[level-membership-for-anesthesiology-category]
7 Molecular and Genetic Cardiovascular Medicine
The past decades have witnessed what may be termed a revolution in the biomedical sciences, as molecular and genetic methodologies have suddenly jumped onto the clinical scene. Molecular biology originated in the 1950s, its birth most commonly identified with the description of the structure of deoxyribonucleic acid (DNA) by Watson and Crick.1 For many years after, it was practiced almost exclusively in the research laboratory. Much of this research involved the laborious process of cloning: the identification of DNA molecules encoding specific proteins. Although at the time most people in the field realized that these advances would one day be of immense importance to clinical medicine, the exact place they would take was unclear.
The machinery behind the cardiac rhythm: ion channels
The cardiac action potential results from the flow of ions through ion channels, which are the membrane-bound proteins that form the structural machinery behind cardiac electrical excitability. In response to changes in electrical potential across the cell membrane, ion channels open and allow the passive flux of ions into or out of the cell along their electrochemical gradients. This flow of charged ions results in a current, which will alter the cell membrane potential toward the potential at which the electrochemical gradient for the ion is zero and is called the equilibrium potential (E) for the ion. Depolarization of the cell could, in principle, result from an inward cation current or an outward anion current; for repolarization, the reverse is true. In excitable cells, action potentials are mainly caused by the flow of cation currents. Membrane depolarization results principally from the flow of Na+ down its electrochemical gradient (ENa is around +50mV), whereas repolarization results from the outward flux of K+ down its electrochemical gradient (EK is around −90 mV). Opening and closing of ion channels selective for a single ion result in an individual ionic current. The integrated activity of many different ionic currents, each activated over precisely regulated potential ranges and at different times in the cardiac cycle, results in the cardiac action potential. Ion channels are usually highly (but not uniquely) selective for a single ion (e.g., K+ channels, Na+ channels). Channels may rectify, that is, pass current in one direction across the membrane more easily than the other. Electrical and chemical stimuli, which lead to opening and closing of the channel, cause a conformational change in the channel molecule (gating). The rate of change of channel conformation (gating kinetics) may be rapid, in which case the channel will open (activate) almost immediately (e.g., Na+ channels), or relatively slowly, which will result in a delay in channel activation (e.g., delayed rectifier K+ channels). After activation, ion channels may stay open until closed by another stimulus (e.g., repolarization of the membrane) or may close (inactivate) in the face of a continued stimulus. Inactivated channels will usually not reopen on repeat stimulation until they have recovered from inactivation (Box 7-1).

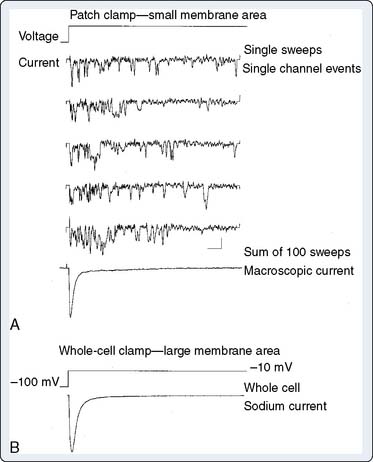
Patch Clamping
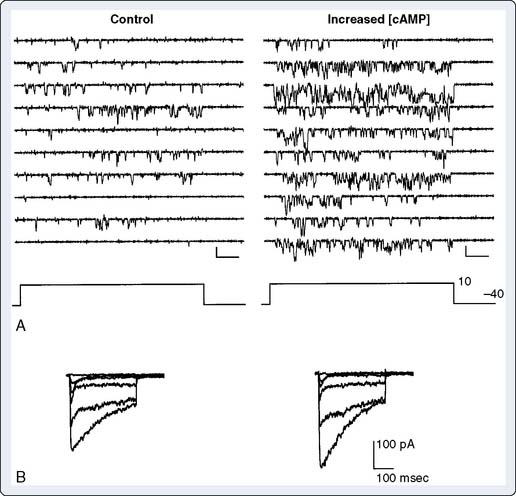
The development of the voltage clamp technique in the early 1950s and its application to multicellular preparations of cardiac muscle allowed identification of the major ionic currents that underlie the cardiac action potential. The whole-cell currents recorded by the technique were smooth waveforms derived from summation of the activity of thousands of ion channels, and many different patterns of events at the single-channel level, arising from more than one molecular specie, can summate to produce identical whole-cell current waveforms. Patch clamping allows resolution of events at the single-channel level. In this technique, small patches of cell membrane (< 1 μm2) are isolated electrically and physically in the tip of a glass micropipette.2 Single-channel events can then be resolved because there are only a few ion channel molecules present in the patch. Current flowing across the patch typically jumps between well-defined values corresponding to sudden opening or closing of the ion-conducting pore (Figure 7-1A). The whole-cell current will be the sum of the currents through all of the individual channels in the cell membrane; summation of the current flowing through a single channel during repeated stimuli will reproduce the macroscopic whole-cell current (see Figures 7-1A and B). As channel opening and closing in response to a stimulus are a stochastic phenomena, the regulation of ion flow, whether resulting from a change in membrane potential or from interaction with regulator molecules, is usually achieved by increasing the probability that the channel will be open. Thus, in Figure 7-1, which shows records from human cardiac muscle Na+ channels, the channels open (activate) a few milliseconds after depolarization because the probability that the channel will be open increases. Similarly, as the channels spontaneously close (inactivate), the open probability decreases.

Electrical Events Underlying the Cardiac Action Potential
Figure 7-2 shows a diagram of a cardiac action potential with a summary of the ionic currents flowing during each phase. As far as has been determined, the probable molecular identity of the ion channels that underlie these currents is also given. This section examines the biophysical properties of these currents; subsequent sections focus on possible molecular mechanisms underlying the biophysical phenomena.
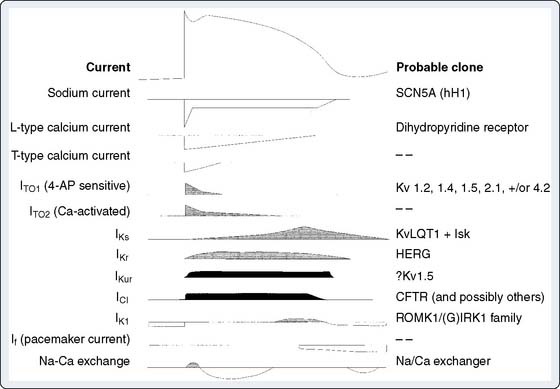
Figure 7-2 Ionic currents underlying the cardiac action potential.
(From Roden DM, Lazzarra R, Rosen R, et al, for the SADS Foundation Task Force on LQTS: Multiple mechanisms in the long QT syndrome: Current knowledge, gaps and future directions. Circulation 94:1996, 1996.)
The Resting Membrane Potential and the Role of IK1
The resting cardiac membrane potential is maintained close to the equilibrium potential for potassium (EK) by a background, inwardly rectifying, highly selective K+ current (IK1). Under physiologic conditions, the EK is around −90 mV, and displacement of the membrane potential from this value should result in an inward or outward current through IK1, thus returning the membrane potential toward EK. However, although IK1 passes significant inward current at potentials negative to EK, at positive potentials, the channels display inward rectification (i.e., they pass inward current more easily than outward current), which limits outward current flow and reduces the tendency for IK1 to hyperpolarize the membrane. This has obvious significance for an excitable cell; inward rectification3 allows the initiation of action potentials and limits cellular K+ loss during the action potential. IK1 is large in ventricular cells, smaller in atrial cells, and almost absent in nodal tissue.4 This explains why ventricular cells rest near EK and have a high threshold for excitation, atrial cells have a more positive resting potential, and nodal cells have no defined resting potential.
Phase 0: Rapid Upstroke of the Cardiac Action Potential
The rapid upstroke of the cardiac action potential (phase 0) is caused by the flow of a large inward Na+ current (Ina) (Box 7-2).5 INa is activated by depolarization of the sarcolemma to a threshold potential of −65 to −70 mV. INa activation, and hence the action potential, is an all-or-nothing response. Subthreshold depolarizations have only local effects on the membrane. After the threshold for activation of fast Na+ channels is exceeded, Na+ channels open (i.e., INa activates) and Na+ ions enter the cell down their electrochemical gradient. This results in displacement of the membrane potential toward the equilibrium potential for Na+ ions, around +50 mV. INa activation is transient, lasting at most 1 to 2 msec because, simultaneous with activation, a second, slightly slower conformational change in the channel molecule occurs (inactivation), which closes the ion pore in the face of continued membrane depolarization (see Figures 7-1A and B). The channel cannot open again until it has recovered from inactivation (i.e., regained its resting conformation), a process that requires repolarization to the resting potential for a defined period. Thus, the channels cycle through three states: resting (and available for activation), open, and inactivated. Although the channel is inactivated, it is absolutely refractory to repeated stimulation. Stimuli that occur during recovery from inactivation will result in opening of fewer Na+ channels (because not all have recovered), and the resulting action potential will have a reduced maximal rate of depolarization and reduced conduction velocity. Na+ channels do not need to open to become inactivated. If the resting membrane potential depolarizes for a time, inactivation will occur in some channels and subsequent stimulation will result in an action potential of reduced amplitude and conduction velocity.
Phase 1: Early Rapid Repolarization
The early rapid repolarization phase of the action potential, which follows immediately after phase 0, results both from rapid inactivation of the majority of the Na+ current and from activation of a transient outward current (ITO), carried mainly by K+ ions. On depolarization of the membrane, ITO opens rapidly, over about 20 msec, before spontaneously inactivating. ITO comprises two separate currents: the rapidly inactivating ITO1, which is activated by depolarization and blocked by 4-aminopyridine, and the slowly inactivating ITO2, which is activated by elevated intracellular Ca++ (possibly explaining the observation that action potential duration tends to decrease with rapid heart rates and hypercalcemia)6,7 (Figure 7-3).
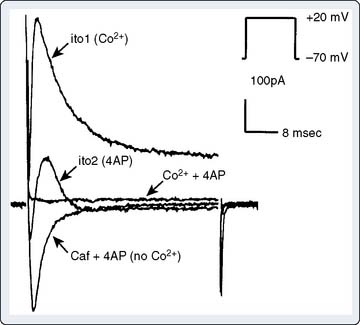
In addition to its effect on phase 1, ITO, in combination with the delayed rectifier potassium currents (IKr and IKs) and IK1, also contributes to membrane repolarization. Arrhythmogenic prolongation of the action potential in myocardial cells recovered from patients with myocardial hypertrophy,8 congestive cardiomyopathy,6 and from the border zone of myocardial infarction in animals9 appears to result from depression of ITO.
Phases 2 and 3: Plateau Phase and Final Rapid Repolarization
The action potential plateau and final rapid repolarization are mediated by a balance between the slow inward current and outward, predominantly K+ current. During the plateau phase, membrane conductance to all ions falls and very little current flows. Potassium conductance is low because of inward rectification of IK1 (i.e., inward current passes more easily than outward current), so little outward current flows despite the large outward electrochemical gradient for K+ ions and the delayed onset of the outwardly rectifying K+ currents (IKs, IKr, and IKur). The resulting small outward current is balanced by inward current, predominantly through L-type Ca++ channels (ICa-L), but also via a slowly inactivating population of Na+ channels, and a small inward flux of chloride (Cl–) ions, possibly carried by the cardiac variant of the ATP-dependent channel (abnormalities of which underlie cystic fibrosis).10,11 Phase 3, regenerative rapid repolarization, results from time-dependent inactivation of L-type Ca++ current and increasing outward current through delayed rectifier K+ channels. The net membrane current becomes outward and the cell repolarizes.
Slow Inward Ca++ Current
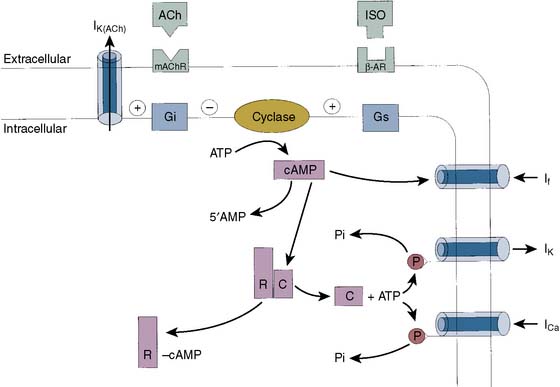
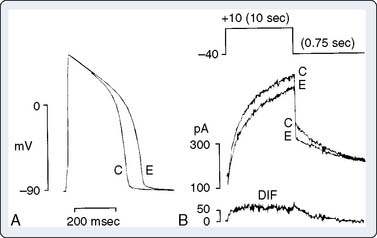
The slow inward current (ICa-L) is activated by depolarization of the cell to potentials less negative than −40 to −50 mV. In ventricular and atrial myocytes and in Purkinje fibers, ICa-L is activated by the regenerative depolarization caused by INa during phase 0 of the action potential. ICa-L does not contribute significantly to phase 0 because, in comparison with INa, it activates much more slowly (over about 10 msec) and is smaller in amplitude. ICa-L also inactivates slowly and, therefore, contributes the major inward current during the plateau of the action potential. ICa-L flows through L-type (long-lasting) Ca++ channels, which are sensitive to block by dihydropyridines (e.g., nifedipine), and activation of contraction is related to the magnitude of the resulting calcium influx.12 Gating of ICa-L is generally similar to INa in that channel opening and closing are dependent on membrane potential and time. Ca++ channels are also, importantly, dynamically regulated by the autonomic nervous system.13 β agonists activate ICa-L (and hence increase myocardial contractility) indirectly by activating adenylyl cyclase via a guanosine triphosphate (GTP)–binding protein, Gs (Figure 7-4). The resulting increase in intracellular cyclic adenosine monophosphate (cAMP) activates protein kinase A (PKA), which phosphorylates the Ca++ channel. Phosphorylated channels open in response to membrane depolarization; nonphosphorylated channels do not, so the effect of β-adrenergic stimulation is to increase the number of functional channels. The electrophysiologic effect of this is illustrated in Figure 7-5, which shows enhancement of the slow inward current by increase of adenosine monophosphate (AMP) level in single-channel Ca++ channels and intact cells. β-Adrenergic effects on ICa-L are antagonized by acetylcholine, which, in myocardial cells, activates M2 muscarinic receptors and inhibits adenylyl cyclase through activation of the GTP-binding protein Gi.
Delayed Rectifier K+ Currents
Delayed rectifier K+ channels are present in all cardiac myocytes. They open slowly (over 200 to 300 msec) after depolarization of the membrane to the plateau level (−10 mV and greater), producing a K+-selective outward current, IK. IK does not inactivate on prolonged depolarization (unlike INa and ICa-L), and the channels close on repolarization of the membrane. Unlike IK1, IK displays outward rectification; that is, it passes outward current more easily than inward current. This is the expected behavior for a K+-selective channel because both the concentration and electrical gradients for potassium are outward. Thus, for any depolarizing displacement of membrane potential from EK, the driving force will be larger in an outward direction. Similar to ICa-L, IK is under autonomic control (see Figure 7-4). β-Adrenergic stimulation enhances IK by a mechanism similar to that of the enhancement of ICa-L, thus ensuring repolarization of the cell in the face of increased inward Ca++ current.14
Three components of IK, carried by different channel molecules, can be distinguished. A rapidly activating component, IKr, is blocked by the compound E4031 (a Class III antiarrhythmic agent), which leaves a slower activating component, IKs, unaffected.15 This is illustrated in Figure 7-6, which also emphasizes the importance of IK in the regulation of repolarization, and hence of action potential duration. A third component, the ultra-rapidly activated delayed rectifier, IKur, can be distinguished in atrial (but not ventricular) myocytes.16 This additional repolarizing current explains, in part, the enhancement of repolarization in atrial myocardium when compared with ventricle and Purkinje fibers.
Repolarization in Different Cardiac Tissue Types
Phase 3 repolarization in atrium and pacemaker tissues, but not in ventricular myocardium, is also enhanced by the presence of a large outward repolarizing K+ current (IK[ACh]).17,18 This potential independent current is activated indirectly by stimulation of muscarinic (M2-type) receptors by acetylcholine or purinergic (A-type) receptors by adenosine.19 This channel is potential independent and is activated via binding of an activated, membrane-bound GTP-binding protein (Gi), as discussed later.20
There is variability in action potential duration between cells in normal ventricle.21 A gradient in action potential duration exists across the myocardium (from epicardium to endocardium), and specialized midmyocardial cells (M cells) have been identified, which exhibit prolongation of action potential duration at slow stimulation rates, possibly as a result of a decrease in IKs.
Phase 4: Diastolic Depolarization and If
Phase 4 diastolic depolarization, or normal automaticity, is a normal feature of cardiac cells in the sinus and atrioventricular (AV) nodes, but subsidiary pacemaker activity is also observed in the His-Purkinje system and in some specialized atrial and ventricular myocardial cells (see Chapter 4). Pacemaker discharge from the sinus node normally predominates because the rate of diastolic depolarization in the sinoatrial (SA) node is faster than in other pacemaker tissues. Pacemaker activity results from a slow net gain of positive charge, which depolarizes the cell from its maximal diastolic potential to threshold.
Pacemaker cells in the sinus node are relatively depolarized, with a maximal diastolic potential of −60 to −70 mV and a threshold potential of −40 mV. Rapid regenerative depolarization (phase 0) is dependent on opening of T-type and then L-type Ca++ channels. Repolarization is dependent on activation of delayed rectifier K+ channels, and the maximum diastolic potential is around −80 mV. Pacemaker channels are activated by hyperpolarization to this potential and produce a slow inward Na+ current, If. This flows against slowly inactivating delayed rectifier K+ currents and results in diastolic depolarization.22 Because the current is nonselective among cations, its reversal potential lies between EK and ENa, at around −10 mV, and activation of If will tend to depolarize the cell toward this value. Similar to ICa-L, If is under autonomic control (see Figure 7-4) through GTP-dependent binding proteins Gs and Gi, which regulate cAMP production by adenylyl cyclase.23,24 β-Adrenergic stimulation shifts the voltage dependence of activation of If to more depolarized potentials, so for any hyperpolarizing stimulus, more If will be activated and diastolic depolarization will be enhanced. Acetylcholine has the opposite effect (Figure 7-7).


