[level-membership-for-basic-science-category]
Modified-release oral drug delivery
Emma L. McConnell and Abdul W. Basit
Chapter contents
Modified-release oral drug delivery
Sites of action for modified-release dosage forms and biopharmaceutical considerations
Designing a modified-release formulation: factors to consider
Key points
• Modified release can refer to extended, sustained, controlled, delayed or gastro-resistant release.
• Drugs can be targeted to the colon by using bacterial enzymes to initiate drug release.
Modified-release oral drug delivery
Administration of medicines by the oral route can seem like the most straightforward option for patients. It is the most commonly used route, with more than 70% of all medicines being delivered in this way. Oral medicines are easy to administer, improve patient compliance and are cheaper than some of the alternatives (e.g. injections). Most medicines administered by the oral route provide what is known as ‘immediate-release’ drug delivery or ‘conventional’ drug delivery. A common example would be the use of paracetamol (acetaminophen) for a headache; the tablet or capsule disintegrates quickly in the stomach fluids releasing the drug to provide rapid onset of effect, following absorption in the gastrointestinal tract. However, there are some situations in which this rapid onset is not desirable and a modification of the drug release pattern (or profile) is necessary to slow it down or make the drug’s effects last longer (e.g. for 24 hours). These more advanced oral drug delivery formulations are often referred to as oral modified-release (MR) drug delivery systems.
Modified-release drug delivery refers to the manipulation or modification of drug release from a dosage form (e.g. tablet, pellet, capsule) with the specific aim of delivering active pharmaceutical ingredients (API) at:
Modified-release drug delivery is a broad term which covers a variety of different approaches. These will be dealt with in further detail throughout this chapter. Briefly, the different types are:
The site of action of each of these systems is shown in Figure 31.1.
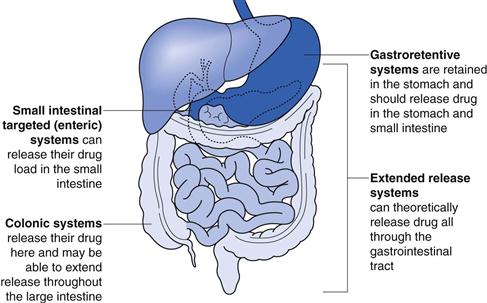
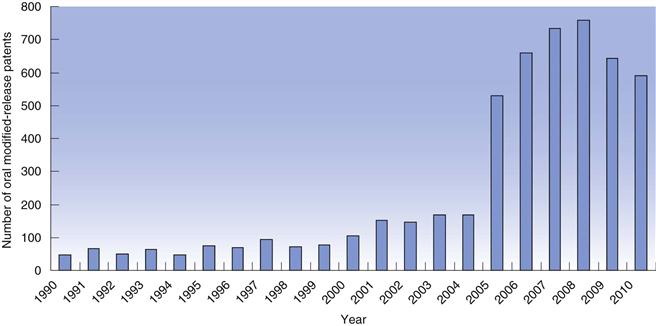
The concept of modified-release dosage forms has been around since the late 1800s when the idea of protecting the stomach from irritant drugs triggered a search for gastro-resistant materials. As knowledge of the gastrointestinal tract increased (pH, bacteria, transit times), the success and scope of the dosage forms targeted to the gastrointestinal tract improved. In recent years, there has been a huge increase in the number of patents filed for modified-release dosage forms (Fig. 31.2) highlighting the intense interest of the pharmaceutical industry in exploiting the benefits of these technologies to improve product performance.
What modified-release drug delivery means for the patient
Keeping drug in the therapeutic range.
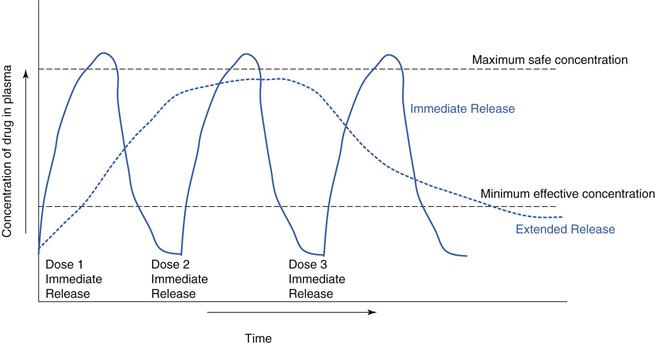
Modified release is often used to improve therapeutic outcomes for a patient relative to an immediate-release medication. For example, a drug which is rapidly absorbed and eliminated can have a steep plasma profile in an immediate-release formulation. An extended-release formulation can keep the drug at therapeutic levels for longer (Fig. 31.3). For many chronic illnesses, symptom breakthrough can occur if the blood concentration falls below the minimum effective concentration e.g. in asthma or depressive illness. This minimum level can also be critical for control of pain, consequently drugs, such as opioid analgesics are often given as extended-release preparations.
Maintaining drug levels overnight.
It is often not acceptable that patients be required to take medications during the night, with consequent loss of sleep. Overnight management of pain in terminally ill patients can be very important to maintain sleep.
Chronotherapy.
Timing the drug release to coincide with when it is required is known as chronotherapy. For example, a modified-release dosage form may be tailored to enable drug release to occur in the morning around the time of wakening, when symptoms of, for example, arthritis, asthma or allergies are often at their worst. A clinical study has shown that patients with arthritis had a better reduction in morning joint stiffness when they received modified-release prednisolone rather than a conventional dosage form.
Reducing side effects.
