[level-membership-for-neurosurgery-category]
CHAPTER 100 Molecular Genetics and the Development of Targets for Glioma Therapy
The modern-day classic Death Be Not Proud by John Gunther chronicled the events surrounding the diagnosis, treatment, and eventual death of his teenage son Johnny, who died of glioblastoma more than half a century ago.1 The anguish he felt as he watched his son suffer was tempered by the belief that someday, perhaps even soon enough for Johnny, there would be a cure or an effective treatment of this horrible disease.
Molecular Genetics of Cancer: The Origins of Cancer Genetics
Our general understanding of the molecular genetics of cancer pathogenesis has advanced significantly in the past 30 years, but the foundations for these discoveries were laid a century ago in 1910, when Peyton Rous, having just graduated from medical school, discovered that cancer could be transmitted from cell culture to animals by using a cell-free ultrafiltrate containing an avian virus. He surmised that certain elements carried by the virus were the cause of cancer! His work lay dormant for many years and was revived in part by Watson and Crick’s groundbreaking discovery that deoxyribonucleic acid (DNA) polymers contained the code necessary for the transmission and interpretation of genetic information.2 Within years of this seminal discovery there was consensus that the cancer-causing element discovered by Rous was in fact v-src, a viral gene capable of transforming infected cells.3 Harold Varmus and Michael Bishop later proved that a mammalian version of the Rous viral cancer gene, called c-src, also caused tumors in mammals. It is now clear that mammalian genes gone awry are the cause of cancer.
Molecular Genetic Tools and the Study of Cancer
DNA Manipulation Techniques in Cancer
Genetic Manipulation in Vivo
Gene Expression in Mice
To verify that a gene defect is the cause of a disease, it is necessary to evaluate the in vivo biologic consequences of genetic alterations. This can be accomplished by using transgenic or knockout mice. Such genetic manipulation of disease-associated genes results in genetically engineered models (GEMs) of disease.4 Genetic manipulation in mice is now fairly sophisticated, and gene expression can be induced or halted in animals with remarkable temporal and cell-specific specificity.
Transgenic Mice
With this technology, human genes can be transferred to animals and their ability to cause disease ascertained. By linking the gene of interest with a tissue-specific promoter, expression can be restricted to a particular organ or cell type. Transgenic mice can also be used to measure the activity of signal transduction pathways or other molecular functions of a cell. For example, p53 activity can be measured by introducing the consensus sequence for p53 binding upstream of the firefly luciferase gene, thereby inducing transcription of the luciferase gene upon p53 binding. The luciferase gene converts a substrate called luciferin into light, and the photons emitted from the reaction can be measured directly by a camera. Emission of light from living organisms is known as bioluminescence. In the p53/luciferase scenario, mice that produce luciferase after p53 binding will convert luciferin into a bioluminescence signal, and this signal serves as an indirect measurement of the DNA binding activity of p53.5 The potential applications of transgene technology in mice are virtually unlimited, and glioma mouse models are beginning to use transgenic animals to measure the proliferation rates of tumors, measure the status of tumor signal transduction pathways, and study a host of other events that occur during the genesis of glioma.
Conditional Knockouts
Conditional knockout mice are created by introducing agents that selectively remove genetically engineered sequences from mouse chromosomes.4 In one such system, known as Cre-lox, the knockout mice are created as described earlier, except that palindromic loxP sites flank the target gene DNA construct. The Cre recombinase protein identifies loxP sites, and the DNA sequence between flanking loxP sites can be excised by the Cre protein to yield a functional knockout. The advantage of this system is that Cre expression can be spatially restricted to certain cell types through the use of tissue-specific promoters. Moreover, Cre constructs may be engineered so that Cre expression is induced only when certain drugs are administered to an animal expressing the Cre transgene. Such temporal and spatial control of knockout genes enables realistic modeling of carcinogenesis because this system can re-create a multistep biologic situation in which specific genes are deleted or overexpressed at specific junctures during tumor progression.
Somatic Cell Gene Transfer
Somatic cell gene transfer involves the transfer of DNA to nongermline cells, often using retroviruses engineered to contain specific genes.4 The concept of somatic cell gene transfer dates back to the discovery of Rous sarcoma virus (RSV) by Rous. Injection of RSV into the organs of several different types of animals results in the formation of tumors because of transfer of the v-src oncogene to somatic cells. However, RSV-based somatic cell gene transfer has some serious disadvantages: (1) there is no control over the type of cells infected by the virus and (2) the use of RSV somatic cell gene transfer allows the examination of only one oncogene.
One somatic cell gene transfer system that obviates these problems is the RCAS/tv-a system.4 This system combines transgene technology with viral infection to achieve cell-specific, gene-specific somatic cell transfer of a number of different oncogenes. Replication-competent avian leukemia virus (ALV) family splice acceptor (RCAS) vectors can be engineered to contain specific genes. Moreover, the RCAS virus specifically infects cells that express the ALV type a receptor tv-a. Thus, in transgenic animals engineered to express tv-a only in certain cell types, RCAS vectors can be used to deliver oncogenes in a cell type–specific manner.
DNA Hybridization Techniques and Array Platform Technologies
Fluorescence In Situ Hybridization
Cancer, especially in its most malignant forms, progresses by accumulating multiple errors in the genome of affected organisms. Many of the genetic errors occurring in cancer specifically affect genes that normally function to repair damaged DNA. As a result of errors in DNA damage response mechanisms, the entire genome of an affected organism becomes prone to error, thereby leading to a phenomenon called genomic instability. One consequence of genomic instability is that damaged genomic DNA is prone to single- and double-strand breaks. At a chromosomal level, these breaks can result in very large losses, gains, or translocations of chromosomal regions. These chromosomal areas encode genes or regulatory elements of genes involved in cancer initiation and progression, and identification of these abnormal chromosomal regions is important for a general understanding of cancer biology. As a consequence, cytogenetics, which encompasses the study of chromosomal structure, has become a central part of cancer biology.6
Comparative Genomic Hybridization Arrays
Although FISH has significantly advanced our progress in the discovery of abnormal target genes and chromosomes, the technique is imperfect for solid tumors in that it requires preparation of tumor metaphase chromosomal spreads that do not differ markedly from normal tissue. Because advanced solid tumors acquire many chromosomal abnormalities, it is often impossible to interpret tumor chromosomal spreads with accuracy inasmuch as chromosomes in solid tumors are often altered beyond recognition. To combat this problem, a variant of FISH called comparative genomic hybridization (CGH) was invented in the early 1990s.6 CGH obviates the need for chromosome spreads by using differentially labeled test and reference genomic DNA oligonucleotide samples, which can then be applied onto genomic DNA microarrays. CGH arrays permit unprecedented ability to evaluate chromosomal abnormalities in cancer. In particular, this technique is especially sensitive for the identification of changes in DNA copy number, such as the increases in gene copy number that occur at the epidermal growth factor receptor (EGFR) locus in a significant number of glioblastomas.
