[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Molecular Diagnostics in the Clinical Laboratory
After completion of this chapter, the reader will be able to:
1. Describe the structure of DNA, including the composition of a nucleotide, the double helix, strand orientation, and the antiparallel complementary characteristic.
2. Predict the nucleotide sequence of a complementary strand of DNA or RNA given the nucleotide sequence of a DNA template.
3. Explain the relationship between DNA structure and protein production.
4. Explain the relationship between the cell cycle and tumor progression.
5. Discuss the process of DNA replication, including replication origin, replication fork, primase, primer, DNA polymerase, Okazaki fragments, leading strand, and lagging strand.
6. Determine the appropriate patient specimen required for DNA isolation to identify an inherited or somatic mutation.
8. Explain the purpose of polymerase chain reaction (PCR), reverse transcription PCR, and nucleic acid hybridization.
10. Compare and contrast the methods for detecting amplified target DNA: (1) gel electrophoresis using either ethidium bromide or autoradiography, (2) restriction fragment length polymorphism, and (3) probe hybridization techniques such as Southern blotting.
11. Interpret an agarose gel electrophoresis result for the factor V Leiden mutation test and a nucleic acid hybridization result for a B- and T-cell gene rearrangement test.
12. Describe the principle of real-time quantitative PCR.
13. Discuss the use of real-time PCR for monitoring minimal residual disease.
Case Study*
After studying this chapter, the reader should be able to respond to the following case study:
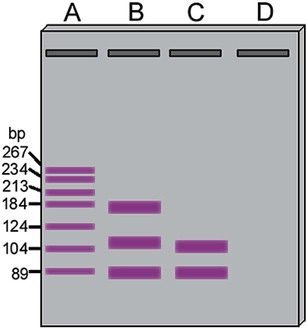
On physical examination, the patient had no evidence of rash or oral ulcers. No petechiae or purpura were noted. He had mild pretibial pitting edema. His right leg measured 36.5 cm at 25 cm distal to the superior aspect of the patella, whereas his left leg measured 33.5 cm in the same location. CBC findings were unremarkable, and both the prothrombin time and activated partial prothrombin time were within the reference ranges. Doppler ultrasonography revealed complete occlusion of the distal superficial femoral vein, anterior tibial vein, and popliteal vein. The diagnosis was DVT without pulmonary emboli. The patient was hospitalized, and a heparin drip was started. The hematologist ordered a factor V Leiden analysis. The initial specimen received was drawn into a red-topped tube. The laboratory scientist requested a redraw using a lavender-topped tube. The patient was still hospitalized, which allowed the collection of a blood specimen in an EDTA tube. Figure 32-1 illustrates the results of the factor V Leiden mutation test initially done by the molecular scientist. The scientist’s supervisor reviewed the gel electrophoresis results and requested that the scientist repeat the analysis. Figure 32-2 represents the repeated analysis.
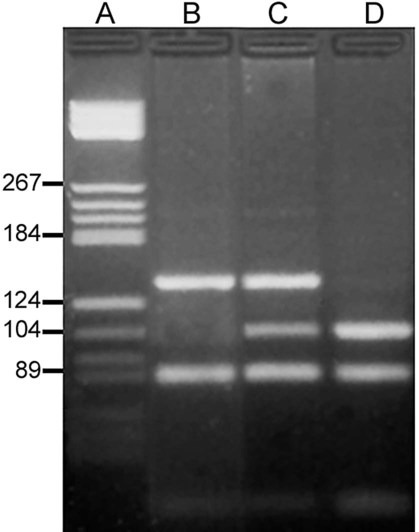
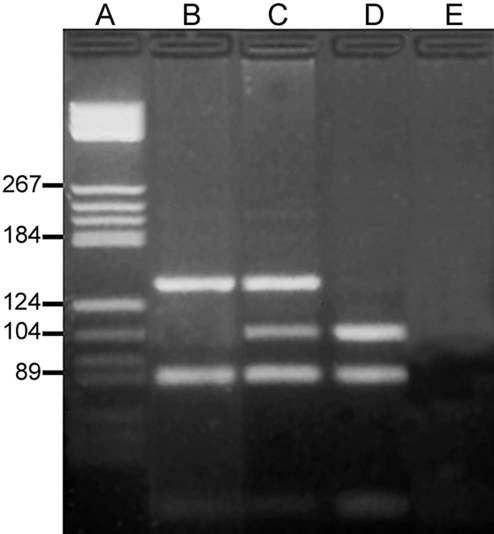
1. What type of specimen is appropriate when analyzing DNA for a hereditary mutation?
2. Examine the first gel electrophoresis result (see Figure 32-1). Are the correct controls present?
3. Examine the second gel electrophoresis result (see Figure 32-2). What band sizes appear in the patient’s sample?
4. What band sizes are expected for an individual who is homozygous for the factor V Leiden mutation, heterozygous for the mutation, and free of the mutation?
5. Why did the laboratory request that the blood sample to be redrawn?
Molecular biology techniques enhance the diagnostic team’s ability to predict or identify an increasing number of diseases in the clinical laboratory. Molecular techniques also enable clinicians to monitor disease progression during treatment and to make accurate prognoses. The short interval required to perform molecular diagnostic tests and analyze their results is an additional positive aspect of this type of testing, resulting in more efficient patient management, especially in cases of infection. The three main areas of hematopathologic molecular testing include detection of chromosomal translocations in hematologic malignancies (Box 32-1) and inherited hematologic disorders (Box 32-2), identification of hematologically important infectious diseases (Box 32-3), and monitoring of minimal residual disease after cancer treatment.
Structure and Function of Dna
The Central Dogma: DNA to RNA to Protein
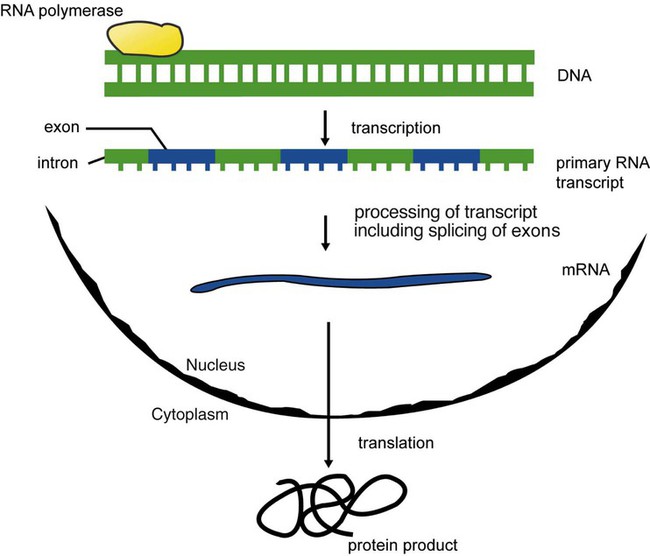
The central dogma in genetics is that information stored in the DNA is replicated to daughter DNA, transcribed to messenger ribonucleic acid (mRNA), and translated into a functional protein (Figure 32-3). This process is essential to carry out cellular functions while preserving a record of the stored information. In eukaryotes, the initial DNA sequence is composed of exons separated by untranslated introns. The introns are enzymatically excised during transcription from DNA to RNA, and the mature mRNA sequence is then translated. Translation is an enzymatic process wherein mRNA three-member base sequences called codons drive the addition of individual amino acids to the growing peptide. The mature protein then carries out its cellular function, which may be structural or may involve recognition, regulation, or enzymatic activity.
The structural units that carry DNA’s message are called genes. The human β-globin gene, part of the hemoglobin molecule, provides a good example of replication and transcription, because it was one of the first sequenced and demonstrates the result of aberrant sequence maintenance. A normal (or wild-type) β-globin gene contains a sequence of bases that code for a β-globin peptide of 146 amino acids. One inherited mutation changes a single DNA base. This is called a point mutation. The mutation occurs in the portion of the sequence that codes for the sixth amino acid of β-globin. The mutation substitutes the amino acid valine for glutamine in the growing peptide. Valine modifies the overall charge, producing a protein that polymerizes in a low-oxygen environment. This leads to sickled erythrocytes, circulatory ischemia, and poor oxygen exchange between blood and tissues.1,2 A mutation in one of the two copies (alleles) of this gene inherited from the parents results in a heterozygous condition, or a sickle cell trait. In a heterozygote, the symptoms of the disease are often unseen or are present only during times of physical stress. If both alleles are mutated, there is overt homozygous sickle cell disease, and the symptoms are severe.
Every active gene is translated. Human somatic cells contain 20,000 to 25,000 genes in 2 meters of DNA.3,4 Significant packing (see Chapter 31) takes place to reduce the volume of the nucleic acid to the size of chromosomes.