Immediate-release formulations can often have a high maximum concentration in the blood (Cmax). If Cmax is above the safety limit of the drug, adverse events may be more likely. Using modified-release formulations to reduce Cmax can reduce the incidence and severity of the side effects of some drugs. Additionally, some drugs, such as potassium chloride can be irritating to the gastrointestinal tract if delivered in an immediate-release bolus. A slow, sustained release is required to minimize the build-up of irritant concentrations.
Improving compliance.
A significant driver to developing a modified-release dosage form comes from trying to achieve once-daily dosing. Once-daily dosing is considered to be more convenient for patients and reduces the risk of missed doses throughout the day.
Treatment of local areas in the gastrointestinal tract.
Some conditions such as inflammatory bowel disease require topical treatment (e.g. with steroids) at the inflamed intestinal surface. Site-specific drug targeting (e.g. to the colon) can deliver the drug directly to its site of action.
What modified-release drug delivery means for the healthcare professional and pharmaceutical industry
Provides doctor, pharmacist and patient choice.
Healthcare professionals will be primarily concerned with the therapeutic advantages outlined above, but increasingly there is concern for personalized medicines and health services. A choice of immediate-release dosage forms and modified-release dosage forms can allow healthcare professionals to tailor treatment to their patients’ needs.
Product life extension.
Improving on current marketed formulations by employing modified-release technologies can sometimes enable pharmaceutical companies to extend a product’s patent life.
Higher development costs.
There are much higher costs for pharmaceutical companies in developing a modified-release formulation compared to a conventional immediate-release dosage form.
Cost savings to healthcare providers.
Cost-savings may be achieved from better disease management.
Sites of action for modified-release dosage forms and biopharmaceutical considerations
The gastrointestinal tract
Biopharmaceutical factors (i.e. the effect of the gastrointestinal physiology and environment on drugs and dosages forms) are considered in more detail in Chapter 19. Here some of the key biological factors that influence the in vivo behaviour of modified-release dosage forms are summarized and discussed. To understand these, the factors limiting drug bioavailability should be noted. The overall process of drug release and absorption will only be as fast as the slowest of many processes. The most common possible rate-limiting steps following oral administration of a solid dosage form are (1) drug release from the dosage form, (2) dissolution of the drug or (3) absorption of drug molecules.
pH
The stomach generally has a low pH and is therefore acidic. Gastro-resistant coated dosage forms are designed to be acid resistant. Some patients can have a higher stomach pH due to age, disease or ethnic origin which can affect dosage form disintegration and dissolution. This can result in premature drug release and/or dose dumping (dose dumping is the release of all the drug in one bolus).
Gastrointestinal pH generally increases in the small intestine, due to bicarbonate secretion. This is often used as a trigger for small intestinal drug delivery via gastro-resistant coating. The pH gradually increases to a maximum of about pH 7 at the ileo-caecal junction. In the colon, the pH drops slightly due to the production of short chain fatty acids by bacteria here, but gradually rises again distally. In some people, the pH does not get as high as pH 7 (and this may change from day to day). Therefore, if a polymer is used which dissolves at pH 7 (see below), then there is a good chance that the dosage form using this polymer will not dissolve, leaving a tablet intact and the patient without their dose. This has been observed in the clinic with some patients with ulcerative colitis.
Transit time
The time that a dosage form spends in the stomach, small intestine and colon can be critical for some modified-release systems. In the fasted state, the stomach will empty a non-disintegrating (i.e. non-immediate release) dosage form within 1–2 hours (via a clearing motility mechanism known as the migrating myoelectric complex). Ingestion of food delays this mechanism, and modified-release dosage forms can sometime be trapped in the stomach as long as food is present.
The small intestine is the site of absorption for most drugs and although the transit time of a dosage form through this region is normally around 3–4 hours, it can actually be highly variable (from 0.5–9 hours has been recorded). A modified-release dosage form which releases drug very slowly needs to take into account that it may only be at its site of absorption for a few hours.
The colon has a very variable transit time (1–72 hours). Often modified-release dosage forms reach the colon (as they may not have disintegrated in the stomach or small intestine). How effective they will be at this point depends on whether or not the drug is absorbed in the colon.
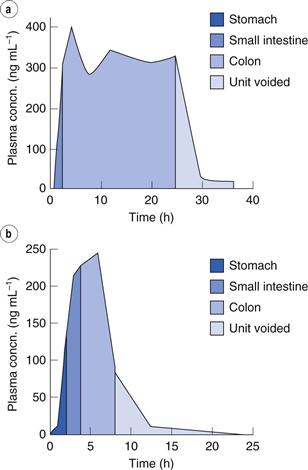
The clinical implications of this are seen in a study in which an OROS (osmotic extended-release system) tablet was administered (see later for more details on this type of device). In one instance it travelled slowly through the intestine and the patient received a suitable dose (i.e. blood levels were adequate and prolonged). In another instance, it travelled through the intestine in less than 10 hours, and very little drug was available to be absorbed by the patient, leaving them with sub-therapeutic blood drug levels (Fig. 31.4).
Fluid
Fluid levels can be highly variable in the stomach, small intestine and colon. In the stomach there may be around 100 mL of total fluid. In the small intestine there is around 50–100 mL of free fluid (i.e. that not bound up with digested material, and thus free to dissolve drugs or dosage forms). The colonic contents can be very viscous with only around 10 mL of free fluid actually available. All modified-release dosage forms require the presence of fluid in order for drug release to occur. Less free liquid is available as the modified-release dosage form travels down the gut.
