Chapter 94 Metabolic Myopathies
Utilization of Bioenergetic Substrates in Exercise
Defects of energy metabolism may profoundly disrupt the function of muscle and other highly energy-dependent tissues, such as brain, nerve, heart, kidney, liver and bowel. The limits of energy utilization in skeletal muscle are set by the adenosine triphosphatases (ATPases) that couple muscle contraction (myosin ATPase) and ion transport (calcium and sodium, potassium ATPases) to the hydrolysis of ATP to adenosine diphosphate (ADP) and inorganic phosphate (Pi) [Kushmerick, 1995]. ADP and Pi in turn activate energy-producing reactions that regenerate ATP. Without this, ATP stores would be exhausted in seconds. The substrates that are used to replenish ATP are determined by the intrinsic properties of these fuels and by the intensity and duration of exercise that modulates fuel selection [Gollnick, 1985; Astrand and Rodahl, 1986]. The creatine kinase reaction and anaerobic glycogenolysis are the major anaerobic sources of ADP phosphorylation. Increases in ADP and adenosine monophosphate (AMP) that occur in heavy exercise are primarily buffered by the coupled adenylate kinase (myokinase), adenylate deaminase (myoadenylate deaminase) reactions. Anaerobic glycogenolysis and phosphocreatine hydrolysis support muscle energy production with 2- to 4-fold higher rates than those supported by oxidative metabolism [Sahlin, 1986]. Anaerobic energy is crucial for rapid bursts of exercise and to fuel the transition from rest to exercise. The acceleration to high rates of energy production occurs instantly for ATP, in less than a second for phosphocreatine, and within seconds for anaerobic glycogenolysis. In contrast, maximal oxidative power requires from 3 minutes (with glycogen as the oxidative substrate) to 30 minutes (for peak fatty acid oxidation). Anaerobic fuels are rapidly depleted and lead to the accumulation of metabolic end products, such as protons and Pi, that promote fatigue.
If exercise needs to be sustained for more than a few minutes, then oxidative phosphorylation is necessary and provides the most abundant source of ATP synthesis. Glycogen is the major endogenous oxidative fuel of skeletal muscle, while blood glucose and free fatty acids (FFA) are the major exogenous fuels. A small percentage of muscle energy needs is supplied by amino acids, predominantly branched chain amino acids, which are oxidized to a limited extent. Oxidative metabolism provides higher yields of ATP per mole of substrate, rising from 2 to 36 for glucose and from 3 to 37 per glycosyl unit of glycogen metabolized anaerobically versus oxidatively. Furthermore, the metabolic end products of oxidative metabolism – namely, CO2 and water – are removed easily from working muscle and do not promote fatigue. The most abundant and critical fuel for the support of prolonged, moderate exercise is lipid. Carbohydrate stores, in the form of muscle and hepatic glycogen and blood glucose (derived mainly from hepatic glycogenolysis), are limited and can support high-intensity exercise for only 1–2 hours. However, carbohydrate, particularly muscle glycogen, is critical for normal oxidative metabolism. Glycogen supports a peak rate of oxidative phosphorylation that is about 2-fold greater than fat. Though incompletely understood, this may be based upon a requirement for glycogen-derived pyruvate to support optimal function of the tricarboxylic acid cycle [Sahlin et al., 1990, 1995; Gibala et al., 1997]. The proportion of carbohydrate relative to lipid oxidation increases progressively as the intensity of the aerobic exercise increases, until carbohydrate is the exclusive fuel of maximum oxidative metabolism [Sahlin, 1986; van Loon et al., 2001]. Second, glycogen is able to accelerate to maximal oxidative power output rapidly compared to other fuels [Sahlin, 1986; Haller and Vissing, 2002]. Third, the molar ratio of ATP produced to O2 consumed is higher for glycogen (6.17) and glucose (5.98) than for fatty acids (5.61) [Rennie and Edwards, 1981]. The importance of this point lies in the fact that peak O2 utilization in healthy humans is limited by O2 delivery [Saltin, 1988].
The primary source of energy for resting muscle is derived from fatty acid oxidation (FAO) [Felig and Wahren, 1975]. At rest, glucose utilization accounts for 10–15 percent of total oxygen consumption [Wahren, 1977]. Both slow- and fast-twitch fibers have similar levels of glycogen content at rest [Essen, 1978]. The choice of the bioenergetic pathway in working muscle depends not only on the type, intensity, and duration of exercise [Essen, 1977; Gollnick et al., 1974], but also on diet and physical conditioning [DiMauro and Haller, 1999]. In the first 5–10 minutes of moderate exercise, high-energy phosphates are used first to regenerate ATP. This is followed by muscle glycogen breakdown, which is indicated by a sharp rise in lactate during the first 10 minutes. Blood lactate levels then drop as muscle triglycerides and blood-borne fuels are used [Felig and Wahren, 1975; Lithell et al., 1979]. After 90 minutes, the major fuels are glucose and FFAs. During 1–4 hours of mild to moderate prolonged exercise, muscle uptake of FFAs increases approximately 70 percent, and after 4 hours, FFAs are used twice as much as carbohydrates.
Box 94-1 Heritable Causes of Myoglobinuria
I Biochemical Abnormality Known
III Abnormal Composition of Sarcolemma
(Modified from Tein I, et al: In Rowland LP, DiMauro S, editors: Handbook of clinical neurology, vol 18[62], Myopathies, Amsterdam, 1992, Elsevier Science Publishers.)
Myoglobinuria
Myoglobinuria is a clinical syndrome, not just a biochemical state [Rowland, 1984]. In the alert patient, myalgia and limb weakness are the most common presenting symptoms. Urine color is usually brownish rather than red, and the urine tests positive for both albumin and heme (a concentration of at least 4 μg/mL). There are few or no red blood cells. Myoglobin can be identified by immunochemical methods. The sarcoplasmic enzymes, including serum creatine kinase (CK), are usually elevated to more than 100 times normal. Inconstant features include hyperphosphatemia, hyperuricemia, and hypocalcemia. If renal failure occurs, serum potassium and calcium levels may rise. If the patient is comatose or if the presenting disorder is one of acute renal failure, there may be no muscle symptoms or signs. Under these conditions, the diagnosis can be made if:
The etiologies of heritable myoglobinuria differ in adults compared with children. In a study of 77 adult patients aged 15–65 years, Tonin et al. [1990] identified the enzyme abnormality in 36 patients (47 percent) as follows: CPT deficiency in 17 patients; glycolytic defects in 15 patients (including PPL in 10, PPL b kinase in 4, and PGK in 1); myoadenylate deaminase in 3; and combined CPT and myoadenylate deaminase in 1. In contrast, in 100 cases of recurrent childhood-onset myoglobinuria, a lower percentage of children have been diagnosed biochemically (24 percent): 16 with CPT deficiency, 1 with SCHAD deficiency, and 7 with various glycolytic defects, including 2 PPL, 1 PGK, 3 PGAM, and 1 LDH deficiency [Tein et al., 1990b]. These children could be divided into two groups – a type I exertional group, in which exertion was the primary precipitating factor (56 cases); and a type II toxic group, in which infection and/or fever and leukocytosis were the primary precipitants (37 cases). The type II toxic childhood group was distinguished from the type I exertional childhood- and adult-onset groups by its etiologies, which were limited to FAO defects, as well as its slight female predominance, which contrasted with the marked male predominance in the latter two groups. The type II toxic group was distinguished further by the earlier age at onset of myoglobinuria, the presence of a more generalized disease (e.g., ictal bulbar signs, seizures, encephalopathy, developmental delay), and a higher mortality rate. Currently, the most common etiology for recurrent myoglobinuria in both adults and in children is CPT II deficiency [Tein et al., 1992].
Glycogenoses
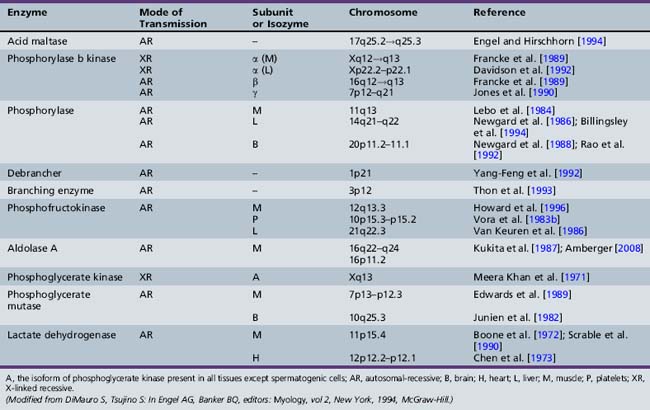
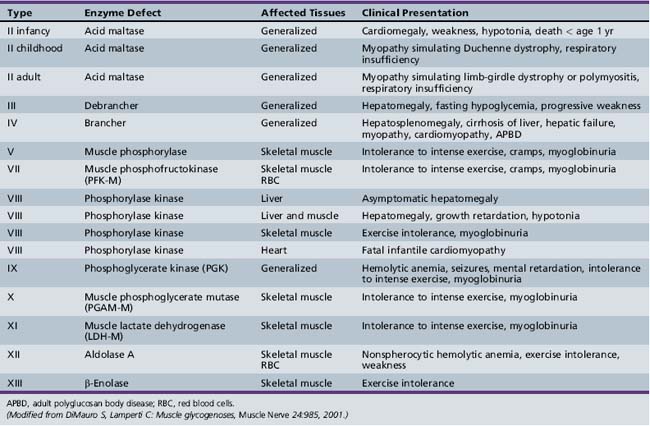
Only glycogenoses affecting skeletal muscle, alone or in association with other tissues, are discussed in this section (Figure 94-1) [DiMauro and Bresolin, 1986]. Molecular genetic analysis has led to the cloning of genes encoding the enzymes involved in most glycogenoses, as well as their chromosomal localization (Table 94-1). All are autosomal-recessive in inheritance, with the exception of PGK, which is X-linked recessive, and PPL b kinase, which may be either. In the glycogenoses, fixed weakness is the primary presentation in acid maltase, debrancher, aldolase, and brancher enzyme deficiencies, whereas exercise intolerance with cramps, myalgia, and recurrent myoglobinuria is the primary presentation in phosphorylase kinase, phosphorylase, phosphofructokinase, phosphoglycerate kinase, phosphoglycerate mutase, β-enolase, and lactate dehydrogenase deficiencies (Table 94-2) [DiMauro and Lamperti, 2001].
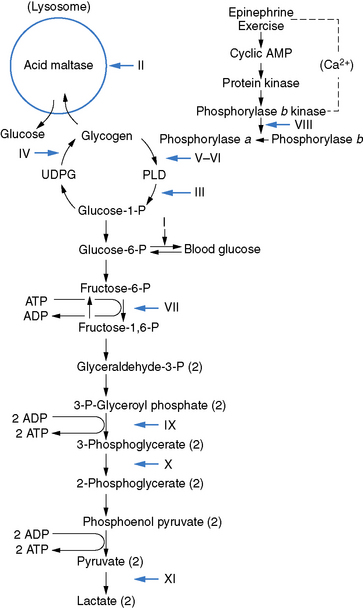
Fig. 94-1 Scheme of glycogen metabolism and glycolysis.
(From DiMauro S, et al: Metabolic myopathies, Am J Med Genet 25:635–651, 1986.)
Pathophysiology
The immediate source of energy for contraction and relaxation is provided by the hydrolysis of ATP. Oxidative phosphorylation provides the largest contribution of energy overall, whereas anaerobic glycolysis plays a relatively minor role, primarily limited to conditions of sustained isometric contraction when blood flow and oxygen delivery to exercising muscles are reduced drastically. The dynamic form of exercise, such as walking or running, is primarily dependent on aerobic glycolysis. Therefore, the pathophysiology of glycogenoses relates more to the impairment of aerobic than anaerobic glycolysis [Lewis and Haller, 1986; Lewis et al., 1991].
The energy substrates used by muscle for aerobic metabolism depend on the type, intensity, and duration of exercise, as well as on physical conditioning and diet. During intense exercise (close to maximal oxygen uptake, or VO2max, in dynamic exercise or maximal force generation in isometric exercise), energy is derived from anaerobic glycolysis, particularly when there is a burst of activity with rapid acceleration to maximal exercise [DiMauro and Tsujino, 1994]. During low-intensity exercise (below 50 percent VO2max), the primary source of energy is derived from blood glucose and FFAs. At higher intensities, the proportion of energy derived from carbohydrate oxidation increases, and glycogen becomes an important fuel. At 70–80 percent of VO2max, the critical energy source is provided by the aerobic metabolism of glycogen, and fatigue occurs when glycogen stores are exhausted [DiMauro and Tsujino, 1994]. Individuals with defective glycolysis/glycogenolysis are most vulnerable during the initial stages of intense exercise, and they must rest soon after beginning exercise because of muscle cramps. However, if they continue to exercise at low intensity, they are able to continue for a longer time. This is known as the second-wind phenomenon, and has been attributed to a metabolic switch from carbohydrate to fatty acid utilization [Felig and Wahren, 1975], and to increased circulation with increased availability of blood glucose from hepatic glycogenolysis (Haller et al., 1985]. A decrease in ATP levels could first cause muscle contracture. Theoretically, a more severe depletion could lead to myoglobinuria [Rowland, 1984], although this has not been proven.
The forearm ischemic exercise test, developed by McArdle [1951], is a useful test for the detection of enzymatic defects in the nonlysosomal glycogenolytic and glycolytic pathways. This test can be performed in cooperative children as young as 6 years of age. A catheter is placed in a superficial antecubital vein, and basal lactate and ammonia levels are obtained without stasis. A sphygmomanometer cuff is then placed above the elbow and inflated above arterial pressure. The patient is asked to squeeze another rolled-up cuff rhythmically to well above 120 mmHg for 1 minute of exercise. This requires constant encouragement from the observer because significant discomfort can occur, even in healthy control subjects. The test should be truncated if the patient develops an acute cramp, as myonecrosis and/or a compartment syndrome may occur in an individual with a glycolytic disorder [Meinck et al., 1982; Lindner et al., 2001]. After 1 minute of exercise, the cuff around the arm is deflated and blood samples are sequentially obtained at 1, 3, 5, 7, 10, and 15 minutes. In healthy subjects there is a 4- to 6-fold increase of lactate over baseline, with the peak occurring at 1–2 minutes post exercise, which declines to baseline values by 15 minutes. This is paralleled by a similar 5-fold or more increase in ammonia, with levels generally peaking at 2–5 minutes after exercise in individuals with normal myoadenylate deaminase levels. In healthy subjects, venous ammonia and lactate are linearly related [Sinkeler et al., 1985]. In individuals with a defect in glycolysis/glycogenolysis, there is an insufficient rise in lactate (less than 2-fold), with a compensatory and exaggerated increase in ammonia, which also indicates sufficient effort on the part of the individual. This exaggerated rise in ammonia is attributable to high cellular levels of ADP, resulting from a combination of blocked glycogenolysis/glycolysis and absent cellular acidosis. An insufficient lactate rise has been demonstrated in PPL, debrancher, PFK, PGK, PGAM, and LDH deficiencies but not in acid maltase or phosphorylase b kinase deficiency. The major limitation of this test is that the rise of venous lactate in individuals who do not have a defect in this pathway is highly dependent on the patient’s ability and willingness to exercise. Therefore, patients in whom lactate levels are low, due to poor effort or to placement of the venous line in other than the median cubital vein, show proportionally blunted ammonia responses.
Given the potential risk of myoglobinuria, alternatives to the traditional ischemic forearm test have been described. One of these employs sustained, intense (70 percent of maximal voluntary contraction, MVC) isometric handgrip exercise [Hogrel et al., 2001]. However, this form of testing is as ischemic as the use of a blood pressure cuff, since the intramuscular pressure in isometric contractions of ≥50 percent MVC completely occludes muscle blood flow. Therefore, any reduction in the incidence of muscle contractures is dependent upon shortening the duration of the test that is also effective in minimizing contractures in the traditional ischemic forearm test. An alternative nonischemic forearm test has been described and involves 30 maximal handgrip contractions in 1 minute without a blood pressure cuff [Kazemi-Esfarjani et al., 2002]. The level of increase in lactate and ammonia in control subjects and the diagnostic sensitivity in patients were similar to the ischemic exercise test. However, in contrast to the ischemic tests, the retained oxidative capacity helps to protect individuals from contractures or significant pain [Kazemi-Esfarjani et al., 2002].
Glycolytic/Glycogenolytic Defects
Acid Maltase Deficiency
Clinical features
Acid maltase deficiency (AMD, glycogenosis type II, GSD type II) results in three very different clinical presentations [DiMauro and Lamperti 2001]:
Slonim et al. [2000] have described a subgroup of patients who share muscle morphological features with the neonatal-onset infantile form patients, but differ from typical Pompe’s disease patients in that the heart is not involved and survival is longer.
Laboratory data
All forms of AMD demonstrate an increase in serum CK. Electromyography (EMG) reveals myopathic abnormalities and may also show fibrillation potentials, positive waves, complex repetitive discharges, and myotonic discharges [DiMauro and Lamperti, 2001]. These are more frequently demonstrated in the paraspinal muscles. The electrocardiogram (EKG) demonstrates characteristic, although not specific, changes, including a short PR interval, giant QRS complexes, and signs of biventricular hypertrophy in infantile AMD. In adults, there is markedly reduced vital capacity.
Pathology
In infantile AMD, glycogen accumulation is evident in all tissues, most notably the heart. There is significant involvement of the anterior horn cells of the spinal cord and of brainstem nuclei, leading to severe weakness and fasciculations. Glycogen accumulation in Schwann cells is seen in peripheral nerve biopsies. There is a vacuolar myopathy on muscle biopsy in all three forms of AMD [DiMauro et al., 1992]. In the infantile form, all muscle fibers contain many vacuoles, which often coalesce into a lacework pattern and contain periodic acid–Schiff (PAS)-positive, diastase-digestible material and stain for acid phosphatase. Electron microscopy reveals excess glycogen either within lysosomal vacuoles or free in the cytoplasm.
Biochemistry and molecular genetics
Although the enzyme defect is generalized and can be documented in lymphocytes [Shanske and DiMauro, 1981] or in cultured skin fibroblasts, biochemical measurement of acid maltase in muscle establishes the diagnosis. It is not entirely clear why symptoms are mainly confined to muscle in the childhood and adult forms. However, differences in clinical expression may relate, in part, to the amounts of residual activity [Mehler and DiMauro, 1977; Reuser et al., 1987; Van Der Ploeg et al., 1988]. Second, genetic heterogeneity has also been demonstrated. In a study of 14 patients, Martiniuk et al. [1990a, 1990b] found that messenger RNA (mRNA) was lacking in 5 of 10 infantile AMD cases and was present in all 4 adult cases, although it was shorter in 2 cases. The defective lysosomal enzyme, α-glucosidase, is encoded by a gene on chromosome 17 [D’Ancona et al. 1979; Weil et al., 1979; Solomon et al., 1979], and over 55 mutations have been identified in patients with the three variants [DiMauro and Lamperti, 2001]. The genotype–phenotype correlation is hard to establish due to the frequent compound heterozygosity. However, there is good correlation between the severity of the mutation and the severity of the clinical phenotype. Therefore, deletions and nonsense mutations are usually associated with the infantile variant, whereas “leaky” mutations, such as the IVS1(-13T>G) splice-site mutations, are associated with the adult-onset variant [Hirschhorn, 1995]. Childhood AMD is often the result of compound heterozygosity [Huie et al., 1998], but has also been associated with at least one homozygous, apparently specific mutation [Adams et al., 1997]. A reliable and rapid first-tier test for screening patients suspected of having Pompe’s disease has been provided, using a fluorometric dried blood spot-based α-glucosidase activity assay [Goldstein et al., 2009]. The measurement of acid α-glucosidase activities in dried blood spots using tandem mass spectrometry has been found suitable for high-throughput analysis and newborn screening for Pompe’s disease [Dajnoki et al., 2008].