Molecular Biology of Ion Channels
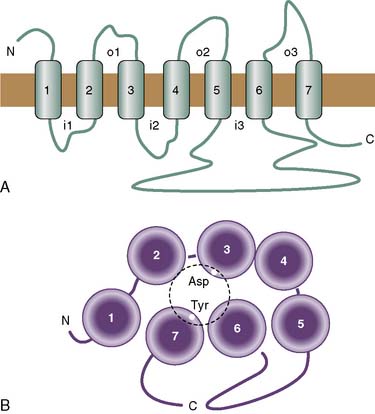
The preceding sections have focused on the electrical events that underlie cardiac electrical excitability, and on the identification of cardiac ionic currents on the basis of their biophysical properties. Here the molecular structures behind these electrical phenomena are reviewed. The first step in understanding the molecular physiology of cardiac electrical excitability is to identify the ion channel proteins responsible for the ionic currents. Figure 7-2 gives the current classification of the ion channel responsible for each of the cardiac ionic currents. There are firm molecular candidates for voltage-gated Na+ and L-type Ca++ channels. Similarly, channel molecules with properties similar to delayed rectifier K+ channels, the 4-aminopyridine–sensitive component of ITO, the inward rectifier IK1, the ligand-gated K+ channel IK(ACh), and the pacemaker current If have been cloned. Figure 7-8 shows diagrams of the predicted membrane topology of some of these channels. Voltage-gated Na+, Ca++, and K+ channels exist as conglomerates of molecules, consisting of a large α subunit and several accessory subunits (labeled β, δ, and γ in Figure 7-8). The α subunit alone is usually sufficient to induce channel activity in biologic membranes, but its activity is modulated by the presence of the accessory subunits. The diagrams in Figure 7-8 were deduced from hydrophobicity analysis of the primary structure of the major channel polypeptides. Regions of the polypeptides predicted to span the membrane are those that contain a high concentration of hydrophobic amino acids, whereas peptides linking these transmembrane sections are hydrophilic. Similarities among the various channels strongly suggest a common evolutionary ancestry. Na+ and Ca++ channel α subunits (see Figures 7-8A and B) are strikingly similar, each consisting of four homologous transmembrane domains (labeled I to IV), linked by cytoplasmic peptides. Each homologous domain contains six linked membrane-spanning segments (labeled S1 to S6). These large polypeptides, containing more than 2000 amino acids, form a tetrameric structure and generate Na+ or Ca++ channel activity in biologic membranes. Voltage-gated K+ channel α subunits, in contrast, are much smaller (see Figure 7-8C) and consist of a single transmembrane domain with six membrane-spanning segments, an arrangement similar to one of the individual domains of Na+ and Ca++ channels. Four molecules are noncovalently linked in the membrane to produce a tetrameric structure, similar to a Na+ or Ca++ channel, to produce K+ channel activity. The structure of the inwardly rectifying K+ channel molecules, IK1 and IK(ACh), is dissimilar to other K+ channels (see Figure 7-8C). The molecules are much less complex, having only two membrane-spanning segments, although these segments share considerable homology with the S5 and S6 segments of the classic voltage-gated K+ channel.
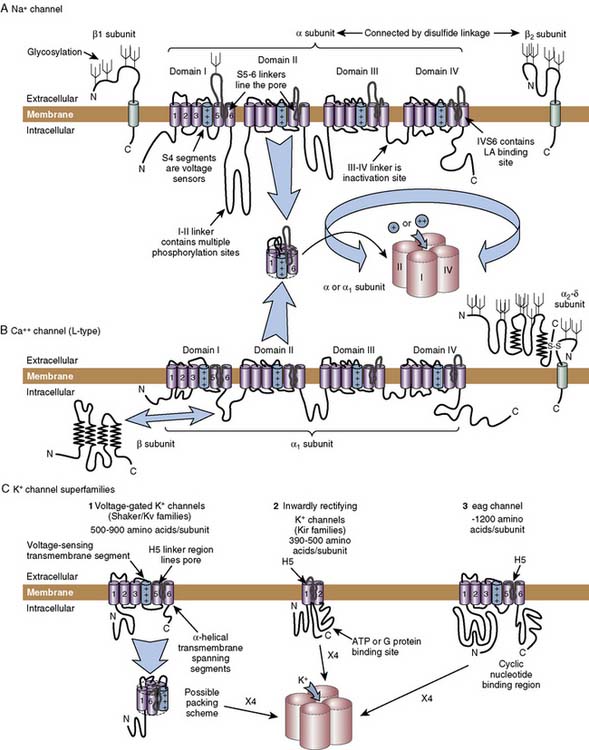
Figure 7-8 Diagrams of ion channel molecular structure.
A, Na+ channel. B, Ca++ channel. C, K+ channels. ATP, adenosine triphosphate.
Molecular Mechanisms
The Voltage Sensor
Channel proteins respond to changes in electrical potential across the cell membrane by conformational changes (gating), which result from electrostatic interactions between charged portions of the molecule and the membrane electric field (Box 7-3). Gating of the channel is associated with a measurable flow of electrical charge through the membrane lipid bilayer (called gating current), as a zone of the molecule rich in electrical charge moves within the membrane.25 This charge movement is linked to opening of the channel pore. The voltage sensor of voltage-dependent ion channels resides in the mobile S4 membrane-spanning segments, α-helical structures unusually rich in positively charged amino acids.26 At rest, each of the positive charges in the S4 segment is balanced by fixed negative charges in other segments of the molecule. The resting membrane potential (negative inside) forces the (mobile) positive charges inward and the fixed negative charges outward. This dynamic equilibrium holds the channel pore closed. On depolarization, the force pulling the positive charge inward is relieved; positive charges (the S4 segments) are repelled outward and assume new partners with the fixed negative membrane charges. This charge movement comprises the gating current. If the depolarizing stimulus is short, repolarization of the membrane is followed by a gating current of equal and opposite magnitude as the S4 segment relaxes to its original position. If the depolarizing stimulus is prolonged, however, the movement of the S4 segments induces a conformational change in the channel molecule, which prohibits easy return to baseline. This conformational change in the channel molecule is manifested as activation (or channel opening), and this is closely coupled to channel closing (or inactivation; see later) in channels that inactivate. Thus, small changes in the membrane electric field cause conformational changes in the channel molecule, which result in opening (and closing) of the channel pore. An S4 segment rich in positive charge is a remarkably consistent feature of voltage-gated ion channels from a variety of different species and with a variety of ion selectivities. The dependence of channel activation on membrane potential is proportional to the density of positive charge in the S4 segment.
Ion Channel Pore and Selectivity Filter
The presence of four homologous domains in voltage-gated Na+ and Ca++ channels suggests that basic ion channel architecture consists of a transmembrane pore surrounded by the four homologous domains arranged symmetrically (see Figure 7-8). The membrane-spanning segments each form an α helix so that the walls of the pore will be derived from α-helical segments from each of the four domains. A pore formed from four such α helices would have limiting dimensions of 3 by 5 Angstrom units, similar to the size inferred for the Na+ channel pore by measurement of the permeability of cations of different sizes.27,28
The selectivity filter is formed by the S5 and S6 membrane-spanning segments of each domain together with their peptide linker.29 As emphasized in Figure 7-8, unlike the hydrophilic extracellular linkers between other membrane-spanning segments, the S5/S6 linker is sufficiently hydrophobic to place it (at least partially) within the membrane lipid bilayer. The channel pore is lined both by the S5/S6 linker and the S5 and S6 membrane-spanning segments. Point mutations in the S5/S6 linker have dramatic effects on channel ion selectivity and reduce channel conductance to its primary ion. Extensive site-directed mutagenesis experiments of the S5/S6 linkers from a variety of channels suggest that they form a funnel that allows the passage of a specific ion into the pore. In Na+ channels, selectivity is imposed by two rings of negatively charged amino acids at the outer mouth of the funnel, which collect Na+ ions for transmission into the cell.27
Channel Inactivation
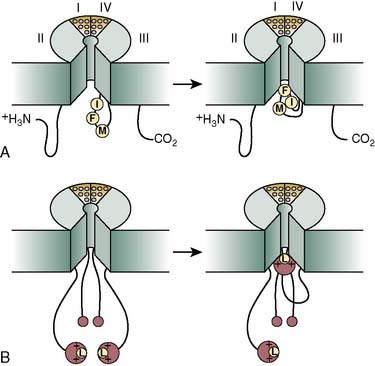
Inactivation gating is the process by which ion channels close in the face of continuing depolarization. Inactivation is characteristic of voltage-gated Na+ and Ca++ channels, as well as the K+ channels underlying ITO. Inactivation begins after activation gating, as a second, slower conformational change in the molecule that halts the ion flux through the channel. Inactivation gating is thus closely coupled to activation gating, and ionic current flows only while both the activation and inactivation gates are open simultaneously. In Na+ channels, the inactivation gate is formed by the intracellular peptide linker between homologous domains III and IV (Figure 7-9A).30 This peptide is postulated to act as a hinged lid, which moves upward to plug the ion pore (and thus halt current flow) shortly after membrane depolarization.26 For the channel to recover from inactivation (i.e., to be ready to open in response to a new depolarizing stimulus), the III/IV linker peptide must resume its resting position, a process that requires hyperpolarization of the membrane to the resting potential for a finite period. Site-directed mutagenesis of the III/IV linker peptide has revealed a trio of hydrophobic amino acid residues (isoleucine, I; phenylalanine, F; and methionine, M), near to the domain III end of the peptide, which are crucial for normal channel inactivation. Replacement of just one of these residues (the phenylalanine) almost completely removes inactivation. These residues are postulated to latch on to a receptor in the channel pore to close the channel. The molecular basis of inactivation in K+ channels is rather different from Na+ channels. Because the four domains of K+ channels are formed by noncovalently linked molecules, there are no interdomain linkers to plug the channel pore. In K+ channels, a picture of an N-terminal ball-and-chain mechanism has emerged (see Figure 7-9B).31 The terminal 20 or so amino acids are very hydrophobic and are postulated to swing up and attach to the open pore. The next few amino acids contain a number of positively charged residues that draw the whole N-terminal end up to the membrane. These two domains act as a ball. The remaining amino acids, up to the beginning of the transmembrane S1 segment, act as a chain. If the chain is made longer, inactivation is slower, and vice versa.

Clinical Correlates
Ion Channels and Antiarrhythmic Drugs
Drug therapy of cardiac arrhythmias would ideally be targeted at an individual ionic current, tailoring the cardiac action potential in such a way that abnormal excitability was reduced but normal rhythmicity was unaffected. This remains an as-yet unrealized goal. The prototype antiarrhythmic agents (e.g., disopyramide and quinidine) have diverse effects on cardiac excitability and, similar to agents introduced more recently, frequently exhibit significant proarrhythmic activity with potentially fatal consequences. In the Cardiac Arrhythmia Suppression Trial (CAST), mortality among asymptomatic postmyocardial infarction patients was approximately doubled by treatment with the potent Na+-channel–blocking agents encainide and flecainide—an effect likely attributable to slowing of conduction velocity with a consequent increase in fatal reentrant arrhythmias.32 The results of the CAST prompted efforts to approach antiarrhythmic drug therapy by prolonging action potential duration (e.g., dofetilide)—a strategy with some support from animal studies, but one that also may cause proarrhythmia through induction of polymorphic ventricular tachycardia (the acquired long QT syndrome [LQTS]).33 Drugs that prolong action potential duration all block IKr, and it is not clear that this therapeutic goal will result in arrhythmia control without induction of clinically significant proarrhythmia. The only drugs currently available that definitely prolong life by reducing fatal arrhythmias are β-blockers (e.g., ISIS-1, 1997), and these agents have no channel-blocking effects.
Ion Channels in Disease
Long QT Syndromes
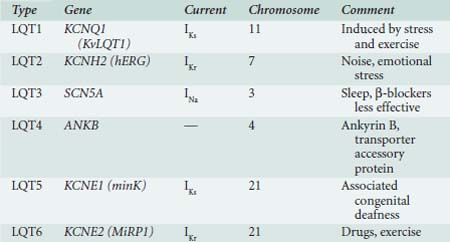
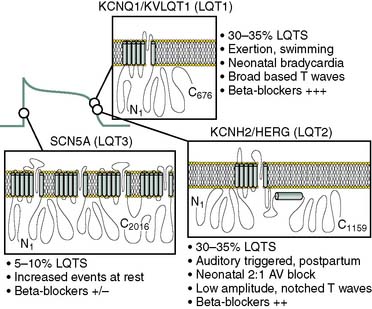
This rare group of ion-channel abnormalities causes abnormal prolongation of the cardiac action potential, resulting in early afterdepolarizations (i.e., oscillations in the action potential during the plateau phase) and death from polymorphic ventricular tachycardia (torsades de pointes type). To date, six LQTS subtypes have been identified on the basis of the affected gene. They are numbered sequentially according to the date of discovery (Table 7-1), and all the loci so far identified encode ion channels except for LQT4. LQTS occurs because of disruption of cardiac repolarization, and in principle this can occur either from enhancement of inward depolarizing current or reduction of outward current. LQT3 is a gain-of-function mutation of the cardiac Na+ channel that results in failure of channel inactivation, most commonly from a deletion of three amino acids from the inactivation gate. LQT1 and LQT2 result from mutations of the delayed rectifier K channels that underlie IKs and IKr, and LQT5 and LQT6 result in reductions of IKr and IKs through mutations in channel accessory subunits. Loss of repolarizing current results in prolongation of the QT interval, and this leads to the syndrome. LQT4 is unique in that it results from a mutation in ankyrin B, an adapter protein that binds to the Na+ pump and Na+/Ca++ exchanger. The resulting effects on cellular Ca++ homeostasis cause ventricular arrhythmias.34 Identification of the molecular substrate for LQTS will allow detection of the disease in asymptomatic carriers and may in the future allow targeted gene therapy; these issues are discussed in more detail later in the Genetic Cardiovascular Medicine section.
Brugada Syndrome
The Brugada syndrome also is a group of ion-channel abnormalities that affect cardiac repolarization and result in cardiac sudden death. It is characterized by incomplete right bundle-branch block and persistent ST-segment elevation in the anterior precordial leads of the electrocardiogram (ECG).35 Cases for which the genotype is available appear to result from channel mutations that reduce depolarizing Na+ current. This results in loss of the action potential dome, an effect that is most marked in the right ventricular epicardium where the transient outward current ITO1 is strongly expressed (hence the ST-segment elevation in the anterior chest ECG leads). Early repolarization of the epicardial action potential results in a transmural repolarization gradient, and this can lead to reentry and sudden cardiac death.36 In more than two thirds of patients with Brugada syndrome, the genetic locus is unknown, and much work remains to be done to elucidate the mechanism of this condition.
Controlling cardiac functioning: receptors

