Chapter 6 Medical treatment of Parkinson disease
Introduction
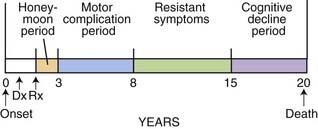
If treatment of Parkinson disease (PD) with levodopa, the most efficacious drug available for this disorder, were uniformly successful and free of complications, there would be no controversies or complexities, and treatment of this disease would be easy. Moreover, treatment with levodopa would begin when the diagnosis of PD was made. But because of levodopa’s propensity to cause motor complications (wearing-off and dyskinesia), which can impair a patient’s quality of life, strategies have been developed to avoid or delay these motor complications. Also, strategies have been developed to overcome these complications once they have appeared. Thus, treatment strategies have evolved to deal with the different phases of the natural history of PD and also the presence of the motor complications. The natural history of PD is one of gradual worsening, not only of the cardinal motor symptoms, but also increasing prominence of nonmotor features; these are discussed in Chapter 8, which covers nonmotor symptoms due to the illness as well as those due to the medication. The natural history of the prototypical person who develops PD is someone who develops the first symptoms around age 60 and lives approximately 20 years, and has clinical problems as illustrated in Figure 6.1. The early stages, including problems of motor complications from medications, are largely treatable. It is the later stages with intractable motor symptoms and dementia that are virtually untreatable at present.
Therapeutic principles
Give priority to any therapies, such as drugs or surgery, that have been established as protective
If any drug could slow the progression of the disease process, it would make sense to use it as soon as the disease is diagnosed. As of this writing, no proven protective or restorative effect of a drug has been demonstrated with certainty. But studies are in progress looking at various agents to determine if they have such an effect. Drugs that have been specifically evaluated in controlled clinical trials for slowing disease progression have been selegiline and tocopherol (Parkinson Study Group, 1989b, 1993a), selegiline alone (Myllyla et al., 1992; Parkinson Study Group, 1996a; Palhagen et al., 1998, 2006), riluzole (Rascol et al., 2003), neuroimmunophilin (NINDS NET-PD Investigators, 2007), coenzyme Q10 (Shults et al., 2002; NINDS NET-PD Investigators, 2007), glial-derived neurotrophic factor (GDNF) (Lang et al., 2006), and rasagiline (Parkinson Study Group, 2004a; Olanow et al., 2009). Two antiapoptic drugs that that were studied in controlled trials were a propargyline that inhibits glyceraldehyde 3-phosphate dehydrogenase (Waldmeier et al., 2000) and an inhibitor of the mixed lineage kinase-3 family that lies upstream of the c-Jun N-terminal kinase signal transduction pathway to apoptotic cell death (Xia et al., 2001). Both studies failed to show benefit (Olanow et al., 2006; Parkinson Study Group PRECEPT Investigators, 2007). Controlled trials with an antibiotic, minocycline, and an energy enhancer, creatine, using a futility design (Tilley et al., 2006), failed to show these drugs to be superior to their comparison placebo group (NINDS NET-PD Investigators, 2006, 2008). The same results were obtained for coenzyme Q10 and neuroimmunophylin (NINDS NET-PD Investigators, 2007). The MAO-B inhibitors, selegiline and rasagiline, have been studied in clinical trials, with positive results suggesting they may modify disease progression, and many neurologists utilize one of these agents at the time of diagnosis, but whether either would delay the long-term, dopa-nonresponsive features is unknown. A discussion of the results of these completed studies is presented in the section “Treatment of early-stage PD.”
Encourage patients to remain active and mobile
PD leads to decreased motivation and increased passivity. An active exercise program, even early in the disease, can often avoid this. Furthermore, such a program involves patients in their own care, allows muscle stretching and full range of joint mobility, and enhances a better mental attitude towards fighting the disease. By being encouraged to take responsibility in fighting the devastations of the disease, the patient becomes an active participant. Physical therapy, which can be implemented in the form of a well-constructed exercise program, is useful in all stages of disease. In early stages, a physical therapy program can instruct the patient in the proper exercises, and the regimen forces the patient to exercise if he or she lacks the motivation to exercise on his or her own. In advanced stages of PD, physical therapy may be even more valuable by keeping joints from becoming frozen, and providing guidance on how best to remain independent in mobility. Therefore, exercise is beneficial in both the early and later stages. It has been shown that PD patients who exercise intensively and regularly have better motor performance (Reuter et al., 1999; Behrman et al., 2000; Craig et al., 2006) and quality of life (Rodrigues de Paula et al., 2006). If exercise is not maintained, the benefit is lost (Lokk, 2000).
A number of basic science studies have discovered that exercise, particularly enriched exercise, can reduce the loss of dopaminergic neurons after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) exposure. When such exercise is initiated shortly after rodents were given experimental lesions of the nigrostriatal dopamine pathway, the result was a significantly less amount of damage to the dopamine pathway (Tillerson et al., 2001, 2002, 2003; Cohen et al., 2003; Bezard et al., 2003; Fisher et al., 2004; Mabandla et al., 2004). The mechanism appears to be the induction of increased trophic factors, such as GDNF (Smith and Zigmond, 2003) and brain-derived neurotrophic factor (BDNF) (Bezard et al., 2003).
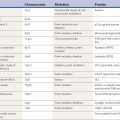
Keep the patient functioning independently as long as possible
There are a number of drugs that have a favorable impact on the clinical features of the disease by reducing its symptoms, but to date none have been shown to stop the progression of the disease. Since PD is a progressive disease and since no medication prevents ultimate worsening, the long-term goal in treating PD is to keep the patient functioning independently for as long as possible. Clearly, if medications that provide symptomatic relief can continue to be effective and without producing adverse effects, this would be excellent. For example, if levodopa therapy could persistently reverse parkinsonian signs and symptoms, we would not have a problem with therapeutic strategy. The difficulty is that 75% of patients have serious complications after 5 years of levodopa therapy (Table 6.1) (Fahn, 1992a), and younger patients (less than 60 years of age) are particularly prone to develop the motor complications of fluctuations and dyskinesias (Quinn et al., 1987; Kostic et al., 1991; Gershanik, 1993; Wagner et al., 1996). Some physicians therefore recommend utilizing dopamine agonists in younger patients, rather than levodopa, when beginning therapy, in an attempt to delay the onset of these problems (Quinn, 1994b; Fahn and Przedborski, 2010). Controlled clinical trials comparing dopamine agonists and levodopa as the initial therapeutic agent have proven that motor complications are less likely to occur with dopamine agonists (Parkinson Study Group, 2000; Rascol et al., 2000; Oertel et al., 2006). But each of these studies also showed that levodopa was more effective in improving parkinsonian symptoms and signs as measured quantitatively by the Unified Parkinson’s Disease Rating Scale (UPDRS) (Fahn and Elton, 1987). But ultimately symptoms develop that are not responsive to levodopa or other dopaminergic agents (Hely et al., 2005, 2008).
Table 6.1 Five major responses to >5 years of levodopa therapy (n = 330 patients)
| No. | % | |
|---|---|---|
| 1.Smooth, good response | 83 | 25 |
| 2.Troublesome fluctuations | 142 | 43 |
| 3.Troublesome dyskinesias | 67 | 20 |
| 4.Toxicity at therapeutic or subtherapeutic dosages | 14 | 4 |
| 5.Total or substantial loss of efficacy | 27 | 8 |
Thirty-six patients had troublesome fluctuations and troublesome dyskinesias.
Data from Fahn S. Adverse effects of levodopa. In Olanow CW, Lieberman AN, eds: The Scientific Basis for the Treatment of Parkinson’s Disease. Carnforth, England: Parthenon Publishing Group, 1992; pp. 89–112.
Individualize therapy
Also, keep in mind that younger patients are more likely to develop motor fluctuations and dyskinesias; older patients are more likely to develop confusion, sleep–wake alterations, and psychosis from medications. We have divided the severity of PD into five stages and describe treatment for each of them except end stage, which is resistant to treatment: early, mild, moderate, advanced, and end stage (Table 6.2).
Table 6.2 Individualizing treatment according to disease severity
|
Mild stage: when PD symptoms and signs begin to interfere with activities, in need of symptomatic treatment
|
Therapeutic choices available for Parkinson disease
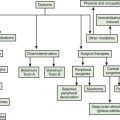
Treatment of patients with PD can be divided into three major categories: physical (and mental health) therapy, medications, and surgery. Physical exercise and physiotherapy were discussed in a previous paragraph. Speech therapy plays a similar role in those with problems of communication. Dysarthria, palilalia, and tachyphemia are difficult to treat, but hypophonia can be overcome by training the patient to shout, known as the Lee Silverman Voice Treatment (Ramig et al., 2001). Psychiatric assistance may be required to handle depression and the social and familial problems that can develop with this chronic, disabling illness. Electroconvulsive therapy (ECT) may have a role in patients with severe, intractable depression; some psychiatrists have been promoting it to help overcome the motor symptoms of PD, but at best ECT provides only short-term motor benefit, and it may not be replicated on repeat treatments. The current practice parameters on treatment of depression, psychosis, and dementia in patients with PD have been summarized in the 2006 report by the American Academy of Neurology Quality Standards Subcommittee (Miyasaki et al., 2006); this topic is covered in Chapter 8.
Neurosurgery for PD is becoming increasingly available as the technique of deep brain stimulation has been developed. This major topic is covered in Chapter 7, and is only mentioned here to be complete in understanding the choices available.
Medications available for Parkinson disease
A great many drugs have been developed for PD. Tables 6.3 through 6.6 classify them according to their mechanisms of action. Selection of the most suitable drugs for the individual patient and deciding when to utilize them in the course of the disease are challenges to the treating clinician. In many of the parkinsonism-plus disorders, the response to treatment is not satisfactory, but the principles for treating PD are the basis for treating these disorders as well. Because PD is a chronic progressive disease, patients require lifelong treatment. Medications and their doses will change over time as adverse effects and new symptoms are encountered. Tactical strategy is based on the severity of the disease.
|
Dopamine precursor: levodopa (combined with carbidopa in immediate-release, extended-release, and dissolvable-in-mouth formulations) (also combined with carbidopa and entacapone)
|
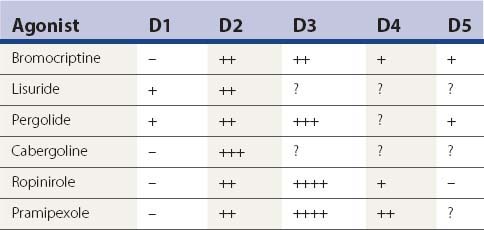
Table 6.5 Nondopaminergic agents for motor symptoms
Table 6.6 Nondopaminergic agents for nonmotor symptoms
| Behavioral |
| Sleep-related |
| Autonomic |
| Gastrointestinal |
Almost all drug trials evaluate a drug’s short-term symptomatic benefit, but the leading unmet need is to stop or slow progression. Dopaminergic medications usually are effective in controlling the early motor symptoms of PD, but ultimately many patients develop new symptoms that do not respond to dopaminergic medication. The Sydney Multicenter Study of Parkinson disease has reported the problems experienced by people who survived 15 years (Table 6.7) (Hely et al., 2005) and 20 years (Table 6.8) (Hely et al., 2008) from diagnosis. The major problems are the development of symptoms that are not responsive to dopaminergic therapy (Tables 6.7 and 6.8), with dementia reaching 81% in 20-year survivors. Only 26% survived 20 years. The standardized mortality ratio, although less than in the pre-levodopa era of 3.0 (Hoehn and Yahr, 1967), was still significantly elevated at 1.86 at 15 years, but was 3.1 between 15 and 20 years. None were employed. Nonmotor symptoms were prevalent.
Table 6.7 Major symptoms after 15 years of PD
| Living in aged care facility – 40% | Employed – 0% |
|---|---|
| Motor symptoms | Nonmotor symptoms |
| Choking – 50% | Symptomatic orthostasis – 35% |
| Falls – 84% | Urinary incontinence – 41% |
| Fractures – 24% | Depression – 50% |
| Motor complications – 95% | Hallucinations – 50% |
| Cognitive decline – 84% | |
| Dementia – 48% |
Data from Hely et al. (2005) on following subjects enrolled in a study comparing bromocriptine and levodopa therapies. Although approximately 95% of subjects have experienced dopa-induced dyskinesia/dystonia and wearing-off, in the majority these symptoms were not disabling. Dyskinesia and dystonia were delayed by early use of bromocriptine, but wearing-off appeared at a similar time once levodopa was added. The most disabling long-term problems of PD relate to the emergence of symptoms that are not improved by levodopa.