Single Nucleotide Polymorphism Genotyping Arrays
Single nucleotide polymorphism (SNP) analysis is a powerful molecular genetic tool capable of identifying changes in DNA copy number, point mutations, and LOH.6 SNPs are nonfunctional point mutations in the genome that occur at a frequency of about 1% in the human genome. They occur at a frequency of one SNP every few hundred base pairs of genomic DNA. Because many SNPs have been sequenced, they can be used to compare the haplotypes of cancer genomes with other nontumor DNA from the same individual. Moreover, because the genomic location of SNPs is known, changes in the SNP genotype of tumor DNA identify genomic regions that can be further analyzed by PCR to detect precise DNA aberrations (point mutations, deletions, insertions, and so on) that underlie the changes in SNP haplotype. Furthermore, because of the availability of microarray platforms, up to 500,000 SNPs can be analyzed in a single experiment, thus allowing excellent genomic resolution.
Gene Expression Arrays
CGH, FISH, and SNP techniques identify changes in genomic DNA. Although these platforms can be used to make inferences about gene expression, they do not provide direct data about the transcription of these genes from DNA to messenger RNA (mRNA). The importance of studying changes in gene expression is aptly represented by the expression pattern of vascular endothelial growth factor (VEGF) during the genesis of glioma. Although the VEGF gene is rarely mutated in glioblastoma multiforme (GBM), VEGF mRNA and protein levels are often markedly elevated, which leads to an increase in tumor angiogenesis and virulence.7
Applying Molecular Genetic Tools to Glioma Analysis
Phillips and colleagues recently used both gene expression and CGH profiling of high-grade gliomas to explore genes that correlate with patient survival.8 Using gene expression microarrays, they identified three gene clusters that correlate with patient survival. Consistent with these findings, CGH analysis of patients with shorter survival showed frequent loss of the phosphatase and tensin homologue from chromosome 10 (PTEN) tumor suppressor locus and gains at the EGFR and phosphatidylinositol-3′-kinase (PI3K) oncogene loci in comparison to patients with longer survival.
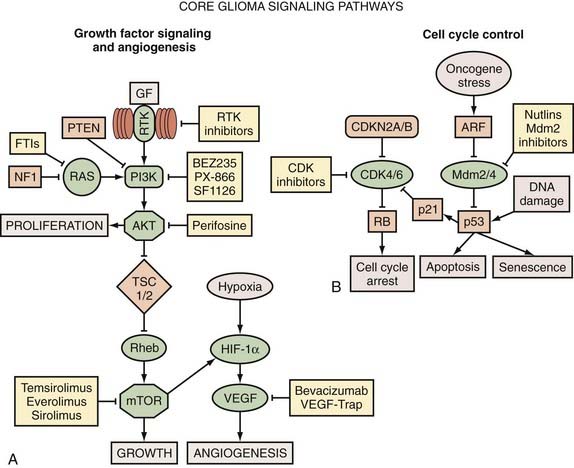
Perhaps the most comprehensive single genetic analysis study of human gliomas to date comes from the efforts of The Cancer Genome Atlas (TCGA), a national initiative whose goal is to identify and catalogue the wide array of genetic aberrations that cause cancer.9 TCGA uses a number of genetic tools, including CGH, SNP genotyping, PCR genotyping, gene expression arrays, microRNA profiling, and DNA methylation arrays, to characterize a cancer specimen. Using 206 GBM samples in a pilot study, the group has identified hitherto unknown genetic aberrations that probably contribute to glioma genesis. In particular, they identified novel deletions of NF1 and PARK2 in glioma samples and characterized NF1 (neurofibromin 1) as a frequent glioma tumor suppressor gene. Beyond discovering individual genetic aberrations in glioma, TCGA characterized a number of systematic, nonrandom patterns that typify genetic aberrations in glioma. For example, the study revealed a tendency for GBM to acquire single mutations in core signaling pathways rather than multiple mutations in a single pathway (Fig. 100-1). One prediction of this model is that aberrations in the receptor tyrosine kinase (RTK) pathway, to use one example, appear to relieve the pressure for accumulating additional RTK pathway aberrations but do not relieve the pressure to acquire errors in the p53 tumor suppressor pathway or in cell cycle progression pathways. This important finding supports the idea that adequate treatment of GBM may require combinations of drugs that simultaneously target components of the three core GBM signaling pathways: growth factor receptor signaling, cell death/apoptosis, and cell cycle progression. In addition to these core signaling pathways, gliomas often use additional proangiogenic and drug efflux mechanisms. Pharmacologic manipulation of these pathways, in addition to the core glioma pathways, might be a necessary part of the treatment regimen for high-grade gliomas.
Other studies, using molecular genetic techniques similar to those used in TCGA, corroborate many of the findings identified in TCGA and a number of other studies of glioma genetics. Parsons and colleagues used direct sequencing, DNA copy number analysis, and gene expression studies to identify mutations in the core pathways just discussed.10 In addition to identifying mutations in these core glioma pathways, they discovered mutations in isocitrate dehydrogenase 1 (IDH1), a gene previously not associated with gliomas, that predicted better overall survival for patients harboring these mutations. Moreover, mutations in IDH1 tend to segregate with favorable GBM characteristics such as younger age and secondary GBM. IDH1 is an enzyme that selectively catalyzes the oxidative decarboxylation of isocitrate to 2-oxoglutarate. The fact that IDH1 is an enzyme raises the intriguing possibility that pharmacologic inhibition could re-create the effects of IDH1 mutations. However, it should be noted that the relationship of IDH1 mutations to the catalytic function of the protein is currently unknown. Because IDH1 is selectively mutated in less aggressive GBMs, elucidation of the biologic function of IDH1 could provide novel therapeutic targets that convert aggressive high-grade gliomas into more differentiated glioma types, such as that found in younger patients. IDH1 mutation and methylguanine methyltransferase (MGMT) promoter methylation are the only individual genetic alterations that predict improved survival in GBM. Overall, these studies highlight recent progress in the discovery of new glioma-associated genes and lay the groundwork for drug discovery.