Treatment
Clinical trials with lysosome-stabilizing agents and activators of glycogenolysis have been unsuccessful in Pompe’s disease [DiMauro et al., 1992]. Recent experimental work with small molecule pharmacological chaperones may hold promise for future clinical trials [Flanagan, 2009]. Enzyme replacement looks promising, particularly for patients with childhood and adult AMD. Recombinant human α-glucosidase, obtained from milk of transgenic rabbits and injected into knockout mice lacking acid maltase, corrected the enzyme defect fully in all tissues but brain [Bijvoet et al., 1999]. Amalfitano et al. [2001] reported the results of a phase I/II open-label, single-dose study of recombinant human α-glucosidase infused intravenously twice weekly in three infants with infantile GSD type II. The results of more than 250 infusions showed that recombinant human α-glucosidase was generally well tolerated. There were steady decreases in heart size and maintenance of normal cardiac function for more than 1 year observed in all three infants. These infants lived past the critical age of 1 year and also show improvements in skeletal muscle function. In a 52-week trial with acid glucosidase alpha, there was marked improvement in the cardiomyopathy, ventilatory function, and overall survival among 18 children under 7 months of age with infantile Pompe’s disease [Kishnani et al., 2009]. Sixteen of the 18 patients enrolled in an extension study continued to receive alglucosidase alpha for up to a total of 3 years and showed reduction in the risk of death by 95 percent, and reduced risk of invasive ventilation or death by 91 percent, compared to an untreated historical control group, over the entire study period. The cardiomyopathy continued to improve and 11 patients learned and sustained substantial motor skills. In another study of late-onset AMD, three patients (aged 11, 16, and 32 years), all wheelchair-dependent and two ventilator-dependent with a history of deteriorating pulmonary function, received 3 years of treatment with weekly infusions of recombinant α-glucosidase from rabbit milk, and had stabilized pulmonary function and reported less fatigue [Winkel et al., 2004]. The youngest and least affected patient showed significant improvement of skeletal muscle strength and function, and, after 72 weeks of treatment, could walk without support. There have also been promising gene therapy results, both in vitro and in vivo, using E1-deleted recombinant adenovirus encoding human α-glucosidase. Transduction of the viral construct into enzyme-deficient human fibroblasts in culture resulted in a dose-dependent return of acid maltase activity, which was localized within lysosomes [Pauly et al., 1998]. Equally encouraging results were obtained by injecting a similar viral construct into the pectoral muscle of acid maltase-deficient Japanese quails that are an avian model of AMD [Tsujino et al., 1998]. Individuals with childhood or adult AMD may require ventilatory assistance. A high-protein diet has improved strength and respiratory function in some but not all patients [DiMauro et al., 1992; Slonim et al., 1983].
Phosphorylase b Kinase Deficiency
Clinical features
Phosphorylase b kinase (PBK) deficiency (glycogenosis type VIII) may be divided into four clinical syndromes [DiMauro and Tsujino, 1994]. These can be differentiated by inheritance and tissue distribution as follows:
Laboratory data
The resting serum CK is variably increased in most patients. The EMG may be normal or may show nonspecific myopathic changes. Muscle biopsy may be normal or show subsarcolemmal accumulations of glycogen, primarily located in type 2B fibers. The PPL stain is normal. There are pools of free, normal-looking glycogen particles on electron microscopy [DiMauro and Tsujino, 1994].
Biochemistry and molecular genetics
PBK participates in glycogen metabolism regulation by acting on the two main enzymes involved in glycogen synthesis and degradation: namely, glycogen synthetase and PPL [Heilmeyer, 1991]. PBK converts PPL from the less active b form to the more active a form, and simultaneously converts glycogen synthetase from a more active, dephosphorylated form to a less active, phosphorylated form. Thus, when glycogen synthesis is turned on, glycogen degradation is turned off, and vice versa. PBK is a multimeric enzyme composed of four different subunits (alpha, beta, gamma, delta) [DiMauro and Tsujino, 1994]. The alpha and beta subunits are regulatory, the gamma subunit is catalytic, and the delta subunit is identical to calmodulin and confers calcium sensitivity to the enzyme [DiMauro and Lamperti, 2001]. In addition, there are two isoforms for the alpha subunit (muscle and liver), which are encoded by genes on the X chromosome. The two isoforms for the gamma subunit (muscle and testis) and the beta subunit are encoded by autosomal genes. Glycogen is normal or moderately increased in patients with myopathy, and the PBK activity is absent or markedly decreased [DiMauro and Tsujino, 1994].
It is not easy to account for the tissue specificity in the different clinical variants. Two distinct genes have been identified. One gene (PHKA1) is on the proximal long arm at Xq13, which encodes a muscle isozyme; the other gene (PHKA2) is on the distal short arm of the X chromosome at Xp22.2–p22.1, which encodes a nonmuscle isozyme, and is in the same region to which the gene for X-linked liver glycogenosis resulting from PBK deficiency has been mapped [Willems et al., 1991]. Tissue-specific isozymes may be the result of alternative RNA splicing. DiMauro and Tsujino [1994] suggest that mutations in tissue-specifically expressed exons of the beta subunit [Harmann et al., 1991] could explain the apparently autosomal-recessive forms of myopathy and fatal infantile cardiomyopathy.
Phosphorylase Deficiency
Clinical features
Myophosphorylase deficiency (type V glycogenosis, or McArdle’s disease) is a rare disease but is an important cause of recurrent myoglobinuria. Exercise intolerance generally starts in childhood, but overt episodes of muscle cramping and myoglobinuria develop later in adolescence. Brief isometric contraction (e.g., lifting heavy objects) and less intense but sustained dynamic exercise (e.g., climbing stairs) are the two primary precipitants. Approximately 50 percent of these patients experience episodes of muscle necrosis and myoglobinuria after exercise, while 27 percent develop acute renal failure. Although there is usually complete functional recovery after the episode of myoglobinuria, about one-third of patients have fixed mild proximal greater than distal weakness, which is seen more commonly in older patients [DiMauro and Bresolin, 1986]. The diagnosis is usually made in the second or third decade of life. There may be a marked variation in the severity of symptoms. In some, progressive weakness may begin late in life (sixth decade), with no history of cramps or pigmenturia [DiMauro and Bresolin, 1986; Pourmand et al., 1983]. The cumulative effect of recurrent muscle damage over the years may explain the appearance of fixed weakness in older individuals. In contrast, there have been four unique childhood cases with severe generalized muscle and respiratory weakness noted at or soon after birth, with death in infancy [DiMauro and Hartlage, 1978; DiMauro and Tsujino, 1994; Milstein et al., 1989]. A hypotonic 1-year-old infant has been described with repeated episodes of hyper-creatine kinase-emia during febrile episodes [Ito et al., 2003].
Laboratory data
Between episodes of myoglobinuria, the serum CK is variably increased in 93 percent of cases. This serves as a differentiating feature from CPT deficiency, in which the resting CK generally is normal. EMG between episodes of myoglobinuria demonstrates fibrillations, myotonic discharges, and positive waves in up to 50 percent of patients, suggesting a mild myopathy [DiMauro and Tsujino, 1994]. There is a lack of cytoplasmic acidification on phosphorus-31 nuclear magnetic resonance (31P-NMR) spectroscopy during aerobic or ischemic exercise, as well as a greater than normal drop of the phosphocreatine/inorganic phosphate ratio [Argov and Bank, 1991; Ross et al., 1981].
Inheritance
There is an unexplained marked male predominance, despite the autosomal-recessive pattern of inheritance. The muscle isoform gene has been localized to chromosome 11 [Lebo et al., 1984]. Two families with apparent autosomal-dominant transmission [Chui and Munsat, 1976; Schimrigk et al., 1967] may be explained on the basis of subsequent generations of homozygotes and manifesting heterozygotes, in whom the residual activity was below a critical threshold [Papadimitriou et al., 1990; Schmidt et al., 1987]. In a third family, Tsujino et al. [1993a] demonstrated that the affected mother was a compound heterozygote carrying two different point mutations in the PPL gene, the unaffected father was heterozygous for a third point mutation, and the three affected children were compound heterozygotes.
Muscle biopsy
There may or may not be focal accumulations of glycogen between myofibrils and in the subsarcolemmal regions on PAS stain. The PPL stain [Takeuchi and Kuriaki, 1955] shows no staining in muscle fibers, in contrast to normal staining of the smooth muscle in the walls of intramuscular vessels. DiMauro and Tsujino [1994] pointed out that a positive histochemical reaction may be seen in McArdle’s disease under the following two conditions:
A false-positive reaction in regenerating fibers is due to expression of a different isozyme in immature muscle cells [DiMauro et al., 1978; Sato et al., 1977]. Accumulation of normal-looking glycogen beta particles under the sarcolemma and between myofibrils and myofilaments may be seen on electron microscopy [DiMauro and Tsujino, 1994].
Biochemical considerations
PPL (alpha-1,4-glucan orthophosphate glycosyl transferase) initiates glycogen breakdown by removing 1,4-glucosyl residues phosphorolytically from the outer branches of the glycogen molecule, with liberation of glucose-1-phosphate until the peripheral chains have been shortened to approximately four glucosyl units. The debranching enzyme catalyzes further breakdown. The combined action of these two enzymes degrades glycogen to glucose-1-phosphate (approximately 93 percent) and glucose (approximately 7 percent). Muscle PPL exists as an active phosphorylated alpha form and a less active dephosphorylated beta form. The muscle PPL activity is undetectable in most or up to 10 percent of normal residual activity in affected patients [DiMauro and Tsujino, 1994]. Glycogen accumulation is moderate (about 2-fold) or normal. The glycogen structure is normal. There is no enzyme protein in muscle immunologically detectable by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE), immunoblot, and enzyme-linked immunosorbent assay (ELISA) in the majority of patients [McConchie et al., 1991; Servidei et al., 1988b]. Normal mature human muscle has a single PPL isozyme, whereas cardiac muscle and brain have three different isozymes on PAGE, including:
Lack of the muscle isozyme therefore should cause partial defects in the heart and brain. A defect of the brain isozyme would be expected to cause heart and brain involvement [DiMauro and Tsujino, 1994].
Pathophysiology
The pathophysiology of exercise intolerance in McArdle’s disease involves a combination of interconnected factors related to impaired ATP production from both aerobic and anaerobic glycolysis. The decrease of ATP generated during anaerobic glycolysis may affect muscle ATPases selectively [James et al., 1996], including the Na+K+-ATPase. This inhibition, together with a decrease in Na+K+ pump numbers, may lead to excessive increases in blood and extracellular potassium, which typically are seen during exercise in McArdle’s disease [Haller et al., 1998a]. These changes can explain, in turn, the sarcolemmal inexcitability during repetitive neural stimulation [Dyken et al., 1967]. Furthermore, the lack of lactic acid production and intracellular acidification during exercise alters the equilibrium of the CK reaction, resulting in an exaggerated rise of ADP during exercise. The excessive intracellular ADP is converted to AMP, inosine monophosphate, ammonia, and adenine nucleotide degradation products, including inosine, hypoxanthine, and uric acid [Mineo et al., 1987]. Premature fatigue may be due to impaired excitation–contraction coupling, mediated by excessive accumulation of ADP [Lewis and Haller, 1991a]. Oxidative metabolism is also affected in McArdle’s disease because the block in glycogenolysis impairs production of pyruvate, which is a major anaplerotic substrate for the Krebs cycle [Sahlin et al., 1995]. In patients with McArdle’s disease, oxygen extraction and maximal oxygen uptake are decreased; these can be restored partially by administration of intravenous glucose [Haller et al., 1985].
Molecular genetics
The three PPL isozyme genes have been cloned, sequenced, and localized to different chromosomes [Billingsley et al., 1994; Lebo et al., 1984; Newgard et al., 1986, 1988; Rao et al., 1992] (see Table 94-1). Tsujino et al. [1993a] identified three different point mutations in the muscle isozyme gene: a C to G mutation in codon 49 of exon 1 (converting an arginine to a stop codon), a G to A mutation in codon 204 of exon 5 (converting a glycine to a serine), and an A to C mutation in codon 542 of exon 14 (converting a lysine to a threonine). In an analysis of 32 patients, 15 were homozygous for the first mutation and 12 were compound heterozygotes [Tsujino et al., 1993a]. More than 100 mutations have been identified in the muscle isozyme gene, which will facilitate carrier identification [DiMauro and Tsujino, 1994; Tsujino et al., 1993a, 1994a, 1994b, 1994c; DiMauro and Lamperti, 2001; Vissing et al., 2009]. These include missense, nonsense, and splice-junction mutations. The most common mutation in Europe and North America is the Arg49Stop, which accounts for 81 percent of the alleles in British patients [Bartram et al., 1993] and 63 percent of alleles in a survey of U.S. patients [El-Schahawi et al., 1996]. The high frequency of this mutation, on at least one allele, has facilitated molecular diagnosis of blood samples from patients with suspected McArdle’s disease. However, it is important to realize that the frequency of different mutations varies in different ethnic groups. The frequency of the Arg49Stop mutation appears to decrease from north to south in Europe, from 81 percent of alleles in the United Kingdom [Bartram et al., 1993] to 56 percent in Germany [Vorgerd et al., 1998] and 32 percent in Italy [Martinuzzi et al., 1996].
To date, there has been no clear genotype–phenotype correlation in McArdle’s disease. For example, the common homozygous Arg49Stop mutation was also present in an infant with the fatal myopathic variant [Tsujino et al., 1993a], and in a child who died of sudden infant death syndrome [El-Schahawi et al., 1997]. This lack of clear genotype–phenotype correlation was also documented in a study of 54 Spanish patients with McArdle’s disease from 40 unrelated families [Martin et al., 2001]. One possible explanation for increased severity of clinical presentation would be the presence of additional gene defects, such as myoadenylate deaminase deficiency [Rubio et al., 1997a; Tsujino et al., 1995a]. Recently, two patients with atypical McArdle’s disease, who carried common mutations on one allele and novel splice mutations in introns 3 and 5 on the other allele, demonstrated minimal phosphorylase activity that ameliorated the typical phenotype by augmenting muscle oxidative capacity [Vissing et al., 2009].
Treatment
A key therapeutic strategy has been the attempt to bypass the metabolic block by providing the muscle with glycolytic substrates [DiMauro and Tsujino, 1994]. The oral administration of glucose or fructose to raise the blood glucose level has been helpful but has resulted in weight gain. Glucagon injections have been impractical, with inconsistent results. Although exercise tolerance was increased by raising the serum FFA concentration through the use of fat emulsions, fasting, and the administration of norepinephrine or heparin, more practical regimens, such as a high-fat, low-carbohydrate diet, have not been effective [DiMauro and Bresolin, 1986]. In a study of intralipid infusion to increase FFA availability during 70 percent maximal oxygen consumption cycle ergometry, FFA levels more than tripled, but heart rate was significantly higher during exercise compared with placebo and glucose infusions trials, suggesting that, although lipids are an important source of fuel for exercising muscle in McArdle’s disease, the maximal rates of fat oxidation appeared limited and could not be increased above physiological normal rates during exercise [Andersen et al., 2009]. It was suggested that the limitation was probably caused by a metabolic bottleneck in the tricarboxylic acid cycle due to impaired glycolytic flux, and that therapies aimed at enhancing fat use in McArdle’s disease should be combined with interventions targeting expansion of the tricarboxylic acid cycle. An alternative strategy was to supply branched-chain amino acids, which are taken up, rather than released, by tissue of patients with McArdle’s disease during exercise [Wahren et al., 1973]. Slonim and Goans [1985] demonstrated an improvement of muscle endurance and strength in a patient with weakness after institution of a high-protein diet. Direct administration of branched-chain amino acids to six patients, however, resulted in impairment rather than improvement of bicycle exercise capacity in 5 of 6 patients, possibly due to a lowering of FFA by the amino acids [MacLean et al., 1998]. In a more recent study, patients with McArdle’s disease were randomized to follow either a carbohydrate-rich or protein-rich diet for 3 days before testing; patients on the carbohydrate-rich diet improved their maximal work capacity and exercise tolerance to submaximal workloads [Andersen and Vissing, 2008]. Aerobic training of four McArdle’s patients improved peak cycle exercise capacity, circulatory capacity, and oxygen uptake [Haller et al., 1998b].
Vitamin B6 is another potential therapeutic aid, as overall body stores of pyridoxal phosphate are depleted in McArdle’s disease due to the lack of enzyme protein to which pyridoxal phosphate is bound [Haller et al., 1983]. One patient has been shown to have a beneficial effect with vitamin B6 supplementation [Phoenix et al., 1998], but further studies need to be done. In addition, oral creatine monohydrate supplementation in a placebo-controlled crossover trial involving nine patients has been shown to alleviate symptoms and increase their capacity for ischemic, isometric forearm exercise [Vorgerd et al., 2000]. Another strategy has been the ingestion of sucrose prior to exercise to increase the availability of glucose [Vissing and Haller, 2003]. In a single-blind, randomized, placebo-controlled crossover study, 12 patients were studied. Ingestion of sucrose prior to exercise improved exercise tolerance and sense of well-being. In another comparative study, ingestion of 37 g of sucrose 5 minutes before exercise had a marked and prolonged effect on exercise tolerance in patients with McArdle’s disease. This was a more sustained effect than that seen with 75 g of sucrose 40 minutes before exercise, probably related to more continuous glucose uptake from the intestine and correspondingly higher circulating glucose levels later during exercise [Andersen et al., 2008].
Finally, a first-generation adenoviral recombinant containing the full-length human myophosphorylase complementary DNA (cDNA) has been efficiently transduced into phosphorylase-deficient sheep and human myoblasts, resulting in restoration of phosphorylase activity [Pari et al., 1999].
Debrancher Deficiency
Clinical features
Debrancher deficiency (glycogenosis type III, GSD type III) is characterized by childhood-onset liver dysfunction with hepatomegaly, growth failure, fasting hypoglycemia, and, infrequently, hypoglycemic seizures. Spontaneous resolution may occur around puberty with normal adult liver function [Smit et al., 1990], although cirrhosis and liver failure may develop later [Fellows et al., 1983]. The myopathy tends to present late in the third or fourth decade in 70 percent of cases and involves primarily distal leg and intrinsic hand muscles. It is slowly progressive and rarely incapacitating. Childhood onset has been documented in 7 of 22 patients. Two patients had exercise intolerance, cramps, and premature fatigue [Murase et al., 1973; Ozand et al., 1967], and 5 others had diffuse weakness and wasting, growth failure, and gross motor delay [Badurska et al., 1970; Slonim et al., 1982]. Myoglobinuria has been reported in one case [Brown, 1986]. Enzymatic deficiency was documented in the muscle of 16 patients, 11 of whom had no weakness [Moses et al., 1986]. However, given the young age of these patients (2–27 years), clinical myopathy may have developed later. Peripheral neuropathy has also been documented [Moses et al., 1986]. Although clinical cardiomyopathy is infrequent, cardiac involvement has been demonstrated in most patients with myopathy [Brunberg et al., 1971; Miller et al., 1972; Moses et al., 1989]. Of interest, Cleary et al. [2002] reported consistent facial features in seven patients with GSD type III, which included midfacial hypoplasia with a depressed nasal bridge and a broad upturned nasal tip, indistinct philtral pillars, and bow-shaped lips with a thin vermillion border. In addition, younger patients had deep-set eyes. Several children had clinical problems, such as persistent otitis media or recurrent sinusitis.
Laboratory data
Glucagon or epinephrine in the fasting state does not increase blood glucose, which indicates liver involvement [DiMauro and Tsujino, 1994]. All patients with myopathy have an increase in serum CK. The EMG is myopathic but also may demonstrate fibrillation activity, and the nerve conduction velocities frequently are decreased [Brunberg et al., 1971; Moses et al., 1986]. Evidence of left ventricular or biventricular hypertrophy may be seen on EKG and echocardiography [Brunberg et al., 1971; Moses et al., 1989; Smit et al., 1990].
Inheritance
There is a male predominance. The gene has been assigned to chromosome 1p21 [Yang-Feng et al., 1992]. Prenatal diagnosis can be made by a qualitative assay based on the persistence of the abnormal polysaccharide in cultured amniocytes exposed to a glucose-free medium [Yang et al., 1990].
Pathology
A severe vacuolar myopathy, which contains PAS-positive material that is digested by diastase, is seen on muscle biopsy. This corresponds to large pools of free and apparently normal glycogen particles by electron microscopy [DiMauro and Tsujino, 1994]. Excessive glycogen is also seen in skin biopsies [Sancho et al., 1990], cultured muscle [Miranda et al., 1981], endomyocardial biopsies [Olson et al., 1984], intramuscular nerves [Powell et al., 1985], Schwann cells and axons of sural nerve [Ugawa et al., 1986], and brain [Hug and Schubert, 1966].
Biochemistry
The debrancher enzyme is a single 160-kDa polypeptide with two distinct catalytic functions. After digestion of glycogen to four glucosyl units by PPL, the residual stubs are removed by the debrancher enzyme in two steps – a transferase and a glucosidase – which are located in different domains of the polypeptide [Bates et al., 1975]. Based on enzymatic and immunologic assays, debrancher deficiency has been classified into three groups [Chen et al., 1987; Ding et al., 1990]:
Molecular genetics
Human debrancher cDNA has been isolated and sequenced [Yang et al., 1992]. A number of mutations have been described in GSD type IIIa [Okubo et al., 1996, 1999, 2000; Parvari et al., 1998; Shen et al., 1996, 1997; Shaiu et al., 2000], as well as in GSD type IIIb [Shen et al., 1996]. It is interesting to note that most patients with the IIIb variant have mutations in exon 3 of the debrancher gene, which are expected to result in truncated proteins [Shen et al., 1996]. Shaiu et al. [2000] reported two frequent mutations, each of which was found in homozygous state in multiple patients, and each of which was associated with a subset of clinical phenotype in those patients with that mutation. The first mutation was a novel point mutation of a single T deletion at cDNA position 3964 (3964 delT), which was first detected in an African American patient who had a severe phenotype and early onset of clinical symptoms. The second mutation was an A to G transition at position −12 upstream of the 3′ splice site of intron 32 (IVS32-12A>G) which was confirmed in a Caucasian GSD IIIa patient presenting with mild clinical symptoms. These two mutations together accounted for more than 12 percent of the molecular defects in the GSD type III patients tested at that time.
