Chapter 12 Megaloblastic Anemias
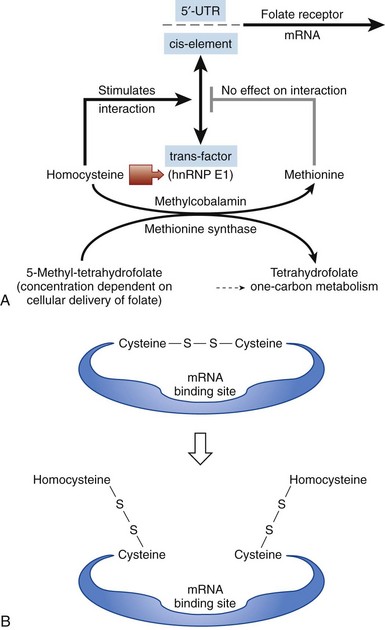
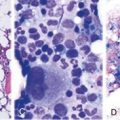
Figure 12-3 MODEL FOR HOW THE CELL SENSES FOLATE DEFICIENCY AND RESPONDS BY UPREGULATING FOLATE RECEPTORS.
(From Tang YS, Khan RA, Zhang Y, et al: Incrimination of heterogeneous nuclear ribonucleoprotein E1 (hnRNP-E1) as a candidate sensor of physiological folate deficiency. J Biol Chem 286:39100, 2011.)

Serum Homocysteine and Methylmalonic Acid Levels in Cobalamin and Folate Deficiencies
The combined use of homocysteine and methylmalonic acid (MMA) levels can differentiate cobalamin from folate deficiency, because most patients with folate deficiency have normal MMA levels, and the remainder have only mild elevations.2 These two tests are useful diagnostically. The abnormally high levels of metabolites return to normal only when the patient receives replacement with the appropriate (deficient) vitamin. A positive response to cobalamin, documented by falling levels of homocysteine and MMA, is evidence of cobalamin deficiency. Conversely, therapy with folate results in a decrease in the isolated homocysteine level if folate deficiency is present.2 Indeed, because several variables that are not related to vitamin deficiency (such as age, mild renal dysfunction) can falsely elevate serum homocysteine and MMA levels, if there is ambiguity, proof of vitamin deficiency would require clear-cut demonstration of a reduction in metabolite levels after specific vitamin supplementation.2,3
Modified Therapeutic Trials
The traditional therapeutic trial using physiologic doses of vitamins (100 mcg of folate or 1 mcg of cobalamin given daily while monitoring the reticulocyte response)1 has given way to a modified therapeutic trial. Rather than making the diagnosis of a deficiency, the intention is often to confirm the clinical suspicion that the patient does not have deficiency. This can be demonstrated by lack of response to full replacement doses of both vitamins (1 mg of folic acid orally for 10 days and 1 mg of cobalamin intramuscularly or subcutaneously daily for 10 days). Clinical scenarios in which such trials may be applicable (after drawing blood for serum cobalamin and folate levels) are as follows:
1. There is a clinical suspicion that the underlying disease is not caused by a vitamin deficiency, but this idea is not supported by results of clinical, morphologic, and biochemical evaluations. Such conditions include anemia with a megaloblastic bone marrow that may be secondary to chemotherapy, myelodysplastic syndromes, or acute leukemia; when time is of the essence in making the diagnosis; when the levels of cobalamin are likely to be falsely abnormal because of these diseases; or when there is underlying dehydration or renal dysfunction that predictably gives falsely high levels of metabolites.
2. In other situations (i.e., pregnancy, acquired immunodeficiency syndrome [AIDS], or alcoholism) with a multifactorial basis for anemia, the response or lack thereof to full replacement doses can eliminate cobalamin or folate deficiency and thereby narrow the (often extensive) differential diagnosis.
3. In instances when severe anemia with megaloblastosis is clinically obvious and so serious that the physician cannot wait for the results of specific tests for deficiency. Full doses of both vitamins are administered, and if there is a response manifested by brisk reticulocytosis by days 5 to 7, retrospective assignment of the deficiency is based on the results of blood samples drawn before beginning the trial.
Table 12-2 Serum Cobalamin: False-Positive and False-Negative Test Results
| FALSELY LOW SERUM COBALAMIN IN THE ABSENCE OF TRUE COBALAMIN DEFICIENCY |
IF, Intrinsic factor; TC, transcobalamin.
*Although a low serum cobalamin level is not synonymous with cobalamin deficiency, 5% of patients with true cobalamin deficiency have low-normal cobalamin levels, a potentially serious problem because the patient’s underlying cobalamin deficiency will progress if uncorrected.
