Chapter 14 Valvular Heart Disease: Replacement and Repair
AORTIC STENOSIS
Clinical Features and Natural History
Aortic stenosis is the most common cardiac valve lesion in the United States. One to 2 percent of the population is born with a bicuspid aortic valve, which is prone to stenosis with aging. Calcific aortic stenosis has several features in common with coronary artery disease (CAD). Both conditions are more common in men, older people, and patients with hypercholesterolemia, and both result in part from an active inflammatory process. There is clinical evidence of an atherosclerotic hypothesis for the cellular mechanism of aortic valve stenosis. There is a clear association between clinical risk factors for atherosclerosis and the development of aortic stenosis: elevated lipoprotein levels, increased low-density lipoprotein (LDL) cholesterol, cigarette smoking, hypertension, diabetes mellitus, increased serum calcium and creatinine levels, and male gender.1 The early lesion of aortic valve sclerosis may be associated with CAD and vascular atherosclerosis. Aortic valve calcification is an inflammatory process promoted by atherosclerotic risk factors.
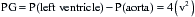
Pathophysiology
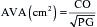
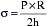
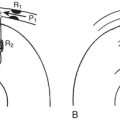
in which P is the intraventricular pressure, R is the inner radius, and h is the wall thickness.
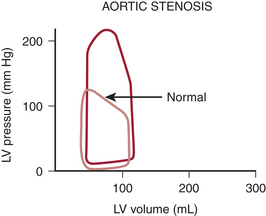
Figure 14-1 shows a typical pressure-volume loop for a patient with aortic stenosis. Two differences from the normal curve are immediately apparent. First, the peak pressure generated during systole is much higher because of the high transvalvular pressure gradient. Second, the slope of the diastolic limb is steeper, reflecting the reduced left ventricular (LV) diastolic compliance that is associated with the increase in chamber thickness. Clinically, this means that small changes in diastolic volume produce relatively large increases in ventricular filling pressure.
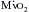 ). The other major determinants of overall
). The other major determinants of overall 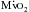 are heart rate, contractility, and, most important, wall tension. Increases in the latter occur as a direct consequence of Laplace’s law in patients with relatively inadequate hypertrophy. The possibility of ischemic contractile dysfunction in the inadequately hypertrophied ventricle arises from increases in wall tension, which directly parallels the imbalance between the elevated peak systolic pressure and the degree of mural hypertrophy. Although there is considerable evidence for “supply-side” abnormalities in the myocardial supply and demand relationship in patients with aortic stenosis, clinical data also support increased
are heart rate, contractility, and, most important, wall tension. Increases in the latter occur as a direct consequence of Laplace’s law in patients with relatively inadequate hypertrophy. The possibility of ischemic contractile dysfunction in the inadequately hypertrophied ventricle arises from increases in wall tension, which directly parallels the imbalance between the elevated peak systolic pressure and the degree of mural hypertrophy. Although there is considerable evidence for “supply-side” abnormalities in the myocardial supply and demand relationship in patients with aortic stenosis, clinical data also support increased 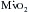 as important in the genesis of myocardial ischemia.
as important in the genesis of myocardial ischemia.Difficulty of Low-Gradient, Low-Output Aortic Stenosis
A subset of patients with severe aortic stenosis, LV dysfunction, and low transvalvular gradient suffers a high operative mortality rate and poor prognosis.2 It is difficult to accurately assess the AVA in this low-flow, low-gradient aortic stenosis because the calculated valve area is proportional to forward SV and because the Gorlin constant varies in low-flow states. Some patients with low-flow, low-gradient aortic stenosis have a decreased AVA as a result of inadequate forward SV rather than anatomic stenosis. Surgical therapy is unlikely to benefit these patients because the underlying pathology is a weakly contractile myocardium. However, patients with severe anatomic aortic stenosis may benefit from valve replacement despite the increased operative risk associated with the low-flow, low-gradient hemodynamic state. Guidelines from the American College of Cardiology (ACC) and American Heart Association (AHA) call for a dobutamine echocardiography evaluation to distinguish patients with fixed anatomic aortic stenosis from those with flow-dependent aortic stenosis with LV dysfunction. Low-flow, low-gradient aortic stenosis is defined for a mean gradient of less than 30 mmHg and a calculated AVA less than 1.0 cm2.
