Chapter 5 Molecular Cardiovascular Medicine
In the past decades we have witnessed what may well be termed a revolution in the biomedical sciences, as molecular methodologies have suddenly become more evident on the clinical scene. Molecular biology originated in the 1950s, its birth most commonly identified with the description of the structure of deoxyribonucleic acid (DNA) by Watson and Crick.1
MACHINERY BEHIND THE CARDIAC RHYTHM: ION CHANNELS
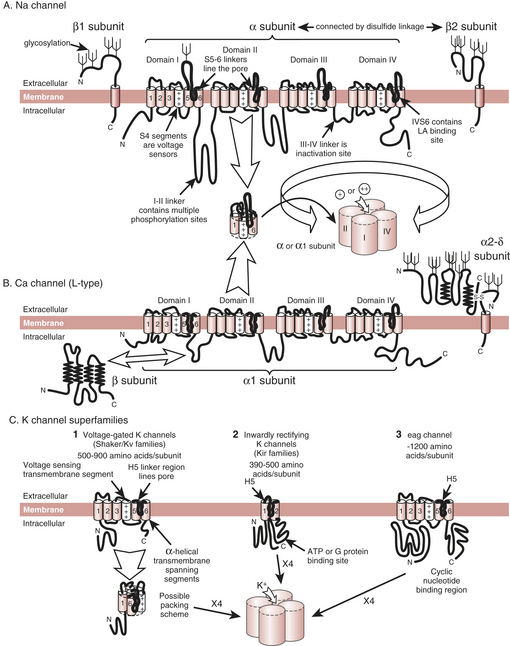
The cardiac action potential results from the flow of ions through ion channels, which are the membrane-bound proteins that form the structural basis of cardiac electrical excitability. In response to changes in electrical potential across the cell membrane, ion channels open to allow the passive flux of ions into or out of the cell along their electrochemical gradients. Ion flux results in a flow of current, which displaces the cell membrane potential toward the potential at which the electrochemical gradient for the ion is zero, called the equilibrium potential (E) for the ion. Depolarization of the cell could, in principle, result from an inward cation current or an outward anion current; for repolarization the reverse is true. In excitable cells, action potentials are mainly caused by the flow of cation currents. Membrane depolarization results principally from the flow of Na+ down its electrochemical gradient (ENa is around +50 mV), whereas repolarization results from the outward flux of K+ down its electrochemical gradient (EK is around −90 mV). Opening and closing of multiple ion channels of a single type result in an individual ionic current. The integrated activity of many different ionic currents, each activated over precisely regulated potential ranges and at different times in the cardiac cycle, results in the cardiac action potential (Box 5-1).
Phase 0: The Rapid Upstroke of the Cardiac Action Potential
The rapid upstroke of the cardiac action potential (phase 0) is caused by the flow of a large inward Na+ current (INa) (Box 5-2). INa is activated by depolarization of the sarcolemma to a threshold potential of −65 to −70 mV. INa activation, and hence the action potential, is an all-or-nothing response. Subthreshold depolarizations have only local effects on the membrane. After the threshold for activation of fast Na+ channels is exceeded, Na+ channels open (i.e., INa activates) and Na+ ions enter the cell down their electrochemical gradient. This results in displacement of the membrane potential toward the equilibrium potential for Na+ ions, around +50 mV. INa activation is transient, lasting at most 1 to 2 ms because, simultaneous with activation, a second, slightly slower conformational change in the channel molecule occurs (inactivation), which closes the ion pore in the face of continued membrane depolarization. The channel cannot open again until it has recovered from inactivation (i.e., regained its resting conformation), a process that requires repolarization to the resting potential for a defined period of time. The channels cycle through three states: resting (and available for activation), open, and inactivated. While the channel is inactivated, it is absolutely refractory to repeated stimulation.
Molecular Biology of Ion Channels
Clinical Correlates
Ion Channels and Antiarrhythmic Drugs
The prototype antiarrhythmic agents, such as disopyramide and quinidine, have diverse effects on cardiac excitability, and these, along with agents introduced more recently, frequently exhibit significant proarrhythmia with potentially fatal consequences. In the Cardiac Arrhythmia Suppression Trial (CAST), mortality among asymptomatic post–myocardial infarction patients was approximately doubled by treatment with the potent Na+ channel-blocking agents encainide and flecainide, an effect that is likely attributable to slowing of conduction velocity with a consequent increase in fatal reentrant arrhythmias.2,3 The only drugs currently available that definitely prolong life by reducing fatal arrhythmias are β-blockers, and these agents have no channel-blocking effects.