Diagnosing Folate Deficiency
When combined with a clinical picture of megaloblastic anemia and additional results of cobalamin levels, the serum folate concentration is the cheapest and most useful initial biochemical test to diagnose folate deficiency2 (see Table 12-1). The serum folate level is highly sensitive to folate intake, and a single hospital meal may normalize it in a patient with true folate deficiency. Rapidly developing nutritional folate deficiency first leads to a decline in the serum folate level below normal (less than 2 ng/mL) in about 3 weeks; it is a sensitive indicator of negative folate balance.1 However, isolated reduction of serum folate level in the absence of megaloblastosis (i.e., false-positive result) occurs in one-third of hospitalized patients with anorexia, after acute alcohol consumption, during normal pregnancy, and in patients on anticonvulsants2; unfortunately, these are the very groups at high risk for folate deficiency and the people who exhibit low serum folate levels when they become folate deficient.1 Conversely, in 25% to 50% of cases (predominantly alcoholics) with folate-deficient megaloblastosis, the serum folate levels may be low normal or borderline (2 to 4 ng/mL).2 The serum folate level alone should never dictate therapy. It is important to consider the clinical picture, peripheral smear, and bone marrow morphology and also to rule out underlying cobalamin deficiency.
Summary of the Clinical Usefulness of Tests for Cobalamin and Folate Deficiencies
Within the clinical context of hematologic or neurologic features that suggest the diagnosis of cobalamin deficiency, if the cobalamin levels are suggestive but not definitive, then the MMA and homocysteine tests are an excellent gold standard test to confirm a clinical diagnosis. Patients with clinical cobalamin deficiency usually have MMA values over 1000 nM and homocysteine values that are over 25 µM. The MMA and homocysteine test results are much more sensitive than cobalamin levels and progressively increase much earlier than the drop in cobalamin levels; one or both metabolites was increased in 99.8% of more than 400 patients with proven cobalamin deficiency.2
Based on the lower costs of serum cobalamin and folate compared with serum MMA and homocysteine levels, it is recommended (see Table 12-1) to first use the cheaper tests that can assist in the diagnosis of cobalamin and folate deficiency.2 Clinicians should also restrict use of serum MMA and homocysteine to patients with borderline cobalamin and folate levels; to patients with existing conditions associated with difficulties in the interpretation of test results; to situations in which cobalamin and folate levels are low, when a high MMA level is useful in confirming cobalamin deficiency (rather than attributing the condition to folate deficiency alone); and to patients with clearly low serum levels but for whom there is an alternative explanation for the findings that caused an unusual serum cobalamin level to be obtained (e.g., a diabetic or alcoholic with peripheral neuropathy, an alcoholic with a high MCV and a low serum cobalamin without anemia). In these cases, serum levels of metabolites can assist in the diagnosis of vitamin deficiency.
Diagnostic algorithms consistently stress the value of clinical data to improve the pretest probability of serum cobalamin and serum folate tests.2 Without detailed clinical information, the combined test results for serum cobalamin, folate, and metabolite (homocysteine and MMA) are not sufficiently unambiguous to diagnose and distinguish cobalamin deficiency from combined cobalamin-plus-folate deficiency. In combined cobalamin-plus-folate deficiency, both vitamins would be needed to restore baseline values, particularly of homocysteine.2
Table 12-3 Clinical Conditions Not to Be Confused With Megaloblastosis
| MACROCYTOSIS* WITHOUT MEGALOBLASTOSIS† |
MCV, Mean corpuscular volume.
*The central pallor that normally occupies about one-third of the normal red blood cell is decreased in macro-ovalocytes. This contrasts with the finding of thin macrocytes, in which the central pallor is increased.
†Although megaloblastosis implies that a bone marrow test has been performed, with the addition of highly sensitive tests for the specific diagnosis of cobalamin and folate deficiency, the need for a bone marrow test is often dictated by the urgency to make the diagnosis.
‡When the Coulter counter readings of a high MCV are not confirmed by looking at the peripheral smear.
