Mitochondrial encephalopathies
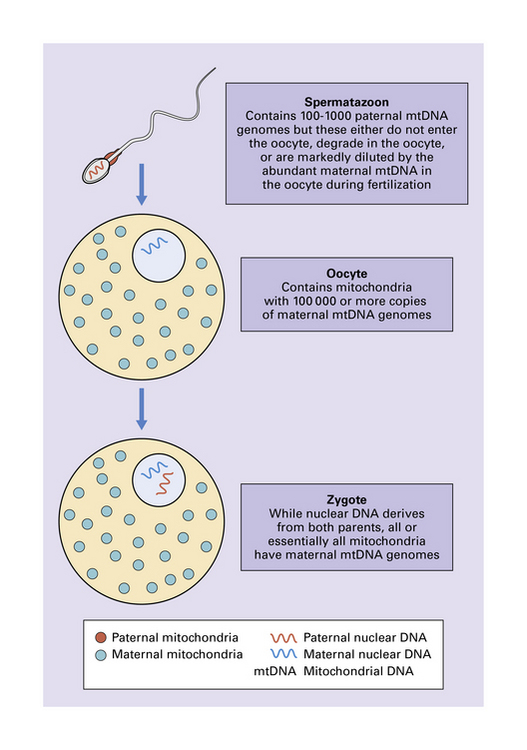
The mitochondrial genome is a circular molecule of DNA (mtDNA) comprising 16 569 base pairs. Each mitochondrion contains 2–10 copies of the genome, which encodes 22 transfer RNAs (tRNAs), two ribosomal RNAs (rRNAs), and 13 subunits of the respiratory chain (Fig. 24.1). Since spermatozoa contribute no or minimal mitochondria during fertilization, mitochondria and their genomes are essentially all maternally inherited (Figs 24.2, 24.3).
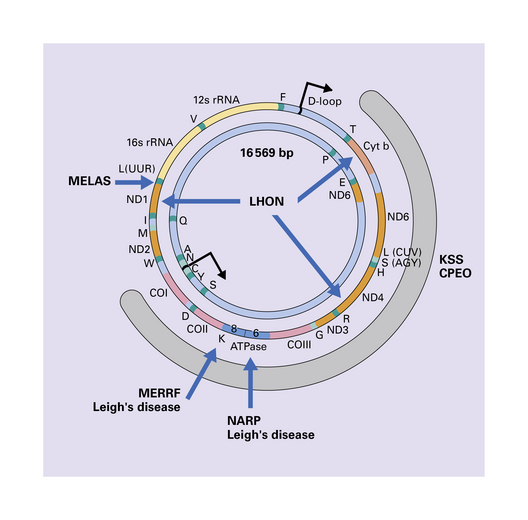
24.1 Diagram of human mtDNA.
The outer and inner circles represent the heavy and light strands respectively. The genes encoding the mitochondrial rRNAs are shown as 12 s and 16 s rRNA, those encoding the reduced form of nicotinamide adenine dinucleotide dehydrogenase (NADH) subunits as ND1–6, the cytochrome oxidase subunits as COI–III, and cytochrome b as Cyt b. Also shown are the genes encoding subunits 6 and 8 of adenosine triphosphatase (ATPase). Conventional single letter amino acid abbreviations are used to indicate the tRNA genes. The broad gray arc shows the region involved to a greater or lesser extent in most deletions of mtDNA. The blue arrows indicate the main sites of point mutations or deletions in mitochondrial disorders that affect the CNS.
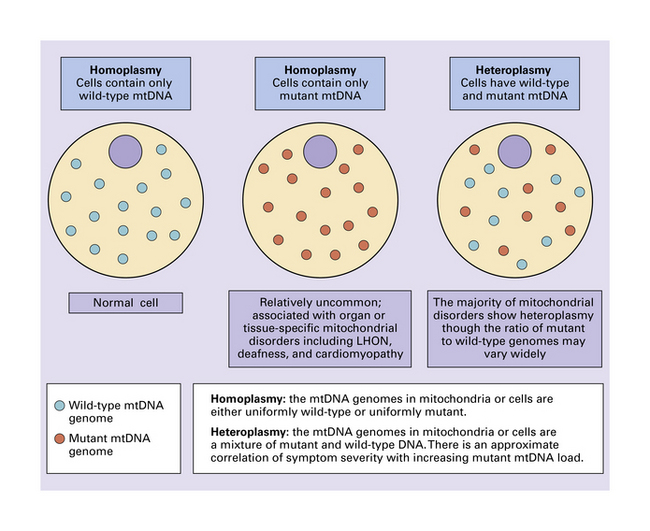
A wide range of disorders affecting the central nervous system (CNS) is attributable to defective mitochondrial function. Most mtDNA defects are heteroplasmic (i.e. cells contain a mixture of mutant and wild-type mtDNA) (Fig. 24.4). As mitochondria are randomly segregated during mitosis, the proportion of mitochondria containing mutant genomes varies from cell to cell. However, the levels of mutated mtDNA tend to be highest in non-dividing cells such as neurons, and skeletal and cardiac muscle. The frequency of mutations and deletions increases with age to a much greater extent in mitochondrial than in nuclear DNA. Several factors probably contribute: much of the damage is mediated by reactive oxygen species that are generated within the mitochondrion itself; there is no ‘redundant’ non-coding mtDNA; mtDNA undergoes progressively more frequent replication (exceeding that of nuclear DNA); the responsible polymerase lacks the proof-reading and repair activities of nuclear DNA polymerases.