Table 6.8 Major symptoms after 20 years of PD
| Living in aged care facility – 48% | Living independently – 1/36 |
|---|---|
| Motor symptoms | Nonmotor symptoms |
| Choking – 48% | Symptomatic orthostasis – 48% |
| Falls – 87% | Urinary incontinence – 71% |
| Fractures – 35% | Depression – 70% |
| Freezing – 81% | Hallucinations – 74% |
| Moderate dysarthria – 81% | Dementia – 83% |
| Excessive daytime sleepiness – 70% |
Data from Hely et al. (2008) on following subjects enrolled in a study comparing bromocriptine and levodopa therapies. The most disabling long-term problems of PD relate to the emergence of symptoms that are not improved by levodopa, particularly dementia.
Dopaminergic agents
Because most of the major motoric symptoms of PD are related to striatal dopamine deficiency (Hornykiewicz, 1966), dopamine replacement therapy is the major medical approach to treating these features of the disease. Table 6.3 lists these dopaminergic drugs. The most powerful drug is levodopa. It is usually administered with a peripheral decarboxylase inhibitor. In Table 6.3, both carbidopa and benserazide are listed as peripheral dopa decarboxylase inhibitors, although in the United States only carbidopa is available. In many other countries, benserazide is also available. Carbidopa/levodopa is marketed as Sinemet or as a generic drug; the combination is available in immediate-release (e.g., Sinemet standard) and extended-release (e.g., Sinemet CR) formulations. The former allows a more rapid “on” and shorter half-life, and the latter allows for a delayed “on” and a slightly longer plasma half-life. Benserazide/levodopa is marketed as standard Madopar and Madopar HBS (for slow release). The peripheral decarboxylase inhibitors potentiate levodopa, allowing about a four-fold reduction of levodopa dosage to obtain the same benefit. Moreover, by preventing the formation of peripheral dopamine, which can act at the area postrema (vomiting center with a lack of a blood–brain barrier), they block the development of nausea and vomiting. If additional carbidopa is needed for patients in whom nausea persists, it can be prescribed, and patients can obtain it from their pharmacy; the additional peripheral decarboxylase inhibitor may overcome the nausea. Keep in mind that levodopa is absorbed only in the proximal small intestine. The slow release of levodopa from the extended-release versions is such that only about two-thirds to three-quarters of levodopa is absorbed per tablet compared to standard Sinemet. This is because some of the levodopa in the slow-dissolving tablet has not been released before the tablet reaches the large intestine. Levodopa is not absorbed from the rectum, so suppository administration is not useful. There is also an immediate-release formulation of carbidopa/levodopa that dissolves in the mouth and is swallowed with saliva, with the trade name of Parcopa. It can be taken without water, which may be an advantage for some patients, e.g., those who have trouble swallowing or who need to be without food or water pre- and postsurgery.
Levodopa is universally accepted as the most effective drug available for symptomatic relief of many of the motor features of PD. If it were uniformly and persistently successful and also free of complications, new strategies utilizing other treatments would not be needed. Unfortunately, 75% of patients have serious complications after 5 years of levodopa therapy (Table 6.1). Fahn (2008) has reviewed the discovery of levodopa as a useful drug and the history of dopamine’s role in PD.
The absorption of levodopa may be increased by eradicating gastric Helicobacter pylori with omeprazole, amoxicillin, and clarithromycin in PD patients documented to be infected with this bacterium (Pierantozzi et al., 2006; Lee et al., 2008). About 50% of the general population is infected with the bacterium.
The question of whether to use levodopa in a patient who has a history of malignant melanoma needs to be considered. Levodopa is an intermediary metabolite in the synthesis of skin melanin, so the concern is whether lurking melanoma cells can be activated by the use of levodopa therapy. A review of the literature does not provide evidence of a definite relationship between treatment with levodopa and the development or reemergence of malignant melanoma (Pfutzner and Przybilla, 1997; Zanetti et al., 2006; Olsen et al., 2007). Epidemiologic studies have shown that people with PD have an increased prevalence of malignant melanoma (Olsen et al., 2006). A clinical trial in which the development of melanoma was a secondary outcome measure showed that patients with PD on the placebo arm of the trial had a much higher rate of developing malignant melanoma than would have been predicted; no association between levodopa therapy and the incidence of melanoma was found (Constantinescu et al., 2007). Yet, it is would seem prudent not to treat with levodopa in a patient with a history of a malignant melanoma if other antiparkinson agents remain effective. Once it becomes necessary to use levodopa to improve quality of life, the patient needs to be informed that he should be observed carefully for changes in or development of new pigmented lesions.
Besides being metabolized by aromatic amino acid decarboxylase (also called dopa decarboxylase), levodopa is also metabolized by catechol-O-methyltransferase (COMT) to form 3-O-methyldopa. Two COMT inhibitors are currently available – tolcapone and entacapone. These agents extend the plasma half-life of levodopa without increasing its peak plasma concentration, and can thereby prolong the duration of action of each dose of levodopa. These drugs are used in conjunction with levodopa to reduce the wearing-off effect, a common motor fluctuation adverse effect of levodopa therapy. The net effect with multiple dosings a day, though, is to elevate the average plasma concentration but smooth out the variations in the concentration. Tolcapone has two potential adverse effects that need to be explained to the patient. The most serious is that a small percentage of patients will develop elevated liver transaminases, and patients need to have baseline and follow-up liver function tests. Death from hepatic necrosis has occurred in three patients who had no liver function surveillance (Watkins, 2000). Entacapone has not shown these hepatic changes. With tolcapone, a small percentage of patients will develop diarrhea, which does not appear until about 6 weeks after starting the drug. The diarrhea can be explosive, so the patient might not have any warning. Entacapone appears not to have these adverse effects. Many clinicians believe that tolcapone is more effective than entacapone in reducing motor fluctuations, but one should not prescribe the former unless the latter has not been effective in relieving wearing-off. Patients on entacapone can be easily switched to tolcapone if the former had less than the desired effect, and a double-blind comparison showed tolcapone to be slightly more effective in reducing the amount of “off” time (Entacapone to Tolcapone Switch Study Investigators, 2007). We advise starting tolcapone at a low dose of 100 mg/day and increasing gradually to 100 mg three times daily.
Elevated total plasma homocysteine, a risk factor for strokes, heart attacks, and dementia, has been found in PD patients using levodopa. The increase of plasma homocysteine with levodopa therapy is thought to be due to the utilization of the methyl group from methionine in the COMT reaction, converting levodopa to 3-O-methyldopa, while converting methionine to homocysteine. A study evaluating the immediate effects of initiating levodopa therapy found a modest elevation of homocysteine and a modest lowering of vitamin B12 levels (O’Suilleabhain et al., 2004). These investigators did not see a reversal with levodopa reduction, agonist treatment, or entacapone treatment. In another study, entacapone also did not reduce homocysteine levels (Nevrly et al., 2010). Another study reported that levodopa treatment does not affect B12 levels, but does reduce folate levels (Lamberti et al. 2005). These investigators found that the addition of COMT inhibitors could reduce the amount of homocysteine, but other investigators did not. Whether the increase in plasma homocysteine with levodopa therapy puts the patient at a greater risk for other medical problems is unknown (Postuma and Lang, 2004).
Adding entacapone to levodopa for patients who are not experiencing motor fluctuations did not add any improvement to motor performance in one study (Olanow et al., 2004), but improved the activities of daily living (ADL) score in another (Brooks and Sagar, 2003). In the FIRST-STEP study, levodopa/carbidopa/entacapone (LCE) 100/25/200 mg three times daily was compared with levodopa/carbidopa (LC) 100/25 mg three times daily in patients with early PD for 39 weeks (Hauser et al., 2009). LCE treatment resulted in slightly better UPDRS Part II activities of daily living (ADL) scores (P = 0.025), but not Part III motor scores.
The concept that intermittent brain levels of levodopa and dopamine contribute to the development of motor complications (see below in discussion of advanced PD) has led to the concept that continuous dopaminergic stimulation may avoid these complications from levodopa. So far, one study (STRIDE-PD) testing this hypothesis has yielded the opposite effect, i.e., an earlier onset of dyskinesias (Stocchi et al., 2010). A total of 747 patients with early PD were randomized to LCE 100/25/200 mg or LC 100/25 mg with flexible dosing to reach 400 mg/day, with a dose 3.5 h apart. The results showed that time to dyskinesia was statistically significantly shorter in LCE-treated patients compared to LC-treated patients. The incidence of dyskinesia during the study period was higher in LCE-treated patients in comparison to the LC group.
The next most powerful drugs, after levodopa, in treating PD symptoms are the dopamine agonists. Of those listed in Table 6.3, bromocriptine, pramipexole, ropinirole, and apomorphine are available in the United States, and these are discussed below. Lisuride, pergolide, cabergoline, rotigotine and piribedil are marketed in some countries. Lisuride is water soluble and can be infused subcutaneously; it has considerable 5-HT agonist activity. Cabergoline is the longest acting and could be taken just once a day (Ahlskog et al., 1996; Hutton et al., 1996); it might prove to be the most important in terms of preventing or reducing the “wearing-off” effect. Piribedil is relatively weak, but has been touted as having an anti-tremor effect. Rotigotine is a dopamine agonist that is utilized as a transdermally applied skin patch (Parkinson Study Group, 2003; Poewe and Luessi, 2005; LeWitt et al., 2007; Poewe et al., 2007; Watts et al., 2007) and was marketed in summer 2007. After the discovery that crystals of rotigotine appear on the patch, the drug was withdrawn from the USA.
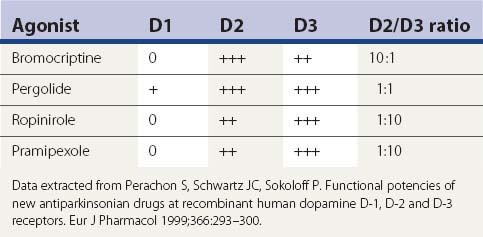
Other than apomorphine and rotigotine, the other dopamine agonists in Table 6.3 are effective orally. Apomorphine needs to be injected subcutaneously or sprayed intranasally. Bromocriptine is the weakest clinically in comparison to the others. Pergolide, pramipexole, and ropinirole appear to be comparable in clinical practice, but some patients will respond better to one than the others. There are some differences between these agonists in their affinity for the dopamine receptor subtypes, as depicted in Tables 6.4 and 6.9. Only apomorphine (strong) and pergolide and lisuride (modest) have agonist activity at the D1 receptor. The activation of the D2 receptor is known to be important in obtaining an anti-PD response, whereas it is unknown how important D3 receptor activation is for improving the anti-PD response. Bromocriptine, pergolide, pramipexole, and ropinirole activate the dopamine D3 as well as the D2 receptor, but their ratios of affinities for these two receptors are different (Table 6.9) (Perachon et al., 1999). All dopamine agonists are less likely to induce dyskinesias compared to levodopa (Schrag et al., 1998). The agonists can be used as adjuncts to levodopa therapy (e.g., Lieberman et al., 1998; Pinter et al., 1999) or as monotherapy (e.g., Kieburtz et al., 1997b; Brooks et al., 1998; Kulisevsky et al., 1998; Rinne et al., 1998a; Sethi et al., 1998). Adverse effects that are more common with dopamine agonists than with levodopa are drowsiness, sleep attacks, confusion, orthostatic hypotension, nausea, and ankle/leg edema associated commonly with erythema (Parkinson Study Group, 2000; Rascol et al., 2000). Edema can spread to involve other areas of the body including the arms and face.