DNA at the Molecular Level
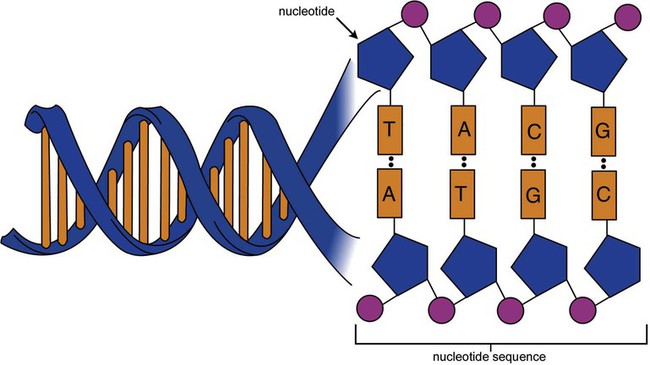
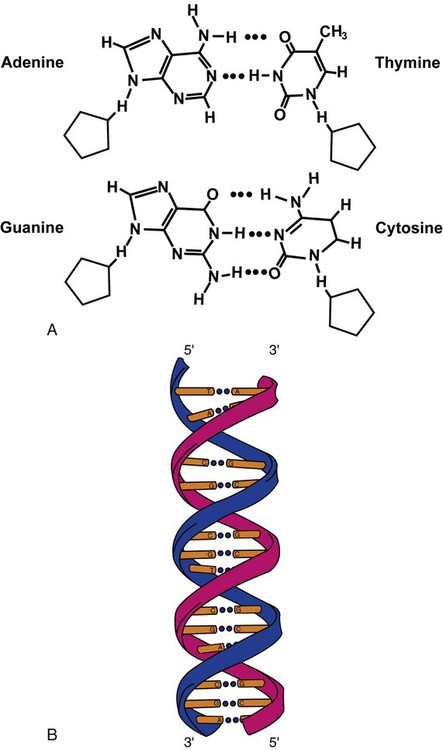
DNA is a duplex molecule composed of two complementary hydrogen-bonded nucleotide strands (Figure 32-4). Deoxyribonucleotides and ribonucleotides are the building blocks of DNA and RNA, respectively. Each nucleotide is composed of a 5-carbon sugar (pentose), a nitrogenous base, and a phosphate group (Figure 32-5). The numbers one prime (1′) to five prime (5′) designate the pentose’s carbons. In DNA, the pentose is a ribose in which the hydroxyl group on the 2′ carbon is replaced by a hydrogen molecule, hence 2′-deoxyribose. In RNA, the 2′ ribose retains the 2′ hydroxyl group. The hydroxyl group present on the 3′ carbon of the sugar is crucial for polymerization of the nucleotide monomers to form the nucleic acid strand.
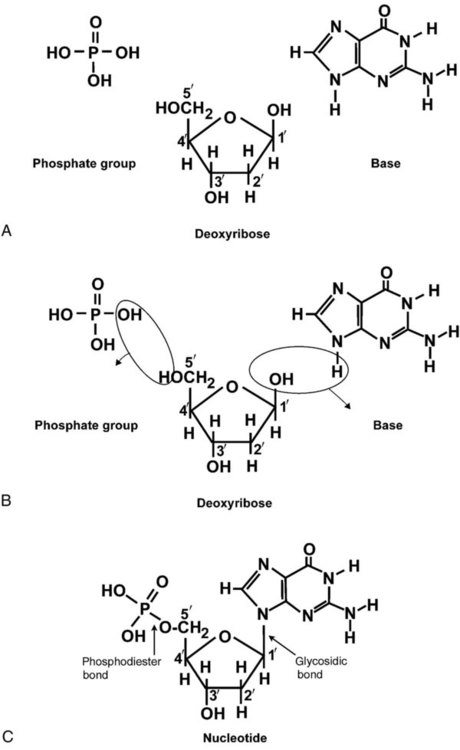
Creation of a phosphodiester bond between the 3′ hydroxyl group of the existing strand and the 5′ α-phosphate of the nucleotide monomer requires the protein enzyme DNA polymerase. This enzyme recognizes the hydroxyl group on the 3′ carbon of the sugar and bonds the 3′ hydroxyl group of one nucleotide with the α-phosphate group of another (Figure 32-6). Polymerization of subsequent nucleotides forms a DNA strand.
DNA consists of two strands that are antiparallel and complementary (Figure 32-7). One strand begins with a phosphate group attached to the 5′ carbon of the first nucleotide oriented to the left and ends with the hydroxyl group on the 3′ carbon of the last nucleotide oriented to the right. This strand is in the 5′-to-3′ direction. The other strand runs in the 3′-to-5′ direction, or antiparallel. The nucleotide sequences composing these strands provide the encoded messages of our genes. Therefore, the addition of nucleotides is highly regulated.
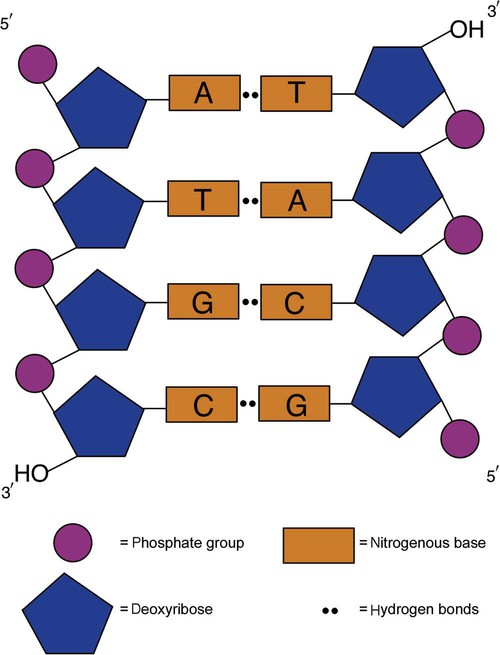
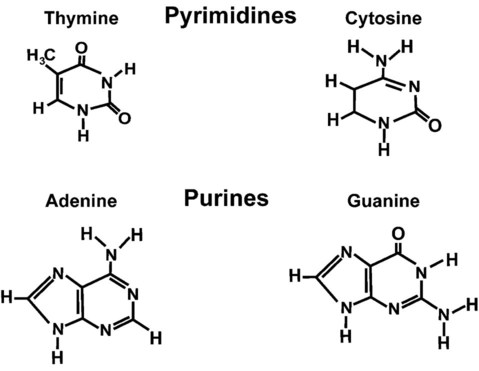
One regulation mechanism arises from the complementary characteristic of the nucleotides. A nucleotide’s identity depends on the type of nitrogenous base present on the template. There are two categories of nitrogenous bases in nucleic acids, purines and pyrimidines (Figure 32-8). The bases adenine (A) and guanine (G) are double-ringed purines, whereas thymine (T) and cytosine (C) are single-ringed pyrimidines. Adenine forms hydrogen bonds at two points with thymine (A:T), whereas guanine forms hydrogen bonds at three points with cytosine (G:C). If a strand has a 5′-CTAG-3′ sequence, the complementary nucleotides on the 3′-to-5′ strand are 3′-GATC-5′. In RNA, the pyrimidine uracil (U) takes the place of thymine and forms hydrogen bonds with adenine. Hydrogen bonds between A:T and G:C hold the strands together (Figure 32-9). RNA is most often single-stranded.
Transcription and Translation
DNA provides a permanent set of instructions. The cellular enzyme RNA polymerase transcribes the code. RNA polymerase recognizes starter sequences called promoters. Promoters lie upstream of coding sequences and bind RNA polymerase to separating DNA strands. The enzyme then slides along the DNA strand 3′ to 5′, “reading” the code and polymerizing (assembling) the complementary ribonucleotides. As the complementary ribonucleotides form hydrogen bonds with the bases of the exposed DNA strand, the RNA polymerase creates phosphodiester bonds to extend the single-stranded primary RNA transcript (Figure 32-10). If the nucleotide sequence of the DNA strand is 3′-CTAG-5′, the primary RNA transcript is 5′-GAUC-3′.
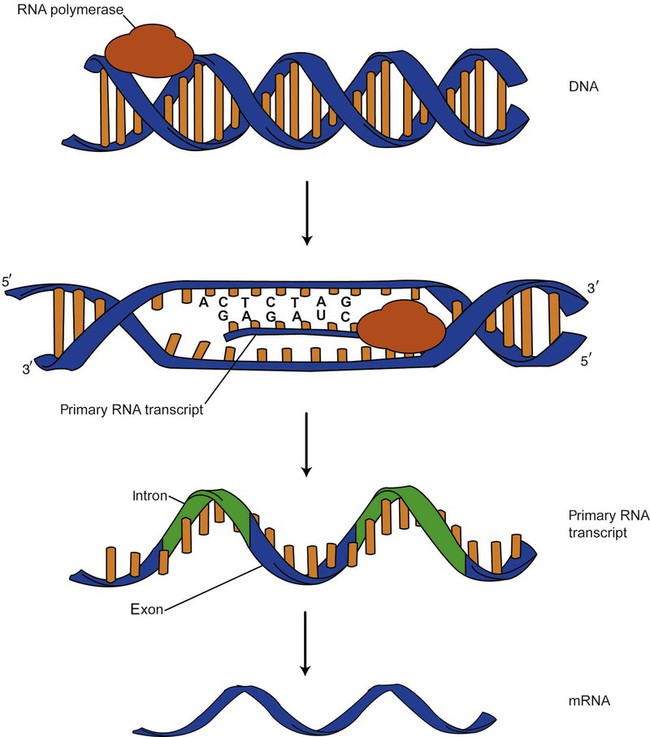
Primary mRNA segments are composed of introns and exons. Introns are untranslated intervening sequences located within the coding portions of genes. Their functions remain unclear, although they may play a role in regulation of gene expression.5 Exons are the sequences that encode the gene product. Before mRNA can serve as a translation template, the introns must be excised from the primary transcript and the exons adjoined. The mature mRNA is completed by the addition of a 5′ cap and a tail of many repeated adenine nucleotides.6 The mRNA leaves the nucleus and enters cytoplasmic ribosomes to be translated.
DNA Replication and the Cell Cycle
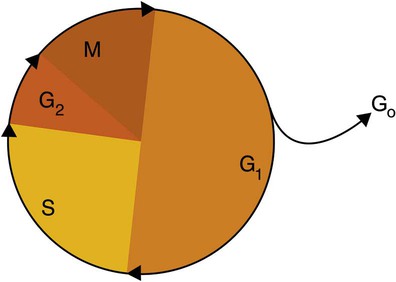
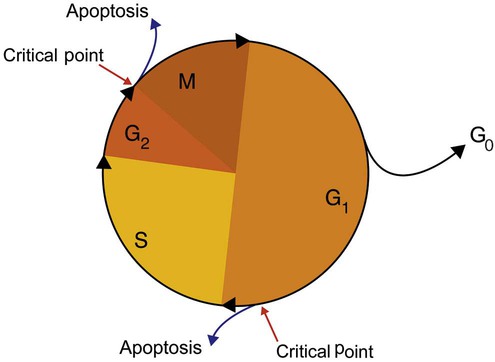
After cells carry out their functions, they either divide via mitosis or die via apoptosis, also called programmed cell death. The cell cycle progresses through a sequence (Figure 32-11). Interphase is made up of the G1, S, and G2 phases. During the G1 phase, the cell grows rapidly and performs its cellular functions. S phase is the synthesis stage, in which DNA is replicated. The G2 phase is the period when the cell produces materials essential for cell division. The M phase refers to mitosis, during which two identical daughter cells are produced, each of which receives one entire set of the DNA that was replicated during S phase. Some cells exit the cell cycle during the G1 phase and enter a phase called G0. Cells in G0 normally do not reenter the cell cycle and remain alive performing their function until apoptosis occurs.
