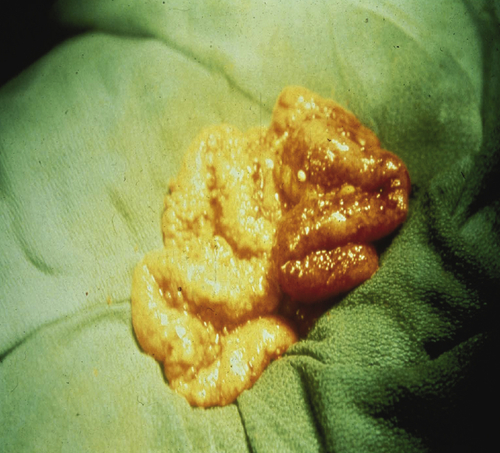
Cellular and Molecular Characteristics of Ovarian Cancer Cells
Heterogeneity of Ovarian Cancers
Cell Proliferation
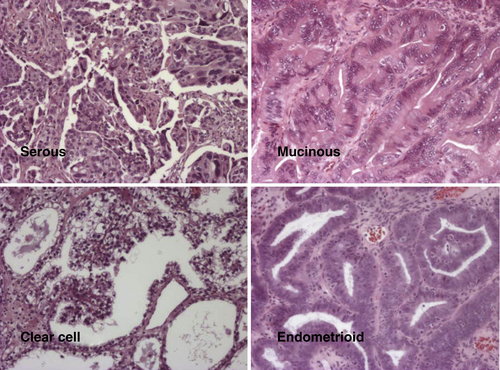
Histotypes
Low and High Grade (Type I and II)
Other Prognostic Subtypes Based on Gene Expression
Immortalization
Genomic Abnormalities in Sporadic Ovarian Cancers
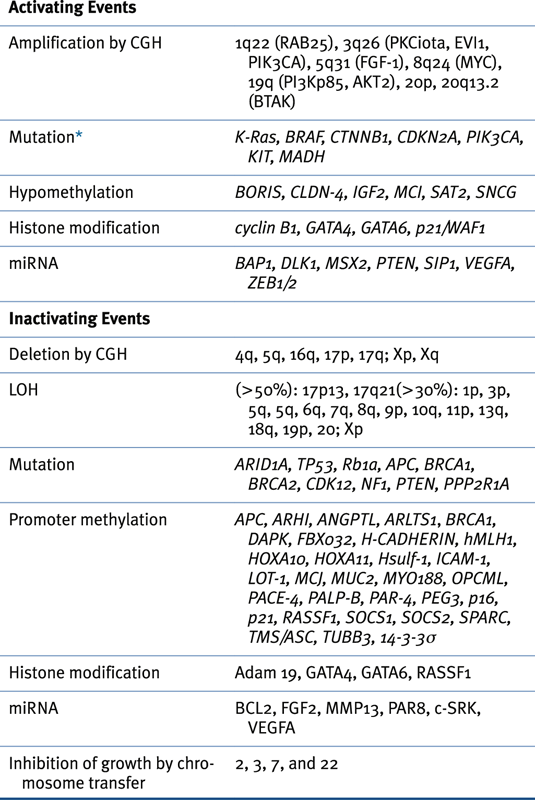
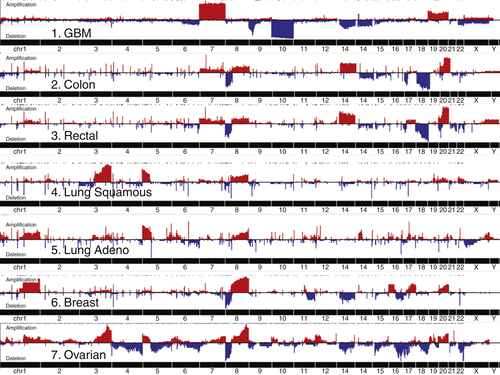
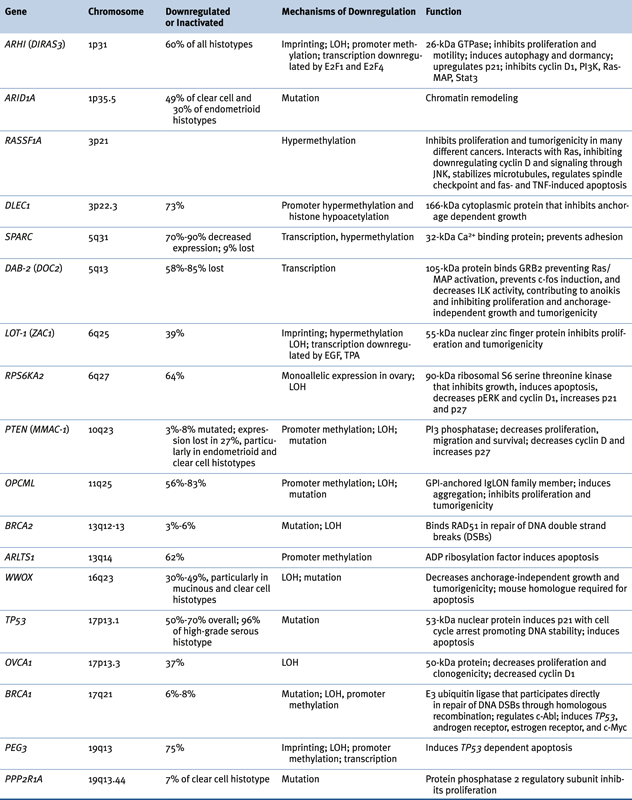
Table 37-3
Oncogenes Associated with Epithelial Ovarian Cancer 14,22
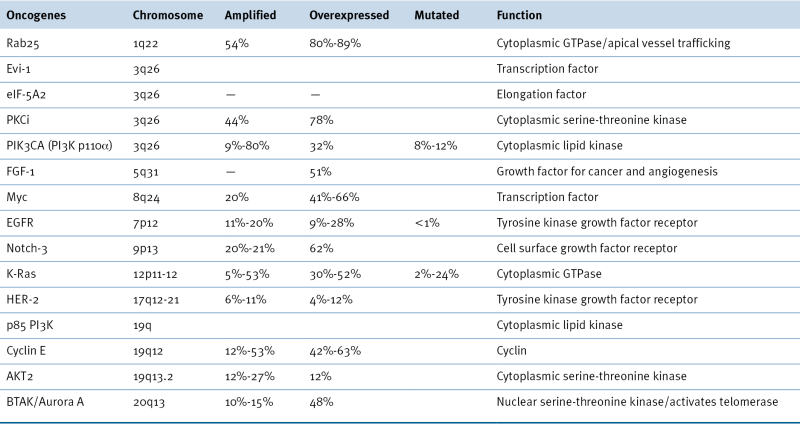
Additional targetable genes with low or high gain of copy number in >20% of high-grade serous ovarian cancers include AKT1, AKT3, CDK2, IL8RB, EPCAM, ERBB3, FGFR2, HDAC4, HSP90AB1, HSP90B1, IGF1, IGFR1, LPAR3, MAP3K6, MAPK15, MAPKAPK2, MAPKAPK5, MECOM, MSTN, MTOR, NCAM1, NOS1, NOS3, PIK3CD, POLB, POLE, RHEB, RICTOR, PPS6KC1, RAPTOR, SKI1, STAT1, STAT4, TERT, TGFB1, TGFB2, TGFBR3, TNFRSF9, VEGFA. 22
Aberrant Signaling
Stem Cells
Animal Models
Interaction of Ovarian Cancer Cells with the Microenvironment
