTP cyclohydrolase 1 gene [Bandmann et al., 1998a, 1996; Furukawa et al., 2000] or the tyrosine hydroxylase gene [Grattan-Smith et al., 2002; Ichinose et al., 1999]. GTP cyclohydrolase 1 is on chromosome 14Q22.1–2 and expression is autosomal-dominant, with variable penetrance [Uncini et al., 2004] that may be greater in girls than in boys [Furukawa et al., 1998]. GTP cyclohydrolase is an enzyme that is important for the synthesis of biopterins and neopterins, which are co-factors for the enzyme tyrosine hydroxylase. The pterins are also co-factors for tryptophan hydroxylase and phenylalanine hydroxylase, although reduced function of these enzymes does not seem to be a cause of symptoms. Rare cases of homozygosity for GTP cyclohydrolase mutations have been reported, with a more severe phenotype [Thony and Blau, 1997].
Significant improvement during a trial with l-DOPA strongly suggests the diagnosis, although a similar response can be seen in juvenile Parkinson’s disease, cerebral palsy, and in other causes of secondary dystonia. The diagnosis can be confirmed by quantitative evaluation of cerebrospinal fluid neurotransmitters and pterins. In particular, biopterin, neopterin, and homovanillic acid (a dopamine metabolite) are low in cerebrospinal fluid. Another test that has been used is the phenylalanine loading test [Ponzone et al., 1994; Saunders-Pullman et al., 2004b; Bandmann et al., 2003; Hyland et al., 1997]. This test depends upon a defect in phenylalanine hydroxylase, which is present in liver and depends on biopterin as a co-factor. To perform the test, 100 mg/kg of phenylalanine is given orally, and the serum phenylalanine to tyrosine ratio is measured at intervals over approximately 6 hours. This test may not be as sensitive and specific as cerebrospinal fluid neurotransmitter metabolite measurement, and it is less commonly used when neurotransmitter analysis is available. There is no common mutation in GTP cyclohydrolase 1 [Nishiyama et al., 2000; Ichinose et al., 1999], so full sequencing of the gene is usually required for genetic diagnosis; sequencing currently detects approximately 60 percent of cases.
Tyrosine hydroxylase deficiency is due to mutations in the tyrosine hydroxylase gene on chromosome 11P15.5 [Royo et al., 2005; Nagatsu and Ichinose, 1996]. Expression is autosomal-recessive, with full penetrance. The onset of the dystonia tends to be earlier, usually in infancy, with more severe symptoms [Furukawa et al., 2001; Brautigam et al., 1999], but mild forms have been reported [Furukawa et al., 2004a]. Examination of the cerebrospinal fluid reveals normal biopterins and neopterins, but low homovanillic acid [Wevers et al., 1999]. Confirmation of the diagnosis requires demonstration of low tyrosine hydroxylase function in fibroblasts or lymphocytes, or sequencing of the tyrosine hydroxylase gene.
In both autosomal-recessive and autosomal-dominant DRD, treatment involves replacement of dopamine using oral l-DOPA [Nutt and Nygaard, 2001]. In particular, the combination of l-DOPA and a peripheral dopa decarboxylase inhibitor, such as carbidopa, usually results in a dramatic improvement in symptoms with minimal side effects [Nutt and Nygaard, 2001; Hwang et al., 2001]. Often very low doses are required, and treatment can be continued throughout life with no adverse events. In particular, the escalation of dose frequently seen in Parkinson’s disease and the development of motor dyskinesias over time do not seem to occur, or occur only rarely. There are reports that a gradual escalation of dose may be needed after several years. Although many children and adults will achieve complete resolution with doses of 100 mg per day or less of l-DOPA, some children have required up to 10 mg/kg per day. Medication is usually divided into two or three doses per day. Other medications for Parkinson’s disease may also be effective, including anticholinergic medication, dopamine agonists, and monoamine oxidate (MAO)-B inhibitors, although these are usually not needed. Peripherally induced side effects of l-DOPA include nausea and hypotension, and these can often be ameliorated by giving an extra dose of carbidopa 30 minutes prior to administration of the l-DOPA-carbidopa combination. Intestinal absorption of l-DOPA is increased by simultaneous carbohydrate intake and decreased by simultaneous protein intake. The prognosis for DRD is excellent with treatment [Nygaard et al., 1991], but untreated children are at risk for severe dystonia with progressive orthopedic deformities. Outcomes in individuals with tyrosine hydroxylase deficiency are less satisfactory and children often remain with developmental delay and motor deficits, despite adequate dopaminergic therapy.
Myoclonus Dystonia
Myoclonus dystonia is a disorder with a combination of both symptoms [Asmus and Gasser, 2004; Lang, 1997]. In some, but not all, families, the disorder is due to a mutation in the epsilon-sarcoglycan gene on chromosome 7Q21 [Nygaard et al., 1999], and this has been designated DYT-11 [Furukawa and Rajput, 2002; Schule et al., 2004; Valente et al., 2003]. The details of the myoclonus will be discussed further in the section on myoclonus. Myoclonus involves the neck, trunk, and arms, and is often alcohol-responsive. Dystonia occurs in approximately half of patients and can be the only manifestation. Dystonia usually manifests itself with torticollis or arm dystonia. In addition, behavioral symptoms and obsessive-compulsive disorder have been associated with gene mutations. Onset is in the juvenile period.
This disorder is autosomal-dominant, although de novo mutations are frequent [Hedrich et al., 2004]. When the epsilon-sarcoglycan gene is inherited from the father, there is approximately 100 percent symptomatic expression, but when inherited from the mother, there is only 10 percent symptomatic expression, suggesting an important role of maternal imprinting [Muller et al., 2002; Grabowski et al., 2003]. It can be diagnosed by sequencing of the epsilon-sarcoglycan gene, and this detects 95 percent of patients in affected families. Treatment is with benzodiazepines, valproate, and alcohol, although chronic use of alcohol frequently leads to dependence, and this has not been proposed as an appropriate long-term therapy. Some reports have suggested the use of gamma-hydroxybutyrate, which has alcohol-like effects and seems to be helpful in small case series [Priori et al., 2000]. There also have been reports of the use of deep brain stimulation in the globus pallidus or ventral intermediate nucleus of the thalamus, with symptomatic relief [Cif et al., 2004]. Children would be expected to have a normal life span, although symptoms do not spontaneously resolve.
Other Identified Primary Dystonias
DYT-12 is the designation for rapid-onset dystonia parkinsonism, which has been associated with the ATP-1A3 gene [de Carvalho Aguiar et al., 2004]. In this disorder, a combination of dystonia and bradykinesia can develop over hours to days. It is autosomal-dominant, with variable penetrance [Brashear et al., 1998]. Once the dystonia has developed, it does not resolve [Dobyns et al., 1993]. Age at onset ranges from 8 to 55 years, but is before 20 years of age in 47 percent, and between 20 and 30 years of age in another 39 percent of affected individuals. Bulbar symptoms are typically worse than appendicular symptoms. Parkinsonism is less prominent than dystonia and usually consists of bradykinesia, postural instability, and hypophonia. Proposed diagnostic criteria consist of:
The disorder does not appear to be progressive, and, once the initial symptoms have developed, they remain at approximately the same level of severity or progress only gradually [Dobyns et al., 1993].
RDP is caused by mutations in the Na+/K+-ATPase α3 subunit (ATP1A3 gene) [de Carvalho Aguiar et al., 2004]. The α3 subunit is only expressed in brain and heart, perhaps indicating a role in electrical excitability. It has been suggested that the reported mutations in RDP impair enzyme activity or stability [Breakefield et al., 2008].
Patients with RDP do not respond consistently to the usual symptomatic treatments for dystonia including l-DOPA [Brashear et al., 2007].
Psychogenic Dystonia
Dystonia as a manifestation of psychiatric disorders is not uncommon. It is most often a form of conversion reaction. Common features of psychogenic disorders include maximal severity at onset, entraining to rhythmic stimuli, distractibility, suggestibility, and a posture or presentation that is otherwise atypical for the disease [Fahn and Williams, 1988; Thomas and Jankovic, 2004; Kirsch and Mink, 2004; Ozekmekci et al., 2003; Pringsheim and Lang, 2003]. Children may not be aware of the psychogenic origin and there may be no history of other psychopathology. There may be no secondary gain. In some cases, psychogenic dystonia occurs in the presence of true dystonia, worsening its apparent severity [Bentivoglio et al., 2002]. It can also appear in families in which other family members have dystonia or other movement disorders. It is essential to exclude organic causes. Surface or needle electromyography can be helpful, as certain patterns of rhythmic contraction sometimes seen in dystonia cannot be mimicked voluntarily [Hallett, 1983], and if these patterns occur, that would be a clue to an organic cause. The primary treatment is a combination of psychotherapy and physical therapy, but this is considered a very primitive psychiatric defense mechanism, and the prognosis for spontaneous resolution is poor [Miyasaki et al., 2003; Feinstein et al., 2001].
Secondary Dystonia
There is a long list of possible secondary causes for dystonia, as shown in Box 68-2. Here we will discuss several of the most important causes. Many of these disorders are discussed in other chapters.
Cerebral Palsy
Cerebral palsy is probably the most common cause of dystonia in childhood. This is due to its relatively high prevalence. Further details about this disorder, including the etiology and details of the symptomatology, can be found in Chapter 69 on cerebral palsy, and we will only summarize the elements contributing to dystonia. Dyskinetic cerebral palsy represents between 6 and 15 percent of all cases of cerebral palsy [SCPE, 2002], and in dyskinetic cerebral palsy, dystonia is usually the primary feature. Dystonia occurs frequently as an associated feature in other forms of cerebral palsy, including tetraplegic and hemiplegic cerebral palsy. Dystonia is usually associated with lesions of the basal ganglia or thalamus [Krageloh-Mann et al., 2002], and these lesions are commonly caused by hypoxic-ischemic injury in the full-term neonate [Volpe, 2000]. A similar syndrome can occur in victims of near-drowning or other forms of asphyxia, as well as in stroke and sometimes with head trauma. Symptoms most often affect the arms, with the legs frequently being affected either by spasticity or by a combination of spasticity and dystonia. There may be associated bradykinesia or choreoathetosis.
Although the dystonia in cerebral palsy presumably is due to a static injury, the symptom can worsen over time. In fact, the onset of dystonia may be many years after the initial injury leading to cerebral palsy [Saint Hilaire et al., 1991].
Kernicterus
Kernicterus occurs due to high bilirubin levels in the perinatal period, but the effects are variable and unpredictable [Shapiro, 2005]. There is probably an increased susceptibility to hyperbilirubinemia with prematurity, hypoxia, or infection [Volpe, 2000]. It has been hypothesized that, in such cases, there is a reduction of the efficacy of the blood–brain barrier, which allows unconjugated bilirubin to penetrate and affect multiple brain regions, including the globus pallidus. Injury to the globus pallidus is thought to be the etiology of the movement disorder [Govaert et al., 2003; Johnston and Hoon, 2000]. The overall severity later in life is quite variable, and symptoms include choreoathetosis, dystonia (which can be progressive), sensorineural hearing loss, and supranuclear gaze palsy [Shapiro, 2005]. In the absence of other associated injury, the intellect is usually normal, and the motor disorder is often the single greatest cause of disability.