Cellular and Molecular Biology of Gliomas
Growth Factors, Receptor Tyrosine Kinases, and Their Signal Transduction Pathways
The most common growth factor aberrations in glioma involve the overproduction of platelet-derived growth factor (PDGF), VEGF, hepatocyte growth factor (HGF), transforming growth factor-α (TGF-α), and TGF-β7 (Table 100-1). These growth factors show increased expression at the mRNA and protein levels, but only rare alterations at the genomic level. Growth factors are secreted by tumor cells and surrounding tissue, where they act in a paracrine and autocrine manner to increase the growth and proliferation of tumor cells and, especially in the case of VEGF and PDGF, induce vascular proliferation in the tumor microenvironment. PDGF is also a powerful glioma oncogene; in animal models, PDGF is sufficient to drive gliomas that are phenotypically indistinguishable from human gliomas.13
| TYPE OF ALTERATION | INCIDENCE (%)* |
|---|---|
| DNA Amplifications(%) | |
| EGFR | ≈20-40 |
| CDK4/6 | ≈14-20 |
| MDM2/4 | ≈11-20 |
| PDGFRA | ≈13 |
| MET | ≈4-20 |
| AKT | ≈2 |
| Homozygous Deletions | |
| CDKN2A/B/C | ≈50 |
| PTEN | <10 |
| NF1 | <10 |
| PARK2 | <10 |
| RB1 | <10 |
| TP53 | <10 |
| Inactivating Mutations | |
| TP53 | ≈35 |
| PTEN | ≈15-26 |
| RB1 | ≈8-10 |
| Activating Mutations | |
| PIK3CA/PIK3R1 | ≈15-18 |
| EGFR† | ≈14-21 |
| ERBB2 | 8 |
| Other Mutations | |
| IDH1 | 11 |
| Gene Expression Changes‡ | |
| VEGF | Overexpressed |
| Survivin | Overexpressed |
| SPARC | Overexpressed |
| Osteopontin (Spp1) | Overexpressed |
| CD44 | Overexpressed |
| IGFBP2 | Overexpressed |
| ITGAV | Overexpressed |
| HGF | Overexpressed |
| PDGFA/B | Overexpressed |
| TGF-α | Overexpressed |
Please see references 7 and 9–12.
* As a percentage of all glioblastoma samples.
† Epidermal growth factor receptor (EGFR)-activating mutations are predominantly EGFRvIII mutations.
‡ Gene expression changes mainly determined by gene expression microarrays.
Growth factors work by extracellular binding of growth factor receptors, which then transmit growth signals to the intercellular compartment. Many growth factor receptors come in the form of RTKs, membrane-bound receptors with cytoplasmic kinase domains. After binding ligands such as PDGF, RTKs dimerize and then become phosphorylated at multiple cytoplasmic residues, thereby enabling binding of adapter proteins and subsequent signal transduction. Unlike their ligands, RTKs display a high frequency of genomic abnormalities in glioma samples. Foremost among these alterations are mutations or amplifications of EGFR, or both, which is altered in approximately 50% of GBMs.9,10 A common EGFR variant, the ligand-independent and constitutively active EGFRvIII variant, contains a deleted extracellular domain and is frequently found in gliomas.14 The PDGF receptor gene PDGFRα is amplified in close to 15% of gliomas, and there are less frequent genomic abnormalities in ERBB2 (an EGFR family RTK) and MET (the receptor for HGF) (see Table 100-1).9
GBMs exhibit frequent genomic alterations in signal transduction pathways downstream of RTKs. Chromosome 10q, which contains PTEN, is frequently deleted in GBMs and high-grade oligodendrogliomas, thereby leading to unrestrained activation of the AKT pathway. The NF1 gene also frequently contains deletions and inactivating mutations; 18% of GBM samples were either mutated or deleted at the NF1 locus in TCGA’s GBM study.9 GBMs also harbor occasional amplification of AKT (AKT3), and PI3K activity is altered through amplification of its regulatory and catalytic domains, PIK3CA and PIK3R1, respectively.9 The net result of these genetic alterations in the RTK/Ras/PI3K pathway in GBMs is a selective growth/proliferation advantage for tumor cells.
Potential Therapeutic Intervention: RTKs, PI3K, AKT, and Ras
Two EGFR inhibitors, erlotinib (Tarceva, Genentech, San Francisco, CA) and gefitinib (Iressa, AstraZeneca, Wilmington, DE), have been used in glioma clinical trials. Unfortunately, these clinical trials did not show a significant reduction in glioma progression.15 Although these results are disappointing, it is possible that the molecular genetic heterogeneity found in gliomas account for some of the response patterns in individual tumors. Highlighting this point are recent studies suggesting that EGFR and PTEN status predict responses to EGFR inhibitors. In one study, gliomas responding to an EGFR inhibitor were EGFRvIII positive and had wild-type PTEN, whereas nonresponding tumors often displayed PTEN deletions.16 Mechanistically, this pattern of response implies that PTEN tumor suppressor loss activates downstream RTK signaling cascades, regardless of the RTK status. Several other promising RTK inhibitors are currently being evaluated for gliomas.
PI3K pathway inhibitors are now available for the treatment of gliomas. In particular, AKT and mTOR, the downstream effectors of PI3K activation, are receiving considerable attention in glioma treatment strategies. Perifosine (KRX-0401, Keryx Biopharmaceuticals, New York, NY), an oral AKT inhibitor, has shown great promise in mouse GEMs and is now in clinical trials for the treatment of malignant gliomas.17 Rapamycin (sirolimus, Wyeth Pharmaceuticals, Collegeville, PA) and CCI-779 (temsirolimus, Wyeth Pharmaceuticals, Collegeville, PA), both mTOR inhibitors, have shown antiglioma activity in vitro and are now being used in clinical trials.
