Chapter 16 Congenital Heart Disease in Adults
Advances in perioperative care for children with congenital heart disease (CHD) over the past several decades has resulted in an ever-increasing number of these children reaching adulthood with their cardiac lesions palliated or repaired. The first paper on adult CHD was published in 1973; the field has grown such that there is now a text devoted to it, and even a specialty society dedicated to it, the International Society for Adult Congenital Cardiac Disease (http://www.isaccd.org).1 There are estimated to be about 32,000 new cases of CHD each year in the United States and 1.5 million worldwide. More than 85% of infants born with CHD are expected to grow to adulthood. It is estimated that there are more than 500,000 adults in the United States with CHD; 55% of these adults remain at moderate to high risk, and more than 115,000 have complex disease.2,3 There are as many adults with CHD as there are children, and the number of adults will only continue to increase. These patients can be seen by anesthesiologists for primary cardiac repair, repair after a prior palliation, revision of repair due to failure or lack of growth of prosthetic material, or conversion of a suboptimal repair to a more modern operation (Box 16-1). In addition, these adults with CHD will be seen for all the other ailments of aging and trauma that require surgical intervention. Although it has been suggested that teenagers and adults can have repair of congenital cardiac defects with morbidity and mortality approaching that of surgery done during childhood, these data are limited and may reflect only a relatively young and acyanotic sampling.4 Other data suggest that, in general, adults older than 50 years of age represent an excessive proportion of the early postoperative mortality encountered, and the number of previous operations and cyanosis are both risk factors. Risk factors for noncardiac surgery include heart failure, pulmonary hypertension, and cyanosis.5
These patients bring with them anatomic and physiologic complexities of which physicians accustomed to caring for adults may be unaware and medical problems associated with aging or pregnancy that might not be familiar to physicians used to caring for children. This problem has led to the establishment of the growing subspecialty of adult CHD. An informed anesthesiologist is a critical member of the team required to care optimally for these patients. A specific recommendation is that noncardiac surgery on adult patients with moderate to complex CHD be done at an adult congenital heart center (regional centers) with the consultation of an anesthesiologist experienced with adult CHD. In fact, one of the founding fathers of the subspecialty wrote: “A cardiac anesthesiologist with experience in CHD is pivotal. The cardiac anesthesiologist and the attending cardiologist are more important than the noncardiac surgeon.”2 Centers may find it helpful to delegate one attending anesthesiologist to be the liaison with the cardiology service to centralize preoperative evaluations and triage of patients to an anesthesiologist with specific expertise in managing patients with CHD, rather than random consultations with generalist anesthesiologists.
GENERAL NONCARDIAC ISSUES WITH LONG-STANDING CONGENITAL HEART DISEASE
A variety of organ systems can be affected by long-standing CHD; these are summarized in Table 16-1. Because congenital cardiac disease can be one manifestation of a multiorgan genetic or dysmorphic syndrome, all patients require a full review of systems and examination.6,7
Table 16-1 Potential Noncardiac Organ Involvement in Patients with Congenital Heart Disease
| Potential Respiratory Implications
• Decreased compliance (with increased pulmonary blood flow or impediment to pulmonary venous drainage)
|
CARDIAC ISSUES
The number of cardiac lesions and subtypes, together with the large number of contemporary and obsolescent palliative and corrective surgical procedures, makes a complete discussion of all CHD impossible. The reader is referred to one of the current texts on pediatric cardiac anesthesia for more detailed descriptions of these lesions, the available surgical repairs, and the anesthetic implications during primary repair.8,9 Some general perioperative guidelines to caring for these patients are offered in Table 16-2.
Table 16-2 General Approaches to Anesthesia for Patients with Congenital Heart Disease
| General |
| The best care for both cardiac and noncardiac surgery in adult patients with CHD is afforded in a center with a multidisciplinary team experienced in the care of adults with CHD and knowledgeable about both the anatomy and physiology of CHD and also the manifestations and considerations specific to adults with CHD. |
| Preoperative |
|
• Review most recent laboratory data, catheterization, and echocardiogram and other imaging data. The most recent office letter from the cardiologist is often most helpful. Obtain and review these in advance.