Timing of Intervention
Echocardiography and exercise testing may identify asymptomatic patients who are likely to benefit from surgery.3 In a study of 58 asymptomatic patients, 21 had symptoms for the first time during exercise testing. Guidelines for AVR in patients with aortic stenosis are shown in Table 14-1.
Table 14-1 Recommendations for the Use of Aortic Valve Replacement in Patients with Aortic Stenosis
| Replacement Indicated |
Adapted from the American Heart Association web site (www.americanheart.org).
Anesthetic Considerations
The foregoing pathophysiologic principles dictate that anesthetic management be based on the avoidance of systemic hypotension, maintenance of sinus rhythm and an adequate intravascular volume, and awareness of the potential for myocardial ischemia (Box 14-1). In the absence of CHF, adequate premedication may reduce the likelihood of undue preoperative excitement, tachycardia, and the resultant potential for exacerbating myocardial ischemia and the transvalvular pressure gradient. In patients with truly critical outflow tract obstruction, however, heavy premedication with an exaggerated venodilatory response can reduce the appropriately elevated LVEDV (and LVEDP) needed to overcome the systolic pressure gradient. In these patients in particular, the additional precaution of administering supplementary oxygen may provide worthwhile insurance.
BOX 14-1 Aortic Stenosis
| Preload: | Increased |
| Afterload: | Increased |
| Goal: | Sinus rhythm |
| Avoid: | Hypotension, tachycardia, bradycardia |
HYPERTROPHIC CARDIOMYOPATHY
Hypertrophic cardiomyopathy (HCM, formerly known as hypertrophic obstructive cardiomyopathy) is a relatively common genetic malformation of the heart with a prevalence of approximately 1 in 500. The hypertrophy initially develops in the septum and extends to the free walls, often giving a picture of concentric hypertrophy. Asymmetric septal hypertrophy leads to a variable pressure gradient between the apical LV chamber and the LV outflow tract (LVOT). The LVOT obstruction leads to increases in LV pressure, which fuels a vicious cycle of further hypertrophy and increased LVOT obstruction.4 Various treatment modalities include β-adrenoceptor antagonists, calcium channel blockers, and surgical myectomy of the septum. For more than 40 years, the traditional standard treatment has been the ventricular septal myotomy-myomectomy of Morrow, in which a small amount of muscle from the subaortic septum is resected. Two new treatment modalities have gained popularity in recent years: dual-chamber pacing and septal reduction (ablation) therapy with ethanol.
Clinical Features and Natural History
Patients vary widely in their clinical presentation. The contribution of echocardiography to the diagnosis has unquestionably increased the number of asymptomatic patients who carry the diagnosis. Most patients with HCM are asymptomatic and have been seen by the echocardiographer because of relatives having clinical disease. Follow-up remains an important problem for cardiologists because sudden death or cardiac arrest may occur as the presenting symptom in slightly more than one half of previously asymptomatic patients.5
Pathophysiology
A consensus exists that the disease is characterized by a wide spectrum of the severity of obstruction. It is totally absent in some patients, may be variable in others, or may be critically severe. Its most distinctive qualities are its dynamic nature (depending on contractile state and loading conditions), its timing (begins early, peaks variably), and its subaortic location. Subaortic obstruction arises from the hypertrophied septum’s encroachment on the systolic outflow tract, which is bounded anteriorly by the interventricular septum and posteriorly by the anterior leaflet of the mitral valve. In most patients with obstruction, exaggerated anterior (i.e., toward the septum) motion of the anterior mitral valve leaflet during systole accentuates the obstruction. The cause of this systolic anterior motion (SAM) is unclear. One possibility is that the mitral valve is pulled toward the septum by contraction of the papillary muscles, whose orientation is abnormal because of the hypertrophic process. Another theory is that vigorous contraction of the hypertrophied septum results in rapid acceleration of the blood through a simultaneously narrowed outflow tract. This could generate hydraulic forces consistent with a Venturi effect whereby the anterior leaflet of the mitral valve would be drawn close to or within actual contact with the interventricular septum (Fig. 14-2). This means that after the obstruction is triggered the mitral valve leaflet is forced against the septum by the pressure difference across the orifice. However, the pressure difference further decreases orifice size and further increases the pressure difference in a time-dependent amplifying feedback loop. This analysis is also consistent with observations that the measured gradient is directly correlated with the duration of mitral-septal contact. There appears to be good correlation between the degree of SAM and the magnitude of the pressure gradient. The SAM-septal contact also underlies the severe subaortic obstruction characteristic of HCM of the elderly, although the narrowing is usually more severe and the contribution of septal movement toward the mitral valve is usually greater.