Cell Cycle Control and Programmed Cell Death
Regulation of the cell cycle and cell fate is critical to normal development. Some mechanisms exist to direct cells to proliferate when appropriate and halt cell division when growth is inappropriate. Other mechanisms instruct cells to terminate when they have suffered extensive damage or when resources are scarce. Several pathways interact to provide cells with control over these important physiologic events, but in cancer, these regulatory controls are often disrupted, which leads to uncontrolled proliferation and inappropriate survival of transformed cells. Cell cycle and cell death regulatory pathways have been extensively investigated, and several critical abnormalities have been identified in gliomas.9
Cell Cycle Abnormalities in Gliomas
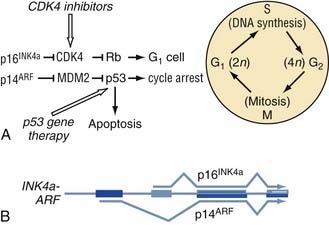
Cell cycle aberrations feature prominently in the genesis of glioma. The most frequent genomic aberration in GBM cell cycle control is the simultaneous deletion of p14ARF and p16INK4. p16INK4, like p21, functions as a CDKI that represses the activity of CDK4. Remarkably, both the p14ARF and p16INK4 genes are encoded by the same genetic locus, CDKN2A, alternative splicing of which gives rise to either p14ARF or p16INK4. Thus, deletion of CDKN2A results in the absence of its two gene products. CDKN2A is deleted in approximately 50% to 60% of all GBMs and is apparently more frequently deleted in primary than in secondary GBMs.9,10 Another genetic locus, CDKN2B, which resides near CDKN2A on chromosome 9p21, encodes for p15INK4 and is almost always deleted along with CDKN2A. The p15 protein inhibits CDK4 and CDK6 function. Inactivation of CDKN2A/CDKN2B or p53 is an important step in glioma genesis because nearly all GBMs feature inactivating mutations at either of these two loci.9,10 In the case of p53, mutations or homozygous deletions occur in approximately 30% to 60% of GBMs. Although previous evidence suggested that p53 alterations were overwhelmingly associated with secondary GBMs, recent data point to an important role for wild-type p53 in the prevention of primary GBMs as well. In addition to these three major tumor suppressors, RB1, the gene for the Rb protein, is homozygously deleted in about 10% of all GBMs.9 Thus, it is clear that loss of cell cycle tumor suppressor genes is a major mechanism for GBM pathogenesis.
Although cell cycle dysregulation in gliomas occurs mainly as a result of tumor suppressor inactivation, genomic aberrations in cell cycle oncogenes are also found. For example, amplification of CDK4 is present in approximately 10% to 20% of GBMs, and a smaller number of GBMs feature amplification of the CDK6 gene.9,10 In addition to the CDKs, many amplifications at the genomic loci of the MDM2 and MDM4 oncogenes have been reported in GBM (Fig. 100-2).
Programmed Cell Death
The balance of cellular proapoptotic and antiapoptotic signals determines cell death decisions. The Bcl family of proteins includes positive and negative regulators of the apoptosis process, with Bcl-2, Bcl-x, and Bcl-xl serving as antiapoptotic members. The proapoptotic function of p53 is carried out by its transcriptional target genes, many of which belong to the proapoptotic arm of the Bcl family. In particular, Bax, p53 upregulated modulator of apoptosis (PUMA), Bak, and NOXA are four Bcl family proteins whose overexpression, induced by p53, causes termination of cells by apoptosis.18 The most frequent p53 mutations in GBM, the “hotspot” mutations, can diminish the ability of p53 to transactivate both p21 and Bax. However, in some GBMs there is emerging evidence that certain p53 mutations specifically incapacitate its proapoptotic functions while sparing its cell cycle arrest functions. In one case of grade II astrocytoma, a germline point mutation in one allele of p53 led to decreased induction of Bax but maintained the ability to induce p21. However, after progression to GBM, the tumor acquired additional point mutations in p53 that rendered the tumor cells incapable of inducing either p21 or Bax. Hence, gliomas gain a selective growth advantage by incapacitating the proapoptotic functions of p53 in addition to its cell cycle arrest properties, and it appears that both functions can hasten the progression of glioma.19
Potential Therapeutic Intervention: Cell Cycle and Apoptosis
Conditional expression of wild-type TP53 in tumor cells has shown remarkable decreases in tumor growth in animal models, thus suggesting that p53 gene therapy can diminish cancer growth. In addition, TP53 gene therapy has been applied successfully to glioma cells in culture. As such, the idea of gene therapy held tremendous promise for the treatment of gliomas in the 1990s; however, clinical trials conducted earlier this decade involving adenoviral transfer of the p53 gene have been disappointing.20 The failure of tumor suppressor gene therapies may be purely technical, and better success may be attained if higher rates of viral infection are achieved in tumor cells. One alternative approach for targeting tumor cells would be to eradicate them by using replicative viruses that selectively replicate in cells lacking certain tumor suppressors.
Glioma Cancer Stem Cells and Resistance to Treatment
Overview
Until recently, the prevailing view was that cancer cells are simply terminally differentiated cells that acquire mutations and the capacity for dysregulated growth. A more recent model of carcinogenesis, the CSC model, has begun to challenge this older paradigm. The CSC model argues that the most tumorigenic of all cancer cells are those that resemble normal stem cells. Thus, the CSC is a type of multipotent tumor cell capable of giving rise to the more differentiated populations in the bulk of the tumor. In human gliomas, some evidence suggests that certain glioma CSCs can be identified by their concurrent expression of the stem cell marker nestin and the transmembrane glycoprotein CD133. CD133+ cells isolated from GBMs are capable of generating GBMs in xenograft models.21 Moreover, although 100,000 CD133− cells were incapable of generating xenograft GBMs, only 100 CD133+ cells were required to produce GBMs that phenocopy the original human GBM. This evidence suggests that at least in a subset of GBMs, CD133+ cells may be tumor-initiating CSCs.
Whether glioma CSCs originate from normal neural stem cells or whether they represent the acquisition of an undifferentiated character by mature brain cells is currently unknown. What is known from genetic analysis of GBMs is that they display a significant increase in gene products usually associated with normal neural progenitor and stem cells. Proteins such as Olig2, nestin, and CD133, which are usually associated with immature stem cells, are enriched in GBM. Indeed, a subset of GBM, the proneural group, can be classified primarily by its ability to produce genes normally associated with neuronal progenitor cells.8 The similarities between glioma CSCs and normal stem cells create a quandary: how do we develop therapies that target the stem-like properties of GBM without destroying our own normal neural stem cells? The answer to this question might be to target GBM CSCs where they live, in the perivascular niche of aggressive brain tumors.
Cancer Stem Cells, the Perivascular Niche, and Radioresistance
Normal neural stem cells reside in a stem cell niche in the subventricular zone (SVZ) of the lateral ventricles. Here, information from the microenvironment and surrounding vasculature provides survival and other cues to the resident stem cells. Perhaps the most intriguing property of SVZ stem cells is their close proximity to a vascular plexus enveloping the entire SVZ. Not surprisingly, GBM and medulloblastoma CSCs also reside next to blood vessels (Fig. 100-3).11 This perivascular property of CSCs implies that tumor vascular proliferation provides critical cues to CSCs and probably helps them maintain their multipotent status.