|
Aortopulmonary Shunts
Depending on their age, adult patients may have had one or more of several aortopulmonary shunts to palliate cyanosis during childhood. These are shown in Figure 16-1. Although life saving, there were considerable shortcomings of these shunts in the long term. All were inherently inefficient, because some of the oxygenated blood returning through the pulmonary veins to the left atrium and ventricle would then return to the lungs through the shunt, thus volume loading the ventricle. It was difficult to quantify the size of the earlier shunts such as the Waterston (side-to-side ascending aorta to right pulmonary artery) and Potts (side-to-side descending aorta to left pulmonary artery). If too small, the patient was left excessively cyanotic; if too large, there was pulmonary overcirculation with the risk of developing pulmonary vascular disease. The Waterston, in fact, could on occasion result in a hyperperfused, hypertensive ipsilateral pulmonary artery and a hypoperfused contralateral pulmonary artery as it could direct flow to the ipsilateral side. There were also surgical issues when complete repair became possible. Takedown of Waterston shunts often required a pulmonary arterioplasty to correct deformity of the pulmonary artery at the site of the anastomosis, and the posteriorly located Potts anastomoses could not be taken down from a median sternotomy. Patients with a classic Blalock-Taussig shunt almost always lack palpable pulses on the side of the shunt. Even if there is a palpable pulse (from collateral flow around the shoulder), blood pressure obtained from that arm will be artifactually low. Even after a modified Blalock-Taussig shunt (using a piece of Gore-Tex tubing instead of an end-to-side anastomosis of the subclavian and pulmonary arteries), there can be a blood pressure disparity between the arms. To ensure a valid measurement, preoperative blood pressure should be measured in both arms (Table 16-3).
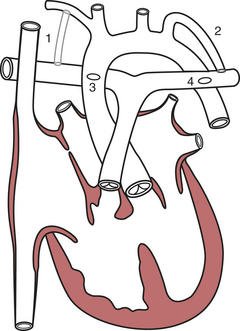
| Waterston | Ascending aorta → right pulmonary artery | No longer done |
| Potts | Descending aorta → left pulmonary artery | No longer done |
| Classic Blalock-Taussig | Subclavian artery → ipsilateral pulmonary artery | No longer done |
| Modified Blalock-Taussig | Gore-Tex tube subclavian artery → ipsilateral pulmonary artery | Current |
| Central shunt | Gore-Tex tube ascending aorta → main pulmonary artery | Current |
Atrial Septal Defect and Partial Anomalous Pulmonary Venous Return
There are several anatomic types of ASD. The most common type, and if otherwise undefined the presumptive type, is the secundum type located in the midseptum. The primum type at the lower end of the atrial septum is a component of the endocardial cushion defects, the most primitive of which is the common atrioventricular canal. The sinus venosus type, high in the septum near the entry of the superior vena cava, is almost always associated with partial anomalous pulmonary venous return, most frequently drainage of the right upper pulmonary vein to the low superior vena cava. An uncommon atrial septal-type defect is when blood passes from the left atrium to the right via an unroofed coronary sinus. Only secundum defects are considered, although the natural histories of all of the defects are similar (Box 16-2).
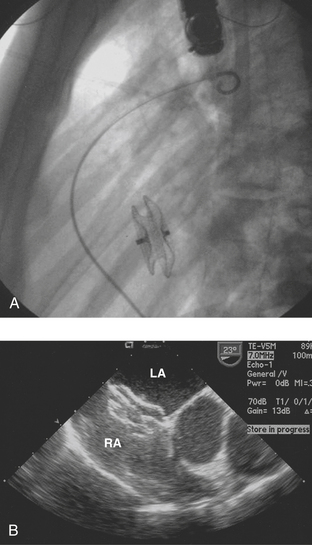
Late closure of the defect, after 5 years of age, has been associated with incomplete resolution of right ventricular dilation. Left ventricular dysfunction has been reported in some patients having defect closure in adulthood, and closure particularly in middle age may not prevent the development of atrial tachyarrhythmias or stroke.10 Survival of patients without pulmonary vascular disease has been reported to be best if operated on before 24 years of age, intermediate if operated on between 25 and 41 years of age, and worst if operated on thereafter. However, more recent series have shown that even at ages over 40, surgical repair provides an overall survival and complication-free benefit compared with medical management.11 Surgical morbidity in these patients is primarily atrial fibrillation, atrial flutter, or junctional rhythm. Current practice is to close these defects in adults in the catheterization laboratory via transvascular devices if anatomically practical (Fig. 16-2). For example, there needs to be an adequate rim of septum around the defect to which the device can attach. Device closure is inappropriate if the defect is associated with anomalous pulmonary venous drainage. The indications for closure are the same as for surgical closure.12
Coarctation of the Aorta
Nonrepaired coarctation of the aorta in the adult brings with it significant morbidity and mortality. Mortality is 25% by age 20, 50% by age 30, 75% by age 50, and 90% by age 60. Left ventricular aneurysm, rupture of cerebral aneurysms, and dissection of a postcoarctation aneurysm all contribute to the excessive mortality. Left ventricular failure can occur in patients over 40 with nonrepaired lesions. If repair is not undertaken early, there is incremental risk for the development of premature coronary atherosclerosis. Even with surgery, coronary artery disease remains the leading cause of death 11 to 25 years after surgery. Coarctation is accompanied by a bicuspid aortic valve in the majority of patients. Although endocarditis of this abnormal valve is a lifelong risk, these valves often do not become stenotic until middle age or later. Coarctation can also be associated with mitral valve abnormalities (Box 16-3).