Fluid composition (beyond pH) is also important. The presence of ions, fats, enzymes and salts can all affect drug release mechanisms from modified-release dosage forms. For example, fats may slow down release from swelling matrix systems, meaning that required blood levels may not be achieved as quickly in their presence. Sugars can sometimes disrupt controlled-release gels.
Designing a modified-release formulation: factors to consider
There are several decisions that need to be taken when designing a modified-release formulation. Assuming it has been established that a drug is a suitable candidate for modified-release drug delivery, the following points should be considered:
Single-unit dosage form or multiple-unit dosage form
A modified-release formulation can be designed as a single-entity (usually a tablet) (Fig. 31.5b). Single-unit tablets are sometimes known as monolithic dosage forms. A single-unit dosage form is advantageous from a manufacturing standpoint, as it can often be manufactured using conventional techniques, such as compaction and film coating. There may be some biopharmaceutical disadvantages to tablet formulations however. For example, as they do not disintegrate in the stomach, the dosage form could become trapped in the stomach for a long time (with food). For drugs targeted to the small or large intestine, this could prevent them reaching their site of action. Multiple-unit systems (e.g. pellets or granules filled into a hard capsule shell, Fig. 31.5a) may have more reproducible gastric emptying and have a reduced risk of dose dumping. However these can be more difficult to manufacture (requiring extrusion spheronization or drug loading onto seed cores) and to scale-up.


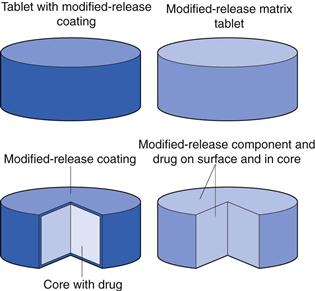
Matrix formulation or coated formulation
The release of an active pharmaceutical ingredient can be modified by two main methods (Figure 31.6). Firstly, the release modifying ingredients can be incorporated throughout the matrix of the dosage form wherein the whole dosage form encompasses the modified-release element. The second option is the application of a modified-release coating to a dosage form, wherein the drug is usually contained in the core and is released through, or via the dissolution of, the MR coat. There are slight deviations from these two techniques however, as will be seen in later sections, for example with osmotic systems.
Type of release rate
Two basic mechanisms can control the rate and extent of drug release. These are (1) dissolution of the active drug component and (2) diffusion of dissolved species. There are four processes operating in a modified-release dosage form to facilitate this:
Given the multi-step process of drug release from modified-release dosage forms, and the complex gastrointestinal environment, it is understandable that precisely controlling drug release is difficult. However, there are various release patterns that are desirable (Table 31.1).
Table 31.1
Release patterns for modified release
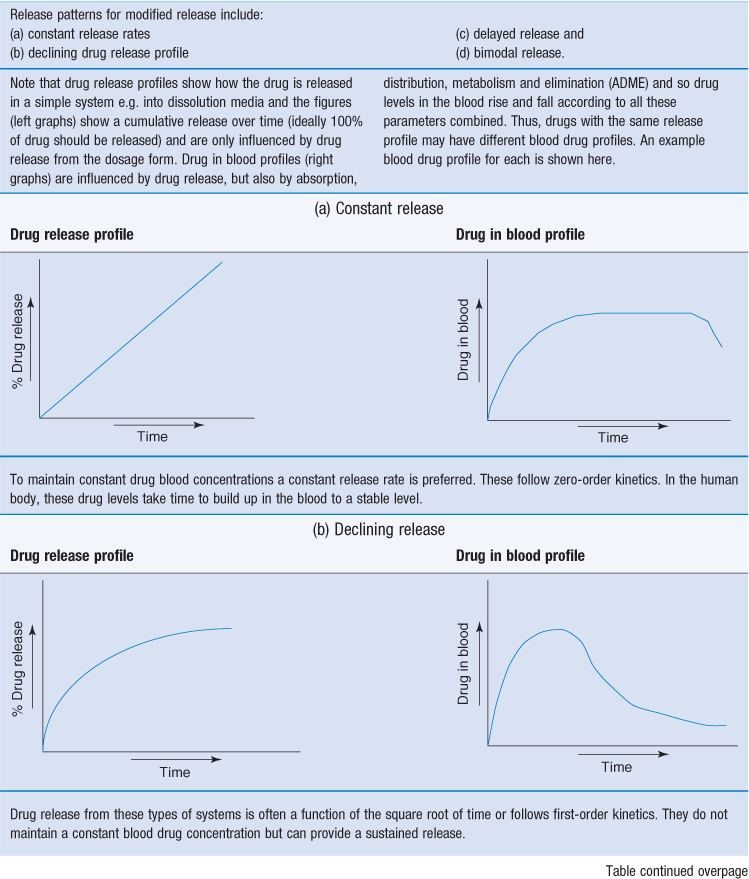
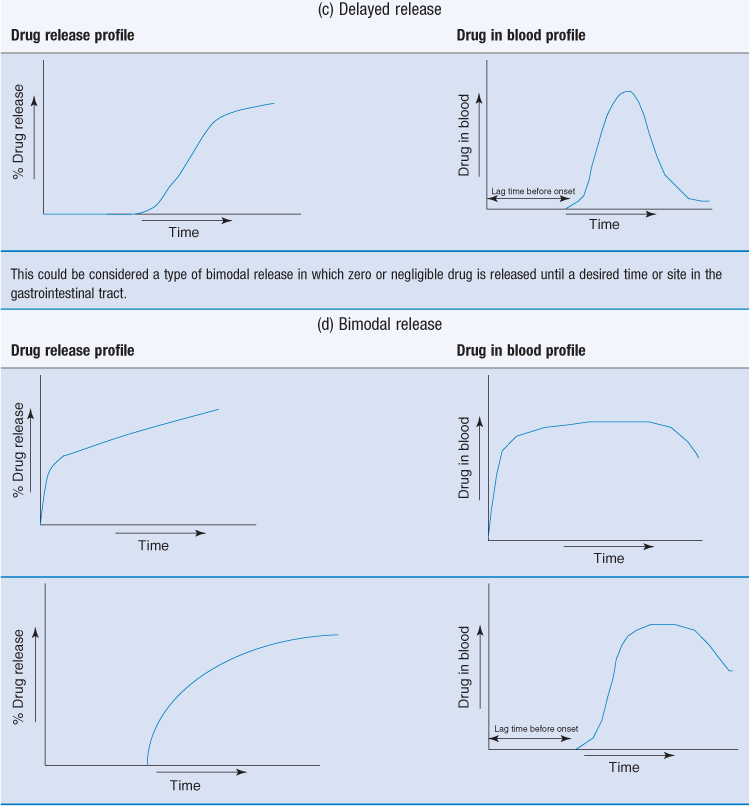
Extended release dosage forms often aim to achieve an initial primer dose of drug to achieve prompt therapeutic response. This primer dose could be an immediate-release layer or coating.