Table 12-4 Beneficial Effects of Homocysteine-Lowering Therapy on Nonhematopoietic Systems
| USING FOLIC ACID, COBALAMIN, PYRIDOXINE SUPPLEMENTATION (GRADE A STUDIES) |
|
Reduction in the progression of carotid intima media thickness5,* (a surrogate marker of early subclinical arteriosclerosis) |
| USING FOLIC ACID SUPPLEMENTATION (GRADE A STUDIES) |
| BENEFICIAL EFFECTS OF FOLIC ACID FORTIFICATION OF FOOD (POPULATION-BASED STUDIES) |
|
Reduction in NTDs15–17 (anencephaly, spina bifida, encephalocele, meningocele, iniencephaly) Reduction in cleft lip with or without cleft palate18 Reduction in severe congenital heart disease18,19 (endocardial cushion defects, conotruncal defects) Reduction in congenital pyloric stenosis, stenosis of the ureteropelvic junction, limb reduction defects18 Reduction in stroke mortality20 Decreased risk for preterm births,21 low birth weight, and small-for-gestational-age babies |
NTD, Neural tube defect.
*Paper with randomized controlled trial data; grade A studies.
Miscellaneous Megaloblastic Anemias Not Caused by Cobalamin or Folate Deficiency
Drugs That Perturb Folate Metabolism
Ethanol. Although beer has a higher folate content than other alcoholic beverages, alcoholism may lead to neglect of healthy dietary practices in favor of alcohol. Patients who have one nutritious meal each day tend to stave off the eventual development of folate deficiency. Alcohol consumption leads to a relatively rapid (2- to 4-day) fall in serum folate levels. Excess alcohol consumption is possibly the most common cause of folate deficiency in the United States.1
Anticonvulsants can induce NTD, and consensus guidelines have stressed the importance of ensuring that pregnant women2 and children22–25 with epilepsy be prescribed folates together with anticonvulsants. Whereas folates protect against spontaneous abortion,26 folic acid supplementation of women receiving antiepileptic drugs, which are known to interfere with folate absorption, also led to a significant reduction of spontaneous abortion.27 Now there is new clinical data on phenytoin-induced gingival hyperplasia, which is a cosmetically undesirable side effect that affects a large percentage of patients, usually between 2 and 6 months of initiating therapy. A recent randomized controlled trial among children 6 to 15 years of age who were initiated on phenytoin has provided incontrovertible evidence that taking folic acid 0.5 mg daily can largely prevent phenytoin-induced gingival hyperplasia14; whereas 88% in the placebo group developed gingival hyperplasia, only 21% in the folic acid group developed this side effect. The data from this paper provides more “ammunition” to encourage young women on antiepileptic drugs to keep taking folic acid to prevent them from getting cosmetically unsightly gingival hyperplasia (particularly if reducing the risk for having a baby with NTD is too nebulous a concept for them). The only caveat is that before initiating long-term folic acid supplements, the cobalamin status must be normalized.
Diagnostic Bone Marrow Aspiration
Is bone marrow aspiration always necessary to diagnose cobalamin- or folate-deficient megaloblastosis? With the addition of highly sensitive serum tests for the specific diagnosis of cobalamin and folate deficiency, the need for a bone marrow test is often dictated by the urgency to diagnose megaloblastosis (with results available in an hour). For example, in the case of florid hematologic disease with or without neurologic disease suggestive of cobalamin or folate deficiency, bone marrow aspiration carried out as soon as possible is invaluable in assisting the rapid diagnosis of megaloblastosis. However, in the outpatient setting, when the patient has a characteristic peripheral smear, or for a patient with a primary neuropsychiatric presentation, a case can be made to initiate the sequence of diagnostic tests without bone marrow aspiration by proceeding with measurement of serum levels of vitamins or metabolites (see Table 12-1). In a pregnant patient with pancytopenia with macro-ovalocytes, hypersegmented PMNs, and reticulocytopenia with a history of noncompliance with prenatal supplements (and no neurologic findings suggestive of cobalamin deficiency), bone marrow aspiration may not be necessary to initiate therapy for a strong presumptive diagnosis of folate deficiency. If there is no evidence of response within 10 days, bone marrow aspiration is indicated.
1 Antony AC. Megaloblastic anemias. In: Hoffman R, Benz EJ, Jr., Shattil SJ, et al, eds. Hematology: Basic principles and practice. ed 1. New York: Churchill-Livingstone; 1991:392.
2 Antony AC. Megaloblastic anemias. In: Hoffman R, Benz EJ, Jr., Shattil SJ, et al, eds. Hematology: Basic principles and practice. ed 4. Philadelphia: Elsevier Churchill Livingstone; 2005:519.
3 Antony AC. Prevalence of cobalamin (vitamin B-12) and folate deficiency in India—audi alteram partem. Am J Clin Nutr. 2001;74:157.