Receptors
Receptors are grouped in several broad classes, the protein tyrosine kinase receptors and the G-protein–coupled receptors (GPCRs) being the most important ones. The protein tyrosine kinase receptors are large molecular complexes that incorporate phosphorylating enzyme activity in the intracellular segment. Ligand binding induces activation of this enzyme activity. Because phosphorylation is one of the major mechanisms of cellular regulation (see, for example, the phosphorylation of the Ca++ channel described earlier), such receptors can have a variety of cellular effects (Box 7-4). GPCRs are much smaller than protein tyrosine kinase receptors. Ligand binding results in activation of an associated protein (G protein) that subsequently influences cellular processes. The receptors discussed in this section all belong to the GPCR superfamily, and the properties of this class are discussed in some detail.
All of these have similar molecular characteristics. They are generally several hundred to 1000 amino acids in length and contain 7 stretches of 20 to 25 hydrophobic amino acids. These hydrophobic domains are thought to form α helices and traverse the membrane, thus anchoring the receptor to the cell (Figure 7-10). For this reason, the family is often referred to as the “seven transmembrane” family. Although crystallographic data are not yet available for the clinically relevant GPCRs, it is thought that the seven-transmembrane domains arrange in a funnel-like structure, the inside of which forms the ligand-binding domain. The intracellular domains, particularly the third intracellular loop and the C terminus, bind to the G protein.

G Proteins
Several classes of heterotrimeric G proteins exist, indicated by subscripts (Box 7-5). The classic types are Gs and Gi, which stimulate and inhibit, respectively, the enzyme adenylate cyclase, thereby leading to changes in cytoplasmic cAMP concentrations. Gq proteins (and Go in brain) activate phospholipase C (PLC) and thereby induce the generation of inositol-1,4,5-triphosphate (IP3) and diacylglycerol (DAG) from phosphatidylinositol bisphosphate (PIP2). IP3 acts on its own receptor/channel complex on intracellular Ca++ storage sites and induces release of Ca++ from these sites, thereby increasing intracellular Ca++ concentrations. DAG activates protein kinase C (PKC), leading to phosphorylation of a variety of targets (including the receptors that initiated the cascade). In recent years, cloning efforts have shown each of these classes of G proteins to consist of a number of members, but their functional differences are as yet incompletely defined.

Adrenergic Receptors and Signaling Pathways
Adrenergic Receptors
The two main subtypes of β-adrenergic receptors are the β1 and β2 subclasses. A β3 subtype exists as well, but its role in the cardiovascular system is unclear37; its most important role is in fat cells. Both β1 and β2 receptors are present in the heart, and both contribute to the increased contractility induced by catecholamine stimulation (this is different from the situation in vascular muscle, where β-adrenergic stimulation induces relaxation). Under normal conditions, the relative ratio of β1 to β2 receptors in heart is approximately 70:30, but as discussed later, this ratio can be changed dramatically by cardiac disease.
Structurally, as well as functionally, the various β-adrenergic receptors are closely related. Both couple to Gs proteins and, as described earlier, thereby activate adenylate cyclase, leading to increased intracellular levels of cAMP. Some differences in their intracellular signaling are likely, however. For example, it has been suggested that β2 receptors couple more effectively than β1 receptors and induce greater changes in cAMP levels.38 In addition to their effect on cAMP signaling, β receptors may couple to myocardial Ca channels.39 However, these additional actions are species specific, and care should be taken in extrapolating animal data to humans.
In the heart, the primary subtype present is α1. Activation of these receptors leads to a modest increase in cardiac contractility.40
Regulation of β-Receptor Functioning
Whereas β-receptor stimulation allows the dramatic increases in cardiac output of which the human heart is capable, it is clearly intended to be a temporary measure. Prolonged adrenergic stimulation has highly detrimental effects on the myocardium; the pronounced increases in cAMP levels are followed by increases in intracellular Ca++ concentration, reductions in RNA and protein synthesis, and finally, cell death. Thus, β-receptor modulation is best viewed as part of the “fight-or-flight” response: beneficial in the short term, but detrimental if depended on for too long. Cardiac failure, in particular, has been shown to be associated with prolonged increases in adrenergic stimulation—even to the extent that norepinephrine “spillover” from cardiac nerve endings can be detected in the blood of patients in heart failure.41
One mechanism for decreasing β-receptor functioning is the downregulation (i.e., decrease in density) of receptors. In cardiac failure, receptor levels are reduced up to 50%. β1 receptors downregulate more than β2 receptors do, resulting in a change in the β1:β2 ratio. As mentioned earlier, the normal ratio is approximately 4:1; in the failing heart, it is approximately 3:2.42 Various molecular mechanisms exist for this downregulation. In the long term, receptors are degraded and permanently removed from the cell surface. In the short term, receptors can be temporarily removed from the cell membrane and “stored” in intracellular vesicles, where they are not accessible by an agonist. These receptors are, however, fully functional and can be recycled to the membrane when adrenergic overstimulation has ceased.43
An additional method by which β-adrenergic receptor functioning can be modified is through phosphorylation of the agonist-occupied receptor by a specific β-adrenergic receptor kinase (β-ARK, which can itself be activated by βγ subunits).44 Phosphorylation by this kinase allows binding to the receptor of a protein, β-arrestin, that inhibits receptor functioning.45 In addition, the β receptor can be phosphorylated by PKA, which itself can be activated by several other receptors. The detrimental actions of adrenergic overstimulation can also be modified by activation of muscarinic receptors, as discussed in the next section.

Muscarinic Receptors and Signaling Pathways
Muscarinic Acetylcholine Receptors
The second major receptor type involved in cardiac regulation is the muscarinic receptor. Although five subtypes of muscarinic receptors exist, only one of these (M2) is present in cardiac tissue. Most of these muscarinic receptors are present on the atria. Indeed, it was thought until recently that there was no vagal innervation of the ventricles, but this view turns out to be incorrect. The ventricles are innervated by the vagus, and muscarinic receptors are, in fact, present in the ventricles, albeit at lower concentrations than in the atria; the amount of muscarinic receptor protein in atrium is approximately twofold greater than in ventricle (200 to 250 vs. 70 to 100 fmol/mg protein).46 Thus, although the primary function of cardiac muscarinic signaling is heart rate control through actions at the atrial level, vagal stimulation is, in fact, able to directly influence ventricular functioning.
M2-muscarinic receptors couple to Gi proteins, thereby inhibiting adenylate cyclase and decreasing intracellular levels of cAMP. In fact, the M2 receptors have been used as an elegant model to determine the site of G-protein binding to the receptor. Exchanging approximately 20 amino acids of the third intracellular loop (i3) (see Figure 7-10) between M2 and M3 receptors resulted in altered coupling; M2 receptors mutated in this manner were now able to release Ca++ from intracellular IP3-sensitive stores by coupling to Gq.47
In the impulse-generating system of the heart, a more important signaling mechanism than changes in cAMP is opening of an inwardly rectifying K+ channel (KACh) in the plasma membrane. The coupling between M2 receptor and KACh is performed by GK, a member of the Gi class of G proteins. Interestingly, it is not the α subunit of GK that activates KACh, but rather the βγ subunit.48 As discussed later, cardiac adenosine receptors couple to this channel as well.
Regulation of Muscarinic Acetylcholine Receptors
Unfortunately, these compensatory mechanisms may not be available in the old heart. Increased age is accompanied by changes in cardiac muscarinic receptor expression that might make it more difficult for the heart to respond to adrenergic stress. In senescent rats, muscarinic receptor density was decreased by approximately 50%.49 Adrenergic receptor levels also were decreased, but to a lesser extent. As a result, the adrenergic/muscarinic receptor ratio is increased, from 0.29 in young adults to 0.42 in senescent animals. Although the physiologic implications of these changes are not well known, the data at least suggest that the muscarinic systems in the aged heart might not be as well prepared to react to prolonged adrenergic stimulation, such as observed in hypertension and cardiac failure.
With the exception of age, muscarinic receptors are little affected in settings that profoundly modify β-receptor expression. There are, for example, no consistent data supporting changes in muscarinic receptor expression in hypertension, cardiac failure, or ischemic heart disease.50,51 Therefore, imbalances between adrenergic and muscarinic stimulation might occur in each of these situations.

Regulation of G-Protein Functioning
In view of the profound changes in GPCR expression that occur in various disease states, the expression and function of G proteins in cardiovascular disease have been studied with interest. Gs proteins appear unchanged in cardiac failure, both in expression level and in function. With Gi proteins, the situation is more interesting, because Giα is considered to have a secondary role in addition to its inhibition of adenylate cyclase. Under normal conditions, Gi is present in greater amounts than Gs. Activation of receptors coupled to Gi would, therefore, lead to the release of a large number of free βγ subunits. These could combine with any free Gsα, thereby making it unavailable for activation of adenylate cyclase.52 In addition, these βγ units can enhance the phosphorylation of β receptors by β-ARK.53 In failing human cardiac tissue, the amount of Gi (as assessed by ADP-ribosylation by pertussis toxin) is increased.54 Although this would be expected to make muscarinic signaling more efficacious (thereby helping counteract the adrenergic overstimulation), it has been difficult to correlate these changes with alterations in adenylate cyclase functioning. It is similarly unclear whether the reported increases in Gi levels are a result of increased mRNA expression or increased stability of the G protein itself.
The catalytic subunit of adenylate cyclase appears little influenced by cardiac disease. Pressure overload is the only situation in which a consistent decrease in its activity has been observed.55

Other Receptors
Although less is understood about the role and regulation of these receptors than about their adrenergic and cholinergic counterparts, it is clear that some are affected by cardiovascular disease. For example, VIP receptors are downregulated by 70% in idiopathic dilated cardiomyopathy (DCM), whereas histamine receptors are unaffected.56 In general, receptors coupled to Gi show little alteration in expression and function during disease states, whereas Gs-coupled receptors are affected more profoundly.

Clinical Correlates
Understanding of the role of adenosine in cardiac regulation has expanded significantly over the past years. Its established use as an antiarrhythmic compound and its probable role in cardiac preconditioning are two examples of clinical advances resulting from this increase in understanding. Adenosine acts through a GPCR, activating several intracellular signaling systems. This section discusses the molecular aspects of adenosine signaling, as well as their clinical implications. More detailed recent reviews on the topic are available.57,58

Adenosine Signaling
Although adenosine can be generated by several pathways, in the heart, it is usually found as a dephosphorylation product of AMP.59 Because AMP accumulation is a sign of a low cellular energy charge, an increased adenosine concentration is a marker of unbalanced energy demand and supply; thus, ischemia, hypoxemia, and increased catecholamine concentrations are all associated with increased adenosine release.60 Adenosine is rapidly degraded by various pathways, both intracellularly and extracellularly. As a result, its half-life is extremely short, on the order of 1 second.61 Therefore, it is not only a marker of a cardiac “energy crisis,” but its concentrations will fluctuate virtually instantly with the energy balance of the heart; it provides a real-time indication of the cellular energy situation.
The A1 adenosine receptor couples to (at least) two intracellular signaling systems. Both of these actions are mediated by G proteins of the Gi class. The first intracellular system is one already encountered: the KACh channel. Presumably, through the same GK protein, adenosine activates this channel in the same way as does M2 muscarinic stimulation, and the cardiac electrophysiologic effects of acetylcholine and adenosine are therefore quite similar. The specific effect of adenosine depends on the cardiac tissue studied because KACh expression varies with location. As has been discussed, whereas the channel is present in large amounts in the atrial conduction system and atrial myocardium, it is virtually absent in the ventricle. Therefore, in the unstimulated heart, adenosine shortens the atrial action potential, decreases atrial refractoriness and decreases atrial contractile force, but it is almost without effect on the ventricle.62
The second intracellular signaling system activated is a Gi protein that inhibits adenylate cyclase. As cAMP levels are quite low under resting conditions, this mechanism plays little role until cAMP concentrations are increased by adrenergic stimulation of the heart. Therefore, cAMP-mediated cardiac actions of adenosine are observed only under conditions of adrenergic drive. Because the adenylate cyclase system is present throughout the heart, its effects are widespread; L-type Ca++ channel functioning is diminished (by inhibiting cAMP-induced phosphorylation of the channel) in atrium as well as ventricle, resulting in decreased inotropy and shortening of the action potential.63
Antiarrhythmic Actions of Adenosine
From these molecular actions of adenosine, its clinical effects easily can be deduced. The antiarrhythmic actions are largely a result of its activation of KACh. Recalling the tissue distribution of KACh, it could be anticipated that adenosine will be much more effective in the treatment of supraventricular arrhythmias than ventricular arrhythmias, and such is indeed the case. Because of its negative chronotropic effects on the atrial conduction system, the compound is most effective in treating supraventricular tachycardias that contain a reentrant pathway involving the AV node. The efficacy of adenosine in terminating such tachycardias has been reported as greater than 90%.64 In contrast, it is consistently ineffective in tachycardias not involving the AV node.65
Most ventricular tachycardias are insensitive to adenosine. The only exception is again easily deduced from the compound’s molecular mechanism of action; a rare form of exercise- or catecholamine-induced ventricular tachycardia responds promptly to adenosine.66 Presumably, in this setting, adenosine-mediated inhibition of adenylate cyclase counteracts the stimulatory effects of catecholamines.
Adenosine and Myocardial Preconditioning
Myocardial preconditioning is the phenomenon in which brief exposure of the myocardium to ischemic conditions will allow it to withstand a subsequent, more prolonged, exposure. The phenomenon has received much attention because its application in the clinical setting might allow the heart to better withstand, for instance, the insults of cardiac surgery. Thus, the mechanisms of this effect were investigated in the hope that they might be activated directly, without the need for ischemia. A variety of mechanisms might account for preconditioning, as has been reviewed recently.58,67
A G-protein–linked K+ channel located on the mitochondrial membrane, the mitochondrial KATP channel (mitoKATP), is a key mediator in myocardial preconditioning. Opening of this channel using compounds that selectively open mitoKATP without affecting other cellular KATP channels68 has been shown to have a protective effect, whereas inhibition of the channel (with the channel blocker glibenclamide) increases ischemic damage to the myocardium. For example, abolishment by glibenclamide of preconditioning could be shown in a study measuring ST-segment changes and cardiac pain in patients undergoing balloon angioplasty.69 Similar findings have been observed in various animal studies. KATP, like KACh, belongs to the class of G-protein–coupled inward rectifier-type channels; its structure is indicated in Figure 7-10. In contrast with KACh, however, it appears to be modulated primarily by changes in intracellular ATP derivatives. For example, intracellular increases in ATP levels have a direct inhibitory effect on the channel. In addition, KATP can be modulated by PKC, which phosphorylates the channel. PKC can be activated directly by ischemic conditions or by activation of α-adrenergic and adenosine receptors. In addition, adenosine also may be able to influence behavior of KATP in the same manner that it regulates KACh: through a Gi protein.70 The mechanisms by which mitoKATP opening is protective are still a focus of investigation. Potential beneficial effects involve inhibition of mitochondrial Ca++ uptake, regulation of mitochondrial volume, and modulation of the generation of reactive oxygen species. Of interest from the anesthesiologist’s point of view are observations that volatile anesthetics induce preconditioning by similar mechanisms. For example, sevoflurane preconditions human myocardium against hypoxia through activation of KATP channels and activation of A1-adenosine receptors.71 Similarly, lidocaine was shown to induce protection against inflammatory stimulation of endothelial and vascular smooth muscle cells by affecting mitoKATP.72
Anesthetic actions
Volatile anesthetics are administered in extremely high concentrations as compared with most other pharmacologic agents. Most commonly used drugs are administered in doses that result in micromolar blood concentrations. This reflects the fact that these compounds are sufficiently potent so that a half-maximal effect on the site of action requires only low micromolar concentrations. Anesthetics, in contrast, require low millimolar concentrations in blood to be effective, almost a thousand times as much. Although they commonly are referred to as “potent agents,” they are certainly not very potent as compared with other pharmacologic compounds. As a result, it is not surprising that they have a wide range of actions in addition to their primary effect site (which, of course, is still not completely defined). In addition, volatile anesthetics are lipophilic compounds, and if, under laboratory conditions, concentrations are increased even further, they can be found to interact with almost any preparation exhibiting a certain degree of lipophilicity. As a result, data exist demonstrating interactions between volatile anesthetics and virtually every component of the cardiovascular system. The issue, then, is not with which channels and receptors the anesthetics interact, but which of all these interactions are clinically important, and this has been difficult to determine. A first test that should be applied, of course, is that of reasonable concentrations; if effects are not observed at 1 to 2 minimal alveolar concentration (MAC) equivalents, it is unlikely that an interaction has clinical relevance. As many experiments are performed at temperatures below 37°C, temperature correction of anesthetic solubility is an important issue. It is also important that actions are shown in several models. Effects observed in an isolated system may not necessarily be reproduced in an organ or whole-animal model; in that case, the relevance of the finding is in doubt. This issue has been reviewed in some detail.73