Whereas the overall incidence of GSD type III in the United States is about 1 in 100,000 live births, it is unusually frequent among North African Jews in Israel (prevalence of 1 in 5400, carrier prevalence 1 in 35) [Parvari et al., 1997]. Another ethnic group with a high prevalence of GSD type III (1 in 3600 with a carrier frequency of 1 in 30) are individuals from the Faroe Islands [Santer et al., 2001]. The population of 45,000 of this small archipelago in the North Atlantic has its roots in colonization by the Norwegians in the 8th century and throughout the Viking age. It was concluded that, due to a founder effect, the Faroe Islands have the highest prevalence of GSD type IIIa worldwide.
Treatment
Fasting hypoglycemia should be avoided in infants and young children with debrancher deficiency through the implementation of frequent feedings and nocturnal gastric glucose infusions and uncooked cornstarch [Fernandes, 1990]. A marked improvement was noted in a 7-year-old child with severe weakness and wasting, following institution of a high-protein diet overnight for 6 months [Slonim et al., 1982]. However, a 6-month high-protein diet had no significant effect in an adult with myopathy and distal wasting [DiMauro and Tsujino, 1994].
Phosphofructokinase Deficiency
Clinical features
Muscle PFK deficiency (Tarui’s disease) (glycogenosis type VII) has been reported in more than 100 patients [Toscano and Musumeci, 2007]. Exercise intolerance with cramps and compensated hemolysis is the main clinical feature, often associated with hyperuricemia. Fixed weakness of late adult onset was prominent in three patients. Myoglobinuria has been documented in 10 of 25 patients. Six children have been reported with early severe myopathy, with or without contractures [Amit et al., 1992; DiMauro and Tsujino, 1994]. One boy also had a cardiomyopathy and died at 21 months of age [Amit et al., 1992]. None of the infantile cases had hemolysis, suggesting different molecular etiologies. Other associated multisystem signs in infants or very young children have included seizures, cortical blindness, corneal opacifications, and cardiomyopathy [DiMauro and Lamperti, 2001; Al-Hassnan et al., 2007]. Four patients had hemolysis without myopathy, and two had normal or only mildly decreased muscle activity and about half-normal erythrocyte PFK activity. This was attributed to instability of the muscle-specific subunit of PFK [Etiemble and Kahn, 1976; Kahn et al., 1975]. Minor clinical differences from McArdle’s disease include more common reports of nausea and vomiting during exercise-induced crises of myalgia, cramps, weakness, and lower frequency of attacks of myoglobinuria [DiMauro and Lamperti, 2001]. As PFK deficiency blocks the metabolism of both muscle glycogen and blood glucose, Haller and Vissing [2004] studied 5 individuals with muscle PFK deficiency and 29 patients with McArdle’s disease during continuous cycle exercise; they found that no PFK-deficient patient developed a spontaneous second-wind phenomenon under conditions that consistently produced one in patients with McArdle’s disease, and concluded that the ability to metabolize blood glucose was critical to the development of a typical spontaneous second wind.
Laboratory data
The serum CK level is usually increased in patients with muscle disease. Hemolysis is indicated by an increased serum bilirubin and moderate reticulocytosis [DiMauro and Tsujino, 1994]. Most patients have an increase in uric acid. EMG may be normal or myopathic. 31P-NMR spectroscopy shows a different pattern from that obtained in McArdle’s disease because, in PFK deficiency, as well as in other defects of distal glycolysis, glycolytic intermediates accumulate in muscle as phosphorylated monoesters and can be detected as a distinct spectral peak [Argov and Bank, 1991].
Inheritance
The gene encoding the muscle (M) subunit is located on chromosome 12 [Howard et al., 1996].
Muscle biopsy
Diagnosis can be made by a negative histochemical PFK stain [Bonilla and Schotland, 1970]. The definitive diagnosis comes from the biochemical analysis, but only if the muscle specimen is snap-frozen at the time of biopsy, as PFK is highly labile and its activity declines rapidly in specimens kept at room temperature or on wet ice; this is why partial PFK deficiencies with residual activities above 10 or 20 percent of normal should be considered skeptically [DiMauro and Lamperti, 2001]. An important differentiating feature from McArdle’s disease is the presence of an abnormal polysaccharide in some cases, particularly older patients [Hays et al., 1981]. This abnormal polysaccharide has the characteristics of polyglucosan and stains intensely with the PAS reaction, but is resistant to diastase digestion; ultrastructurally, it is composed of finely granular and filamentous material, similar to the storage material seen in branching enzyme deficiency and in Lafora’s disease [DiMauro and Lamperti, 2001].
Biochemistry
PFK (ATP:d-fructose 1-phosphotransferase) is a tetrameric enzyme under the control of three structural loci that encode three distinct subunits: namely, M (muscle), L (liver), and P (platelet) [Vora, 1983; Vora et al., 1983a]. The three subunits show variable expression in different tissues. Mature human muscle expresses only the M subunit, whereas erythrocytes express both the M and L subunits, thereby containing five isozymes. In the typical form of PFK deficiency, genetic defects of the M subunit cause total lack of activity in muscle and partial enzyme deficiency in erythrocytes, where the homotetramer L4 is responsible for the residual activity. Other tissues, such as liver, that express predominantly the L subunit are not affected.
Pathophysiology
The functional consequences of PFK deficiency are similar to those observed in McArdle’s disease and are related to the inability of muscle to generate pyruvate [DiMauro and Tsujino, 1994]. With the ischemic exercise test, venous lactate fails to rise [Tarui et al., 1965]. However, PFK deficiency differs significantly from PPL deficiency because of the inability of PFK-deficient muscle to use glucose. Due to this inability, maximal oxygen uptake has been obtained by increasing FFA availability [Haller and Lewis, 1991]. Exercise intolerance is not decreased by glucose administration, which is potentially harmful because it reduces the concentration of FFAs and ketones. This explains why high-carbohydrate meals exacerbate exercise intolerance in patients with PFK deficiency, a situation referred to as the “out-of-wind” phenomenon [Haller and Lewis, 1991]. The excessive exercise-induced degradation of muscle purine nucleotides likely accounts for the hyperuricemia seen in PFK, PPL, and debrancher-deficient patients [Mineo et al., 1987]. Finally, in PFK deficiency, as in McArdle’s disease, exercise triggers increases in heart rate, cardiac output, and blood flow, which are exaggerated relative to the muscle capacity to use oxygen [Lewis et al., 1991].
Molecular genetics
The genes encoding the subunits M, P, and L have been assigned to chromosomes 12, 10, and 21 (see Table 94-1) [Van Keuren et al., 1986; Vora et al., 1982, 1983b]. The M subunit cDNA has been isolated and sequenced [Nakajima et al., 1987], and evidence for tissue-specific alternative mRNA splicing has been documented [Nakajima et al., 1990]. At least 20 mutations responsible for muscle PFK deficiency have now been identified [DiMauro et al., 1995; Sherman et al., 1994; Tsujino et al., 1994d; DiMauro and Lamperti, 2001]. Raben and Sherman [1995] tabulated 15 disease-inducing mutations of the muscle PFK gene and several polymorphisms. These included splicing defects, frameshifts, and missense mutations in patients from six different ethnic backgrounds, supporting genetic heterogeneity of the disease. This disorder appeared to be particularly prevalent among individuals of Ashkenazi Jewish descent. The most frequent mutation in this group was an exon 5 splicing defect that accounted for approximately 68 percent of mutant alleles in the Ashkenazim. Patients with muscle PFK deficiency diagnosed in the United States have been of Ashkenazi Jewish origin, and the several mutations identified in them differ from those described in the non-Ashkenazi Italian or Japanese patients [DiMauro et al., 1995].
Treatment
Glucose is not an alternative substrate in PFK deficiency. A high-protein diet may be beneficial, although this has not been tried [DiMauro and Tsujino, 1994]. A 2-year old boy with the severe infantile form of PFK deficiency, including arthrogryposis multiplex congenita, respiratory insufficiency, slowed motor nerve conduction velocities, and abnormal electroencephalogram (EEG), had a significant improvement in strength, EMG features, and EEG pattern on the ketogenic diet [Swoboda et al., 1997]. Unfortunately, the child died of complications of pneumonia at 35 months.
Phosphoglycerate Kinase Deficiency
Clinical features
PGK deficiency (glycogenosis type IX) presents with nonspherocytic hemolytic anemia and dysfunction of the central nervous system [Boivin et al., 1974; Valentine et al., 1960]. Neurologic problems have included behavioral abnormalities, mental retardation, seizures, and strokes. In hemizygous males, the severe hemolytic anemia manifests soon after birth with jaundice, splenomegaly, and hemoglobinuria. Childhood mortality has been reported in four members of one family [Valentine et al., 1960]. Heterozygous females may be normal or may have mild chronic hemolytic anemia. Myopathic features have included myopathy in males [DiMauro et al., 1981a, 1983a; Rosa et al., 1982; Tonin et al., 1993; Morimoto et al., 2003], with exercise intolerance, cramps, and myoglobinuria. Twenty PGK variants with reduced PGK activity have been identified; myopathy was present in eight of these variants [Tsujino et al., 1995b]. Spiegel et al. [2009] reported an 18-year-old man of Arab Bedouin descent with onset of muscle cramps and recurrent myoglobinuria at age 7 years, having elevated CK, without hemolytic anemia or brain dysfunction, in whom PGK deficiency was documented in muscle and erythrocytes due to a novel mutation, T378P. This was the ninth case presenting with isolated myopathy, whereas most other patients show nonspherocytic hemolytic anemia alone or associated with brain dysfunction and a few patients have myopathy and brain involvement.
Inheritance
PGK is located on the long arm of the X chromosome (see Table 94-1). The defect is expressed in fibroblasts, and prenatal diagnosis is possible [DiMauro and Tsujino, 1994].
Muscle biopsy
PAS stain for glycogen was mildly increased in one patient. Electron microscopy demonstrated glycogen accumulation in all patients [DiMauro and Tsujino, 1994].
Biochemistry
Human PGK is a single polypeptide encoded on the X chromosome for all tissues except spermatogenic cells [DiMauro and Tsujino, 1994]. The complete amino-acid sequence of normal human PGK has been determined [Huang et al., 1980a, 1980b], and mutations have been identified. Patients with hemoglobinuria and those with myopathy demonstrate genetic heterogeneity in enzyme activities. PGK mutants associated with myopathy have been named Creteil [Rosa et al., 1982], New Jersey [DiMauro et al., 1983a], Alberta [Tonin et al., 1993], Hamamatsu [Sugie et al., 1989, 1998], Shizuoka [Fujii et al., 1992], and North Carolina [Tsujino et al., 1994e]. As PGK is a monomer, there are no tissue-specific isoforms, with the exception of PGK2, which is confined to spermatogenic cells; therefore, the defect should be expressed in all tissues. In three patients with myopathy, PGK was significantly decreased in erythrocytes, leukocytes, platelets, fibroblasts, and muscle culture [DiMauro et al., 1983a; Rosa et al., 1982; Tonin et al., 1993].
Pathophysiology
It is difficult to explain the lack of clinical myopathy in many patients with hemolytic anemia, and, conversely, the selective involvement of muscle in other patients [DiMauro and Tsujino, 1994]. However, one patient with PGK Shizuoka has been described with chronic hemolysis and myoglobinuria [Fujii et al., 1992], and a second patient has been described with hemolytic anemia, central nervous system dysfunction, and myopathy [Sugie et al., 1994]. Systematic correlation of molecular genetic, biochemical, and clinical data in future patients may provide insight into the explanation for the striking clinical heterogeneity of PGK deficiency [Tsujino et al., 1995b].
Molecular genetics
A full-length cDNA for the normal PGK gene on the X chromosome has been isolated [Michelson et al., 1983], and genetic heterogeneity has been documented [Tonin et al., 1993]. Seven missense mutations and one splice-junction mutation have been identified in eight patients, three who had myopathy [Fujii et al., 1992; Tsujino et al., 1994e, 1995b; Morimoto et al., 2003]. The testicular isozyme, PGK2, is encoded by a gene on chromosome 19 [DiMauro and Lamperti, 2001].
Phosphoglycerate Mutase Deficiency
Clinical features
PGAM deficiency (glycogenosis type X) has been reported in four men and two women [DiMauro et al., 1981b; DiMauro and Tsujino, 1994]. Exercise intolerance, cramps, and myoglobinuria are the main clinical features.
Muscle biopsy
A variable increase in the PAS reaction has been demonstrated on histochemical examination, and electron microscopy has confirmed mild glycogen accumulation in most cases [DiMauro and Tsujino, 1994].
Biochemistry
PGAM catalyzes the conversion of 3-phosphoglycerate to 2-phosphoglycerate, and is very active in normal human muscle [DiMauro et al., 1981b, 1982]. The glycogen concentration of muscle was normal in four patients and moderately increased in two [DiMauro and Tsujino, 1994]. Insufficient in vitro lactate production has been demonstrated in three patients during anaerobic glycolysis [DiMauro et al., 1981b; Servidei and DiMauro, 1989]. The residual PGAM activity varied from 2.1 to 6 percent of normal. Human PGAM is a dimeric enzyme that consists of different proportions of a slow-migrating muscle (MM) isozyme, a fast-migrating brain (BB) isozyme, and an intermediate hybrid (MB) form in different tissues [Omenn and Cheung, 1974; Omenn and Hermodson, 1975]. The electrophoretic pattern of normal adult human muscle PGAM reveals a marked predominance of the MM band, with only faint BB and MB bands [DiMauro et al., 1981b; DiMauro et al., 1982; Omenn and Cheung, 1974; Omenn and Hermodson, 1975]. The only other tissues containing substantial amounts of the M subunit are heart and sperm, although there is no evidence of cardiomyopathy or male infertility in PGAM deficiency [DiMauro and Lamperti, 2001]. In cardiac muscle, the MM isozyme predominates, although all three bands are seen [DiMauro et al., 1981b; Omenn and Hermodson, 1975]. In most other tissues, such as brain, liver, leukocytes, and erythrocytes, PGAM-BB is the only isozyme on electrophoresis [Omenn and Cheung, 1974; Omenn and Hermodson, 1975].
Pathophysiology
Normal lactate production was demonstrated in one patient during maximal aerobic exercise [Kissel et al., 1985]. 31P-NMR studies revealed exercise-induced accumulation of PME and mild intracellular acidosis [Argov et al., 1987].
Molecular genetics
A full-length cDNA [Shanske et al., 1987] and the genomic clone for PGAM-M [Tsujino et al., 1989] have been isolated and sequenced, and the gene has been localized to chromosome 7 [Edwards et al., 1989]. Tsujino et al. [1993b] identified three distinct point mutations in five patients with PGAM-M deficiency. A full-length cDNA has been identified for PGAM-B [Sakoda et al., 1988]. The gene has been localized to chromosome 10 [Junien et al., 1982].
Lactate Dehydrogenase Deficiency
Clinical features
LDH deficiency (glycogenosis type XI) has been reported in two young men with exercise intolerance and recurrent myoglobinuria [Bryan et al., 1990; Kanno et al., 1980]. It has also been reported in an asymptomatic woman through blood screening [Maekawa et al., 1984]. In addition to muscle symptoms, three affected Japanese women suffered from dystocia, necessitating cesarian sections, and a few patients had dermatologic problems [Kanno and Maekawa, 1995]. Takayasu et al. [1991] reported a 16-year-old Japanese girl who had desquamating erythematosquamous lesions, primarily on the extensor surface of the extremities. The epidermis of the diseased skin and scalp hair follicles were virtually devoid of LDH activity.
Laboratory data
The forearm ischemic exercise test demonstrated an abnormally low increase in venous lactate, contrasting with an excessive increase in pyruvate, in all three patients studied. The muscle biopsy was normal in one patient [Bryan et al., 1990].
Biochemistry
LDH is a tetrameric enzyme composed of two subunits, M (or A) and H (or B), which result in five isozymes: the two homotetramers, M4 and H4, and the three hybrid forms. The M subunit-containing isozymes predominate in skeletal muscle. The H subunit-containing isozymes predominate in heart and other tissues [DiMauro and Tsujino, 1994].
Molecular genetics
The gene encoding LDH-M has been assigned to chromosome 11 [Boone et al., 1972]. Scrable et al. [1990] demonstrated that LDH-M is located in band 11p15.4. The gene encoding LDH-H has been assigned to chromosome 12 [Chen et al., 1973]. Five mutations have been described that lead to muscle LDH deficiency [DiMauro et al., 1995; Maekawa et al., 1990, 1991, 1994].
Branching Enzyme Deficiency
Clinical features
Branching enzyme (brancher) deficiency (glycogenosis type IV) presents in infancy as a rapidly progressive disease dominated by liver dysfunction with hepatosplenomegaly and cirrhosis, death from hepatic failure or gastrointestinal bleeding supervening by 4 years of age [DiMauro and Tsujino, 1994]. Other clinical features include hypotonia, muscle wasting, and contractures [Fernandes and Huijing, 1968; Zellweger et al., 1972]. Cardiomyopathy has also been documented in several children [Farrans et al., 1966; Servidei et al., 1987]. Isolated myopathy has been reported in six patients: three siblings with delayed motor development and chronic proximal weakness [Reusche et al., 1992], a child with myopathy and hepatic involvement [Bruno et al. 1999], and two adults in whom proximal weakness had started at 26 and 49 years of age [Bornemann et al., 1996; Ferguson et al., 1983]. Schroder et al. [1993] reported a case of juvenile type IV glycogenosis, with total branching enzyme deficiency in skeletal muscle and liver, in a male who presented with severe myopathy, dilated cardiomyopathy, heart failure, dysmorphic features, and subclinical neuropathy; the patient died at age 19 of a sudden cardiac death. Alegria et al. [1999] reported hydrops fetalis as a presenting manifestation of GSD type IV. The infant was delivered by cesarean section at 34 weeks and had generalized edema, severe hypotonia, and arthrogryposis of the lower limbs at birth, and died at 4 days of age. Cox et al. [1999] reported three sibling fetuses who were shown to have type IV GSD by pathologic and biochemical studies; they had onset of hydrops, limb contractures, and akinesia in the early second trimester.
This disease shows considerable clinical heterogeneity. The neuromuscular presentation of GSD type IV can be distinguished by age at onset into four groups: perinatal, presenting as fetal akinesia deformation sequence and perinatal death; congenital, with hypotonia, neuronal involvement, and death in early infancy; childhood, with myopathy or cardiomyopathy; and adult, with isolated myopathy or adult polyglucosan body diseases [Bruno et al., 2004; 2007]. Brancher deficiency has been identified in the leukocytes of two Israeli patients with adult polyglucosan body disease [Lossos et al., 1991]. Adult polyglucosan body disease has been described in approximately 15 patients and is characterized by progressive upper and lower motor neuron involvement, sensory loss, sphincter problems, neurogenic bladder, and in some cases, dementia [Bruno et al., 1993; Cafferty et al., 1991]. Burrow et al. [2006] reported a 30-month-old girl with GSD type IV, who had stable congenital hypotonia, gross motor delay, and severe fibro-fatty replacement of the muscles, but no hepatic or cardiac involvement, secondary to compound heterozygosity for two missense mutations in the glycogen branching enzyme gene.
Laboratory data
Carbohydrate tolerance tests and the response of blood glucose to glucagon or epinephrine are normal in patients with liver disease [DiMauro and Tsujino, 1994]. The serum CK is inconsistently increased. Echocardiography showed dilatation and impaired shortening of the left ventricular ejection fraction in one patient [Servidei et al., 1987]. Sensorimotor axonopathy has been demonstrated in patients with adult polyglucosan body disease [Bruno et al., 1993; Cafferty et al., 1991].
Muscle biopsy
Muscle biopsy demonstrates deposits of basophilic, intensely PAS-positive material, which is only partially sensitive to diastase digestion. Electron microscopy reveals storage material consisting of filamentous and finely granular material, often associated with normal-looking glycogen beta particles [DiMauro and Tsujino, 1994]. The abnormal polysaccharide is found in skin, liver, muscle, heart, and the central nervous system. In adult polyglucosan body disease, polyglucosan bodies have been found in both gray and white matter in the processes of neurons and astrocytes. In sural nerve biopsies, the bodies are in the axoplasm, primarily of myelinated fibers and in Schwann cells [Cafferty et al., 1991].
Biochemistry
Branching enzyme catalyzes the last step in glycogen biosynthesis by attaching a short glucosyl chain in an alpha-1,6-glucosidic link to a naked peripheral chain of nascent glycogen [DiMauro and Tsujino, 1994]. The polysaccharide that accumulates has longer than normal peripheral chains and fewer branching points than normal glycogen.