The most effective treatment is prevention of neonatal hyperbilirubinemia [Bhutani et al., 2004; Rubaltelli, 1998; Blackmon et al., 2004; Stevenson et al., 2004]. Treatment of symptomatic cases is often quite difficult. Many children receive cochlear implants for the hearing loss. There is only a slight benefit to anticholinergic medications, valproic acid [Kulkarni, 1992], benzodiazepines, or botulinum toxin. Some children have benefited from intrathecal baclofen. In general, treatment of this disorder is poorly effective. Life span is shortened in severely affected children due to pulmonary complications.
Pantothenate Kinase-Associated Neurodegeneration
PKAN is a member of the group of diseases now referred to as neuronal brain iron accumulation [Bertrand, 2002a, b]. Symptoms of PKAN include progressive dystonia, dysarthria, rigidity, ballism, choreoathetosis, spasticity, dementia, and pigmentary retinal degeneration [Swaiman, 1991]. In its later stages, this disorder has characteristic ballistic flinging movements of arms and legs, as well as involuntary and repetitive tongue protrusion. The limb and tongue movements may lead to injury, requiring restraint of the arms and legs, and dental extraction [Sheehy et al., 1999]. There is a gradual progression over years, with loss of ambulation within 5–15 years of onset. The initial symptoms usually occur in either the childhood or the juvenile years.
PKAN has been divided into the characteristic type with presentation in the first decade (average onset at 3 years), and an atypical form with onset in the second decade, which seems to have slower progression and lower severity [Thomas et al., 2004]. Dystonia in PKAN usually starts in the leg, but sometimes the earliest presenting sign is visual loss. There is often associated bradykinesia. The rate of progression is more rapid with younger onset. The overall incidence is estimated to be approximately 3 cases per million population. The severity of dementia is variable and difficult to quantitate, as the severity of the movement disorder makes assessment of cognitive function in later stages difficult.
The gene responsible for PKAN is pantothenate-kinase-2 (PANK-2) on chromosome 20P13-P12.3 [Zhou et al., 2001; Hayflick, 2003a]. Inheritance is autosomal-recessive. Pantothenate kinase is responsible for synthesis of coenzyme A, and the neurologic abnormalities may be due to a combination of factors, including accumulation of upstream metabolites causing iron chelation, as well as decreased availability of coenzyme A for fatty acid metabolism [Hayflick, 2003b; Gordon, 2002]. The pathology is characterized by rust-colored iron pigmentation of the globus pallidus and substantia nigra pars reticulata. Injury to the globus pallidus, with consequent abnormalities of outflow patterns, may be responsible for the movement disorder [Mink, 2001b]. In addition, there are axonal spheroids similar to those seen in neuroaxonal dystrophy [Gordon, 2002].
MRI shows a characteristic “eye of the tiger” sign. This sign consists of a dark globus pallidus internus on T2 imaging, with a bright region in the center of the globus pallidus that is thought to be due to central necrosis (Figure 68-4). The dark signal is due to iron accumulation [Hayflick et al., 2001], although deposition of iron may be secondary to other metabolic deficits rather than the primary cause of symptoms [Koeppen and Dickson, 2001]. When the full eye of the tiger sign is present in childhood, 100 percent of such cases have the PANK-2 mutation [Hayflick et al., 2003]. Similarly, 100 percent of children with PANK-2 mutations show the eye of the tiger sign at some point during the illness [Trimble, 2003]. Children with clinical features of PKAN but without the eye of the tiger sign only have a 50 percent chance of being positive for the PANK-2 mutation. Ophthalmologic examination and electroretinogram may detect presymptomatic retinopathic changes and early visual loss [Swaiman, 1991]. Acanthocytes are sometimes, but not always, present, and there may be a low level of prebetalipoprotein. This is similar to what is seen in the HARP syndrome (hypoprebetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and pallidal degeneration). Some families with HARP syndrome have been shown to have the PANK-2 mutation, and therefore it is not known whether or not this is a separate disorder or a variant of PKAN [Houlden et al., 2003]. It is important to test for other disorders that can present with similar generalized dystonia in childhood, including HD, neuronal ceroid-lipofuscinosis, and hexosaminidase A deficiency (Tay–Sachs disease).
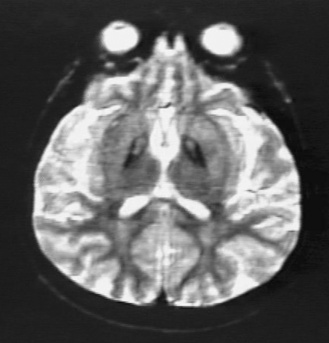
Treatment is symptomatic. Some children receive benefit from benzodiazepines, anticholinergic medications, botulinum toxin, baclofen, or intrathecal baclofen. It is unlikely that any of these treatments slows the progression of the underlying disease, but several case reports of treatment with pallidotomy or deep brain stimulation of the globus pallidus internus suggest possibility of symptomatic improvement [Umemura et al., 2004; Justesen et al., 1999]. The prognosis is universally poor, with death from medical complications usually within 10–20 years of the onset in the typical form; in the atypical form, survival may be longer.
Lesch–Nyhan Disease
Lesch–Nyhan disease is a progressive neurodegenerative disorder that initially presents with hypotonia and developmental delay in the first year of life. It progresses over several years, and can mimic athetotic cerebral palsy, with ballism, choreoathetosis, axial dystonia, and spasticity [Nyhan, 1968a, b, 1972]. There is a striking prevalence of self-mutilation behavior, including tongue-biting, head-banging, biting the fingers and lips, and thrusting arms or legs against objects [Lee et al., 2002; Robey et al., 2003]. There are also compulsive behaviors, and slowly progressive dementia is thought to be characteristic [Matthews et al., 1999], although more recent reports cast doubt on whether dementia is always severe [Schretlen et al., 2005; Jinnah et al., 2006]. The behavioral disorder seems to be unique, with adult patients describing compulsions to injure themselves or to behave inappropriately [Schretlen et al., 2005; Jinnah et al., 2006]. There is an increase in uric acid in both the blood and the urine, and this leads to symptoms of gout in advanced cases, although gout is less common in children. Both arthritis and renal stones have been reported, but usually in older patients.
The responsible gene is HPRT-1 (hypoxanthine-guanine phosphoribosyltransferase) [Mak et al., 2000]. Deficiency of HPRT-1 can be detected in fibroblasts and leukocytes. The disorder is X-linked recessive, and the incidence of symptomatic cases is approximately 1 in 400,000 boys. MRI shows only nonspecific atrophy.
Treatment is symptomatic. Allopurinol may alleviate signs of gout and prevent renal calculi [Hiraishi et al., 1987; Brock et al., 1983]. Behavioral programs can be used to reduce self-mutilation [Olson and Houlihan, 2000]. Benzodiazepines, carbamazepine, and possibly selective serotonin reuptake inhibitors can be helpful for the behavior disorder [Roach et al., 1996; Saito and Takashima, 2000; Cusumano et al., 2001]. Tetrabenazine or l-DOPA may be helpful to treat the dystonia [Jankovic et al., 1988]. Naltrexone has been attempted to reduce self-mutilation, with only limited success. In some cases, restraint and removal of dentition are required in order to prevent self-mutilation, and patients who can communicate often will request to be placed in restraint in order to reduce self-injury. The injurious behavior seems to become worse in times of stress, and avoidance of stressful situations may be helpful. A single case report suggests the possibility of treatment with deep brain stimulation [Taira et al., 2003]. The prognosis is poor. Most children never walk, and there is progression of symptoms and dementia over several years. Survival is possible into the second or third decade.
Spinocerebellar Ataxia
Spinocerebellar ataxia is discussed in Chapter 67. However, it is worth mentioning here that spinocerebellar ataxia type 3 (Machado–Joseph disease) has a characteristic presentation, with a dystonic-rigid or parkinsonian syndrome. This is referred to as type 1 Machado–Joseph disease, with type 2 presenting more typically with ataxia and spasticity. The type 1 dystonic-rigid form tends to present more commonly in children, and can resemble juvenile HD [Jardim et al., 2001a, b]. There is often progressive external ophthalmoplegia and bulging eyes, superimposed on a nuclear or supranuclear gaze palsy. Presentation can be as early as 5 years of age, although it is more common for disease to present in the second decade or later. Machado–Joseph disease is due to a CAG triplet repeat on chromosome 14Q24.3–Q31, and children with 53–86 repeats will be symptomatic. Higher numbers of repeats are associated with earlier onset and greater severity. Inheritance is autosomal-dominant. Treatment is primarily symptomatic, although some cases have responded to treatment with l-DOPA early in the course.
Organic Acidemias
A particularly striking presentation that often occurs in infancy, but which can occur later in childhood, has been referred to as infantile bilateral striatal necrosis (IBSN) [Straussberg et al., 2002; Basel-Vanagaite et al., 2004]. This can occur with many metabolic disorders, but also seems to occur in the context of acute infections, particularly with mycoplasma, and it has been reported in children with PANK-2 mutations. Most often, the cause is unknown. There have been some rare familial cases. This disorder is characterized by a rapid onset of dystonia and dyskinesia, sometimes associated with chorea [Mito et al., 1986; Roytta et al., 1981], and the MRI often shows evidence of irreversible striatal and sometimes pallidal injury on diffusion-weighted, T2, or gadolinium-enhanced images [Mito et al., 1986; Roytta et al., 1981].
Early diagnosis of a possible metabolic defect is essential, and treatment of any precipitating infectious process should be initiated rapidly. Once the process has started, usually only symptomatic treatment is available. There have been a few reported cases of a biotin-responsive type of striatal necrosis that has been thought to be due to an abnormality of biotin transport, and such cases may have rapid and effective resolution when treated with biotin [Straussberg et al., 2002]. There have been reported cases of trauma being the inciting event, but whether or not trauma by itself is able to do this, or whether another associated metabolic abnormality is required, is not known.
Fahr’s Disease
Fahr’s disease is characterized by basal ganglia calcification, as well as calcification of other gray-matter structures, including cerebellar nuclei and punctate calcifications in thalamus and sometimes cortex [Oliveira et al., 2004]. This is usually an adult-onset disease, but in rare cases it can occur in the second decade of life. When it does, it is characterized by microcephaly, hypertonia, and choreoathetosis. The etiology is often unknown, but in some cases disease is due to hypoparathyroidism, and therefore parathyroid function should be checked. It is inherited as an autosomal-dominant trait, with reduced penetrance in most families, and appears to be slowly progressive. Diagnosis is based on evidence of calcification detected by head computed tomography (CT) [Oliveira et al., 2004; Koller and Klawans, 1980; Koller et al., 1979]. Treatment is supportive, unless a specific disorder of parathyroid hormone can be found, in which case specific treatment will be available.