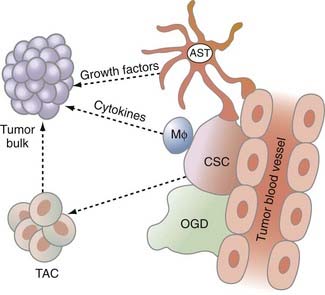
FIGURE 100-3 The glioma tumor microenvironment. Chemoresistant and radioresistant cancer stem cells (CSCs) exist in a perivascular niche with astrocytes (ASTs), macrophages (MΦ), and oligodendrocytes (OGDs). CSCs give rise to transit-amplifying cells (TACs) in the tumor, which then give rise to the tumor bulk. Please see Gilbertson and Rich.11
Stem-like cells, in addition to possessing high tumorigenic potential, are also emerging as the population of tumor cells with the highest resistance to current GBM treatment regimens. Recent evidence suggests that CD133+ xenograft GBMs are more resistant to radiation than are their CD133− counterparts. According to one study, the radiation resistance mechanism of CD133+ cells involves activation of the DNA damage checkpoint kinases Chk1 and Chk2. These kinases are involved in postreplication DNA damage repair. During glioma formation, Chk1- and Chk2-induced DNA damage repair might obviate fatal DNA damage in tumor cells and promote survival despite the genotoxic stress induced by radiation.22
Another radiation resistance mechanism used by stem-like cells is to temporarily arrest proliferation after radiotherapy because rapidly proliferating cells are the primary targets of radiation-induced cell death. In medulloblastomas, there is evidence that CSCs, residing in the tumor perivascular niche, briefly stop proliferating. These nestin-expressing cells respond to radiation by inducing cell cycle arrest, but unlike cells from the bulk of the tumor, these cells do not die and instead reenter the cell cycle 72 hours after irradiation.23 Whatever the mechanisms for their radiation resistance, it is likely that certain undifferentiated cells survive radiation and contribute to repopulation of the tumor bulk, thus highlighting the importance of a therapeutic approach that targets them.
Glioma Angiogenesis and Invasion
Angiogenesis
Without a blood supply to carry oxygen and nutrients to rapidly proliferating cells, tumors would essentially be self-limited. During early neoplastic growth, the same capillaries that serve surrounding normal tissues feed tumor cells. As tumors grow and require additional blood supply for survival, they send out signals to neighboring vessels that instruct endothelial cells to proliferate and form new vessels. The endothelial cells that make up these blood vessels are not neoplastic themselves; they simply respond to recruitment messages from the tumor cells. Angiogenesis is the process by which tumor cells and endothelial cells communicate to give rise to new blood vessels. Not surprisingly, the language of this communication requires growth factor–growth factor receptor interactions and signal transduction. Both tumor cells and endothelial cells produce a number of positive regulators of angiogenesis, including VEGF, a powerful angiogenic growth factor that was briefly discussed earlier. In addition to VEGF, other proangiogenic factors such as basic fibroblast growth factor (bFGF), interleukin-6 (IL-6) and IL-8, hypoxia inducible factor-1α (HIF-1α), and members of the angiopoietin family are produced by tumors. At the same time, tumor cells often decrease the production of negative regulators of angiogenesis, especially thromobospondins, certain interferons, and endostatins.12
Malignant gliomas frequently exhibit a so-called angiogenic switch that occurs during the transition from low-grade to high-grade tumors and is marked by intense proliferation of blood vessels in the tumors. One major regulator of angiogenic switch is HIF-1α, which is induced by hypoxic regions of the tumor and serves as a transcription factor for many angiogenic targets, especially VEGF and VEGF receptors (VEGFRs).12 VEGFRs are RTKs and, on binding VEGF, induce a series of signal transduction pathways leading to endothelial cell proliferation, increases in vascular permeability, and promotion of tumor cell invasion. Other events occurring during the transition from low-grade to high-grade tumor, such as loss of the PTEN tumor suppressor, activation of the PI3K pathway, activation of Ras, and activation of other RTKs, promote vascular proliferation via VEGF-VEGFR–independent mechanisms. The importance of tumor angiogenesis is now being reviewed anew, especially given the relationship of CSCs to perivascular regions in most GBMs.
Invasion
At a molecular level, gliomas produce a large number of proteins that function to aid glioma cell propagation from the tumor bulk to more distant sites within the brain. GBMs use cell adhesion molecules whose function is to interact with the ECM, extracellular proteases that break down the ECM, and promigratory/cytoskeletal proteins that help cells migrate from one region of the brain to the next. Chief among these proinvasion molecules is the cell adhesion receptor CD44. CD44 is a transmembrane glycoprotein for the ECM components osteopontin (OP) and hyaluronic acid (HA).12 OP and HA binding of CD44 causes cells to become more motile by rearranging the actin cytoskeleton and forming cell protrusions known as lamellipodia. Other transmembrane proteins in glioma cells, particularly those of the integrin family, induce cytoskeletal rearrangements and increase cell motility on binding ECM components. CD44, OP, HA, and members of the integrin family are significantly upregulated in gene expression studies of GBM.
Invasive GBM cells must overcome an ECM that is normally restrictive to cell migration. To achieve significant tumor cell migration, GBMs engage in an active ECM remodeling program that uses ECM proteases. Four protein families are particularly important for glioma-induced ECM remodeling: the zinc-dependent matrix metalloproteinase (MMP) family, a family of cysteine proteases called cathepsins, the α disintegrin and metalloproteinase (ADAM) family, and the urokinase-type plasminogen activator receptor (uPAR). These proteins all play important roles in degrading protein components of the ECM, which allows the ECM to loosen and thereby enhances glioma cell migration through the ECM. Glioma gene expression studies confirm the hyperactivity of these pathways in GBMs, and the levels of these genes show positive correlation with glioma grade.12
Potential Therapeutic Intervention: Angiogenesis and Invasion
Given the close physiologic relationship between angiogenesis and invasion, it is hard to interrupt one of these processes without affecting the other. For example, because integrins are expressed on both endothelial cells and tumor cells, treatments aimed at integrins will probably disrupt the interaction of both cell types with the ECM. In the case of endothelial cells, angiogenesis would be inhibited, whereas tumor cells treated with anti-integrin therapies should have limited invasive and proliferative properties. Cilengitide (Merck, Whitehouse Station, NJ), an αvβ3/5 integrin inhibitor, has shown promising results in early clinical trials of malignant glioma.12 Because αvβ3 and αvβ5 integrin are expressed on both tumor and endothelial cells, it is possible that the antineoplastic mechanism of the molecule is related to inhibition of tumor cell migration, endothelial cell migration, and endothelial cell proliferation. Many other agents that specifically target microenvironment factors in malignant gliomas are being developed, and these agents will be an important part of integrated approaches to the treatment of glioma.