Aneurysms at the site of coarctation repair can develop years later, and restenosis as well can develop in adolescence or adulthood. Repair includes resection of the coarctation and end-to-end reanastomosis. Because this sometimes resulted in recoarctation when done in infancy, for many years a common repair was the Waldhausen or subclavian flap operation, in which the left subclavian artery is ligated and the proximal segment opened and rotated as a flap to open the area of the coarctation. Aneurysms in the area of repair are a particular concern in adolescents and adults after coarctectomy. Persistent systemic hypertension is common after coarctation repair. Adult patients require continued periodic follow-up for hypertension. A pressure gradient of 20 mmHg or more (less in the presence of extensive collaterals) is an indication for treatment. Recoarctation can be treated surgically or by balloon angioplasty with stenting.13 Surgical repair of recoarctation or aneurysm in adults is associated with increased mortality and can be associated with significant intraoperative bleeding. It requires lung isolation for optimal surgical exposure and placement of an arterial catheter in the right arm.
Eisenmenger’s Syndrome
Eisenmenger physiology is compatible with survival into adulthood. However, reported rates of survival after diagnosis vary, probably based on the relatively long life expectancy and variability in the time of diagnosis. Cantor and coworkers reported median survival to 53 years but with wide variation. Others reported survival of 80% at 10 years after diagnosis and 42% at 25 years; or survival of 77% at 5 years and 58% at 10 years. Syncope, increased central venous pressure, and arterial desaturation to less than 85% are all associated with poor short-term outcome.14,15 Most deaths are sudden cardiac deaths. Other causes of death include heart failure, hemoptysis, brain abscess, thromboembolism, and complications of pregnancy and noncardiac surgery. These patients face potentially significant perioperative risks. Findings of Eisenmenger’s syndrome are summarized in Table 16-4.
Patent Ductus Arteriosus
Beyond the neonatal period, spontaneous closure of a PDA is uncommon. The risk of a long-standing moderate to large PDA is volume overloading of the left atrium and left ventricle with the risk of development of pulmonary vascular disease. With time, the ductus can become calcified or aneurysmally dilated with a risk of rupture. Ductal calcification or aneurysm increases the risk of surgery, which rarely requires cardiopulmonary bypass. Nonrepaired, the natural history is for one third of patients to die of heart failure, pulmonary hypertension, or endocarditis by 40 years of age and two thirds by age 60. Although small PDAs are of no hemodynamic consequence, even small PDAs carry relatively high endocarditis risk. Surgical closure should be considered for all adults with PDA, and transvascular closure by means of one of several devices is possible.16
Tetralogy of Fallot
The classic description of tetralogy of Fallot includes (1) a large, nonrestrictive malaligned VSD, with (2) an overriding aorta, (3) infundibular pulmonic stenosis, and (4) consequent right ventricular hypertrophy, all derived from an embryonic anterocephalad deviation of the outlet septum. However, there is a spectrum of disease, with more severe defects including stenosis of the pulmonary valve, stenosis of the pulmonary valve annulus, or stenosis and hypoplasia of the pulmonary arteries in the most severe cases. Pentalogy of Fallot refers to the addition of an ASD. With advances in genetics, up to one third or more of cases of tetralogy have been ascribed to one of several genetic abnormalities, including trisomy 21, the 22q11 microdeletion, the genes NKX 2.5 and FOG 2.4, and others. Tetralogy of Fallot is the most common cyanotic lesion encountered in the adult population. Nonrepaired or nonpalliated, approximately 25% of patients survive to adolescence, after which the mortality is 6.6% per year. Only 3% survive to age 40.17 Unlike children, teenagers and adults with tetralogy do not develop “tet spells.” Long-term survival with a good quality of life is expected after repair. The 32- to 36-year survival has been reported to be 85% to 86%, although symptoms, primarily arrhythmias and decreased exercise tolerance, occur in 10% to 15% at 20 years after the primary repair (Box 16-4).