Interactions with Channels: Ca++ Channels
Anesthetic actions on cardiac Ca++ channels have been studied in a variety of models. The original observations that halothane blocked Ca++ flux into heart cells date back approximately 30 years,74 and much specific information has been gained since. In particular, voltage-clamp and patch-clamp studies have contributed significantly to the understanding of the interactions between anesthetics and Ca++ channels, and have elegantly described the effects of anesthetics on electrophysiologic behavior. However, it is not straightforward to assign the observed electrophysiologic effects to a molecular substrate. This issue is only beginning to be addressed with the use of recombinant technology.
Almost all volatile anesthetics inhibit L-type Ca++ channels.75,76 Inhibition is modest, approximately 25% to 30% at 1 MAC anesthetic, but certainly sufficient to account for the physiologic changes induced by the anesthetics. Volatile anesthetics decrease peak current and, in addition, tend to increase the rate of inactivation.77 Hence maximal Ca++ current is depressed, and duration of Ca++ current is shortened. Together, these actions significantly limit the Ca++ influx into the cardiac myocyte. However, some specific actions may depend on the particular compound studied. In the presence of β-adrenergic stimulation, halothane, but not sevoflurane, is associated with a long-lasting enhancement of Ca++ channel function that may contribute to its proarrhythmic effects.78 Xenon is without effect on cardiac Ca++ channels, explaining, in large part, its lack of effect on myocardial contractility. Other types of Ca++ channels have different sensitivities. Neuronal (N-type) channels have been shown to be quite resistant to volatile anesthetics. T-type channels in general tend to be much more sensitive than L-type channels; at clinical concentrations of most volatile anesthetics, T currents are inhibited 50% or more.
The effects of volatile anesthetics on cardiac Ca++ channels can be modulated greatly by concurrent interactions of the compounds on other cardiac signaling systems. As discussed later, volatile anesthetics inhibit function of several types of muscarinic acetylcholine receptor systems. Because Ca channel function can be inhibited by muscarinic signaling, as discussed earlier, clinicians would anticipate additional interactions when this system is exposed to volatile anesthetics, and such is the case. Halothane and isoflurane further inhibited currents through Ca++ channels when either volatile anesthetic was applied after inhibition produced by prior muscarinic stimulation. However, when muscarinic receptors were stimulated after administration of volatile anesthetic, its effect was reduced. Thus, whereas volatile anesthetics directly inhibit L-type channels, they also interfere with channel modulation by GPCR.79
Not only volatile but also injected anesthetics have been reported to inhibit cardiac L-type Ca++ channels in some models. However, the concentrations used generally exceed those used in clinical practice. Thiopental and methohexital block L-type Ca++ currents.76,80 Similarly, propofol has been reported to inhibit these channels, but at concentrations well beyond the clinical range.

Interactions with Receptors: Muscarinic Receptors
Aronstam et al81 published a series of articles reporting the effect of various anesthetics on muscarinic receptor binding. A summary of their findings has appeared in print as well. Several studies investigated the effect of halothane on agonist and antagonist binding.82,83 The conclusions drawn from this work were: (1) the anesthetic enhanced antagonist binding by slowing the rate of ligand dissociation, and (2) the anesthetic inhibited agonist binding (by 48%, using 10% halothane).
The site of action of these effects was studied in more detail by investigating the interactions of anesthetics with G-protein functioning.84 As discussed earlier, G proteins are activated by GDP-GTP exchange. An intrinsic, but remarkably slow, enzyme activity hydrolyzes GTP back to GDP, thereby inactivating the G protein after several seconds. While active, the G protein is no longer able to couple to the receptor, and uncoupled receptors exhibit a decreased affinity for their agonist. This decrease in activity can be induced in the experimental setting by including a nonhydrolyzable analog of GTP [such as Gpp(NH)p] in the reaction mixture, resulting in irreversibly activated G proteins. This effect is known as the GTP shift in receptor affinity. Halothane shifts the Gpp(NH)p concentration–response relation to the right. In other words, a greater concentration of the GTP analog was necessary to induce a similar decrease in agonist binding. In the absence and presence of 5% halothane, the half-maximal inhibitory concentration (IC50) values for the inhibitory effect on agonist binding of Gpp(NH)p were 0.7 and 83 μM, respectively—a 100-fold difference. These findings were interpreted as an ability of halothane to stabilize high-affinity G-protein–receptor complexes.
Therefore, it appears that halothane affects receptor binding, as well as receptor–G-protein interaction. In a subsequent study, the effect of the anesthetic on G-protein functioning—its ability to hydrolyze bound GTP—was investigated. Halothane was found not to inhibit binding of radiolabeled GTP analog to G proteins.82 However, the anesthetic completely blocked the stimulation of G-protein GTPase activity induced by acetylcholine, with a half-maximal effect at the clinically relevant halothane concentration of 0.3 mM. The site of this effect was not determined. Largely similar findings were obtained with other anesthetics. Halothane also has been shown to interfere with G-protein–mediated Ca++ sensitization in airway smooth muscle by inhibiting G proteins.85 More recently, however, the interaction between halothane and purified, recombinant Gi was investigated. In contrast with the findings described earlier, no effect of the anesthetic on Gi protein function was found.86 Hence the earlier results might have been contaminated by interactions with other G-protein subtypes that are less relevant to muscarinic M2-receptor signaling. The conclusion to be drawn from these studies is that anesthetics may interfere with several components of the muscarinic signaling pathway. However, the following should be kept in mind: (1) most times a mixture of muscarinic receptor subtypes was studied; (2) the anesthetics were administered in relatively high, and clinically unequal, concentrations; and (3) anesthetic effects on functional properties of the receptor–G-protein unit were not specifically addressed. Magyar and Szabo,87 using the patch-clamp technique, have investigated the effects of halothane (0.9 mM) and isoflurane (0.8 mM) on acetylcholine (10 μM)-induced activation of the muscarinic K+ channel in frog atrial myocytes. They found that if anesthetic and agonist were administered at the same time, a reduction in the peak K+ current was observed, which was greater with halothane than with isoflurane. However, pretreatment with the anesthetic was found to have a significant, time-dependent, additional effect: 25-minute exposure to halothane restored the peak and significantly increased the steady-state current, whereas exposure to isoflurane decreased both. As equilibration of the anesthetic with the direct signaling pathway should be complete within milliseconds to seconds, the prolonged time course of these effects appears to indicate that additional intracellular pathways (such as PKC-mediated phosphorylation) are involved. Exposure of the membrane patches to the nonhydrolyzable GTP analog GTPγS, thereby irreversibly activating GK, prolonged the current. When single-channel measurements were performed, halothane was found to enhance the frequency of channel opening without significantly affecting the single-channel conductance. Isoflurane was without effect. Therefore, halothane affects the signaling pathway downstream of the muscarinic receptor in this model. Whether this action is on the G protein or on the channel itself cannot be conclusively determined from these studies. When the time considerations are taken into account, it appears likely that the initial action of halothane on the system is inhibiting, whereas that of isoflurane is less so. After prolonged exposure, halothane is found to enhance signaling, whereas isoflurane inhibits it. These results are probably due to various effects of the anesthetics on intracellular systems that modify the signaling properties of the muscarinic pathway. In addition, halothane, but not isoflurane, has a direct activating effect downstream of the muscarinic receptor. The latter is consistent with findings of halothane effect on Ca++ channels mentioned earlier.

Genetic Cardiovascular Medicine
Over the past few decades, considerable progress has been made in the identification and understanding of the genetic basis of cardiovascular disease. More than 40 cardiovascular disorders are now known to be caused directly by gene defects. These disorders, spanning all aspects of cardiovascular disease and affecting all parts of the heart structure, can be divided into two groups (Box 7-6). Monogenic disorders are (usually rare) Mendelian disorders for which single gene changes are implicated in the disease process and which usually exhibit characteristic inheritance patterns. Examples include familial hypercholesterolemia, hypertrophic cardiomyopathies (HCM), DCMs, and the LQTSs.88 More commonly, however, multiple genes influence the disease process by enhancing disease susceptibility or by augmenting the impact of environmental risk factors. The genetic component in those multigenic disorders comprises a collection of gene variants such as single nucleotide mutations, referred to as single nucleotide polymorphisms (SNPs). Each individual SNP may have a modest effect on the quantity or function of a translated protein product. However, when individual SNPs aggregate and interact with environmental risk factors, they may have a major impact on disease biology. Common diseases postulated to follow this paradigm include coronary artery disease (CAD), hypertension, and atherosclerosis.89
This section discusses the current status of genetic diagnosis of monogenic and complex cardiovascular disorders, as well as the main techniques used to perform such diagnostic testing (Box 7-7).

Monogenic Cardiovascular Disorders
Great progress has been made in the identification and characterization of the disease genes specific to Mendelian cardiovascular diseases,88,90 but several factors complicate these investigative efforts. Locus heterogeneity (many genes causing the same disease) is one of those.91 The channelopathies, encompassing the LQTS and related genetic arrhythmogenic disorders, are accounted for by more than 10 genes. Whereas HCM genes MYH7, MYBPC3, and TNNT2 (encoding the β-myosin heavy chain, cardiac myosin-binding protein C, and cardiac troponin T genes, respectively) account for 70% to 80% of HCM as diagnosed by molecular genetic approaches,92 an additional 19 genes have been implicated to cause the HCM phenotype.93 In addition, more than 20 genes have been implicated in DCM.91 A second, related factor complicating genetic diagnosis is that not all disease loci have been identified, which diminishes the sensitivity of molecular testing. Whereas it has been estimated that a mutation causing LQTS can be identified in up to three quarters of index patients, this is not true for other cardiovascular single-gene disorders. For example, only 30% to 60% of the genetic causes of HCM94 and only 20% to 30% of genetic causes for DCM have been identified.90,91 Further complicating diagnostic strategies is allelic heterogeneity (many mutations within a single gene). As an example, MYH7 consists of 40 exons encoding the β-myosin heavy chain protein, and 194 mutations in this structure have been reported to be associated with HCM. For diagnostic purposes, this necessitates sequencing the entire coding sequence and intron/exon boundaries of each gene, making sequencing a time- and effort-intensive undertaking.
Methodologies for Identifying Mutations: Sequencing and Microarrays
Techniques for identifying gene sequences have evolved from the classic Southern blotting procedure into highly automated systems that can rapidly screen hundreds of thousands of gene sequences. Southern blotting, invented in the mid-1970s, is based on the transfer to nitrocellulose of DNA molecules separated on gels and their subsequent identification with DNA probes. It allows detection of small mutations, as well as large deletions, duplications, and gene rearrangements. The principle was expanded and automated in the development of DNA microarrays (Figure 7-11), which consist of series of thousands of microscopic spots of DNA oligonucleotides, each containing tiny amounts of a specific DNA sequence. The arrays are printed on a platform, usually a glass slide. A DNA probe, encoding a specific sequence, is then used to bind to and identify homologous nucleic acids, as detected by fluorophore-labeled targets whose signal is proportional to the relative abundance of nucleic acid sequences in the target. This makes it possible to examine the relative abundance of thousands of genes at any given time. Newer chip-based sequencing methods, designed to improve speed and efficiency, enable simultaneous sampling of 25,000 genes (Figure 7-12).
Clinical Applications
The ability of these techniques to identify diseases before they become clinically manifest allows preventive treatment as summarized in Table 7-2. For example, implantable cardioverters-defibrillators (ICDs) can prevent sudden cardiac death, improve quality of life, and prolong the time to cardiac transplantation (or avoid it altogether) in patients with genetic cardiomyopathies and arrhythmias95–97 (Figure 7-13). Medical therapy may ameliorate the progression of genetic DCM. Prospective identification of those patients yet asymptomatic, but at greater risk for development of the disease, enables close surveillance and early intervention. Knowledge of the LQTS genotype may be used to tailor the treatment plan (Figure 7-14). For instance, LQT3 patients benefit less from β-blockers, so a lower threshold for ICD implantation is advised in LQT3 patients.98 In addition, the triggers for malignant arrhythmias have been found to differ based on gene involved, and, for example, patients with LQT1 can be asked to avoid vigorous activities such as exercise and competitive sports.99

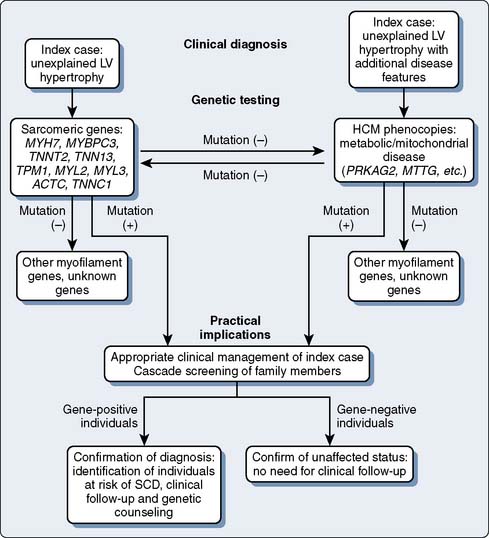
Figure 7-13 Proposed sequence of genetic testing for patients with hypertrophic cardiomyopathy.
HCM, hypertrophic cardiomyopathy; LV, left ventricular; SCD, sudden cardiac death. (From Keren A, Syrris P, McKenna WJ: Hypertrophic cardiomyopathy: The genetic determinants of clinical disease expression. Nat Clin Pract Cardiovasc Med 5:158–168, 2008.)