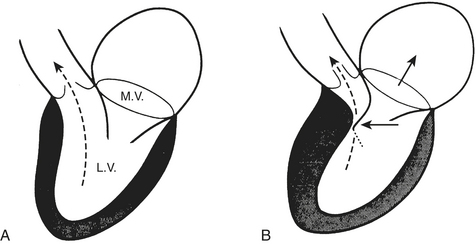
 , specifically the increased overall muscle mass and the high systolic wall tension generated by the ventricle’s ejection against the dynamic subaortic obstruction. However, as in patients with aortic stenosis, there is also evidence of a compromise in myocardial oxygen supply.
, specifically the increased overall muscle mass and the high systolic wall tension generated by the ventricle’s ejection against the dynamic subaortic obstruction. However, as in patients with aortic stenosis, there is also evidence of a compromise in myocardial oxygen supply.Anesthetic Considerations
Priorities in anesthetic management are to avoid aggravating the subaortic obstruction while remaining aware of the derangements in diastolic function that may be somewhat less amenable to direct pharmacologic manipulation (Box 14-2). It is therefore necessary to maintain an appropriate intravascular volume while avoiding direct or reflex increases in contractility or heart rate. The latter goals can be achieved with a deep level of general anesthesia and the associated direct myocardial depression. Regardless of the specific technique, the preservation of an adequate CPP, using vasoconstrictors rather than inotropes, is necessary to avoid myocardial ischemia. Heavy premedication is advisable with a view to avoiding anxiety-induced tachycardia or a reduction in ventricular filling. Chronic β-blockade or calcium channel blockade, or both, should be continued up to and including the day of surgery. These medications should be restarted immediately after surgery, particularly in those patients undergoing noncardiac surgery.
BOX 14-2 Hypertrophic Cardiomyopathy
| Preload: | Increased |
| Afterload: | Increased |
| Goal: | Myocardial depression |
| Avoid: | Tachycardia, inotropes, vasodilators |
AORTIC REGURGITATION
Pathophysiology
Chronically, aortic regurgitation results in a state of LV volume and pressure overload. Progressive volume overloading from aortic regurgitation increases end-diastolic wall tension (i.e., ventricular afterload) and stimulates the serial replication of sarcomeres, producing a pattern of eccentric ventricular hypertrophy.6 This dilation of the ventricle, in accordance with Laplace’s law, also elevates the systolic wall tension, stimulating some concentric hypertrophy. This process of eccentric hypertrophy results in the greatest absolute degrees of cardiomegaly seen in valve disease. End-diastolic volume may be three to four times normal, and very high cardiac outputs can be sustained.
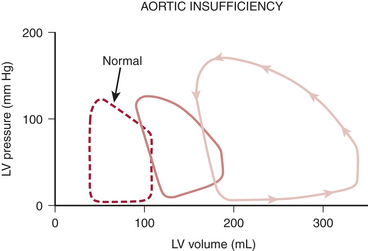
Figure 14-3 shows the pressure-volume loops for acute and chronic aortic regurgitation. In the chronic form, the diastolic pressure-volume curve is shifted far to the right. This permits a tremendous increase in LVEDV with minimal change in filling pressure, a property frequently described as high diastolic compliance.
 , angina can occur in one third of patients with severe aortic regurgitation, even in the absence of CAD. Patients with chronic aortic regurgitation may be at risk for myocardial ischemia caused by hypertrophy-induced abnormalities of the coronary circulation. The increase in total myocardial mass can increase baseline
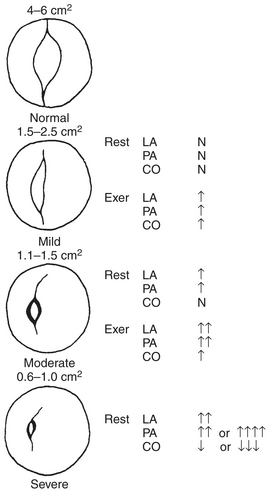
, angina can occur in one third of patients with severe aortic regurgitation, even in the absence of CAD. Patients with chronic aortic regurgitation may be at risk for myocardial ischemia caused by hypertrophy-induced abnormalities of the coronary circulation. The increase in total myocardial mass can increase baseline 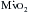 , and there is evidence that total coronary blood flow (CBF), although increased, fails to keep pace with the increase in myocardial mass. Evidence suggests that the insidious development of contractile dysfunction may in part have an ischemic basis.