Molecular genetics
Several mutations have been identified in patients with different clinical presentations [Bao et al., 1996; Bruno et al., 1999]. One of these – namely, Tyr329Ser – was present in seven patients with adult polyglucosan body disease [Lossos et al., 1998]. It was concluded that adult polyglucosan body disease represents an allelic variant of GSD type IV. In a non-Ashkenazi patient with adult polyglucosan body disease, Ziemssen et al. [2000] identified compound heterozygosity for the R515H mutation and the R524Q mutation in the glycogen branching enzyme gene, both of which had been found in patients with GSD type IV. The findings confirmed that adult polyglucosan body disease and GSD IV are allelic disorders. In a series of 8 patients with GSD type IV with four different neuromuscular presentations – namely, 3 with fetal akinesia deformation sequence, 3 with congenital myopathy, 1 with juvenile myopathy, and 1 with combined myopathic and hepatic features – nine novel mutations were identified, including nonsense, missense, deletion, insertion, and splice-junction mutations [Bruno et al., 2004]. The three cases with fetal akinesia deformation sequence were homozygous, whereas all the other cases were compound heterozygotes.
Treatment
A number of strategies have been used, including administration of glucagon; long-term use of steroids; a high-protein, low-carbohydrate diet; and administration of fungal alpha-1,4-glucosidase and alpha-1,6-glucosidase. These strategies have been ineffective [DiMauro and Tsujino, 1994]. Liver transplantation was thought to be beneficial in ten children [Selby et al., 1991], although one child developed intractable cardiomyopathy 2 years after transplantation [Sokal et al., 1992].
Aldolase A Deficiency (Glycogenosis Type XII)
Clinical features
This disorder was discovered in 1996 in a 4½-year-old boy who presented with episodic exercise intolerance and weakness following febrile illnesses [Kreuder et al., 1996]. This boy was considered to have myoglobinuria, even though his highest serum CK was 6480 U/L (normal <60 U/L). This boy also had proximal muscle wasting with weakness and episodes of nonspherocytic hemolytic anemia and jaundice.
Biochemistry
Biochemical assays revealed a profound reduction in muscle and red cell aldolase levels and a decrease in the thermostability of the residual enzyme. The enzyme is a tetramer of identical 40,000-dalton subunits. Vertebrates have three aldolase isozymes that are distinguished by their electrophoretic and catalytic properties [Charlesworth, 1972].
Molecular genetics
The aldolase A gene has been mapped to chromosome 16q22–q24 and two pseudogenes were found on chromosomes 3 and 10 [Kukita et al., 1987; Serero et al., 1988]. The patient described by Kreuder et al. [1996] was found to be homozygous for germline mutation in which a negatively charged glutamic acid is changed to a positively charged lysine at residue 206, a residue that is highly conserved within the subunit interface region. Subsequently, Amberger [2008] mapped the ALDOA gene to chromosome 16p11.2, based on an alignment of the ALDOA sequence with the genomic sequence.
Fatty Acid Oxidation Disorders
Historical Background
Fatty acid oxidation (FAO) disorders are an important group of diseases because they potentially are rapidly fatal and a source of major morbidity. They present with a spectrum of clinical disorders, including recurrent myoglobinuria, progressive lipid storage myopathy, neuropathy, progressive cardiomyopathy, recurrent hypoglycemic hypoketotic encephalopathy or Reye-like syndrome, seizures, and mental retardation. All of the currently known conditions are inherited as autosomal-recessive traits. Thus there is frequently a family history of sudden infant death syndrome in siblings. Early recognition, prompt institution of therapy, appropriate preventive measures, and, in certain cases specific therapy, may be life-saving and may decrease long-term morbidity significantly, particularly with respect to central nervous system sequelae. There are now at least 25 enzymes and specific transport proteins in the pathway of β-oxidation, and defects in 18 of them have been recognized as causing disease in humans. With the significant advances in biomedical technology, there has been a rapid increase in the number of subsequently diagnosed cases. For example, in a study that examined 410 consecutive neonatal blood-spot cards in a northern English population for the most common mutation for medium-chain acyl-CoA dehydrogenase (MCAD) deficiency (substitution of a guanine for an adenine at basepair 985 in the MCAD gene), the data suggested a heterozygote carrier state of 1 in 68 for this particular mutation, with a projected homozygote frequency of approximately 1 in 18,500 births [Blakemore et al., 1991; Gregersen et al., 1991]. The incidence for MCAD deficiency was even higher, at 1 in 8930 live births in a more recent survey from the Pennsylvania newborn screening program using tandem mass spectrometry, followed with confirmation through molecular analysis for several common mutations [Ziadeh et al., 1995].
The delay in recognition of these disorders can be attributed to three major factors. First, FAO does not play a major role in energy production until relatively late in fasting. Thus affected individuals may remain clinically silent until they are exposed to periods of fasting beyond 12 hours or to prolonged exercise. In a number of cases, the acute decompensation may be precipitated by an intercurrent infection with fasting, vomiting, and shivering thermogenesis. Second, routine laboratory tests, such as urinary ketones, may not demonstrate the defect in FAO, unless the blood and urine samples are obtained at the time of the acute episode. If they are obtained after the child has received intravenous glucose therapy or has recovered from the acute illness, the defect may be missed. Third, there has been a rapid expansion in the methodologies to identify defects in FAO. For example, methods to identify abnormal fatty acid metabolites in the urine using gas chromatography/mass spectrometry (GCMS) have only been available since the mid-1970s. The identification of certain characteristic organic acid profiles in the acute urine sample has been seminal in the diagnosis of the probable site of defect. Further separation and identification of the individual serum acylcarnitine esters have been facilitated by electrospray ionization-tandem mass spectrometry (ESI-MS/MS) and isotopic exchange high-performance liquid chromatography [Kerner and Bieber, 1983; Millington et al., 1992; Fingerhut et al., 2009]. They are particularly helpful in the diagnosis of long-chain FAO disorders because they overcome the problem of the renal threshold effect, whereby long-chain acylcarnitines are selectively reabsorbed at the renal carnitine transporter site at the expense of free carnitine. This limits the urinary excretion and detection of long-chain acylcarnitines. Serum acylcarnitine measurements also overcome the problem of the poor solubility of long-chain fatty acids in urine.
Fasting Adaptation
Fats constitute the most important and efficient fuel for oxidative metabolism and are the largest reserve of fuel in the body. As liver glycogen stores are depleted within a few hours of a meal and because there are no reserve stores of protein in the body, fatty acids become the predominant substrate for oxidation quite early in fasting [Stanley, 1987]. In adults, fatty acids provide approximately 80 percent of caloric requirements after a 24-hour period of fasting, which increases to 94 percent during more prolonged fasting. Fatty acids serve three major functions. First, the partial oxidation of fatty acids by the liver produces ketones, which are an important auxiliary fuel for almost all tissues and particularly for the brain. As the blood–brain barrier prevents the direct use of long-chain fatty acids by the brain, ketones provide an important mechanism to spare glucose oxidation and proteolysis during prolonged fasting [Cahill, 1970]. Second, fatty acids serve as a major fuel for cardiac and skeletal muscle. Exercising muscle, during mild to moderate prolonged exercise, and resting muscle depend primarily on FAO [Essen, 1978; Felig and Wahren, 1975; Gollnick et al., 1974]. Third, the high rates of hepatic gluconeogenesis and ureagenesis needed for maintaining fasting homeostasis are sustained by the production of energy (ATP), reducing equivalents, and metabolic intermediates derived from FAO [Hale and Bennett, 1992].
Increased Susceptibility of the Child
There are several reasons for an increased risk of problems with fasting adaptation in infants and young children [Aynsley-Green, 1982; Bier et al., 1977; Pildes et al., 1973]. First, given the large ratio of surface area to body mass, basal energy needs in the infant are high to maintain body temperature. Shivering thermogenesis, which maintains body temperature, is highly dependent on efficient FAO. Second, infants have a larger brain compared with body size. The developing brain is highly dependent on glucose and has a high rate of metabolism [Hale and Bennett, 1992]; therefore, infants and children show an even earlier activation of FAO, with hyperketonemia within 12–24 hours of fasting. Third, there is lower activity of several key enzymes involved in energy production in the infant compared with the older child and adult, leading to further impairment of the infant’s ability to maintain glucose homeostasis [Pagliara et al., 1973].
Normal Pathway of Fatty Acid Oxidation
The pathway of mitochondrial FAO is outlined in Figure 94-2 [DiMauro et al., 1992]. FFAs are liberated from adipocytes during fasting and are transported to other tissues either as triglyceride-rich lipoproteins or bound to serum albumin [Stremmel et al., 1986]. Once across the plasma membrane, short- (e.g., 4-carbon) and medium-chain (e.g., 8-carbon) fatty acids of less than 10 carbon atoms are able to cross the outer and inner mitochondrial membranes as free acids to enter the mitochondrial matrix. They then are activated into their CoA esters for intramitochondrial β-oxidation. The mitochondrial membrane is impermeable to long-chain (e.g., 16-carbon) fatty acids. Therefore, long-chain fatty acids diffuse or are transported to the outer mitochondrial membrane and to the endoplasmic reticulum, where they are activated by conversion to their CoA thioesters by long-chain acyl-CoA synthetase [Schulz, 1985]. The inner mitochondrial membrane is impermeable to CoA and its derivatives. Therefore, the long-chain acyl-CoA first must be converted into its acylcarnitine form (e.g., palmitoylcarnitine), with release of free CoA by CPT I, which is located on the inner side of the outer mitochondrial membrane [Murthy and Pande, 1987]. The palmitoylcarnitine is then translocated across the inner mitochondrial membrane by carnitine:acylcarnitine translocase [Pande, 1975]. In the mitochondrial matrix, palmitoylcarnitine, in the presence of free CoA, is then converted back to palmitoyl-CoA and carnitine by CPT II on the inner side of the inner mitochondrial membrane.
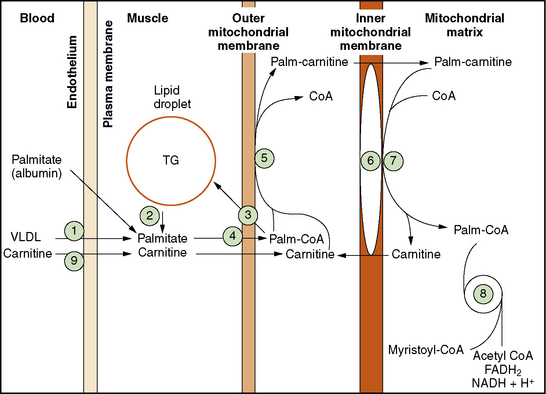
Fig. 94-2 Schematic representation of long-chain fatty acid metabolism in muscle.
(From DiMauro S, et al: In Rowland LP, DiMauro S, editors: Handbook of clinical neurology, vol 18[62], Myopathies, Amsterdam, 1992, Elsevier Science Publishers.)
There are four sequential steps in mitochondrial β-oxidation, which is composed of chain length-specific enzymes. Originally, it was held that the complete intramitochondrial oxidation of long-chain fatty acids required a minimum of nine enzymes, including the three genetically distinct acyl-CoA dehydrogenases (short-, medium-, and long-chain) [Ikeda et al., 1983], at least two enoyl-CoA hydratases (crotonase and long-chain enoyl-CoA hydratase) [Fong and Schulz, 1981], two l-3-hydroxyacyl-CoA dehydrogenases (short- and long-chain) [El-Fakhri and Middleton, 1982], and two 3-ketoacyl-CoA thiolase (acetoacetyl-CoA thiolase and generalized 3-ketoacyl-CoA thiolase) [Middleton, 1975]. Further characterization has revealed the presence of an additional very long-chain acyl-CoA dehydrogenase enzyme [Izai et al., 1992; Kelley, 1992], an acyl-CoA dehydrogenase 9 (ACAD9) that demonstrates maximal activity with unsaturated long-chain acyl-CoAs [He et al., 2007], and of a trifunctional enzyme, which combines the activities of the long-chain enoyl-CoA hydratase, long-chain-l-3-hydroxyacyl-CoA dehydrogenase (LCHAD), and the long-chain thiolase enzymes [Carpenter et al., 1992; Hashimoto, 1992; Luo et al., 1993]. Despite a significant overlap of substrate specificity, it appears that ACAD9 and VLCAD are unable to compensate for each other in patients with either deficiency [He et al., 2007]. Studies of the tissue distribution and gene regulation of ACAD9 and VLCAD identify the presence of two independently regulated functional pathways for long-chain fat metabolism, indicating that these two enzymes are likely to be involved in different physiological functions [He et al., 2007]. ACAD9 is required for the assembly of mitochondrial complex [Nouws et al., 2010]. The trifunctional protein is a heterocomplex of four alpha and four beta subunits, which are encoded by two nuclear genes. The alpha subunit contains the long-chain enoyl-CoA hydratase and LCHAD activities, and the beta subunit contains the long-chain 3-ketoacyl-CoA thiolase activity [Kamijo et al., 1993]. There are two different biochemical phenotypes of trifunctional protein deficiency [Jackson et al., 1992; Kamijo et al., 1994]. The first includes deficiency of all three enzyme activities of the trifunctional protein, with loss of both alpha and beta subunits by immunoblotting studies. The second has isolated LCHAD deficiency with preservation of the long-chain enoyl-CoA hydratase and 3-ketoacyl-CoA thiolase activities and normal amounts of alpha- and beta-subunit proteins.
With each complete cycle of intramitochondrial β-oxidation, a two-carbon fragment is cleaved, and an acetyl-CoA moiety is released. Most tissues, such as muscle and heart, oxidize acetyl-CoA for energy production via the tricarboxylic acid cycle. In liver, approximately 90 percent of the hepatic acetyl-CoA is converted into ketones via the coordinated action of acetyl-CoA acetyltransferase, β-hydroxy β-methylglutaryl-CoA lyase, and β-hydroxybutyrate dehydrogenase [Reed and Lane, 1975]. The ketones are then exported for final oxidation by other tissues, such as brain.
Alternative Pathways for Fatty Acid Metabolism
Mitochondrial β-oxidation accounts for the majority of FAO under normal circumstances. The peroxisome may contribute up to 20 percent of total cellular FAO, under conditions of prolonged fasting, through ω-oxidation leading to the formation of dicarboxylic acids [Krahling et al., 1978]. The specific pattern of DCA in serum or urine may be useful in the identification of specific inborn errors of FAO. When there is impairment in mitochondrial β-oxidation, three additional mechanisms may be important. These include the deacylation of CoA by thioesterases and the conjugation of acyl groups to glycine and to carnitine [Kolvraa and Gregersen, 1986; Roe et al., 1985]. Measurement of acylcarnitines and acylglycines is also diagnostically useful in the identification of genetic defects in FAO [Baretz et al., 1976; Millington, 1986; Rinaldo et al., 1988].
Clinical and Biochemical Features of Identified Defects
Common Features of Fatty Acid Oxidation Disorders
Hale and Bennett [1992] suggest that there are at least four clinical and laboratory features that should lead the clinician to suspect a genetic defect in FAO:
Metabolic decompensation in association with fasting
Children with FAO defects are most prone to decompensation, within the context of depleted glycogen and glucose reserves, during conditions that place stress on the FAO pathway for fuel generation. These conditions include prolonged exercise (particularly after 1 hour of mild to moderate aerobic exercise), fasting, infection with vomiting, and cold-induced shivering thermogenesis. In cold exposure, ketogenesis is stimulated in normal individuals [Johnson et al., 1961], and shivering, which is an involuntary form of muscle activity, is also dependent on long-chain FAO [Bell and Thompson, 1979]. After an overnight fast, children are most likely to be found comatose in the early morning hours. During infection, there may be an added problem with vomiting and decreased oral intake. Children may also present with a Reye-like syndrome. Infants and younger children are at greater risk during fasting because of their decreased abilities for fasting adaptation. Prolonged fasting for an infant of less than 1 year of age would be 6–10 hours, compared with 12 hours for a child between 1 and 4 years of age [Hale and Bennett, 1992].
Involvement of fatty acid oxidation-dependent tissues
Tissues such as skeletal muscle, heart, and liver have high energy demands and are therefore dependent on efficient FAO. When there is a defect in hepatic ketogenesis, glucose becomes the only available fuel and thus becomes rate-limiting under conditions of FAO stress when glycogen and glucose stores have been depleted. As a result, FFAs, which are liberated during fasting and cannot be metabolized because of the block, may be stored in the cytosol as triglycerides. This produces a progressive lipid storage myopathy with weakness, a hypertrophic and/or dilatative cardiomyopathy, and a fatty liver. Increased content of short- or medium-chain fatty acids and, in particular, their dicarboxylic metabolites, from compensatory ω-oxidation, may cause a variety of secondary biochemical abnormalities, including an impairment of gluconeogenesis, β-oxidation, and the citric acid cycle [Corkey et al., 1988; Tonsgard and Getz, 1985; Tonsgard, 1986], leading to a further decrease in cellular ATP production. In addition, in the long-chain FAO disorders, which may have recurrent episodes of acute muscle breakdown or myoglobinuria (e.g., CPT II, VLCAD, trifunctional enzyme deficiencies), the accumulation of long-chain fatty acids and long-chain acylcarnitines may have detergent-like actions on muscle membranes. Excessive amounts of palmitoyl-CoA and palmitoylcarnitine have detergent properties on isolated canine myocytic sarcolemmal membranes and potentiate free radical-induced lipid membrane peroxidative injury in ischemia [Mak et al., 1986]. Long-chain acylcarnitines also activate calcium channels in cardiac [Inoue and Pappano, 1983; Spedding, 1985] and smooth muscle myocytes [Spedding, 1985; Spedding and Mir, 1987]. They may thus potentiate the increase in cytosolic calcium associated with arrhythmogenesis, as seen in ischemic myocardium [Lee et al., 1987].
Alterations in plasma and tissue concentrations of carnitine
In most cases of intramitochondrial β-oxidation defects, the total carnitine concentration is decreased (<50 percent of normal), and the acylcarnitine fraction is increased (>50 percent esterified; normal is 10–25 percent in the fed state and 30–50 percent in the fasted state) [Hale and Bennett, 1992]. In the intramitochondrial β-oxidation disorders, the excessive acyl-CoAs that accumulate proximal to the metabolic block may be converted into acylcarnitines by chain length-specific carnitine acyltransferases [Bremer, 1983]. These acylcarnitines, when filtered through the kidneys, compete with free carnitine at the renal tubular reabsorptive site. As the longer chain-length acylcarnitines have an increasingly higher affinity for the carnitine transporter than free carnitine [Stanley et al., 1991], the free carnitine will be excreted, leading to a decrease in free carnitine in the serum. In the case of the plasmalemmal carnitine transporter defect, the total carnitine is markedly reduced (e.g., <5 percent of normal) and the esterified fraction is normal [Tein et al., 1990a] because the transport defect, which is also expressed in kidney, leads to a decreased renal threshold for carnitine reabsorption.
Additional Laboratory Findings
During acute catabolic crises, other biochemical derangements may be noted. A modest hyperammonemia (100–200 μmol/L), accompanied by 3- to 5-fold elevation of liver transaminases, may be documented during the Reye-like syndrome presentation [Hale and Bennett, 1992]. In acute myoglobinuria, there are marked increases in the sarcoplasmic enzymes, including serum CK, which may rise higher than 100,000 IU/L (normal <250 IU/L). The serum concentrations of amino acids (especially taurine), creatinine, potassium, phosphate, and urate may also be increased [Tein et al., 1990b]. These changes may have deleterious renal and cardiac effects, thereby further exaggerating the damage [Knochel, 1976]. Lactic acidosis may also be noted during the acute catabolic presentations. This may reflect either poor tissue perfusion or inhibition of critical enzymes, such as pyruvate carboxylase, by accumulated metabolites [Corkey et al., 1988]. Urine organic acid screening may demonstrate unusual or excessive amounts of organic acids, which may be diagnostic of a specific block in β-oxidation.
Specific Features of Individual Genetic Defects
For most of the defects in FAO, with the exception of MCAD, the carnitine uptake (OCTN2 transporter), classic late-onset CPT II, VLCAD, LCHAD, α˜electron transfer flavoprotein (ETF), ETF-CoQ, and HMG-CoA lyase deficiencies, there are fewer than 25 reported cases in the literature, although more cases may have been diagnosed. Thus, the clinical and laboratory characteristics presented here are based on a review of these cases and are summarized in Table 94-3 [Tein, 1995, 2000a; Rinaldo et al., 2002].