Genetically Engineered Models of Glioma and Preclinical Trials
Several glioma GEMs are currently in use. Detailed descriptions of each of these models is beyond the scope of this chapter, but this topic has been reviewed in detail elsewhere.4 GEMs require the manipulation of gene expression in vivo by using knockout or transgenic mice, somatic cell gene transfer, or combinations of such manipulations to achieve carcinogenesis in mice. Overexpression of oncogenes can be achieved by somatic cell gene transfer or by mice that are transgenic for certain oncogenes. Conversely, loss of tumor suppressor genes can be modeled by using mice that exhibit germline knockout of tumor suppressor genes or mice in which tumor suppressor loss can be conditionally induced. GEMs are important because they allow appropriate in vivo modeling of gliomas and also because they can be used for preclinical trials of potential glioma therapies. GEMs also appear to have advantages over xenograft models, particularly in view of the fact that GEMs more effectively recapitulate the resistance to therapy seen in human gliomas more effectively than xenografts do.4
Astrocytoma Models
GEMs confirm the importance of many genetic alterations seen in human astrocytoma. Central nervous system–specific overexpression of oncogenes such as RTKs and their downstream signal transduction partners can result in astrocytoma. Fourteen percent of transgenic mice expressing the RSV oncogene v-src under control of the glial fibrillary acidic protein (GFAP) promoter eventually form astrocytomas of the brain and spinal cord. Similarly, in mice that express a constitutively active form of Ras from the GFAP promoter, malignant astrocytomas develop by 9 months.4
GEMs also capture the effects of tumor suppressor loss in astrocytoma. Compound heterozygous loss of Nf1 and TP53 causes high-grade astrocytomas whose penetrance is partially dependent on the mouse strain. Similarly, conditional knockout of Nf1 in p53-null mice produces malignant astrocytomas that appear to arise from the SVZ.24 Loss of cell cycle pathway tumor suppression can also result in high-grade astrocytoma. For example, expression of the pRb-inactivating SV40 T antigen T121 in GFAP-positive cells leads to high-grade astrocytomas.
The RCAS/tv-a system is capable of inducing astrocytomas with histologic fidelity to human astrocytoma and relatively short latency. Astrocytomas can be formed in mice with activated Akt and Ras (RCAS-Akt Myr Δ 11-60 and RCAS-RasG(12)D) delivery to nestin-positive, tv-a–positive (Ntv-a) mice.25 Combined RCAS-RasG12 with loss of the PTEN tumor suppressor also results in astrocytoma in mice. Interestingly, these same RCAS-oncogene combinations in GFAP-positive, tv-a–positive (Gtv-a) mice did not result in astrocytoma formation, thus suggesting that specific cell types require specific genetic alterations for astrocytoma formation.
Oligodendroglioma Models
Human oligodendrogliomas express high levels of PDGF ligand and PDGFRα. Therefore, it is not surprising that overexpression of PDGF ligand results in oligodendrogliomas in a number of mouse models. One well-studied GEM of PDGF overexpression in gliomas is the RCAS/tv-a system, where RCAS-PDGFB–injected Ntv-a mice acquire oligodendrogliomas with high penetrance and a latency of 3 weeks to a few months, depending on the tumor suppressor loss status of the mice.4 Notably, RCAS-PDGFB–infected Gtv-a mice result in mixed oligoastrocytomas, again reflecting cell-type and gene-alteration specificities in the formation of different glial tumors.
EGFR activation also has a role in the formation of oligodendrogliomas. GEMs of oligodendroglioma can be created in mice that have activated viral EGFR homologue v-erbB expression driven by the glial promoter S100β. These mice usually form low-grade oligodendrogliomas. However, like RCAS/tv-a oligodendrogliomas, tumor grade can be increased by heterozygous deletion of p53 or Ink4a/Arf.4
Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756-760.
Bennett MR. Mechanisms of p53-induced apoptosis. Biochem Pharmacol. 1999;58:1089-1095.
Brandes AA, Franceschi E, Tosoni A, et al. Epidermal growth factor receptor inhibitors in neuro-oncology: hopes and disappointments. Clin Cancer Res. 2008;14:957-960.
Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061-1068.
Chi A, Norden AD, Wen PY. Inhibition of angiogenesis and invasion in malignant gliomas. Expert Rev Anticancer Ther. 2007;7:1537-1560.
Ekstrand AJ, Longo N, Hamid ML, et al. Functional characterization of an EGF receptor with a truncated extracellular domain expressed in glioblastomas with EGFR gene amplification. Oncogene. 1994;9:2313-2320.
Fomchenko EI, Holland EC. Platelet-derived growth factor–mediated gliomagenesis and brain tumor recruitment. Neurosurg Clin N Am. 2007;18:39-58. viii
Fulci G, Ishii N, Maurici D, et al. Initiation of human astrocytoma by clonal evolution of cells with progressive loss of p53 functions in a patient with a 283H TP53 germ-line mutation: evidence for a precursor lesion. Cancer Res. 2002;62:2897-2905.
Gilbertson RJ, Rich JN. Making a tumour’s bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer. 2007;7:733-736.
Gunther J. Death Be Not Proud. New York: Harper Perennial Modern Classics; 1949.
Hambardzumyan D, Becher OJ, Rosenblum MK, et al. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche One pathway, the tumor suppressor following radiation in medulloblastoma in vivo. Genes Dev. 2008;22:436-448.
Hamstra DA, Bhojani MS, Griffin LB, et al. Real-time evaluation of p53 oscillatory behavior in vivo using bioluminescent imaging. Cancer Res. 2006;66:7482-7489.
Hoelzinger DB, Demuth T, Berens ME. Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. J Natl Cancer Inst. 2007;99:1583-1593.
Holland EC. Mouse Models of Human Cancer. New York: John Wiley & Sons; 2004.
Holland EC, Celestino J, Dai C, et al. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000;25:55-57.
Mellinghoff IK, Wang MY, Vivanco I, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012-2024.
Momota H, Nerio E, Holland EC. Perifosine inhibits multiple signaling pathways in glial progenitors and cooperates with temozolomide to arrest cell proliferation in gliomas in vivo. Cancer Res. 2005;65:7429-7435.
Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807-1812.
Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157-173.