, and there is evidence that total coronary blood flow (CBF), although increased, fails to keep pace with the increase in myocardial mass. Evidence suggests that the insidious development of contractile dysfunction may in part have an ischemic basis.Anesthetic Considerations
Intraoperative monitoring should include an ECG system with the capability of monitoring a lateral precordial lead, because ischemia is a potential hazard (Box 14-3). For most valvular procedures, a PA catheter provides very useful information. A PA catheter allows determination of basal filling pressures and cardiac output, which is particularly useful in chronic aortic regurgitation given the potential unreliability of the clinical history and EF. Equally important is the ability to accurately monitor ventricular preload and cardiac output response to pharmacologic interventions. The aggressive use of vasodilators is often appropriate therapy perioperatively for the failing ventricle, but their use can compromise the preload to which the ventricle has chronically adjusted. Concurrent preload augmentation, guided by the pulmonary artery diastolic pressure or PCWP, may be crucial to optimize cardiac output when afterload is pharmacologically manipulated. The other requirement for a PA catheter is to allow for pacing when it is anticipated. The deleterious effects of significant bradycardia in aortic regurgitation have been described. In patients who arrive in the operating room with heart rates less than 70 or in patients for whom rapid epicardial pacing may be difficult to establish (e.g., redo operations), placement of a pacing wire is probably indicated. Typically, only a ventricular wire would be appropriate; it is more reliable than atrial pacing and, in aortic regurgitation, the atrial contribution to ventricular diastolic volume usually is not essential. Capturing the ventricle with a PA-based transvenous wire can be difficult because of the very large ventricular cavity size in patients with chronic aortic regurgitation.
BOX 14-3 Aortic Regurgitation
| Preload: | Increased |
| Afterload: | Decreased |
| Goal: | Augmentation of forward flow |
| Avoid: | Bradycardia |
MITRAL REGURGITATION
Clinical Features and Natural History
Because it can be caused by a wide variety of disease processes, the natural history of mitral regurgitation is quite variable. Even among patients with acute-onset disease, the clinical course depends on the mechanism of regurgitation and the response to treatment. For instance, patients presenting with acute, severe mitral regurgitation due to a ruptured papillary muscle have a dismal outcome without surgery. However, the clinical course of acute mitral regurgitation due to endocarditis could be favorable if the patient responds well to antibiotic therapy. Although those with chronic mitral regurgitation usually enter an initial, often asymptomatic, compensated phase, the time course for progression to LV dysfunction and symptomatic heart failure is unpredictable. The literature reflects the wide variability in the natural history of mitral regurgitation, with published 5-year survival rates for patients with mitral regurgitation of 27% to 97%.7
Pathophysiology
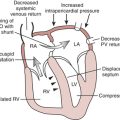
With the eventual decline in LV systolic function, patients enter a decompensated phase. Progressive LV dilatation increases wall stress and afterload, causing further deterioration in LV performance, mitral annular dilatation, and worsening of the mitral regurgitation. LV end-systolic pressure increases. The increased LV filling pressures result in elevation of LA pressures and, given time, pulmonary vascular congestion, pulmonary hypertension, and RV dysfunction. In addition to fatigue and weakness, patients with decompensated, chronic mitral regurgitation may also report dyspnea and orthopnea. It is difficult to predict when a patient with mitral regurgitation is likely to decompensate clinically. The progression of disease in any given patient depends on the underlying cause of mitral regurgitation, its severity, the response of the left ventricle to volume overload, and possibly the effect of medical management.8
Surgical Decision Making
In part because of improved surgical techniques, the operative mortality rate for patients with organic mitral regurgitation who are younger than 75 years is about 1% in some centers. Besides preservation of the subvalvular apparatus, valve repair represents another surgical technique associated with improved postoperative outcome.9 Although not applicable to all patients, such as those with advanced rheumatic disease, the popularity of valve repairs continues to grow. Studies indicate numerous benefits associated with mitral repair. For instance, after accounting for baseline characteristics, patients who undergo mitral repair instead of replacement experience lower operative mortality and better long-term survival largely because of improved postoperative LV function. The survival benefit that accompanies valve repair is also observed among patients undergoing combined valve and coronary artery surgery. Valve repair does not increase the likelihood of reoperation when compared with replacement. Although originally used most often for posterior leaflet disease, surgeons now routinely repair anterior mitral leaflets with good success. When repairing anterior leaflet prolapse, surgeons may insert artificial chordae. The approach to flail or prolapsing posterior mitral leaflet segments often involves resection of a portion of the leaflet. In addition to resecting a portion of the leaflet and plicating the redundant tissue, an annuloplasty ring is often placed to reduce mitral orifice size and return the annulus to a more anatomic shape. Some surgeons favor a flexible, partial, posterior annuloplasty band, which may allow improved systolic contraction of the posterior annulus and better postoperative LV function.