This can then be followed by a slow release of the remaining drug (the maintenance dose) which should sustain the blood levels in the therapeutic range. First-order release kinetics can be used to achieve the primer dose in a rapid fashion. Dose maintenance will follow zero-order kinetics.
Another example of bimodal release is delayed release followed by extended release.
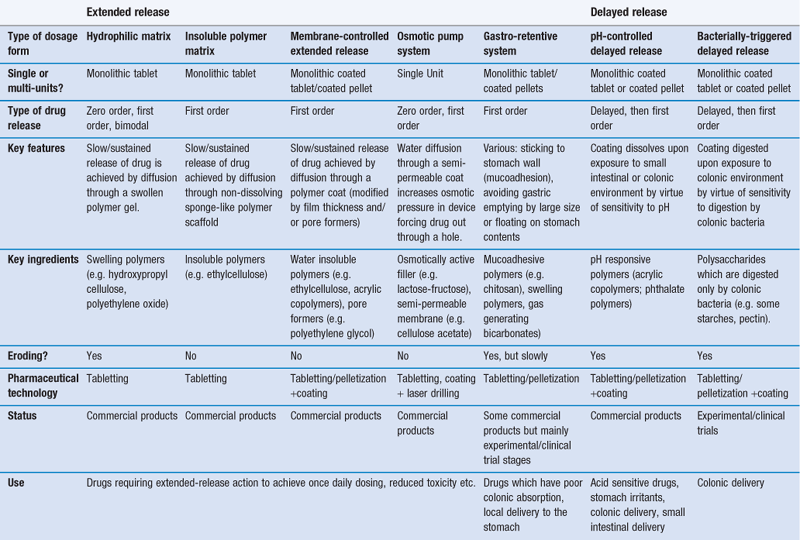
There are various strategies which have been adopted in an attempt to control and manipulate drug release patterns. These are summarized in Table 31.2 and are discussed in more detail below.
Extended release
Before an extended-release dosage form is developed, the suitability of the drug in question should be considered. The solubility of a drug in aqueous media and the intestinal permeability of the drug are key considerations when assessing whether a drug may be suitable for modified release. There are three potential rate-limiting steps in the bioavailability of drug from a dosage form:
Drugs are categorized according to steps 2 and 3. The Biopharmaceutics Classification System of drugs (see Chapter 21 for details) classifies drugs into four categories:
• Type I: high solubility, high permeability
• Type II: high solubility, low permeability
High solubility and high permeability drugs (Class I) are most suitable for extended-release delivery (ideally by passive diffusion). These properties mean that drug release from dosage forms can be the rate-limiting step in the process and this can then be tailored by the dosage form design. For drugs with low solubility (<1 mg/ml), the low rate of dissolution can already give some inherent sustained-release behaviour of the pure drug molecule, and dissolution of drug particles in the gut can be the rate-limiting step. After drug release and dissolution have occurred, absorption must then occur. Drugs with low permeability (<0.5 × 10−6 mm s−1 through CaCo-2 tissue culture [see Chapter 21]) are unlikely to be suitable for extended-release preparations. This is because they are already rate-limited in their absorption. Class IV drugs have low solubility and low permeability and these are the most challenging to formulate as modified-release products.
Other considerations as to the suitability of a drug for extended release include how quickly a drug is eliminated once in the blood stream. The most suitable drugs may have relatively short half-lives (t1/2 = 4–6 hours). Drugs with long half-lives may achieve pseudo-sustained release blood levels despite being formulated as immediate release, whereas shorter half-lives may need very high doses to maintain blood levels.
Dose is another factor to consider. To limit the size of the dosage form, the potency of the drug in the modified-release form can be critical. Up to 1000 mg potency tablets are available in extended-release formulations, but this is only achieved by using very large tablets, which may not always be acceptable for some patient populations (especially paediatric or geriatric patients, Chapter 43).