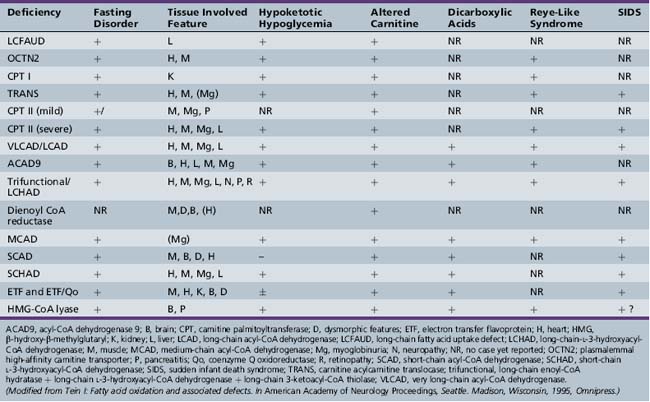
There are several differentiating features. In the defects involving the transport of fatty acids into the mitochondria (carnitine uptake [OCTN2] defect, CPT I, carnitine-acylcarnitine translocase, CPT II), there generally is not an associated abnormal dicarboxylicaciduria. However, this has been reported in one case of severe infantile CPT II deficiency [Elpeleg et al., 1993] and, on one occasion, during an acute metabolic decompensation in an individual with infantile CPT I deficiency, who presented with recurrent Reye-like syndrome [Poll-The et al., 1992]. The carnitine uptake (high-affinity carnitine transporter OCTN2) defect, severe CPT II, and carnitine acylcarnitine translocase defects involve both skeletal and cardiac muscle, and present in infancy or early childhood. The OCTN2 defect is characterized by a progressive hypertrophic and/or dilatative cardiomyopathy, which pathologically resembles endocardial fibroelastosis, limb-girdle myopathy, and recurrent hypoketotic hypoglycemic encephalopathy [Tripp et al., 1981; Tein et al., 1990a]. Importantly, this defect is exquisitely responsive to high-dose oral carnitine supplementation, which reverses the cardiomyopathy and myopathy [Stanley et al., 1991; Tein et al., 1990a]. Renal tubular acidosis has been documented in one case of CPT I deficiency [Falik-Borenstein et al., 1992]. At this time, the only human defect that has been identified for CPT I deficiency is the hepatic and renal CPT1A [Bennett, 2009], which presents with fasting-induced hepatic failure (Reye-like encephalopathy) and hypoketotic hypoglycemia. Notably, the P479L DNA change that was initially described in an Inuit [Brown et al., 2001] has been identified in a large number of Alaskan Inuit and Canadian First Nations individuals [Greenberg et al., 2009]. Of note, these groups have higher than normal rates of infant deaths, and a significant number of newborns identified in the Alaskan newborn screening program have this DNA variant [Gillingham et al., 2006]. It is uncertain whether this is a pathological mutation or an adaptive polymorphism, as the amino-acid substitution removes any inhibition of CPT 1A by malonyl-CoA [Brown et al., 2001], theoretically allowing an individual to generate energy slowly and continuously via ketone body production from fatty acids, even when not fasted, which may be an adaptive advantage in the Inuit during the long winter months when food is scarce and provided there are ample fat stores generated during the summer season [Greenberg et al., 2009]. Fasting in the absence of adequate fat stores, as in the infant, may lead to the high infant mortality rate. Of note, one individual with atypical symptoms and homozygosity for the P479L change, had approximately 20 percent normal L-CPT1 activity and presented with adult-onset recurrent muscle cramps and vomiting, with elevated serum CK (2733 U/L) [Brown et al., 2001].
Classic adult CPT II deficiency is characterized by adolescent-onset recurrent episodes of acute myoglobinuria precipitated by prolonged exercise or fasting, in which power between episodes is normal and in which lipid accumulation in muscle is noted only under conditions of fasting and prolonged exercise [DiMauro and Papadimitriou, 1986]. Fasting ketogenesis generally is normal in this condition, although it may be delayed, and there is no fixed cardiomyopathy, although arrhythmias may occur secondary to hyperkalemia and hypocalcemia during the acute myoglobinuric crises. Statistically speaking, classic CPT II deficiency is the most common cause of recurrent myoglobinuria in children and in adults [Tonin et al., 1990; Tein et al., 1990b]. Cases of severe infantile CPT II deficiency have also been described as presenting with recurrent Reye-like syndrome, hepatomegaly with hypoketotic hypoglycemia and elevated liver aminotransferase, cardiomegaly with cardiac arrhythmias and elevated serum CK, and evidence of lipid storage in the heart, skeletal muscle, liver, and kidney [Demaugre et al., 1991; Elpeleg et al., 1993; Hug et al., 1991]. In these cases, the activity of CPT II in cultured skin fibroblasts was less than 10 percent of control values, in contrast to the 25 percent residual activity documented in classic CPT II deficiency [Demaugre et al., 1991], which may explain the differences in severity of the clinical phenotype. Even more severe mutations may lead to a lethal neonatal form of CPT II deficiency [Isackson et al., 2008].
In addition to the plasmalemmal high-affinity carnitine transporter (OCTN2) defect, there is another plasma membrane FAO disorder: namely, long-chain fatty acid transport/binding defect [Al-Odaib et al., 1998]. This defect in the active transport of long-chain FFAs across the plasma membrane has been described in two young boys, who presented with acute liver failure with hyperammonemia, hyperbilirubinemia, coagulopathy, and mild encephalopathy with no evidence of cardiac or skeletal myopathy, and who required liver transplantation. One boy had documented hypoketotic hypoglycemia. The uptake of oleic acid (C18:1) in skin fibroblasts from the patients was lower than in controls. The features that were atypical of an FAO defect included the absence of fatty liver with low concentrations of long-chain FFAs and elevated carnitine concentrations in liver homogenate, due to defective transport of long-chain FFAs.
The intramitochondrial β-oxidation defects (SCAD, MCAD, LCAD, VLCAD, ETF, ETF-CoQ, SCHAD, M/SCHAD, trifunctional/LCHAD, MCKAT (Medium-chain 3-Ketoacyl CoA thiolase), 2,4-dienoyl-CoA reductase, HMG-CoA synthase, HMG-CoA lyase deficiencies) share many features, although there are differentiating characteristics. Those cases previously attributed to LCHAD deficiency are now known to be related to a defect in the trifunctional enzyme that combines the activities of the long-chain enoyl-CoA hydratase, long-chain l-3-hydroxyacyl-CoA dehydrogenase, and long-chain 3-ketoacyl-CoA thiolase enzymes [Hashimoto, 1992; Sims et al., 1995], though isolated LCHAD deficiency appears to be more common than trifunctional protein deficiency [Rinaldo et al., 2002]. A number of previously diagnosed cases of LCAD deficiency are now believed to be attributable instead to VLCAD deficiency [Aoyama et al., 1995b; Ogilvie et al., 1994; Yamaguchi et al., 1993]. Cardiac involvement (cardiomegaly, cardiomyopathy) is more commonly found in the long-chain defects (e.g., LCAD, VLCAD, trifunctional) [Glasgow et al., 1983; Hale et al., 1985, 1990a, 1990b; Strauss et al., 1995; Tein et al., 1995; Laforet et al., 2009], although it has also been documented in one case of SCHAD deficiency [Tein et al., 1991] and one case of SCAD deficiency [Tein et al., 1999a]. Cardiomyopathy has been documented in 17 of 24 reported cases of trifunctional enzyme deficiency with presumed or proved marked deficiency of the LCHAD component [Tein et al., 1995].
Chronic skeletal muscle weakness is seen in all of the intramitochondrial β-oxidation defects, but less commonly in MCAD [Stanley et al., 1990] and HMG-CoA lyase deficiency [Gibson et al., 1988]. A unique case of SCAD deficiency has been reported in a 13-year-old girl with progressive external ophthalmoplegia, ptosis, cardiomyopathy, and multicore myopathy [Tein et al., 1999a]. Additional cases of SCAD deficiency with multicore myopathy have now been identified [Tein et al., 2008]. Recurrent myoglobinuria has been reported with LCAD [Hale et al., 1990a], VLCAD [Ogilvie et al., 1994; Voermans et al., 2006], trifunctional protein/LCHAD [Dionisi-Vici et al., 1991; Tein et al., 1995], SCHAD [Tein et al., 1991], ACAD9 [He et al., 2007], MCKAT [Kamijo et al., 1997], and MCAD [Ruitenbeek et al., 1995] deficiencies. In a long-term follow-up study of 13 patients with VLCAD deficiency, all patients exhibited exercise intolerance and recurrent myoglobinuria triggered by strenuous exercise, fasting, cold, or fever, with a mean age at onset of 10 years [Laforet et al., 2009]. Muscle biopsies showed mild lipid accumulation in four patients. Perinatal onset of VLCAD deficiency has been reported in a male neonate with fetal distress, neonatal asphyxia, and transient hyper-creatine kinase-emia (8400 IU/L), followed by repeated episodes of myoglobinuria 1–2 times/year during infancy and early childhood [Korematsu et al., 2009]. Ohashi et al. [2004] showed that immunohistochemistry is an effective and useful diagnostic method for the detection of VLCAD deficiency that facilitated the identification of 13 new Japanese patients with VLCAD deficiency. Recurrent myoglobinuria has been reported in trifunctional protein deficiency beginning as young as 7 months of age [Scheuerman et al., 2009]. A first episode of myoglobinuria has also been described in an adult with MCAD deficiency [Ruitenbeek et al., 1995].
Three cases of ACAD9 deficiency have been reported [He et al., 2007]. Patient 1 was a 14-year-old, previously well boy who died of a Reye-like episode and cerebellar stroke, triggered by a mild viral illness and ingestion of aspirin. Patient 2 was a 10-year-old girl who first presented at age 4 months with recurrent episodes of acute liver dysfunction and hypoglycemia with otherwise minor illnesses. Patient 3 was a 4.5-year-old girl who died of cardiomyopathy, and whose sibling died of cardiomyopathy at age 21 months. Mild chronic neurological dysfunction was reported in all three patients.
All intramitochondrial enzyme defects demonstrate hepatic dysfunction at the time of catabolic decompensation. Persistent dysfunction has been documented in trifunctional enzyme [Hagenfeldt et al., 1990], severe CPT II [Elpeleg et al., 1993], and LCAD/VLCAD [Hale et al., 1985] deficiencies. Importantly, isolated LCHAD deficiency in children may also be associated with severe maternal illness in the heterozygous mother occurring during pregnancies with affected fetuses who have a common mutation (G1528C, E474Q) in one or both alleles [Wilcken et al., 1993; Ibdah et al., 1998b; Sims et al., 1995; Treem et al., 1996; Tein, 2000b]. The maternal illnesses include the acute fatty liver of pregnancy (AFLP) syndrome; the hypertension or hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome; and hyperemesis gravidarum. In AFLP syndrome, the mother may suffer fulminant liver failure and death. In both AFLP and HELLP syndromes, there is microvesicular fatty infiltration of the maternal liver. In addition to the recurrent episodes of myoglobinuria, trifunctional protein/LCHAD deficiency may present with progressive pigmentary retinopathy [Tyni et al., 1998; 2004] and motor-sensory neuropathy [Spiekerkoetter et al., 2004]. Thus, individuals with the myoneuropathic variant of trifunctional protein/LCHAD deficiency may mimic the spinal muscular atrophy phenotype. There appear to be three major phenotypes of trifunctional protein/LCHAD deficiency with variable overlap: a hepatic form with Reye-like syndrome that is associated with AFLP; a neonatal cardiomyopathic form; and a less common, milder myoneuropathic form. These phenotypes appear to correlate with specific genotypes and may be a reflection of the severity of the mutation [Sims et al., 1995; Isaacs et al., 1996; Ibdah et al., 1998a, 1999; Bennett, 2009].
Maternal complications in pregnancy are not limited to carriers of LCHAD mutations. Patients with deficiencies of CPT I, MCAD, and SCAD were also born to mothers who developed liver disease during their pregnancies [Matern et al., 2001a; Innes et al., 2000; Nelson et al., 2000; Walter, 2000]. Therefore, all babies born following pregnancies that are complicated by AFLP or HELLP syndrome should be considered at risk for having a defect in β-oxidation. A further complication of pregnancy includes placental floor infarction in the pregnancy of a fetus with LCHAD deficiency [Matern et al., 2001b].
SCAD deficiency is a clinically heterogeneous disorder in which the clinical phenotype varies from fatal metabolic decompensation, including intermittent metabolic acidosis and neonatal hyperammonemia, to infantile-onset lipid storage myopathy with failure to thrive, developmental delay, hypotonia, seizures, and hyperreflexia [Bhala et al., 1995]; to more subtle, later-onset progressive myopathy, including multicore myopathy [Tein et al., 1999a, 2008]; to asymptomatic individuals [Ribes et al., 1998]. The characteristic metabolites of ethylmalonic and methylsuccinic acids in SCAD deficiency may also be due to the presence of one of two relatively common variants of SCAD (625G>A and 511C>T), which predispose to excessive ethylmalonic acid production and also are present in normal populations. These common variants may represent disease susceptibility variations when occurring in combination with other genetic and/or environmental factors, especially fever [Gregersen et al., 1998; Corydon et al., 2001]. They encode proteins with nearly normal enzymatic activity at physiological conditions in vitro; however, SCAD enzyme function is impaired at increased temperature and the tendency to misfold increases under conditions of cellular stress [Pedersen et al., 2008]. We have demonstrated further that vulnerability to oxidative stress may contribute to the pathophysiology of SCAD deficiency in patients homozygous for the common variant 625G>A, 625A/319T compound heterozygous patients, and patients homozygous for the pathogenic 319C>T and 1138C>T mutations [Zolkipli et al., 2009]. Furthermore, the c.319C>T ACADS gene mutation presents with clinical heterogeneity and appears to be a candidate founder mutation in the Ashkenazim [Tein et al., 2008]. The challenge with SCAD deficiency is to elucidate whether ACADS gene variations are disease-associated, especially when combined with other genetic/cellular/environmental factors, such as fever, which may act synergistically [Gregersen et al., 2008].
Multiple acyl-CoA dehydrogenase deficiency or glutaric acidemia type II is due to defects of the electron transfer flavoprotein (ETF) or ETF-ubiquinone oxidoreductase (ETF-Qo) which are nuclear encoded proteins through which electrons from flavoprotein acyl-CoA dehydrogenases, dimethylglycine dehydrogenase, and sarcosine dehydrogenase enter the respiratory chain [Frerman and Goodman, 1989]. These patients can be divided into three groups, each consistent within a family, and which have been designated as:
The first two groups are sometimes said to have severe multiple acyl-CoA dehydrogenase deficiency (MADD) and the third group to have mild MADD. Neonatal-onset patients with congenital anomalies often are premature, presenting within the first 24–48 hours of life with severe hypoglycemia, hypotonia, hepatomegaly, metabolic acidosis, and an odor, similar to sweaty feet. The infants may have enlarged kidneys and facial dysmorphism (high forehead, low-set ears, hypertelorism, hypoplastic mid-face, etc.), rocker-bottom feet, muscular defects of the anterior abdominal wall, and anomalies of the external genitalia (hypospadias, chordee). Most of these patients die with in the first week of life. In others, congenital anomalies are not noted, but renal cysts are found at necropsy. The infants without congenital anomalies usually develop hypotonia, tachypnea, hepatomegaly, hypoglycemia, metabolic acidosis, and a “sweaty feet” odor within the first few days of life, many in the first 24 hours. The few infants with this form who have survived beyond the first week of life, due to prompt diagnosis and treatment, have died within a few months, usually with severe cardiomyopathy. A few other infants have been hypoglycemic in the newborn period and later developed typical Reye-like episodes; they have survived longer. The late-onset type II glutaric acidemia (GA II) is extremely variable in age at presentation and course of the disease. One infant presented at 7 weeks of age with intermittent episodes of vomiting, hypoglycemia, and acidosis, whereas another individual was asymptomatic during childhood and presented in adult life with episodic vomiting, hypoglycemia, hepatomegaly, and proximal myopathy. Some individuals have had progressive lipid storage myopathy with carnitine deficiency. A 12-year-old girl presented with nausea, vomiting, and depression, followed by progressive muscle weakness, elevated CK, and lipid storage myopathy, which responded well to riboflavin, l-carnitine, and a high-calorie, fat-reduced diet [Ishii et al., 2009]. For the riboflavin-responsive-MADD, the challenge is to find the connection between variations in the ETF dehydrogenase gene and the observed deficiency of a number of different mitochondrial dehydrogenases, as well as deficiency of FAD (flavin adenine dinucleotide) and coenzyme Q10 [Gregersen et al., 2008]. The isolated myopathic form of coenzyme Q10 deficiency is caused by mutations in the ETF dehydrogenase gene and can be treated with CoQ10 and riboflavin supplementation [Gempel et al., 2007]. These patients present with exercise intolerance, fatigue, proximal myopathy, and high serum CK, with lipid storage myopathy, subtle signs of mitochondrial myopathy, and severely decreased activities of respiratory chain complexes I and II+III, moderately decreased complex IV, and significantly decreased CoQ10 in muscle. Late-onset GA II and the myopathic form of CoQ10 deficiency may be allelic diseases.
SCHAD deficiency has only been reported rarely. It is also now known as medium- and short-chain l-3-hydroxyacyl-CoA dehydrogenase (M/SCHAD) deficiency, given its broader chain-length specificity [Kobayashi et al., 1996]. Furthermore, it has been argued that it should be called HAD deficiency to distinguish it from the SCHAD enzyme, also known as 17-beta-hydroxysteroid dehydrogenase type 10, that is not a member of the HAD family but instead belongs to the short-chain dehydrogenase/reductase superfamily that acts on a wide spectrum of substrates, including steroids, cholic acids, and fatty acids, with a preference for short-chain methyl-branched acyl-CoAs [Yang et al., 2005]. Three distinct clinical and biochemical phenotypes have been described. One girl presented with recurrent myoglobinuria, myopathy, cardiomyopathy, and hypoketotic hypoglycemic encephalopathy, and died at 16 years of age [Tein et al., 1991]. SCHAD activity was deficient in muscle but not in cultured skin fibroblasts, though these fibroblasts demonstrated marked microvesicular steatosis when cultured in a glucose-free medium to simulate fasting stress [Renaud et al., 2002]. Other tissues were not examined enzymatically. A second phenotype presented with ketotic hypoglycemia, and SCHAD deficiency was described in isolated mitochondria from cultured skin fibroblasts, but no other tissues were studied [Bennett et al., 1996]. A third group of patients presented with fatal infantile hepatic involvement and steatosis, and SCHAD activity was deficient in liver but normal in muscle and fibroblasts [Bennett et al., 1999]. However, Western blot analysis of SCHAD revealed an identical truncated protein in both liver and muscle from one patient, suggesting that SCHAD is similar in liver and muscle and that the normal activity in muscle may be due to other enzymes with C4 activity. Autopsy findings revealed marked steatosis and a muscle pattern consistent with spinal muscular atrophy in one patient. Mutation analysis of the SCHAD gene in patients with SCHAD deficiency has been inconclusive, suggesting that other proteins, in addition to M/SCHAD, may be necessary for enzymatic activity and that these proteins may have a tissue-specific distribution. Apparent disease-causing mutations have been described in a patient with fulminant hepatic failure that required transplantation [O’Brien et al., 2000]. Hyperinsulinism with hypoglycemia has also been reported in SCHAD deficiency [Eaton et al., 2003; Molven et al., 2004]. The pathophysiology of hyperinsulinism in M/SCHAD deficiency is not yet understood fully; however, it is becoming increasingly evident that it results from an interaction of the SCHAD protein with glutamate dehydrogenase [Filling et al., 2008].
Only one patient with 2,4-dienoyl-CoA reductase deficiency in muscle and liver has been reported, who presented with neonatal hypotonia, microcephaly, a small cardiac ventriculoseptal defect, and dysmorphism, and who died at 4 months of age of respiratory acidosis, with biventricular hypertrophy at autopsy [Roe et al., 1990]. The clinical significance of the biochemical abnormalities, therefore, cannot be confirmed until additional patients have been identified. In addition, one patient with medium-chain 3-ketoacyl-coenzyme A thiolase deficiency has been described in a Japanese male neonate, who died at 13 days of age after presenting at 2 days of age with vomiting, dehydration, metabolic acidosis, liver dysfunction, and myoglobinuria [Kamijo et al., 1997].
There are several disorders of ketone body production. Deficiency of 3-hydroxy 3-methylglutaryl-CoA lyase (HMG-CoA lyase), which is also active in the metabolism of leucine, presents with hypoketotic hypoglycemia with hyperammonemia and acidosis [Mitchell and Fukao, 2001]. Succinyl-coenzyme A:3-ketoacid coenzyme A transferase (SCOT) functions in conjunction with mitochondrial acetoacetyl-coenzyme A thiolase to generate ketones in extrahepatic tissues. SCOT deficiency presents as persistent ketonuria in the first 1–2 years of life, while acetoacetyl-coenzyme A thiolase deficiency presents with variable clinical symptoms and exaggerated ketoacidosis in response to minor physiological stresses [Fukao et al., 1995; 1997; Kassovskabratinova et al., 1996; Pretorius et al., 1996]. HMG-CoA synthetase deficiency has been reported in a child who presented with fasting hypoketotic hypoglycemia [Thompson et al., 1997].
The current remaining challenges in mitochondrial FAO defects, such as MCAD, riboflavin-responsive multiple acyl-CoA dehydrogenase, and SCAD deficiencies, are summarized well by Gregersen et al. [2008].