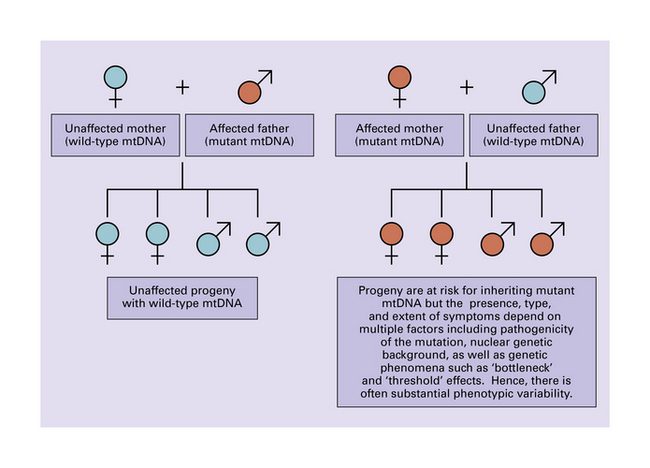
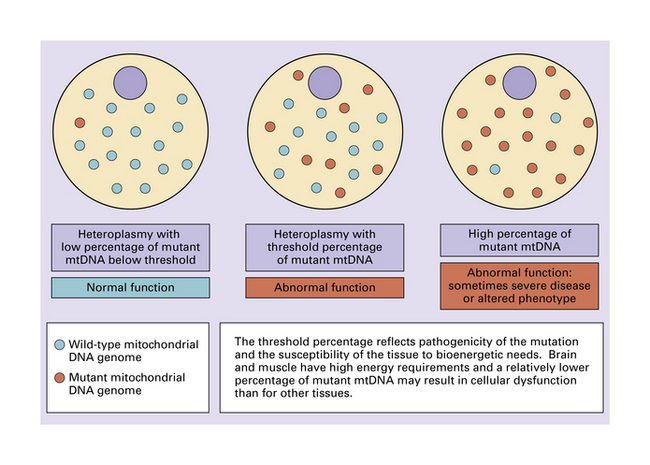
Tissues with a high energy demand, such as neural tissue and muscle, are particularly susceptible to impairment of mitochondrial function (Fig. 24.5). Probably because relatively few mitochondria (and therefore mitochondrial genomes) are transmitted by a mother to her progeny during embryogenesis, pronounced shifts in the degree of mitochondrial heteroplasmy can occur from generation to generation (Figs 24.6, 24.7).
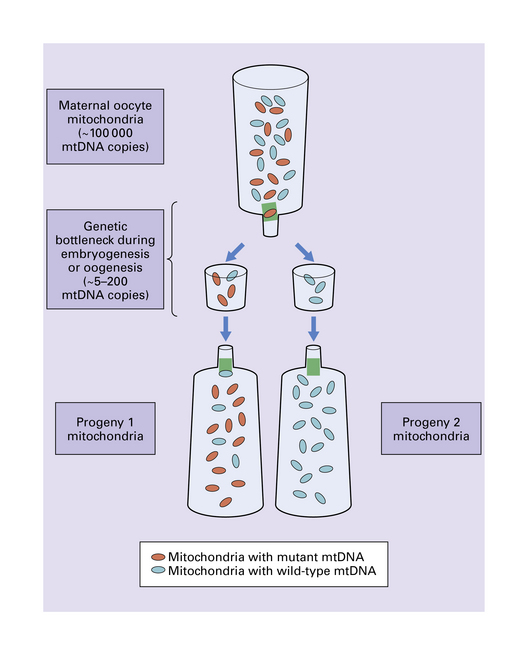
24.6 Mitochondrial genetic bottleneck effect.
Rapid and marked shifts in degree of mitochondrial heteroplasmy can occur in one generation. A mitochondrial genetic bottleneck that permits only a small subset of maternal mtDNA genomes to be effectively transmitted to progeny could account for these incidences of sudden genetic drift. In this figure, the genetic bottleneck effect has resulted in a drastic shift to a state of wild-type homoplasmy for progeny 2. Given that the maternal mitochondria are approximately 50% mutant in this example, progeny homoplasmy would be unlikely if there was simply a random distribution of mtDNA genomes alone. Without the bottleneck effect, accumulated maternal mitochondrial mutations would always be transmitted in the germline. Over generations, additional accumulated mutations would result inexorably in non-functional mitochondria, non-viable oocytes or progeny, and species extinction. With random mitotic segregation, which may have a greater role in somatic cells, genetic drift is likely to be less pronounced. In contrast, the bottleneck genetic effect has the theoretical evolutionary advantage of potentially producing some progeny that have no mutant mtDNA. In the short term, deleterious mutations may accumulate in some oocytes or progeny, e.g. progeny 1, but these would be selected against.
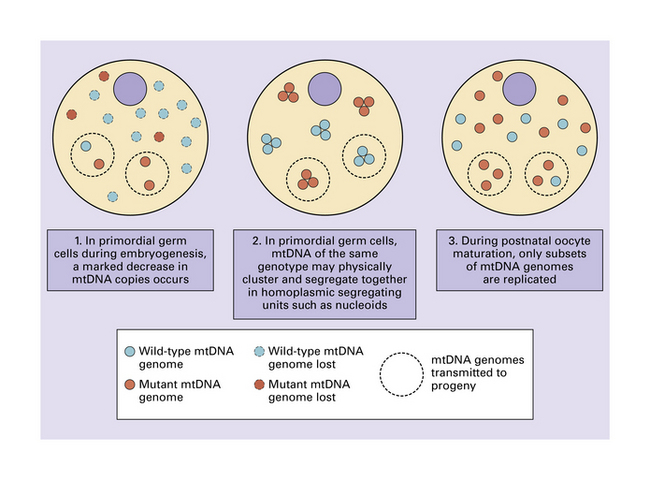
24.7 Possible mechanisms for the mitochondrial genetic bottleneck.
Whether one or all of these mechanisms contribute to the putative genetic bottleneck remains under investigation.