Hydrophilic matrix systems
Hydrophilic matrix systems can also be referred to as swellable soluble matrices. They are used for extended (sustained) release. Drug is mixed with a water-swellable, hydrophilic polymer (usually along with some other excipient materials) and compressed into a tablet. The polymer is usually in the form of a powder or granule, and tablets will be manufactured by direct compression or roller compaction (dry granulation processes). The resulting tablet has drug material interspersed between polymer particles. On exposure to fluid, the polymer material in the tablet starts to swell, producing a gel matrix. The gel can then allow drug release by dissolution of the gel and the drug trapped within it or erosion of the gel and release and dissolution of drug particles trapped within it.
The rate at which water can diffuse through the tablet – and later through the hydrated gel – affects the drug release rate. The rate of hydration is affected by the structure of the gel. Hydrophilic gels can be regarded as a network of interlinked/interspersed polymer stands. In the interstitial spaces between the strands is a continuous phase through which water and drug may diffuse. The interstices connect together to form a tortuous pathway through the gel. The tortuosity of this pathway is therefore critical for drug release. This can be affected by using polymers of different molecular weights or by using cross-linked gels, and so release rate can be modified by these factors. Increasing the polymer concentration can also make the ‘pathways’ fewer, and slows down drug release.
Polymers, such as hydroxypropyl methylcellulose or polyethylene oxide (which are commonly used for modified-release matrix systems), do not actually form true gels and are better described as forming very viscous solutions. Their structure is more dynamic than true gels (e.g. cross-linked alginic acid) as the chains can move relative to one another, so the interstitial continuum is not fixed. The mechanism of drug release is depicted in Figure 31.7. Diffusion-based release mechanisms usually follow zero-order or first-order kinetics (assuming sink conditions in the gastrointestinal tract and sufficient fluid) but additional erosion of the matrix due to gastrointestinal motility and hydrodynamics can complicate the true in vivo release rate. Often polymer type and concentration are used to control drug release, which can be tailored (faster and slower) as required (Fig. 31.8).
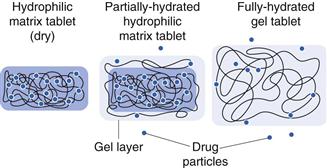
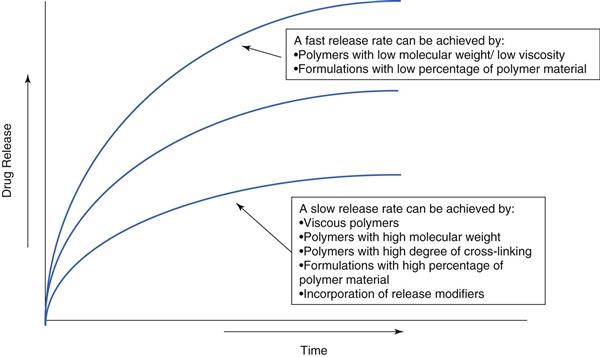
Hydrophilic matrix systems would generally be selected where a sustained drug release is required. They have the advantage of using standard safe excipients, use standard technologies, are well-established and can attain high drug loads. They do have the risk of ‘food effects’, whereby different blood profiles are attained in the fed and fasted states. This often results from the challenge of the gastrointestinal environment which is variable with respect to fluid, food and transit. These can be challenging factors for a hydrophilic matrix tablet.
Insoluble polymer matrix
These are far less commonly used than their water-soluble/swellable counterparts. They consist of an inert matrix system in which drug is embedded in an inert polymer. Their structure has been likened to that of a sponge. If drug molecules were interspersed throughout a sponge and water was applied, drug could leach out via the water filled channels (Fig. 31.9). In contrast to hydrophilic matrices, these systems stay intact throughout the gastrointestinal tract.
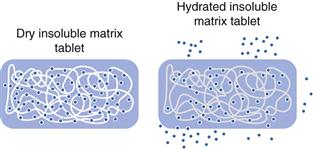
Drug release rate from insoluble polymer matrices is controlled by the pore size and number of pores, and tortuosity of the matrix. Pore-forming agents can be added to increase tortuosity and facilitate drug release. The release mechanism will also depend greatly on how the drug is dispersed within the system (dissolved, molecularly dissolved, or dispersed). The drug release does not follow zero-order kinetics; drug release decreases with time due to the increasing distance drug molecules have to travel to reach the surface of the device.
Like their hydrophilic counterparts, insoluble matrices represent a relatively simple concept which uses standard tabletting technology. However, they can also suffer some food effects, in particular related to rapid transit through the small intestine, or entrapment in the stomach in the fed state.
Membrane-controlled systems
Membrane–controlled delivery systems differ from the matrix formulations in that the rate-controlling part of the system is a membrane through which the drug must diffuse, rather than diffusing through the whole matrix. Generally, drug is concentrated in the core, and must traverse a polymeric membrane or film which slows down the release rate. Important criteria for such a dosage form are that the drug should not diffuse in the solid state. Upon exposure to an aqueous environment, water should be able to diffuse into the system and form a continuous phase through which drug diffusion and release can occur (Fig. 31.10). Drug release through a membrane is controlled by the thickness and the porosity of the membrane, as well as the solubility of the drug in the gastrointestinal fluids.
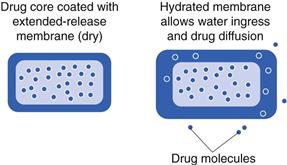
The biopharmaceutical considerations of transit and fluid are much the same as for monolithic matrix tablets. However, membrane-controlled drug delivery systems may be more likely to be in the form of pellets than in monolithic systems. Pellets and tablets have different biopharmaceutical considerations. For example, tablets are more likely to become trapped in the stomach if administered with food (especially with a high calorie meal). Pellets can get trapped too, but there is an improved chance of fortuitous emptying through the pyloric sphincter. Pellets will tend to distribute through the small intestine. They are also at less risk of dose dumping; if a tablet coating fails, then the whole dose can be dumped; with a pellet formulation, the disruption of one pellet coating may release only a small fraction of the total drug dose.