Having several dopamine agonists to choose from allows the opportunity to find one that is better tolerated as well as one that might have more effect. Adverse effects may be the deciding factor as to which drug a patient will do best on. Unfortunately, all these drugs can induce confusion and hallucinations in elderly patients. Leg edema occurs in some patients, usually after a few years. Pramipexole and ropinirole, and other dopaminergics as well, though with probably less frequency, can cause sleepiness and sleep attacks. This could be dangerous for the patient who drives an automobile, and motor vehicle accidents have occurred when patients fell asleep at the wheel (Frucht et al., 1999; Ferreira et al., 2000; Hoehn, 2000; Schapira, 2000). So when deciding to place a patient on pramipexole or ropinirole, the physician should determine the extent of the driving to be done by the patient, and warn the patient about this potential hazard. Short trips, e.g., 10 minutes or so, should be without risk. Should sudden falling asleep occur in any non-driving activity, this event can serve as a warning against driving or else it would be best to taper and even discontinue these medications if driving is necessary. Dopamine agonists also are more likely to induce impulse control problems, such as gambling, hypersexuality, shopping, and binge eating (see Chapter 8) (Weintraub et al., 2010).
Monoamine oxidase (MAO) inhibitors offer mildly effective symptomatic benefit. Type B MAO inhibitors eliminate concern about the “cheese effect” that can occur with type A inhibitors and a high tyramine meal. Although there is debate about possible protective benefit with selegiline, it does have mild symptomatic effects when used alone (Parkinson Study Group, 1993a, 1996a, 1996b) and also potentiates levodopa when used in combination with it (Lees, 1995). A more thorough discussion of selegiline’s possible protective effect is presented below in the section entitled “Selegiline, rasagiline, and antioxidants.” Selegiline has a mild ameliorating effect for mild “wearing-off” from levodopa (Golbe et al., 1988). Zydis selegiline is a form of selegiline that dissolves in the mouth and is absorbed through the oral mucosa, avoiding first pass metabolism in the liver (Waters et al., 2004). This preparation of selegiline, formulated in a freeze-dried tablet that contains a fast dissolving selegiline (Zelapar), has been approved by the Food and Drug Administration in 2006 for clinical use (Clarke and Jankovic, 2006).
Like selegiline, rasagiline is another irreversible type B MAO inhibitor with mild symptomatic benefit (Rabey et al., 2000; Parkinson Study Group, 2002a, 2004a) and with a similar chemical structure; both are propargylamine compounds. Rasagiline is available for use in both early and advanced stages of PD and has a good safety record (Goetz et al., 2006). Both the TEMPO trial and the subsequent larger ADAGIO trial using a delayed-start design showed that starting earlier with rasagiline allows a better clinical outcome than starting later (Parkinson Study Group, 2004a; Olanow et al., 2009). A more thorough discussion of rasagiline’s possible protective effect is presented below in the section entitled “Selegiline, rasagiline, and antioxidants.”
Lazabemide is another type B MAO inhibitor, but is a reversible inhibitor. It shows the same symptomatic effect in PD (Parkinson Study Group, 1993b) as does selegiline and rasagiline. It is not known whether it has a neuronal rescue effect. Lazabemide is not commercially available in the United States. In contrast to selegiline, neither lazabemide nor rasagiline is metabolized to methamphetamine. Type B MAO inhibitors should not require a tyramine-restricted diet, provided that the dose remains no higher than the FDA-authorized dose. Higher doses will begin to inhibit MAO type A, and could cause severe hypertension (the so-called “cheese effect”) if the diet contained too much tyramine. A controlled tyramine challenge showed that rasagiline up to 2 mg/day did not induce a significant blood pressure or pulse change when tyramine was added (deMarcaida et al., 2006). Inhibitors of both type A and type B MAO would offer greater inhibition of dopamine oxidation in the brain and thus the combination would theoretically be more capable of reducing oxidative stress as well as providing more symptomatic effect (Fahn and Chouinard, 1998). But tranylcypromine and phenelzine (both nonselective inhibitors of types A and B MAO) cannot be taken in the presence of levodopa therapy because of the “cheese effect,” and even in the absence of levodopa, patients on these drugs need to adhere to a reduced tyramine diet (Gardner et al., 1996).
We will return to discuss MAOIs and antioxidants below in their possible role in treating early-stage PD. Next, we will review the nondopaminergic drugs that are useful in treating PD, both the motor problems (Table 6.5) and the nonmotor problems (Table 6.6).
Nondopaminergic agents for motor symptoms
Nondopaminergic agents (Table 6.5) are also useful to treat motoric PD symptoms, particularly antimuscarinic drugs (commonly referred to as anticholinergics), which have been widely used since the 1950s, but these are much less effective than the dopaminergic agents, including amantadine. Antimuscarinic drugs have been thought to be somewhat helpful in reducing all symptoms of PD, but they have found special favor in reducing the severity of tremor. But because of sensitivity to memory impairment and hallucinations in the elderly population, antimuscarinic drugs should usually be avoided in patients over the age of 70 years. The antihistaminics, tricyclics, and cyclobenzaprine (Flexeril) have milder anticholinergic properties that make them useful in PD, particularly in older patients who should not take the stronger anticholinergics.
Amantadine, listed in Table 6.3 as a dopaminergic agent, is listed also in Table 6.5 because it has antiglutamatergic effects; this property might account for its usefulness in reducing choreic dyskinesias induced by levodopa (Rajput et al., 1997; Metman et al., 1998a). Dextromethorphan is another antiglutamatergic agent, and it has been found effective in reducing the severity of dyskinesias by 50% (Metman et al., 1998b). Another useful class of drugs is the benzodiazepines to reduce anxiety, and thereby decrease parkinsonian tremor that is exacerbated by stress. Diazepam is usually well tolerated and does not exacerbate parkinsonian symptoms, whereas chlordiazepoxide can (Schwarz and Fahn, 1970). Lorazepam and alprazolam are other useful benzodiazepine agents; the latter has the added benefits of being short-acting and having antidepressant effects. The muscle relaxants listed in Table 6.5 might help in treating “off” and peak-dose dystonias. Because oxidative stress appears to play a role in the pathogenesis of PD, high doses of antioxidant vitamins have been tried for patients with PD. The DATATOP study showed that tocopherol by itself has no effect, but the combination of ascorbate and tocopherol may be more effective than either of these two vitamins alone (Fahn, 1992b; Yoshikawa, 1993). Ascorbate has proven effective in blocking degeneration of nerve cells in vitro induced by levodopa (Mena et al., 1993; Mytilineou et al., 1993; Pardo et al., 1993, 1995; Lai and Yu, 1997). Adenosine A2A receptors are located on GABA neurons in the striatum and antagonize the effect of dopamine on these neurons (Benarroch, 2008). Antagonizing adenosine A2A receptors has a behavioral effect similar to enhancing dopaminergic transmission. One of these receptor antagonists, istradefylline, has undergone clinical trials for patients with motor fluctuations (Bara-Jimenez et al., 2003; Hauser et al., 2003, LeWitt et al., 2008; Stacy et al., 2008), but the results were mixed, with insufficient relief of fluctuations while enhancing dyskinesias. Another adenosine A2A receptor antagonist, preladenant, is currently undergoing clinical trials.
Nondopaminergic agents for nonmotor symptoms
Many nonmotor problems are commonly present in patients with PD; these are discussed in more detail in Chapter 8. But a list of the common drugs used for these nonmotor symptoms is provided in Table 6.6, and a brief explanation of some of the drugs used is provided here. Drugs that are available to improve memory in Alzheimer disease may be tried in patients with Parkinson disease who have dementia, whether from diffuse Lewy body disease or from concomitant Alzheimer disease. These drugs are the centrally active cholinesterase inhibitors, donepezil, rivastigmine, and galantamine. Initial concern that they might worsen tremor and bradykinesia have not been borne out, perhaps because dopaminergic agents are also being given in these patients. These drugs have also been reported to be useful in treating levodopa-induced psychosis.
Psychosis induced by levodopa and the dopamine agonists can often be controlled by clozapine and quetiapine without worsening the parkinsonism. Both agents are dibenzodiazepine antipsychotic drugs. They are called atypical antipsychotics because they rarely cause drug-induced parkinsonism. They are relatively selective D4 receptor antagonists, although they have some D2 blocking action, particularly at high doses, because akathisia (Safferman et al., 1993; Friedman, 1993), acute dystonic reaction (Kastrup et al., 1994; Thomas et al., 1994), and tardive dyskinesia (Dave, 1994) have been associated with them. Clozapine is the most effective agent in treating levodopa-induced psychosis in patients with PD without aggravating the PD (Friedman and Lannon, 1990; Pfeiffer et al., 1990; Kahn et al., 1991; Factor and Brown, 1992; Greene et al., 1993; Pinter and Helscher, 1993; Factor et al., 1994; Diederich et al., 1995; Rabey et al., 1995; Factor and Friedman, 1997; Ruggieri et al., 1997; Friedman et al., 1999; Pollak et al., 2004). But weekly monitoring of white blood cells is necessary with clozapine to prevent irreversible agranulocytosis that can occur rarely with clozapine; this allow a timely discontinuation of this drug when a drop of leukocytes is observed. Because of this need for weekly blood counts, quetiapine is a useful, although somewhat less effective substitute for clozapine, and it is now the drug of first choice. Both clozapine and quetiapine are given at bedtime because of their soporific effect. Olanzapine is an effective antipsychotic, but the dose needs to be kept small because it can worsen PD (Jimenez-Jimenez et al., 1998). There is a window of dosing with olanzapine by which psychosis can be reduced without increasing parkinsonism.
Various sleep problems are encountered in PD. Excessive drowsiness can occur after a dose of levodopa or dopamine agonist. Modafinil can sometimes help to overcome this problem. Insomnia needs to be treated, otherwise quality of life suffers and daytime sleepiness is enhanced. Hypnotics, such as zolpidem and benzodiazepines, can be safely used in PD. Quetiapine and clozapine often allow a good night’s sleep, and can be utilized even in the absence of psychosis. Acting out dreams, so-called REM-sleep behavior disorder, is not uncommon and is usually treated with clonazepam at bedtime. Restless legs syndrome (RLS) and periodic movements in sleep are quite common in patients with PD. If the dopaminergic agent they are taking is ineffective, then an opioid such as propoxyphene, tramadol or oxycodone can be effective. These should be administered an hour or so before the usual onset of these symptoms. Because dopaminergic medication can augment an existing restless legs syndrome, it is reasonable to consider that these agents might also induce it to develop, when previously it did not exist. Thus, restless legs could be considered a complication of dopaminergic medications in PD patients who develop RLS after starting these medications. In a survey of 447 consecutive Korean patients with PD, 16.3% had RLS (Lee et al., 2009). Multivariate logistic regression analysis revealed that the duration of antiparkinson therapy was the most significant factor contributing to the development of RLS in patients with PD, and this supports the notion that medications are likely a causative factor. RLS and its treatment is covered more thoroughly in Chapters 8 and 23.
One of the most common complaints by patients with PD is constipation. This symptom can be a factor of both the disease and the medications used to treat PD. A high fiber diet, including dried fruits, is often sufficient to relieve constipation. The “rancho recipe” is given in Chapter 8. If that is not effective, one can try the standard laxatives or polyethylene glycol (MiraLax). Nausea can be a complication of dopamine agonists and levodopa. Domperidone, a peripheral dopamine receptor blocker, is effective. Because domperidone is not available in the United States, trimethobenzamide (Tigan) can be tried. Sialorrhea is due to infrequent and inadequate spontaneous swallowing of saliva. Peripherally active peripheral antimuscarinics such as propantheline and glycopyrrolate can be quite effective. Injecting botulinum toxin into the parotid glands may benefit some patients (Racette et al., 2003).
Treatment of early-stage Parkinson disease
The earliest stage of PD begins when the symptoms are first noticed and the diagnosis is made. At this stage, symptoms are mild, and there is no threat to the patient’s activities. The designation of “early stage” lasts until the symptoms begin to become troublesome to the patient, and intervention with symptomatic medications is needed. All symptomatic drugs can induce side effects, and if a patient is not troubled by mild symptoms socially or occupationally, the introduction of these drugs can be delayed until symptoms become more pronounced. The clinician needs to discuss this choice with the patient and his/her family. Most neurologists do not use levodopa or other potent antiparkinson agents when the diagnosis is first established and the disease presents with no threat to physical, social, or occupational activities (Fahn, 1991, 1999; Fahn et al., 1996).
Because symptomatically beneficial medications are not needed and because there is no proven neuroprotective treatment, patients in the early, recently diagnosed stage of PD are excellent candidates for participating in a clinical trial in which a placebo is one of the treatment arms. A literature review of clinical trials related to neuroprotection in PD has been conducted by Fahn and Sulzer (2004) and by the Quality Standards Subcommittee of the American Academy of Neurology (Suchowersky et al., 2006). Another elective option is to use one of the drugs described in this section for which hints of neuroprotection have been demonstrated in controlled clinical trials.