In disorders with an increased level of oxidative stress, the age-related accumulation of mutations in mtDNA tends to be exacerbated. Some studies have suggested that this may impair oxidative phosphorylation and contribute to cell death in neurodegenerative diseases such as Parkinson’s disease and Alzheimer’s disease. (These neurodegenerative diseases are covered in Chapters 28 and 31). Several other diseases are either due to mutations in nuclear genes that encode mitochondrial proteins (e.g. Alpers syndrome, Menkes syndrome, and a range of defects of fatty acid, amino acid, and pyruvate metabolism), or cause secondary mitochondrial dysfunction (e.g. Batten’s disease). These too are considered separately, in Chapters 6 and 7.
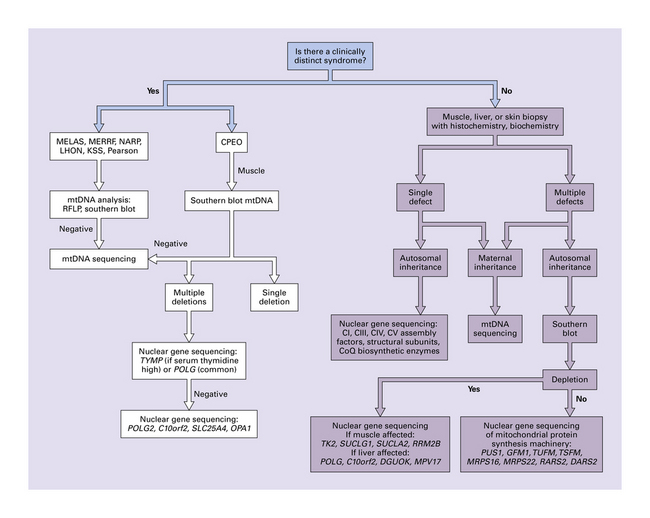
This chapter covers only those mitochondrial diseases that involve the CNS and are due to mutations of mtDNA. In most of the diseases that are included in this chapter, the mutation in mtDNA is the primary genetic defect, but the chapter also covers a few disorders in which abnormalities in mtDNA are secondary to mutations of nuclear DNA (Table 24.1). Although Leigh syndrome can be caused by point mutations or deletions of mtDNA, it more often results from mutations in nuclear DNA, and the etiology, clinical and pathologic findings are therefore described in Chapter 6. An algorithm for genetic analysis of mitochondrial disorders is shown in Figure 24.8. Mitochondrial disorders that do not involve the CNS are not considered in this book.

MITOCHONDRIAL MYOPATHY, ENCEPHALOPATHY, LACTIC ACIDOSIS, AND STROKE-LIKE EPISODES (MELAS)
 ETIOLOGY OF MELAS
ETIOLOGY OF MELAS In over 80% of cases, MELAS is caused by an A-to-G transition at nucleotide 3243 in the mitochondrial tRNALeu(UUR) gene. This transition is within the region of termination of transcription of the mitochondrial rRNAs, but whether this is relevant to the genesis of MELAS lesions is uncertain.
In over 80% of cases, MELAS is caused by an A-to-G transition at nucleotide 3243 in the mitochondrial tRNALeu(UUR) gene. This transition is within the region of termination of transcription of the mitochondrial rRNAs, but whether this is relevant to the genesis of MELAS lesions is uncertain.
• This same mutation can also cause Kearns–Sayre syndrome (KSS), at least some features of which are often present (see below).
• When only a relatively low proportion of mtDNA is mutated at nucleotide 3243, patients may develop adult-onset, non-insulin-dependent diabetes mellitus (type I diabetes) with or without deafness (DMDF).
 MELAS can also be caused by a T-to-C transition at nucleotide 3271 in the tRNALeu(UUR) gene.
MELAS can also be caused by a T-to-C transition at nucleotide 3271 in the tRNALeu(UUR) gene.

 The lesions of MELAS probably result from defective mitochondrial metabolism, in which case strictly-speaking they are not infarcts in that the necrosis is not due to ischemia/oligemia or hypoxia. Some researchers have suggested that the stroke-like lesions are due to impaired cerebral blood flow resulting from an accumulation of mitochondria within the cerebrovascular endothelium and smooth muscle cells. However, the stroke-like lesions have been shown to be associated with hyperemia rather than reduced perfusion.
The lesions of MELAS probably result from defective mitochondrial metabolism, in which case strictly-speaking they are not infarcts in that the necrosis is not due to ischemia/oligemia or hypoxia. Some researchers have suggested that the stroke-like lesions are due to impaired cerebral blood flow resulting from an accumulation of mitochondria within the cerebrovascular endothelium and smooth muscle cells. However, the stroke-like lesions have been shown to be associated with hyperemia rather than reduced perfusion.