Differentiating Laboratory Features
Analysis of the fatty acid intermediates in the serum or urine of affected children may suggest the site of defect, depending on the chain-length specificity and the species type of the intermediates. These intermediates often require specialized biomedical technology, which may require referral to specialized metabolic laboratories. Particularly useful is the assessment of plasma total and free carnitine, serum acylcarnitines, urine acylcarnitines and urine acylglycines, and organic acids. These intermediates may be absent at times when the child is metabolically stable and receiving adequate supplies of glucose because this would reduce the stress on the FAO pathway. They are best detected in the serum and urine of children during acute catabolic crises or during times of fasting. Specific intermediates characteristic for individual defects are shown in Table 94-4 [Tein, 1995].
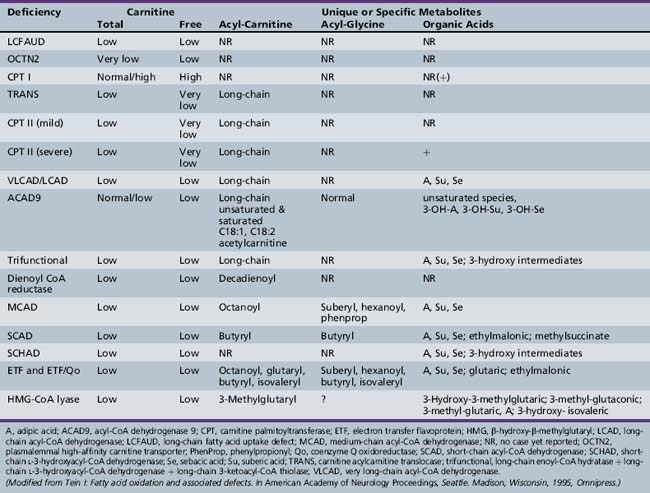
Carnitine
FAO disorders are associated with a decrease in plasma total carnitine concentration (<30 μmol/L; normal is 40–60 μmol/L). The carnitine uptake defect has the lowest concentrations, which are usually less than 5 percent of control values [Stanley et al., 1991]. In the intramitochondrial β-oxidation defects, the plasma total carnitine concentrations vary between 10 and 50 percent of normal. In contrast, in CPT I deficiency, the total plasma carnitine concentration may be normal or increased because the esterification of palmitate to carnitine is defective [Stanley et al., 1992].
In most FAO defects, with the exception of the carnitine uptake and CPT I defects, there is an increase in the ratio of esterified carnitine to total carnitine. This reflects the esterification to carnitine of the excessive acyl-CoAs that accumulate proximal to the block in β-oxidation. Estimates of the amount of acylcarnitines are based on the difference between the free and total carnitine measurements. Under normal conditions, the esterified carnitine is 10–25 percent of total in the fed state, and 30–50 percent of total in the fasted state [Hale and Bennett, 1992]. The carnitine esters can be further separated on the basis of the acid insolubility of long-chain acylcarnitine esters [Hale and Bennett, 1992]. Increased amounts of long-chain acylcarnitines are found in defects of long-chain FAO [Glasgow et al., 1983]. Further separation and identification of the individual acylcarnitine esters have been facilitated by fast atom bombardment-tandem mass spectrometry and isotopic exchange high-performance liquid chromatography [Kerner and Bieber, 1983; Millington et al., 1992]. Profiles of specific serum acylcarnitines have been useful in the diagnosis of certain defects (e.g., octanoyl-carnitine in MCAD deficiency) [Millington, 1986]. They are particularly helpful in the diagnosis of long-chain FAO disorders because they overcome the problem of the renal threshold effect, whereby long-chain acylcarnitines are selectively reabsorbed at the renal carnitine transporter site at the expense of free carnitine. This limits the urinary excretion and detection of long-chain acylcarnitines. Serum acylcarnitine measurements also overcome the problem of the poor solubility of long-chain fatty acids in urine. A novel approach that has been successfully applied to skin fibroblasts for the diagnosis of various FAO defects involves probing the β-oxidation pathway by incubating the cells in the presence of a stable isotopically labeled long-chain fatty acid and then analyzing the produced acylcarnitine intermediates by tandem mass spectrometry [Nada et al., 1995a, 1995b]. This method has also been used for the successful prenatal diagnosis of MCAD and VLCAD deficiencies in cultured amniocytes [Nada et al., 1996].
Furthermore, ESI-MS/MS is used increasingly in newborn screening programs from dried blood spots, which can be used to detect FAO disorders, carnitine cycle disorders, and organic acidurias. Stored dried blood spots are also a valuable source for postmortem investigations to elucidate the cause of sudden infant death syndrome. In a systematic study of the stability of acylcarnitines and free carnitine during storage, it was found that, at −18°C, acylcarnitines are stable for at least 330 days [Fingerhut et al., 2009]. If stored for prolonged periods (>14 days) at room temperature, acylcarnitines are hydrolyzed to free carnitine and the corresponding fatty acids. The velocity of decay is logarithmic and depends on the chain length of the acylcarnitines. Short-chain acylcarnitines hydrolyze faster than long-chain acylcarnitines. It was concluded that stored filter cards should be used for retrospective quantitation of acylcarnitines only if appropriate correction for sample decay during storage is applied. Free carnitine was found to increase on storage but could be quantitated reliably under standardized derivatization conditions. Furthermore, OCTN2 deficiency could be diagnosed reliably by examining acylcarnitine profiles [Fingerhut et al., 2009].
A novel tandem mass spectrometry method for rapid confirmation of medium- and very long-chain acyl-CoA dehydrogenase deficiency in newborns has been developed using liquid chromatography tandem mass spectrometry (LC-MS/MS) to measure MCAD- or VLCAD-catalyzed production of enoyl-CoA and hydroxyacyl-CoA in human lymphocytes [ter Veld et al., 2009].
Dicarboxylic Acids
Dicarboxylic acids (DCAs) (adipic, suberic, sebacic acids) are found in all identified intramitochondrial β-oxidation defects [Gregersen et al., 1982a]. Hale and Bennett [1992] point out that there are several limitations to the value of these compounds in the recognition of FAO defects:
The site of defect may be suggested by the organic acid pattern. For example, children with LCAD/VLCAD deficiency excrete primarily medium- and long-chain-saturated DCA, in contrast to children with trifunctional enzyme deficiency, who excrete almost equimolar amounts of the saturated and the 3-hydroxydicarboxylic acids [Hale et al., 1990a, 1990b]. However, toxic reactions with acetaminophen and intrinsic liver disease may also present with urinary excretion of 3-hydroxy compounds [Pollitt, 1990]. Further advances in stable-isotope dilution mass spectrometry have improved the ability to quantitate metabolites in very small quantities [Gregersen et al., 1986] in plasma or urine. Acylglycines that are consistently excreted in small quantities in the urine do not appear to have the same limitations of DCA. Useful glycine metabolites, therefore, have been identified for several defects, including MCAD, SCAD, ETF, and ETF-CoQ oxidoreductase deficiencies [Rinaldo et al., 1988].
Diagnostic Approaches and Screening Methods
Fasting Studies
If the important samples have not been taken during an acute catabolic event, a fasting study may be considered to distinguish an FAO defect from the other etiologies of hypoglycemia. However, if a fasting study is to be undertaken, it must be done under carefully controlled hospital conditions, with continuous monitoring, and by physicians who are knowledgeable with respect to hypoglycemia, hypopituitarism, hyperinsulinism, and FAO disorders. It is thought by some authorities that fasting studies should not be performed in children with FAO disorders, because diagnostic fasting may precipitate an acute metabolic crisis, leading to further morbidity or death. It has been suggested instead that loading tests with carnitine or phenylpropionate can be used to aid in diagnosis. Hale and Bennett [1992] have outlined the pros and cons of each method. The specific advantages of a fasting study are the following:
Other Studies
Fatty acid oxidation studies
Provided the defect is expressed in cultured skin fibroblasts, a useful screening tool is the measurement of the oxidation rates of [1-14C]-labeled palmitate (C16), octanoate (C8), and butyrate (C4) and deuterium-labeled oleic acid in the fibroblasts to establish the chain-length specificity of the defect [Rhead, 1990; He et al., 2007]. Another useful screening test to suggest an underlying defect in FAO is to culture skin fibroblasts in a glucose-free medium, which will simulate fasting and exacerbate microvesicular steatosis in the fibroblasts expressing a genetic defect in FAO [Renaud et al., 2002]. Another method has been developed for quantitative acylcarnitine profiling by ESI-MS/MS in human skin fibroblasts, using unlabeled palmitic acid as substrate; this has revealed pathognomonic acylcarnitine profiles in a variety of short-, medium-, and long-chain FAO defects [Okun et al., 2002]. In addition, this method delineates different variants of MCAD deficiency, e.g., mild and classical. A rapid method for acylcarnitine profiling has also been described [Tyni et al., 2002], using stable isotopically labeled palmitate and intact mitochondria prepared from fresh-biopsied muscle homogenates, which reduces the delay in diagnosis related to tissue culture.
Enzymatic assays
Depending on the suspected site of defect, a direct enzymatic assay may then be performed for the specific enzyme (Table 94-5) [Tein, 1995]. These assays can be performed in cultured skin fibroblasts or in biopsied muscle specimens for CPT I, CPT II, SCAD, LCAD, SCHAD, and trifunctional enzyme deficiencies, among others. However, certain of these defects may be tissue-specific, such as muscle SCHAD deficiency [Tein et al., 1991]; in this case, the defect is expressed in biopsied muscle and not in fibroblasts, and therefore may be missed unless the affected tissue is examined. Carnitine acylcarnitine translocase deficiency [Pande et al., 1993] and MCAD deficiency can be measured in cultured skin fibroblasts. Evidence for a defect in ETF or ETF-CoQ relies on demonstration of a combined deficiency in the activities of the SCAD, MCAD, and LCAD enzymes. CPT II deficiency has been rapidly diagnosed prenatally by enzyme assay in 10 mg of chorionic villus sampling at the 11th week of gestation, in combination with haplotyping using polymorphic markers linked to the CPT II gene [Vekemans et al., 2003].
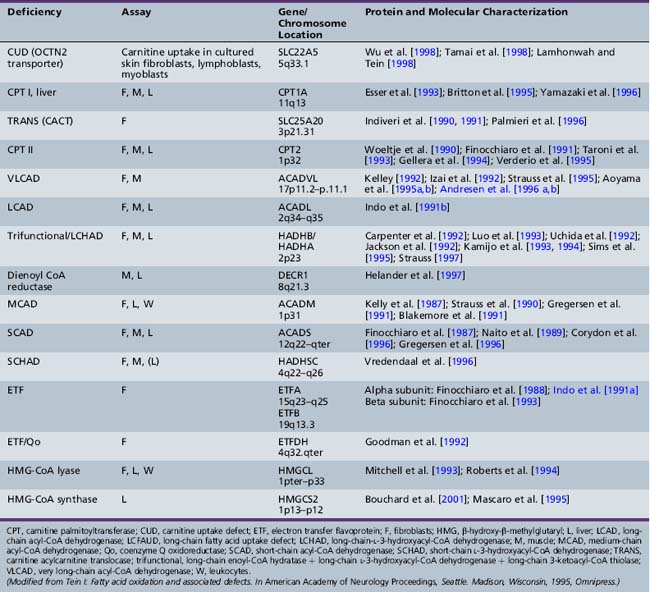
Carnitine uptake studies
For the carnitine uptake defect or OCTN2 deficiency, diagnosis is confirmed by in vitro studies of carnitine uptake in cultured skin fibroblasts. This study demonstrates negligible or minimal uptake of carnitine in the affected homozygote or compound heterozygote patients, thereby precluding the calculation of Km and Vmax values [Tein et al., 1990a; Treem et al., 1988; Lamhonwah et al., 2002]. This supports the concept that primary carnitine deficiency is due to a defect in the specific high-affinity, plasmalemmal carrier-mediated carnitine transporter (OCTN2). Heterozygotes demonstrated normal Km values but reduced Vmax values ranging from 13 to 44 percent of control values [Stanley et al., 1991; Tein et al., 1990a], suggesting that the heterozygotes in this series had a reduced number of normally functioning transporters. This is the most sensitive study for detection of the carrier state, as serum carnitine concentrations in the heterozygotes may be normal. Negligible carnitine uptake has also been demonstrated in the cultured lymphoblasts [Tein and Xie, 1996] and myoblasts [Pons et al., 1997] of affected children.
Molecular studies
Molecular characterization of the specific defects includes Western blotting to determine whether the defects are crossreacting material-positive, suggesting a kinetic deficiency, or whether they are crossreacting material-negative, suggesting a decrease in the production of the affected protein. Western blotting has also been used in the determination of the amounts of the alpha and beta subunits of ETF [Finocchiaro et al., 1990]. A number of the enzymes (e.g., CPT I, CPT II, VLCAD, LCAD, SCAD, MCAD, trifunctional protein) and transporters (e.g., high-affinity plasmalemmal carnitine OCTN2 transporter) have now been cloned (see Table 94-4). This has led to the discovery of specific mutations resulting in the defective activity, which has given rise to the development of specific molecular probes. These probes can now be used for the precise and rapid detection of certain specific defects. Specific mutations have been documented for CPT II [Taroni et al., 1993; Verderio et al., 1995], VLCAD [Andresen et al., 1996a, 1996b; Souri et al., 1996; Strauss et al., 1995], MCAD [Blakemore et al., 1991; Gregersen et al., 1991], SCAD [Corydon et al., 1996; Gregersen et al., 1996; Naito et al., 1990], trifunctional protein [Sims et al., 1995; Strauss, 1997], and carnitine OCTN2 transporter defects [Lamhonwah et al., 2002], among others. The types of mutations may include missense mutations, small amino acid deletions and insertions, truncating mutations, nonsense mutations, out-of-frame deletions and insertions, and splice-site mutations [Gregersen et al., 2001]. The most straightforward application of mutation analysis is to confirm the diagnosis in suspected patients, particularly in the context of family studies and for prenatal/preimplantation analysis.
Treatment
Due to the small number of patients in any given institution, it is difficult to evaluate any single treatment regimen systematically. However, the mainstay of therapy is the avoidance of precipitating factors, particularly prolonged fasting. General treatment strategies are discussed next. Recently, treatment recommendations in long-chain FAO defects have been made by a consensus panel of 21 FAO investigators, based on observations in 75 patients with long-chain FAO defects from 18 metabolic centers in Central Europe [Spiekerkoetter et al., 2009a].
Avoidance of Precipitating Factors
Avoidance of precipitating factors, such as prolonged fasting, prolonged aerobic exercise (30 minutes), and cold exposure leading to shivering thermogenesis, is key. Suggested maximal fasting periods (during stable metabolic conditions) from a European consensus panel are as follows: neonates, 3 hours, also at night; under 6 months, 4 hours, also at night; over 6 months, 4 hours in the daytime, 6(–8) hours at night in well-nourished infants; 12 months to 3 years, 4 hours in the daytime, 10(–12) hours at night; 4–7 years, 4 hours during the day, 10(–12) hours at night [Spiekerkoetter et al., 2009a]. These fasting periods apply under healthy, steady-state conditions, when, on a normocaloric diet, glycogen stores can be built up during the day and used for glucose production during longer fasting intervals at night. In the event of progressive lethargy or obtundation, or an inability to take oral feedings because of vomiting, the child should be taken immediately to the emergency room for intravenous glucose therapy. Intravenous glucose should be provided at rates sufficient to prevent fatty acid mobilization (8–10 mg/kg/min) [Hale and Bennett, 1992]. This regimen should be continued until the catabolic cascade has been reversed and the child is able to take oral feedings again. It is wise to avoid prolonged exercise (i.e., over 30 minutes) because, during this time, there is increased fat mobilization. A high-carbohydrate load before exercise is advisable, with a rest period and repeat carbohydrate loading at 15 minutes. Avoidance of cold exposure to limit shivering thermogenesis is essential.
High-Carbohydrate, Low-Fat Diet
In general, it is advisable to institute a high-carbohydrate, low-fat diet with frequent feedings throughout the day, which would be commensurate with the nutritional needs of the child, given his or her age. This goal is best achieved with the aid of a dietitian, aiming toward approximately 60 percent of calories from carbohydrate sources, 15 percent from protein, and approximately 25–30 percent from fat. Long-chain fat intake in healthy infants is 40–45 percent of total energy and in children of school age it is 30–35 percent, according to German/Austrian/Swiss Nutrition Societies 2000 [Spiekerkoetter et al., 2009a, b]. Monitoring of essential fatty acid levels is important to ensure that the child is receiving adequate essential fatty acids; these may require supplementation. For long-chain FAO defects, dietary long-chain fat should be restricted. Augmentation of the diet with essential fatty acids (at 1–2 percent of total energy intake) is used to reduce the risk of essential fatty acid deficiency [Gillingham et al., 1999; Uauy et al., 1989; Solis and Singh, 2002; Spiekerkoetter et al., 2009a, b]. Flaxseed, canola, walnut, soy, wheatgerm, or safflower oils can be used for this purpose.
An older child should have three regular meals per day with three equidistantly placed intermeal snacks, including a bedtime snack. In younger children, oral or nasogastric tube administration of an appropriate formula is indicated. In HMG-CoA lyase deficiency, a high-carbohydrate, low-fat, low-protein diet with leucine restriction should be implemented [Gibson et al., 1988].
Uncooked Corn Starch
Routine enrichment of the diet with glucose polymers or cornstarch is not recommended, but is part of oral prophylactic and therapeutic emergency management [Spiekerkoetter et al., 2009a]. To delay the onset of fasting overnight, the nightly institution of uncooked corn starch, in doses similar to those used in the treatment of glycogen storage disease (1–2 g/kg body weight/day as a single night-time dose), will prolong the postabsorptive state and delay fasting [Dionisi-Vici et al., 1991]. Cornstarch provides a sustained release source of glucose, thereby preventing hypoglycemia and lipolysis [Fernandes and Smit, 2001]. Cornstarch is usually initiated at 8 months of age when pancreatic enzymes first are able to function at full capacity for appropriate absorption [Hayde and Widhalm, 1990]. The initial recommended doses are generally 1.0 g/kg/day, which can be gradually increased to 1.5–2.0 g/kg/day by age 2 years, as needed [Fernandes and Smit, 2001]. This may lead to undesirable weight gain.
Specific measures for individual FAO disorders include the following.
Medium-chain triglyceride oil
Medium-chain triglyceride oil as a nutritional source could be useful in long-chain FAO disorders because the medium-chain fatty acids would circumvent the block in long-chain FAO and thereby facilitate ATP production from the remainder of the patent FAO pathway. The medium-chain triglyceride oil could be started at a dose of 0.5 g/kg/day, divided in three daily doses, and could be increased up to 1 or 1.5 g/kg/day as tolerated. The major side effect is diarrhea. The usefulness of this approach, however, may be limited because excess medium-chain triglyceride would ultimately be stored as long-chain fats in adipocytes. The success of medium-chain triglyceride oil supplementation in LCHAD deficiency has been variable [Tein et al., 1995]. When a high percentage of energy from fat is provided by medium-chain triglyceride oil, patients are at risk for essential fatty acid deficiency and their diet should therefore be supplemented with essential fatty acid (1–2 percent of total energy intake) [Solis and Singh, 2002].
Riboflavin
Certain cases of the multiple acyl-CoA dehydrogenase deficiencies (e.g., ETF or ETF-CoQ-linked deficiencies) are responsive to riboflavin supplementation [Gregersen et al., 1982b]. The dosage is approximately 50 mg 3 times a day for infants and young children, and 100 mg 3 times a day for older children.
Carnitine
The essential indication for carnitine therapy is the carnitine transporter (OCTN2) defect, which is characterized by carnitine-responsive cardiomyopathy and very low plasma and tissue concentrations of carnitine (generally below 5 percent of normal) [Stanley et al., 1991; Tein et al., 1990a]. There was a dramatic improvement in the cardiomyopathy and myopathy in all 22 patients treated with high-dose oral carnitine supplementation within the first few weeks, as well as a reduction of heart size toward normal within a few months of therapy. In addition, three children with significant failure to thrive before therapy demonstrated a marked improvement in growth after therapy [Tein et al., 1990a]. Of 19 patients treated with carnitine therapy for 5–20 years, 18 continued to be healthy [Stanley et al., 1991; Cederbaum et al., 2002]. Thus, in the carnitine transporter defect, high-dose oral carnitine supplementation at 100 mg/kg/day in four divided daily doses is critical and life-saving, significantly reversing the pathology in this otherwise progressive and lethal disease. Furthermore, if a child is diagnosed prospectively from birth, early carnitine therapy from birth has been shown to prevent the development of the clinical phenotype [Lamhonwah et al., 2002].