Neuroacanthocytosis
Neuroacanthocytosis has also been referred to as chorea acanthocytosis or Levine–Critchley syndrome. It is characterized by spiky projections on erythrocytes (Figure 68-5), although such projections are often seen in non-neurologic disease, including hepatic disease. Acanthocytes are also seen in abetalipoproteinemia (Bassen–Kornzweig disease) [Bohlega et al., 1998], vitamin E deficiency, hypoprebetalipoproteinemia, HARP syndrome, and with abnormal Kell blood group antigens (McLeod’s syndrome) [Rampoldi et al., 2002; Danek et al., 2001; Mehndiratta et al., 2000; Dotti et al., 2000]. Onset is usually in the adult years, but onset as early as 8 years of age has been reported. Symptoms in childhood resemble juvenile HD, with dystonia and an akinetic-rigid syndrome, as well as a severe and progressive tic disorder [Kutcher et al., 1999; Yamamoto et al., 1982]. Presentation in adults is more typically with chorea or psychiatric illness [Dixit et al., 1993; Bruneau et al., 2003; Robinson et al., 2004]. There is frequent tongue protrusion and difficulty eating, and severe dysarthria is often a presenting feature. Some children and adults show areflexia, impulsive behavior, and, occasionally, mild dementia.
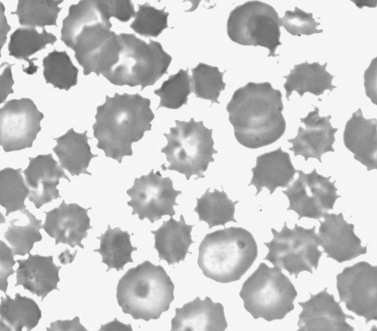
Testing for this disease requires examination of a fixed smear of blood under the microscope, stressed by mixing 1:1 with normal saline, and this requires an experienced and knowledgeable technician. A level of more than 3 percent acanthocytes is diagnostic. MRI shows caudate atrophy [Kutcher et al., 1999; Okamoto et al., 1997]. PET scanning shows decreased fluoro-dopa uptake and decreased raclopride binding, suggesting both pre- and postsynaptic disorders of striatal function [Brooks et al., 1991; Peppard et al., 1990]. Treatment is symptomatic. Benzodiazepines are often helpful. When chorea is present, neuroleptics are helpful, but this is probably less useful in childhood. There has been an isolated report of improvement with calcium channel blockers.
Medication-Induced Dystonia
Medications can cause dystonia, and this is most commonly seen with neuroleptic medications. In particular, there is a higher rate of acute dystonic reactions in children than in adults, and this has been reported to be as high as 10–20 percent in some series [Spina et al., 1993; Ayers and Dawson, 1980; Olsen et al., 2000]. Acute dystonic reactions are easily treated with intravenous diphenhydramine (1 mg/kg, usually given intravenously) or other anticholinergics [Dahiya and Noronha, 1984]. Other types of acute reactions to neuroleptics, such as akathisia, can be more difficult to treat, but in some cases respond to benzodiazepines. Acute dystonic reactions often present with uncontrollable tongue movements, opisthotonus, and neck hyperextension or torticollis. It should be noted that medications not normally classed with the neuroleptics, including antiemetic medications such as metoclopramide [Cowan, 1982; Lu and Chu, 1988; Renwick, 1990], domperidone [Madej, 1985], and prochlorperazine [Leeman, 1965], are able to produce the full range of neuroleptic side effects. It is likely that any medication that either blocks or depletes central catecholamines has the potential to cause an acute dystonic reaction [McCann et al., 1990; Burke et al., 1985].
Tardive dystonia and tardive dyskinesia are particularly worrisome consequences of neuroleptic use because symptoms may not resolve following discontinuation of the causative medication [Burke et al., 1982]. Tardive syndromes and their treatment are described in a separate section below.
Other Disorders Causing Secondary Dystonia
Aromatic l-amino acid decarboxylase deficiency (ALAD) leads to dopamine agonist-responsive dystonia. In this disorder, conversion of l-DOPA to dopamine is impaired, and therefore administration of l-DOPA will not be helpful [Swoboda et al., 2003, 1999]. While this disorder does respond to dopamine agonists, due to the combination of severe autonomic and sleep disturbances, treatment with serotonergic medication is often required as well [Swoboda et al., 1999]. Very few cases have been reported. This disorder is detectable by a characteristic pattern of cerebrospinal fluid neurotransmitter metabolites. Symptoms include dysautonomia, sleep disturbances, eye movement disturbances, and severe generalized dystonia, usually with onset in the first year of life. It appears to be autosomal-recessive.
Juvenile Parkinson’s disease can present with a picture very similar to DRD, with a combination of progressive dystonia and bradykinesia, and this will be described further in the next section. HD in children often presents with a rigid-akinetic syndrome and severe dystonia, as described above. Pontocerebellar hypoplasia type 2 is characterized by infantile onset of a combination of extrapyramidal symptoms, dyskinesia, and dystonia [Grosso et al., 2002; Grellner et al., 2000; Coppola et al., 2000].
Alternating hemiplegia is characterized by episodes of hemiplegia or dystonia that can last for hours to days and then resolve [Bassi et al., 2004; Saltik et al., 2004]. Transmission appears to be recessive, although in some cases there is autosomal-dominant transmission [Kanavakis et al., 2003]. Cases with dominant transmission must be distinguished from familial hemiplegic migraine. There is usually a slowly progressive generalized dystonia between episodes [Swoboda et al., 2004]. Treatment for alternating hemiplegia is with calcium channel blockers; in particular, flunarizine (5–15 mg/day) may reduce the frequency and severity of attacks. Symptomatic treatment, including anticholinergic medication, amantadine, and l-DOPA, may help in some cases [Sone et al., 2000; Sasaki et al., 2001].
Folate-transporter deficiency is characterized by baseline hypotonia and developmental delay, with episodes of hyperkinetic dystonia and chorea that can last minutes to days and which are sometimes triggered by changes in sleep, or by intercurrent illness [Moretti et al., 2005; Ramaekers and Blau, 2004]. Untreated, this disorder results in severe developmental delay and microcephaly. Primary folate transporter deficiency is probably due to a genetic alteration of the folate transporter across the blood–brain barrier. More commonly, folate transporter deficiency is acquired, and recent reports suggest the occurrence of either blocking or binding antibodies [Ramaekers et al., 2007, 2005]. Testing is performed by measurement of cerebrospinal fluid folate levels, and testing for blocking or binding antibodies in serum is possible. Oral treatment with folinic acid or methyl-tetrahydrofolate can lead to improvement of symptoms in some children [Ramaekers et al., 2005]. Immune therapy has been attempted, with variable success, in antibody-mediated cases, and concerns for cross-reactivity have led to the recommendation to avoid cow’s milk products [Ramaekers et al., 2008].
Dopamine-transporter deficiency is a newly reported disorder in which the presynaptic reuptake of dopamine in the striatum is impaired [Kurian, 2010]. The disorder is due to mutations in the dopamine transporter (DAT) gene, and symptoms include severe dystonia, hypotonia, bradykinesia, and developmental delay, with onset in the first year of life. Some children exhibit spontaneous hyperthermic spells associated with rigidity and rhabdomyolysis. Cerebrospinal fluid shows dramatic elevation of homovanillic acid, and serum prolactin levels may be decreased, particularly during hyperthermic episodes. No specific treatment has been found, although some children have responded to l-DOPA or dopamine agonists.
Treatments for Dystonia
Because DRD can mimic cerebral palsy, it has been recommended that all children with unexplained dystonia or cerebral palsy be given a trial of l-DOPA [Nygaard et al., 1988; Jan, 2004]. l-DOPA may be helpful in some children, even with secondary dystonia [Brunstrom et al., 2000]. The required dose of l-DOPA is very low in DRD (typically 50–100 mg per day), but may be much higher in juvenile Parkinson’s disease and secondary causes of dystonia, including cerebral palsy. Dosages as high as 10 mg/kg/day divided three times per day are sometimes needed. Common side effects of l-DOPA include nausea, vomiting, and diarrhea. Orthostatic hypotension occurs rarely in children. l-DOPA must be combined with a peripheral decarboxylase inhibitor, such as carbidopa, in order to increase central uptake and decrease peripheral side effects. Commercial preparations with a ratio of l-DOPA to carbidopa of 4:1 are typically used. Peripheral side effects can often be reduced by an additional dose of the peripheral decarboxylase inhibitor given one half-hour before the l-DOPA dose. l-DOPA competes for absorption with neutral amino acids, and therefore absorption will be slowed and the peak effect will be less when it is taken with a protein meal. Conversely, absorption rate is increased, the peak effect is greatest, and the side effects may be greatest when it is taken with carbohydrates. l-DOPA and anticholinergic medications are frequently used together. The appropriate dose of l-DOPA is difficult to determine, and it needs to be titrated gradually over a period of 2 weeks to 2 months. Rapid titration is possible, but the purpose of gradual titration is to find the optimal dose. Excessive doses of l-DOPA will usually cause dyskinesias or worsen dystonia, and often an intermediate dose is most effective. Rapid withdrawal can lead to worsening of dystonia and there are rare reports of symptoms similar to neuroleptic malignant syndrome following rapid withdrawal in adults.
Anticholinergic medication is very effective in acute dystonic reactions, as described above. It is partly effective in a subset of children with chronic dystonia [Hoon et al., 2001], and trihexyphenidyl remains the best studied centrally active antidystonia medication [Burke et al., 1986; Burke and Fahn, 1983; Greene et al., 1988]. Its mechanism of action for dystonia is not known, although it is presumed to have an effect on the large cholinergic interneurons of the striatum. Its anticholinergic effects would be expected to be widespread throughout basal ganglia and cortex [Fahn, 1987]. It is often necessary to proceed to very high doses: 1 mg/kg/day and, in some cases, higher [Fahn, 1983a, b]. In younger children, lower doses may be sufficient, but children will tolerate the high doses better than adults. In particular, adults and older children are more likely to develop confusion or behavior changes on high doses. The effectiveness may be greater in young children, and speech and hand function seems to improve the most [Hoon et al., 2001]. The dose must be increased gradually, and it may often take 3–4 months to achieve an appropriate dosage. Appropriate initial dosage is 0.05–0.1 mg/kg/day or less. The medication is usually divided three times per day and given during the day. Side effects are similar to those of other anticholinergic medications. Behavior changes are not uncommon, and deficits in attention have been reported in adult trials. Anticholinergic medications can worsen chorea and possibly choreoathetosis. The benefit on dystonia is often delayed, and there may be no apparent benefit for several months after starting the medication. At high doses, abrupt withdrawal could potentially be dangerous, and there are rare reports of neuroleptic malignant syndrome following abrupt withdrawal of high doses of trihexyphenidyl in adults. Other anticholinergic medications may work as well, but have not been extensively tested.