MACROSCOPIC APPEARANCES
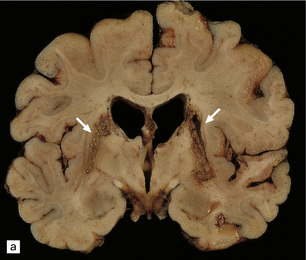
Scattered foci of necrosis and cavitation are usually evident in the cerebral cortex (Fig. 24.9). These infarct-like lesions may be of varying ages. Although they can occur in any part of the cerebral cortex, the occipital lobe is often most severely affected. The basal ganglia, thalamus, and cerebellum are much less commonly involved.
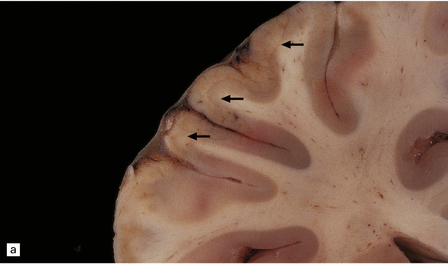
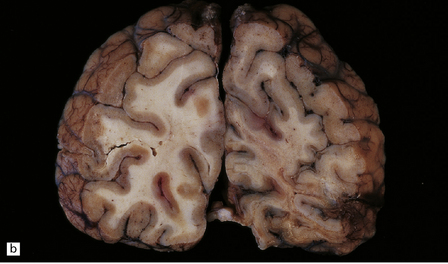
24.9 Macroscopic appearance of brain in MELAS.
Infarct-like lesions of varying ages. (a) Recent lesions (arrows) in the frontal cortex. The distribution of necrosis in these lesions differs from that associated with hypoxia/ischemia in that the crests of the gyri are more extensively affected than the depths of sulci, and the deep laminae are largely spared. The occipital lobes (b) are often severely affected in this case by lesions of differing ages. The older lesions have produced shrinkage and brown discoloration of the cortical ribbon.
 MELAS
MELAS




MICROSCOPIC APPEARANCES
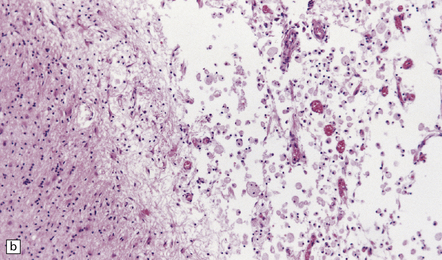
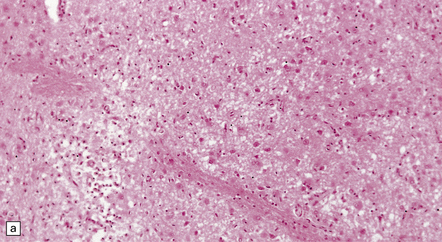
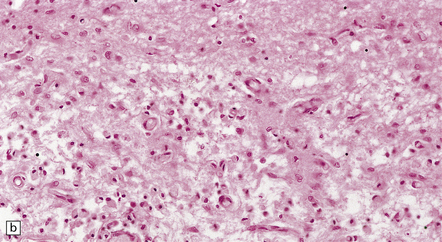
The necrotic foci are histologically indistinguishable from infarcts of various ages (Fig. 24.10), although their distribution does not correspond to that of any particular arterial perfusion territory (Fig. 24.10, see also Fig. 24.9) and the crests of gyri are often more extensively affected than the depths of sulci. As with infarcts, there is capillary proliferation around and within the lesions. In many cases there is also mineralization in and around the walls of blood vessels in the globus pallidus, and occasionally in the cerebral white matter, thalamus, and dentate nucleus. This may be associated with fibrous thickening of the affected blood vessels and severe narrowing of the lumen. There may be spongy vacuolation of cerebral and cerebellar white matter, but this is unusual. Histology, histochemistry, and electron microscopy of skeletal muscle reveal ‘ragged-red’ fibers containing accumulations of morphologically abnormal mitochondria, some with paracrystalline inclusions. There are also accumulations of mitochondria in the endothelium and smooth muscle of intramuscular blood vessels (Fig. 24.10).