Osmotic systems
Osmotic pump systems are another form of membrane-controlled release drug delivery system, but work in a different way to that described previously. A drug is included in a tablet core which is water soluble, and which will dissolve (or suspend) the drug in the presence of water. The tablet core is coated with a semi-permeable membrane which will allow water to pass into the core. As the core components dissolve, a hydrostatic pressure builds up and forces (pumps) drug solution (or suspension) through a hole drilled in the coating (Fig. 31.11). The rate at which water is able to pass through the membrane and how quickly the drug solution (or suspension) can pass out of the hole govern the rate of drug release. The orifice needs to be small enough to prevent diffusion, but large enough to minimize hydrostatic pressure (600 µm to 1 mm diameter is normal). The orifice can be made by laser drilling, indentations in tablet (not fully covered by coating) or the use of leachable substances (pore formers).
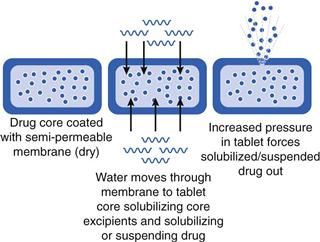
The rate at which the drug solution/suspension is forced out can be modified by changes in the viscosity of the solution formed inside the system. The essential difference between an osmotic pump system and a ‘classic’ membrane-controlled system is that for the osmotic pump, only one diffusion process is required (in this case ‘water in’).
Osmotic pump systems require exposure to sufficient fluid in order to build up an internal osmotic pressure. This will depend on fluid levels in the gut. Like any other non-disintegrating dosage form, they are reliant on being at the site of drug absorption for sufficient time to release their drug load. For example, if the osmotic system was designed to have drug release over 12 hours, it needs to be in the stomach and intestine for this amount of time, otherwise drug release will be incomplete.
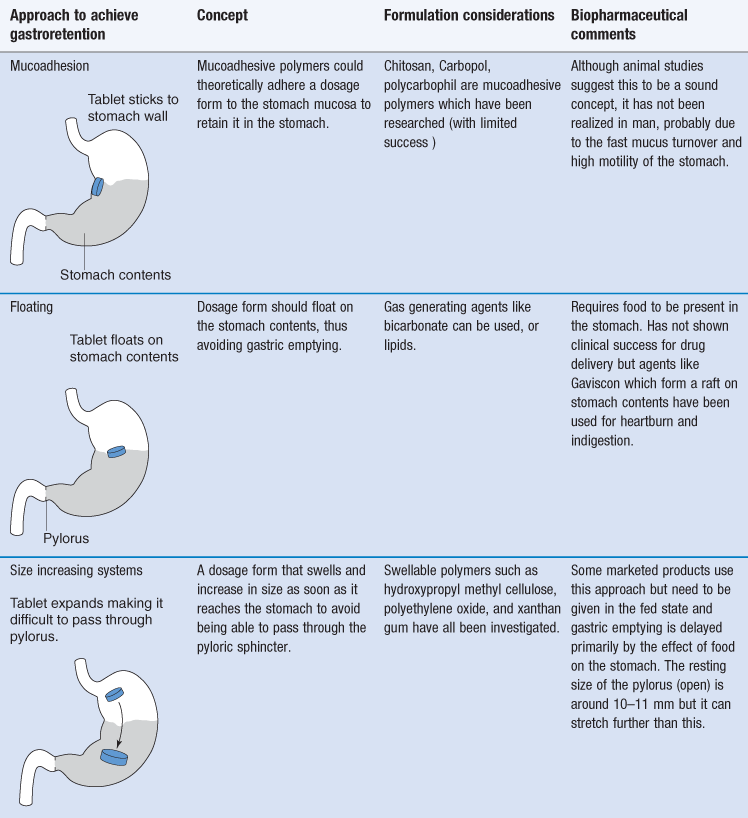
Gastroretention
Gastroretention is the mechanism by which a dosage form is retained in the stomach, generally for the purposes of improving drug delivery. It has been proposed as a mechanism by which drug absorption in the upper gastrointestinal tract can be maximized. Gastroretentive approaches to drug delivery aim to overcome the physiological mechanisms in the stomach which would normally enable gastric emptying, so that a modified-release dosage form is retained for longer in the stomach. Drugs which may benefit from gastroretention include those for local action in the stomach (e.g. to treat H. pylori), drugs which have a narrow absorption window in the small intestine and drugs which are degraded in the colon.
Several approaches have been investigated, but none deliver true gastroretention. The approaches which have been used to try to achieve gastroretention are very varied and are summarized in Table 31.3. Success with gastroretention has been limited, mainly due to the challenge presented by the stomach and gastric emptying which is incredibly difficult to overcome by formulation methods alone.
Delayed release
Gastro-resistant coatings
The concept here is similar to that of membrane-controlled extended release, except that the membrane is designed to disintegrate or dissolve at a pre-determined point. The most common trigger for delayed release coatings is pH. Gastro-resistant coatings are polymer coatings which are insoluble at low pH, but are soluble at higher pH (e.g. somewhere between pH 5–7 depending on the polymer). The drug release rate is controlled by its exposure to the correct pH (Fig. 31.12). Generally, this will require sufficient time to allow full coat dissolution. This approach is most commonly used for releasing drug in the small intestine. A similar concept can be used for the colon. The highest pH in the gastrointestinal tract is generally at the ileo-caecal junction, just before the colon. Here the pH can be around pH 7. Using polymers which dissolve at pH 7 should theoretically dissolve a dosage form here and release drug as the device moves into the colon. This approach has been used to deliver anti-inflammatory medications including budesonide, beclometasone and mesalazine to treat ulcerative colitis in the large intestine.