DNA replication during the S phase requires a complex orchestration of events; this discussion focuses on those events that are exploited for molecular diagnostic testing. Contained within the double-stranded DNA helix are multiple origins of replication. At each origin, the enzyme helicase disrupts and untwists the hydrogen bonds, separating the DNA strands and producing two replication forks. Here a deoxyribonucleotide (deoxynucleotide triphosphate, or dNTP) polymerizes to form new complementary strands (Figure 32-12). DNA replication occurs bidirectionally from the two replication origin sites. Each DNA strand in the replication fork serves as a template for the formation of a daughter or complementary strand through the activity of DNA polymerase.7 The DNA polymerase substrate is the free hydroxyl group located on the 3′ carbon of a deoxyribonucleotide. DNA polymerase recognizes the group and catalyzes the joining of the complementary deoxyribonucleotide. DNA is read 3′ to 5′ by DNA polymerase, and the complementary strand is synthesized 5′ to 3′.
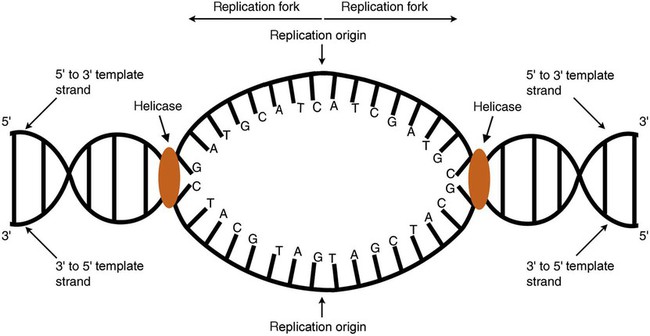
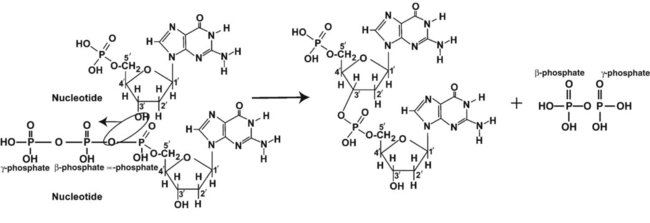
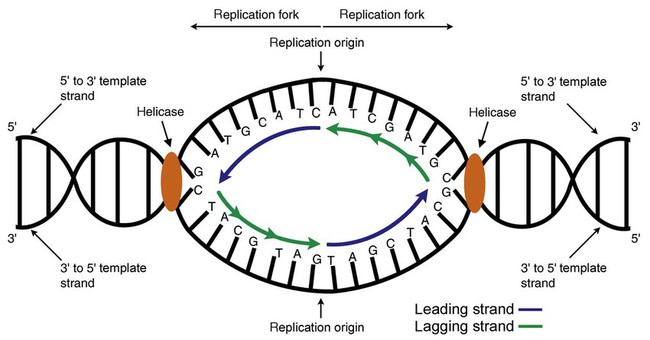
A primer provides the free 3′ hydroxyl group required for DNA polymerase activity. Primers are short nucleotide polymers complementary to the template. The hybridization of the primer to the template requires the enzyme primase. At the replication origin, primase joins a primer to the 3′ end of the 5′-to-3′ (top) template strand (Figure 32-13). Then DNA polymerase recognizes the free hydroxyl group on the 3′ carbon of the last nucleotide in the primer and catalyzes the formation of phosphodiester bonds between the correct complementary nucleotide triphosphate and the primer, releasing the β- and γ-phosphate groups. DNA polymerase continues adding deoxyribonucleotides along the replication fork, going to the left of the replication origin, producing the complementary strand called the leading strand.
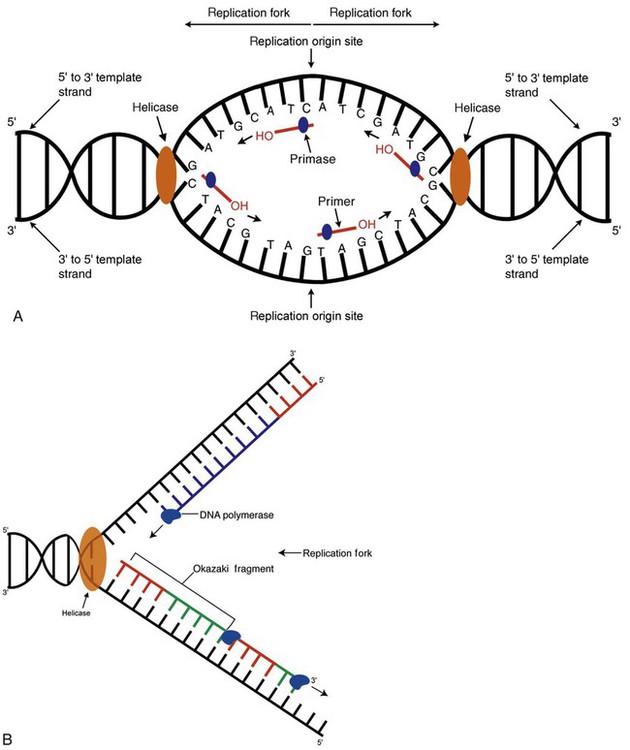
DNA polymerase not only joins nucleotides, it also degrades the RNA primers and fills in the correct complementary deoxyribonucleotides. Because the replication of the lagging strand produces many small fragments, it is called discontinuous replication, and the fragments are called Okazaki fragments. Finally, the enzyme ligase joins the discontinuous fragments. The replication fork to the right (downstream) is replicated in the same fashion, although the lagging strand is now formed complementary to the top (5′-to-3′) strand, and the leading strand is formed from the 3′-to-5′ strand; the opposite of the situation described occurs for the left replication fork (Figure 32-14).
The cell cycle is highly regulated. At certain critical points within the cycle, decisions are made to continue or begin cell death via apoptosis. This decision may depend on the state of the DNA replicated (Figure 32-15). Normally, the cell detects errors made during replication and either corrects them or begins apoptosis. This prevents the persistence of daughter cells with genetic errors. If the sensing molecules fail, cell division may continue. Debilitating mutations that mediate cell cycle control may result in tumor formation.
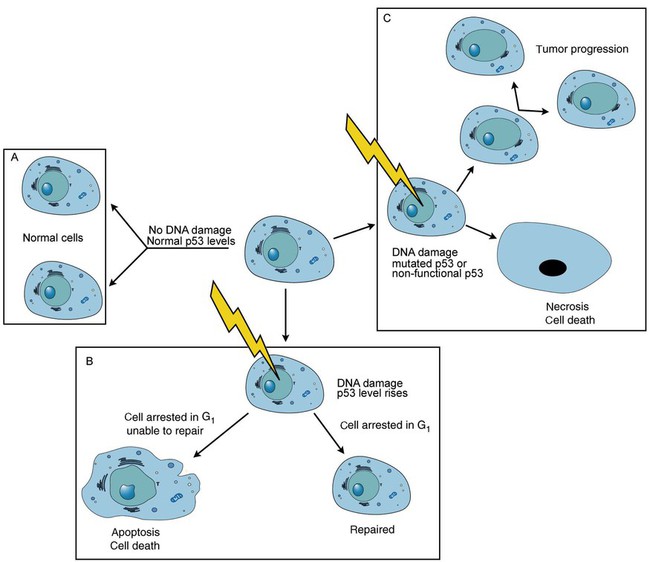
One protein responsible for signaling damaged DNA is p53, a tumor suppressor protein. Damaged cells with increased p53 arrest cell division at G1, which allows time for DNA repair (Figure 32-16). Cells with mutant p53 are unable to arrest cells in G1; they continue the process of cell division with damaged DNA.8–10 If the cell can repair the DNA damage, the cell cycle continues. If the cell damage is too severe, the cell undergoes apoptosis. Hematologic malignancies, such as 21% of chronic myelogenous leukemias (CML),11–13 23% of chronic lymphocytic leukemias (CLL),14–16 and 17% of acute lymphoblastic leukemias (ALL),17–19 are associated with a p53 mutation or deletion (see Box 32-3). In summary, DNA synthesis and accurate cell cycle control demand that the integrity of the nucleotide sequence be maintained during DNA replication.
Nucleic Acid Isolation (Extraction)
Isolating DNA from Clinical Specimens
If a delay in the molecular testing is necessary, the isolated DNA sample can be stored at −80° C indefinitely. Similar procedures are used to extract DNA from dispersed cells from bone marrow and needle aspirate specimens.20
DNA from tissue suspected of being cancerous can be isolated from formalin-fixed, paraffin-embedded tissue sections mounted on glass microscope slides. Tissue is obtained from the entire section or from a portion of the section by microdissection, either by scraping or by laser. The tissue is degraded by an enzyme called proteinase K to break open the cells and release the DNA. The sample is then heated to 94° C for several minutes to inactivate the proteinase K and to degrade other proteins, and the DNA is purified and precipitated as previously discussed.21 In addition to paraffin-embedded samples, fresh or frozen tissue samples are appropriate for DNA isolation. Quickly thawing and mincing the frozen tissue prepares the sample for DNA isolation. The minced tissue is mixed with an extraction buffer to release the DNA from the cells; it is then purified and precipitated as described earlier.