When dystonia is associated with increased tone, botulinum toxin has been shown to be very effective [Boyd et al., 2001; Graham et al., 2003]. It may be effective in directly reducing the tone in the hypertonic muscles, but may also be effective in changing the overall pattern of dystonia. In particular, it is sometimes observed that botulinum toxin injection into one muscle will relax other muscles of the same limb. Injection is often performed with electromyographic guidance, and although some practitioners will use general anesthesia and electrical stimulation for localization, many have now chosen to inject with only local anesthesia. The procedure should not be performed more frequently than every 3 months, due to the risk of build-up of neutralizing antibodies. In some cases, repeated doses of botulinum toxin every 3 months are needed but, in other cases, short-term treatment can be effective. Phenol or alcohol nerve blocks and dantrolene have not been commonly used in dystonia.
Baclofen has been used and may be effective in some cases, although its mechanism of effectiveness in dystonia is not known. Intrathecal baclofen can reduce tone in generalized dystonia if the catheter is placed at cervical levels [Albright et al., 1996, 1998, 2001; Butler and Campbell, 2000; Ford et al., 1998]. There is a high rate of complications, up to 35 percent in some series [Campbell et al., 2002]. Complications can be serious, and can include meningitis. Overdoses are rare, but pump failure or catheter disconnection with acute and possibly life-threatening baclofen withdrawal can occur. Withdrawal needs to be treated with a combination of oral baclofen and intravenous diazepam, as well as surgical reimplantation of the catheter as soon as possible. For this reason, many clinicians recommend that children with intrathecal baclofen pumps remain within 30 minutes of an appropriately experienced emergency room at all times.
Neurosurgical intervention is reserved for the most severe cases, but the possibility that earlier surgical intervention may slow progression of the disease or lead to a longer disease-free interval has been raised. Pallidotomy and thalamotomy have been in use for many years, with some success [Gros et al., 1976a, b; Vercueil, 2003]. More recently, deep brain stimulation has become available, and multiple targets have been attempted, including the ventrolateral thalamus [Thompson et al., 2000; Ghika et al., 2002], internal globus pallidus [Coubes et al., 2000; Tronnier and Fogel, 2000; Vercueil et al., 2001], and, most recently, subthalamic nucleus [Deep-brain stimulation for Parkinson’s Disease Study Group, 2001; Benabid et al., 2001]. The advantage of deep brain stimulation is the ability to control the current delivery and the ability to select one or a combination of up to four electrodes through an external programmer. The stimulation wire is implanted using a combination of stereotactic neurosurgical techniques and usually microelectrode recording for precise localization. The lead is connected to a pacemaker implanted in the chest. For generalized dystonia, usually both sides need to be implanted. The full effect of ablative surgery or stimulation implantation is often not seen for many months, and there are reports of continued improvement up to 1 year or longer after pallidotomy [Lin et al., 2001]. Surgical procedures in the globus pallidus internus seem to be particularly effective in DYT-1 and other primary dystonias [Eltahawy et al., 2004; Coubes et al., 2004], [Vercueil et al., 2002; Ford, 2004; Vitek et al., 1998; Lozano et al., 1997], and these are cases in which earlier intervention may be most effective [Coubes et al., 2000]. The secondary causes of dystonia may also respond [Ghika et al., 2002; Rakocevic et al., 2004], but surgery seems to be less effective for secondary dystonia [Cif et al., 2003], [Coubes et al., 2002; Krack and Vercueil, 2001; Teive et al., 2001]. The largest case series suggests up to 50 percent effectiveness in children with secondary dystonia, but up to 80 percent effectiveness in children with primary dystonia. Complications are essentially those of the implantation procedure. Infections do occur, but these are rare outside of the immediate perioperative period. Batteries will need to be replaced every 3–5 years, and this requires a much simpler surgical procedure. The long-term efficacy of deep brain stimulation is not known. Whether there is a difference in programming parameters between children and adults is likewise not known.
Treatment of Dyskinesia and Hyperkinetic Dystonia
Hyperkinetic disorders are more difficult to treat and, although they represent a positive symptom, it is often difficult to remove the effects of dyskinetic movements through botulinum toxin due to the likely need to paralyze the affected muscle completely. Trihexyphenidyl can often make dyskinesia worse, as can l-DOPA. There is some effectiveness of clonazepam and valproate. Tetrabenazine [Jankovic and Orman, 1988; Jankovic and Beach, 1997], reserpine, and amantadine have been tried, but there are no consistent reports of benefit. Whether hyperkinetic dystonia and dyskinesia respond to pallidotomy or deep brain stimulation is not currently known. In Parkinson’s disease, pallidotomy does seem to improve dyskinesias, but the mechanism of dyskinesia in this case may be very different from the hyperkinetic symptoms seen in children, and there are not yet adequate data to guide therapy.
Tremor
Classification of Tremor
Tremor has been described as “a rhythmical, involuntary, oscillatory movement of a body part” [Deuschl et al., 1998]. It can be classified based on the time of greatest severity as rest, postural, action, or intention tremor. Rest tremor is defined as tremor involving a body part that is inactive and supported against gravity. It may improve during movement. Rest tremor is most commonly associated with other signs of parkinsonism, but may occur in isolation. Postural tremor is worse when a child attempts to sustain a posture against gravity, such as with the arms outstretched in front or with the hands near the nose. It may improve with movement or with rest supported against gravity. Action tremor is worse during active movement, but postural or rest components may be present as well. Intention tremor is a specific form of tremor associated with cerebellar disorders, in which the tremor becomes worse as the arm approaches an endpoint or target, and there is great difficulty achieving target accuracy. Tremor can affect the head [DiMario, 2000], extremities, trunk, or voice.
Tremor can be described in terms of its frequency. Tremor associated with Parkinson’s disease or parkinsonism is between 4 and 6 Hz [Findley et al., 1981], while essential tremor is more likely at 5–8 Hz [Elble, 2000], and physiologic tremor has two components at 8–12 Hz and at 20–25 Hz [Makabe and Sakamoto, 2002; Takanokura et al., 2002]. In diagnosing tremor, it is important to distinguish it from dystonic tremor and myoclonus. Dystonic tremor, as described above, tends to be less regular, and often exhibits a null point. Myoclonus can be rhythmic and can be a form of tremor, but the rapid shocklike quality distinguishes it from tremor. Electromyography may be helpful in distinguishing myoclonic-type tremor from other forms of tremor, due to the short bursts of less than 50 milliseconds and correlation with scalp potentials that are often seen with myoclonus [Shibasaki et al., 1986; Shibasaki, 2000]. Myoclonic tremor is also more likely to have a burst-and-release (saw-tooth) pattern rather than a symmetric back-and-forth reciprocal pattern [Sanger et al., 2010].








































Primary Tremor
Etiologies of primary tremor are shown in Box 68-3. Familial essential tremor is an autosomal-dominant disorder with variable penetrance and severity. It can present in childhood and often is slowly progressive over many years [Jankovic et al., 2004; Louis et al., 2001; Paulson, 1976]. It is rarely disabling in childhood, except in extreme cases. Tremor is primarily postural, but can also be worsened with action. Tremor with holding a cup or handwriting are often some of the earliest complaints, but head, trunk, and voice can be affected [Jankovic, 2000]. There is no specific treatment. In adults, there may be a statistical association between essential tremor and Parkinson’s disease [Yahr et al., 2003].
Task-specific tremor is rare in children; when it does occur, it most often seems to involve handwriting. Whether task-specific tremors are a variant of dystonic tremor with task specificity, or whether this is a form of essential tremor is not known [Rosenbaum and Jankovic, 1988]. It is important to examine the child for other evidence of dystonia either in the affected limb or elsewhere. Medical treatment is usually ineffective, but occupational therapy may be helpful to develop compensatory strategies.
Physiological tremor is normally present and becomes particularly noticeable when a muscle is exerting high levels of force [Makabe and Sakamoto, 2002; Takanokura et al., 2002]. The distinguishing characteristic of physiologic tremor is that it occurs or worsens only when the child is carrying a heavy object or otherwise needs to contract the muscle against high resistance. Some children have an enhanced physiologic tremor that can become noticeable but is only rarely bothersome. There is no effective treatment for enhanced physiological tremor, but progression is probably rare.
Tremor is a frequent manifestation seen in psychogenic movement disorders. As noted in the section on psychogenic dystonia, psychogenic tremor likely represents a conversion reaction, and may reflect relatively serious psychopathology with consequent poor prognosis [Miyasaki et al., 2003; Kim et al., 1999]. Psychogenic tremor can often be distinguished from organic bases of tremor by entrainment (i.e., synchronization) to the examiner. In particular, when a child is asked to make a rhythmic movement with the unaffected hand or foot at a rhythm different from the tremor, often the tremor will entrain to the rhythmic movement, or the rhythmic movement will entrain to the tremor, such that either the tremor becomes irregular during accurate performance of the rhythmic movement, or performance of the rhythmic movement is impossible in the apparently unaffected limb [Zeuner et al., 2003]. Similarly, distraction using mathematical tasks or other difficult cognitive tasks can often change the nature or frequency of a physiologic tremor, but this would not be expected to occur for an organic tremor. However, the severity (but usually not the frequency) of an organic tremor can worsen in the context of a cognitive performance task if it is worsened by stress.
Secondary Tremor
There are a large number of secondary causes of tremor, and many of these are listed in Box 68-3. It is important to exclude Wilson’s disease. This disorder is characterized by a wing-beating tremor that often involves the adductors of the arm, and is shown when the child is asked to abduct the arms with elbows bent [Stremmel et al., 1991; Saito, 1987]. The tremor is primarily postural but may occur at rest. Other forms of tremor may occur in individuals with Wilson’s disease. Other symptoms include dystonia, rigidity, dysarthria, and psychiatric disorders. The defect is due to a mutation in the ATP7B copper-transport atp-ase gene on 13q14.3, which leads to reduced biliary excretion of copper and consequent accumulation of copper in multiple tissues. Symptom onset is between 6 and 45 years of age. Diagnosis of Wilson’s disease is made by noting a low serum ceruloplasmin and elevated excretion of copper in a 24-hour urine collection with measurement of the copper to creatinine ratio. In patients with neurological symptoms, slit-lamp examination almost always reveals Kayser–Fleischer rings. Overt or occult liver disease is often, but not always, present. Because Wilson’s disease is treatable, it is very important to assess patients with tremor for this disorder. Common treatments include zinc acetate, trientine, or penicillamine, although if disease is not detected early, then liver transplantation may be needed [Roberts and Schilsky, 2003].
Rubral tremor describes a large-amplitude, rhythmic, but partly ballistic movement that often affects the arms and can be unilateral. It is associated with attempts at movement and is therefore an action tremor. In some cases, it can be difficult to distinguish chorea from rubral tremor, although rubral tremor is much less common. It is associated with lesions in the thalamus [Tan et al., 2001], midbrain [Leung et al., 1999], pons, or cerebellar peduncles, and not necessarily the red nucleus, despite its name. Rubral tremor is usually nonprogressive but can be severely disabling.