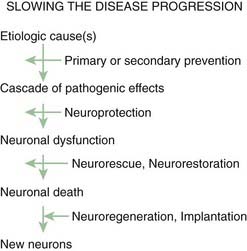
One should keep in mind that the generic label neuroprotection can be divided into at least three different classes of action: slowing the pathogenetic cascade that leads to cell death so that the natural history of the disease is less progressive (neuroprotection), restoring injured dysfunctional neurons (neurorescue, neurorestoration), and replacing dead neurons (neuroregeneration) (Fig. 6.2). In this section we discuss the rationale and results of clinical trials for neuroprotection of PD.
Selegiline, rasagiline, and antioxidants
The first controlled clinical trial for the purpose of evaluating medications as neuroprotective agents for PD was the DATATOP (Deprenyl and Tocopherol Antioxidative Therapy of Parkinsonism) study (Parkinson Study Group, 1989a, 1989b). Deprenyl (selegiline) is an irreversible noncompetitive inhibitor of type B MAO with a long duration of action (MAO-B inhibition half-life of 40 days (Fowler et al., 1994)). Selegiline was tested along with the antioxidant alpha-tocopherol (vitamin E), in a 2 × 2 design. Patients were enrolled in the study early in the course of the illness, and did not require symptomatic therapy. They were placed on selegiline (5 mg twice daily), alpha-tocopherol (1000 IU twice daily), the combination, or double placebo, with approximately 200 subjects in each of the four treatment arms. The primary endpoint was the need for dopaminergic therapy. The study showed that tocopherol had no effect in delaying parkinsonian disability, but selegiline delayed symptomatic treatment by 9 months (Fig. 6.3) (Parkinson Study Group, 1993a). It also reduced the rate of worsening of the UPDRS by half (Table 6.10). Other investigators conducted other studies testing selegiline, showing similar results (Myllyla et al., 1992; Palhagen et al., 1998).
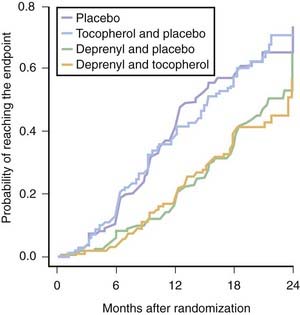
Table 6.10 Average annual rate of decline in UPDRS scores (results are mean ± SD)
| Treatment | Total UPDRS |
|---|---|
| Placebo | 14.02 ± 12.32 |
| Tocopherol | 15.16 ± 16.12 |
| Selegiline | 7.00 ± 10.76 |
| Tocopherol and selegiline | 7.28 ± 11.11 |
| P value | <0.001 |
From Parkinson Study Group. Effects of tocopherol and deprenyl on the progression of disability in early Parkinson’s disease. N Engl J Med 1993;328:176–183. © 1993 Massachusetts Medical Society. All rights reserved.
Because selegiline has a mild symptomatic effect that is long lasting (Parkinson Study Group, 1993a), one could explain its ability to delay progression of disability entirely on this symptomatic effect. In favor of some neuroprotective effect is that after 2 months of washout of the drug, patients had slightly milder PD than did those on placebo (Parkinson Study Group, 1993a). But because of selegiline’s very long duration of action as an inhibitor of MAO-B (Parkinson Study Group, 1995), this observation could represent an insufficient washout period. Furthermore, selegiline’s benefit in delaying the introduction of levodopa gradually diminishes over time (Parkinson Study Group, 1993a), with the best results occurring in the first year of treatment. The odds ratio increased from 0.35 for the first 6 months, to 0.38 in the second 6 months, to 0.77 in the third 6 months, and to 0.86 after 18 months. Follow-up of DATATOP subjects showed that placebo-treated subjects fared better than selegiline-treated subjects when the drug was reintroduced after a 2-month washout period and that the two groups were identical in developing levodopa complications (Parkinson Study Group, 1996a, 1996b). The net understanding by the year 2000 was that there is no convincing evidence that selegiline delayed the need for levodopa because of any protective effect; all results could be those of a drug with a continuing mild symptomatic benefit.
On the other hand, basic scientific research was finding that in animal models, tiny doses of selegiline have a neuronal rescue effect (Tatton, 1993). This effect is not via its MAO inhibitor mechanism of action, but is believed to be due to enhanced protein synthesis of a neurotrophic agent, which is antagonized by amphetamine. Ultimately this finding led to investigation of other agents for their rescue effect, resulting in the discovery of a propargyline drug that was tested in a clinical trial (Waldmeier et al., 2000, 2006).
When the DATATOP study was evaluated to better understand the development of freezing of gait, it was discovered that the group that was treated with selegiline had a statistically significantly decreased risk for developing freezing (Fig. 6.4) (Giladi et al., 2001b). It could not be discerned whether this benefit was because of selegiline’s mild symptomatic benefit or because of some unknown neuroprotection effect. Whichever it was, the authors concluded that one should consider using selegiline in patients who are likely to develop freezing of gait (absence of tremor, gait involvement as the initial symptom).
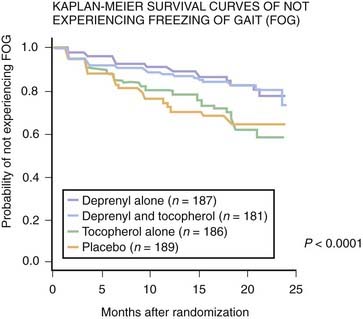
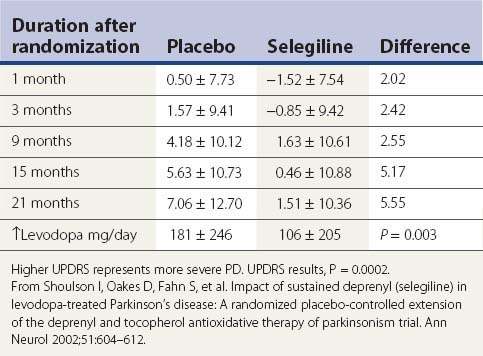
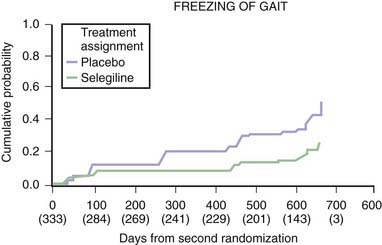
Based on the BLIND-DATE study, it now appears that the decreased risk of freezing of gait with selegiline is not simply from its symptomatic effect as an enhancer of dopamine. The investigators of the DATATOP study, while continuing to follow their subjects, carried out a re-randomization in a controlled trial (called the BLIND-DATE study). A total of 368 subjects who were now on both selegiline and levodopa therapy agreed to be randomized to either selegiline or placebo, while remaining on levodopa. The results were dramatic. The subjects on selegiline required a lower dosage of levodopa, had a slower rate of worsening of symptoms and signs of PD (Table 6.11), and had less freezing of gait (Fig. 6.5) (Shoulson et al., 2002). These results support the view that selegiline does provide some neuroprotective effect or else it has a symptomatic effect separate from dopamine. The possibility that this benefit is derived from an anti-apoptotic effect rather than its antioxidative effect is discussed below.
Table 6.11 Change in total UPDRS after second randomization to either selegiline or placebo while taking levodopa
A similar study was carried out by Palhagen and colleagues (2006), who followed patients for at least 7 years after they entered a controlled clinical trial evaluating selegiline versus placebo in those with early, untreated PD. Then, when any subject required symptomatic therapy, open-label levodopa was added, while maintaining the blind on selegiline versus placebo. During the 7 years of follow-up from the start of the study, the selegiline-treated group had a statistically significantly slower rate of worsening of clinical signs and symptoms as measured by UPDRS scores. Like the Shoulson and colleagues (2002) study mentioned above, this also shows the added benefit that selegiline provides in slowing clinical symptoms. Whether this can be attributed to a neuroprotective effect or to a symptomatic effect that does not appear to be through dopamine is undetermined by the two studies.
The safety of selegiline was raised, though, in an open-label clinical trial in the United Kingdom (Lees, 1995). The use of selegiline when combined with levodopa was reported to be associated with a higher mortality rate than was seen in the patients assigned to levodopa treatment alone. Analysis of this result by others found a number of flaws in the study to refute this conclusion (Olanow et al., 1996). The UK investigators followed up their report with a more detailed analysis of the cause of death (Ben-Shlomo et al., 1998). The excess mortality in the selegiline + levodopa group was greatest in the third and fourth year of treatment. The cause of the increase in deaths showed the excess to be from PD only, and to occur particularly in patients with dementia and a history of falls. No significant differences in mortality were found for revised diagnosis, disability rating scores, autonomic or cardiovascular events, other clinical features, or drug interactions. Other studies with selegiline have failed to find any excess mortality from the combination treatment with levodopa (Myllyla et al., 1997; Aaltonen et al., 1998; Olanow et al., 1998). After being followed by the Parkinson Study Group for an average of 8.2 years, the subjects in the DATATOP study showed no difference in mortality between the groups assigned to treatment with selegiline, tocopherol, or placebo; the death rate averaged 2.1% per year (Parkinson Study Group, 1998), much lower than in the UK study.
A meta-analysis of 17 controlled clinical trials involving type B MAO inhibitors found that no significant difference in mortality existed between patients on type B MAO inhibitors and control patients (Ives et al., 2004). The analysis also found that subjects randomized to type B MAO inhibitors had significantly better total scores, motor scores, and ADL scores on the UPDRS at 3 months compared with patients taking placebo; they were also less likely to need additional levodopa or to develop motor fluctuations. No difference existed between the two groups in the incidence of side effects or withdrawal of patients.
High-dosage vitamin E has also been suggested to increase mortality, but analysis of the DATATOP cohort followed for up to 13 years failed to find any difference in mortality between the groups on vitamin E and the group on placebo (Marras et al., 2005).
In a more recent analysis of retrospective observational data from Scotland (Donnan et al., 2000) comparing PD patients with a comparable control population, the patients with PD had a higher rate of mortality than those without PD (rate ratio (RR) 1.76; 95% confidence interval (CI) 1.11–2.81). There was significantly greater mortality in monotherapy (RR = 2.45, 95% CI 1.42–4.23) relative to the comparators, adjusting for previous cardiovascular drug use and diabetes. However, there was no significant difference in mortality in patients with PD who received combination therapy of selegiline with levodopa and other drugs in relation to the comparators (RR = 0.92, 95% CI 0.37–2.31). Thus, from this study, selegiline did not increase the mortality rate, whether used as monotherapy or in combination with levodopa. In fact, levodopa monotherapy had the highest mortality rate.
Mortality in PD patients was also determined in a multicenter European study (Berger et al., 2000). As in the Scotland study (Donnan et al., 2000), the mortality rate was twice that of a controlled population (RR 2.3; 95% CI 1.8–3.0). The risk for death in men with PD (RR 3.1; 95% CI 2.1–4.4) was higher than that in women with PD (RR 1.8; 95% CI 1.2–5.1). Women with PD had a fivefold higher risk of living in a care facility than men with PD.