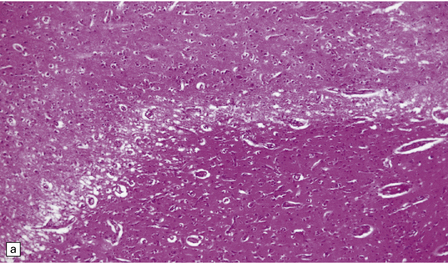
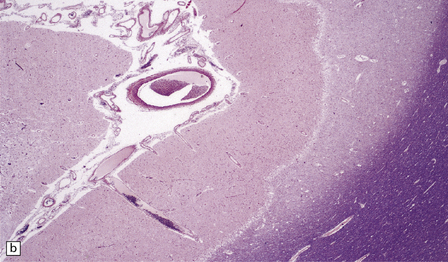
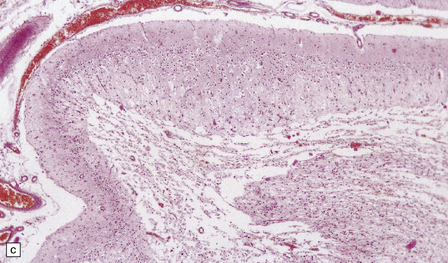
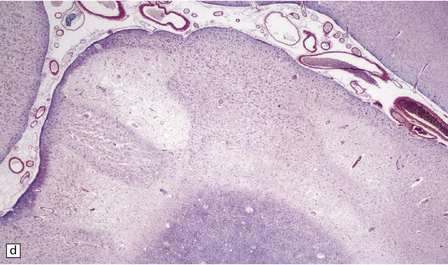
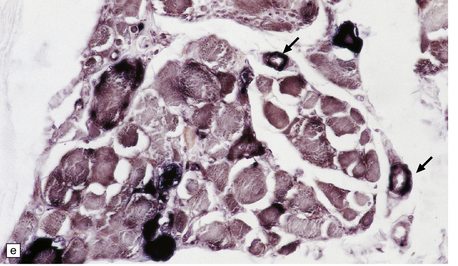
24.10 Histology of MELAS.
(a) Infarct-like acute lesion in MELAS. This section is through the edge of the lesion. (b) The boundary between the acutely affected superficial cortex and the intact deeper cortex has a serpiginous shape. (c) Chronic lesion, which is partly cavitated. (d) ‘Punched-out’ chronic lesions within cortex that is gliotic and depleted of neurons. (e) Skeletal muscle in MELAS. Demonstration of abnormal accumulations of mitochondria in paraffin sections of muscle from a patient with MELAS by in situ hybridization with probes to mitochondrial rRNA. An abnormally strong signal is present not only in several muscle fibers but also in some of the blood vessels (arrows).
MYOCLONIC EPILEPSY WITH RAGGED-RED FIBERS (MERRF)
 ETIOLOGY OF MERFF
ETIOLOGY OF MERFF


 MERRF
MERRF

MACROSCOPIC AND MICROSCOPIC APPEARANCES
The brain usually appears unremarkable macroscopically, but there may be obvious brown discoloration and shrinkage of the dentate nuclei (Fig. 24.11) and inferior olives. Microscopy reveals neuronal loss and gliosis, which is most severe in the dentate nuclei (Fig. 24.11), inferior olivary nuclei, substantia nigra, red nuclei, and basal ganglia (Fig. 24.11). The gracile and cuneate nuclei and Clarke’s column in the spinal cord may also be affected. Blood vessels in the basal ganglia and cerebral white matter may show mineralization. Skeletal muscle reveals histologic and histochemical changes of mitochondrial myopathy, with ‘ragged-red’ fibers containing accumulations of morphologically abnormal mitochondria, some with paracrystalline inclusions.
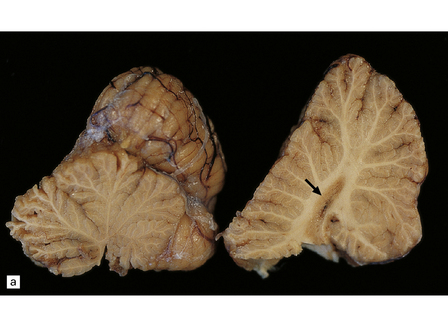
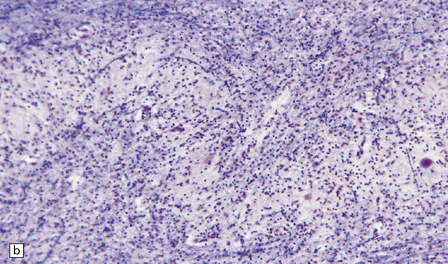
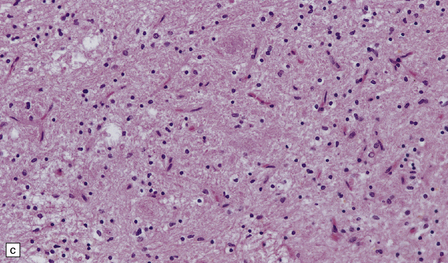
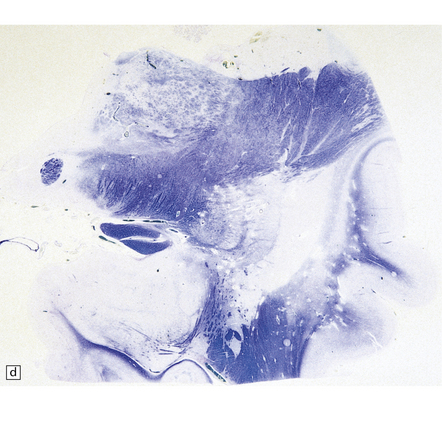
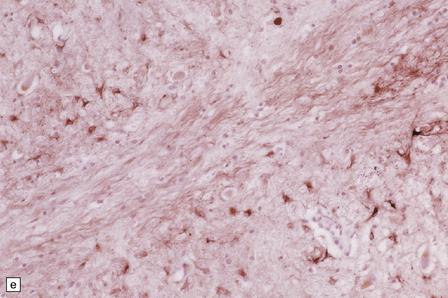
24.11 MERRF.
(a) On macroscopic examination, the dentate nucleus (arrow) appears shrunken and shows slight brown discoloration. (b) Histology reveals loss of neurons from the ribbon of the nucleus and of myelinated fibers from the hilum. (c) Higher magnification of the ribbon of the dentate nucleus shown in (b). (d) Marked pallor of myelin staining in the putamen in a case of MERRF. (Courtesy of Professor F Scaravilli, Institute of Neurology, London, UK.) (e) The putamen is gliotic, as demonstrated using immunohistochemistry for glial fibrillary acidic protein.
The findings in Leigh syndrome, which is also occasionally caused by an A-to-G transition at nucleotide 8344 in the mitochondrial tRNALys gene, are considered in Chapter 6. Other point mutations in mtDNA that can cause Leigh syndrome, include T8993G, T8993C and T9176C mutations in the gene for ATPase 6.