Ion Channels in Disease
Elucidation of the molecular mechanisms of the cardiac action potential is beginning to directly affect patient management, particularly in patients with inherited genetic abnormalities of ion channels leading to cardiac sudden death. Two groups of diseases serve to illustrate this point—the LQT syndrome and the Brugada syndrome. An understanding of the molecular mechanism of cardiac electrical excitability is also leading to the emergence of gene therapies and stem cell therapies that may in the future allow manipulation of cardiac rhythm and function.4,5
CONTROLLING CARDIAC FUNCTIONING: RECEPTORS
Receptors
Receptors are grouped into several broad classes, the protein tyrosine kinase receptors and the G protein–coupled receptors (GPCRs) being the most important ones. The protein tyrosine kinase receptors are large molecular complexes. Ligand binding induces activation of a phosphorylating enzyme activity in the intracellular segment of the molecule. Because phosphorylation is one of the major mechanisms of cellular regulation, such receptors can have a variety of cellular effects (Box 5-3). In contrast, GPCRs are much smaller. Ligand binding results in activation of an associated protein (G protein) that subsequently influences cellular processes.
The heart and blood vessels express a variety of GPCRs. The β-adrenergic and muscarinic acetylcholine receptors are those most important for regulation of cardiac functioning, but a number of others play relevant modulatory roles. These include the α-adrenergic, adenosine A1, adenosine triphosphate (ATP), histamine-2 (H2), vasoactive intestinal peptide (VIP), and angiotensin II receptors (Fig. 5-2).
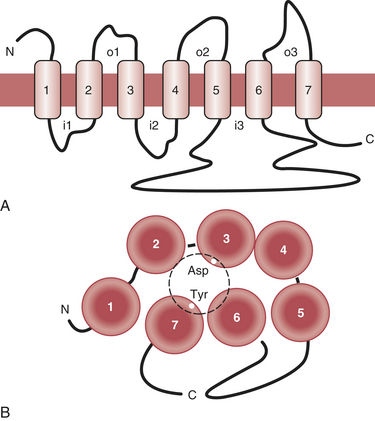
Adrenergic Receptors and Signaling Pathways
Regulation of β-Receptor Functioning
Although β-receptor stimulation allows the dramatic increases in cardiac output of which the human heart is capable, it is clearly intended to be a temporary measure. Prolonged adrenergic stimulation has highly detrimental effects on the myocardium: The pronounced increases in cAMP levels are followed by increases in intracellular Ca2+ concentration, reductions in RNA and protein synthesis, and finally cell death. Thus, β-receptor modulation is best viewed as part of the “fight or flight” response—beneficial in the short term but detrimental if depended on too long. Cardiac failure, in particular, has been shown to be associated with prolonged increases in adrenergic stimulation, even to the extent that norepinephrine “spillover” from cardiac nerve endings can be detected in the blood of patients in heart failure.6
One mechanism for decreasing β-receptor functioning is the downregulation (i.e., decrease in density) of receptors. In cardiac failure, receptor levels are reduced up to 50%. β1-Receptors downregulate more than do β2-receptors, resulting in a change in the β1:β2 ratio. As mentioned earlier, the normal ratio is approximately 70:30; in the failing heart, it is approximately 3:2. Various molecular mechanisms exist for this downregulation. Some of them, particularly in the longer term, are degradation and permanent removal of receptors from the cell surface. In the short term, receptors can be temporarily removed from the cell membrane and “stored” in intracellular vesicles, where they are not accessible to agonists. These receptors are, however, fully functional and can be recycled to the membrane when adrenergic overstimulation has ceased.7
Muscarinic Receptors and Signaling Pathways
Muscarinic Acetylcholine Receptors
The second major receptor type involved in cardiac regulation is the muscarinic receptor. Although five subtypes of muscarinic receptors exist, only one of these (m2) is present in cardiac tissue. Most of these muscarinic receptors are present on the atria. Indeed, it was thought until recently that there was no vagal innervation of the ventricles, but this view turns out to be incorrect. The ventricles are innervated by the vagus, and muscarinic receptors are, in fact, present in the ventricles, albeit at lower concentrations than in the atria; the amount of muscarinic receptor protein in atrium is approximately twofold greater than in ventricle (200 to 250 vs. 70 to 100 fmol/mg protein). Thus, although the primary function of cardiac muscarinic signaling is heart rate control through actions at the atrial level, vagal stimulation is, in fact, able to directly influence ventricular functioning.8
Clinical Correlate: Adenosine Signaling and Cardiac Function
Understanding of the role of adenosine in cardiac regulation has expanded significantly over the past years. Its established use as an antiarrhythmic compound and its probable role in cardiac preconditioning are two examples of clinical advances resulting from this increase in understanding. Adenosine acts through a GPCR, activating several intracellular signaling systems.9–11
Adenosine Signaling
Although adenosine can be generated by several pathways, in the heart it is usually found as a dephosphorylation product of AMP.12 Because AMP accumulation is a sign of a low cellular energy charge, an increased adenosine concentration is a marker of unbalanced energy demand and supply; thus, ischemia, hypoxemia, and increased catecholamine concentrations are all associated with increased adenosine release. Adenosine is rapidly degraded by various pathways, both intracellularly and extracellularly. As a result, its half-life is extremely short, on the order of 1 second. Therefore, it is not only a marker of a cardiac “energy crisis” but its concentrations will fluctuate virtually instantly with the energy balance of the heart; it provides a real-time indication of the cellular energy situation.