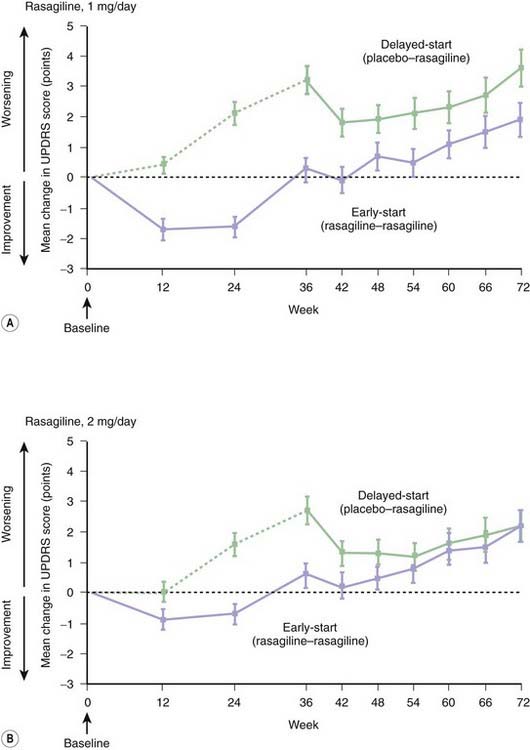
Rasagiline, based on the delayed-start studies, TEMPO (Parkinson Study Group, 2004a), and ADAGIO (Olanow et al., 2009), also can reduce the rate of clinical worsening in patients with early PD. But there were inconsistent outcomes between these two studies. In TEMPO, the 2 mg dose of rasagiline had a superior result compared to the 1 mg dose. In ADAGIO, only the 1 mg dose was superior to placebo; the 2 mg dose was no better than placebo (Fig. 6.6). Starting a symptomatically effective drug early does not automatically lead to a reduced rate of clinical worsening as tested by the delayed-start design. For example, pramipexole, an effective dopaminergic agent, does not give a superior clinical result if started early compared to starting it later (Schapira et al., 2009). Thus, there would appear to be a special property of the MAO inhibitors to be able to delay clinical worsening in a delayed-start study.
As mentioned above, the dose of selegiline and rasagiline should not exceed their specificity as selective type B inhibitors of MAO. Selegiline greater than 10 mg/day and rasagiline greater than 2 mg/day will also inhibit type A MAO. A woman given rasagiline at 4 mg/day in the presence of levodopa therapy developed the serotonin syndrome of hyperpyrexia, confusion, agitation and episodic periods of unconsciousness (Fernandes et al., 2011). Because selegiline is metabolized to amphetamine and methamphetamine, insomnia could develop, and one should avoid taking it late in the day. It may be necessary to limit selegiline to 5 or 10 mg in the morning only if insomnia is a problem. Male impotence is not common with MAO inhibitors. In the presence of levodopa, MAO inhibitors potentiate levodopa’s effect, and lower doses of levodopa can usually be achieved (Lees, 1995; Shoulson et al., 2002). Selegiline does not prevent the development of levodopa-induced complications of fluctuations and dyskinesias (Parkinson Study Group, 1996b). Selegiline decreases the risk of patients developing freezing of gait (Giladi et al., 2001b; Shoulson et al., 2002). It is not clear if rasagiline has this ability. Interestingly, type A MAO inhibitors, but not type B MAO inhibitors, have been shown to reduce stress-induced freezing behavior in rats (Maki et al., 2000).
The DATATOP study showed that selegiline inhibits MAO activity by about 20% in the CNS (Parkinson Study Group, 1995). Because the original premise for the DATATOP study was that selegiline might be neuroprotective by inhibiting MAO (reducing formation of hydrogen peroxide and thereby decreasing oxidant stress), the CSF analysis of homovanillic acid indicates that selegiline is a poor inhibitor of CNS MAO. This finding could explain the lack of success of selegiline as a powerful neuroprotective agent. Whether a more potent inhibitor of MAO could be more successful remains to be determined. In the meantime, it is reasonable for patients to consider an inhibitor of both types A and B, as possibly augmenting inhibition of MAO in the brain. Such MAO inhibitors can induce the “cheese effect,” so the MAO inhibitor diet, avoiding dietary tyramine, needs to be utilized. Such MAO inhibitors can be used only in the absence of levodopa because the combination will create marked blood pressure fluctuations.
Because the oxidant stress hypothesis is widely held as one of the pathogenic mechanisms in PD (Graham et al., 1978; Cohen, 1983, 1986; Fahn, 1989; Fornstedt et al., 1990; Olanow, 1990, 1992; Jenner, 1991; Fahn and Cohen, 1992; Jenner et al., 1992a, 1992b; Zigmond et al., 1992; Spencer et al., 1995; Alam et al., 1997), the use of a combination of antioxidants seems a reasonable approach.
Fahn has used tranylcypromine (Parnate), an irreversible inhibitor of both types A and B MAO, along with high dosages of antioxidants (Fahn and Chouinard, 1998). As measured by cerebrospinal fluid concentration of the metabolite of dopamine, selegiline just partially inhibits dopamine oxidation, reducing hydrogen peroxide formation by only 20% (Parkinson Study Group, 1995), whereas tranylcypromine inhibits by 75% (Fahn et al., 1998). Using tranylcypromine requires the patient to be placed on an MAO inhibitor diet, which is not onerous (Gardner et al., 1996), and if adhered to, avoids the “cheese effect.” In the presence of an irreversible type A MAO inhibitor, tyramine cannot be deaminated in the gut. The absorption of tyramine results in the release of norepinephrine from sympathetic nerve terminals, thereby raising blood pressure, and potentially creating a hypertensive crisis (“cheese effect”). Some patients can develop intracerebral hemorrhage during an episode of such a crisis. The usual dose of tranylcypromine is 10 mg three times daily, but doses up to 60 mg per day can be used. Insomnia and male impotence are fairly common adverse effects that would require shifting the times of the dosages from the evening hours or reducing or discontinuing the drug. A side benefit is the lifting of any existing depression. Levodopa cannot be given in the presence of an inhibitor of type A MAO because the combination produces a volatile blood pressure. Meperidine (Demerol) and antidepressants such as tricyclics and selective serotonin uptake inhibitors are also to be avoided because of the potential for psychiatric and autonomic reactions (“serotonin syndrome”) that could be fatal.
The antioxidants ascorbate (vitamin C) and tocopherol (vitamin E) are recommended solely on the basis of the oxidant stress hypothesis of the pathogenesis of PD. Although the DATATOP trial showed that tocopherol by itself is ineffective in slowing down the progression of PD, a combination of ascorbate and tocopherol potentiates the antioxidant efficacy of both (Yoshikawa, 1993; Hamilton et al., 2000). This combination of antioxidants in early PD patients has been used since 1979 and has not produced any harmful effects (Fahn, 1992b). The dosages gradually reached in four divided doses are 3000 mg per day of ascorbate and 3200 IU per day of d-alpha-tocopherol. Coenzyme Q10 and vitamin E need each other as antioxidants (Kagan et al., 2000). There is evidence that the natural form of tocopherol (d-alpha) achieves higher blood levels than the synthetic racemic (d,l-alpha) tocopherol (Acuff et al., 1994).
Riluzole
Glutamate is the major excitatory neurotransmitter in the CNS and can induce excitotoxicity. A slow excitotoxic process has been proposed by Beal (1998) to be a possible mechanism of cell death in PD. Riluzole impairs glutamatergic neurotransmission by blocking voltage-dependent sodium channel currents. In experimental animal models of PD, riluzole was found to have neuroprotective effects (Benazzouz et al., 1995; Barneoud et al., 1996; Boireau et al., 2000; Obinu et al., 2002). However, in controlled clinical trials in patients with early PD, riluzole was not found to be effective as a neuroprotective agent (Jankovic and Hunter, 2002; Rascol et al., 2003).
Providing trophic factors
Glial-derived neurotrophic factor (GDNF) promotes the survival of DA neurons (Burke et al., 1998), DA neuron neurite outgrowth, and quantal size (the amount of DA released per synaptic vesicle exocytic event) (Pothos et al., 1998). When GDNF was injected into the midbrain of primates rendered parkinsonian by MPTP, there was improvement of the parkinsonian features (Gash et al., 1996). Moreover, DA concentration in the substantia nigra (SN) was increased on the injected side and the nigral DA neurons were 20% larger with an increased fiber density. In a subsequent study, primates received infusions of GDNF into a lateral ventricle (Grondin et al., 2002). This approach also showed restoration of the nigrostriatal dopaminergic system and improved the motor function in rhesus monkeys. The functional improvements were associated with pronounced upregulation and regeneration of nigral DA neurons and their processes innervating the striatum. However, in a randomized, double-blind, placebo-controlled trial of infusing GDNF into the lateral ventricle of patients with PD, there was no clinical improvement (Nutt et al., 2003). Nausea, anorexia, and vomiting were common, hours to several days after injections of GDNF. Weight loss occurred in the majority of subjects receiving 75 µg or larger doses. Paresthesias, often described as electric shocks (Lhermitte sign), were common in GDNF-treated subjects.
One subsequent open-label study in five patients with PD showed that infusing GDNF directly into the putamen improved motor performance, and that there was increased FDOPA uptake on PET scans in some of the patients (Gill et al., 2003). However, a subsequent larger placebo-controlled trial failed to show clinical improvement although FDOPA uptake did increase (Lang et al., 2006).
Another approach of delivering GDNF directly into the brain was successfully achieved in primates using lentoviral vectors containing the gene for producing GDNF (Kordower et al., 2000). Lenti-GDNF was injected into the striatum and SN of rhesus monkeys that had been treated 1 week previously with MPTP. Lenti-GDNF reversed functional deficits and completely prevented nigrostriatal degeneration. Long-term gene expression (8 months) was seen in intact monkeys that were given this treatment.
A novel nonimmunosuppressive immunophilin ligand, GPI-1046 (henceforth called neuroimmunophilin), was found to have trophic activity, including regenerative sprouting from spared nigrostriatal dopaminergic neurons following MPTP toxicity in mice or 6-hydroxydopamine toxicity in rats (Steiner et al., 1997). Since then, there have been reports supporting a regenerative effect by neuroimmunophilins (Guo et al., 2001) and with a proposed mechanism of increasing glutathione in the brain (Tanaka et al., 2001, 2002). On the other hand, there have been many reports that failed to find such benefits in various animal models of PD, including primates (Harper et al., 1999; Bocquet et al., 2001; Emborg et al., 2001; Eberling et al., 2002). One controlled clinical trial testing neuroimmunophilin in patients was unsuccessful, and a subsequent larger and longer one also failed to show benefit.
Enhancing mitochondria and energy function
Coenzyme Q10 is the electron acceptor for mitochondrial complexes I and II and is also a potent antioxidant. Complex I activity was found to be affected by MPTP, and subsequently found to be selectively decreased postmortem in SN in patients with PD (Schapira et al., 1990). Coenzyme Q10 is reduced in the mitochondria (Shults et al., 1997) and in sera of patients with PD (Matsubara et al., 1991). Oral supplementation of coenzyme Q10 in rats resulted in increases of coenzyme Q10 in cerebral cortex mitochondria (Matthews et al., 1998). A controlled clinical pilot trial of coenzyme Q10 was undertaken in 80 patients with early PD. They were randomized into four equal arms and were assigned 300 mg/day, 600 mg/day, 1200 mg/day, or placebo and followed up to 16 months (Shults et al., 2002). There was a positive trend (P = 0.09) for a linear relationship between the dosage and the mean change in the total UPDRS score. The highest dose group (total UPDRS change of +6.69) was statistically less than the UPDRS change of +11.99 for the placebo group (Fig. 6.7). The change in UPDRS for the lower doses showed no significant difference from the placebo group. There was a slower decline in the change of all three components of the UPDRS scores in the 1200 mg/day group, with the greatest effect in Part II (the subjective ADL component) (Fig. 6.8). This raises the question of whether patients on 1200 mg/day of coenzyme Q10 might simply feel better rather than having an objective improvement of their motoric features of PD. After 1 month of treatment, there was improvement of the Part II UPDRS (ADL) score in the 1200 mg/day group of −0.66, compared to worsening in the placebo group of +0.52. This wash-in effect supports the concern that there might be a “feel good” response from coenzyme Q10 rather than a neuroprotective effect. Also, it should be noted that those who were treated with the 1200 mg/day failed to show a delay in the need for dopaminergic therapy. Of course, the study was not powered for a modest effect, and the study investigators urged caution in interpretation of the results until a larger study could be conducted and evaluated. A futility trial showed little difference over time between coenzyme Q10 and placebo in the clinical progression of PD (NINDS NET-PD Investigators, 2007). Also, coenzyme Q10 showed no symptomatic benefit (Storch et al., 2007).
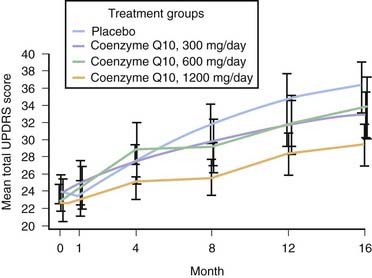
Figure 6.7 Change in total UPDRS with different dosages of coenzyme Q10.