Isolating RNA from Clinical Specimens
RNA isolation poses greater technical challenges than DNA isolation. Ubiquitous ribonucleases (RNases) degrade RNA. These enzymes are the body’s primary defense against pathogens and are found on mammalian epidermal surfaces; therefore, they contaminate all laboratory surfaces.22 Clinical laboratories that isolate RNA must be RNase free, which necessitates costly precautions and decontamination steps.23
The steps of RNA isolation are (1) RNA release by cell lysis, (2) RNase inhibition with strong chemical agents such as urea or guanidine isothiocyanate, (3) protein and DNA removal, and (4) RNA precipitation. In step 3, extraction is performed using phenol at a pH of 4, chloroform, and isoamyl alcohol. These separate the DNA and protein into the organic phase, while the RNA remains in the aqueous phase. RNA resists acidic pH, whereas DNA is readily depurinated, because acid cleaves the bond between the purine base and the deoxyribose sugar. Therefore, acidic phenol preferentially isolates and preserves RNA while the genomic DNA (all the DNA) is partitioned along with contaminating proteins, lipids, and carbohydrates. As with DNA, precipitating the RNA from the aqueous phase requires the addition of salt to neutralize the charge of the phosphodiester backbone and ethanol to make the nucleic acid insoluble.24,25
Amplification of Nucleic Acids
Polymerase Chain Reaction for Amplifying DNA
PCR is the principal technique in the clinical molecular laboratory. PCR is an enzyme-based method for amplifying a target sequence to allow its detection from a small volume of material.26 Sickle cell anemia results from a single β-globin nucleotide substitution (point mutation) in which an adenine replaces a thymine. Detecting this mutation from among 6 billion nucleotides would be like finding a needle in a haystack if only a few cells were assessed. When millions of β-globin copies are produced, however, the mutation is easily detected.
As with natural DNA replication, PCR amplification requires primers. In testing for the sickle cell mutation, for example, selected primers flank (i.e., bind on either side of) the β-globin gene sequence containing the mutation. The total base pair (bp) length of the primer sequences plus the target sequence can vary, but is 110 bp for the β-globin gene, a typical sequence length for many mutation sites (Figure 32-17).27 Besides primers, the PCR master mix reagents include a heat-insensitive DNA polymerase called Taq polymerase, isolated from the thermophilic bacterium Thermus aquaticus, and the deoxyribonucleotides deoxyadenosine triphosphate (dATP), deoxythymidine triphosphate (dTTP), deoxyguanosine triphosphate (dGTP), and deoxycytidine triphosphate (dCTP) in a magnesium buffer.
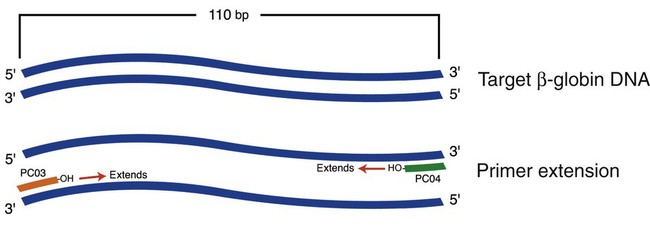
The DNA is first denatured at 95°C, which separates the strands; then cooled to the primer annealing temperature of 40° to 60°C; then warmed to 72°C to promote Taq polymerase–promoted chain extension, in which nucleotides are added to the primers (Figure 32-18). The annealing temperature is optimized for each set of primers. A thermocycler is used to accurately produce and monitor the rapid temperature changes.
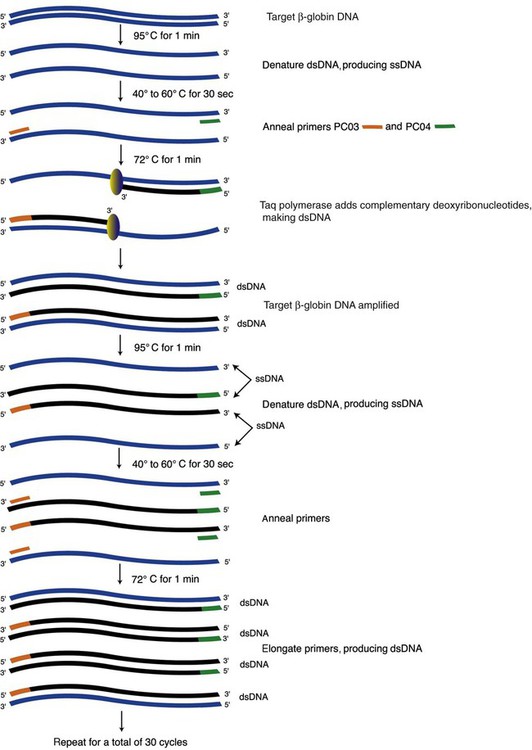
Once the double-stranded DNA is denatured, one primer anneals to the 5′-to-3′ strand and the other to the 3′-to-5′ strand. Both primers possess a free 3′ hydroxyl group. The Taq polymerase recognizes this hydroxyl group, reads the template, and catalyzes formation of the phosphodiester bond joining the first complementary deoxyribonucleotide to the primer. The polymerase continues down the template strand at 1000 nucleotides per second, extending the complementary strand to eventually produce a complete daughter strand that continues to the 3′ end of the template.28 This completes one PCR cycle. In the second cycle, the temperature changes are repeated, and the first-cycle product becomes the template for a daughter strand. After the second cycle, the daughter strand is bounded by the primer sequences at the 5′ and 3′ ends, producing a fragment of DNA of the desired length. In 25 to 40 subsequent cycles, this DNA of specific length and sequence, called an amplicon, is reproduced millions of times.29,30
Commercial kits contain primer sets that have been tested for annealing specificity, but care must be taken to use the optimal annealing temperature. Even if the primer is properly designed, it can anneal to nonidentical regions if the annealing temperature is too low. Laboratory researchers who design primers look to digital algorithms such as the Basic Local Alignment Sequence Tool (BLAST).31 Further, complementarities must be avoided between the primers themselves to prevent hybridization to one another, which forms undesirable primer dimers.
Controls are essential. The three controls required for PCR are the negative, positive, and “no-DNA” controls. All three are included in each run. The negative control consists of DNA known to lack the sequence of interest; the positive control contains the target sequence. Comparison of the bands in the patient specimen electrophoretic lanes to bands in the negative and positive control lanes determines whether the target DNA sequence is present in the patient’s DNA. The no-DNA control detects master mix contamination. A band in the no-DNA lane indicates DNA contamination, which renders the entire test result unreliable.32
Reverse Transcription Polymerase Chain Reaction for Amplifying RNA
Some hematology analyses require mRNA. Genetically altered mRNA sequences translate to an altered protein. For instance, the Philadelphia chromosome (Ph′), carrying the mutation t(9;22)(q34;q11.2), is present in 95% of CML cases plus 20% of adult ALL and 5% of pediatric ALL cases, and in rare instances in acute myeloid leukemia.33,34 Ph′ results from a reciprocal translocation of the ABL (Ableson) gene on chromosome 9 to the breakpoint cluster region (BCR) of chromosome 22, producing a BCR/ABL hybrid (Figure 32-19).35–37 Transcription of BCR/ABL produces a chimeric mRNA made up of fragments from both the BCR and ABL genes. Translation generates a fusion protein, tyrosine kinase, that alters normal cell cycle control, which results in unrestrained cell proliferation.38 RT-PCR is performed to detect the chimeric mRNA, thus the mRNA template is preferred to DNA. Although the mutation is present at the DNA level, the position at which the two chromosome sections join is variable, whereas the chimeric mRNA is always the same. Also, the DNA includes untranslated introns, which make the chimera too long to replicate. Physiologic mRNA excision and splicing yields a much shorter target that is more easily amplified.
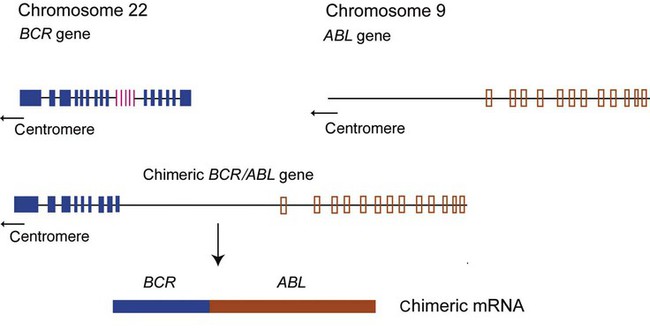
In RT-PCR, the reverse transcriptase enzyme produces complementary DNA (cDNA) from mRNA present in a total RNA sample extracted from patient blood cells (Figure 32-20). PCR subsequently amplifies the cDNA. The RT-PCR master mix includes an oligo(dT), random, or specific primer; reverse transcriptase; deoxyribonucleotides; primers; the mRNA template; and Taq polymerase.