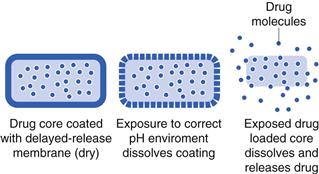
Gastro-resistant coating of formulations has two functions: i) to protect the stomach from the drug or ii) to protect acid-sensitive drugs from the stomach environment. They can be used to eliminate drug release in the stomach in order to target the small intestine or colon. Although the concept of gastro-resistant coating has been used for many years, and there are many products on the market, there are some shortcomings based on the fact that gastrointestinal pH is not always predictable and reproducible in vivo. For instance, in patients with a higher than normal stomach pH (achlorydia), there is a risk of premature drug release.
Colonic drug delivery
Colonic drug delivery can be achieved by the utilization of pH responsive polymers e.g. Eudragit S which dissolves at around pH 7 to target the colon. Targeting the colon is difficult as a tablet or pellet may only be in the region of highest pH (at the ileocaecal junction) for a short time, and the target pH (often pH 7) may not be reached. This can lead to dosage form failure (i.e. it does not disintegrate and is passed intact in the stools, consequently, not releasing the drug).
An alternative approach to this is the use of the gut bacteria as a trigger for drug release. A coating is prepared from a material which is insoluble in the gastrointestinal fluids (e.g. ethylcellulose), but it will also contain a component that can be digested only by colonic bacteria (not by pancreatic enzymes). An example of a material that can be used is the polysaccharide known as ‘resistant starch’. This type of starch can only be broken down by bacterial enzymes in the colon. When the dosage form reaches the colon, the starch component of the coat is digested and dissolves, leaving pores through which drug can be released (Fig. 31.13).
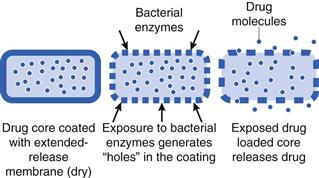
Bacterially triggered systems tend to be more reproducible in terms of consistent drug release than pH responsive system. However, there may be some patient populations in which gastrointestinal microorganism (microbiota) levels are affected by disease, and the effect on such modified-release drug delivery systems is not fully known. Being a relatively recent development, this technology has also not advanced as far clinically as pH-responsive colonic drug delivery, and is still in the experimental and clinical testing stages. Other new systems have also been proposed which combine both Eudragit S as the polymer and resistant starch to give a dual release mechanism (i.e. release is triggered by both the pH change and the bacteria). This is said to ensure rapid and consistent drug release.
Conclusions
Modified-release drug delivery can be broken down into two main categories: delayed release and extended release. Within these, there are different strategies and formulation techniques which can be employed to meet the desired treatment criteria. Furthermore, the release mechanisms can be combined to give bimodal release e.g. delayed and sustained release. Choosing which approach is most suitable will depend on the drug molecule, the type of release that is required, and the condition it is treating. There is no ‘one-size fits all’ solution for tailoring drug release, and significant resources have to be employed to develop these successfully. Continued research into this area and understanding of the biopharmaceutical implications are necessary to develop quality products to improve patient treatment.
Reference
1. Drug delivery to the large intestine and rectum Washington N, Washington C, Wilson CG, eds. Physiological Pharmaceutics: Barriers to Drug Absorption. 2nd edn 2001.
Bibliography
1. Davis SS. Formulation strategies for absorption windows. Drug Discovery Today. 2005;10:249–257.
2. McConnell EL, Fadda HM, Basit AW. Gut instincts: explorations in intestinal physiology and drug delivery. International Journal of Pharmaceutics. 2008;364:213–226.
3. McConnell EL, Liu F, Basit AW. Colonic treatments and targets: issues and opportunities. Journal of Drug Targeting. 2009;17:335–363.
4. Varum FJO, Merchant HA, Basit AW. Modified-release formulations in motion: the relationship between gastrointestinal transit and drug absorption. International Journal of Pharmaceutics. 2010;395:26–36.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Modified-release oral drug delivery
Emma L. McConnell and Abdul W. Basit
Chapter contents
Modified-release oral drug delivery
Sites of action for modified-release dosage forms and biopharmaceutical considerations
Designing a modified-release formulation: factors to consider
Key points
• Modified release can refer to extended, sustained, controlled, delayed or gastro-resistant release.
• Drugs can be targeted to the colon by using bacterial enzymes to initiate drug release.
Modified-release oral drug delivery
Administration of medicines by the oral route can seem like the most straightforward option for patients. It is the most commonly used route, with more than 70% of all medicines being delivered in this way. Oral medicines are easy to administer, improve patient compliance and are cheaper than some of the alternatives (e.g. injections). Most medicines administered by the oral route provide what is known as ‘immediate-release’ drug delivery or ‘conventional’ drug delivery. A common example would be the use of paracetamol (acetaminophen) for a headache; the tablet or capsule disintegrates quickly in the stomach fluids releasing the drug to provide rapid onset of effect, following absorption in the gastrointestinal tract. However, there are some situations in which this rapid onset is not desirable and a modification of the drug release pattern (or profile) is necessary to slow it down or make the drug’s effects last longer (e.g. for 24 hours). These more advanced oral drug delivery formulations are often referred to as oral modified-release (MR) drug delivery systems.