Treatment of Tremor
The first goal in treatment of tremor is to exclude other possible causes of rhythmic movement, including myoclonus, dystonia, seizures, or cerebellar dysfunction. Any underlying metabolic or hormonal cause must be corrected. Essential tremor is treated with propranolol, primidone, benzodiazepines (clonazepam may be particularly effective), or gabapentin [Jankovic et al., 2004; Paulson, 1976; Rajput et al., 2004; Gironell et al., 1999]. If tremor is caused or worsened by anxiety, benzodiazepines or a serotonin reuptake inhibitor may be helpful. Tremor due to “stage-fright,” social phobia, or panic disorder often responds to propranolol.
These same medications can be tried in other forms of tremor, but usually there is minimal responsiveness. Rubral tremor is notoriously refractory to all attempts at intervention, although some cases will respond to l-DOPA [Yuill, 1980; Findley and Gresty, 1980], clonazepam [Jacob and Pratap Chand, 1998], or thalamic deep brain stimulation [Nikkhah et al., 2004].
Parkinsonism
Bradykinesia can be difficult to detect when hypertonia is present, since hypertonia may be interpreted as the reason for slow movement. In parkinsonian syndromes in children, bradykinesia is likely to be a more disabling symptom than rigidity. Rigidity can be difficult to distinguish from other causes of hypertonia, including dystonia or spasticity. The absence of a spastic catch or fixed posture distinguishes rigidity from spasticity. The absence of overflow movements or a fixed abnormal posture helps to distinguish rigidity from dystonia, although both may exhibit co-contraction [Sanger et al., 2003]. Arm rigidity is often suspected by observing decreased arm swing during walking, and this is most easily seen when it is asymmetric. Rigidity can be brought out by facilitation maneuvers, such as having the child open and close one fist while the resistance to movement at the opposite elbow is tested.
In adults, the most common cause of parkinsonism is Parkinson’s disease. In children, parkinsonism is uncommon but can be a manifestation of several different disorders, as discussed below [Riederer and Foley, 2002]. It is often associated with a dopaminergic deficit, and since dopaminergic deficits can produce dystonia in children, dystonia may be present as well [Yokochi, 2000].
Parkinson’s Disease
Primary causes of parkinsonism in children are rare [Pranzatelli et al., 1994]. DRD may be a relatively common cause compared to others, and has been described above. Juvenile Parkinson’s disease presents with leg dystonia in younger children, and bradykinesia and rigidity in older children. It rarely demonstrates tremor. It most commonly presents in young adults, and childhood or juvenile onset is rare. Many cases are due to mutations in the Parkin gene, labeled PARK-2, on chromosome 6Q25.2–27 [Ruiz-Linares, 2004; Huynh et al., 2003]. This is autosomal-recessive, and can be contrasted with the autosomal-dominant inheritance of some forms of adult parkinsonism associated with mutations in the alpha-synuclein gene. The autosomal-dominant form does not appear to occur in childhood. Other genes have been identified in specific families [Bentivoglio et al., 2001; DeStefano et al., 2002; Valente et al., 2002; West et al., 2002a, b, 2003]. The pathology of Parkin-associated Parkinson’s disease does not show Lewy bodies, although there is neuronal loss and neurofibrillary tangles. Fluoro-dopa PET scanning can show decreased uptake, suggesting a presynaptic abnormality consistent with degeneration of the nigro-striatal pathways, rather than a metabolic defect in the synthesis of dopamine, as seen in DRD [Khan et al., 2002; Pal et al., 2002].




































Secondary Parkinsonism
Secondary causes of parkinsonism are shown in Box 68-4. These include infarcts, particularly in the striatum or pallidum, as well as the ceroid-lipofuscinoses, in which a combination of bradykinesia and visual loss leads to a very striking hesitancy of gait [Nijssen et al., 2002]. PKAN, as described above, can cause bradykinesia. Similarly, spinocerebellar ataxia, Rett’s syndrome, HD, and Wilson’s disease can be causes of bradykinesia. Parkinsonism can be associated with acute encephalitis or postencephalitic parkinsonism. The widespread epidemic of encephalitic parkinsonism (von Economo’s disease) that occurred between 1916 and 1930 has not recurred, and the term encephalitis lethargica should probably be reserved for patients who were victims of that epidemic. There are reported cases of a similar syndrome following streptococcal illness [Vincent, 2004; Dale et al., 2004; Kiley and Esiri, 2001]. Whether or not the original epidemic of encephalitis lethargica was due to influenza virus remains in question [Lo et al., 2003; Jellinger, 2001].
Several medications and toxins are known to cause parkinsonism [Pranzatelli et al., 1994]. This is a common side effect of tetrabenazine and reserpine, and can be seen with neuroleptic treatment as well. The chemical MPTP, which is a synthetic methamphetamine derivative used for illicit recreational purposes, led to several cases of very rapid onset of irreversible parkinsonism, but also has become very important in research into the pathogenesis and treatment of parkinsonian syndromes [Eldridge and Rocca, 1985; Burns et al., 1985].
Treatment of Parkinsonism
In adults and children with Parkinson’s disease, long-term treatment with dopamine will eventually lead to dyskinesias and freezing episodes that can limit its use. Some authors have advocated “dopamine-sparing” strategies that involve the early use of dopamine agonists, such as pergolide, pramipexole, or ropinirole, or of dopamine breakdown inhibitors, such as entacapone or selegiline [Jenner, 2003]. This approach has not been studied in children.
Amantadine may be used to treat dyskinesias associated with acute or chronic use of l-DOPA [Furukawa et al., 2004b], and it has some antiparkinsonian effects as well [Paci et al., 2001; Crosby et al., 2003]. Anticholinergic medication will improve parkinsonism, but it is more commonly used to treat associated dystonia. Deep brain stimulation in the internal globus pallidus or subthalamic nucleus has met with success in ameliorating symptoms of adult Parkinson’s disease [Peppe et al., 2004; Germano et al., 2004]. Whether similar neurosurgical procedures would be effective in juvenile parkinsonism is not known.
Myoclonus
Myoclonus consists of very brief, abrupt, involuntary, nonsuppressible, jerky contractions (or interruption of contraction), involving a single muscle or muscle group. The rapidity of these movements warrants the descriptor “shocklike,” as if an electrical shock had just been applied to the peripheral nerve innervating the muscle [Marsden et al., 1983]. Myoclonus can be rhythmic, in which case it often appears tremorlike. However, in true tremor the movement oscillates with near-equal amplitude around a midpoint, while in myoclonus the movement has more of a “saw-tooth” character. In some cases, myoclonus can be elicited by a sensory stimulus (reflex myoclonus; the most common example is the acoustic startle response in infancy) or volitional movement (action myoclonus). It is present in normal (associated with sleep, exercise, or anxiety) and pathological situations, both epileptic and nonepileptic. Epileptic myoclonus is discussed elsewhere. We limit our focus here to nonepileptic myoclonus.
Classification of Myoclonus
Several different schemes have been used to classify myoclonus, using clinical, anatomical, or etiological criteria. Perhaps the simplest classification is based upon when the myoclonus occurs. Thus, myoclonus may occur at rest, with action, or in response to a sensory stimulus (reflex myoclonus). In adults, it is helpful to classify based on presumed neuroanatomical location, e.g., cerebral cortex, thalamus, brainstem, or spinal cord. The neurophysiology of myoclonus has been reviewed elsewhere [Caviness and Brown, 2004; Shibasaki and Hallett, 2004]. In children, etiological classification is the most useful, as summarized in Box 68-5.











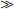 juvenile)
juvenile)
































Physiologic and Developmental Myoclonus
Physiologic myoclonus is that which occurs in normal individuals in specific settings. It includes such entities as hiccups, sleep starts, and sleep myoclonus. Sleep starts, also known as hypnic or hypnagogic myoclonus, occur with sleep initiation [Montagna, 2004]. They are often accompanied by a sense of falling. Sleep starts are normal physiologic phenomena and no treatment is required. Sleep myoclonus (nocturnal myoclonus) is also a part of normal sleep physiology. It is thought to result from paradoxical excitation during rapid eye movement (REM) sleep, due to transient failure of brainstem inhibition [Montagna, 2004]. Sleep myoclonus in older children is more fragmentary than neonatal sleep myoclonus and tends to persist throughout life. No treatment is required.
Benign myoclonus may occur in association with specific developmental stages. Benign neonatal sleep myoclonus and benign myoclonus of early infancy are discussed in the section entitled “Transient Developmental Movement Disorders,” below. Myoclonus can also occur with fever in otherwise normal children [Dooley and Hayden, 2004]. The myoclonic jerks may be quite frequent, but they are self-limited, ceasing when the fever resolves. Febrile myoclonus may be more common in younger children. No treatment is required.
Essential Myoclonus
Essential myoclonus is a relatively mild condition, starting in the first or second decade. It is inherited as an autosomal-dominant trait with incomplete penetrance [Quinn, 1996; Fahn and Sjaastad, 1991]. It is usually slowly progressive for a few years after onset and then stabilizes. The myoclonus may fluctuate slightly over the years, or may show mild spontaneous improvement. The myoclonus usually responds to ethanol, but the magnitude of benefit varies across individuals, and ethanol is not useful as a long-term treatment. The most common mutation identified is in the epsilon-sarcoglycan gene (7q21–q31). Mutations in this gene have also been associated with myoclonus-dystonia syndrome (see above). Many patients with essential myoclonus also have dystonia, and it appears that the two clinical syndromes may have the same underlying genetic basis [Nygaard et al., 1999; Quinn, 1996]. Some family members may have only myoclonus, and others may have only dystonia, while yet others may have both myoclonus and dystonia. Essential myoclonus may respond to benzodiazepines, primidone, or propranolol.
Symptomatic Myoclonus
Myoclonus can by symptomatic of many disorders, as listed in Box 68-5. Many of these conditions are discussed elsewhere in this text. In this section, we discuss specific conditions in which myoclonus can be the primary feature.
Myoclonic Encephalopathy of Infancy (Opsoclonus-Myoclonus Syndrome)
Myoclonic encephalopathy is an uncommon acquired disorder of late infancy or childhood. It was first described by Kinsbourne and often is referred to as “Kinsbourne’s syndrome” [Kinsbourne, 1962]. It has also been called infantile polymyoclonia [Dyken and Kolar, 1968] and opsoclonus-myoclonus syndrome. The clinical features of the syndrome include acute or subacute onset of rapid, “dancing” eye movements and myoclonic jerking of the limbs and trunk. The eye movements (opsoclonus) are usually conjugate, but can be dysconjugate. As the disorder progresses, there is increased severity of the truncal myoclonus, which often prevents child from standing or walking. The myoclonus usually ceases during sleep, but may persist. The opsoclonus may also persist during sleep. In some children, the myoclonus is stimulus-sensitive and it usually increases with action. The marked increase of myoclonus with action may make it difficult to distinguish from ataxia. Many children with this condition have prominent irritability.