Loss of Adhesion, Epithelial-Mesenchymal Transition, Invasion, and Metastasis
Angiogenesis
Immunologic and Inflammatory Factors
Cytokines and Chemokines
Immunosuppression
Humoral Immune Response
Cellular Immune Response
Conclusion
Acknowledgments
1. Cancer statistics . CA Cancer J Clin . 2013 ; 63 : 11 – 30 .
2. Molecular pathogenesis of epithelial ovarian cancer . In: Mendelsohn J. , Howley P. , Israel M. , Gray J.W. , Thompson C.B. , eds. The Molecular Basis of Cancer . 3rd ed . Philadelphia, Pa : Saunders-Elsevier ; 2008 : 441 – 454 .
3. Shih IeM. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer—shifting the paradigm . Hum Pathol . 2011 ; 42 : 918 – 931 .
4. Lessons from BRCA: the tubal fimbria emerges as the origin of pelvic serous cancer . Clin Med Res . 2007 ; 5 : 35 – 44 .
5. Spectrum of mutation and frequency of allelic deletion of the p53 gene in ovarian cancer . J Natl Cancer Inst . 1993 ; 85 : 1513 – 1519 .
6. Natural history and detection of ovarian cancer . In: Sciarra J.W. , ed. Gynecology and Obstetrics, Vol iv: Oncology . Philadelphia, PA : Harper & Row ; 1988 : 2 .