Pulkkanen KJ, Yla-Herttuala S. Gene therapy for malignant glioma: current clinical status. Mol Ther. 2005;12:585-598.
Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396-401.
Speicher MR, Carter NP. The new cytogenetics: blurring the boundaries with molecular biology. Nat Rev Genet. 2005;6:782-792.
Stehelin D, Varmus HE, Bishop JM, et al. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature. 1976;260:170-173.
Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171:737-738.
Zhu Y, Guignard F, Zhao D, et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 2005;8:119-130.
1 Gunther J. Death Be Not Proud. New York: Harper Perennial Modern Classics; 1949.
2 Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171:737-738.
3 Stehelin D, Varmus HE, Bishop JM, et al. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature. 1976;260:170-173.
4 Holland EC. Mouse Models of Human Cancer. New York: John Wiley & Sons; 2004.
5 Hamstra DA, Bhojani MS, Griffin LB, et al. Real-time evaluation of p53 oscillatory behavior in vivo using bioluminescent imaging. Cancer Res. 2006;66:7482-7489.
6 Speicher MR, Carter NP. The new cytogenetics: blurring the boundaries with molecular biology. Nat Rev Genet. 2005;6:782-792.
7 Hoelzinger DB, Demuth T, Berens ME. Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. J Natl Cancer Inst. 2007;99:1583-1593.
8 Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157-173.
9 Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061-1068.
10 Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807-1812.
11 Gilbertson RJ, Rich JN. Making a tumour’s bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer. 2007;7:733-736.
12 Chi A, Norden AD, Wen PY. Inhibition of angiogenesis and invasion in malignant gliomas. Expert Rev Anticancer Ther. 2007;7:1537-1560.
13 Fomchenko EI, Holland EC. Platelet-derived growth factor–mediated gliomagenesis and brain tumor recruitment. Neurosurg Clin N Am. 2007;18:39-58. viii
14 Ekstrand AJ, Longo N, Hamid ML, et al. Functional characterization of an EGF receptor with a truncated extracellular domain expressed in glioblastomas with EGFR gene amplification. Oncogene. 1994;9:2313-2320.
15 Brandes AA, Franceschi E, Tosoni A, et al. Epidermal growth factor receptor inhibitors in neuro-oncology: hopes and disappointments. Clin Cancer Res. 2008;14:957-960.
16 Mellinghoff IK, Wang MY, Vivanco I, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012-2024.
17 Momota H, Nerio E, Holland EC. Perifosine inhibits multiple signaling pathways in glial progenitors and cooperates with temozolomide to arrest cell proliferation in gliomas in vivo. Cancer Res. 2005;65:7429-7435.
18 Bennett MR. Mechanisms of p53-induced apoptosis. Biochem Pharmacol. 1999;58:1089-1095.
19 Fulci G, Ishii N, Maurici D, et al. Initiation of human astrocytoma by clonal evolution of cells with progressive loss of p53 functions in a patient with a 283H TP53 germ-line mutation: evidence for a precursor lesion. Cancer Res. 2002;62:2897-2905.
20 Pulkkanen KJ, Yla-Herttuala S. Gene therapy for malignant glioma: current clinical status. Mol Ther. 2005;12:585-598.
21 Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396-401.
22 Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756-760.
23 Hambardzumyan D, Becher OJ, Rosenblum MK, et al. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008;22:436-448.
24 Zhu Y, Guignard F, Zhao D, et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 2005;8:119-130.
25 Holland EC, Celestino J, Dai C, et al. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000;25:55-57.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 100 Molecular Genetics and the Development of Targets for Glioma Therapy
The modern-day classic Death Be Not Proud by John Gunther chronicled the events surrounding the diagnosis, treatment, and eventual death of his teenage son Johnny, who died of glioblastoma more than half a century ago.1 The anguish he felt as he watched his son suffer was tempered by the belief that someday, perhaps even soon enough for Johnny, there would be a cure or an effective treatment of this horrible disease.
Molecular Genetics of Cancer: The Origins of Cancer Genetics
Our general understanding of the molecular genetics of cancer pathogenesis has advanced significantly in the past 30 years, but the foundations for these discoveries were laid a century ago in 1910, when Peyton Rous, having just graduated from medical school, discovered that cancer could be transmitted from cell culture to animals by using a cell-free ultrafiltrate containing an avian virus. He surmised that certain elements carried by the virus were the cause of cancer! His work lay dormant for many years and was revived in part by Watson and Crick’s groundbreaking discovery that deoxyribonucleic acid (DNA) polymers contained the code necessary for the transmission and interpretation of genetic information.2 Within years of this seminal discovery there was consensus that the cancer-causing element discovered by Rous was in fact v-src, a viral gene capable of transforming infected cells.3 Harold Varmus and Michael Bishop later proved that a mammalian version of the Rous viral cancer gene, called c-src, also caused tumors in mammals. It is now clear that mammalian genes gone awry are the cause of cancer.
Molecular Genetic Tools and the Study of Cancer
DNA Manipulation Techniques in Cancer
Genetic Manipulation in Vivo
Gene Expression in Mice
To verify that a gene defect is the cause of a disease, it is necessary to evaluate the in vivo biologic consequences of genetic alterations. This can be accomplished by using transgenic or knockout mice. Such genetic manipulation of disease-associated genes results in genetically engineered models (GEMs) of disease.4 Genetic manipulation in mice is now fairly sophisticated, and gene expression can be induced or halted in animals with remarkable temporal and cell-specific specificity.
Transgenic Mice
With this technology, human genes can be transferred to animals and their ability to cause disease ascertained. By linking the gene of interest with a tissue-specific promoter, expression can be restricted to a particular organ or cell type. Transgenic mice can also be used to measure the activity of signal transduction pathways or other molecular functions of a cell. For example, p53 activity can be measured by introducing the consensus sequence for p53 binding upstream of the firefly luciferase gene, thereby inducing transcription of the luciferase gene upon p53 binding. The luciferase gene converts a substrate called luciferin into light, and the photons emitted from the reaction can be measured directly by a camera. Emission of light from living organisms is known as bioluminescence. In the p53/luciferase scenario, mice that produce luciferase after p53 binding will convert luciferin into a bioluminescence signal, and this signal serves as an indirect measurement of the DNA binding activity of p53.5 The potential applications of transgene technology in mice are virtually unlimited, and glioma mouse models are beginning to use transgenic animals to measure the proliferation rates of tumors, measure the status of tumor signal transduction pathways, and study a host of other events that occur during the genesis of glioma.