It is uncommon to encounter an adolescent or adult with nonrepaired tetralogy. However, it can be encountered in immigrants or in patients whose anatomic variation was considered to be inoperable when they were children. In tetralogy, the right ventricle “sees” the obstruction from the pulmonic stenosis. Pulmonary vascular resistance is typically normal to low. Right-to-left shunting is unaffected by attempts at modulating pulmonary vascular resistance. Shunting is minimized, however, by pharmacologically increasing systemic vascular resistance. Increases in the inotropic state of the heart increase the dynamic obstruction at the right ventricular infundibulum and worsen right-to-left shunting. β-Blockers are often used to decrease inotropy. Although halothane was the historic anesthetic of choice in children with tetralogy due to its myocardial depressant effects and ability to maintain systemic vascular resistance, current practice is to use sevoflurane, without undue consequence from a reduction in systemic vascular resistance. Anesthetic induction in adults can easily be achieved with narcotics.18
Some patients require repair of pulmonic stenosis by placement of a transannular patch, with obligate residual pulmonary insufficiency. Isolated mild to moderate pulmonary insufficiency is generally well tolerated, but in the long term it can contribute to right ventricular dysfunction with a risk of ventricular tachycardia and sudden death. Atrial tachyarrhythmias occur in about one third of adults late after repair and can contribute to late morbidity.19 The development of atrial flutter or atrial reentrant tachycardia is often a harbinger of hemodynamic compromise. The substrate is usually an atrial surgical scar and the trigger is atrial dilation, such as from tricuspid insufficiency with right ventricular dysfunction. The mechanism for the development of ventricular arrhythmias is presumably the same, namely dilation superimposed on surgical scar.
Sudden death or ventricular tachycardia requiring treatment can occur in up to 5.5% of postoperative patients over 30 years, often years postoperatively. The foci for these arrhythmias are typically in the right ventricular outflow tract in the area that has had surgery, and they can be ablated in the catheterization laboratory. Older age at repair, severe left ventricular dysfunction, postoperative right ventricular hypertension from residual or recurrent outflow tract obstruction, wide-open pulmonary insufficiency, and prolongation of the QRS are all predictors of sudden death.20 Premature ventricular contractions and even nonsustained ventricular tachycardia are not rare but do not seem to be associated with sudden death, making appropriate treatment options difficult. It has been suggested that QRS prolongation to longer than 180 milliseconds is a risk factor. This marker, although highly sensitive, has a low positive predictive value. The impact of this risk factor in the current group of younger patients who have not had ventriculotomies is unclear, as their initial postoperative QRS durations are shorter than in patients who had a right ventriculotomy.
Most adult patients require reoperation to repair the right ventricular outflow tract or to insert or replace a valve in the pulmonic position. Other reasons for reoperation include repair of an outflow tract aneurysm at the site of a patch, repair of a residual VSD, or repair of an incompetent tricuspid valve. These patients often have diminished right ventricular diastolic compliance and require higher-than-normal central venous pressure. Postoperative management includes minimizing pulmonary vascular resistance and maintaining central venous pressure. Patients often require treatment post-bypass with an inotrope and afterload reduction.21
Transposition of the Great Arteries (D-Transposition)
Very-long-term outcome after the arterial switch procedure is still not known. It does appear that there is essentially no mortality after 5 years postoperatively, and late surgical reintervention is mostly due to supravalvular pulmonic stenosis.22 Although many of these children have abnormal resting myocardial perfusion, up to 9% can have evidence of exercise-induced myocardial ischemia. The implication for the development of premature coronary artery disease in adulthood is not known, and there is also some concern about the ultimate function of the neoaortic valve.