4 Sato Y, Honda Y, Iwamoto J, et al. Effect of folate and mecobalamin on hip fractures in patients with stroke: A randomized controlled trial. JAMA. 2005;293:1082.
5 Hodis HN, Mack WJ, Dustin L, et al. High-dose B vitamin supplementation and progression of subclinical atherosclerosis: A randomized controlled trial. Stroke. 2009;40:730.
6 Christen WG, Glynn RJ, Chew EY, et al. Folic acid, pyridoxine, and cyanocobalamin combination treatment and age-related macular degeneration in women: The Women’s Antioxidant and Folic Acid Cardiovascular Study. Arch Intern Med. 2009;169:335.
7 Smith AD, Smith SM, de Jager CA, et al. Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: A randomized controlled trial. PLoS One. 2010;5:e12244.
8 Wang X, Qin X, Demirtas H, et al. Efficacy of folic acid supplementation in stroke prevention: A meta-analysis. Lancet. 2007;369:1876.
9 Durga J, van Boxtel MP, Schouten EG, et al. Effect of 3-year folic acid supplementation on cognitive function in older adults in the FACIT trial: A randomised, double blind, controlled trial. Lancet. 2007;369:208.
10 Durga J, Verhoef P, Anteunis LJ, et al. Effects of folic acid supplementation on hearing in older adults: A randomized, controlled trial. Ann Intern Med. 2007;146:1.
11 Czeizel AE, Dudas I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N Engl J Med. 1992;327:1832.
12 Berry RJ, Li Z, Erickson JD, et al. Prevention of neural-tube defects with folic acid in China: China-U.S. Collaborative Project for Neural Tube Defect Prevention. N Engl J Med. 1999;341:1485.
13 Grosse SD, Collins JS. Folic acid supplementation and neural tube defect recurrence prevention. Birth Defects Res A Clin Mol Teratol. 2007;79:737.
14 Arya R, Gulati S, Kabra M, et al. Folic acid supplementation prevents phenytoin-induced gingival overgrowth in children. Neurology. 2011;76:1338.
15 Honein MA, Paulozzi LJ, Mathews TJ, et al. Impact of folic acid fortification of the US food supply on the occurrence of neural tube defects. JAMA. 2001;285:2981.
16 De Wals P, Tairou F, Van Allen MI, et al. Reduction in neural-tube defects after folic acid fortification in Canada. N Engl J Med. 2007;357:135.
17 Sayed AR, Bourne D, Pattinson R, et al. Decline in the prevalence of neural tube defects following folic acid fortification and its costbenefit in South Africa. Birth Defects Res A Clin Mol Teratol. 2008;82:211.
18 Czeizel AE. Periconceptional folic acid and multivitamin supplementation for the prevention of neural tube defects and other congenital abnormalities. Birth Defects Res A Clin Mol Teratol. 2009;85:260.
19 Ionescu-Ittu R, Marelli AJ, Mackie AS, et al. Prevalence of severe congenital heart disease after folic acid fortification of grain products: Time trend analysis in Quebec. Canada. BMJ. 2009;338:b1673.
20 Yang Q, Botto LD, Erickson JD, et al. Improvement in stroke mortality in Canada and the United States, 1990 to 2002. Circulation. 2006;113:1335.
21 Bukowski R, Malone FD, Porter FT, et al. Preconceptional folate supplementation and the risk of spontaneous preterm birth: A cohort study. PLoS Med. 2009;6:e1000061.
22 Huemer M, Ausserer B, Graninger G, et al. Hyperhomocysteinemia in children treated with antiepileptic drugs is normalized by folic acid supplementation. Epilepsia. 2005;46:1677.
23 Vilaseca M, Monros E, Artuch R, et al. Anti-epileptic drug treatment in children: Hyperhomocysteinaemia, B-vitamins and the 677C–>T mutation of the ethylenetetrahydrofolate reductase gene. Eur J Paediatr Neurol. 2000;4:269.
24 Apeland T, Mansoor M, Pentieva K, et al. The effect of B-vitamins on hyperhomocysteinemia in patients on antiepileptic drugs. Epilepsy Res. 2002;51:237.
25 Kruman I, Culmsee C, Chan S, et al. Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J Neurosci. 2000;20:6920.
26 George L, Mills JL, Johansson AL, et al. Plasma folate levels and risk of spontaneous abortion. JAMA. 2002;288:1867.
27 Pittschieler S, Brezinka C, Jahn B, et al. Spontaneous abortion and the prophylactic effect of folic acid supplementation in epileptic women undergoing antiepileptic therapy. J Neurol. 2008;255:1926.