Antiarrhythmic Actions of Adenosine
From these molecular actions of adenosine, its clinical effects can easily be deduced. The antiarrhythmic actions are largely a result of its activation of KACh. Remembering the tissue distribution of KACh, it could be anticipated that adenosine will be much more effective in the treatment of supraventricular arrhythmias than ventricular arrhythmias, and such is indeed the case. Because of its negative chronotropic effects on the atrial conduction system, the compound is most effective in treating supraventricular tachycardias that contain a reentrant pathway involving the atrioventricular node. The efficacy of adenosine in terminating such tachycardias has been reported as greater than 90%. In contrast, it is consistently ineffective in tachycardias not involving the atrioventricular node.13,14
ANESTHETIC ACTIONS
Interactions with Channels: Ca2+ Channels
Anesthetic actions on cardiac Ca2+ channels have been studied in a variety of models. The original observations that halothane blocked Ca2+ flux into heart cells date back to the 1970s, and much specific information has been gained since then.15
Almost all volatile anesthetics inhibit L-type Ca2+ channels.16 Inhibition is modest (25% to 30% at 1 MAC anesthetic) but certainly sufficient to account for the physiologic changes induced by the anesthetics. Volatile anesthetics decrease peak current and, in addition, tend to increase the rate of inactivation. Hence, maximal Ca2+ current is depressed and duration of Ca2+ current is shortened. Together, these actions significantly limit the Ca2+ influx into the cardiac myocyte.
Not only volatile, but also injected, anesthetics have been reported to inhibit cardiac L-type Ca2+ channels in some models. However, the concentrations used generally exceed those used in clinical practice. Thiopental and methohexital block L-type Ca2+ currents. Similarly, propofol has been reported to inhibit these channels, but at concentrations well beyond the clinical range.17
SUMMARY
1. Watson J.A., Crick F.H.C. Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid. Nature. 1953;171:737.
2. Kaupp U.B., Seifert R. Molecular diversity of pacemaker ion channels. Annu Rev Physiol. 2001;63:235.
3. Echt D.S., Liebson P.R., Mitchell L.B., et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N Engl J Med. 1991;324:781.
4. Mohler P.J., Schott J.J., Gramolini A.O., et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634.
5. Antzelevitch C., Brugada P., Brugada J., et al. Brugada syndrome: A decade of progress. Circ Res. 2002;91:1114.
6. Hasking G.J., Esler M.D., Jennings G.L., et al. Norepinephrine spillover to plasma in patients with congestive heart failure: Evidence of increased overall and cardiorenal sympathetic nervous activity. Circulation. 1986;73:615.
7. Harding S.E., Brown L.A., Wynne D.G., et al. Mechanisms of beta-adrenoceptor desensitisation in the failing human heart. Cardiovasc Res. 1994;28:1451.
8. Deighton N.M., Motomura S., Borquez D., et al. Muscarinic cholinoceptors in the human heart: Demonstration, subclassification, and distribution. Naunyn Schmiedebergs Arch Pharmacol. 1990;341:14.
9. Belardinelli L., Shryock J.C., Song Y., et al. Ionic basis of the electrophysiological actions of adenosine on cardiomyocytes. FASEB J. 1995;9:359.
10. Shen W.K., Kurachi Y. Mechanisms of adenosine-mediated actions on cellular and clinical cardiac electrophysiology. Mayo Clin Proc. 1995;70:274.
11. Murphy E. Primary and secondary signaling pathways in early preconditioning that converge on the mitochondria to produce cardioprotection. Circ Res. 2004;94:7.
12. Schutz W., Schrader J., Gerlach E. Different sites of adenosine formation in the heart. Am J Physiol. 1981;240:H963.
13. Rankin A.C., Brooks R., Ruskin J.N., McGovern B.A. Adenosine and the treatment of supraventricular tachycardia. Am J Med. 1992;92:655.
14. diMarco J.P., Sellers T.D., Lerman B.B., et al. Diagnostic and therapeutic use of adenosine in patients with supraventricular tachyarrhythmias. J Am Coll Cardiol. 1985;6:417.
15. Bosnjak Z.J., Supan F.D., Rusch N.J. The effects of halothane, enflurane, and isoflurane on calcium current in isolated canine ventricular cells. Anesthesiology. 1991;74:340.
16. Fassl J., Halaszovich C.R., Huneke R., et al. Effects of inhalational anesthetics on L-type Ca2+ currents in human atrial cardiomyocytes during beta-adrenergic stimulation. Anesthesiology. 2003;99:90.
17. Ikemoto Y., Yatani A., Arimura H., Yoshitake J. Reduction of the slow inward current of isolated rat ventricular cells by thiamylal and halothane. Acta Anaesthesiol Scand. 1985;29:583.