Anesthetic Considerations
Patients who present to the operating room with mitral regurgitation may differ significantly with respect to duration of disease, symptoms, hemodynamic stability, ventricular function, and involvement of the right heart and pulmonary circulation (Box 14-4). For instance, a patient presenting with severe mitral regurgitation due to acute papillary muscle rupture may enter the operating room in cardiogenic shock with pulmonary congestion requiring intra-aortic balloon pump (IABP) augmentation. Another patient with a newly diagnosed flail posterior mitral leaflet may enter the surgical suite with relatively preserved LV function and no symptoms whatsoever. In the latter patient, the compliance of the left atrium may have prevented pulmonary vascular congestion, pulmonary hypertension, and RV dysfunction. Despite the differences in presentation, the general management goals remain similar and include maintenance of forward cardiac output and reduction in the mitral regurgitant fraction. The anesthesiologist must also seek to optimize RV function, in part by avoiding increases in pulmonary vascular congestion and pulmonary hypertension. Various degrees of intervention are needed to achieve these hemodynamic management goals depending on the patient’s presentation.
BOX 14-4 Mitral Regurgitation
| Preload: | Increased |
| Afterload: | Decreased |
| Goal: | Mild tachycardia, vasodilation |
| Avoid: | Myocardial depression |
Intraoperative TEE provides invaluable information during the surgical correction of mitral regurgitation. It reliably identifies the mechanism of mitral regurgitation, thereby guiding the surgical approach,10 and it objectively demonstrates the size and function of the cardiac chambers. TEE can readily identify the cause of hemodynamic derangements, facilitating proper intervention. For instance, the appearance of SAM of the mitral apparatus immediately after valve repair allows the anesthesiologist to intervene with volume infusion and medications such as esmolol or phenylephrine as appropriate. In rare circumstances when hemodynamically significant SAM persists despite these interventions, the surgeon may elect to further repair or even replace the mitral valve. TEE also identifies concomitant pathology that may warrant surgical attention, such as atrial level shunts and additional valve disease.
MITRAL STENOSIS
Clinical Features and Natural History
A decades-long asymptomatic period characterizes the initial phase of rheumatic mitral stenosis. Symptoms rarely appear until the normal mitral valve area of 4 to 6 cm2 (Fig. 14-4) has been reduced to 2.5 cm2 or less. When the mitral valve area reaches 1.5 to 2.5 cm2, symptoms usually occur only in association with exercise or other conditions, such as fever, pregnancy, or atrial fibrillation, that lead to an increase in heart rate or cardiac output. After the mitral valve area falls below 1.5 cm2, symptoms may develop at rest. Some patients are able to remain asymptomatic for long periods by gradually reducing their level of activity. Patients with mitral stenosis commonly report dyspnea as their initial symptom, a finding reflective of elevated LA pressure and pulmonary congestion. In addition to dyspnea, patients may report palpitations that signal the onset of atrial fibrillation. Systemic thromboembolization occurs in 10% to 20% of patients with mitral stenosis and does not appear to be correlated with the mitral valve area or LA size. Chest pain that simulates angina is present in a small number of patients with mitral stenosis and may result from RV hypertrophy rather than CAD.
Pathophysiology
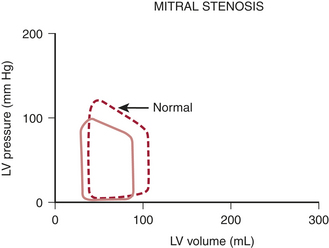
Mitral stenosis results in diminished LV preload reserve. As seen in the pressure-volume loop in Figure 14-5, LVEDV and LVEDP are reduced with an accompanying decline in SV. Controversy exists, however, regarding the contractile state of the left ventricle in these patients. Limited preload may contribute to a reduced EF in some of these patients. However, the observation that LV contractile impairment persists postoperatively in some patients suggests that other causes of LV dysfunction may exist. Rheumatic myocarditis has been reported, although its role in producing LV contractile dysfunction is uncertain.