In the intramitochondrial β-oxidation defects with secondary carnitine deficiency, the results of carnitine therapy have been highly variable and insufficiently evaluated. Theoretically, carnitine has been given to limit the intracellular concentrations of potentially toxic acyl-CoA intermediates within the cell through transesterification and thereby to liberate CoA, which is a critical intracellular co-factor [Hale and Bennett, 1992]. However, there has been no objective prospective study to prove that carnitine administration has had a beneficial effect. In one study of a patient with MCAD deficiency, Stanley et al. [1990] demonstrated that the associated carnitine deficiency was not the cause of the defect in FAO, as shown by the lack of effect of carnitine replacement on fasting ketogenesis. After 3 months of oral carnitine therapy, this patient had no increase in plasma ketones and still became ill and hypoglycemic after 14 hours of fasting. Furthermore, there is increasing evidence to suggest that carnitine administration may have deleterious effects in the long-chain FAO disorders. In these disorders, there is an accumulation of long-chain acyl-CoAs proximal to the metabolic block, which, upon esterification, become long-chain acylcarnitines. Excessive palmitoylcarnitine may have detergent effects on membranes and arrhythmogenic effects, as previously discussed [Inoue and Pappano, 1983; Lee et al., 1987; Mak et al., 1986; Spedding, 1985; Spedding and Mir, 1987]. This field warrants further investigation. In view of potential adverse effects, supplementation at times of severe metabolic derangement should be avoided.
Specific therapies for lchad/trifunctional protein deficiency
Oral prednisone has been shown to lead to a dramatic reversal of the limb-girdle myopathy and marked reduction in the episodic myoglobinuria in one boy with the myoneuropathic form of LCHAD deficiency [Tein et al., 1995]. Several children with LCHAD deficiency who had associated pigmentary retinopathy were shown to have a deficiency of the n-3-polyunsaturated fatty acid, docosahexaenoic acid (DHA), and subsequent supplementation with DHA led to some improvement in visual function [Gillingham et al., 1997; Harding et al., 1999]. Furthermore, the daily oral administration of a cod liver oil extract containing high amounts of DHA led to a marked clinical and electrophysiological recovery of the progressive peripheral sensorimotor axonopathy in one boy with the myoneuropathic form of LCHAD deficiency [Tein et al., 1999b]. The dose of DHA supplementation recommended by a European consensus panel for trifunctional protein complex, including LCHAD, deficiency is 60 mg/day in children weighing less than 20 kg, and 120 mg/per day in children of over 20 kg body weight [Spiekerkoetter et al., 2009a, b]. Higher amounts of DHA were used to reverse the neuropathy [Tein et al., 1999b].
Triheptanoin
Use of the anaplerotic odd-chain triglyceride, triheptanoic acid, has been reported to be of value in the therapy of long-chain FAO defects [Roe et al., 2002]. In three patients with very long-chain acyl-CoA dehydrogenase deficiency, who were fed controlled diets in which the fat component was switched from medium-even-chain triglycerides to triheptanoin, this treatment led rapidly to clinical improvement that included the resolution of chronic cardiomyopathy, myoglobinuria, and muscle weakness for more than 2 years in one child, and resolution of myoglobinuria and weakness in the others.
Peroxisome proliferator-activated receptor agonists
The peroxisome proliferator-activated receptor agonists (PPARs) have been shown to upregulate mitochondrial FAO [Lee et al., 2003; Feige et al., 2006]. Three different isoforms of these nuclear receptors have been described in mammalian cells – namely, PPARα, PPARδ and PPARγ – which exhibit distinct tissue distributions, ligand specificities, and physiological functions [Lee et al., 2003]. Bezafibrate, a hypolipidemic agent, is the only fibrate capable of activating the PPARδ isoform together with PPARα [Peters et al., 2003; Tenenbaum et al., 2005]. It has been found to increase palmitate oxidation in fibroblasts from patients with a typical mild myopathic phenotype of CPT II or VLCAD deficiency, but had no effect in cells from severely affected patients in whom the mutations were severe and likely involved residues that were essential for the architecture of the active site [Djouadi and Bastin, 2008]. One of the reported adverse effects that should be monitored is possible drug-induced increase in serum CK; however, reports of bezafibrate-induced myoglobinuria are rare, and it has only been described in patients with renal insufficiency who tend to accumulate the drug [Monk and Todd, 1987]. Djouadi and Bastin [2008] recommend that FAO tests in patient fibroblasts may offer a reliable way to assess responsiveness to the drug and, with the combination of clinical, biochemical, and molecular analysis, should provide a rational framework for patient stratification in any future clinical bezafibrate trials in CPT II- or VLCAD-deficient patients. The practical applications of bezafibrate require future studies.
Genetics and Presymptomatic Recognition
One approach to screening would be to evaluate populations that are considered at risk for an FAO defect, such as those manifesting with sudden infant death syndrome, “near-miss” sudden infant death syndrome, and Reye’s syndrome. Bennett et al. [1987] have demonstrated that the incidence of dicarboxylic aciduria in an acutely ill pediatric population is approximately 1 in 12,000 and that, in most cases, the organic acid pattern was consistent with a defect in FAO. In another study of 7058 dried postmortem filter-paper blood-spot specimens obtained at autopsy from infants with unexplained causes of death using electrospray tandem MS/MS analysis of acylcarnitine profiles, there were 23 cases of MCAD deficiency, 9 cases of VLCAD deficiency, 8 cases of GA II, 6 cases of CPT II/translocase deficiencies, 4 cases of severe carnitine deficiency, and 4 cases of LCHAD/trifunctional protein deficiency [Chace et al., 2001].
More recently, specific molecular probes have been used to screen for one of the more common defects, MCAD deficiency. In one study, through use of the specific molecular probe for the gene mutation found in more than approximately 60 percent of U.K. children with MCAD deficiency (A985G point mutation), Blakemore et al. [1991] examined 410 consecutive neonatal blood-spot cards in a northern English population. The data suggested that the carrier state for this mutation is 1 in 68, which would correspond to a frequency of approximately 1 in 18,500 births for the homozygous MCAD-deficient state in this particular population group.
Tandem mass spectrometry has made screening possible for most FAO defects, based upon the profiling of acylcarnitine in blood spots [Rinaldo and Matern, 2000]. The inclusion of FAO disorders in newborn screening programs is highly desirable to prevent morbidity and mortality, implement preventative measures and treatment strategies, and contain the cost of care of affected patients. This is now run in several countries, allowing the detection of asymptomatic newborns long before the onset of clinical manifestations [Shekhawat et al., 2005; Wilcken, 2007; Wilcken et al., 2003]. For example, the incidence for MCAD deficiency was 1 in 8930 live births in a survey from the Pennsylvania newborn screening program, using tandem mass spectrometry followed by confirmation through molecular analysis for several common mutations [Ziadeh et al., 1995].
Mitochondrial Encephalomyopathies
Though all of the mitochondrial encephalomyopathies that are expressed in muscle are at potential risk for the development of myopathy and exercise intolerance, the specific defects in which acute episodes of myoglobinuria have been documented to date include defects of complexes I, II, III, and IV activities, as well as multiple mtDNA deletions, mtDNA depletion syndromes, and coenzyme Q10 deficiency. As there is an entire chapter dedicated to the detailed discussion of the individual mitochondrial disorders (see Chapter 37), the following sections will serve to summarize briefly and to highlight the key clinical, morphological, biochemical, genetic, and physiological features of this group of disorders.
Historical Considerations
In 1962, the concept of mitochondrial disease was first introduced when Luft et al. [1962] reported a young Swedish woman who suffered from severe hypermetabolism that was not due to thyroid dysfunction. Muscle biopsy revealed abnormal mitochondria, and biochemical investigation revealed loose coupling of oxidation and phosphorylation in isolated muscle mitochondria. In the 1960s and 1970s, investigation of muscle biopsies led to the recognition of different patterns of mitochondrial changes [Shy and Gonatas, 1964; Shy et al., 1966]. In 1963, the modification of the Gomori trichrome stain by Engel and Cunningham allowed identification of abnormal deposits of mitochondria as ragged red fibers (RRF). In the 1970s and 1980s, systematic biochemical investigation led to a rational biochemical classification of mitochondrial diseases into five main groups. With advances in mitochondrial genetics, which contain their own DNA (mtDNA), a genetic classification subsequently has been proposed, based on nuclear and/or mitochondrial inheritance. Currently, more than 250 pathogenic mtDNA mutations have been identified [Tuppen et al., 2009]. An expanding number of nuclear DNA (nDNA) mutations are also being reported, as the majority of proteins involved in mitochondrial metabolism and maintenance are encoded by the nucleus. Over the last few years, mutations in POLG, the gene coding for the catalytic subunit of the mtDNA polymerase, polγ, have been recognized as a major cause of human disease, possibly accounting for up to 25 percent of all patients with mitochondrial diseases, with variegate phenotypes including fatigue, muscle weakness, and muscle pain [Horvath et al., 2006a; Chinnery and Zeviani, 2008].
Morphologic Considerations
Although the finding of RRF or ultrastructural alterations of mitochondria in muscle biopsy specimens suggests the possibility of a mitochondrial disorder, there are important limitations. These have been articulated by DiMauro [1993], as follows:
In addition, there are defects of the respiratory chain, such as Leigh’s syndrome secondary to cytochrome c oxidase (COX) deficiency, that tend not to have RRF. Although RRF are most often present in transfer RNA (tRNA) encoding mtDNA defects, which affect the respiratory chain, protein-coding defects, such as neurogenic muscle weakness, ataxia, retinitis pigmentosa (NARP) and Leber’s hereditary optic neuropathy (LHON), have only subtle mitochondrial changes, without RRF [Uemura et al., 1987]. Furthermore, the appearance of RRF may also depend on the stage of the disease and on the threshold effect (the percentage of mutant mtDNA). Succinate dehydrogenase (SDH) and COX stains are two other useful histochemical stains. RRF are often COX-negative, although not all COX-negative fibers are RRF [DiMauro, 1993]. COX-negative RRF are seen in patients with progressive external ophthalmoplegia and mtDNA deletions and in MERRF (myoclonus epilepsy with ragged red fibers) syndrome. They are not seen in MELAS (mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes) syndrome, which usually has COX-positive RRF. A global decrease in the activity of COX is usually suggestive of a nuclear mutation in an ancillary protein required for COX assembly, such as SURF1, but can also be associated with some homoplasmic mitochondrial-tRNA defects [McFarland et al., 2002; Taylor et al., 2003]. There may be increased numbers of mitochondria (pleoconial myopathy), increased size (megaconial myopathy), disoriented or rarefied cristae, or osmiophilic or paracrystalline inclusions [DiMauro, 1993] on electron microscopy. The paracrystalline inclusions are deposits of mitochondrial creatine kinase [Stadhouders et al., 1990]. There may also be lipid or glycogen storage on muscle biopsy, signifying a defect of terminal oxidation [Jerusalem et al., 1973].
Clinical Considerations
The prevalence of mtDNA-related disorders has been estimated only recently. In northeastern England, mtDNA defects were the cause of disease in 6.57/100,000 adults of working age [Chinnery et al., 2000]. Overall, it was estimated that 12.48/100,000 individuals in the adult and child population either had mtDNA disease or were at risk of developing mtDNA disease. In western Sweden, the incidence of mitochondrial diseases overall was 1 in 11,000 preschool children [Darin et al., 2001]. Certain mtDNA mutations are relatively common, such as the A3243G MELAS mutation in northern Finland that was estimated to occur in 16.3 per 100,000 in the adult population [Majamaa et al., 1998]. These prevalence rates support the conclusion that mitochondrial diseases are among the most common metabolic disorders, at least in northern Europe [Vu et al., 2002]. More recently, the prevalence of mtDNA point mutations that cause disease is estimated as 1 in 5000–10,000, and the frequency of mtDNA mutations amongst healthy subjects as 1 in 200 [Schaefer et al., 2004; Elliott et al., 2008].
Mitochondrial diseases are clinically heterogeneous. There may be variation in the age at onset, course, and distribution of weakness in pure myopathies [DiMauro, 1993]. On average, the age of onset reflects the level of mutation and the severity of the biochemical defects; however, other considerations, including nuclear genetic and/or environmental factors, can also affect the expression of disease. Additional features may include exercise intolerance and premature fatigue. In clinical classification, there has been controversy between the “lumpers” and the “splitters.” Originally, three specific clinical syndromes were described: Kearns–Sayre, MERRF, and MELAS syndromes, each resulting from three distinct mutations in mtDNA. All three share the features of short stature, dementia, sensorineural hearing loss, lactic acidosis, and RRF [DiMauro, 1993]. The distinct clinical syndromes were more evident in adults, who had milder phenotypes, in contrast to children, who may present with “overlap” syndromes, in which there may be greater disease severity, as supported by the earlier age of onset. Mitochondria and mtDNA are ubiquitous, which explains why every tissue in the body can be affected by mtDNA mutations. The most common presenting clinical features include short stature, sensorineural hearing loss, migraine headaches, ophthalmoparesis, myopathy, axonal neuropathy, diabetes mellitus, hypertrophic cardiomyopathy, and renal tubular acidosis. Additional features may include strokelike episodes, seizures, myoclonus, retinitis pigmentosa, optic atrophy, ataxia, and gastrointestinal pseudo-obstruction. In the case of mtDNA mutations, there may be a diverse spectrum of associated syndromes, even in a single pedigree, because of heteroplasmy and the threshold effect, whereby different tissues harboring the same mtDNA mutation may be affected to different degrees or not at all. Furthermore, the same mutation can cause different syndromes (e.g., the T8993G mutation can cause either NARP or maternally inherited Leigh’s syndrome [MILS]), and different mutations can cause the same phenotype (e.g., A3243G mutation, single deletion, and multiple deletions of the mtDNA can all cause progressive external ophthalmoplegia [PEO]) [Moraes et al., 1993; Vu et al., 2002]. Thus, the diagnosis of mtDNA-related disorders often requires a careful synthesis of the clinical history, signs, mode of inheritance, laboratory data, neuroradiological findings, exercise physiology, muscle biopsy, biochemistry, and molecular genetics.
Biochemical Classification
Mitochondrial encephalomyopathies can be classified into five groups [DiMauro, 1993], according to the area of mitochondrial metabolism specifically affected:
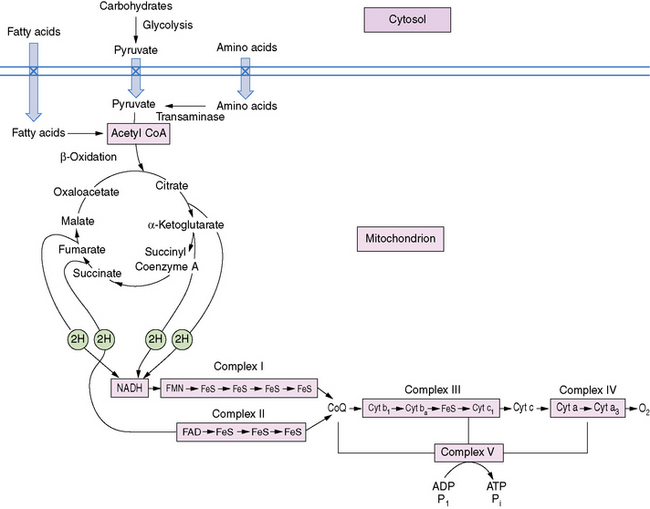
Fig. 94-3 Scheme of mitochondrial metabolism.
(From DiMauro S, DeVivo DC: In Siegel GJ, et al, editors: Basic neurochemistry, ed 4, Molecular, cellular and medical aspects, New York, 1989, Raven Press.)
Limitations of this classification scheme relate to the respiratory chain defects that can result from genetic defects of mtDNA, which usually are heteroplasmic, and to deletions of mtDNA or point mutations in tRNA, which affect mtDNA translation as a whole and may lead to multiple respiratory chain defects to those that can result from genetic defects of nDNA (Figure 94-4) [DiMauro and Schon, 2008].
Fig. 94-4 The mitochondrial respiratory chain showing nDNA-encoded subunits (blue) and mtDNA-encoded subunits.
(Colors correspond to the genes in the map in Figure 94-5.) Protons are pumped from the matrix to the intermembrane space through complexes I, III, and IV, and are pumped back to the matrix through complex V to produce adenosine triphosphate. Coenzyme Q and cytochrome c are electron (e–) transfer carriers. Diseases caused by mutations in mtDNA (above the respiratory chain [RC]) and in nDNA (below the RC) are listed according to the correspondingly affected RC complex. Genes in bold encode RC subunits; those in plain text encode ancillary or assembly proteins. ALS, amyotrophic lateral sclerosis; FBSN, familial bilateral striatal necrosis; IMM, inner mitochondrial membrane; IMS, intermembrane space; LHON, Leber’s hereditary optic neuropathy; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes; MERRF, myoclonic epilepsy with ragged red fibers; MILS, maternally inherited Leigh’s syndrome; NARP, neuropathy, ataxia, retinitis pigmentosa.
(From DiMauro S, Schon E: Mitochondrial disorders in the nervous system, Annu Rev Neurosci 31:91–123, 2008, Fig.2, p.95.)
Genetic Classification
Mitochondria are the only subcellular organelles with their own DNA (mtDNA) [Nass and Nass, 1963] that are capable of synthesizing a vital set of proteins. Human mtDNA is a small (16.5-kb), circular, double-stranded molecule that has been completely sequenced [Anderson et al., 1981]. It encodes 13 structural proteins, all of which are subunits of respiratory chain complexes, as well as 2 ribosomal RNAs (rRNAs) and 22 tRNAs needed for translation.
mtDNA has a number of unique features [DiMauro, 1993]:
In the formation of the zygote, mtDNA is contributed only by the oocyte [Giles et al., 1980]. Therefore, a mother carrying an mtDNA mutation passes it on to all her children, but only her daughters will transmit it to their progeny. If there is a mutation in some mtDNA in the ovum or zygote, this may be passed on randomly to subsequent generations of cells, some of which will receive primarily or exclusively mutant genomes (mutant homoplasmy); others will receive few or no mutant genomes (normal or wild-type homoplasmy); and others will receive a mixed population of mutant and wild-type mtDNAs (heteroplasmy).
Maternal inheritance and heteroplasmy have several important implications [DiMauro, 1993]:
The critical number of mutant mtDNA needed for the threshold effect may vary, depending on the specific vulnerability of the tissue to impairments of oxidative metabolism. It may also vary according to the vulnerability of the same tissue over time, which may increase with age [Moraes et al., 1991a; Ozawa, 1995; Wallace et al., 1988b]. A map of the human mitochondrial genome is shown in Figure 94-5 [DiMauro and Schon, 2008].
Fig. 94-5 The human mitochondrial genome.
(From DiMauro S, Schon E: Mitochondrial disorders in the nervous system, Annu Rev Neurosci 31:91–123, 2008, Fig.1, p.94.)
Although the mtDNA-encoded peptides are functionally important, they represent only a small proportion of the total mitochondrial protein. Of the approximately 80 proteins that make up the respiratory chain, only 13 are encoded by mtDNA. Complexes I, III, IV, and V contain some subunits encoded by mtDNA; seven for complex 1 (ND1–ND4, ND4L, ND5, and ND6), one for complex III (cytochrome b), three for complex IV (COX 1–COX III), and two for complex V (ATPase 6 and ATPase 8) [DiMauro, 2004]. In contrast, complex II, coenzyme Q, and cytochrome c are encoded exclusively by nDNA. Thus, the majority of mitochondrial proteins are encoded by nDNA. The nuclear-encoded proteins are synthesized in the cytoplasm and then imported into mitochondria. This transport of proteins requires a complex series of post-translational events and translocation machinery. Transport involves synthesis of larger precursors in the cytosol, amino terminal leader peptides, which function as address signals and recognize specific mitochondrial membrane receptors. This is followed by translocation across the mitochondrial membrane and cleavage of the leader peptides, with assembly of mature peptides at their final intramitochondrial location [Schatz, 1991].
Therefore, the genetic classification of mitochondrial diseases must take into account defects of nDNA or mtDNA and defects of communication between the two genomes (Box 94-2) [Vu et al., 2002; DiMauro, 2004; DiMauro and Schon, 2008; Rahman and Hanna, 2009; Finsterer et al., 2009]. A summary of the corresponding biochemical defects and the disease phenotypes arising from mutations in certain of the recognized nuclear and mitochondrial genes is shown in Table 94-6 [DiMauro, 2004]. Many nDNA defects have been identified in the past 10 years.
Box 94-2 Genetic Classification of Mitochondrial Respiratory Chain Diseases
Defects of Mitochondrial DNA








Defects of Nuclear DNA (Mendelian Transmission)
(Modified from Vu TH, et al: Mitochondrial diseases, Neurol Clin N Am 20:810, 2002; DiMauro S: Mitochondrial diseases, Biochim Biophys Acta 1658:80, 2004; DiMauro S, Schon EA: Annu Rev Neurosci 31:91–123, 2008; Rahman S, Hanna MG: Diagnosis and therapy in neuromuscular disorders: diagnosis and new treatments in mitochondrial diseases. J Neurol Neurosurg Psychiatry 80:943–953, 2009; Finsterer J, et al: EFNS guidelines on the molecular diagnosis of mitochondrial disorders. Eur J Neurol 16:1255–1264, 2009.)
Due to its small size, screening the entire mitochondrial genome can be undertaken relatively easily for the purposes of identifying rare or novel mtDNA mutations in patients who are suspected of having a mtDNA disease. Denaturing gradient gel electrophoresis [Fischer and Lerman, 1979; Michikawa et al., 1997] and denaturing high-performance liquid chromatography (dHPLC) [van Den Bosch et al., 2000; Oefner and Underhill, 1995] have been available for a number of years, whereas there are emerging technologies with resequencing microarrays, such as the Affymetrix MitoChip 2.0 [Zhou et al., 2006; Hartmann et al., 2009]. Many laboratories continue to sequence the entire mitochondrial genome of 16.5 kb directly in affected tissues [Taylor et al., 2001].