LEBER’S HEREDITARY OPTIC NEUROPATHY (LHON), BILATERAL STRIATAL NECROSIS, AND MULTIPLE SCLEROSIS (MS)-LIKE MITOCHONDRIAL DISEASE
 ETIOLOGY OF LEBER’S HEREDITARY OPTIC NEUROPATHY
ETIOLOGY OF LEBER’S HEREDITARY OPTIC NEUROPATHY
 Over 90% of families with LHON have missense G-to-A mutations at nucleotides 11 778, 3460, or 14 484.
Over 90% of families with LHON have missense G-to-A mutations at nucleotides 11 778, 3460, or 14 484.





• the factors determining the timing and close concordance of onset between the two eyes
• the considerably greater risk of visual loss in males than in females, which may lead to mistaken diagnosis of an X-linked recessive disorder. A recessive X-linked susceptibility gene that works in concert with the LHON mutation has been postulated.
 LHON
LHON LHON usually presents in the third or fourth decade with acute or subacute loss of central vision.
LHON usually presents in the third or fourth decade with acute or subacute loss of central vision.
 Both eyes are affected simultaneously, or sequentially over a period of up to 2 years.
Both eyes are affected simultaneously, or sequentially over a period of up to 2 years.


 If due to the 11 778 mutation or, rarely, the 3460 mutation, LHON may be associated with a multiple sclerosis-like disease (see Chapter 19). The MRI characteristics of this condition are strongly suggestive of foci of demyelination in the cerebral white matter.
If due to the 11 778 mutation or, rarely, the 3460 mutation, LHON may be associated with a multiple sclerosis-like disease (see Chapter 19). The MRI characteristics of this condition are strongly suggestive of foci of demyelination in the cerebral white matter.

NEUROPATHY, ATAXIA, AND RETINITIS PIGMENTOSA (NARP)
T-to-C transition or T-to-G transversion at nucleotide 8993 in the mitochondrial ATPase 6 gene can produce a syndrome of sensory neuropathy, cerebellar ataxia, retinitis pigmentosa (NARP), and mental retardation. These mutations are invariably heteroplasmic and the precise clinical manifestations and severity depend on the percentage of mutant mtDNAs. Scant information is available concerning the histologic changes in the CNS. Cerebellar atrophy with neuronal loss and gliosis has been described. Optic, auditory, and olfactory pathways may show neuronal loss as well. Biopsies of skeletal muscle from patients with NARP do not show ‘ragged-red’ fibers, but contain accumulations of lipid and glycogen within vacuoles. Hypocitrullinemia is a frequent finding. Patients in whom a high proportion of mtDNA is mutated may develop Leigh syndrome, which is considered in Chapter 6. Some authors consider NARP and maternally inherited Leigh syndrome (MILS) to be a continuum.
KEARNS–SAYRE SYNDROME (KSS), AND CHRONIC PROGRESSIVE EXTERNAL OPHTHALMOPLEGIA (CPEO)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
The commonest neuropathologic abnormality in KSS is vacuolation of the white matter in the brain stem, cerebellum, and cerebrum (in decreasing order of frequency). Myelinated fibers in the deep gray nuclei and spinal cord may also be involved. This change is usually not discernible on macroscopic examination, but affected white matter may appear slightly gray (Fig. 24.13). On microscopy the affected white matter has a spongy appearance (Fig. 24.13) due to separation of myelin lamellae by clear vacuoles (Fig. 24.13), which may be elongated in the long axis of the nerve fibers. Occasionally, severe spongiform vacuolation in the brain stem or cerebellum leads to oligodendrocyte degeneration and rather poorly demarcated foci of demyelination.
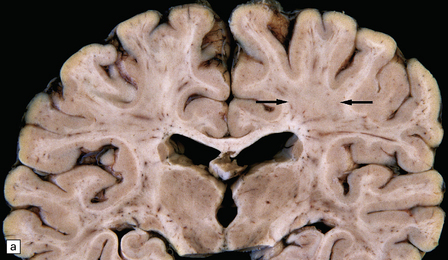
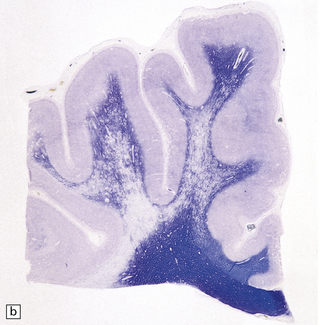
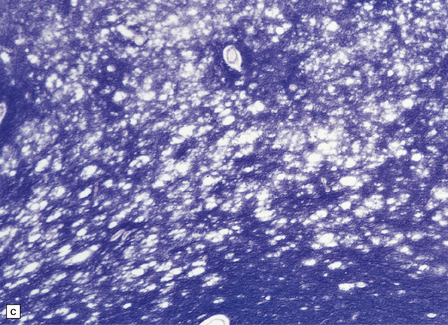
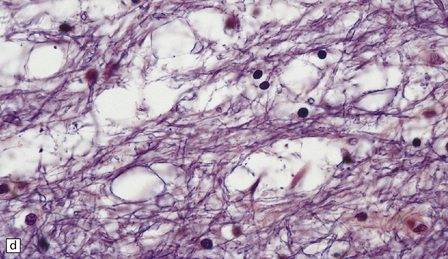
24.13 Kearns–Sayre syndrome: brain.
(a) In this case, the cerebral white matter shows ill-defined gray discoloration (e.g. between arrows). On histology, spongy vacuolation of the white matter (b) and (c), is typical. (d) At higher magnification, some of the vacuoles can be seen to be within myelin sheaths.