Myoclonic encephalopathy has several potential causes. An important association has been made between myoclonic encephalopathy and neuroblastoma [Warrier et al., 1985]. The opsoclonus and myoclonus typically occur before the tumor is diagnosed [Dyken and Kolar, 1968]. The pathophysiology of myoclonic encephalopathy associated with neuroblastoma is unknown, but is presumed to be immune-mediated [Pranzatelli et al., 2004]. Removal of the tumor does not prevent relapse or permanent sequelae. Immunomodulatory therapy can be effective in some patients, but even with treatment, the majority of patients have at least mild neurological sequelae [Hayward et al., 2001].
Approximately half the patients with myoclonic encephalopathy do not have an identifiable neuroblastoma [Tate et al., 2005]. In these instances, the cause is more likely infectious or postinfectious. It has been associated with aseptic meningitis, but more commonly follows a viral infection [Kinsbourne, 1962; Sheth et al., 1995]. There can be elevation of cerebrospinal fluid protein and mild lymphocytic pleocytosis, but the presence of cerebrospinal fluid pleocytosis does not distinguish paraneoplastic from infectious opsoclonus-myoclonus syndrome [Tate et al., 2005; Mitchell and Snodgrass, 1990]. Similar serum antineuronal antibodies have been detected in patients with postinfectious or neuroblastoma-associated opsoclonus-myoclonus syndrome [Connolly et al., 1997].
It is necessary to undertake a careful investigation for occult neuroblastoma in all infants and children with myoclonic encephalopathy, unless the cause is obvious. The symptoms of children with and without neuroblastoma respond to adrenocorticotropic hormone (ACTH) therapy [Tate et al., 2005]. ACTH (40 units/m2/day) has proved highly effective in many instances. Removal of the tumor may lead to symptom resolution, but it may be followed by recurrence.
Dyssynergia Cerebellaris Myoclonica (Ramsay–Hunt Syndrome)
Hunt reported six patients with seizures, action myoclonus, and intention tremor, and termed their condition dyssynergia cerebellaris myoclonica [Hunt, 1921]. One twin, whose progressive neurologic symptoms began in the third decade of life, came to postmortem examination; the other twin experienced myoclonus 2 years before seizures began. Atrophy of the dentate nucleus and hypodense myelin in the superior cerebellar peduncle were evident, as well as disruption of the spinocerebellar tracts. Hunt ascribed the clinical findings to the dentate nucleus pathology. A familial pattern was suggested. A number of patients have been reported since the first description. There is not a single underlying cause for this syndrome and it has been debated whether this is even a unique syndrome.
Dentato-Rubral-Pallido-Luysian Atrophy
Dentato-rubral-pallido-luysian atrophy (DRPLA) is an autosomal-dominant, degenerative disorder of the dentate nucleus, red nucleus, globus pallidus, and subthalamic nucleus [Becher et al., 1997]. Clinical features include myoclonus, cerebellar ataxia, epilepsy, choreoathetosis, and dementia. The disease is caused by a CAG repeat expansion in the DRPLA gene, on chromosome 12p13. It is most common in people of Japanese or Portuguese origin, but has been reported in other groups.
Postanoxic Myoclonus
Lance and Adams described myoclonus as a consequence of severe hypoxia during surgery in adults [Lance and Adams, 1963]. Postanoxic myoclonus is primarily an action or intention myoclonus. It can be disabling and can include negative myoclonus that may lead to falls. Some infants who undergo hypoxic-ischemic brain injury at birth develop continuous myoclonic activity of the face and limbs. This condition has been called polymyoclonus, but may be a variant of the Lance–Adams syndrome.
Treatment of Myoclonus
Myoclonus is often refractory to medical treatment. Cortical myoclonus may respond to benzodiazepines and is commonly treated with clonazepam (although sleep myoclonus may worsen [Reggin and Johnson, 1989]). Valproic acid is sometimes helpful, but it must be used with caution due to the ability to cause tremor as a side effect, with consequent confusion of symptoms. Piracetam has been used for many years to treat myoclonus, with good efficacy. Levetiracetam is a piracetam analog, but does not seem to have equivalent efficacy to piracetam for treatment of myoclonus. There are reports of efficacy with zonisamide in some forms of myoclonus [Kyllerman and Ben-Menachem, 1998; Wallace, 1998]. Posthypoxic myoclonus seems to be particularly responsive to serotonergic medications, including selective serotonin reuptake inhibitors and oral administration of 5-hydroxytryptophan or 5-hydroxytryptamine. Baclofen has been occasionally used, but the mechanism of action is not clear. Carbamazepine can worsen myoclonus.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 68 Movement Disorders
Introduction
Movement disorders are syndromes characterized by impaired voluntary movement, the presence of involuntary movements, or both. There may be impaired targeting and velocity of intended movements, abnormal involuntary movements, abnormal postures, or excessive normal-appearing movements at inappropriate or unintended times. Movement disorders in children include athetosis, chorea, dystonia, myoclonus, parkinsonism, stereotypies, tics, and tremor [Sanger, 2003a]. Movement disorders may be accompanied by weakness, spasticity, hypotonia, ataxia, apraxia [Koski et al., 2002], and other motor deficits, although many authors do not include these accompanying deficits.
Movement disorder terminology has been well defined for adults, but less so for children. Therefore, it is likely that movement disorders are under-reported in children, and that there is inconsistent terminology. Recently, there have been attempts to provide specific definitions of childhood motor disorders [Sanger et al., 2003, 2006, 2010]. According to these definitions, childhood disorders can be divided into three major categories: hypertonic disorders, hyperkinetic disorders, and negative signs. Hypertonic disorders include spasticity, dystonia, and rigidity. Hyperkinetic disorders include chorea, dystonia, athetosis, myoclonus, tremor, stereotypies, and tics. Negative signs include weakness, reduced selective motor control, ataxia, apraxia, and developmental dyspraxia, although we will not discuss these here. Consensus definitions for these terms have been established, although the list is not intended to be exhaustive and disorders of gait, balance, speech, and eye movement are not included. Several important terms, including hypotonia and bradykinesia, have not yet been defined for children. The prevalence in children of different types of disorders is not known, although there have been studies investigating symptoms in certain populations, including children with cerebral palsy.
Characteristic Features of Pediatric Movement Disorders
Movement disorders in children differ from those in adults in several aspects. Perhaps the most important is that movement disorders in childhood are primarily symptoms of other diseases, rather than diseases in themselves [Sanger, 2003a, b]. In adults, dystonia and parkinsonism are usually due to primary dystonia or idiopathic Parkinson’s disease, respectively. However, dystonia or parkinsonism in children are more likely to be features of an underlying static or progressive neurological disorder. Diagnosis in children is complicated by the fact that many symptoms have more than one cause, and any particular underlying pathophysiology may lead to a complex combination of symptoms. The diagnostic work-up in children is guided by symptoms, but the existence of a large class of diseases that can lead to the same set of symptoms often necessitates a broad etiologic work-up. There may be both specific etiologic treatments, as well as symptomatic treatments, both of which may be beneficial in an individual child. In particular, many of the causes of childhood movement disorders do not yet have any specific treatment, yet symptomatic treatment for the resulting movement disorder can be extremely helpful and lead to improvement in quality of life.
Diagnosis of Movement Disorders
Classification of a movement disorder based upon the spatial and temporal pattern is essential for diagnosis. It is also important to define the context in which the movements occur. While it is often helpful to list the characteristics of the movements (Table 68-1), the diagnosis relies on pattern recognition, and the clinician must see the movements. If the movements are not apparent during the neurological examination, repeating the examination at another time or obtaining video recordings of the movements is important to making an accurate diagnosis. The widespread availability of video cameras has substantially improved diagnosis of movement disorders.
Table 68-1 Phenomenological Classification of Movement Disorders
| Movement Disorder | Brief Description |
|---|---|
| Athetosis | Slow, continuous, writhing movements of distal body parts, especially the fingers and hands |
| Chorea/ballism | Chaotic, random, repetitive, brief, purposeless movements. Rapid, but not as rapid as myoclonus. When very large in amplitude and affecting proximal joints, choreic limb movements are often called ballism |
| Dystonia | Repetitive, sustained, abnormal postures and/or movements. Abnormal postures typically have a twisting quality |
| Myoclonus | Sudden, brief, shocklike movements that may be repetitive or rhythmic |
| Parkinsonism | Hypokinetic syndrome characterized by a combination of rest tremor, slow movement (bradykinesia), rigidity, and postural instability |
| Stereotypy | Patterned, episodic, repetitive, purposeless, rhythmic movements |
| Tics | Stereotyped intermittent, sudden, discrete, repetitive, nonrhythmic movements, most frequently involving head and upper body |
| Tremor | Rhythmic oscillation about a central point or position, involving one or more body parts |
When approaching a patient with a movement disorder, it is helpful to address some key questions:
The Role of the Basal Ganglia in Movement Disorders
The basal ganglia are subcortical structures comprising several interconnected nuclei in the forebrain, midbrain, and diencephalon (Figure 68-1). They include the striatum (caudate, putamen, nucleus accumbens), the subthalamic nucleus (STN), the globus pallidus (internal segment [GPi]); external segment [GPe]; and ventral pallidum), and the substantia nigra (pars compacta [SNpc] and pars reticulata [SNpr]). The striatum and STN are the primary input structures of the basal ganglia, receiving excitatory input from cerebral cortex. The GPi and SNpr are the primary output nuclei, sending inhibitory output to thalamus and brainstem targets. Acting through the thalamus, the basal ganglia output influences frontal lobe cortical neurons. By virtue of the inhibitory output from the basal ganglia, conditions associated with destruction of the output nuclei may be associated with unwanted and nonspecific overactivity of thalamocortical and brainstem targets.
Medium spiny striatal neurons contain the inhibitory neurotransmitter GABA and are inhibitory to their targets. In addition, they have peptide neurotransmitters that are co-localized with GABA. Based on the type of neurotransmitters and the predominant type of dopamine receptor they contain, the medium spiny neurons can be divided into two populations. One population contains GABA, dynorphin, and substance P, and primarily expresses D1 dopamine receptors. These neurons project to the basal ganglia output nuclei, GPi, and SNpr, and form the “direct pathway” [Alexander and Crutcher, 1990; Albin et al., 1989; DeLong, 1990]. The second population contains GABA and enkephalin, and primarily expresses D2 dopamine receptors. These neurons project to GPe and form the first limb of the “indirect pathway” [Alexander and Crutcher, 1990; Albin et al., 1989; DeLong, 1990].
The STN receives an excitatory glutamatergic input from many areas of frontal lobes with particularly large inputs from motor areas of cortex. The STN also receives an inhibitory GABA input from GPe. The output from the STN is glutamatergic and excitatory to the basal ganglia output nuclei, GPi and SNpr. This projection forms the second limb of the “indirect pathway” [Alexander and Crutcher, 1990; Albin et al., 1989; DeLong, 1990].