From Shults CW, Oakes D, Kieburtz K, et al. Effects of coenzyme Q(10) in early Parkinson disease – evidence of slowing of the functional decline. Arch Neurol 2002;59(10):1541–1550. © American Medical Association. All rights reserved.
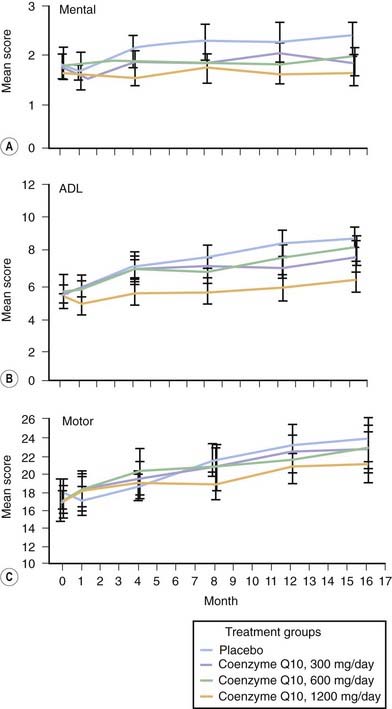
Figure 6.8 Change in the different components of the UPDRS with coenzyme Q10.
From Shults CW, Oakes D, Kieburtz K, et al. Effects of coenzyme Q(10) in early Parkinson disease – evidence of slowing of the functional decline. Arch Neurol 2002;59(10):1541–1550. © American Medical Association. All rights reserved.
Creatine is a guanidine-derived compound that is generated in the body. The creatine/phosphocreatine system functions as an energy buffer between the cytosol and mitochondria (Beal, 2003). Creatine has been proposed to serve as a neuroprotectant in neurodegeneration, and has been tested in a controlled clinical futility trial in early PD. Creatine was found not to be futile and is deserving of a phase III trial (NINDS NET-PD Investigators, 2006, 2008).
Counteracting inflammation
Gliosis and reactive microglia are seen in the substantia nigra of patients with PD, indicating an ongoing inflammatory process. Such changes have also been seen following MPTP (Vila et al., 2001) and rotenone (Betarbet et al., 2000) neurotoxicity. Inflammation is considered to be a secondary effect, but may play an important role in enhancing neurodegeneration by the production of cytokines and prostaglandins. Experimental animal models have shown that treatment with the antibiotic minocycline can reduce the level of degeneration by MPTP (Du et al., 2001; Wu et al., 2002). As a result of these reports, a controlled clinical futility trial testing minocycline was conducted, showing minocycline not to be futile (NINDS NET-PD Investigators, 2006).
Inhibiting apoptosis
Studies on selegiline, in an effort to explain its effectiveness in the DATATOP study, have shown it to have a neuronal rescue effect that is independent of its MAO inhibition (Tatton, 1993). This finding led to the investigation of other agents for their neuronal rescue effect, resulting in the discovery that propargylamines have an anti-apoptotic action. A search for similar compounds but without inhibiting MAO resulted in the discovery of one agent (TCH346) that is anti-apoptotic and that may act by stabilizing glyceraldehyde-3-phosphate dehydrogenase (Waldmeier et al., 2000). This drug was tested in a controlled clinical trial, but was found not to be effective in slowing progression of PD (Waldmeier et al., 2006; Olanow et al., 2006). The propargylamine rasagiline is also anti-apoptotic in laboratory and animal models. It was studied in a delayed-start neuroprotective trial, and the press release in June 2008 stated that the study was successful. The results have not yet been reported in a scientific meeting or publication. Another anti-apoptotic drug, CEP1347, which inhibits mitogen linear kinases was shown to be an effective neuroprotectant in animal models of PD. This drug was tested in a large controlled clinical trial that was stopped early because of lack of effectiveness of the drug (Parkinson Study Group PRECEPT Investigators, 2007).
Dopamine agonists
There are four published trials comparing a dopamine agonist and levodopa in patients with PD who were in need of symptomatic therapy. These compared cabergoline and levodopa (Rinne et al., 1998a) ropinirole and levodopa (Rascol et al., 2000), pramipexole and levodopa (the so-called CALM-PD trial) (Parkinson Study Group, 2000), and pergolide and levodopa (Oertel et al., 2006). The clinical outcomes of these studies are discussed below in “Treatment of mild-stage Parkinson disease.” In this section regarding neuroprotection, the results of the neuroimaging component of these trials are discussed. In the CALM-PD (pramipexole vs. levodopa) trial, the 4-year imaging results show a statistically significant lesser rate of decay of dopamine transporter binding (β-CIT SPECT) (a marker of integrity of nerve terminals of the dopaminergic nigrostriatal fibers) in the striatum in the group originally assigned to pramipexole treatment (Fig. 6.9) (Parkinson Study Group, 2002b). A separate study evaluating FDOPA PET scans, a marker of dopa uptake and dopa decarboxylase activity, showed a similar statistically significant lesser rate of decay of labeling in the striatum in a controlled trial in the group assigned to ropinirole compared to the group assigned to levodopa therapy (Whone et al., 2003).
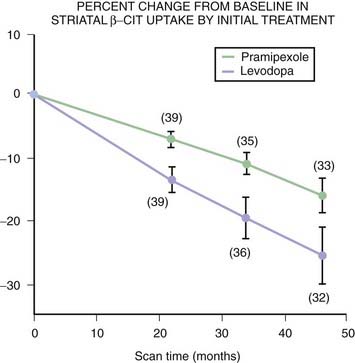
Figure 6.9 Striatal β-CIT SPECT binding in the CALM-PD study.
From Parkinson Study Group. Dopamine transporter brain imaging to assess the effects of pramipexole vs levodopa on Parkinson disease progression. JAMA 2002;287:1653–1661. © American Medical Association. All rights reserved.
Because there was no placebo comparator in either study, interpretation is difficult. Whether dopamine agonists slow the rate of progression of PD, whether levodopa hastens it, and whether both explanations are playing a role, are possibilities. Another possibility would be a pharmacodynamic effect on the dopamine transporter and dopa decarboxylase by either the agonists or levodopa. For example, if levodopa downregulated the dopamine transporter, β-CIT SPECT binding would be reduced. If levodopa downregulated dopa decarboxylase, FDOPA PET binding would be reduced. Short trials of levodopa showed no change in these imaging markers, so there is no evidence that levodopa affects either type of imaging study in such a pharmacologic manner. But a consensus conference concluded that there is insufficient information about the effect of medications on dopaminergic imaging to recommend neuroimaging as a biomarker for disease progression in the presence of medication (Ravina et al., 2005). Without knowing whether the agonists actually slow the rate of progression, it is not possible to recommend the starting treatment on the basis of these results alone. Pramipexole was tested in a delayed-start trial and failed to show any slowing of PD (Schapira et al., 2009).
Inhibiting calcium entry
Substantia nigra pars compacta (SNc) dopamine neurons are autonomously active; that is, they generate action potentials at a clock-like 2–4 Hz in the absence of synaptic input (Surmeier et al., 2005). In this respect, they are much like cardiac pacemakers. Juvenile dopamine-containing neurons in the SNc use sodium influx as the pacemaking mechanisms common to neurons not affected in PD, but the sodium mechanism remains latent in adulthood (C.S. Chan et al., 2007). Instead, the autonomous activity is generated by Ca2+ influx (Mercuri et al., 1994; C.S. Chan et al., 2007; Surmeier, 2007). The SNc dopamine neurons rely on L-type Ca(v)1.3 Ca2+ channels. With increased intracellular calcium, mitochondrial function can be affected with increased demand on oxidative phosphorylation, leading to increased production of reactive oxygen species and eventually cellular damage. As the cells undergo more stress over time, they thus “age faster.” This would be a link with the risk factor of age (Surmeier, 2007). Blocking Ca(v)1.3 Ca2+ channels in adult neurons induces a reversion to the juvenile form of pacemaking. Such blocking (“rejuvenation”) protects these neurons in both in-vitro and in-vivo models of Parkinson disease, pointing to a new strategy that could slow or stop the progression of the disease (C.S. Chan et al., 2007; Surmeier, 2007).
As it turns out, use of calcium channel blockers for treating hypertension has been shown to be associated with less risk for developing PD in two database-mining studies (Becker et al., 2008a; Ritz et al., 2010), but not in a third (Simon et al., 2011). A clinical trial to evaluate the dihydropyridine isradipine, a calcium channel blocker, is currently underway and has been shown to be well tolerated (Simuni et al., 2010).
Statins and NSAIDs
One epidemiologic study in California surveying use of statins in patients with PD and a control population found that statin use was more common in the controls (Wahner et al., 2008), but another study surveying statin use in the UK, did not find any difference between patients with PD and controls (Becker et al., 2008b). One data-mining epidemiologic study reported that ibuprofen, but not other nonsteroidal anti-inflammatory drugs (NSAIDs), was associated with a lower risk of developing PD (Gao et al., 2011).
Treatment of mild-stage Parkinson disease
Strategy
The mild stage of PD occurs when the signs and symptoms of the illness are beginning to interfere with daily activities or with quality of life. The judgment to initiate symptomatic drug therapy is made in discussions between the patient and the treating physician. According to a survey (Parkinson Study Group, 1989a) the most common problems that clinicians consider important for the decision to initiate symptomatic agents are (1) threat to employability, (2) threat to ability to handle domestic, financial, or social affairs, (3) threat to ability to handle activities of daily living, and (4) appreciable worsening of gait or balance. According to a Norwegian quality of life study (Karlsen et al., 1999), the factors that produce the highest distress for PD patients compared to healthy elderly people are depressive symptoms, self-reported insomnia, and a low degree of independence, measured by the Schwab and England scale. Severity of parkinsonian motor symptoms contributed, but to a lesser extent. A sense of lack of energy was seen in half of the PD patients compared to a fifth of controls, and this could be only partially accounted for by depressive symptoms and the UPDRS motor scores.
The choice of drugs (Tables 6.3 and 6.5) is wide, but the degree of disability and the age (or mental acuity) of the patient are two critical factors. If the delay in initiating symptomatic treatment was so prolonged that the symptoms now threaten employment or endanger falling, one needs to begin levodopa to get a quick response. The advantages of using levodopa when the symptoms are this pronounced, in preference to a dopamine agonist or other medications, are that a therapeutic response is both rapid and virtually guaranteed, because nearly all patients with PD will respond to levodopa and relatively quickly. In contrast, only a minority of patients with severe symptoms will benefit sufficiently from a dopamine agonist given alone, and it takes more time (often months) to build up the dose to adequate levels to discover this. If levodopa is to be utilized, inhibitors of type A MAO must be discontinued. If selegiline (or another selective type B MAO inhibitor) was the MAO inhibitor that was utilized, this drug can be continued. A type A MAO inhibitor can be used safely with dopamine agonists.
If the symptoms are not severe enough to require levodopa and the patient is younger than 60 (younger than 70 if the patient is mentally young), we prefer to employ a dopa-sparing strategy to avoid as long as possible the development of levodopa-induced dyskinesias and motor fluctuations (mainly the wearing-off effect). These complications are more likely to occur in younger patients (Quinn et al., 1987; Kostic et al., 1991; Gershanik, 1993; Wagner et al., 1996). The choices are dopamine agonists, amantadine, and anticholinergics. Tranylcypromine can be continued in the presence of any of these drugs. Dopamine agonists are the most potent antiparkinsonian agents among this group of drugs. Four-year results of the pramipexole versus levodopa trial reveal that levodopa is clinically more potent, but is also much more likely to induce dyskinesias and clinical fluctuations (Holloway and Parkinson Study Group, 2004). For patients older than 70 years or those with any cognitive decline, employ levodopa as the initial therapy. Not only is there less need for a dopa-sparing strategy in these elderly patients, they are more susceptible to confusion, psychosis, or drowsiness from other antiparkinson drugs, including dopamine agonists. Levodopa provides the greatest benefit for the lowest risk of these adverse effects compared to the other drugs.
Rationale for dopa-sparing strategy in young patients
As was mentioned earlier, younger patients (less than 60 years of age) are particularly prone to develop the motor complications of fluctuations and dyskinesias (Quinn et al., 1987; Kostic et al., 1991; Gershanik, 1993; Wagner et al., 1996). Some physicians therefore recommend utilizing dopamine agonists, rather than levodopa, in younger patients when beginning therapy, in an attempt to delay the onset of these problems (Quinn, 1994b; Fahn and Przedborski, 2010; Montastruc et al., 1999). But others prefer starting with levodopa (Weiner, 1999). A conference on this topic failed to produce a consensus (Agid et al., 1999).