Multigenic Cardiovascular Disorders
Methodologies for Multigenic Genetic Screening: Linkage Analysis, Whole-Genome Association, and Gene Expression Profiling
Linkage analysis is a nonbiased and powerful approach for identifying causative genes underlying complex diseases.100 The analysis is conducted after a priori identification of potential candidate genes or their chromosomal locations. It then looks for DNA markers that cosegregate with the disease phenotype between affected family members at a rate that is statistically greater than random chance. The chromosomal region containing the DNA markers can then be examined in further detail to look for potential candidate genes. Several successful studies have been performed using CAD as a model.101–103
Genetic association studies are performed to determine whether a genetic variant is associated with a disease or trait; if association is present, a particular allele, genotype, or haplotype of a polymorphism or polymorphism(s) will be seen more often than expected by chance in an individual carrying the trait. Thus, a person carrying one or two copies of a high-risk variant is at increased risk for development of the associated disease or having the associated trait. Using modern genomic technologies, researchers can now assay hundreds and thousands of SNPs simultaneously in a single individual using “SNP chips,” the principle of which is the same as previously described for gene arrays. Two recent studies performed whole-genome association to identify SNPs associated with the development and progression of myocardial infarctions.104,105
Gene expression profiling (functional genomics) is another approach to identify genes and pathways that contribute to the development and progression of complex cardiovascular diseases such as coronary atherosclerosis. This approach analyzes disease in relevant tissues to look for changes in the abundance of transcribed genes or messenger RNA (mRNA) that correlate with a disease state, clinical outcome, or therapeutic response.106,107
Clinical Application
As in single-gene disorders, genetic information is used in multigenic disorders to establish disease susceptibility by defining a person’s individual risk beyond currently available clinical and laboratory assessment tools. Several commercially available tests fall in this category, including tests for susceptibility to atrial fibrillation108 and myocardial infarction.104,105 Higher risk individuals, once identified, can then receive more intensive preventive medical treatments to delay or prevent disease development. However, there are several issues to consider before using genetic testing in this set of patients.109 The first concern is the interpretation of results. Unlike single-gene disorders, the results of a positive or negative test provide no concrete diagnostic or prognostic information. A positive test simply means that a patient has an increased risk for development of the disorder—it is not a near certainty. Similarly, a negative test does not guarantee that the patient will not ultimately develop the disease. A second issue is the clinical interventions that should be administered given the test results. For example, if genotyping indicates that the individual is at risk for a future myocardial infarction, should intensive medical therapies be prescribed? Approaches such as blood pressure reduction, augmented antiplatelet therapy, and aggressive cholesterol lowering have all been shown to substantially reduce risk for CAD and are considered safe, but universal administration of these therapies is financially unfeasible and would lead to unavoidable side effects. Theoretically, with the use of more precise genomic information, it may be possible to identify disease-susceptible individuals for whom intensive prevention is cost effective, but this remains to be proven. Will the benefits of interventions applied in response to a positive genomic test outweigh the risks for unnecessary treatment and financial costs and result in durable and cost-effective improvements in health? On the other hand, given that there is no guarantee of a disease-free future, will a negative test lead to a false sense of security and encourage the maintenance or resumption of prior deleterious behaviors? Further evaluation of the utility of these tests through carefully designed disease outcome trials is needed. Finally, potentially problematic legal and ethical issues surround the clinical genetic testing for complex diseases. Although the results of the test provide no concrete diagnostic or prognostic information, they may have potentially significant effects on health insurance premiums or employment.
Another application of genetic testing in multigenic disorders lies in the development of new diagnostic and prognostic tools. This can be exemplified by its use in cardiac transplantation, in which it can identify subtle changes in peripheral blood mononuclear cells, allowing the detection of organ rejection much earlier than traditional histopathology.110 In this example, genetic testing can be thought of as an extension of currently available biochemical and histopathologic approaches that aids in clinical decision making or determining prognosis by virtue of providing more detailed molecular phenotype information. Unfortunately, there are currently no genetic tests that meet these criteria with respect to more common disorders such as CAD.
Finally, an additional way to translate new genetic information into clinical practice is through the identification of targets for drug development or more precise indications for currently used medications. This is a straightforward idea, but a number of hurdles need to be overcome. The primary problem is to provide convincing evidence that a gene contributes to disease development and progression. In the case of linkage studies, it is sometimes difficult to determine the dominant candidate genes. Often, these studies identify chromosomal regions associated with disease that are 10 cM (roughly 10 million nucleotides) in length. A region of this size may contain 100 to 300 potential genes, making it challenging to identify the causal gene(s). For whole-genome association studies, an associated SNP is not necessarily functionally relevant and may simply be tightly linked with the real (but unidentified) causal gene. In some cases, the SNP may not even lie within a known or putative gene. For candidate genes identified by functional genomic studies, the difficulty is in determining whether gene expression levels are altered because the genes are contributing to disease biology or whether the genes are altered as a consequence of the disease process. There are success stories, however. The information on genes obtained from genome-wide association studies has been used successfully to study the variations in response to some critical drugs such as warfarin. Warfarin has a narrow therapeutic window and shows large variations in dose requirements between patients. Patients with CYP29*2 and CYP29*3 allele variants seem to require lower doses of warfarin to achieve an optimal state of anticoagulation.111 In 2005, the U.S. Food and Drug Administration (FDA) changed the label of warfarin to point out the potential relevance of genetic information to prescribing decisions.112

Perioperative Genomics in Cardiac Surgery
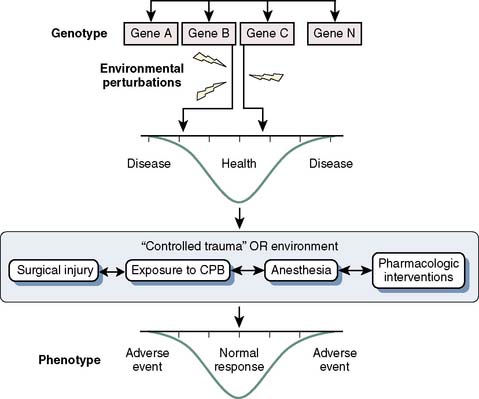
Despite advances in surgical, anesthetic, and cardioprotective strategies, the incidence of perioperative adverse events in cardiac surgery continues to be significant and is associated with reduced short- and long-term survival.113 Because all surgical patients are exposed to perturbations that potentially activate inflammation, coagulation, and other stress-related pathways, but only a subset experience adverse perioperative events (even after controlling for coexistent disease), a genetic component is likely to be involved114–116 (Figure 7-15). Perioperative genomics is a new field that uses functional genomic approaches to discover underlying biologic mechanisms that explain why similar patients have dramatically different outcomes after surgery.117
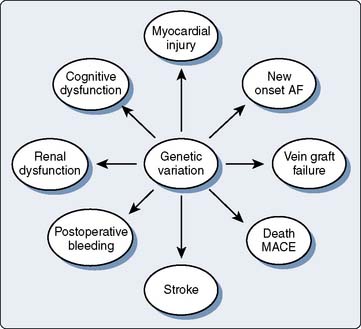
Genetic variants have been found for adverse events such as myocardial ischemia,118 postoperative arrhythmias,119 vein graft restenosis,118 renal compromise,120 neurocognitive dysfunction,121 stroke,122 and death,118 as well as more systemic outcomes such as bleeding,123 thrombosis,124 inflammatory responses, and severe sepsis125–128 (Figure 7-16). The Duke Perioperative Genomics investigative team initiated a prospective study (the Perioperative Genomics And Safety Study, US [PEGASUS]) in 2001. In recent years, broader multi-institutional perioperative genomics groups have been formed in the United States (PeriGReN) and internationally (iPEGASUS). The goal of the PEGASUS group is to use genetic variability to determine which individuals are at risk for adverse events after surgery. As an example of the power of perioperative genetic/genomic approaches, the PEGASUS group recently examined the perioperative myocardial injury/infarction (PMI) end point by genotyping 48 polymorphisms from 23 candidate genes in a prospective cohort of 434 patients undergoing elective cardiac surgery with cardiopulmonary bypass.129 After adjusting for multiple comparisons and clinical risk factors, three polymorphisms were found as independent predictors of PMI. These represent SNPs in genes encoding IL-6 and 2 adhesion molecules, intercellular adhesion molecule 1 (ICAM-1) and E-selectin. In contrast, one mutation (SELE98G>T) decreased the risk for PMI in the study. The investigators concluded that functional genetic variants in cytokine and leukocyte-endothelial interaction pathways are independently associated with severity of myonecrosis after cardiac surgery.
Do these genetic/genomic studies result in practical information capable of facilitating therapeutic interventions designed to improve outcome? A study of the effects of a 5-lipoxygenase-activating protein (FLAP) inhibitor on biomarkers associated with increased risk for myocardial infarction demonstrated that, by defining at-risk patients using two genes in the leukotriene pathway, it can be predicted who will respond to targeted drug therapy.130 Use of the FLAP inhibitor DG-031 in patients with these variants leads to significant and dose-dependent suppression of biomarkers associated with increased risk for myocardial infarction events. Although this study did not take place during surgery, similar principles would be expected to be operational in the perioperative period. These findings may soon translate into prospective risk assessment incorporating genomic profiling of markers important in inflammatory, thrombotic, vascular, and neurologic responses to perioperative stress, with implications ranging from individualized additional preoperative testing and physiologic optimization, to perioperative decision making, options for monitoring approaches, and critical care resource utilization.

Gene Therapy
Replacement of genes is by now a routine technique in laboratory cell cultures, and can be performed with reasonable ease in animal models such as transgenic animals. Application in the clinical setting has been difficult, however. Obviously, it is most times not necessary to replace a gene in every cell of the body. In cystic fibrosis, for example, a disease in which gene therapy is probably closest to succeeding, the main target tissue will be the pulmonary system. The main issue then becomes the delivery of the functional gene to the target tissue in such a manner that it will be consistently expressed at adequate levels. A variety of experimental techniques exists. Cells can be removed from the patient’s body, changed in culture, and then be returned to the patient. Alternatively, the gene can be incorporated in an otherwise innocuous virus that then is used to infect the patient’s tissues. Both approaches show promise, but the technical difficulties indicate that this is still a considerable time away from routine clinical application. Nonetheless, several clinical trials are in progress.131,132
Inhibition of gene expression by antisense treatment is similarly full of promise.133–135 The technique is based on the fact that mRNA can no longer be translated efficiently into protein when bound to a complementary strand of nucleic acid, a so-called antisense strand. Thus, by introducing specific antisense DNA into cells, expression of a selected gene product can be inhibited. The problem here, again, is efficient targeting of the tissue of interest. Remarkably, cells are able to take up antisense material from the extracellular environment without degrading it, but the material still has to be brought in contact with the tissue of interest. A second technical problem involves the stability of the antisense DNA. Because most diseases for which this technique seems suitable require constant inhibition of gene expression over long periods, the antisense construct has to be highly stable, and this degree of stability has not been consistently achieved yet.
Somatic Gene Therapy for Cardiac Arrhythmias
Somatic gene therapy means addition of genes as either DNA or RNA to cells other than eggs or sperm, with the goal of treating or preventing a disease. Potential delivery strategies include replication-deficient viruses, and physicochemical techniques such as liposomes and direct injection. Viral transfection of an inhibitory G protein into the AV node of pigs has been used to control the ventricular response to atrial fibrillation.136 The rationale for this strategy was that overexpression of an inhibitory G protein would, in effect, create a localized β receptor blockade. The success of this strategy may point to human trials in the future.
Stem Cell Therapy
Stem cells are immature tissue precursor cells that are undifferentiated and can differentiate into specialized cells including cardiomyocytes and cardiac rhythm-generating tissue. Stem cells may be isolated from the fetus, but limited supply and ethical considerations have mandated a search for a more easily accessible source. Skeletal myoblasts, endothelial progenitor cells, and adult mesenchymal stem cells have all been used. There are early reports of the use of stem cells for cellular cardiomyoplasty in patients with end-stage heart failure after myocardial infarction, but there remain concerns about incorporation of the cells into the myocardium and the possibility of arrhythmogenesis.137 Prospective randomized trials are awaited. Human mesenchymal stem cells transfected with HCN2, a pacemaker channel, have been incorporated into canine myocardium. These cells have been shown to form gap junctions with the myocardium and to generate rhythmic activity after AV nodal blockade.138 These experiments offer the distant prospect of a biologic cardiac pacemaker.