Modified-release drug delivery refers to the manipulation or modification of drug release from a dosage form (e.g. tablet, pellet, capsule) with the specific aim of delivering active pharmaceutical ingredients (API) at:
Modified-release drug delivery is a broad term which covers a variety of different approaches. These will be dealt with in further detail throughout this chapter. Briefly, the different types are:
The site of action of each of these systems is shown in Figure 31.1.
The concept of modified-release dosage forms has been around since the late 1800s when the idea of protecting the stomach from irritant drugs triggered a search for gastro-resistant materials. As knowledge of the gastrointestinal tract increased (pH, bacteria, transit times), the success and scope of the dosage forms targeted to the gastrointestinal tract improved. In recent years, there has been a huge increase in the number of patents filed for modified-release dosage forms (Fig. 31.2) highlighting the intense interest of the pharmaceutical industry in exploiting the benefits of these technologies to improve product performance.
What modified-release drug delivery means for the patient
Keeping drug in the therapeutic range.
Modified release is often used to improve therapeutic outcomes for a patient relative to an immediate-release medication. For example, a drug which is rapidly absorbed and eliminated can have a steep plasma profile in an immediate-release formulation. An extended-release formulation can keep the drug at therapeutic levels for longer (Fig. 31.3). For many chronic illnesses, symptom breakthrough can occur if the blood concentration falls below the minimum effective concentration e.g. in asthma or depressive illness. This minimum level can also be critical for control of pain, consequently drugs, such as opioid analgesics are often given as extended-release preparations.
Maintaining drug levels overnight.
It is often not acceptable that patients be required to take medications during the night, with consequent loss of sleep. Overnight management of pain in terminally ill patients can be very important to maintain sleep.
Chronotherapy.
Timing the drug release to coincide with when it is required is known as chronotherapy. For example, a modified-release dosage form may be tailored to enable drug release to occur in the morning around the time of wakening, when symptoms of, for example, arthritis, asthma or allergies are often at their worst. A clinical study has shown that patients with arthritis had a better reduction in morning joint stiffness when they received modified-release prednisolone rather than a conventional dosage form.
Reducing side effects.
Immediate-release formulations can often have a high maximum concentration in the blood (Cmax). If Cmax is above the safety limit of the drug, adverse events may be more likely. Using modified-release formulations to reduce Cmax can reduce the incidence and severity of the side effects of some drugs. Additionally, some drugs, such as potassium chloride can be irritating to the gastrointestinal tract if delivered in an immediate-release bolus. A slow, sustained release is required to minimize the build-up of irritant concentrations.
Improving compliance.
A significant driver to developing a modified-release dosage form comes from trying to achieve once-daily dosing. Once-daily dosing is considered to be more convenient for patients and reduces the risk of missed doses throughout the day.
Treatment of local areas in the gastrointestinal tract.
Some conditions such as inflammatory bowel disease require topical treatment (e.g. with steroids) at the inflamed intestinal surface. Site-specific drug targeting (e.g. to the colon) can deliver the drug directly to its site of action.
What modified-release drug delivery means for the healthcare professional and pharmaceutical industry
Provides doctor, pharmacist and patient choice.
Healthcare professionals will be primarily concerned with the therapeutic advantages outlined above, but increasingly there is concern for personalized medicines and health services. A choice of immediate-release dosage forms and modified-release dosage forms can allow healthcare professionals to tailor treatment to their patients’ needs.
Product life extension.
Improving on current marketed formulations by employing modified-release technologies can sometimes enable pharmaceutical companies to extend a product’s patent life.
Higher development costs.
There are much higher costs for pharmaceutical companies in developing a modified-release formulation compared to a conventional immediate-release dosage form.
Cost savings to healthcare providers.
Cost-savings may be achieved from better disease management.
Sites of action for modified-release dosage forms and biopharmaceutical considerations
The gastrointestinal tract
Biopharmaceutical factors (i.e. the effect of the gastrointestinal physiology and environment on drugs and dosages forms) are considered in more detail in Chapter 19. Here some of the key biological factors that influence the in vivo behaviour of modified-release dosage forms are summarized and discussed. To understand these, the factors limiting drug bioavailability should be noted. The overall process of drug release and absorption will only be as fast as the slowest of many processes. The most common possible rate-limiting steps following oral administration of a solid dosage form are (1) drug release from the dosage form, (2) dissolution of the drug or (3) absorption of drug molecules.
pH
The stomach generally has a low pH and is therefore acidic. Gastro-resistant coated dosage forms are designed to be acid resistant. Some patients can have a higher stomach pH due to age, disease or ethnic origin which can affect dosage form disintegration and dissolution. This can result in premature drug release and/or dose dumping (dose dumping is the release of all the drug in one bolus).
Gastrointestinal pH generally increases in the small intestine, due to bicarbonate secretion. This is often used as a trigger for small intestinal drug delivery via gastro-resistant coating. The pH gradually increases to a maximum of about pH 7 at the ileo-caecal junction. In the colon, the pH drops slightly due to the production of short chain fatty acids by bacteria here, but gradually rises again distally. In some people, the pH does not get as high as pH 7 (and this may change from day to day). Therefore, if a polymer is used which dissolves at pH 7 (see below), then there is a good chance that the dosage form using this polymer will not dissolve, leaving a tablet intact and the patient without their dose. This has been observed in the clinic with some patients with ulcerative colitis.
Transit time
[/not-level-membership-for-basic-science-category]