7. Role of vascular endothelial growth factor in ovarian cancer: Inhibition of ascites formation by immunoneutralization . Am J Pathol . 1998 ; 153 : 1249 – 1256 .
8. Ovarian carcinoma diagnosis . Cancer . 2000 ; 89 : 2068 – 2075 .
9. Clonal origin of epithelial ovarian carcinoma: analysis by loss of heterozygosity, p53 mutation, and X-chromosome inactivation . J Natl Cancer Inst . 1984 : 1793 – 1798 .
10. Targeted therapy for epithelial ovarian cancer: current status and future prospects . Int J Gynecol Cancer . 2003 ; 13 : 701 – 734 .
11. Patterns of gene expression in different histotypes of epithelial ovarian cancer correlate with those in normal fallopian tube, endometrium and colon . Clin Cancer Res . 2005 ; 11 : 6116 – 6126 .
12. Lineage infidelity of epithelial ovarian cancers is controlled by HOX genes that specify regional identity in the reproductive tract . Nat Med . 2005 ; 11 : 531 – 537 .
13. Early events in the pathogenesis of epithelial ovarian cancer . J Clin Oncol . 2008 ; 26 : 995 – 1005 .
14. The biology of ovarian cancer: new opportunities for translation . Nat Rev Cancer . 2009 ; 9 : 415 – 428 .
15. Minireview: Human ovarian cancer: biology, current management and paths to personalized therapy . Endocrinology . 2012 ; 153 : 1593 – 1602 .
16. Rethinking ovarian cancer: recommendations for improving outcomes . Nat Rev Cancer . 2011 ; 11 : 719 – 725 .
17. Recurrent low-grade serous ovarian carcinoma is relatively chemoresistant . Gynecol Oncol . 2009 ; 114 : 48 – 52 .
18. Gene expression profiles of serous, endometrioid, and clear cell subtypes of ovarian and endometrial cancer . Clin Cancer Res . 2005 ; 11 : 6422 – 6430 .
19. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma . Science . 2010 ; 330 : 228 – 231 .
20. ARID1A mutations in endometriosis-associated ovarian carcinomas . N Engl J Med . 2010 ; 363 : 1532 – 1543 .
21. Mutation of FOXL2 in granulosa cell tumors of the ovary . N Engl J Med . 2009 ; 360 : 2719 – 2729 .
22. . Integrated genomic analyses of ovarian carcinoma . Nature . 2011 ; 474 : 609 – 615 .
23. Dissecting PI3Kness: the complexity of personalized therapy for ovarian cancer . Cancer Discov . 2012 ; 2 : 16 – 18 .
24. Personalizing therapy for ovarian cancer: BRCAness and beyond . J Clin Oncol . 2010 ; 28 : 3545 – 3548 .
25. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome . Clin Cancer Res . 2008 ; 14 : 5198 – 5208 .
26. Prognostically relevant gene signatures of high grade serous ovarian cancer . J Clin Invest . 2013 ; 123 : 517 – 525 .
27. Disruption of the retinoblastoma pathway by small interfering RNA and ectopic expression of the catalytic subunit of telomerase lead to immortalization of human ovarian surface epithelial cells . Oncogene . 2007 ; 26 : 1492 – 1498 .
28. Dicer, drosha and outcomes in patients with ovarian cancer . N Engl J Med . 2008 ; 359 : 2641 .
29. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers . N Engl J Med . 2012 ; 366 : 234 – 242 .
30. Minireview: Epigenetic changes in ovarian cancer . Endocrinology . 2009 ; 150 : 4003 – 4011 .
31. NOEY2 (ARHI), an imprinted putative tumor suppressor gene in ovarian and breast carcinomas . Proc Natl Acad Sci USA . 1999 ; 96 : 214 – 219 .
32. Biochemistry and biology of ARHI (DIRAS3), an imprinted tumor suppressor gene whose expression is lost in ovarian and breast cancers . In: Balch W.E. , Der C. , Hall A. , eds. Methods in Enzymology, Regulators and Effectors of Small GTPases. Part D. Ras Proteins . New York, NY : Academic Press ; 2006 : 455 .
33. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells . J Clin Invest . 2008 ; 118 : 3917 – 3929 .
34. SIK2 is a centrosome kinase required for bipolar mitotic spindle formation that provides a potential target for therapy in ovarian cancer . Cancer Cell . 2010 ; 18 : 109 – 121 .
35. The Hippo pathway transcriptional co-activator YAP is an ovarian cancer oncogene . Oncogene . 2011 ; 30 : 2810 – 2822 .