Choice of drug when employing a dopa-sparing strategy
Dopamine agonists
The dopamine agonists are the group of agents that is next most powerful in reducing the symptoms of PD after levodopa therapy. Thus, they are a good choice. Based on a clinical trial, there is no evidence that they provide neuroprotection (Schapira et al., 2009a). Perhaps the main reason many patients are started with this class of drugs, is that they are less likely to induce dyskinesias and motor fluctuations (Rinne et al., 1998a; Parkinson Study Group, 2000; Rascol et al., 2000; Holloway and Parkinson Study Group, 2004).
Rinne (1989a, 1989b) first proposed that early use of the dopamine agonists would reduce the likelihood of developing complications from chronic levodopa therapy. However, the Rinne reports were on retrospective analyses, using historical rather than contemporary controls. In one double-blind study, Weiner and colleagues (1993) could not confirm Rinne’s findings. However, in another controlled trial, Montastruc and colleagues (1994) reported that there were fewer motor complications in patients who started on bromocriptine, to which levodopa was later added. Studies of dopamine agonists as primary monotherapy in early PD have shown that, even with sustained treatment, drug-induced dyskinesias rarely develop, but that monotherapy is successful for more than 3 years in only about 30% of all PD patients (Poewe, 1998). In addition to this benefit, the dopamine agonists are the most powerful antiparkinson medications after levodopa. Therefore, if one wants to use dopa-sparing strategies, one should choose among the dopamine agonists.
The first double-blind study comparing an agonist with levodopa was with cabergoline. A smaller percentage of patients in the cabergoline group developed motor fluctuations (22%) versus 34% on levodopa (P < 0.02) (Rinne et al., 1998a). Controlled clinical trials comparing ropinirole (Rascol et al., 2000), pramipexole (Parkinson Study Group, 2000), and pergolide (Oertel et al., 2006) have shown that starting treatment with a dopamine agonist is less likely than treatment with levodopa to induce dyskinesias. On the other hand, these studies all showed that levodopa is more potent and improves UPDRS scores more than the agonists did. Moreover, the agonists are more likely to produce hallucinations and sedation than levodopa.
Post hoc analysis revealed that starting treatment with ropinirole (Rascol et al., 2006) or pramipexole (Kieburtz et al., 2006) delays levodopa-induced dyskinesias by delaying the start of levodopa therapy. A 10-year follow-up of the ropinirole trial showed that motor complications remained fewer in those subjects who started on ropinirole, but there was no difference in UPDRS. (Hauser et al., 2007). Of course, all subjects were taking levodopa at that time. A similar finding was found in the 6-year follow-up of the CALM-PD subjects (initiating levodopa or pramipexole) (Parkinson Study Group CALM Cohort Investigators, 2009).
Ergots rarely can induce red, inflamed skin (St Anthony’s fire), which is reversible on discontinuing the drug. They also have the potential (although rare) with long-term use to induce fibrosis: retroperitoneal, pleuropulmonary, and pericardial (Pfitzenmeyer et al., 1996; Ling et al., 1999; Shaunak et al., 1999). Pergolide has also been seen in association with fibroproliferative changes in heart valves, initially reported in three patients (Pritchett et al., 2002). Since then there have been other reports (Baseman et al., 2004; Horvath et al., 2004; Van Camp et al., 2004). The frequency of this complication is still being resolved. The reports of this complication raised new concerns, and questions as to whether pergolide should be used as a drug for PD unless other dopamine agonists have been unsatisfactory in terms of benefits or adverse effects (Agarwal et al., 2004). Now echocardiograms performed on patients taking pergolide have revealed a much higher prevalence, about 33%, of restrictive valvulopathies (Van Camp et al., 2004). This indicates that all patients on pergolide need to undergo echocardiography. Fortunately, the valvulopathy is reversible in some patients if pergolide is discontinued. If this ergoline can cause this problem, then it is possible that the other ergoline agonists can do likewise. It seems prudent to utilize non-ergot dopamine agonists rather than starting pergolide on other patients. With the publication of larger studies (Peralta et al., 2006; Zanettini et al., 2007; Dewey et al., 2007) and the analysis of the United Kingdom General Practice Research Database for cardiac valve regurgitation showing pergolide and cabergoline to increase its incidence rate (Schade et al., 2007), the FDA issued a strong warning, and the pharmaceutical company discontinued manufacturing pergolide in 2007.
Heart valves contain 5-HT2B receptors. Drugs like fenfluramine that activate these receptors can induce thickening of the heart valves. Roth (2007) reviewed the effect on 5-HT receptors by dopamine agonists. The only two dopamine agonists that are also potent 5-HT2B agonists are pergolide and cabergoline, and these are the only ones that were significantly associated with cardiac valve disease (Schade et al., 2007). Lisuride, which is an agonist at 5-HT2A and 5-HT2C receptors, but not at 5-HT2B receptors, was not associated with valve disease. Although regurgitation was reported with bromocriptine, restrictive valvulopathy was not (Tan et al., 2009).
Pramipexole and ropinirole, as was mentioned earlier, appear more readily to produce drowsiness and sleep attacks in which patients fall asleep without warning, including while driving, although there have now been rare incidences of sleep attacks with all dopamine agonists and levodopa (Frucht et al., 1999; Hoehn, 2000; Schapira, 2000; Ferreira et al., 2000). The Epworth Sleepiness Scale is not predictive as to which patient may develop a sleep attack (Hobson et al., 2002). Korner and colleagues (2004) sent a questionnaire to 12 000 patients and received responses from 63%, 42% of whom reported that they had experienced sudden onset of sleep; 10% of these had not experienced sleepiness before their first sleep attack. Predicting factors were nonergoline dopamine agonists, age less than 70 years, and disease duration less than 7 years. Modafinil has successfully been used to prevent sleep attacks (Hauser et al., 2000).
Adverse effects that are more common with agonists than with levodopa are orthostatic hypotension, nausea (because nausea from levodopa is blocked by carbidopa), drowsiness, hallucinations, and leg edema. All agonists have a propensity to produce ankle and leg edema (Tan and Ondo, 2000), which is not an early problem, but tends to occur after a few years of treatment. In a retrospective review of developing pedal edema with pramipexole treatment, there was no relationship between dose of pramipexole and incidence and severity of pedal edema. The risk of development of pedal edema was 7.7% in the first year after initiation of pramipexole therapy, with more rapid development of edema among those with a history of coronary artery disease (Kleiner-Fisman and Fisman, 2007). The edematous skin is often red, and some clinicians, unaware of this adverse effect, assume that there is a deep vein thrombosis. The edema and redness persists unless the drug is stopped. Diuretics are only partially effective in relieving the edema. Whether it is dangerous to continue to allow the edema to persist is not known. But continued use of the agonist can eventually result in discolored, indurated skin in the lower part of the legs where the edema was located. The tight skin prevents edema from accumulating there, so the edema is seen above the induration. Substituting one agonist for another may occasionally allow the edema to dissipate, but more often than not, once the edema has occurred, it persists in the presence of other dopamine agonists as well. The only satisfactory treatment of the edema is to discontinue the agonist, and substitute levodopa, which does not cause this problem. The edema of the legs that is induced by dopamine agonists resembles that induced by amantadine.
A new adverse effect was reported in 2010 by Rabinak and Nirenberg, who described the “dopamine agonist withdrawal syndrome (DAWS).” This was defined as a cluster of physical and psychological symptoms that correlate with dopamine agonist withdrawal, causing clinically significant distress or social/occupational dysfunction, and are refractory to levodopa and other PD medications, and cannot be accounted for by other clinical factors. Symptoms of DAWS resembled those of other drug withdrawal syndromes and included anxiety, panic attacks, agoraphobia, depression, dysphoria, diaphoresis, fatigue, pain, orthostatic hypotension, and drug cravings. Some of those with DAWS needed to be restarted on the dopamine agonist.
An experimental study in MPTP-treated primates showed that treatment with dopamine agonists (ropinirole and bromocriptine in this study) significantly less likely to cause severe dyskinesias than treatment with levodopa (Pearce et al., 1998). The investigators titrated dosages that produced similarly increased locomotion and improved motor disability. However, these investigators also showed that an agonist will elicit comparable dyskinesia once levodopa priming has occurred, and they therefore recommended early use of dopamine agonists. Controlled clinical trials comparing ropinirole (Rascol et al., 2000), pramipexole (Parkinson Study Group, 2000), and pergolide (Oertel et al., 2006) have shown that starting treatment with a dopamine agonist is less likely than treatment with levodopa to induce dyskinesias.
Each of the dopamine agonists easily induces orthostatic hypotension, particularly when the drug is first introduced (Kujawa et al., 2000). After that period, this complication is much less common. Therefore it is best to start with a tiny dose (bromocriptine 1.25 mg at bedtime; pergolide 0.05 mg at bedtime; pramipexole 0.125 mg at bedtime) for the first 3 days, and then switch from bedtime to daytime dosing for the remainder of the first week. Ropinirole can be started at 0.25 mg three times daily for the first week. The daily dose can be increased gradually (bromocriptine 1.25 mg every week; pergolide 0.125 mg weekly; pramipexole 0.125 mg every 2 days for 10 days, then 0.125 mg per day weekly; ropinirole 0.5 mg/day twice weekly), building the dosage up on a four times a day dosing schedule until benefit or a dose around 40 mg/day (bromocriptine), 4–6 mg/day (pergolide and pramipexole), or 24 mg/day (ropinirole) is reached. Use the lowest dose that provides adequate benefit.
Bromocriptine appears to be the weakest of the four agonists, yet it can induce psychotoxicity just as readily as the other agonists, if not more so. That is why bromocriptine is not used as much and therefore not presented in Table 6.12. The choice between the other three drugs is one of personal preference and experience, and perhaps should be based on adverse effects because the benefits are similar. Some patients may benefit from all three equally; some may get adverse effects from one, but not the others. By having these three drugs available, the clinician has the ability to switch from one to another should one of them not be tolerated. Switching can be done rapidly using a ratio of 1:1:5 for pergolide:pramipexole:ropinirole, without having to build up the dose of the new drug from a much lower level. If the response is less than satisfactory, and it is desired to maintain the dopa-sparing strategy for as long as possible, one can then add amantadine or an anticholinergic (see below). If none of these agents is helpful or tolerated, the patient moves into the next stage of illness, the stage in which levodopa is required. Nausea and vomiting are other potential side effects that would limit the usefulness of the dopamine agonists. These symptoms are usually avoided by increasing the dose slowly. The peripherally-acting dopamine antagonist domperidone will block these gastrointestinal side effects. The usual dose is 10–20 mg thrice daily. Even if a dopamine agonist is effective, many patients require the addition of levodopa within a year or two.