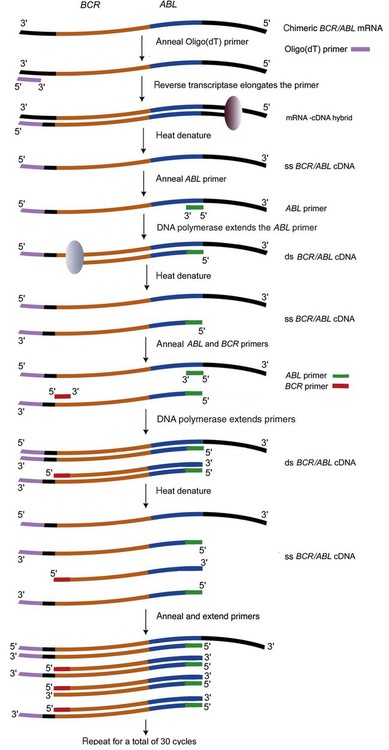
The first step employs reverse transcriptase and a specialized primer to produce an RNA-cDNA hybrid. The primer, called oligo(dT), is a series of thymine nucleotides. Most mRNAs possess a string of adenine nucleotides on the 3′ end called the polyA tail. The oligo(dT) primer anneals to the polyA tail of mRNA. Reverse transcriptase recognizes the hydroxyl group on the last nucleotide of the primer and reads the mRNA template strand, then adds the correct complementary deoxyribonucleotide. Reverse transcriptase continues up the mRNA template strand, joining the complementary deoxyribonucleotides to the growing cDNA strand to form the mRNA-cDNA hybrid. Subsequently, heat denaturation breaks the hydrogen bonds between the mRNA-cDNA hybrid, separating the two strands. The cDNA strand then acts as a template for replication by DNA polymerase. In the next step, the single-stranded cDNA is amplified as in DNA-based PCR using primers specific for a target sequence in the BCR gene and ABL gene. DNA polymerase extends the primers, forming a double-stranded cDNA of the target chimeric gene. The cycling continues, resulting in millions of copies of the BCR/ABL sequence.39,40
Detection of Amplified DNA
Gel Electrophoresis
Nucleic acid phosphate groups confer a net negative charge. Consequently, in electrophoresis, the rate at which DNA fragments (amplicons) pass through gels is proportional to their mass only and, unlike proteins, not their relative charge. DNA fragment mass is a function of the length in base pairs (bp) or kilobase pairs (kb, 1000 × bp). Fragments are sieved through an agarose or polyacrylamide gel matrix by passing a current through the gel as it is bathed in a buffered conducting salt solution. Electrophoresis gel pore diameter is a function of gel concentration. The pores of an agarose gel are larger than the pores of a polyacrylamide gel. When larger fragments (500 bp to 50 kb) are to be separated, an agarose gel is most effective. For smaller DNA fragments (5 to 1000 bp), a polyacrylamide gel is used.41
DNA fragments (amplicons), controls, and a mass marker or ladder are pipetted into the sample wells near the anode. Electrical current moves the negatively charged fragments toward the positive electrode. Smaller fragments move faster (farther) than larger fragments. The ladder, composed of fragments of known masses, measured in base pairs or kilobase pairs, runs alongside the sample and control lanes. Amplicon masses of the sample and control are determined by comparing their bands with the bands of the ladder (Figure 32-21).
For autoradiography, deoxyribonucleotides that are incorporated during the PCR elongation step, usually the adenine nucleotide, are conjugated to a radioactive α-phosphate group ([α32P]dATP). After electrophoresis, x-ray film is placed over the dried gel. The radioactivity in the amplified DNA fragments exposes the film, producing a banding pattern that is interpreted by the laboratory scientist (Figure 32-22).42
Restriction Endonuclease Methods
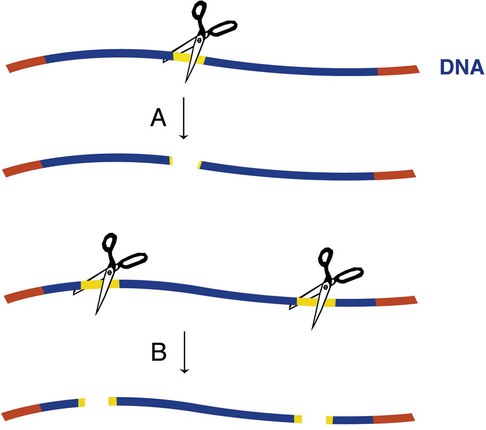
One method to determine whether an amplified target DNA fragment contains a mutation of interest uses enzymes called restriction endonucleases (also known as restriction enzymes). These enzymes are produced naturally in bacteria and are so named because they restrict foreign (phage) DNA from entering and destroying the bacterium. Each restriction enzyme recognizes a specific nucleotide sequence and cuts both strands of the target DNA at the sequence, producing restriction fragments. Recognition sequences can be 4 to 15 nucleotides long. There are hundreds of commercially available restriction endonucleases, which allows recognition of many sequences. The number of restriction fragments produced depends on the number of restriction sites present in the amplified target.43,44 Enzyme action at one restriction site produces two restriction fragments, action at two restriction sites produce three restriction fragments, and so on (Figure 32-23). A restriction enzyme detects even a single base substitution, because the mutation alters its sequence and prevents digestion at the site.
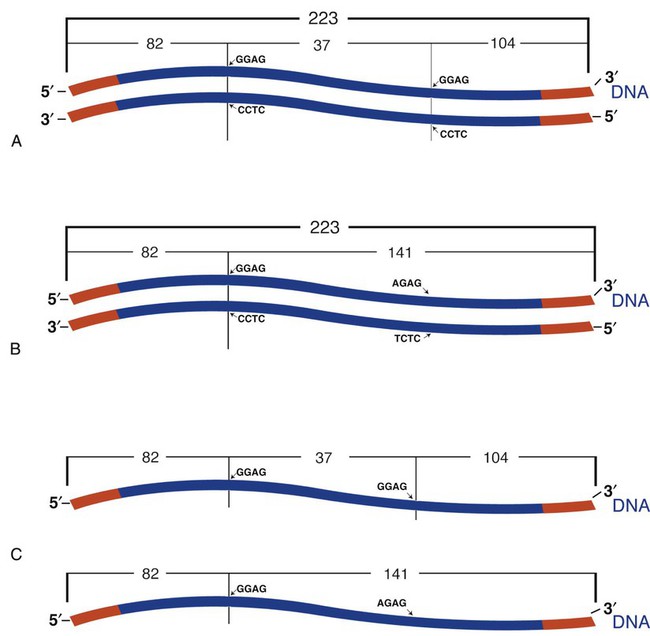
A restriction fragment length polymorphism (RFLP) is a mutation or polymorphism-induced change in the position of the restriction fragment. The factor V Leiden mutation is an excellent example of RFLP. Individuals possessing this mutation have an increased risk of venous thrombosis. The factor V Leiden mutation results from the replacement of guanine with adenine at position 1691 (G1691A) of the coagulation factor V gene.45,46 The mutation alters a site normally detected and cut by the restriction enzyme Mnl1. The wild-type (normal) factor V amplicon is 223 bp long with two Mnl1-specific sites. After PCR and incubation with Mnl1, the wild-type amplicon is cut to three restriction fragments, separable using polyacrylamide gel (Figure 32-24). The fragments are 37, 82, and 104 bp long. The mutant gene generates only two fragments with lengths of 82 and 141 bp. A sample from an individual homozygous for the wild-type gene generates the three anticipate fragments; 37, 82, and 104 bp. A sample from an individual homozygous for the factor V Leiden mutation possesses two copies of the mutated factor V gene and generates only two bands, 82 and 141 bp. A sample from a heterozygous individual possesses one normal and one mutated factor V gene and produces four bands of lengths 37, 82, 104, and 141 bp (see Figure 32-1).
Nucleic Acid Hybridization and Southern Blotting
Another detection method employs a nucleic acid probe designed to hybridize (base-pair) to a selected sequence. Nucleic acid hybridization may be combined with restriction enzyme digestion to make identifications on an electrophoresis gel.47,48
Dr. Edwin Southern developed the classic Southern blot hybridization, which was originally performed using isolated DNA without amplification, but is now performed using PCR-amplified DNA. The Southern blot procedure, which takes 3 days to perform, is now confined to research applications, but pointed the way to RFLP and probe hybridization technology. Restriction endonuclease EcoRI cuts DNA at many sites (Figure 32-25). Gel electrophoresis separates the fragments. The sample is then acid-depurinated to “nick” the fragments. Sodium hydroxide next denatures the DNA, producing single-stranded DNA without changing its nucleotide sequence or two-dimensional position on the gel. The single-stranded DNA is transferred to a nitrocellulose filter by electric current or capillary action, so that the nitrocellulose filter reflects the banding pattern. The DNA is permanently affixed to the nitrocellulose filter by baking and UV cross-linking.49,50
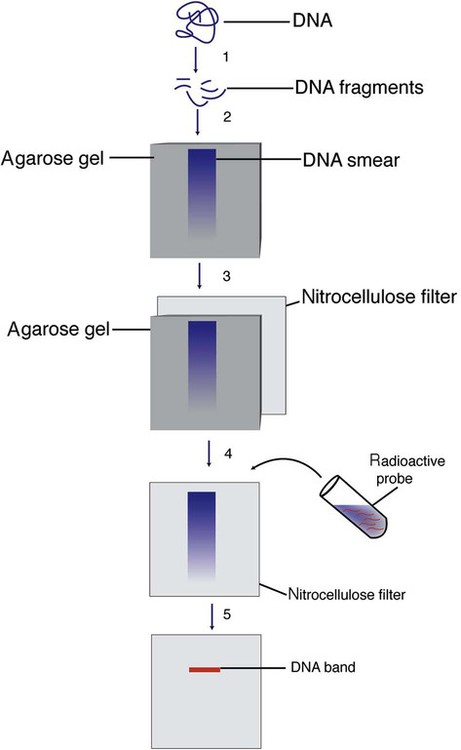
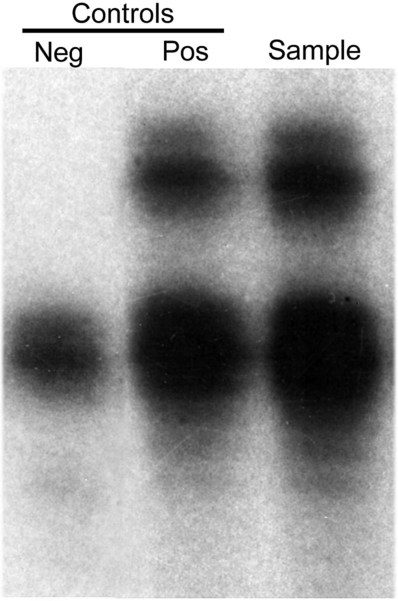
In the classic Southern blot procedure, detection of the band containing the sequence of interest requires a radioactive or enzyme (horseradish peroxidase or alkaline phosphatase)–conjugated, single-stranded probe complementary to the target sequence. The probe hybridizes to the target DNA, leftover unhybridized probe is washed off, and the hybridized bands are visualized by autoradiography. Southern blot analysis may also be performed using a fluorescently labeled probe instead of a radioactive one.51,52
A time-honored application of the Southern blot technique is detection of B-cell immunoglobulin heavy chain gene or T-cell receptor (TCR) gene rearrangement. Each immunoglobulin heavy chain or TCR gene possesses a unique sequence, the result of somatic rearrangement. Rearrangement joins distinct gene segments from pooled clusters with unique sequences. Thus, each B cell and its progeny produce a specific antibody, and each T cell and its progeny possess a specific cell receptor.53–55 Lymphoproliferative disorders arise from a malignant transformation of a B or T cell. The malignant cells, no longer under the control of cell division regulatory proteins, divide uncontrollably, forming a malignant clone. All the cells within the clone contain the same gene rearrangement; thus the cells are monoclonal. Gene rearrangement analysis detects the monoclonal population and determines whether it is a B-cell or T-cell lymphoproliferative disorder.