 ETIOLOGY OF KSS AND CPEO
ETIOLOGY OF KSS AND CPEO







 KEARNS–SAYRE SYNDROME
KEARNS–SAYRE SYNDROME

Histologic, histochemical, and electron microscopic examination of skeletal muscle usually reveals ‘ragged-red’ fibers (Fig. 24.14) containing accumulations of morphologically abnormal mitochondria, some with paracrystalline inclusions (Fig. 24.14).
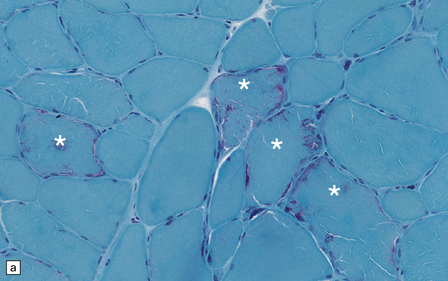
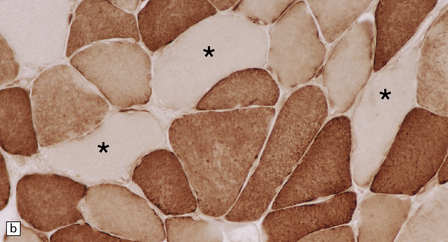
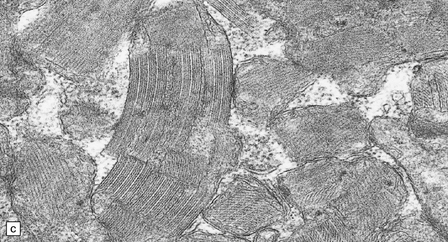
MYONEUROGASTROINTESTINAL ENCEPHALOPATHY (MNGIE) SYNDROME
The differential diagnosis includes familial visceral myopathy with external ophthalmoplegia (also known as oculogastrointestinal muscular dystrophy), and intestinal dysplasia with neuronal intranuclear inclusions (see Table 24.2). Intestinal pseudo-obstruction has also been reported in association with MELAS. Allogeneic hematopoietic stem cell transplantation has shown promise as a therapy.
Table 24.2
The diagnosis of mitochondrial disorders
Important investigations
In many cases coming to autopsy, the precise genetic abnormality will not have been determined.
If a mitochondrial disorder it is advisable to:
• Sample skeletal muscles for histology and, if possible, enzyme histochemistry and electron microscopy.
Findings suggestive of mitochondrial disorder
The morphologic findings on examination of the CNS may suggest or indicate a mitochondrial disorder although without genetic analysis precise classification is often difficult. There is extensive overlap between the neuropathologic findings in different mitochondrial disorders. Features suggestive of a mitochondrial disorder include:
• Intramyelinic edema or spongy vacuolation of white matter (but see also differential diagnosis on p. 525).
• Extensive calcification in the basal ganglia or cerebral white matter, thalamus, dentate nucleus, and brain stem (but see Chapter 1 and Fahr’s disease, on p. 469).
• Neuronal loss and gliosis involving the dentate nucleus, inferior olives, striatum, and gracile and cuneate nuclei. Differential diagnoses include Friedreich’s ataxia and other system degenerations (see Chapters 27, 29, and 30).
• Rarefaction and coarse vacuolation of gray matter with preservation of neurons, particularly in the striatum, brain stem, and dentate nucleus (see also Wernicke’s encephalopathy on p. 453 and Leigh’s disease on p. 150).
HEPATOCEREBRAL FORM OF MITOCHONDRIAL DNA DEPLETION SYNDROME
This rare disorder has been associated with mutations in the DGUOK, C10orf2, POLG, and MPV17 genes. DGUOK codes for mitochondrial deoxyguanosine kinase which phosphorylates purine deoxyribonucleosides. C10orf2 encodes a DNA helicase with a crucial role in mtDNA replication. POLG encodes a DNA polymerase subunit and also plays a role in DNA replication. MPV17 encodes a mitochondrial inner membrane protein important for the metabolism of reactive oxygen species. Depletion of mtDNA in some tissues may reach 98%. It presents in infancy or childhood with severe myopathy, liver failure, and encephalopathy. The CNS may show the neuropathologic changes of liver failure (see Chapter 22). In a personal case, this syndrome was also associated with neuropathologic abnormalities resembling those in Leigh’s disease (Fig. 24.15).
REFERENCES
Carling, P.J., Cree, L.M., Chinnery, P.F. The implications of mitochondrial DNA copy number regulation during embryogenesis. Mitochondrion.. 2011;11:686–692.
DiMauro, S., Mitochondrial, D.N.A. medicine. Biosci Rep.. 2007;27:5–9.
Filosito, M., Tomelleri, G., Tonin, P., et al. Neuropathology of mitochondrial diseases. Biosci Rep.. 2007;27:23–30.
Finsterer, J., Harbo, H.F., Baets, J., European Federation of Neurological Sciences. EFNS guidelines on the molecular diagnosis of mitochondrial disorders. Eur J Neurol. 2009;16:1255–1264.
Friedman, S.D., Shaw, D.W., Ishak, G., et al. The use of neuroimaging in the diagnosis of mitochondrial disease. Dev Disabil Res Rev.. 2010;16:129–135.