36. Identification of FGFR4 as a potential therapeutic target for advanced-stage high grade serous ovarian carcinoma . Clin Cancer Res . 2013 ; 19 : 809 – 820 .
37. Roles of LPA in ovarian cancer development and progression . Future Oncol . 2009 ; 5 : 1659 – 1673 .
38. Ovarian cancer stem cell markers: prognostic and therapeutic stem cell markers . Cancer Lett . 2012 ; 322 : 1 – 7 .
39. Ovarian surface epithelium at the junction area contains a cancer-prone stem cell niche . Nature . 2013 ; 495 : 241 – 245 .
40. Minireview: Animal models and mechanisms of ovarian cancer development . Endocrinology . 2012 ; 153 : 1585 – 1592 .
41. Cell adhesion in ovarian cancer . Cancer Treat Res . 2009 ; 149 : 297 – 318 .
42. Microenvironmental regulation of ovarian cancer metastases . Cancer Treat Res . 2009 ; 149 : 319 – 334 .
43. Ovarian cancer development and metastasis . Am J Pathol . 2010 ; 177 : 1059 – 1064 .
44. More than a biomarker: CA125 may contribute to ovarian cancer pathogenesis . Gynecol Oncol . 2011 ; 121 : 429 – 430 .
45. A radioimmunoassay using a monoclonal antibody to monitor the course of epithelial ovarian cancer . N Engl J Med . 1983 ; 309 : 883 – 887 .
46. Commentary: CA125 and the detection of recurrent ovarian cancer: a reasonably accurate biomarker for a difficult disease . Cancer . 2010 ; 116 : 2850 – 2853 .
47. Effect of screening on ovarian cancer mortality: the prostate, lung, colon and ovarian (PLCO) cancer screening randomized trial . JAMA . 2011 ; 302 : 2295 – 2303 .
48. Sensitivity and specificity of multimodal and ultrasound screening for ovarian cancer, and stage distribution of detected cancers: results of the prevalence screen of the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS) . Lancet Oncol . 2009 ; 10 : 327 – 340 .
49. Development of a multimarker assay for detection of ovarian cancer . J Clin Oncol . 2010 ; 28 : 2159 – 2166 .
50. Ligand independent activation of c-Met by fibronectin and α5β1 integrin regulates ovarian cancer invasion and metastasis . Oncogene . 2010 ; 30 : 1566 – 1576 .
51. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian cancer . Nat Med . 2006 ; 12 : 939 – 944 .
52. Src activation by β-adrenoreceptors is a key switch for tumour metastasis . Nat Commun . 2013 ; 4 : 1403 .
53. Mechanisms of human tumor metastasis studied in patients with peritoneovenous shunts . Cancer Res . 1984 ; 44 : 3584 .
54. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumour growth . Nat Med . 2011 ; 17 : 1498 – 1503 .
55. Expression of angiogenesis-related genes and progression of human ovarian carcinomas in nude mice . J Natl Cancer Inst . 1998 ; 90 : 447 – 454 .
56. Targeting angiogenesis in ovarian cancer . Cancer Treat Rev . 2012 ; 38 : 272 – 283 .
57. Moving beyond VEGF for anti-angiogenesis strategies in gynecologic cancer . Current Pharm Des . 2012 ; 18 : 2713 – 2719 .
58. A phase 3 trial of bevacizumab in ovarian cancer . N Engl J Med . 2011 ; 365 : 2484 – 2496 .
59. Incorporation of bevacizumab in the primary treatment of ovarian cancer . N Engl J Med . 2011 ; 365 : 2473 – 2483 .
60. Regulation of tumor angiogenesis by EZH2 . Cancer Cell . 2010 ; 18 : 185 – 197 .
61. The inflammatory cytokine tumor necrosis factor-alpha generates and autocrine tumor-promoting network in epithelial ovarian cancer cells . Cancer Res . 2007 ; 67 : 585 – 592 .
62. Paraneoplastic thrombocytosis in ovarian cancer . N Engl J Med . 2012 ; 366 : 610 – 618 .
63. Ovarian cancer creates a suppressive microenvironment to escape immune elimination . Gynecol Oncol . 2010 ; 117 : 366 – 372 .
64. Immunotherapy of ovarian cancer: What’s next? J Clin Oncol . 2011 ; 29 : 925 – 933 .
65. Abagovomab: an anti-idiotypic CA-125 targeted therapeutic agent for ovarian cancer . Immunotherapy . 2011 ; 3 : 153 – 162 .