Assessment of Mitral Stenosis
As for patients with mitral regurgitation, echocardiography represents the diagnostic modality of choice for patients with suspected mitral stenosis.11 Two-dimensional and Doppler echocardiographic techniques are able to accurately and noninvasively measure the transvalvular pressure gradient and mitral valve area. Because the pressure gradient varies with the flow rate and diastolic period, the assessment of mitral stenosis severity ideally should be based on the measured or calculated mitral valve area. Echocardiographic methods used to obtain mitral valve area include the pressure half-time technique, the continuity equation, planimetry of the valve orifice, and Proximal Iso velocity Surface Area (PISA) analysis. Other invaluable information obtained during an echocardiographic study includes the size and function of the ventricles and an estimation of the pulmonary artery pressure.
Surgical Decision Making
Percutaneous mitral commissurotomy (PMC) allows a less invasive, catheter-based approach to mitral stenosis. First reported by Inoue in 1984,12 clinicians worldwide perform PMC more than 10,000 times each year. The technique of PMC involves directing a balloon-tipped catheter across the stenotic mitral valve. Specifically designed balloons allow sequential inflation of the distal and proximal portions of the balloon, ensuring correct positioning across the mitral valve before the middle portion of the device is inflated to split the fused commissures. Patient selection for PMC requires careful echocardiographic evaluation.
Anesthetic Considerations
Several important goals should guide the anesthetic management of patients with significant mitral stenosis. First, the anesthesiologist should seek to prevent tachycardia and treat it promptly if it develops in the perioperative period (Box 14-5). Maintenance of LV preload without exacerbation of pulmonary vascular congestion represents a second management goal. Third, anesthesiologists should avoid factors that aggravate pulmonary hypertension and impair RV function.
BOX 14-5 Mitral Stenosis
| Preload: | Normal or increased |
| Afterload: | Normal |
| Goal: | Controlled ventricular response |
| Avoid: | Tachycardia, pulmonary vasoconstriction |
TRICUSPID REGURGITATION
Clinical Features and Natural History
Surgical tricuspid disease is caused by a structural defect in the valve apparatus or is a functional lesion. Functional tricuspid regurgitation is far more common and usually results from RV overload and tricuspid annular dilation. Left-sided valvular disease, usually mitral regurgitation, is most commonly responsible. Functional tricuspid incompetence can also result from mitral stenosis, aortic regurgitation or stenosis, or from isolated pulmonary hypertension. When mitral regurgitation is severe enough to warrant valve repair or replacement, tricuspid regurgitation may be present in 30% to 50% of patients.13
Tricuspid regurgitation may also be caused by structural defects as in rheumatic valve disease, carcinoid syndrome, endocarditis, Epstein’s anomaly, or trauma.14 In rheumatic disease, histologic involvement of the tricuspid valve may occur in 46% of patients, but it is rarely clinically severe, and in these cases, the valve is usually also stenotic. Tricuspid regurgitation has also been described in association with CAD as a result of ischemia, infarction, or rupture of the RV papillary muscles.
Surgical Decision Making
In structural tricuspid insufficiency, the decision to repair or replace the valve is straightforward. The same cannot be said of functional tricuspid regurgitation. Because most functional cases are the consequence of left-sided valve lesions with RV overload, the tricuspid regurgitation usually improves significantly after the aortic or mitral valve is repaired or replaced, typically at least one grade. It can be unclear in the operating room whether addition of a tricuspid procedure to the left-sided valve surgery is indicated. In this situation, intraoperative TEE plays an essential role. If the tricuspid regurgitation is severe in the pre-CPB assessment, tricuspid valve surgery is almost always performed.14 However, the evidence is less clear when the regurgitation is graded as moderate. Some surgeons choose to repair the tricuspid with moderate regurgitation, but others advocate observation.15 It is common with moderate or moderate to severe tricuspid regurgitation, in the context of left-sided valve surgery, to complete the left-sided procedure and then reassess the tricuspid valve with TEE when the heart is full and ejecting. If the regurgitation remains more than moderate after the left-sided valve is fixed, many surgeons then do the tricuspid procedure. If the regurgitation is moderate or less, the appropriate surgical course may remain unclear. Some patients having left-sided valve procedures must return to the operating room in the future for tricuspid surgery. When this occurs, the morbidity and mortality rates are probably significantly increased over what would have been experienced were the tricuspid valve fixed at the time of the aortic or mitral valve procedure. Decision making in functional tricuspid regurgitation is made more complicated by the inability to rigorously quantify the severity of the regurgitation and RV dysfunction.