, MITOMAP: A human mitochondrial genome database. 2011. Available at: www.mitomap.org.
OMIM, Online Mendelian inheritance in man. OMIM®. Baltimore, MD: McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University; 2011. Available at: http://omim.org/.
Sarnat, H.B., Marín-García, J. Pathology of mitochondrial encephalomyopathies. Can J Neurol Sci.. 2005;32:152–166.
Tucker, E.J., Compton, A.G., Thorburn, D.R. Recent advances in the genetics of mitochondrial encephalopathies. Curr Neurol Neurosci Rep.. 2010;10:277–285.
Chinnery, P.F., Howell, N., Lightowlers, R.N., et al. MELAS and MERRF. The relationship between maternal mutation load and the frequency of clinically affected offspring. Brain.. 1998;121:1889–1894.
Finsterer, J. Management of mitochondrial stroke-like-episodes. Eur J Neurol.. 2009;16:1178–1184.
Ito, H., Mori, K., Kagami, S. Neuroimaging of stroke-like episodes in MELAS. Brain Dev.. 2011;33:283–288.
Love, S., Nicoll, J.A., Kinrade, E. Sequencing and quantitative assessment of mutant and wild-type mitochondrial DNA in paraffin sections from cases of MELAS. J Pathol.. 1993;170:9–14.
Nicoll, J.A., Moss, T.H., Love, S., et al. Clinical and autopsy findings in two cases of MELAS presenting with stroke-like episodes but without clinical myopathy. Clin Neuropathol.. 1993;12:38–43.
Sproule, D.M., Kaufmann, P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann N Y Acad Sci.. 2008;1142:133–158.
Brinckmann, A., Weiss, C., Wilbert, F., et al. Regionalized pathology correlates with augmentation of mtDNA copy numbers in a patient with myoclonic epilepsy with ragged-red fibers (MERRF-syndrome). PLoS One.. 2010;5:e13513.
DiMauro, S., Hirano, M., Kaufmann, P., et al. Clinical features and genetics of myoclonic epilepsy with ragged red fibers. Adv Neurol.. 2002;89:217–229.
Wu, S.B., Ma, Y.S., Wu, Y.T., et al. Mitochondrial DNA mutation-elicited oxidative stress, oxidative damage, and altered gene expression in cultured cells of patients with MERRF syndrome. Mol Neurobiol.. 2010;41:256–266.
LHON, bilateral striatal necrosis, and MS-like mitochondrial disease
Finsterer, J. Leigh and Leigh-like syndrome in children and adults. Pediatr Neurol.. 2008;39:223–235.
Huoponen, K. Leber hereditary optic neuropathy: clinical and molecular genetic findings. Neurogenetics.. 2001;3:119–125.
Kirches, E. LHON: Mitochondrial mutations and more. Curr Genomics.. 2011;12:44–54.
Man, P.Y., Turnbull, D.M., Chinnery, P.F. Leber hereditary optic neuropathy. J Med Genet.. 2002;39:162–169.
Palace, J. Multiple sclerosis associated with Leber’s Hereditary Optic Neuropathy. J Neurol Sci.. 2009;286:24–27.
Yu-Wai-Man, P., Griffiths, P.G., Hudson, G., et al. Inherited mitochondrial optic neuropathies. J Med Genet.. 2009;46:145–158.
Fryer, A., Appleton, R., Sweeney, M.G., et al. Mitochondrial DNA 8993 (NARP) mutation presenting with a heterogeneous phenotype including ‘cerebral palsy’. Arch Dis Child.. 1994;71:419–422.
Gelfand, J.M., Duncan, J.L., Racine, C.A., et al. Heterogeneous patterns of tissue injury in NARP syndrome. J Neurol.. 2011;258:440–448.
Parfait, B., de Lonlay, P., von Kleist-Retzow, J.C., et al. The neurogenic weakness, ataxia and retinitis pigmentosa (NARP) syndrome mtDNA mutation (T8993G) triggers muscle ATPase deficiency and hypocitrullinaemia. Eur J Pediatr.. 1999;158:55–58.
Rojo, A., Campos, Y., Sánchez, J.M., et al. NARP-MILS syndrome caused by 8993 T > G mitochondrial DNA mutation: a clinical, genetic and neuropathological study. Acta Neuropathol (Berl).. 2006;111:610–616.
Sgarbi, G., Casalena, G.A., Baracca, A., et al. Human NARP mitochondrial mutation metabolism corrected with alpha-ketoglutarate/aspartate: a potential new therapy. Arch Neurol.. 2009;66:951–957.
Almousa, R., Charlton, A., Rajesh, S.T., et al. Optimizing muscle biopsy for the diagnosis of mitochondrial myopathy. Ophthal Plast Reconstr Surg.. 2009;25:366–370.
Barthelemy, C., Ogier de Baulny, H., Diaz, J., et al. Late-onset mitochondrial DNA depletion: DNA copy number, multiple deletions, and compensation. Ann Neurol.. 2001;49:607–617.
Brockington, M., Alsanjari, N., Sweeney, M.G., et al. Kearns-Sayre syndrome associated with mitochondrial DNA deletion or duplication: a molecular genetic and pathological study. J Neurol Sci.. 1995;131:78–87.
Greaves, L.C., Yu-Wai-Man, P., Blakely, E.L., et al. Mitochondrial DNA defects and selective extraocular muscle involvement in CPEO. Invest Ophthalmol Vis Sci.. 2010;51:3340–3346.