According to the most common model of basal ganglia function, there are several distinct routes through which information flows from the cerebral cortex to the basal ganglia output nuclei. The two most direct are the disynaptic, inhibitory, “direct pathway” from cortex to striatum to GPi and SNpr, and the disynaptic, excitatory, “hyperdirect pathway” [Nambu et al., 2000] from cortex to STN to GPi and SNpr. The “direct pathway” is inhibitory to GPi and SNpr; the “hyperdirect pathway” is excitatory to GPi and SNpr. The “hyperdirect” pathway is the fastest route through the basal ganglia. There are several indirect routes, but the most important is the “indirect pathway” from cortex to striatum to GPe to STN to GPi and SNpr. Organization of these pathways confers a pattern of fast, powerful, and relatively broad excitation via the hyperdirect pathway, and more focused inhibition via the direct pathway (Figure 68-2) [Nambu et al., 2000; Parent and Hazrati, 1993; Mink, 1996]. The indirect pathway confers additional focus.
Output from basal ganglia to thalamocortical circuits appears to be segregated anatomically and possibly functionally. Thalamic targets of GPi and SNpr project, in turn, to frontal lobe, with the strongest output going to motor areas. The basal ganglia motor circuit has a somatotopic organization, with separate representation of different body parts maintained throughout the basal ganglia. There also appears to be relative segregation of motor from nonmotor basal ganglia circuits [Hoover and Strick, 1993].
A popular model of basal ganglia dysfunction in movement disorders was developed in the late 1980s (Figure 68-3) [Albin et al., 1989, 1995; DeLong, 1990]. In simple terms, the model proposes that hypokinetic disorders (e.g., parkinsonism) can be distinguished from hyperkinetic movement disorders (e.g., chorea, dystonia, tics), based on the magnitude and pattern of the basal ganglia output neurons in GPi and SNpr [Wichmann and DeLong, 1996]. Because basal ganglia output neurons are inhibitory to thalamus and the PPA, their function is analogous to a braking mechanism, such that increased activity inhibits and decreased activity facilitates motor pattern generators in cerebral cortex and brainstem [Mink, 1996]. As described above, the anatomic organization of the basal ganglia confers a pattern of focused facilitation and surround inhibition of motor mechanisms in thalamocortical and brainstem circuits (see Figure 68-2). The normal function of this organization is selectively to facilitate desired movements and to inhibit potentially competing movements [Mink, 1996, 2003].
Basal Ganglia Pathophysiology in Movement Disorders
Lesions in the striatum produce variable results that depend in part on the location of the lesion, in part on the lesion mechanism, and in part on what is measured. Lesions in the caudate nucleus most commonly cause behavioral disturbance, such as abulia, but may also cause chorea or dystonia. Lesions in the putamen are likely to produce movement deficits and typically cause dystonia (most common) or parkinsonism [Bhatia and Marsden, 1994].
A unilateral STN lesion is the classic cause of hemiballism, but smaller-amplitude chorea can also result from STN lesions [Carpenter and Carpenter, 1951]. When bilateral, STN lesions cause bilateral chorea or ballism. Most commonly, the chorea or hemiballism resulting from STN lesions involves the lower extremities more than the upper extremities, and can persist for days to months. The intensity of hemiballism usually diminishes over time. Globus pallidus lesions usually involve GPe and GPi, GPe and putamen, or GPi and SNpr. Such lesions can cause dystonia, parkinsonism, or both [Mink, 1996; Bhatia and Marsden, 1994; Heidenreich et al., 1988]. Chorea rarely accompanies globus pallidus lesions. Focal lesions of SNpr can cause involuntary eye movements [Hikosaka and Wurtz, 1985]. Lesions of SNpc dopamine neurons cause depletion of dopamine in the striatum, resulting in parkinsonism, dystonia, or both [Calne et al., 1997].
Focal basal ganglia lesions are uncommon in children. Movement disorders in children are more likely to result from global dysfunction of basal ganglia circuits. The model shown in Figure 68-3 has been used to explain the basis for hypokinetic and hyperkinetic movement disorders, but does not distinguish among the different hyperkinetic movement disorders. However, based on what is known about these disorders and the underlying neural circuitry, specific models have been proposed [Mink, 2003]. Diseases affecting the basal ganglia cause movement disorders that can be understood as failure to facilitate desired movements (e.g., Parkinson’s disease), failure to inhibit unwanted movements (e.g., chorea, dystonia, tics), or both [Albin et al., 1989; DeLong, 1990; Mink, 1996, 2003]. The involuntary movements of chorea, dystonia, and tics differ in important spatial and temporal characteristics that reflect important pathophysiologic differences. More extensive discussion of the neural basis of these disorder is contained in Mink [2003].
Etiology of Movement Disorders in Children
The causes of pediatric movement disorders are extensive. The most common cause of secondary disorders is likely to be cerebral palsy, with a prevalence of 2 per 1000. However, cerebral palsy itself represents a constellation of injuries and symptoms, and there is a wide range of types of injury, localization, and combinations of symptoms [SCPE, 2000]. Cerebral palsy can be associated with almost all forms of childhood movement disorders, and despite the lack of an on-going destructive process, the clinical picture may change during development. The diagnosis and management of cerebral palsy is complex and is discussed in Chapter 69.
Specific types of movement disorder may represent dysfunction of particular localized regions of the central nervous system [Sanger, 2003b]. Ataxia most likely occurs due to injury to the cerebellum or its afferent and efferent pathways [Taroni and DiDonato, 2004]. Bradykinesia most likely occurs with injury to the substantia nigra or striatum, leading to presynaptic or postsynaptic failure of dopaminergic transmission [Albin et al., 1989]. Chorea typically occurs with injury to the subthalamic nucleus, but it also can occur with widespread cerebral injury (e.g., following encephalitis). Dystonia most likely involves injury to the basal ganglia, but cortical or cerebellar abnormalities cannot be excluded as contributors [Hallett, 1998; Sanger, 2003b]. Myoclonus most likely involves cortical, brainstem, or spinal injury to gray matter [Caviness and Brown, 2004]. Localization of tremor depends upon the type, with some forms involving cerebellar or brainstem circuits [Uddin and Rodnitzky, 2003; Jankovic et al., 2004]. Tic disorders probably involve an abnormality of the basal ganglia, but cortical mechanisms may also contribute [Mink, 2001a].
Classification of Childhood Movement Disorders
The first step toward diagnosing and treating a movement disorder is to define the disorder (see Table 68-1). Individual names of movement disorders can refer to neurological signs or to neurological syndromes or diseases, which can cause some confusion. In this section, we will limit the definitions to the neurological signs to which they refer. In subsequent sections, we will present a more complete discussion of the syndromes and diseases in which the different signs are seen, either alone or in combination with other signs.
The most common movement disorders in the pediatric population are tic disorders. The prevalence of tic disorders in schoolchildren may be as high as 25 percent [Snider et al., 2002]. Most tic disorders are not severe, and the level of morbidity is low. Nevertheless, because of the lack of association with other neurological symptoms, it is not unreasonable to consider tic disorders as a primary disorder in idiopathic cases. There are rare exceptions in which tics are due to secondary disorders, such as brain injury or neuroacanthocytosis.
Dystonia is a relatively prevalent disorder in children. This most likely is due to its occurrence in cerebral palsy. Dystonia as the primary symptom in cerebral palsy is less common, but dystonia as a complicating factor, particularly in the upper extremities of children with cerebral palsy otherwise characterized by weakness or spasticity, is probably more common. The incidence and classification of cerebral palsy are discussed in Chapter 69.
Choreoathetosis is a term that has been applied to a specific subset of children with a dyskinetic form of cerebral palsy. This term is not generally used in adults. It is not known whether or not choreoathetosis represents a form of dystonia, chorea, or an entirely different disorder [Turny et al., 2004], but a recent consensus definition [Sanger et al., 2010] suggests that it is most appropriately considered to be a combination of chorea (random-appearing, more rapid movements) and athetosis (slower, flowing movements without intervening periods of rest). Certainly, choreoathetotic cerebral palsy (also commonly termed dyskinetic cerebral palsy) is an easily recognized syndrome, and since many of these children also possess some degree of dystonia as well as chorea, choreoathetosis may simply be the expression of a combination of other movement disorders [Morris et al., 2002a]. Spasticity and weakness are very common in childhood motor disorders, again due to the higher prevalence of cerebral palsy.
Chorea
Chorea describes an apparently random, nonrhythmic, purposeless set of movements of either distal or proximal muscles that appears to flow from one muscle or muscle group to another without any pattern. The causes of chorea and chorealike movements in childhood are summarized in Box 68-1. Chorea occurs at rest and with action, and gives the child a “fidgety” appearance and the inability to remain still. It is associated with motor impersistence (for example, the inability to maintain the tongue extended). Chorea may worsen or improve with voluntary movement, but even very severe chorea may not prevent accurate voluntary movement for some children, suggesting that compensatory mechanisms exist. Many individuals with chorea will incorporate the involuntary movements into a voluntary movement in order to mask the movements. However, the involuntary movements can lead to significant disability, and some children will injure themselves or others due to rapid, ballistic, flinging movements of the arms or legs. Tone is normal or reduced in pure chorea but, in children, chorea may occur in the presence of hypertonic disorders, including dystonia. As with most movement disorders, chorea disappears in sleep but it may be at its worst when the child is drowsy. The term “choreiform” is often used to describe the minimal twitching or “piano-playing” movements seen in many normal young children when arms are extended during the neurological exam. The movements of chorea are briefer than the sustained muscle contractions seen in dystonia and are longer in duration than the “shocklike” movements of myoclonus [Marsden et al., 1983].
Box 68-1 Causes of Chorea in Childhood
Drugs/Toxins
Huntington’s Disease
Huntington’s disease (HD) is transmitted as an autosomal-dominant trait, caused by a trinucleotide repeat expansion of the IT-15 gene on chromosome 4 [Li et al., 1993]. This disorder is characterized by a combination of dystonia, chorea, myoclonus, behavioral abnormalities, ataxia, and, ultimately, dementia. When HD begins in childhood, the typical presentation of the movement disorder is dystonia and rigidity, and not chorea. Seizures are not infrequently the first manifestation of HD in childhood. This has been referred to as the Westphal variant of HD [Quinn and Schrag, 1998; Topper et al., 1998]. The age of onset is earlier for children with a higher number of repeats [Langbehn et al., 2004]. There tends to be amplification of the number of repeats, particularly when it is transmitted from father to child, and therefore in most children with juvenile HD disease is inherited from the father and involves repeat lengths that are significantly higher than those seen in adults.
Diagnosis is based upon identification of the trinucleotide repeat sequence. Although at least 38 repeats are usually required for the occurrence of symptoms in adults, a larger number of repeats would be expected when symptoms present in childhood [Quinn and Schrag, 1998; Langbehn et al., 2004]. Magnetic resonance imaging (MRI) shows atrophy of the heads of the caudate nucleus and, in later stages, generalized cerebral and cerebellar atrophy.