INNOVATIONS IN VALVE REPAIR
Aortic Valve Repair
Over the past several years there has been a major shift from valve replacement to valve repair in degenerative mitral valve disease. The same has not been true of the aortic valve, in part because the valve disease is different in most patients, but also because the high flow and pressure conditions across the aortic valve might make repair more prone to failure. That said, aortic valve repair is being increasingly done as an appropriate patient population is being defined. Valve repair for aortic regurgitation has found broader use when regurgitation is associated with dissection or dilation of the aortic root, but isolated valve repair has been less common.16 A growing body of data suggests that aortic valve repair may offer advantages over valve replacement in younger individuals with aortic insufficiency caused by bicuspid valves. In contrast to aortic valve replacement, this eliminates the need for anticoagulation for a mechanical valve and may delay the need for reoperation if a tissue valve has been placed. When regurgitation occurs with a bicuspid valve, the insufficiency is usually caused by retraction or prolapse of the conjoined cusp and repair consists of a triangular incision to shorten and elevate that cusp to improve apposition. Although very long-term follow-up has not been reported, in a large series from the Mayo Clinic with a mean follow-up of 4.2 years, late failure of the repair requiring reoperation occurred in 14 of 160 consecutive patients, with most of that failure occurring from repairs done in the first decade of the 15-year experience.
Techniques for Mitral Valve Repair
Mitral Leaflet Repair
Alfieri and associates showed that mitral regurgitation could be improved using an edge-to-edge technique by which mitral valve leaflets are brought together by a central suture. This surgical approach led to a catheter-based technology, which results in edge-to-edge repair by apposing the edges of a regurgitant mitral valve. The Evalve device consists of a catheter-mounted clip that is threaded using a femoral and trans-septal approach.17 With the use of general anesthesia and echocardiographic and fluoroscopic guidance, the clip is placed to achieve apposition of the central portion of the anterior and posterior leaflets. If the severity of mitral regurgitation is not reduced, the device can be opened and repositioned.
Percutaneous Mitral Annuloplasty
The second type of approach to functional mitral regurgitation is alteration of the mitral annulus, as might occur with a traditional open mitral repair; however, with the percutaneous approach the annulus is downsized by an extracardiac restraint.18 With this technique, interventional cardiologists percutaneously thread a wire from the venous system into the right atrium and the coronary sinus. The relationship between the coronary sinus and posterior leaflet allows downsizing of the septal-lateral diameter from this position. Under echocardiographic guidance, tension is applied to cinch the mitral annulus smaller and an anchor is deployed to maintain position.
Altering Ventricular Anatomy to Reduce Mitral Regurgitation
The third approach to closed mitral valve repair consists of altering the geometry of the lateral and septal LV walls to bring the valve leaflets together. The commercial Coapsys device has entered clinical trials. This device consists of anterior and posterior epicardial pads connected by a cord. With an open chest, the cord is placed transventricularly in a subvalvular position and the tension on the cord is adjusted before the opposing epicardial pad is fixed in place.19 This effectively brings the ventricular walls together and in doing so improves leaflet coaptation. TEE is used to optimize cord length and pad positioning. In contrast to the leaflet-based and annular-based approaches, the Coapsys approach is surgical, requiring an open chest but not CPB. The position of the epicardial vessels and the relationship of the submitral apparatus could pose significant risk, but the device has been used successfully in animal models and preliminary clinical trials.