1 Watson J.D., Crick F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171:737-738.
2 Hamill O.P., Marty A., Neher E., et al. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85-100.
3 Sakmann B., Trube G. Voltage-dependent inactivation of inward-rectifying single-channel currents in the guinea-pig heart cell membrane. J Physiol. 1984;347:659-683.
4 Hume J.R., Uehara A. Ionic basis of the different action potential configurations of single guinea-pig atrial and ventricular myocytes. J Physiol. 1985;368:525-544.
5 Moorman J.R. Sodium channels. In: Yaksh T., et al, editors. Anesthesia: Biologic foundations. Philadelphia: Lippincott-Raven Publishers; 1997:145.
6 Coraboeuf E., Carmeliet E. Existence of two transient outward currents in sheep cardiac Purkinje fibers. Pflugers Arch. 1982;392:352-359.
7 Hiraoka M., Kawano S. Calcium-sensitive and insensitive transient outward current in rabbit ventricular myocytes. J Physiol. 1989;410:187-212.
8 Li Q., Keung E.C. Effects of myocardial hypertrophy on transient outward current. Am J Physiol. 1994;266:H1738-H1745.
9 Lue W.M., Boyden P.A. Abnormal electrical properties of myocytes from chronically infarcted canine heart. Alterations in Vmax and the transient outward current. Circulation. 1992;85:1175-1188.
10 Attwell D., Cohen I., Eisner D., et al. The steady state TTX-sensitive (“window”) sodium current in cardiac Purkinje fibres. Pflugers Arch. 1979;379:137-142.
11 Grant A.O., Starmer C.F. Mechanisms of closure of cardiac sodium channels in rabbit ventricular myocytes: Single-channel analysis. Circ Res. 1987;60:897-913.
12 Tsien R.W., Bean B.P., Hess P., et al. Mechanisms of calcium channel modulation by beta-adrenergic agents and dihydropyridine calcium agonists. J Mol Cell Cardiol. 1986;18:691-710.
13 Reuter H. Calcium channel modulation by neurotransmitters, enzymes and drugs. Nature. 1983;301:569-574.
14 Bennett P., McKinney L., Begenisich T., Kass R.S. Adrenergic modulation of the delayed rectifier potassium channel in calf cardiac Purkinje fibers. Biophys J. 1986;49:839-848.
15 Sanguinetti M.C., Jurkiewicz N.K. Two components of cardiac delayed rectifier K+ current. differential sensitivity to block by class III antiarrhythmic agents. J Gen Physiol. 1990;96:195-215.
16 Wang Z., Fermini B., Nattel S. Sustained depolarization-induced outward current in human atrial myocytes. Evidence for a novel delayed rectifier K+ current similar to Kv1.5 cloned channel currents. Circ Res. 1993;73:1061-1076.
17 Trautwein W., Taniguchi J., Noma A. The effect of intracellular cyclic nucleotides and calcium on the action potential and acetylcholine response of isolated cardiac cells. Pflugers Arch. 1982;392:307-314.
18 Giles W., Noble S.J. Changes in membrane currents in bullfrog atrium produced by acetylcholine. J Physiol. 1976;261:103-123.
19 Ragazzi E., Wu S.N., Shryock J., Belardinelli L. Electrophysiological and receptor binding studies to assess activation of the cardiac adenosine receptor by adenine nucleotides. Circ Res. 1991;68:1035-1044.
20 Pfaffinger P.J., Martin J.M., Hunter D.D., et al. GTP-binding proteins couple cardiac muscarinic receptors to a K channel. Nature. 1985;317:536-538.
21 Liu D.W., Gintant G.A., Antzelevitch C. Ionic bases for electrophysiological distinctions among epicardial, midmyocardial, and endocardial myocytes from the free wall of the canine left ventricle. Circ Res. 1993;72:671-687.
22 DiFrancesco D. The onset and autonomic regulation of cardiac pacemaker activity: Relevance of the f current. Cardiovasc Res. 1995;29:449-456.
23 DiFrancesco D., Tortora P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature. 1991;351:145-147.
24 Yatani A., Okabe K., Codina J., et al. Heart rate regulation by G proteins acting on the cardiac pacemaker channel. Science. 1990;249:1163-1166.
25 Armstrong C.M., Bezanilla F. Currents related to movement of the gating particles of the sodium channels. Nature. 1973;242:459-461.
26 Catterall W.A. Molecular properties of voltage gated ion channels in the heart. In: Fozzard H.A., Haber E., Jennings R., editors. The heart and cardiovascular system—scientific foundations. New York: Raven Press; 1992:945-962.
27 Lipkind G.M., Fozzard H.A. A structural model of the tetrodotoxin and saxitoxin binding site of the Na+ channel. Biophys J. 1994;66:1-13.
28 Hille B. The permeability of the sodium channel to organic cations in myelinated nerve. J Gen Physiol. 1971;58:599-619.
29 Tomaselli G.F., Backx P.H., Marban E. Molecular basis of permeation in voltage-gated ion channels. Circ Res. 1993;72:491-496.
30 Armstrong C.M., Bezanilla F., Rojas E. Destruction of sodium conductance inactivation in squid axons perfused with pronase. J Gen Physiol. 1973;62:375-391.
31 Korn S.J., Trapani J.G. Potassium channels. IEEE Trans Nanobioscience. 2005;4:21-33.
32 Echt D.S., Liebson P.R., Mitchell L.B., et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N Engl J Med. 1991;324:781-788.
33 Riera A.R., Uchida A.H., Ferreira C., et al. Relationship among amiodarone, new class III antiarrhythmics, miscellaneous agents and acquired long QT syndrome. J Cardiol. 2008;15:209-219.
34 Mohler P.J., Schott J.J., Gramolini A.O., et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634-639.
35 Brugada P., Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20:1391-1396.
36 Benito B., Brugada R., Brugada J., Brugada P. Brugada syndrome. Prog Cardiovasc Dis. 2008;51:1-22.
37 Rozec B., Gauthier C. Beta3-adrenoceptors in the cardiovascular system: Putative roles in human pathologies. Pharmacol Ther. 2006;111:652-673.
38 Bristow M.R., Hershberger R.E., Port J.D., et al. Beta 1- and beta 2-adrenergic receptor-mediated adenylate cyclase stimulation in nonfailing and failing human ventricular myocardium. Mol Pharmacol. 1989;35:295-303.
39 Lipsky R., Potts E.M., Tarzami S.T., et al. Beta-adrenergic receptor activation induces internalization of cardiac Cav1.2 channel complexes through a beta-arrestin 1-mediated pathway. J Biol Chem. 2008;283:17221-17226.
40 Bristow M.R., Minobe W., Rasmussen R., et al. Alpha-1 adrenergic receptors in the nonfailing and failing human heart. J Pharmacol Exp Ther. 1988;247:1039-1045.
41 Hasking G.J., Esler M.D., Jennings G.L., et al. Norepinephrine spillover to plasma in patients with congestive heart failure: Evidence of increased overall and cardiorenal sympathetic nervous activity. Circulation. 1986;73:615-621.
42 Harding S.E., Brown L.A., Wynne D.G., et al. Mechanisms of beta adrenoceptor desensitisation in the failing human heart. Cardiovasc Res. 1994;28:1451-1460.
43 Maisel A.S., Ziegler M.G., Carter S., et al. In vivo regulation of beta-adrenergic receptors on mononuclear leukocytes and heart. Assessment of receptor compartmentation after agonist infusion and acute aortic constriction in guinea pigs. J Clin Invest. 1988;82:2038-2044.
44 Chen C.Y., Dion S.B., Kim C.M., Benovic J.L. Beta-adrenergic receptor kinase. Agonist-dependent receptor binding promotes kinase activation. J Biol Chem. 1993;268:7825-7831.
45 Lohse M.J., Benovic J.L., Codina J., et al. Beta-arrestin: A protein that regulates beta-adrenergic receptor function. Science. 1990;248:1547-1550.
46 Deighton N.M., Motomura S., Borquez D., et al. Muscarinic cholinoceptors in the human heart: Demonstration, subclassification, and distribution. Naunyn Schmiedebergs Arch Pharmacol. 1990;341:14-21.
47 Lechleiter J., Hellmiss R., Duerson K., et al. Distinct sequence elements control the specificity of G protein activation by muscarinic acetylcholine receptor subtypes. EMBO J. 1990;9:4381-4390.
48 Logothetis D.E., Kurachi Y., Galper J., et al. The beta gamma subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature. 1987;325:321-326.
49 Swynghedauw B., Besse S., Assayag P., et al. Molecular and cellular biology of the senescent hypertrophied and failing heart. Am J Cardiol. 1995;76:2D-7D.
50 Schmitz W., Boknik P., Linck B., Muller F.U. Adrenergic and muscarinic receptor regulation and therapeutic implications in heart failure. Mol Cell Biochem. 1996;157:251-258.
51 Bohm M., Gierschik P., Jakobs K.H., et al. Increase of Gi alpha in human hearts with dilated but not ischemic cardiomyopathy. Circulation. 1990;82:1249-1265.
52 Fleming J.W., Wisler P.L., Watanabe A.M. Signal transduction by G proteins in cardiac tissues. Circulation. 1992;85:420-433.
53 Pitcher J.A., Inglese J., Higgins J.B., et al. Role of beta gamma subunits of G proteins in targeting the beta-adrenergic receptor kinase to membrane-bound receptors. Science. 1992;257:1264-1267.
54 Feldman A.M., Jackson D.G., Bristow M.R., et al. Immunodetectable levels of the inhibitory guanine nucleotide-binding regulatory proteins in failing human heart: Discordance with measurements of adenylate cyclase activity and levels of pertussis toxin substrate. J Mol Cell Cardiol. 1991;23:439-452.
55 Bristow M.R., Minobe W., Rasmussen R., et al. Beta-adrenergic neuroeffector abnormalities in the failing human heart are produced by local rather than systemic mechanisms. J Clin Invest. 1992;89:803-815.
56 Hershberger R.E., Anderson F.L., Bristow M.R. Vasoactive intestinal peptide receptor in failing human ventricular myocardium exhibits increased affinity and decreased density. Circ Res. 1989;65:283-294.
57 Mustafa S.J., Morrison R.R., Teng B., Pelleg A. Adenosine receptors and the heart: Role in regulation of coronary blood flow and cardiac electrophysiology. Handb Exp Pharmacol. 2009;193:161-188.
58 Murphy E., Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581-609.
59 Schutz W., Schrader J., Gerlach E. Different sites of adenosine formation in the heart. Am J Physiol. 1981;240:H963-H970.
60 Schrader J. Metabolism of adenosine and sites of production in the heart. In: Berne R.M., Rall T.W., Rubio R., editors. Regulatory functions of adenosine. Martinus Nijhoff: The Hague; 1983:133.
61 Moser G.H., Schrader J., Deussen A. Turnover of adenosine in plasma of human and dog blood. Am J Physiol. 1989;256:C799-C806.
62 Visentin S., Wu S.N., Belardinelli L. Adenosine-induced changes in atrial action potential: Contribution of Ca and K currents. Am J Physiol. 1990;258:H1070-H1078.
63 Dobson J.G.Jr. Mechanism of adenosine inhibition of catecholamine-induced responses in heart. Circ Res. 1983;52:151-160.
64 Innes J.A. Review article: Adenosine use in the emergency department. Emerg Med Australas. 2008;20:209-215.
65 diMarco J.P., Sellers T.D., Lerman B.B., et al. Diagnostic and therapeutic use of adenosine in patients with supraventricular tachyarrhythmias. J Am Coll Cardiol. 1985;6:417-425.
66 Shen W.K., Hamill S.C., Cardiac arrhythmias. Mayo clinic practice of cardiology. ed 3; 1996. CV Mosby. St. Louis
67 Huffmyer J., Raphael J. Physiology and pharmacology of myocardial preconditioning and postconditioning. Semin Cardiothorac Vasc Anesth. 2009;13:5-18.
68 Sato T., Sasaki N., Seharaseyon J., et al. Selective pharmacological agents implicate mitochondrial but not sarcolemmal K(ATP) channels in ischemic cardioprotection. Circulation. 2000;101:2418-2423.
69 Tomai F., Crea F., Gaspardone A., et al. Ischemic preconditioning during coronary angioplasty is prevented by glibenclamide, a selective ATP-sensitive K+ channel blocker. Circulation. 1994;90:700-705.
70 Kirsch G.E., Codina J., Birnbaumer L., Brown A.M. Coupling of ATP-sensitive K+ channels to A1 receptors by G proteins in rat ventricular myocytes. Am J Physiol. 1990;259:H820-H826.
71 Yvon A., Hanouz J.L., Haelewyn B., et al. Mechanisms of sevoflurane-induced myocardial preconditioning in isolated human right atria in vitro. Anesthesiology. 2003;99:27-33.
72 de Klaver M.J., Buckingham M.G., Rich G.F. Lidocaine attenuates cytokine-induced cell injury in endothelial and vascular smooth muscle cells. Anesth Analg. 2003;97:465.
73 Huneke R., Fassl J., Rossaint R., Luckhoff A. Effects of volatile anesthetics on cardiac ion channels. Acta Anaesthesiol Scand. 2004;48:547-561.
74 Porsius A.J., van Zwieten P.A. Influence of halothane on calcium movements in isolated heart muscle and in isolated plasma membranes. Arch Int Pharmacodyn Ther. 1975;218:29-39.
75 Bosnjak Z.J., Supan F.D., Rusch N.J. The effects of halothane, enflurane, and isoflurane on calcium current in isolated canine ventricular cells. Anesthesiology. 1991;74:340-345.
76 Yamakage M., Hirshman C.A., Croxton T.L. Inhibitory effects of thiopental, ketamine, and propofol on voltage-dependent Ca2+ channels in porcine tracheal smooth muscle cells. Anesthesiology. 1995;83:1274-1282.
77 Pancrazio J.J. Halothane and isoflurane preferentially depress a slowly inactivating component of Ca2+ channel current in guinea-pig myocytes. J Physiol. 1996;494:91-103.
78 Fassl J., Halaszovich C.R., Huneke R., et al. Effects of inhalational anesthetics on L-type Ca2+ currents in human atrial cardiomyocytes during beta-adrenergic stimulation. Anesthesiology. 2003;99:90-96.
79 Kamatchi G.L., Durieux M.E., Lynch C.3rd. Differential sensitivity of expressed L-type calcium channels and muscarinic M(1) receptors to volatile anesthetics in Xenopus oocytes. J Pharmacol Exp Ther. 2001;297:981-990.
80 Ikemoto Y., Yatani A., Arimura H., Yoshitake J. Reduction of the slow inward current of isolated rat ventricular cells by thiamylal and halothane. Acta Anaesthesiol Scand. 1985;29:583-586.
81 Aronstam R.S., Dennison R.L.Jr. Anesthetic effects on muscarinic signal transduction. Int Anesthesiol Clin. 1989;27:265-272.
82 Aronstam R.S., Anthony B.L., Dennison R.L.Jr. Halothane effects on muscarinic acetylcholine receptor complexes in rat brain. Biochem Pharmacol. 1986;35:667-672.
83 Dennison R.L.Jr, Anthony B.L., Narayanan T.K., Aronstam R.S. Effects of halothane on high affinity agonist binding and guanine nucleotide sensitivity of muscarinic acetylcholine receptors from brainstem of rat. Neuropharmacology. 1987;26:1201-1205.
84 Krnjevic K., Puil E. Halothane suppresses slow inward currents in hippocampal slices. Can J Physiol Pharmacol. 1988;66:1570-1575.
85 Kai T., Jones K.A., Warner D.O. Halothane attenuates calcium sensitization in airway smooth muscle by inhibiting G-proteins. Anesthesiology. 1998;89:1543-1552.
86 Streiff J., Jones K., Perkins W.J., et al. Effect of halothane on the guanosine 5× triphosphate binding activity of G-protein alpha subunits. Anesthesiology. 2003;99:105-111.
87 Magyar J., Szabo G. Effects of volatile anesthetics on the G protein-regulated muscarinic potassium channel. Mol Pharmacol. 1996;50:1520-1528.
88 Robin N.H., Tabereaux P.B., Benza R., Korf B.R. Genetic testing in cardiovascular disease. J Am Coll Cardiol. 2007;50:727-737.
89 Nabel E.G. Cardiovascular disease. N Engl J Med. 2003;349:60-72.
90 Cowan J., Morales A., Dagua J., Hershberger R.E. Genetic testing and genetic counseling in cardiovascular genetic medicine: Overview and preliminary recommendations. Congest Heart Fail. 2008;14:97-105.
91 Burkett E.L., Hershberger R.E. Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2005;45:969-981.
92 Ho C.Y., Seidman C.E. A contemporary approach to hypertrophic cardiomyopathy. Circulation. 2006;113:e858-e862.
93 Bos J.M., Poley R.N., Ny M., et al. Genotype-phenotype relationships involving hypertrophic cardio-myopathy-associated mutations in titin, muscle LIM protein, and telethonin. Mol Genet Metab. 2006;88:78-85.
94 Van Driest S.L., Ommen S.R., Tajik A.J., et al. Yield of genetic testing in hypertrophic cardiomyopathy. Mayo Clin Proc. 2005;80:739-744.
95 Nishimura R.A., Holmes D.R.Jr. Clinical practice. Hypertrophic obstructive cardiomyopathy. N Engl J Med. 2004;350:1320-1327.
96 Semsarian C. CSANZ Cardiovascular Genetics Working Group: Guidelines for the diagnosis and management of hypertrophic cardiomyopathy. Heart Lung Circ. 2007;16:16-18.
97 Maron B.J., McKenna W.J., Danielson G.K., et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation task force on clinical expert consensus documents and the European Society of Cardiology Committee for practice guidelines. J Am Coll Cardiol. 2003;42:1687-1713.
98 Priori S.G., Napolitano C., Schwartz P.J., et al. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA. 2004;292:1341-1344.
99 Schwartz P.J., Priori S.G., Spazzolini C., et al. Genotype-phenotype correlation in the long-QT syndrome: Gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89-95.
100 Ott J., Bhat A. Linkage analysis in heterogeneous and complex traits. Eur Child Adolesc Psychiatry. 1999;3:43-46.
101 Wang L., Fan C., Topol SE., et al. Mutation of MEF2A in an inherited disorder with features of coronary artery disease. Science. 2003;302:1578-1581.
102 Broeckel U., Hengstenberg C., Mayer B., et al. A comprehensive linkage analysis for myocardial infarction and its related risk factors. Nat Genet. 2002;30:210-214.
103 Hauser E.R., Crossman D.C., Granger C.B., et al. A genomewide scan for early-onset coronary artery disease in 438 families: The GENECARD study. Am J Hum Genet. 2004;75:436-447.
104 Helgadottir A., Thorleifsson G., Manolescu A., et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491-1493.
105 McPherson R., Pertsemlidis A., Kavaslar N., et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488-1491.
106 Bell J. Predicting disease using genomics. Nature. 2004;429:453-456.
107 Tuomisto T.T., Binder B.R., Yla-Herttuala S. Genetics, genomics and proteomics in atherosclerosis research. Ann Med. 2005;37:323-332.
108 Gudbjartsson D.F., Arnar D.O., Helgadottir A., et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. 2007;448:353-357.
109 Rockhill B., Kawachi I., Colditz G.A. Individual risk prediction and population-wide disease prevention. Epidemiol Rev. 2000;22:176-180.
110 Horwitz P.A., Tsai E.J., Putt M.E., et al. Detection of cardiac allograft rejection and response to immunosuppressive therapy with peripheral blood gene expression. Circulation. 2004;110:3815-3821.
111 Cooper G.M., Johnson J.A., Langaee T.Y., et al. A genome-wide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood. 2008;112:1022-1027.
112 FDA releases final guidance for pharmacogenomic data. Pharmacogenomics. 2005;6:209.
113 Newby L.K., Alpert J.S., Ohman E.M., et al. Changing the diagnosis of acute myocardial infarction: Implications for practice and clinical investigations. Am Heart J. 2002;144:957-980.
114 Fox A.A., Shernan S.K., Body S.C. Predictive genomics of adverse events after cardiac surgery. Semin Cardiothorac Vasc Anesth. 2004;8:297-315.
115 Stuber F., Hoeft A. The influence of genomics on outcome after cardiovascular surgery. Curr Opin Anaesthesiol. 2002;15:3-8.