The appropriate specimens for gene rearrangement analysis are bone marrow, blood, or tissue from the tumor site. After DNA extraction, restriction enzymes EcoRI, BamHI, and HindIII cut the DNA into restriction fragments. Agarose gel electrophoresis separates restriction fragments. The bands are then depurinated, denatured, and transferred to a nitrocellulose filter. Probes hybridize the nucleotide sequences in either B- or T-cell genes, and autoradiography visualizes the banding pattern. The presence of a distinct band represents a monoclonal cell population. A polyclonal population, consisting of cells with different gene rearrangements, appears as a smear with no distinct banding pattern.56–58 Gene rearrangement analysis provides physicians with an important tool to diagnose and monitor the progress of lymphoproliferative disorders.
Hybridization Labels
Labels are visualizing molecules conjugated to, or incorporated into, probes or primers. Radioactivity is incorporated by introducing isotope-labeled nucleotides during probe synthesis. Radioactivity is detected by applying the sample to photographic film, which is an example of the use of a direct label (Figure 32-26). Direct labeling means that the molecule can be visualized without the addition of a reporter system, whereas indirect labels require additional molecules and reactions to be visualized. For health and disposal reasons, the use of radioactive isotopes is restricted in the clinical setting, but alternative labeling techniques and detection kits are commercially available. For instance, fluorogenic acridinium ester molecules may be incorporated as a direct label. Sodium tetraborate solution, containing 1% Triton X-100, degrades acridinium ester on unhybridized probes, whereas base pairing of the probe to its target protects the ester.59 The bound ester is detected by a brief emission of light after addition of hydrogen peroxide.
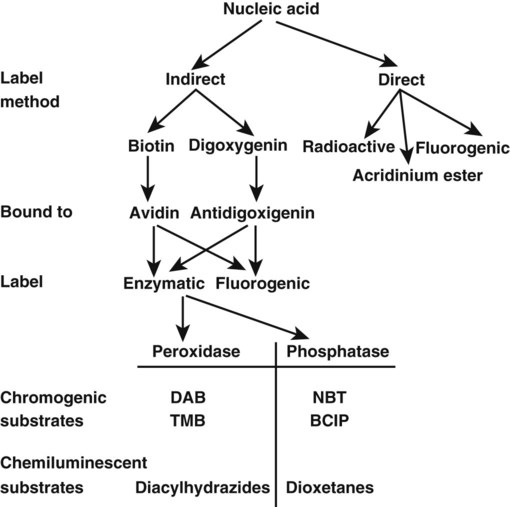
Biotin and digoxigenin are two commonly used indirect labels. Biotin-labeled probes bind avidin. Avidin is in turn linked to a fluorescent label or to a detectable enzyme (see Figure 32-26). Digoxigenin-labeled probes are bound by an antidigoxigenin antibody conjugated to a fluorescent molecule or enzyme. Alkaline phosphatase or horseradish peroxidase are the enzymes most commonly used in indirect labeling systems. Their activity cleaves substrates to produce visible pigments or chemiluminesence.60–62
DNA Sequencing
The ability to read the sequence of the nucleic acid has been just as important as PCR in the development of molecular biology.63 A combination of these two important techniques (cycle sequencing) has made DNA sequencing more efficient and its analysis less subjective. In cycle sequencing, the order of the nucleotide bases is determined after amplification.64 Cycle sequencing is applied in molecular testing to assess amplified sequences for insertions, deletions, or mutations, such as coagulation factor V Leiden mutation or β-globin mutation.
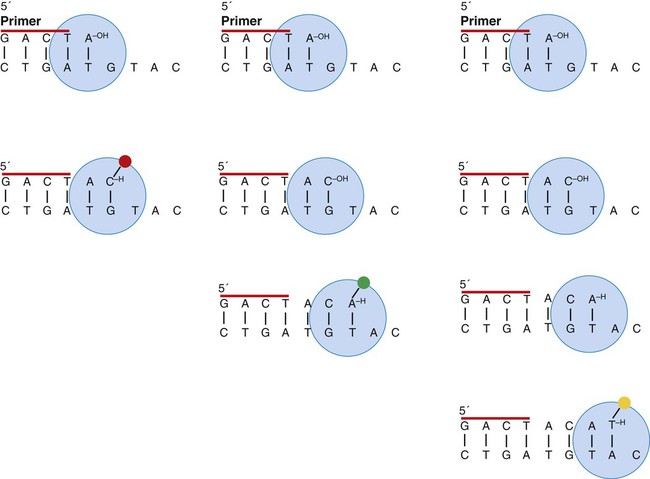
Cycle sequencing is based on dideoxynucleotide terminator sequencing.65 The addition of nucleotides to a growing polymer requires a 3′ hydroxyl group on the last added nucleotide and a triphosphate group on the 5′ end of the next nucleotide to be added. If a nucleotide lacks the 3′ hydroxyl group, it can be incorporated but cannot be added to, so the fragment terminates at the “defective” base (Figure 32-27). If small concentrations of dideoxyadenosine triphosphate, dideoxycytosine triphosphate, dideoxyguanine triphosphate, and dideoxythymine triphosphate each are included in the PCR master mix, over a number of cycles a series of fragments that terminate at each successive base is produced. This is called a nested series of fragments. The dideoxynucleotides are fluorescently labeled to produce a different color for each base, and the fragments are subjected to capillary electrophoresis. The fragments with their labels pass through the beam of a detecting laser one by one, with their order based on their length, which allows their sequence to be read. Because DNA is double-stranded, two PCR primers would produce two series of nested fragments and the detector would read two bases at each position. To avoid this, the PCR reaction is done with one primer, which produces single-sided PCR.
Real-Time Polymerase Chain Reaction
In contrast to standard or end-point PCR, real-time quantitative PCR measures the change in nucleic acid amplification as replication progresses using fluorescent marker dyes.66 The time interval, expressed as the number of replication cycles, required to reach a selected fluorescence threshold is proportional to the copy number of target molecules in the extracted sample.67 Typical targets in this technique include viruses, bacteria, and tumor cells with discernible somatic mutations. Real-time thermocyclers, such as the LightCycler (Roche Applied Science, Indianapolis, IN), first developed in 1993, record the dynamic (“delta”) rise in fluorescence intensity during PCR. Real-time thermocyclers assay multiple samples from various sources in replicate concurrently with internal standards to achieve a clinical accuracy hitherto unattainable using phenotypic or cultural assay techniques.68
The key to real-time quantitative PCR is fluorescence resonance energy transfer (FRET).69 Probes or primers are labeled with fluorophores designed to fluoresce only upon binding their selected nucleic acid sequences. The unbound fluorophore possesses two active sites: a fluorescing reporter site and a quencher site separated by the selected base sequence. When unbound, the quencher site draws energy from the reporter site and thereby extinguishes its fluorescence. Upon binding, the quencher site becomes physically removed from the vicinity of the reporter, which permits the reporter site to fluoresce. As cycling proceeds and more amplicons are produced, the fluorescent signal grows until it exceeds the threshold. SYBR green, which nonspecifically binds the minor groove of double-stranded DNA, exhibits FRET. TaqMan (Applied Biosystems, Carlsbad, CA) is the prototype FRET-based specific probe, and scores of patented competitors, exploiting a variety of creative physical properties, are available to the operator.70
Minimal Residual Disease in Leukemia
Real-time quantitative PCR provides the opportunity to measure minimal residual disease in leukemia, a key indicator of treatment efficacy, clinical remission, and prognosis.71,72 Currently, chemotherapy, radiation therapy, and peripheral blood stem cell transplantation reduce leukemic cells to levels undetectable first by visual bone marrow smear or peripheral blood film review and later by flow cytometry assay.73 This is called minimal residual disease, but despite the seeming disappearance of leukemic cells as recorded by these phenotypic assays, 1010 malignant cells may continue to reside undetected in blood, marrow, or lymphatic tissue.74 Real-time quantitative PCR identifies residual cells and helps guide the types and intensity of therapy with the goal of “molecular” remission. Subsequent to remission, periodic real-time quantitative PCR assays may be used to detect early relapse and drug resistance, enabling the hematologist to initiate follow-up therapy.75
Real-time quantitative PCR may detect a single malignant cell within a population of a million cells, providing unparalleled sensitivity. Manufacturers compete to develop series of FRET-labeled probes that are comprehensive, speedy, and specific for various hematologic diseases.76 Current applications include detection of BCR/ABL (Ph′) in CML and some acute leukemias (see Figure 31-19); JAK2 (“just another kinase” or Janus kinase) in the myeloproliferative neoplasms polycythemia vera and essential thrombocythemia; the t(15;17)(q22;q21) mutation in acute promyelocytic leukemia; and gene rearrangement in lymphatic neoplasms.77
Real-time quantitative PCR may also exploit gene rearrangement in lymphomas or lymphocytic leukemias to test for minimal residual disease. Because the rearranged base sequence varies depending on the individual patient’s malignant clone, the patient’s blood must first be collected and the tumor cell DNA cloned and sequenced.78 Thereafter, primers and probes may be designed (and labeled with SYBR green or FRET probes) to replicate and detect the patient’s sequence.79 This process is called allele-specific oligonucleotide PCR.