Percutaneous Valve Replacement
Although surgery, particularly for aortic valve disease, has expanded to include a much older population in recent years, there remains a subset of patients for whom cardiac surgery may entail unacceptable risks. For this population, less invasive techniques such as percutaneous aortic valve replacement are being developed, with the initial clinical experience with percutaneous replacement having been reported.20
It is likely that many of these technologies will come to clinical trial and will demonstrate various degrees of efficacy. However, it is unclear what the long-term benefits of these less invasive interventions will be or how they will compare with each other or with traditional surgical approaches. Some may find use in high-risk patients as temporizing procedures, in place of reoperations, or in conjunction with percutaneous approaches for coronary disease.21 These technologies will get better, and there will be pressure for clinical application. The most important factors will be case selection and long-term follow-up of outcomes; otherwise, these innovations and others like them will add markedly to the burden of health care costs without clear social benefit. For any of the percutaneous valves to succeed, they must demonstrate long-term successful clinical outcomes similar to the excellent results seen with mechanical or tissue valves for more than 10 to 15 years.22
SUMMARY
1. Rajamannan N.M., Gersh B., Bonow R.O. Calcific aortic stenosis: From bench to the bedside—emerging clinical and cellular concepts. Heart. 2003;89:801.
2. Grayburn P.A., Eichhorn E.J. Dobutamine challenge for low-gradient aortic stenosis. Circulation. 2002;106:763.
3. Carabello B.A. Clinical practice. Aortic stenosis. N Engl J Med. 2002;346:677.
4. Roberts R., Sigwart U. New concepts in hypertrophic cardiomyopathies: II. Circulation. 2001;104:2249.
5. Nishimura R.A., Holmes D.R. Hypertrophic obstructive cardiomyopathy. N Engl J Med. 2004;350:1320.
6. Enriquez-Sarano M., Tajik A.T. Clinical practice: Aortic regurgitation. N Engl J Med. 2004;351:1539.
7. Enriquez-Sarano M., Avierinos J.F., Messika-Zeitoun D., et al. Quantitative determinants of the outcome of asymptomatic mitral regurgitation. N Engl J Med. 2005;352:875.
8. Carabello B.A. The pathophysiology of mitral regurgitation. J Heart Valve Dis. 2000;9:600.
9. Enriquez-Sarano M., Schaff H.V., Orszulak T.A., et al. Valve repair improves the outcome of surgery for mitral regurgitation: A multivariate analysis. Circulation. 1995;91:1022.
10. Shapira Y., Vaturi M., Weisenberg D., et al. Impact of intraoperative transesophageal echocardiography in patients undergoing value replacement. Ann Thorac Surg. 2004;78:579.
11. Popovic A.D., Stewart M., Stewart M. Echocardiographic evaluation of valvular stenosis: The gold standard for the next millennium? Echocardiography. 2001;18:59.
12. Iung B., Vahanian A. The long-term outcome of balloon valvuloplasty for mitral stenosis. Curr Cardiol Rep. 2002;4:118.
13. Mueller X.M., Tevaearai H.T., Stumpe F., et al. Tricuspid valve involvement in combined mitral and aortic valve surgery. J Cardiovasc Surg. 2001;42:443.
14. Messika-Zeitoun D., Thomson H., Bellamy M., et al. Medical and surgical outcome of tricuspid regurgitation caused by flail leaflets. J Thorac Cardiovasc Surg. 2004;128:296.
15. Shatapathy P., Aggarwal B.K., Kamath S.G. Tricuspid valve repair: A rational alternative. J Heart Valve Dis. 2000;9:276.
16. Minakata K., Schaff H.V., Zehr K.J., et al. Is repair of aortic valve regurgitation a safe alternative to valve replacement? J Thorac Cardiovasc Surg. 2004;127:645.
17. Block P.C. Percutaneous mitral valve repair for mitral regurgitation. J Int Cardiol. 2003;16:93.
18. Kaye D.M., Byrne M., Alferness C., et al. Feasibility and short-term efficacy of percutaneous mitral annular reduction for the therapy of heart failure-induced mitral regurgitation. Circulation. 2003;108:1795.
19. Inoue M., McCarthy P.M., Popovic Z.B., et al. The Coapsys device to treat functional mitral regurgitation: In vivo long-term canine study. J Thorac Cardiovasc Surg. 2004;127:1068-discussion 1076.
20. Cribier A., Eltchaninoff H., Tron C., et al. Early experience with percutaneous transcatheter implantation of heart valve prosthesis for the treatment of end-stage inoperable patients with calcific aortic stenosis. J Am Coll Cardiol. 2004;43:698.
21. Byrne J.G., Leacche M., Unic D., et al. Staged initial percutaneous coronary intervention followed by valve surgery (“hybrid approach”) for patients with complex coronary and valve disease. J Am Coll Cardiol. 2005;45:14.
22. Rahimtoola S.H. The year in valvular heart disease. J Am Coll Cardiol. 2006;47:427.