Conditional Knockouts
Conditional knockout mice are created by introducing agents that selectively remove genetically engineered sequences from mouse chromosomes.4 In one such system, known as Cre-lox, the knockout mice are created as described earlier, except that palindromic loxP sites flank the target gene DNA construct. The Cre recombinase protein identifies loxP sites, and the DNA sequence between flanking loxP sites can be excised by the Cre protein to yield a functional knockout. The advantage of this system is that Cre expression can be spatially restricted to certain cell types through the use of tissue-specific promoters. Moreover, Cre constructs may be engineered so that Cre expression is induced only when certain drugs are administered to an animal expressing the Cre transgene. Such temporal and spatial control of knockout genes enables realistic modeling of carcinogenesis because this system can re-create a multistep biologic situation in which specific genes are deleted or overexpressed at specific junctures during tumor progression.
Somatic Cell Gene Transfer
Somatic cell gene transfer involves the transfer of DNA to nongermline cells, often using retroviruses engineered to contain specific genes.4 The concept of somatic cell gene transfer dates back to the discovery of Rous sarcoma virus (RSV) by Rous. Injection of RSV into the organs of several different types of animals results in the formation of tumors because of transfer of the v-src oncogene to somatic cells. However, RSV-based somatic cell gene transfer has some serious disadvantages: (1) there is no control over the type of cells infected by the virus and (2) the use of RSV somatic cell gene transfer allows the examination of only one oncogene.
One somatic cell gene transfer system that obviates these problems is the RCAS/tv-a system.4 This system combines transgene technology with viral infection to achieve cell-specific, gene-specific somatic cell transfer of a number of different oncogenes. Replication-competent avian leukemia virus (ALV) family splice acceptor (RCAS) vectors can be engineered to contain specific genes. Moreover, the RCAS virus specifically infects cells that express the ALV type a receptor tv-a. Thus, in transgenic animals engineered to express tv-a only in certain cell types, RCAS vectors can be used to deliver oncogenes in a cell type–specific manner.
DNA Hybridization Techniques and Array Platform Technologies
Fluorescence In Situ Hybridization
Cancer, especially in its most malignant forms, progresses by accumulating multiple errors in the genome of affected organisms. Many of the genetic errors occurring in cancer specifically affect genes that normally function to repair damaged DNA. As a result of errors in DNA damage response mechanisms, the entire genome of an affected organism becomes prone to error, thereby leading to a phenomenon called genomic instability. One consequence of genomic instability is that damaged genomic DNA is prone to single- and double-strand breaks. At a chromosomal level, these breaks can result in very large losses, gains, or translocations of chromosomal regions. These chromosomal areas encode genes or regulatory elements of genes involved in cancer initiation and progression, and identification of these abnormal chromosomal regions is important for a general understanding of cancer biology. As a consequence, cytogenetics, which encompasses the study of chromosomal structure, has become a central part of cancer biology.6
Comparative Genomic Hybridization Arrays
Although FISH has significantly advanced our progress in the discovery of abnormal target genes and chromosomes, the technique is imperfect for solid tumors in that it requires preparation of tumor metaphase chromosomal spreads that do not differ markedly from normal tissue. Because advanced solid tumors acquire many chromosomal abnormalities, it is often impossible to interpret tumor chromosomal spreads with accuracy inasmuch as chromosomes in solid tumors are often altered beyond recognition. To combat this problem, a variant of FISH called comparative genomic hybridization (CGH) was invented in the early 1990s.6 CGH obviates the need for chromosome spreads by using differentially labeled test and reference genomic DNA oligonucleotide samples, which can then be applied onto genomic DNA microarrays. CGH arrays permit unprecedented ability to evaluate chromosomal abnormalities in cancer. In particular, this technique is especially sensitive for the identification of changes in DNA copy number, such as the increases in gene copy number that occur at the epidermal growth factor receptor (EGFR) locus in a significant number of glioblastomas.
Single Nucleotide Polymorphism Genotyping Arrays
Single nucleotide polymorphism (SNP) analysis is a powerful molecular genetic tool capable of identifying changes in DNA copy number, point mutations, and LOH.6 SNPs are nonfunctional point mutations in the genome that occur at a frequency of about 1% in the human genome. They occur at a frequency of one SNP every few hundred base pairs of genomic DNA. Because many SNPs have been sequenced, they can be used to compare the haplotypes of cancer genomes with other nontumor DNA from the same individual. Moreover, because the genomic location of SNPs is known, changes in the SNP genotype of tumor DNA identify genomic regions that can be further analyzed by PCR to detect precise DNA aberrations (point mutations, deletions, insertions, and so on) that underlie the changes in SNP haplotype. Furthermore, because of the availability of microarray platforms, up to 500,000 SNPs can be analyzed in a single experiment, thus allowing excellent genomic resolution.
Gene Expression Arrays
CGH, FISH, and SNP techniques identify changes in genomic DNA. Although these platforms can be used to make inferences about gene expression, they do not provide direct data about the transcription of these genes from DNA to messenger RNA (mRNA). The importance of studying changes in gene expression is aptly represented by the expression pattern of vascular endothelial growth factor (VEGF) during the genesis of glioma. Although the VEGF gene is rarely mutated in glioblastoma multiforme (GBM), VEGF mRNA and protein levels are often markedly elevated, which leads to an increase in tumor angiogenesis and virulence.7
Applying Molecular Genetic Tools to Glioma Analysis
Phillips and colleagues recently used both gene expression and CGH profiling of high-grade gliomas to explore genes that correlate with patient survival.8 Using gene expression microarrays, they identified three gene clusters that correlate with patient survival. Consistent with these findings, CGH analysis of patients with shorter survival showed frequent loss of the phosphatase and tensin homologue from chromosome 10 (PTEN) tumor suppressor locus and gains at the EGFR and phosphatidylinositol-3′-kinase (PI3K) oncogene loci in comparison to patients with longer survival.
Perhaps the most comprehensive single genetic analysis study of human gliomas to date comes from the efforts of The Cancer Genome Atlas (TCGA), a national initiative whose goal is to identify and catalogue the wide array of genetic aberrations that cause cancer.9 TCGA uses a number of genetic tools, including CGH, SNP genotyping, PCR genotyping, gene expression arrays, microRNA profiling, and DNA methylation arrays, to characterize a cancer specimen. Using 206 GBM samples in a pilot study, the group has identified hitherto unknown genetic aberrations that probably contribute to glioma genesis. In particular, they identified novel deletions of NF1 and PARK2 in glioma samples and characterized NF1
[/not-level-membership-for-neurosurgery-category]