SUMMARY
1. Gatzoulis M., Webb G.D., Daubeney P.E.F. Diagnosis and Management of Adult Congenital Heart Disease. Philadelphia: Churchill Livingstone, 2003.
2. Perloff J.K., Warnes C.A. Challenges posed by adults with repaired congenital heart disease. Circulation. 2001;103:2637.
3. Warnes C.A., Liberthson R., Danielson G.K., et al. Task force 1: The changing profile of congenital heart disease in adult life. J Am Coll Cardiol. 2001;37:1170.
4. Andropoulos D.B., Stayer S.A., Skjonsby B.S., et al. Anesthetic and perioperative outcome of teenagers and adults with congenital heart disease. J Cardiothorac Vasc Anesth. 2002;16:731.
5. Warnes C.A. The adult with congenital heart disease: Born to be bad? J Am Coll Cardiol. 2005;46:1-8.
6. Kamphuis M., Ottenkamp J., Vliegen H.W., et al. Health-related quality of life and health status in adult survivors with previously operated complex congenital heart disease. Heart. 2002;87:356.
7. Vonder Muhll I., Cumming G., Gatzoulis M.A. Risky business: Insuring adults with congenital heart disease. Eur Heart J. 2003;24:1595.
8. Lake C.L., Booker P.D. Pediatric Cardiac Anesthesia, 4th edition, Philadelphia: Lippincott-Williams and Wilkins, 2004.
9. Andropoulos D.B., Stayer S.A., Russell I.A. Anesthesia for Congenital Heart Disease. Armonk, NY: Futura; 2004.
10. Gatzoulis M.A., Freeman M.A., Siu S.C., et al. Atrial arrhythmia after surgical closure of atrial septal defects in adults. N Engl J Med. 1999;340:839.
11. Attie F., Rosas M., Granados N., et al. Surgical treatment for secundum atrial septal defects in patients >40 years old. A randomized clinical trial. J Am Coll Cardiol. 2001;38:2035.
12. Du Z.D., Hijazi Z.M., Kleinman C.S., et al. Comparison between transcatheter and surgical closure of secundum atrial septal defect in children and adults: Results of a multicenter nonrandomized trial. J Am Coll Cardiol, 39. 2002:1836.
13. Hamdan M.A., Maheshwari S., Fahey J.T., Hellenbrand W.E. Endovascular stents for coarctation of the aorta: Initial results and intermediate-term follow-up. J Am Coll Cardiol. 2001;38:1518.
14. Vongpatanasin W., Brickner M.E., Hillis L.D., Lange R.A. The Eisenmenger syndrome in adults. Ann Intern Med. 1998;128:745.
15. Cantor W.J., Harrison D.A., Moussadji J.S., et al. Determinants of survival and length of survival in adults with Eisenmenger syndrome. Am J Cardiol. 1999;84:677.
16. Fisher R.G., Moodie D.S., Sterba R., Gill C.C. Patent ductus arteriosus in adults-long-term follow-up: Nonsurgical versus surgical treatment. J Am Coll Cardiol. 1986;8:280.
17. Nollert G., Fischlein T., Bouterwek S., et al. Long-term survival in patients with repair of tetralogy of Fallot: 36-Year follow-up of 490 survivors of the first year after surgical repair. J Am Coll Cardiol. 1997;30:1374.
18. Russell I.A., Miller Hance W.C., Gregory G., et al. The safety and efficacy of sevoflurane anesthesia in infants and children with congenital heart disease. Anesth Analg. 2001;92:1152.
19. Harrison D.A., Siu S.C., Hussain F., et al. Sustained atrial arrhythmias in adults late after repair of tetralogy of Fallot. Am J Cardiol. 2001;87:584.
20. Abd El Rahman M.Y., Abdul-Khaliq H., Vogel M., et al. Relation between right ventricular enlargement, QRS duration, and right ventricular function in patients with tetralogy of Fallot and pulmonary regurgitation after surgical repair. Heart. 2000;84:416.
21. Heggie J., Poirer N., Williams R.G., Karski J. Anesthetic considerations for adult cardiac surgery patients with congenital heart disease. Semin Cardiothorac Vasc Anesth. 2003;7:141.
22. Losay J., Touchot A., Serraf A., et al. Late outcome after arterial switch operation for transposition of the great arteries. Circulation. 2001;104:I-121.