Infectious Disease Load
Real-time quantitative PCR can detect and quantitate a number of blood-borne viruses: hepatitis B and C viruses, human papillomavirus, CMV, Epstein-Barr virus, and HIV.80 Human bacterial pathogens such as β-hemolytic streptococcus from throat swabs, anaerobes from wound swabs, and bacteria from urine or other body fluids can be detected within hours of collection. Antibacterial therapy can be initiated based upon the speedy results of molecular susceptibility testing. Real-time quantitative PCR is the reference method for detection and quantification of methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant enterococcus, and opportunistic Clostridium difficile. Molecular diagnostic techniques are effecting in identifying and monitoring malarial and other blood-borne parasites. The challenge to primer and probe developers is to select sequences that are specific enough to avoid false positives caused by nonpathogenic strains, sensitive enough to positively identify infectious strains, and flexible enough to remain effective as pathogenic microorganisms mutate and evolve.
Summary
• DNA directs cell function as described by the central dogma.
• DNA retains the genetic code and reproduces itself through replication.
• The genetic code is transcribed from DNA to mRNA.
• During transcription, untranscribed DNA introns are excised.
• The mRNA transports code from the nucleus to the cytoplasm, where it is translated in cytoplasmic ribosomes.
• Translation depends upon tRNA, small RNA molecules designed to transport and add amino acid to growing peptide chains in cytoplasmic ribosomes.
• DNA consists of a five-carbon sugar (deoxyribose), a phosphate group, and a nitrogenous base. The bases are either purines or pyrimidines.
• The purines in DNA are adenine (A) and guanine (G)
• The pyrimidines in DNA are thymine (T) and cytosine (C).
• DNA is a double-stranded molecule held together by hydrogen bonding between the bases, A:T and G:C.
• DNA is denatured by heat, which breaks the hydrogen bonds to produce single-stranded DNA.
• DNA strands are antiparallel.
• The tumor suppressor protein p53 detects damaged DNA and either halts cell division for DNA repair or begins the process of apoptosis.
• RNA is a single-stranded molecule that contains the sugar ribose instead of the deoxyribose found in DNA and the pyrimidine uracil in place of thymine.
• RNA polymerase recognizes a sequence of deoxyribonucleotides called the promoter within DNA.
• RNA polymerase separates the DNA strands and begins adding ribonucleotides, forming an initial RNA transcript consisting of introns and exons.
• Proteins function as structural components of the cell, as enzymes involved in metabolism or regulation, as receptors to regulate cellular functions, or as antibodies for the immune system.
• Mutation within a gene ultimately alters the protein produced, which often affects the function of the protein.
• Four areas of hematopathologic molecular diagnostic testing include:
Detection of somatic mutations in hematologic malignancies
Detection of inherited hematologic and thrombotic disorders
• Blood, bone marrow, tissue biopsy samples, and fine-needle aspirates are specimens used for DNA and RNA isolation.
• DNA is amplified by in vitro PCR and RT-PCR.
• Amplified DNA is detected by:
• Molecular testing allows more sensitive assessment of therapeutic efficacy and the presence of residual diseased cells.
• Molecular testing permits clinicians to make more accurate therapeutic and prognostic decisions.
• Real-time quantitative PCR allows assessment of the level of disease remaining, not just its presence or absence.
Review Questions
1. If the DNA nucleotide sequence is 5′-ATTAGC-3′, then the mRNA sequence transcribed from this template is:
2. Cells with damaged DNA and mutated or nonfunctioning p53:
a. Are arrested in G1 and the DNA is repaired
b. Continue to divide, which leads to tumor progression
3. To start DNA replication, DNA polymerase requires an available 3′ hydroxyl group found on the:
4. Ligase joins Okazaki fragments of the:
5. A 40-year-old patient enters the hospital with a rare form of cancer caused by faulty cell division regulation. This cancer localized in the patient’s spleen. An ambitious laboratory developed a molecular test to verify the type of cancer present. This molecular test would require patient samples taken from which two tissues?
a. Abnormal growths found on the skin and in the bone marrow
b. Normal splenic tissue and cancerous tissue
6. One main difference between PCR and RT-PCR is that:
b. PCR uses reverse transcriptase to elongate the primers
7. Which one of the following statements about gel electrophoresis is false?
a. The gel is oriented in the chamber with the wells at the positive terminal.
b. A buffer solution is required to maintain the electrical current.
c. The matrix of a polyacrylamide gel is tighter than that of an agarose gel.
d. The larger DNA fragments will be closest to the wells of the gel.
8. Autoradiography of DNA is the:
a. Detection of radioactive deoxyribonucleotides
b. Exposure of the gel to UV light
9. In a test for B-cell gene rearrangement, the pattern seen in the patient’s sample is a smear. This smear indicates that:
a. A monoclonal population of B cells exists
b. A polyclonal population of B cells exists
10. Which of the following statements about minimal residual disease is true?
a. Clinical remission of hematologic cancers can be determined by molecular techniques such as PCR and flow cytometry.
b. Real-time quantitative PCR–determined copy number of BCR/ABL rearrangements will always be lower in molecular remission than in clinical remission.
c. Qualitative PCR that uses a known copy number of a target sequence is of use in determining minimal residual disease levels.
d. Minimal residual disease assessment can aid physicians in making treatment decisions but does not yet offer insights into prognosis.
*This case was provided by George A. Fritsma, MS, MLS, manager, The Fritsma Factor: Your Interactive Hemostasis Resource, http://www.fritsmafactor.com, sponsored by Precision BioLogic, Inc., Cambridge, Nova Scotia.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Molecular Diagnostics in the Clinical Laboratory
After completion of this chapter, the reader will be able to:
1. Describe the structure of DNA, including the composition of a nucleotide, the double helix, strand orientation, and the antiparallel complementary characteristic.
2. Predict the nucleotide sequence of a complementary strand of DNA or RNA given the nucleotide sequence of a DNA template.
3. Explain the relationship between DNA structure and protein production.
4. Explain the relationship between the cell cycle and tumor progression.
5. Discuss the process of DNA replication, including replication origin, replication fork, primase, primer, DNA polymerase, Okazaki fragments, leading strand, and lagging strand.
6. Determine the appropriate patient specimen required for DNA isolation to identify an inherited or somatic mutation.
8. Explain the purpose of polymerase chain reaction (PCR), reverse transcription PCR, and nucleic acid hybridization.
10. Compare and contrast the methods for detecting amplified target DNA: (1) gel electrophoresis using either ethidium bromide or autoradiography, (2) restriction fragment length polymorphism, and (3) probe hybridization techniques such as Southern blotting.
11. Interpret an agarose gel electrophoresis result for the factor V Leiden mutation test and a nucleic acid hybridization result for a B- and T-cell gene rearrangement test.
12. Describe the principle of real-time quantitative PCR.
13. Discuss the use of real-time PCR for monitoring minimal residual disease.
Case Study*
After studying this chapter, the reader should be able to respond to the following case study:
On physical examination, the patient had no evidence of rash or oral ulcers. No petechiae or purpura were noted. He had mild pretibial pitting edema. His right leg measured 36.5 cm at 25 cm distal to the superior aspect of the patella, whereas his left leg measured 33.5 cm in the same location. CBC findings were unremarkable, and both the prothrombin time and activated partial prothrombin time were within the reference ranges. Doppler ultrasonography revealed complete occlusion of the distal superficial femoral vein, anterior tibial vein, and popliteal vein. The diagnosis was DVT without pulmonary emboli. The patient was hospitalized, and a heparin drip was started. The hematologist ordered a factor V Leiden analysis. The initial specimen received was drawn into a red-topped tube. The laboratory scientist requested a redraw using a lavender-topped tube. The patient was still hospitalized, which allowed the collection of a blood specimen in an EDTA tube. Figure 32-1 illustrates the results of the factor V Leiden mutation test initially done by the molecular scientist. The scientist’s supervisor reviewed the gel electrophoresis results and requested that the scientist repeat the analysis. Figure 32-2 represents the repeated analysis.
1. What type of specimen is appropriate when analyzing DNA for a hereditary mutation?
2. Examine the first gel electrophoresis result (see Figure 32-1). Are the correct controls present?
3. Examine the second gel electrophoresis result (see Figure 32-2). What band sizes appear in the patient’s sample?
4. What band sizes are expected for an individual who is homozygous for the factor V Leiden mutation, heterozygous for the mutation, and free of the mutation?
5. Why did the laboratory request that the blood sample to be redrawn?
Molecular biology techniques enhance the diagnostic team’s ability to predict or identify an increasing number of diseases in the clinical laboratory. Molecular techniques also enable clinicians to monitor disease progression during treatment and to make accurate prognoses. The short interval required to perform molecular diagnostic tests and analyze their results is an additional positive aspect of this type of testing, resulting in more efficient patient management, especially in cases of infection. The three main areas of hematopathologic molecular testing include detection of chromosomal translocations in hematologic malignancies (Box 32-1) and inherited hematologic disorders (Box 32-2), identification of hematologically important infectious diseases (Box 32-3), and monitoring of minimal residual disease after cancer treatment.