Physiologic Considerations
Standard exercise physiology tests, such as cycle ergometers or treadmills, can be used to detect alterations of oxidative metabolism [Haller et al., 1989; Lewis and Haller, 1991b]. Maximal oxygen uptake is the most useful indicator of a patient’s capacity for oxidative metabolism [DiMauro, 1993]. Typical physiologic responses in patients with defects in oxidative metabolism are as follows [Haller et al., 1989, 1991; Taivassalo et al., 2003]:
Aerobic forearm exercise provides an easily performed screening test that sensitively detects impaired O2 use and accurately assesses the severity of oxidative impairment in patients with mitochondrial myopathy and exercise intolerance. In a study of 13 patients with mitochondrial myopathy and exercise intolerance, the exercise venous PO2 paradoxically rose from 27.2 ± 4.0 mmHg to 38.2 ± 13.3 mmHg, whereas the PO2 fell from 27.2 ± 4.2 mmHg to 24.2 ± 2.7 mmHg in healthy subjects [Taivassalo et al., 2002]. The range of elevated venous PO2 during forearm exercise in the patients (32–82 mmHg) correlated closely with the severity of oxidative impairment, as assessed during cycle exercise. Impaired oxygen extraction by exercising muscles also can be detected by near-infrared spectroscopy, which measures the degree of deoxygenation of hemoglobin [Bank et al., 1998].
Mitochondrial function in muscle in vivo can be evaluated quantitatively using 31P-NMR [Radda et al., 1995]. The ratio of phosphocreatine (PCr) to inorganic phosphate (Pi) can be measured in muscle at rest, during exercise, and during recovery. In patients with mitochondrial dysfunction, PCr/Pi ratios are lower than normal at rest, decrease excessively during exercise, and return to baseline values more slowly than normal [Argov and Bank, 1991].
Defects of mtDNA
Defects in Mitochondrial Protein Synthesis
mtDNA rearrangements: single deletions and duplications
Single deletions of mtDNA have been associated with three conditions that are usually sporadic [DiMauro and Hirano, 2003]:
Duplications of mtDNA can occur in isolation or together with single deletions, and have been documented in patients with Kearns–Sayre syndrome or with diabetes mellitus and deafness [DiMauro, 2004]. Duplications and duplications/deletions are rare and are usually transmitted by maternal inheritance.
Kearns–Sayre Syndrome
The clinical criteria for Kearns–Sayre syndrome are:
plus at least one of the following: heart block, cerebellar ataxia, or a cerebrospinal fluid protein level above 100 mg/dL. Other relatively common but nonspecific features include dementia, sensorineural hearing loss, optic disk pallor, and multiple endocrine abnormalities (short stature, diabetes, hypoparathyroidism) [DiMauro, 1993; Tuppen et al., 2010]. Ragged red fibers and variable COX-negative fibers are seen on muscle biopsy. The prognosis is poor, despite pacemaker placement. The course is progressively downhill, with death by the third or fourth decade. Renal tubular acidosis [Eviatar et al., 1990; Goto et al., 1990b] and Lowe’s syndrome [Moraes et al., 1991b] are unusual clinical variants. A few overlapping cases, such as two children with Kearns–Sayre syndrome and strokelike episodes [Zupanc et al., 1991], have been described. The genetic basis of Kearns–Sayre syndrome is attributable to mtDNA deletions in the great majority of cases [Holt et al., 1989; Moraes et al., 1989].
Sporadic Progressive External Ophthalmoplegia with Ragged Red Fibers
Sporadic progressive external ophthalmoplegia with ragged red fibers is a clinically benign condition, characterized by adolescent- or young adult-onset ophthalmoplegia, ptosis, and proximal limb weakness, which is slowly progressive and compatible with a relatively normal life. The muscle biopsy shows ragged red and COX-negative fibers. Approximately 50 percent of patients with progressive external ophthalmoplegia have mtDNA deletions [Moraes et al., 1989]. The family history is negative.
In terms of therapy, one woman with adult-onset chronic progressive external ophthalmoplegia, ptosis, mild proximal weakness, and muscle mtDNA deletion, who was treated with 7 months of oral lipoic acid, demonstrated an increase in brain energy availability and skeletal muscle performance, as shown by in vivo 31P-NMR spectroscopy [Barbiroli et al., 1995].
Deletions of mtDNA: Molecular Biology
Deletions of mtDNA range from approximately 2 to 8.5 kb and are primarily confined to an 11-kb region [DiMauro, 1993]. Regardless of clinical presentation, 30–40 percent of all deletions are identical. They span 4977 basepairs, from the ATPase 8 gene to the ND 5 gene [Holt et al., 1989; Shoffner et al., 1989; Tanaka et al., 1989]. These deletions can be divided into two main classes [Mita et al., 1990]. The distribution and relative abundance of mtDNA deletions in different tissues appear to determine the clinical phenotype [DiMauro, 1993].
mtDNA point mutations
MERRF Syndrome
MERRF syndrome is characterized by myoclonus, seizures, mitochondrial myopathy, and cerebellar ataxia. Less common signs include dementia, optic atrophy, spasticity, hearing loss, and peripheral neuropathy. Inheritance is maternal; however, expression depends on the percentage of mutant mtDNA. Non-neurologic manifestations include re-entrant atrioventricular tachycardias, such as Wolff–Parkinson–White syndrome, and multiple lipomas [Tuppen et al., 2010]. The highly specific, although not exclusive, point mutation is an A8344G in the tRNALys gene of mtDNA [Shoffner et al., 1990]. This facilitates identification of patients independently of the severity of their clinical phenotype. Genetic heterogeneity is suggested by the absence of the mutation in four patients with typical MERRF syndrome [Hammans et al., 1991; Zeviani et al., 1991]. Other identified mutations in the tRNALys gene that have been associated with MERRF syndrome are the T8356C and G8363A point mutations [DiMauro, 2004]. One patient with MERRF syndrome has been identified with a mutation in a different tRNA: namely, G611A in the tRNAPhe gene [Mancuso et al., 2004a]. Although the genotype–phenotype correlation of the 8344A>G mutation is tighter than that of the other mtDNA mutations, it also causes phenotypes as different as Leigh’s syndrome, isolated myoclonus, familial lipomatosis, or myopathy [Finsterer et al., 2009].
Onset of MERRF syndrome may be in childhood or in adulthood, and the course may be slowly progressive or rapidly downhill. Wallace et al. [1988b] have suggested that there may be a hierarchy of vulnerability of different organs in which the brain suffers first, followed by muscle and heart. However, variations in the sequence of organ involvement may occur from patient to patient [Lombes et al., 1989; Tsairis et al., 1973]. Muscle biopsy shows ragged red fibers, although exceptions occur [Hammans et al., 1991]. There are two populations of mitochondria: those that have normal COX activity and immunoreactivity for subunit II, and those with decreased COX activity and COX II immunoreactivity [Lombes et al., 1989]. Neuropathologic findings include neuronal loss and gliosis affecting, in particular, the dentate nucleus and inferior olivary complex, with some dropout of Purkinje cells and neurons of the red nucleus. There is pallor of the posterior columns of the spinal cord [DiMauro, 1993]. Muscle biochemistry has shown variable defects of complex III [Berkovic et al., 1989; Morgan-Hughes et al., 1982], complexes II and IV [Berkovic et al., 1989], complexes I and IV [Wallace et al., 1988b], or complex IV alone [Lombes et al., 1989]. Combined partial defects of all complexes with subunits encoded by mtDNA have been suggested by studies using human cell lines completely devoid of mtDNA (rho0 cells) [Chomyn et al., 1991; King and Attardi, 1989]. There is no specific therapy, although CoQ10 appeared to benefit a mother and daughter with the MERRF mutation [Wallace et al., 1991].
MELAS Syndrome
This clinical syndrome is characterized by:
In one series, onset was before 15 years of age in 62 percent of patients, and hemianopia or cortical blindness was the most common manifestation [Ciafaloni et al., 1992]. Inheritance is maternal, and there is a highly specific, although not exclusive, point mutation at nt 3243 in the tRNALeu(UUR) gene of mtDNA [Goto et al., 1990a; Kobayashi et al., 1990] which is present in approximately 80 percent of cases [Finsterer et al., 2009]. Other point mutations have also been described. Cerebrospinal fluid protein was abnormally increased in approximately 50 percent of patients [Ciafaloni et al., 1992]. Basal ganglia calcifications were demonstrated in 6 of 22 patients, and dilatative or hypertrophic cardiomyopathy in 4 [Ciafaloni et al., 1992]. Ragged red fibers are usually, but not always, seen on muscle biopsy [Ciafaloni et al., 1992]. Mitochondrial accumulations have been demonstrated in smooth muscle cells of intramuscular vessels [Sakuta and Nonaka, 1989] and of brain arterioles [Ohama et al., 1987], and in epithelial cells and blood vessels of the choroid plexus [Ohama and Ikuta, 1987]. Muscle complex I deficiency has been demonstrated in many cases [Kobayashi et al., 1987; Koga et al., 1988]; however, multiple defects have also been demonstrated involving complexes I, III, and IV [Ciafaloni et al., 1992]. As the number of mutant genomes is lower in blood than in muscle [Ciafaloni et al., 1992], the mutation may not be detectable in blood cells from some patients in whom it is detectable in muscle. In a study of 50 patients with MELAS and the A3243G mutation in tRNALeu (UUR), clinical myopathy was present in 50 percent of patients, but manifested most commonly in the fifth decade, and varied in severity and distribution, including mild to moderate limb weakness, ptosis, and ophthalmoplegia. The 3243A>G mutation may also cause maternally inherited progressive external ophthalmoplegia, Kearns–Sayre syndrome, maternally inherited diabetes and deafness, Leigh’s syndrome, cluster headache, isolated myopathy, cardiomyopathy, renal failure, or pancreatitis [Finsterer et al., 2009]. Two siblings with the T3271C mutation in tRNALeu (UUR) that typically causes MELAS had congenital distal arthrogryposis [McPherson and Zabel, 2006]. This is the second most frequent mtDNA mutation for MELAS [Finsterer et al., 2009]. Other mtDNA genes mutated in MELAS syndrome include the tRNAPhe, tRNAVal, tRNALys, COXII, COXIII, ND1, ND5, ND6 or rRNA genes [Finsterer et al., 2009].
The prognosis in patients with the full syndrome is poor. Therapeutic trials have included corticosteroids [Shapira et al., 1975] and CoQ10 [Yamamoto et al., 1987]. In some patients with severe lactic acidosis, the use of dichloroacetate to lower serum lactate levels has led to significant biochemical [De Stefano et al., 1995] or clinical improvement [De Vivo, personal communication, 1992; Saijo et al., 1991]; however, this has not occurred in all cases. Furthermore, peripheral neuropathy is an important complication of chronic dichloroacetate therapy and should be avoided [Spruijt et al. 2001, Kaufmann et al., 2006].
A long-standing controversy in the mitochondrial field has related to the role of apoptosis in the pathogenesis of mitochondrial myopathies. In a study of muscle biopsies, all showing ragged red fibers from 16 patients with large-scale mtDNA deletions and four with the A3243G MELAS mutation, apoptotic features were present in a small proportion of muscle fibers in 16 of 20 biopsies and apoptosis occurred only in ragged red fibers where there was mitochondrial proliferation with high abundance of mutations [Aure et al., 2006].
More recently, attention has been paid to the effects of l-arginine therapy in MELAS syndrome, particularly in the alleviation and prevention of strokelike episodes [Koga et al., 2005, 2006, 2007], which may also have implications for muscle perfusion. l-arginine, as a precursor of nitric oxide in the reaction catalyzed by nitric oxide synthase, is thought to reduce cerebral ischemic damage by a variety of mechanisms: increasing the rate of cerebral microcirculation by nitric oxide-dependent vasodilatation [Morikawa et al., 1994]; decreasing cytotoxic damage by superoxide and hydrogen peroxide radicals [Wink et al., 1993]; and inhibiting leukocyte–endothelial adherence [Gidday et al., 1998]. It has been shown that mean plasma concentrations of l-arginine and l-citrulline (immediate precursor for arginine synthesis) were lower in both acute and interictal phases of MELAS patients compared to controls [Koga et al., 2005]. Concentrations of l-arginine in the acute phase were also significantly lower than in the interictal phase. Plasma nitric oxide metabolite concentrations were lower in the acute phase of MELAS than in controls, whereas in the interictal phase they were higher than in controls. With intravenous l-arginine treatment in the acute phase of a strokelike episode, all symptoms suggesting stroke improved dramatically, possibly due to enhancement of the microcirculation with reduction of tissue injury from ischemia, and the concentrations of lactate, pyruvate, l-arginine, l-citrulline, and nitric oxide metabolites returned to interictal phase levels within 24 hours. With oral l-arginine supplementation (0.15–0.3 g/kg/day) for 18 months in six MELAS patients, the frequency and severity of symptoms caused by the stroke decreased dramatically; no patient suffered a major strokelike episode, including hemiconvulsion or hemiparesis, but only headache or teichopsia in this time period [Koga et al., 2005]. An improvement in cerebral hypoperfusion, as measured by statistical parametric mapping-single-photon emission computed tomography (SPM-SPECT) scanning, was demonstrated in one patient at the third strokelike episode 2 hours after intravenous l-arginine (0.5 g/kg),which correlated with a dramatic improvement in symptoms [Koga et al., 2006]. In a second patient, SPM-SPECT showed a region of hypoperfusion. Following treatment with daily oral l-arginine (0.15–0.4 g/kg/day), repeat SPM-SPECT performed 60 days after initiation of therapy showed a significant improvement in the previously insufficient regional cerebral blood flow and the patient suffered no strokelike episodes for 2 years after oral administration of l-arginine [Koga et al., 2006]. In further work, these investigators measured flow-mediated vasodilatation (FMD) by brachial artery diameter responses to hyperemic flow (endothelium-dependent) before and after 2 hours of intravenous l-arginine (0.5 g/kg), using high-resolution ultrasound, and demonstrated an improvement in the formerly reduced FMD values in MELAS patients but no change in controls. FMD values after 2 years of oral l-arginine (0.15–0.3 g/kg/day) were improved compared to original values and returned to control levels in all MELAS patients, correlating with normalized plasma l-arginine levels. The vascular sequelae in MELAS syndrome are not limited solely to the cerebral system. There has been some evidence to suggest that intramuscular blood vessel pathology is also contributory [Sakuta and Nonaka, 1989; Hasegawa et al., 1991]. Moreover, l-arginine may increase microperfusion in muscle capillaries, which may improve muscle performance and exercise capacity [Ohta et al., 2007] if vascular flow has been rate-limiting.
Defects of Protein-Coding Genes
ATPase 6 mutation
These maternally inherited disorders present with either Leigh’s syndrome or developmental delay, retinitis pigmentosa, dementia, seizures, ataxia, proximal weakness, and sensory neuropathy, and are caused by a point mutation at nt 8993 within the ATPase 6 gene [Holt et al., 1990]. The severity of disease presentation appears to correlate with the percentage of mutant mtDNA. When the degree of heteroplasmy is moderate (around 70 percent), the clinical expression is NARP, a subacute or chronic disease of young adults; but when the degree of heteroplasmy is very high (about 90 percent), the clinical expression is a rapidly progressive encephalopathy of infancy or childhood – Leigh’s syndrome [Tatuch et al., 1992]. MILS is a more severe infantile encephalopathy with characteristic symmetric lesions in the basal ganglia and the brainstem. MILS patients present with dysmorphism, developmental delay, epilepsy, strokelike episodes, dystonia, ophthalmoparesis, myopathy, or polyneuropathy [Finsterer et al., 2009]. NARP and MILS may coexist in the same family. There are two recognized mutations: namely, T8993G and T8993C, with the T to G transversion being both clinically and biochemically more deleterious than the T to C change [Vázquez-Memije et al., 1998].
Leber’s hereditary optic neuroretinopathy
LHON is characterized by the onset, usually between 18 and 30 years of age, of acute or subacute loss of vision resulting from severe bilateral optic atrophy. There is a marked male predominance [Newman and Wallace, 1990]. The classic ophthalmologic features include circumpapillary telangiectatic microangiopathy and pseudoedema of the optic disc. Associated features may include cardiac conduction abnormalities (pre-excitation syndrome), cerebellar ataxia, hyperreflexia, and peripheral neuropathy. About a dozen different mtDNA point mutations in structural genes have been associated with LHON, but only three appear to be pathogenic, even when present in isolation. A homoplasmic G to A transition at nt 11778, which causes the replacement of a highly conserved arginine residue by histidine at position 340 of the ND4 protein of complex 1, was first described in 11 American pedigrees [Wallace et al., 1988a]. Another point mutation was found, in which a G to A transition at nt 3460 in the ND1 gene was identified in three Finnish families [Huoponen et al., 1991]. A third pathogenic mutation – namely, T14484C in ND6 – has been identified [Carelli, 2002]. All three mutations affect genes of complex I. Predominance of affected men (approximately 50 percent of males but only approximately 20 percent of females have optic atrophy) has been attributed to the influence of the nuclear gene on the X chromosome [DiMauro, 1993]. Complex I deficiency was demonstrated in platelets from four affected members of an Australian family in which the molecular genetic defect was not documented [Parker et al., 1989].
Interestingly, point mutations in mtDNA protein-coding genes often escape the rules of mitochondrial genetics, in that they affect single individuals and single tissues, and most commonly involve skeletal muscle [DiMauro et al., 2003]. For example, patients with exercise intolerance, myalgia, and, occasionally, recurrent myoglobinuria, may have isolated defects of complex I, complex III, or complex IV, due to pathogenic mutations in genes encoding ND subunits, COX subunits, and, in particular, cytochrome b [Andreu et al., 1999]. The lack of maternal inheritance and the involvement of muscle alone would suggest that the mutations arose de novo in myogenic stem cells after germ-layer differentiation (“somatic mutations”) [DiMauro, 2004]. mtDNA COX gene mutations are important causes of exercise intolerance and myoglobinuria. Three young adults have been reported with life-long exercise intolerance, exertional myalgia, and at least one episode of myoglobinuria. All three cases were sporadic, harboring presumably somatic, heteroplasmic mutations confined to skeletal muscle and affecting COX 1 (G6708A) [Kollberg et al., 2005], COX II (T7989C) [McFarland et al., 2004], and COX III (T9789C) [Horvath et al., 2005].
In complex I deficiency, mutations in ND1 and ND4 have been associated with pure myopathy [Hays et al., 2006]. Mutation in ND2 (G4810A) may cause severe exercise intolerance, followed by mild ptosis and progressive external ophthalmoplegia [Pulkes et al., 2005].
Management of mtDNA Disease
Despite significant advances over the last decade in our understanding of mitochondrial genetics and function and the pathogenesis of mtDNA diseases, effective treatment options are limited. The administration of various pharmacologic and biochemical agents, including vitamins, co-factors, metabolites, antioxidants, and electron acceptors to correct or to bypass the underlying respiratory chain defect, have met with limited success [Chinnery et al., 2006]. The current focus is on developing mutation- and disease-specific therapies to ameliorate symptoms and prevent complications. Two main approaches that are being actively pursued are exercise therapy and gene therapy [Tuppen et al., 2010]. Gene therapy is largely in the experimental stages in cellular and animal models.
The primary aim of exercise training is to improve physical capacity and quality of life in patients with high levels of heteroplasmic mtDNA mutations in muscle. Endurance training for up to 14 weeks has been found to improve OXPHOS oxidative phosphorylation function with no significant concomitant shift in mutated mtDNA levels in muscle, and cessation of training leads to a loss of physiological adaptations [Taivassalo and Haller, 2005; Jeppesen et al., 2006; Taivassalo et al., 2006]. This training has been found to be well tolerated and safe. Long-term follow-up training studies are currently under way to determine whether changes in mtDNA mutation load may occur. Resistance training protocols have also been promising. This approach is based on the stimulation of muscle regeneration in response to fiber injury. Satellite cells, which are undifferentiated myogenic cells, are known to proliferate in response to damage caused by concentric (shortening) or eccentric (lengthening) muscle contractions. In individuals with high levels of mutated mtDNA in mature muscle fibers but low or undetectable levels in satellite cells, activation of these stem cells, with subsequent incorporation of the wild-type satellite mtDNA into mature muscle, may help to shift heteroplasmy toward the wild-type, leading to a recovery of muscle function (known as “gene shifting”) [Taivassalo et al., 1999]. Trials of 12 weeks of resistance training in patients with single, large-scale mtDNA deletions have shown increased satellite cell numbers with concurrent improvements in muscular strength and oxidative capacity, as shown by a decrease in the proportion of COX-negative fibers [Murphy et al., 2008]. Further work is needed to determine the optimal exercise paradigms.