116 Ziegeler S., Tsusaki B.E., Collard C.D. Influence of genotype on perioperative risk and outcome. Anesthesiology. 2003;99:212-219.
117 Donahue B.S., Balser J.R. Perioperative genomics. Venturing into uncharted seas. Anesthesiology. 2003;99:7-8.
118 Zotz R.B., Klein M., Dauben H.P., et al. Prospective analysis after coronary-artery bypass grafting: Platelet GP IIIa polymorphism (HPA-1b/PIA2) is a risk factor for bypass occlusion, myocardial infarction, and death. Thromb Haemost. 2000;83:404-407.
119 Gaudino M., Andreotti F., Zamparelli R., et al. The -174G/C interleukin-6 polymorphism influences postoperative interleukin-6 levels and postoperative atrial fibrillation. Is atrial fibrillation an inflammatory complication? Circulation. 2003;108:195-199.
120 Stafford-Smith M., Podgoreanu M., Swaminathan M., et al. Association of genetic polymorphisms with risk of renal injury after coronary bypass graft surgery. Am J Kidney Dis. 2005;45:519-530.
121 Mathew J.P., Rinder C.S., Howe J.G., et al. Platelet PlA2 polymorphism enhances risk of neurocognitive decline after cardiopulmonary bypass. Multicenter study of perioperative ischemia (McSPI) research group. Ann Thorac Surg. 2001;71:663-666.
122 Grocott H.P., White W.D., Morris R.W., et al. Genetic polymorphisms and the risk of stroke after cardiac surgery. Stroke. 2005;36:1854-1858.
123 Welsby I.J., Podgoreanu M.V., Phillips-Bute B., et al. Genetic factors contribute to bleeding after cardiac surgery. J Thromb Haemost. 2005;3:1206-1212.
124 Donahue BS., Gailani D., Higgins M.S., et al. Factor V leiden protects against blood loss and transfusion after cardiac surgery. Circulation. 2003;107:1003-1008.
125 Burzotta F., Iacoviello L., Di Castelnuovo A., et al. Relation of the -174 G/C polymorphism of interleukin-6 to interleukin-6 plasma levels and to length of hospitalization after surgical coronary revascularization. Am J Cardiol. 2001;88:1125-1128.
126 Grocott H.P., Newman M.F., El-Moalem H., et al. Apolipoprotein E genotype differentially influences the proinflammatory and anti-inflammatory response to cardiopulmonary bypass. J Thorac Cardiovasc Surg. 2001;122:622-623.
127 Roth-Isigkeit A., Hasselbach L., Ocklitz E., et al. Inter-individual differences in cytokine release in patients undergoing cardiac surgery with cardiopulmonary bypass. Clin Exp Immunol. 2001;125:80-88.
128 Galley H.F., Lowe P.R., Carmichael R.L., Webster N.R. Genotype and interleukin-10 responses after cardiopulmonary bypass. Br J Anaesth. 2003;91:424-426.
129 Podgoreanu M.V., White W.D., Morris R.W., et al. Inflammatory gene polymorphisms and risk of postoperative myocardial infarction after cardiac surgery. Circulation. 2006;114:I275-I281.
130 Hakonarson H., Thorvaldsson S., Helgadottir A., et al. Effects of a 5-lipoxygenase-activating protein inhibitor on biomarkers associated with risk of myocardial infarction: A randomized trial. JAMA. 2005;293:2245-2256.
131 Alexander B.L., Ali R.R., Alton E.W., et al. Progress and prospects: Gene therapy clinical trials (part 1). Gene Ther. 2007;14:1439-1447.
132 Aiuti A., Bachoud-Levi AC., Blesch A., et al. Progress and prospects: Gene therapy clinical trials (part 2). Gene Ther. 2007;14:1555-1563.
133 Aboul-Fadl T. Antisense oligonucleotides: The state of the art. Curr Med Chem. 2005;12:2193-2214.
134 Nath RK., Xiong W., Humphries AD., Beri R. Treatment with antisense oligonucleotide reduces the expression of type I collagen in a human-skin organ-wound model: Implications for antifibrotic gene therapy. Ann Plast Surg. 2007;59:699-706.
135 Takeshima Y., Yagi M., Wada H., et al. Intravenous infusion of an antisense oligonucleotide results in exon skipping in muscle dystrophin mRNA of Duchenne muscular dystrophy. Pediatr Res. 2006;59:690-694.
136 Donahue J.K., Heldman A.W., Fraser H., et al. Focal modification of electrical conduction in the heart by viral gene transfer. Nat Med. 2000;6:1395-1398.
137 Lee M.S., Makkar R.R. Stem-cell transplantation in myocardial infarction: A status report. Ann Intern Med. 2004;140:729-737.
138 Potapova I., Plotnikov A., Lu Z., et al. Human mesenchymal stem cells as a gene delivery system to create cardiac pacemakers. Circ Res. 2004;94:952-959.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
7 Molecular and Genetic Cardiovascular Medicine
The past decades have witnessed what may be termed a revolution in the biomedical sciences, as molecular and genetic methodologies have suddenly jumped onto the clinical scene. Molecular biology originated in the 1950s, its birth most commonly identified with the description of the structure of deoxyribonucleic acid (DNA) by Watson and Crick.1 For many years after, it was practiced almost exclusively in the research laboratory. Much of this research involved the laborious process of cloning: the identification of DNA molecules encoding specific proteins. Although at the time most people in the field realized that these advances would one day be of immense importance to clinical medicine, the exact place they would take was unclear.
The machinery behind the cardiac rhythm: ion channels
The cardiac action potential results from the flow of ions through ion channels, which are the membrane-bound proteins that form the structural machinery behind cardiac electrical excitability. In response to changes in electrical potential across the cell membrane, ion channels open and allow the passive flux of ions into or out of the cell along their electrochemical gradients. This flow of charged ions results in a current, which will alter the cell membrane potential toward the potential at which the electrochemical gradient for the ion is zero and is called the equilibrium potential (E) for the ion. Depolarization of the cell could, in principle, result from an inward cation current or an outward anion current; for repolarization, the reverse is true. In excitable cells, action potentials are mainly caused by the flow of cation currents. Membrane depolarization results principally from the flow of Na+ down its electrochemical gradient (ENa is around +50mV), whereas repolarization results from the outward flux of K+ down its electrochemical gradient (EK is around −90 mV). Opening and closing of ion channels selective for a single ion result in an individual ionic current. The integrated activity of many different ionic currents, each activated over precisely regulated potential ranges and at different times in the cardiac cycle, results in the cardiac action potential. Ion channels are usually highly (but not uniquely) selective for a single ion (e.g., K+ channels, Na+ channels). Channels may rectify, that is, pass current in one direction across the membrane more easily than the other. Electrical and chemical stimuli, which lead to opening and closing of the channel, cause a conformational change in the channel molecule (gating). The rate of change of channel conformation (gating kinetics) may be rapid, in which case the channel will open (activate) almost immediately (e.g., Na+ channels), or relatively slowly, which will result in a delay in channel activation (e.g., delayed rectifier K+ channels). After activation, ion channels may stay open until closed by another stimulus (e.g., repolarization of the membrane) or may close (inactivate) in the face of a continued stimulus. Inactivated channels will usually not reopen on repeat stimulation until they have recovered from inactivation (Box 7-1).
Patch Clamping
The development of the voltage clamp technique in the early 1950s and its application to multicellular preparations of cardiac muscle allowed identification of the major ionic currents that underlie the cardiac action potential. The whole-cell currents recorded by the technique were smooth waveforms derived from summation of the activity of thousands of ion channels, and many different patterns of events at the single-channel level, arising from more than one molecular specie, can summate to produce identical whole-cell current waveforms. Patch clamping allows resolution of events at the single-channel level. In this technique, small patches of cell membrane (< 1 μm2) are isolated electrically and physically in the tip of a glass micropipette.2 Single-channel events can then be resolved because there are only a few ion channel molecules present in the patch. Current flowing across the patch typically jumps between well-defined values corresponding to sudden opening or closing of the ion-conducting pore (Figure 7-1A). The whole-cell current will be the sum of the currents through all of the individual channels in the cell membrane; summation of the current flowing through a single channel during repeated stimuli will reproduce the macroscopic whole-cell current (see Figures 7-1A and B). As channel opening and closing in response to a stimulus are a stochastic phenomena, the regulation of ion flow, whether resulting from a change in membrane potential or from interaction with regulator molecules, is usually achieved by increasing the probability that the channel will be open. Thus, in Figure 7-1, which shows records from human cardiac muscle Na+ channels, the channels open (activate) a few milliseconds after depolarization because the probability that the channel will be open increases. Similarly, as the channels spontaneously close (inactivate), the open probability decreases.
Electrical Events Underlying the Cardiac Action Potential
Figure 7-2 shows a diagram of a cardiac action potential with a summary of the ionic currents flowing during each phase. As far as has been determined, the probable molecular identity of the ion channels that underlie these currents is also given. This section examines the biophysical properties of these currents; subsequent sections focus on possible molecular mechanisms underlying the biophysical phenomena.
Figure 7-2 Ionic currents underlying the cardiac action potential.
(From Roden DM, Lazzarra R, Rosen R, et al, for the SADS Foundation Task Force on LQTS: Multiple mechanisms in the long QT syndrome: Current knowledge, gaps and future directions. Circulation 94:1996, 1996.)
The Resting Membrane Potential and the Role of IK1
The resting cardiac membrane potential is maintained close to the equilibrium potential for potassium (EK) by a background, inwardly rectifying, highly selective K+ current (IK1). Under physiologic conditions, the EK is around −90 mV, and displacement of the membrane potential from this value should result in an inward or outward current through IK1, thus returning the membrane potential toward EK. However, although IK1 passes significant inward current at potentials negative to EK, at positive potentials, the channels display inward rectification (i.e., they pass inward current more easily than outward current), which limits outward current flow and reduces the tendency for IK1 to hyperpolarize the membrane. This has obvious significance for an excitable cell; inward rectification3 allows the initiation of action potentials and limits cellular K+ loss during the action potential. IK1 is large in ventricular cells, smaller in atrial cells, and almost absent in nodal tissue.4 This explains why ventricular cells rest near EK and have a high threshold for excitation, atrial cells have a more positive resting potential, and nodal cells have no defined resting potential.
Phase 0: Rapid Upstroke of the Cardiac Action Potential
The rapid upstroke of the cardiac action potential (phase 0) is caused by the flow of a large inward Na+ current (Ina) (Box 7-2).5 INa is activated by depolarization of the sarcolemma to a threshold potential of −65 to −70 mV. INa activation, and hence the action potential, is an all-or-nothing response. Subthreshold depolarizations have only local effects on the membrane. After the threshold for activation of fast Na+ channels is exceeded, Na+ channels open (i.e., INa activates) and Na+ ions enter the cell down their electrochemical gradient. This results in displacement of the membrane potential toward the equilibrium potential for Na+ ions, around +50 mV. INa activation is transient, lasting at most 1 to 2 msec because, simultaneous with activation, a second, slightly slower conformational change in the channel molecule occurs (inactivation), which closes the ion pore in the face of continued membrane depolarization (see Figures 7-1A and B). The channel cannot open again until it has recovered from inactivation (i.e., regained its resting conformation), a process that requires repolarization to the resting potential for a defined period. Thus, the channels cycle through three states: resting (and available for activation), open, and inactivated. Although the channel is inactivated, it is absolutely refractory to repeated stimulation. Stimuli that occur during recovery from inactivation will result in opening of fewer Na+ channels (because not all have recovered), and the resulting action potential will have a reduced maximal rate of depolarization and reduced conduction velocity. Na+ channels do not need to open to become inactivated. If the resting membrane potential depolarizes for a time, inactivation will occur in some channels and subsequent stimulation will result in an action potential of reduced amplitude and conduction velocity.
Phase 1: Early Rapid Repolarization
The early rapid repolarization phase of the action potential, which follows immediately after phase 0, results both from rapid inactivation of the majority of the Na+ current and from activation of a transient outward current (ITO), carried mainly by K+ ions. On depolarization of the membrane, ITO opens rapidly, over about 20 msec, before spontaneously inactivating. ITO comprises two separate currents: the rapidly inactivating ITO1, which is activated by depolarization and blocked by 4-aminopyridine, and the slowly inactivating ITO2, which is activated by elevated intracellular Ca++ (possibly explaining the observation that action potential duration tends to decrease with rapid heart rates and hypercalcemia)6,7 (Figure 7-3).
In addition to its effect on phase 1, ITO, in combination with the delayed rectifier potassium currents (IKr and IKs) and IK1, also contributes to membrane repolarization. Arrhythmogenic prolongation of the action potential in myocardial cells recovered from patients with myocardial hypertrophy,8 congestive cardiomyopathy,6 and from the border zone of myocardial infarction in animals9 appears to result from depression of ITO.
Phases 2 and 3: Plateau Phase and Final Rapid Repolarization
The action potential plateau and final rapid repolarization are mediated by a balance between the slow inward current and outward, predominantly K+ current. During the plateau phase, membrane conductance to all ions falls and very little current flows. Potassium conductance is low because of inward rectification of IK1 (i.e., inward current passes more easily than outward current), so little outward current flows despite the large outward electrochemical gradient for K+ ions and the delayed onset of the outwardly rectifying K+ currents (IKs, IKr, and IKur). The resulting small outward current is balanced by inward current, predominantly through L-type Ca++ channels (ICa-L), but also via a slowly inactivating population of Na+ channels, and a small inward flux of chloride (Cl–) ions, possibly carried by the cardiac variant of the ATP-dependent channel (abnormalities of which underlie cystic fibrosis).10,11 Phase 3, regenerative rapid repolarization, results from time-dependent inactivation of L-type Ca++ current and increasing outward current through delayed rectifier K+ channels. The net membrane current becomes outward and the cell repolarizes.
Slow Inward Ca++ Current
The slow inward current (ICa-L) is activated by depolarization of the cell to potentials less negative than −40 to −50 mV. In ventricular and atrial myocytes and in Purkinje fibers, ICa-L is activated by the regenerative depolarization caused by INa during phase 0 of the action potential. ICa-L does not contribute significantly to phase 0 because, in comparison with INa, it activates much more slowly (over about 10 msec) and is smaller in amplitude. ICa-L also inactivates slowly and, therefore, contributes the major inward current during the plateau of the action potential. ICa-L flows through L-type (long-lasting) Ca++ channels, which are sensitive to block by dihydropyridines (e.g., nifedipine), and activation of contraction is related to the magnitude of the resulting calcium influx.12 Gating of ICa-L is generally similar to INa in that channel opening and closing are dependent on membrane potential and time. Ca++ channels are also, importantly, dynamically regulated by the autonomic nervous system.13 β agonists activate ICa-L (and hence increase myocardial contractility) indirectly by activating adenylyl cyclase via a guanosine triphosphate (GTP)–binding protein, Gs (Figure 7-4). The resulting increase in intracellular cyclic adenosine monophosphate (cAMP) activates protein kinase A (PKA), which phosphorylates the Ca++ channel. Phosphorylated channels open in response to membrane depolarization; nonphosphorylated channels do not, so the effect of β-adrenergic stimulation is to increase the number of functional channels. The electrophysiologic effect of this is illustrated in Figure 7-5, which shows enhancement of the slow inward current by increase of adenosine monophosphate (AMP) level in single-channel Ca++ channels and intact cells. β-Adrenergic effects on ICa-L are antagonized by acetylcholine, which, in myocardial cells, activates M2 muscarinic receptors and inhibits adenylyl cyclase through activation of the GTP-binding protein Gi.
Delayed Rectifier K+ Currents
Delayed rectifier K+ channels are present in all cardiac myocytes. They open slowly (over 200 to 300 msec) after depolarization of the membrane to the plateau level (−10 mV and greater), producing a K+-selective outward current, IK. IK does not inactivate on prolonged depolarization (unlike INa and ICa-L), and the channels close on repolarization of the membrane. Unlike IK1, IK displays outward rectification; that is, it passes outward current more easily than inward current. This is the expected behavior for a K+-selective channel because both the concentration and electrical gradients for potassium are outward. Thus, for any depolarizing displacement of membrane potential from EK, the driving force will be larger in an outward direction. Similar to ICa-L, IK is under autonomic control (see Figure 7-4). β-Adrenergic stimulation enhances IK by a mechanism similar to that of the enhancement of ICa-L, thus ensuring repolarization of the cell in the face of increased inward Ca++ current.14