Structure and Function of Dna
The Central Dogma: DNA to RNA to Protein
The central dogma in genetics is that information stored in the DNA is replicated to daughter DNA, transcribed to messenger ribonucleic acid (mRNA), and translated into a functional protein (Figure 32-3). This process is essential to carry out cellular functions while preserving a record of the stored information. In eukaryotes, the initial DNA sequence is composed of exons separated by untranslated introns. The introns are enzymatically excised during transcription from DNA to RNA, and the mature mRNA sequence is then translated. Translation is an enzymatic process wherein mRNA three-member base sequences called codons drive the addition of individual amino acids to the growing peptide. The mature protein then carries out its cellular function, which may be structural or may involve recognition, regulation, or enzymatic activity.
The structural units that carry DNA’s message are called genes. The human β-globin gene, part of the hemoglobin molecule, provides a good example of replication and transcription, because it was one of the first sequenced and demonstrates the result of aberrant sequence maintenance. A normal (or wild-type) β-globin gene contains a sequence of bases that code for a β-globin peptide of 146 amino acids. One inherited mutation changes a single DNA base. This is called a point mutation. The mutation occurs in the portion of the sequence that codes for the sixth amino acid of β-globin. The mutation substitutes the amino acid valine for glutamine in the growing peptide. Valine modifies the overall charge, producing a protein that polymerizes in a low-oxygen environment. This leads to sickled erythrocytes, circulatory ischemia, and poor oxygen exchange between blood and tissues.1,2 A mutation in one of the two copies (alleles) of this gene inherited from the parents results in a heterozygous condition, or a sickle cell trait. In a heterozygote, the symptoms of the disease are often unseen or are present only during times of physical stress. If both alleles are mutated, there is overt homozygous sickle cell disease, and the symptoms are severe.
Every active gene is translated. Human somatic cells contain 20,000 to 25,000 genes in 2 meters of DNA.3,4 Significant packing (see Chapter 31) takes place to reduce the volume of the nucleic acid to the size of chromosomes.
DNA at the Molecular Level
DNA is a duplex molecule composed of two complementary hydrogen-bonded nucleotide strands (Figure 32-4). Deoxyribonucleotides and ribonucleotides are the building blocks of DNA and RNA, respectively. Each nucleotide is composed of a 5-carbon sugar (pentose), a nitrogenous base, and a phosphate group (Figure 32-5). The numbers one prime (1′) to five prime (5′) designate the pentose’s carbons. In DNA, the pentose is a ribose in which the hydroxyl group on the 2′ carbon is replaced by a hydrogen molecule, hence 2′-deoxyribose. In RNA, the 2′ ribose retains the 2′ hydroxyl group. The hydroxyl group present on the 3′ carbon of the sugar is crucial for polymerization of the nucleotide monomers to form the nucleic acid strand.
Creation of a phosphodiester bond between the 3′ hydroxyl group of the existing strand and the 5′ α-phosphate of the nucleotide monomer requires the protein enzyme DNA polymerase. This enzyme recognizes the hydroxyl group on the 3′ carbon of the sugar and bonds the 3′ hydroxyl group of one nucleotide with the α-phosphate group of another (Figure 32-6). Polymerization of subsequent nucleotides forms a DNA strand.
DNA consists of two strands that are antiparallel and complementary (Figure 32-7). One strand begins with a phosphate group attached to the 5′ carbon of the first nucleotide oriented to the left and ends with the hydroxyl group on the 3′ carbon of the last nucleotide oriented to the right. This strand is in the 5′-to-3′ direction. The other strand runs in the 3′-to-5′ direction, or antiparallel. The nucleotide sequences composing these strands provide the encoded messages of our genes. Therefore, the addition of nucleotides is highly regulated.
One regulation mechanism arises from the complementary characteristic of the nucleotides. A nucleotide’s identity depends on the type of nitrogenous base present on the template. There are two categories of nitrogenous bases in nucleic acids, purines and pyrimidines (Figure 32-8). The bases adenine (A) and guanine (G) are double-ringed purines, whereas thymine (T) and cytosine (C) are single-ringed pyrimidines. Adenine forms hydrogen bonds at two points with thymine (A:T), whereas guanine forms hydrogen bonds at three points with cytosine (G:C). If a strand has a 5′-CTAG-3′ sequence, the complementary nucleotides on the 3′-to-5′ strand are 3′-GATC-5′. In RNA, the pyrimidine uracil (U) takes the place of thymine and forms hydrogen bonds with adenine. Hydrogen bonds between A:T and G:C hold the strands together (Figure 32-9). RNA is most often single-stranded.
Transcription and Translation
DNA provides a permanent set of instructions. The cellular enzyme RNA polymerase transcribes the code. RNA polymerase recognizes starter sequences called promoters. Promoters lie upstream of coding sequences and bind RNA polymerase to separating DNA strands. The enzyme then slides along the DNA strand 3′ to 5′, “reading” the code and polymerizing (assembling) the complementary ribonucleotides. As the complementary ribonucleotides form hydrogen bonds with the bases of the exposed DNA strand, the RNA polymerase creates phosphodiester bonds to extend the single-stranded primary RNA transcript (Figure 32-10). If the nucleotide sequence of the DNA strand is 3′-CTAG-5′, the primary RNA transcript is 5′-GAUC-3′.
Primary mRNA segments are composed of introns and exons. Introns are untranslated intervening sequences located within the coding portions of genes. Their functions remain unclear, although they may play a role in regulation of gene expression.5 Exons are the sequences that encode the gene product. Before mRNA can serve as a translation template, the introns must be excised from the primary transcript and the exons adjoined. The mature mRNA is completed by the addition of a 5′ cap and a tail of many repeated adenine nucleotides.6 The mRNA leaves the nucleus and enters cytoplasmic ribosomes to be translated.
DNA Replication and the Cell Cycle
After cells carry out their functions, they either divide via mitosis or die via apoptosis, also called programmed cell death. The cell cycle progresses through a sequence (Figure 32-11). Interphase is made up of the G1, S, and G2 phases. During the G1 phase, the cell grows rapidly and performs its cellular functions. S phase is the synthesis stage, in which DNA is replicated. The G2 phase is the period when the cell produces materials essential for cell division. The M phase refers to mitosis, during which two identical daughter cells are produced, each of which receives one entire set of the DNA that was replicated during S phase. Some cells exit the cell cycle during the G1 phase and enter a phase called G0. Cells in G0 normally do not reenter the cell cycle and remain alive performing their function until apoptosis occurs.
DNA replication during the S phase requires a complex orchestration of events; this discussion focuses on those events that are exploited for molecular diagnostic testing. Contained within the double-stranded DNA helix are multiple origins of replication. At each origin, the enzyme helicase disrupts and untwists the hydrogen bonds, separating the DNA strands and producing two replication forks. Here a deoxyribonucleotide (deoxynucleotide triphosphate, or dNTP) polymerizes to form new complementary strands (Figure 32-12). DNA replication occurs bidirectionally from the two replication origin sites. Each DNA strand in the replication fork serves as a template for the formation of a daughter or complementary strand through the activity of DNA polymerase.7 The DNA polymerase substrate is the free hydroxyl group located on the 3′ carbon of a deoxyribonucleotide. DNA polymerase recognizes the group and catalyzes the joining of the complementary deoxyribonucleotide. DNA is read 3′ to 5′ by DNA polymerase, and the complementary strand is synthesized 5′ to 3′.
A primer provides the free 3′ hydroxyl group required for DNA polymerase activity. Primers are short nucleotide polymers complementary to the template. The hybridization of the primer to the template requires the enzyme primase. At the replication origin, primase joins a primer to the 3′ end of the 5′-to-3′ (top) template strand (Figure 32-13). Then DNA polymerase recognizes the free hydroxyl group on the 3′ carbon of the last nucleotide in the primer and catalyzes the formation of phosphodiester bonds between the correct complementary nucleotide triphosphate and the primer, releasing the β- and γ-phosphate groups. DNA polymerase continues adding deoxyribonucleotides along the replication fork, going to the left of the replication origin, producing the complementary strand called the leading strand.
DNA polymerase not only joins nucleotides, it also degrades the RNA primers and fills in the correct complementary deoxyribonucleotides. Because the replication of the lagging strand produces many small fragments, it is called discontinuous replication, and the fragments are called Okazaki fragments. Finally, the enzyme ligase joins the discontinuous fragments. The replication fork to the right (downstream) is replicated in the same fashion, although the lagging strand is now formed complementary to the top (5′-to-3′) strand, and the leading strand is formed from the 3′-to-5′ strand; the opposite of the situation described occurs for the left replication fork (Figure 32-14).
The cell cycle is highly regulated. At certain critical points within the cycle, decisions are made to continue or begin cell death via apoptosis. This decision may depend on the state of the DNA replicated (Figure 32-15). Normally, the cell detects errors made during replication and either corrects them or begins apoptosis. This prevents the persistence of daughter cells with genetic errors. If the sensing molecules fail, cell division may continue. Debilitating mutations that mediate cell cycle control may result in tumor formation.
One protein responsible for signaling damaged DNA is p53, a tumor suppressor protein. Damaged cells with increased p53 arrest cell division at G1, which allows time for DNA repair (Figure 32-16). Cells with mutant p53 are unable to arrest cells in G1; they continue the process of cell division with damaged DNA.8–10 If the cell can repair the DNA damage, the cell cycle continues. If the cell damage is too severe, the cell undergoes apoptosis. Hematologic malignancies, such as 21% of chronic myelogenous leukemias (CML),11–13 23% of chronic lymphocytic leukemias (CLL),14–16 and 17% of acute lymphoblastic leukemias (ALL),17–19 are associated with a p53 mutation or deletion (see Box 32-3). In summary, DNA synthesis and accurate cell cycle control demand that the integrity of the nucleotide sequence be maintained during DNA replication.
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