In teenagers, the initial presentation of HD may be with psychiatric illness and, in particular, major depression [Tost et al., 2004]. Dystonia or chorea usually supervenes later. The pathology includes basal ganglia, cerebellar, and cortical degeneration [Vonsattel et al., 1985; de la Monte et al., 1988; Myers et al., 1988]. The origin of the chorea has been hypothesized to be a primary loss of medium spiny cells in the striatum that bear D2-like receptors [Deng et al., 2004]. In children, there may be a relatively symmetric loss of D1-bearing and D2-bearing medium spiny neurons, which could account for the primarily dystonic rather than choreic presentation [Augood et al., 1997; Albin et al., 1990].
Treatment of Huntington’s chorea in adults is typically based upon modification of the chorea or myoclonus. Since the chorea has been presumed to be due to loss of medium spiny neurons in the indirect pathway, neuroleptic medications have been most frequently used in adults [Bonelli and Hofmann, 2004; Bonelli et al., 2004]. Neuroleptics would be expected to compensate partially for early loss of indirect pathway neurons by blocking the inhibitory effects of dopamine and disinhibiting the remaining indirect pathway neurons. There is much less experience in children, for whom dystonia and rigidity may be greater contributors to disability. Other medications that have been used successfully in HD include tetrabenazine [Asher and Aminoff, 1981; Ondo et al., 2002], clonazepam [Thompson et al., 1994], and valproic acid [Grove et al., 2000]. There is currently no treatment that modifies the progression of the disease.
Ataxia-Telangiectasia
Despite the name of this disorder, ataxia is not always the presenting finding, and ataxia may be a much less prominent early symptom than upper-extremity chorea. However, the chorea is usually less disabling than the ataxia. Other symptoms associated with ataxia-telangiectasia include dystonia, which is often proximal [Bodensteiner et al., 1980]. Ataxia-telangiectasia (Louis–Bar syndrome) is most commonly due to mutations in the ATM (ataxia-telangiectasia mutated) gene on 11q22–23 [Chun and Gatti, 2004; Coutinho et al., 2004; Gatti et al., 1988]. Mutation in this gene leads to decreased inhibition of cell cycling in the context of injury to nuclear DNA, and this leads to an accumulation of mutations at known “hot spots,” including translocations between regions on chromosome 7 and 14 known to be involved with immune function [Farina et al., 1994]. Therefore, one of the cardinal features of this disorder is a history of frequent sinopulmonary infections [Centerwall and Miller, 1958], which requires very close attention to pulmonary function and aggressive treatment of pulmonary infections at all ages. In later stages, hematologic malignancy may occur [Stankovic et al., 1998]. Children with ataxia-telangiectasia may be particularly vulnerable to injury from ionizing radiation and certain chemotherapy agents, and this must be taken into consideration in planning any treatment for malignancy.
The cause of the cerebellar degeneration is not known [Farina et al., 1994], but the pathology shows a characteristic loss of Purkinje cells, such that, by late stages in the disease, Purkinje cells are often completely absent. The cause of the chorea is not known, and basal ganglia pathology is minimal. Eye movement disorders are well described, and include an inability to suppress, as well as an inability to initiate, saccades [Lewis et al., 1999; Bundey, 1994]. The inability to initiate saccades leads to a characteristic finding of oculomotor apraxia, in which a child will use thrusts of the head in order to position the eyes on the target. Diagnosis is based upon laboratory testing, including decreased immunoglobulin (Ig) G2 and IgA, abnormalities in T-cell subsets, elevated α-fetoprotein, lymphocyte radiosensitivity, and decreased radiation-induced mitotic suppression [Stray-Pedersen et al., 2004]. Treatment of this disorder is primarily supportive, although there are isolated reports of dystonic symptoms responding to trihexyphenidyl or l-DOPA. The chorea generally is mild and does not require specific treatment. There is no known treatment for the ataxia.
Ataxia-oculomotor apraxia is an autosomal-recessive disorder with similar motor symptoms, caused in some cases by mutations in the aprataxin gene AOA-1 [Tranchant et al., 2003; Shimazaki et al., 2002; Moreira et al., 2001]. This disorder commonly presents with chorea, ataxia, and oculomotor apraxia [Sano et al., 2004]. It is most prevalent in Japan and Europe. AOA-1 does not seem to be involved with cell cycle regulation, and therefore the DNA breakage, immune abnormalities, and malignancies seen in ataxia-telangiectasia do not occur. These patients have hypoalbuminemia [Shimazaki et al., 2002]. AOA-2 is a similar autosomal-recessive disorder due to mutations in Senataxin (SETX). Like ATM, Senataxin is involved with DNA repair, although mutations are not associated with increased radiosensitivity [Suraweera et al., 2009]. Clinical features include ataxia, oculomotor apraxia, peripheral neuropathy, and, more rarely, chorea or dystonia [Criscuolo et al., 2006; Le Ber et al., 2004]. The long-term prognosis of AOA-1 and AOA-2 is considerably better than for ataxia-telangiectasia.
Other Genetic Choreas
Other genetic causes of chorea include familial benign hereditary chorea [Kleiner-Fisman et al., 2003; Breedveld et al., 2002a], which is an autosomal-dominant disorder that, in some cases, is due to mutations in the TITF-1 gene on chromosome 14 [Breedveld et al., 2002b]. Affected family members have normal or near-normal intelligence, and chorea is often the only complaint, although mild truncal dystonia and gait ataxia have been reported [Fernandez et al., 2001]. The severity of chorea can vary between different affected family members. It may be accompanied by hypothyroidism, pulmonary disease, or both in some family members with TITF-1 mutations. Symptomatic treatment may yield some mild benefit.
Neuroacanthocytosis is also a potential cause of chorea [Marson et al., 2003], although classification as a primary or secondary chorea is not universally agreed upon.
Sydenham’s Chorea
Of the secondary causes of childhood chorea, Sydenham’s chorea is the most common [Jordan and Singer, 2003; Jummani and Okun, 2001; Garvey and Swedo, 1997]. It has onset weeks or months after an acute infection with group A beta-hemolytic streptococcus (GABHS) and is one of the cardinal symptoms of rheumatic fever. Symptoms may persist for weeks or months, but chorea almost always resolves spontaneously within 6 months [Jordan and Singer, 2003]. There have been rare reported cases of continued or recurring symptoms [Faustino et al., 2003]. The chorea typically involves the distal musculature, initially of one hand and then of both, with a “piano-playing” pattern, but other forms of chorea, including ballism, have been observed. Large, generalized body movements in some patients previously inspired the term “St. Vitus’ dance.”
Anti-basal ganglia antibodies (ABGA) are found in some children [Church et al., 2002] and it has been hypothesized that production of these antibodies is triggered due to molecular mimicry by streptococcal antigens [Loiselle and Singer, 2001; Kirvan et al., 2003; Singer et al., 2003; Goldenberg et al., 1992]. ABGA can be detected in cerebrospinal fluid [Singer et al., 2003], and anti-streptolysin (ASLO) antibodies can be detected in serum. However, the high prevalence of positive ASLO titers in the general population means that both acute and convalescent ASLO titers must be measured in order to probe for an acute infection. The clinical situation often provides a strong indication for the diagnosis, but if other neurological symptoms are present or there is doubt about etiology, a more complete work-up may be needed, including tests for thyroid function, toxins, metabolic disorders, or encephalitis.
Sydenham’s chorea usually does not require treatment, although the acute streptococcal infection should be treated. All children diagnosed with Sydenham’s chorea, even in cases of isolated chorea, should be treated with penicillin, both acutely for treatment and long-term for prophylaxis, according to American Academy of Pediatrics guidelines [American Academy of Pediatrics, 2006].
Penicillin or an acceptable alternative is effective as secondary prevention of recurrent chorea, but more importantly reduces the likelihood that future GABHS infections will cause carditis and permanent valvular damage. Current recommendations in the United States are for prophylactic treatment until age 21 years, regardless of the age of chorea onset. Valproic acid, carbamazepine, or neuroleptics may have some symptomatic benefit in more severe or persistent cases [Pena et al., 2002; Genel et al., 2002; Kulkarni and Anees, 1996]. Treatment with immune suppressant medication, including corticosteroids or intravenous immunoglobulin preparations, has been studied, but the natural history, with spontaneous resolution of symptoms, makes interpretation of efficacy in open clinical trials difficult [Garvey and Swedo, 1997; Green, 1978; Cardoso et al., 2003]. One randomized, blinded, placebo-controlled study showed that a 4-week, 2-mg/kg daily oral dose of prednisone, followed by a taper, reduced duration of chorea and accelerated the reduction in symptoms. Weight gain was substantial by the end of 2 months, and long-term outcome including recurrence rates was not different between groups [Paz et al., 2006]. There are sometimes associated obsessive-compulsive or behavioral symptoms [Moore, 1996; Wilcox and Nasrallah, 1988], and these symptoms are often managed with selective serotonin reuptake inhibitors.
The prognosis for the movement disorder is excellent, and complete resolution occurs in most cases. Recurrence is rare and is sometimes associated with recurrence of other symptoms of rheumatic fever [Korn-Lubetzki et al., 2004]. There appears to be a higher risk of chorea gravidarum in women with a previous history of Sydenham’s chorea [Korn-Lubetzki et al., 2004].
The rapid onset of Sydenham’s chorea has raised the question of whether an autoimmune mechanism could be responsible for other acute-onset movement disorders. This has led to the hypothesis of pediatric autoimmune neuropsychiatric disorders associated with Streptococcus (PANDAS) [March, 2004; Trifiletti and Packard, 1999]. PANDAS is characterized by an abrupt or explosive onset of tics, obsessive-compulsive behavior, and a movement disorder (usually chorea) that is temporally associated with streptococcal infection [Snider and Swedo, 2004; Chmelik et al., 2004; Pavone et al., 2004; Swedo, 2002]. Symptoms may persist, with a relapsing and remitting course. The existence of this disorder has been debated [Singer and Loiselle, 2003] [Swedo et al., 2004; Kurlan and Kaplan, 2004; Kurlan, 2004], and ABGA have not been shown to be elevated [Singer et al., 2004]. It has not been possible to transmit the disorder passively to animals by inoculation with antibodies from affected humans [Loiselle et al., 2004]. Nevertheless, there have been an increasing number of case reports of explosive onset of neurobehavioral disorders, including tic disorders, chorea, and obsessive-compulsive disorder following streptococcal infections, and therefore the hypothesis merits further investigation. It is not clear that there is any specific treatment, although some authors advocate immune modulation, long-term antibiotics, or tonsillectomy [Green, 1978], [Araujo et al., 2002; Heubi and Shott, 2003; van Toorn et al., 2004; Leonard and Swedo, 2001; Gebremariam, 1999].
Medication-Induced Chorea
Chorea is frequently associated with administration of medications, and any child with chorea needs to be carefully examined for the possibility of iatrogenic causes. In particular, treatment of other movement disorders with anticholinergic medication, such as trihexyphenidyl, can precipitate chorea, even at relatively low doses [Nomoto et al., 1987]. Antiepileptic medications, including carbamazepine and phenytoin, have been associated with precipitation of chorea, and in children with diffuse brain injury, sedating medications can sometimes precipitate chorea.