Three components of IK, carried by different channel molecules, can be distinguished. A rapidly activating component, IKr, is blocked by the compound E4031 (a Class III antiarrhythmic agent), which leaves a slower activating component, IKs, unaffected.15 This is illustrated in Figure 7-6, which also emphasizes the importance of IK in the regulation of repolarization, and hence of action potential duration. A third component, the ultra-rapidly activated delayed rectifier, IKur, can be distinguished in atrial (but not ventricular) myocytes.16 This additional repolarizing current explains, in part, the enhancement of repolarization in atrial myocardium when compared with ventricle and Purkinje fibers.
Repolarization in Different Cardiac Tissue Types
Phase 3 repolarization in atrium and pacemaker tissues, but not in ventricular myocardium, is also enhanced by the presence of a large outward repolarizing K+ current (IK[ACh]).17,18 This potential independent current is activated indirectly by stimulation of muscarinic (M2-type) receptors by acetylcholine or purinergic (A-type) receptors by adenosine.19 This channel is potential independent and is activated via binding of an activated, membrane-bound GTP-binding protein (Gi), as discussed later.20
There is variability in action potential duration between cells in normal ventricle.21 A gradient in action potential duration exists across the myocardium (from epicardium to endocardium), and specialized midmyocardial cells (M cells) have been identified, which exhibit prolongation of action potential duration at slow stimulation rates, possibly as a result of a decrease in IKs.
Phase 4: Diastolic Depolarization and If
Phase 4 diastolic depolarization, or normal automaticity, is a normal feature of cardiac cells in the sinus and atrioventricular (AV) nodes, but subsidiary pacemaker activity is also observed in the His-Purkinje system and in some specialized atrial and ventricular myocardial cells (see Chapter 4). Pacemaker discharge from the sinus node normally predominates because the rate of diastolic depolarization in the sinoatrial (SA) node is faster than in other pacemaker tissues. Pacemaker activity results from a slow net gain of positive charge, which depolarizes the cell from its maximal diastolic potential to threshold.
Pacemaker cells in the sinus node are relatively depolarized, with a maximal diastolic potential of −60 to −70 mV and a threshold potential of −40 mV. Rapid regenerative depolarization (phase 0) is dependent on opening of T-type and then L-type Ca++ channels. Repolarization is dependent on activation of delayed rectifier K+ channels, and the maximum diastolic potential is around −80 mV. Pacemaker channels are activated by hyperpolarization to this potential and produce a slow inward Na+ current, If. This flows against slowly inactivating delayed rectifier K+ currents and results in diastolic depolarization.22 Because the current is nonselective among cations, its reversal potential lies between EK and ENa, at around −10 mV, and activation of If will tend to depolarize the cell toward this value. Similar to ICa-L, If is under autonomic control (see Figure 7-4) through GTP-dependent binding proteins Gs and Gi, which regulate cAMP production by adenylyl cyclase.23,24 β-Adrenergic stimulation shifts the voltage dependence of activation of If to more depolarized potentials, so for any hyperpolarizing stimulus, more If will be activated and diastolic depolarization will be enhanced. Acetylcholine has the opposite effect (Figure 7-7).
Molecular Biology of Ion Channels
The preceding sections have focused on the electrical events that underlie cardiac electrical excitability, and on the identification of cardiac ionic currents on the basis of their biophysical properties. Here the molecular structures behind these electrical phenomena are reviewed. The first step in understanding the molecular physiology of cardiac electrical excitability is to identify the ion channel proteins responsible for the ionic currents. Figure 7-2 gives the current classification of the ion channel responsible for each of the cardiac ionic currents. There are firm molecular candidates for voltage-gated Na+ and L-type Ca++ channels. Similarly, channel molecules with properties similar to delayed rectifier K+ channels, the 4-aminopyridine–sensitive component of ITO, the inward rectifier IK1, the ligand-gated K+ channel IK(ACh), and the pacemaker current If have been cloned. Figure 7-8 shows diagrams of the predicted membrane topology of some of these channels. Voltage-gated Na+, Ca++, and K+ channels exist as conglomerates of molecules, consisting of a large α subunit and several accessory subunits (labeled β, δ, and γ in Figure 7-8). The α subunit alone is usually sufficient to induce channel activity in biologic membranes, but its activity is modulated by the presence of the accessory subunits. The diagrams in Figure 7-8 were deduced from hydrophobicity analysis of the primary structure of the major channel polypeptides. Regions of the polypeptides predicted to span the membrane are those that contain a high concentration of hydrophobic amino acids, whereas peptides linking these transmembrane sections are hydrophilic. Similarities among the various channels strongly suggest a common evolutionary ancestry. Na+ and Ca++ channel α subunits (see Figures 7-8A and B) are strikingly similar, each consisting of four homologous transmembrane domains (labeled I to IV), linked by cytoplasmic peptides. Each homologous domain contains six linked membrane-spanning segments (labeled S1 to S6). These large polypeptides, containing more than 2000 amino acids, form a tetrameric structure and generate Na+ or Ca++ channel activity in biologic membranes. Voltage-gated K+ channel α subunits, in contrast, are much smaller (see Figure 7-8C) and consist of a single transmembrane domain with six membrane-spanning segments, an arrangement similar to one of the individual domains of Na+ and Ca++ channels. Four molecules are noncovalently linked in the membrane to produce a tetrameric structure, similar to a Na+ or Ca++ channel, to produce K+ channel activity. The structure of the inwardly rectifying K+ channel molecules, IK1 and IK(ACh), is dissimilar to other K+ channels (see Figure 7-8C). The molecules are much less complex, having only two membrane-spanning segments, although these segments share considerable homology with the S5 and S6 segments of the classic voltage-gated K+ channel.
Figure 7-8 Diagrams of ion channel molecular structure.
A, Na+ channel. B, Ca++ channel. C, K+ channels. ATP, adenosine triphosphate.
Molecular Mechanisms
The Voltage Sensor
Channel proteins respond to changes in electrical potential across the cell membrane by conformational changes (gating), which result from electrostatic interactions between charged portions of the molecule and the membrane electric field (Box 7-3). Gating of the channel is associated with a measurable flow of electrical charge through the membrane lipid bilayer (called gating current), as a zone of the molecule rich in electrical charge moves within the membrane.25 This charge movement is linked to opening of the channel pore. The voltage sensor of voltage-dependent ion channels resides in the mobile S4 membrane-spanning segments, α-helical structures unusually rich in positively charged amino acids.26 At rest, each of the positive charges in the S4 segment is balanced by fixed negative charges in other segments of the molecule. The resting membrane potential (negative inside) forces the (mobile) positive charges inward and the fixed negative charges outward. This dynamic equilibrium holds the channel pore closed. On depolarization, the force pulling the positive charge inward is relieved; positive charges (the S4 segments) are repelled outward and assume new partners with the fixed negative membrane charges. This charge movement comprises the gating current. If the depolarizing stimulus is short, repolarization of the membrane is followed by a gating current of equal and opposite magnitude as the S4 segment relaxes to its original position. If the depolarizing stimulus is prolonged, however, the movement of the S4 segments induces a conformational change in the channel molecule, which prohibits easy return to baseline. This conformational change in the channel molecule is manifested as activation (or channel opening), and this is closely coupled to channel closing (or inactivation; see later) in channels that inactivate. Thus, small changes in the membrane electric field cause conformational changes in the channel molecule, which result in opening (and closing) of the channel pore. An S4 segment rich in positive charge is a remarkably consistent feature of voltage-gated ion channels from a variety of different species and with a variety of ion selectivities. The dependence of channel activation on membrane potential is proportional to the density of positive charge in the S4 segment.
Ion Channel Pore and Selectivity Filter
The presence of four homologous domains in voltage-gated Na+ and Ca++ channels suggests that basic ion channel architecture consists of a transmembrane pore surrounded by the four homologous domains arranged symmetrically (see Figure 7-8). The membrane-spanning segments each form an α helix so that the walls of the pore will be derived from α-helical segments from each of the four domains. A pore formed from four such α helices would have limiting dimensions of 3 by 5 Angstrom units, similar to the size inferred for the Na+ channel pore by measurement of the permeability of cations of different sizes.27,28
The selectivity filter is formed by the S5 and S6 membrane-spanning segments of each domain together with their peptide linker.29 As emphasized in Figure 7-8, unlike the hydrophilic extracellular linkers between other membrane-spanning segments, the S5/S6 linker is sufficiently hydrophobic to place it (at least partially) within the membrane lipid bilayer. The channel pore is lined both by the S5/S6 linker and the S5 and S6 membrane-spanning segments. Point mutations in the S5/S6 linker have dramatic effects on channel ion selectivity and reduce channel conductance to its primary ion. Extensive site-directed mutagenesis experiments of the S5/S6 linkers from a variety of channels suggest that they form a funnel that allows the passage of a specific ion into the pore. In Na+ channels, selectivity is imposed by two rings of negatively charged amino acids at the outer mouth of the funnel, which collect Na+ ions for transmission into the cell.27
Channel Inactivation
Inactivation gating is the process by which ion channels close in the face of continuing depolarization. Inactivation is characteristic of voltage-gated Na+ and Ca++ channels, as well as the K+ channels underlying ITO. Inactivation begins after activation gating, as a second, slower conformational change in the molecule that halts the ion flux through the channel. Inactivation gating is thus closely coupled to activation gating, and ionic current flows only while both the activation and inactivation gates are open simultaneously. In Na+ channels, the inactivation gate is formed by the intracellular peptide linker between homologous domains III and IV (Figure 7-9A).30 This peptide is postulated to act as a hinged lid, which moves upward to plug the ion pore (and thus halt current flow) shortly after membrane depolarization.26 For the channel to recover from inactivation (i.e., to be ready to open in response to a new depolarizing stimulus), the III/IV linker peptide must resume its resting position, a process that requires hyperpolarization of the membrane to the resting potential for a finite period. Site-directed mutagenesis of the III/IV linker peptide has revealed a trio of hydrophobic amino acid residues (isoleucine, I; phenylalanine, F; and methionine, M), near to the domain III end of the peptide, which are crucial for normal channel inactivation. Replacement of just one of these residues (the phenylalanine) almost completely removes inactivation. These residues are postulated to latch on to a receptor in the channel pore to close the channel. The molecular basis of inactivation in K+ channels is rather different from Na+ channels. Because the four domains of K+ channels are formed by noncovalently linked molecules, there are no interdomain linkers to plug the channel pore. In K+ channels, a picture of an N-terminal ball-and-chain mechanism has emerged (see Figure 7-9B).31 The terminal 20 or so amino acids are very hydrophobic and are postulated to swing up and attach to the open pore. The next few amino acids contain a number of positively charged residues that draw the whole N-terminal end up to the membrane. These two domains act as a ball. The remaining amino acids, up to the beginning of the transmembrane S1 segment, act as a chain. If the chain is made longer, inactivation is slower, and vice versa.
Clinical Correlates
Ion Channels and Antiarrhythmic Drugs
Drug therapy of cardiac arrhythmias would ideally be targeted at an individual ionic current, tailoring the cardiac action potential in such a way that abnormal excitability was reduced but normal rhythmicity was unaffected. This remains an as-yet unrealized goal. The prototype antiarrhythmic agents (e.g., disopyramide and quinidine) have diverse effects on cardiac excitability and, similar to agents introduced more recently, frequently exhibit significant proarrhythmic activity with potentially fatal consequences. In the Cardiac Arrhythmia Suppression Trial (CAST), mortality among asymptomatic postmyocardial infarction patients was approximately doubled by treatment with the potent Na+-channel–blocking agents encainide and flecainide—an effect likely attributable to slowing of conduction velocity with a consequent increase in fatal reentrant arrhythmias.32 The results of the CAST prompted efforts to approach antiarrhythmic drug therapy by prolonging action potential duration (e.g., dofetilide)—a strategy with some support from animal studies, but one that also may cause proarrhythmia through induction of polymorphic ventricular tachycardia (the acquired long QT syndrome [LQTS]).33 Drugs that prolong action potential duration all block IKr, and it is not clear that this therapeutic goal will result in arrhythmia control without induction of clinically significant proarrhythmia. The only drugs currently available that definitely prolong life by reducing fatal arrhythmias are β-blockers (e.g., ISIS-1, 1997), and these agents have no channel-blocking effects.
Ion Channels in Disease
Long QT Syndromes
This rare group of ion-channel abnormalities causes abnormal prolongation of the cardiac action potential, resulting in early afterdepolarizations (i.e., oscillations in the action potential during the plateau phase) and death from polymorphic ventricular tachycardia (torsades de pointes type). To date, six LQTS subtypes have been identified on the basis of the affected gene. They are numbered sequentially according to the date of discovery (Table 7-1), and all the loci so far identified encode ion channels except for LQT4. LQTS occurs because of disruption of cardiac repolarization, and in principle this can occur either from enhancement of inward depolarizing current or reduction of outward current. LQT3 is a gain-of-function mutation of the cardiac Na+ channel that results in failure of channel inactivation, most commonly from a deletion of three amino acids from the inactivation gate. LQT1 and LQT2 result from mutations of the delayed rectifier K channels that underlie IKs and IKr, and LQT5 and LQT6 result in reductions of IKr and IKs through mutations in channel accessory subunits. Loss of repolarizing current results in prolongation of the QT interval, and this leads to the syndrome. LQT4 is unique in that it results from a mutation in ankyrin B, an adapter protein that binds to the Na+ pump and Na+/Ca++ exchanger. The resulting effects on cellular Ca++ homeostasis cause ventricular arrhythmias.34 Identification of the molecular substrate for LQTS will allow detection of the disease in asymptomatic carriers and may in the future allow targeted gene therapy; these issues are discussed in more detail later in the Genetic Cardiovascular Medicine section.
Brugada Syndrome
The Brugada syndrome also is a group of ion-channel abnormalities that affect cardiac repolarization and result in cardiac sudden death. It is characterized by incomplete right bundle-branch block and persistent ST-segment elevation in the anterior precordial leads of the electrocardiogram (ECG).35 Cases for which the genotype is available appear to result from channel mutations that reduce depolarizing Na+ current. This results in loss of the action potential dome, an effect that is most marked in the right ventricular epicardium where the transient outward current ITO1 is strongly expressed (hence the ST-segment elevation in the anterior chest ECG leads). Early repolarization of the epicardial action potential results in a transmural repolarization gradient, and this can lead to reentry and sudden cardiac death.36 In more than two thirds of patients with Brugada syndrome, the genetic locus is unknown, and much work remains to be done to elucidate the mechanism of this condition.
Controlling cardiac functioning: receptors
Receptors
Receptors are grouped in several broad classes, the protein tyrosine kinase receptors and the G-protein–coupled receptors (GPCRs) being the most important ones. The protein tyrosine kinase receptors are large molecular complexes that incorporate phosphorylating enzyme activity in the intracellular segment. Ligand binding induces activation of this enzyme activity. Because phosphorylation is one of the major mechanisms of cellular regulation (see, for example, the phosphorylation of the Ca++ channel described earlier), such receptors can have a variety of cellular effects (Box 7-4). GPCRs are much smaller than protein tyrosine kinase receptors. Ligand binding results in activation of an associated protein (G protein)
[/not-level-membership-for-anesthesiology-category]