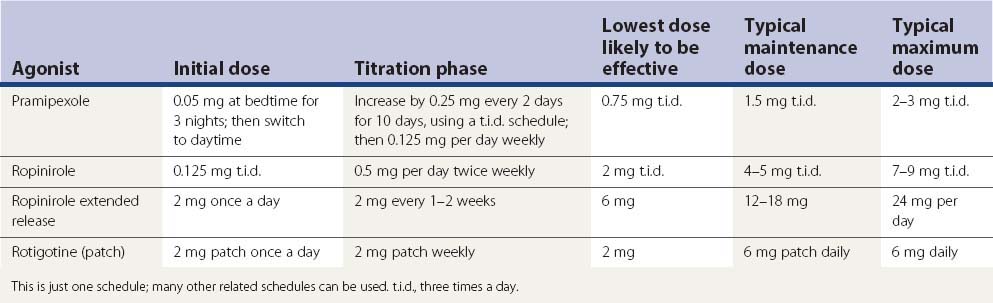
One can quickly switch from one dopamine agonist to another without having to build up the dose slowly for the new one (Canesi et al., 1999). Using a conversion factor, the new agonist can begin at full dosage at the beginning of the day while the current one is suddenly discontinued. A conversion table is provided (Table 6.13), in which pergolide is given at unity (1). Pramipexole is also 1, ropinirole is 5, and bromocriptine is 10. This means that if the dose of pergolide or pramipexole were 2 mg/day, ropinirole and bromocriptine equivalents would be 10 and 20 mg/day, respectively.
| Agonist | Ratio |
|---|---|
| Pergolide | 1 |
| Pramipexole | 1 |
| Ropinirole | 5 |
| Bromocriptine | 10 |
Amantadine
Amantadine is a mild indirect dopaminergic, acting by augmenting dopamine release from storage sites and possibly blocking reuptake of dopamine into the presynaptic terminals. It also appears to have some anticholinergic properties, as well as glutamate receptor blocking activity. In the mild stage of PD it is effective in about two-thirds of patients (Fahn and Isgreen, 1975). A major advantage is that substantial benefit, if it occurs, is seen in a couple of days. Unfortunately, its benefit in more advanced PD is often short-lived, with patients reporting a fall-off effect after several months of treatment in the absence of concomitant levodopa. The mechanism appears to be a depletion of already reduced dopamine stores in the dopaminergic nerve terminals so that the effect of amantadine is exhausted. A common adverse effect is livedo reticularis (a reddish mottling of skin) around the knees; this is not dangerous, although it can be cosmetically a problem for some patients. Occasional adverse effects are ankle edema and visual hallucinosis. Sometimes when the drug is discontinued, there can be a gradual worsening of parkinsonian signs, indicating that the drug has been helpful. The usual dosage is 100 mg twice daily, but sometimes a higher dose (up to 400 mg) may be required.
Amantadine can be useful, not only in the early phases of symptomatic therapy, thereby forestalling the introduction of levodopa or reducing the required dosage of levodopa, but also in the advanced stage of the disease as an adjunctive drug to levodopa and the dopamine agonists. It is also effective in reducing levodopa-induced dyskinesias (Rajput et al., 1997; Metman et al., 1998a; Thomas et al., 2004), probably from its antiglutamatergic activity.
Amantadine is excreted mostly unchanged in the urine, so the dose needs to be reduced in patients with renal impairment. The half-life is long, about 28 hours, so twice daily dosing is adequate. CNS adverse effects of myoclonus, hallucinations, and delirium are associated with very high plasma levels of amantadine, which can be due to overdosage (Fahn et al., 1971) or from impaired renal function (Nishikawa et al., 2009).
Antimuscarinic drugs (anticholinergics)
The anticholinergics are less effective antiparkinson agents than the dopamine agonists. The anticholinergics are estimated to improve parkinsonism by about 20%. Many clinicians find that if tremor is not relieved by an agonist or levodopa, then the addition of an anticholinergic drug is often effective. Sometimes, the anticholinergic can lessen tremor severity even in the absence of levodopa, so clinicians can use such an agent as monotherapy for tremor. If this is not helpful, then continuing to use the drug while a dopamine agonist or levodopa is added can be helpful. Later, if tremor is relieved by the dopaminergic agent, one can try to discontinue the anticholinergic. Commonly used anticholinergics are trihexyphenidyl (Artane) and benztropine mesylate (Cogentin); but there are many others. To minimize adverse effects, start with low doses (trihexyphenidyl 1 mg twice daily; benztropine 0.5 mg twice daily) and increase gradually to 2 mg three times daily for trihexyphenidyl and 1 mg three times daily for benztropine. As would be expected, if anticholinergics lessen parkinsonism, cholinergic agents aggravate parkinsonian symptoms (Duvoisin, 1967), including nicotine (Ebersbach et al., 1999).
There is concern that use of anticholinergic drugs in PD may hasten cognitive decline. In one study, where patients were followed for 8 years, cognitive decline was higher in those who were begun with anticholinergic drugs (Ehrt et al., 2010). Their median decline on the Mini-Mental State Examination (MMSE) was 6.5 points compared with a 1 point decline in those who had not taken such drugs (P = 0.025). The duration of using these drugs also correlated with a greater decline in MMSE scores (P = 0.032).
Another point of view: utilize levodopa as the first drug
There are a number of neurologists who advocate starting with levodopa when symptomatic therapy is needed (Agid, 1998; Weiner, 1999; Factor, 2000), and for the sake of the reader, this point of view should be made known. The argument is that there is no proof that levodopa itself causes either neurotoxicity or the motor complications of dyskinesias and “off” states. Rather, they suggest that it is the severity of the disease that allows these complications to appear with levodopa. Therefore, they prefer to use the most effective drug first when the symptoms are mild in order to provide the highest quality of life. However, despite the ELLDOPA trial (Parkinson Study Group, 2004b), uncertainty about neurotoxicity remains, and until there is a follow-up controlled clinical trial to answer the uncertainties from that trial, the justification can be made that unless a dopamine agonist is not tolerated or effective, levodopa can be delayed until needed (Fahn, 1999).
The ELLDOPA study was a controlled clinical trial evaluating the effect of levodopa on the natural history of PD (Parkinson Study Group, 2004b). Unexpectedly, the clinical results showed that subjects treated with levodopa had less clinical progression 2 weeks after stopping the drug than subjects treated with placebo, and this effect was dose-dependent (Fig. 6.10). But concern has been raised that perhaps the 2-week washout of levodopa was insufficient to eliminate all of its symptomatic benefit. Moreover, the ELLDOPA study showed a discordance between the neuroimaging component and the clinical results. The dopamine transporter binding ligand imaging study was compatible with a more rapid decline of dopamine neurons. But that result has now raised the question as to whether levodopa itself interferes with this binding. Therefore, the interpretation of the ELLDOPA study remains uncertain. On the other hand, a computer simulation of the ELLDOPA results, combined with the DATATOP results, supports the conclusion that levodopa slows disease progression (P.L. Chan et al., 2007). It is of interest that in subsequent clinical trials, such as the PRECEPT study, subjects reach endpoint at a lower level of severity of parkinsonism compared to the DATATOP study which was conducted prior to the ELLDOPA study (Marras et al., 2009). Subjects may feel that starting levodopa sooner is not as serious a threat to their well-being, having seen the ELLDOPA results.
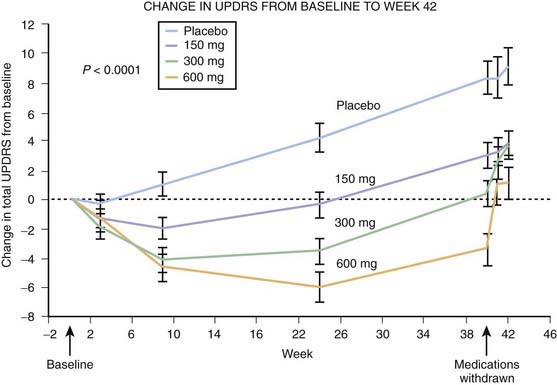
Figure 6.10 Levodopa and the progression of PD.
From Parkinson Study Group. Levodopa and the progression of Parkinson’s disease. N Engl J Med 2004;351(24):2498–2508.
Another argument is that levodopa has delayed mortality, and therefore it should be used early. However, the initial improvement of mortality rates occurred when levodopa was first introduced and most likely was from the improvement of mobility, not from the drug itself. Improved or maintained mobility with another antiparkinson agent would probably be just as effective. Actually, mortality rates have gone back to their earlier increased level, now that levodopa has taken care of the backlog of disabled patients (Clarke, 1995). In a more recent analysis of retrospective data from Scotland (Donnan et al., 2000) comparing PD patients with their treatment assignment, levodopa monotherapy had a higher mortality rate than did selegiline monotherapy or selegiline plus levodopa.
From the DATATOP study, the 387 subjects who reached endpoint, i.e., the need for symptomatic therapy, were placed on levodopa, and their UPDRS scores were reduced by approximately 33% (from approximately 43 units to 29 units) (Fig. 1 in Growdon et al., 1998).
Whether levodopa actually provides a better quality of life (Martinez-Martin, 1998; Glozman et al., 1998) for patients with mild-stage PD remains to be determined, particularly as compared to dopamine agonists in patients with early PD. In one study – the levodopa versus pramipexole controlled clinical trial (Holloway and Parkinson Study Group, 2004) – health-related quality of life (HRQOL) was assessed by three different measures. All three measures resulted in similar profiles over time characterized by initial improvement over the first 3–6 months and followed by a gradual decline in years 2, 3, and 4 (Noyes et al., 2006). The difference in HRQOL between the treatment arms widened in favor of pramipexole in years 3 and 4 for all HRQOL measures used. Because levodopa had a superior result in UPDRS scores, the results suggest that pramipexole and levodopa affect HRQOL via improvement on different domains of well-being: nonmotor effect for pramipexole and mobility improvement for levodopa.
Whether the motor complications that are seen with chronic levodopa therapy in patients with PD are actually caused by long-term levodopa therapy or simply reflect the progression of the disease is unknown and widely debated (de Jong et al., 1987; Quinn et al., 1987; Blin et al., 1988; Roos et al., 1990; Caraceni et al., 1991; Cedarbaum et al., 1991). Advanced disease with altered sensitivity of dopamine receptors is a critical factor, but one does not see these motor complications if the patient was never exposed to levodopa and was treated only with the other antiparkinson agents. In untreated, but advanced PD, levodopa-induced dyskinesias occur shortly after levodopa is started (Onofri et al., 1998). In the parkinsonian states of postencephalitic parkinsonism and MPTP-induced parkinsonism there is rapidly severe depletion of nigral neurons (Bernheimer et al., 1973; Davis et al., 1979; Burns et al., 1983; Langston et al., 1984). These patients may develop dyskinesias and fluctuations within weeks to months after starting levodopa (Calne et al., 1969; Sacks et al., 1970; Duvoisin et al., 1972; Sacks, 1974; Langston et al., 1983; Langston and Ballard, 1984; Ballard et al., 1985). But if patients with those diseases had been treated with dopamine agonists instead of levodopa, it is likely that those motor complications would not have occurred.
If one chooses to use levodopa in the mild-stage disease, there is evidence to suggest keeping the dose as low as possible (Poewe et al., 1986; Lesser et al., 1979). One strategy is to build the dosage from carbidopa/levodopa 25/100 mg to 50/200 mg three times daily and then add a dopamine agonist if the patient needs more symptomatic relief. (For older patients, stay with levodopa and increase that if more medication is needed – see the next section.)
Treatment of moderate-stage Parkinson disease
Moderate-stage PD is when the disability is beyond the scope of efficacy of dopamine agonists, amantadine, anticholinergics, and a MAO-B inhibitor; treatment with levodopa is necessary to control symptoms. The rule of thumb is to utilize the lowest dosage that brings about adequate symptom reversal, not the highest dosage that the patient can tolerate, in an attempt to avoid response fluctuations and dyskinesias. This recommendation seems contra to the ELLDOPA results (Parkinson Study Group, 2004b), which suggests that higher doses may offer more neuroprotection. But interpretation of that study is difficult because a longer duration of benefit may exceed the 2-week washout period, and the imaging results suggest the opposite interpretation, namely that levodopa may enhance loss of dopamine nerve terminals. Until the uncertainties are clarified, we are left with the reality that levodopa dosage contributes to the development of motor complications, as recorded in the ELLDOPA study.
Carbidopa/levodopa is marketed in both immediate release (Sinemet and generics) and extended release (Sinemet CR and generic carbidopa/levodopa ER) tablets; the latter provides a longer plasma half-life and lower peak plasma levels of levodopa compared to standard Sinemet. Unfortunately, Sinemet CR has not been shown to avoid the development of response fluctuations. A 5-year study in 618 dopa-naive patients compared Sinemet CR and standard Sinemet therapy. There was no difference between the two groups in the development of either fluctuations or dyskinesias (Block et al., 1997; Koller et al., 1999). An Italian study found that using small, divided doses during the day is more likely to lead to loss of the long duration response (Zappia et al., 2000).
Immediate release carbidopa/levodopa is available in 10/100, 25/100, and 25/250 mg tablets. Because of the desire to have at least 75 mg per day of carbidopa, one should start with the 25/100 mg tablets when the drug is introduced. An increase by 25/100 mg per day per week until three times daily dosing is achieved is often adequate. A 25/100 mg three times daily dosing is the most common plateau schedule neurologists aim for (Fahn and Mazzoni, 2006). Not every symptom of PD responds equally well. Bradykinesia and rigidity respond best, while tremor can be more resistant. If a response is seen, but with symptoms later returning or worsening, increasing to 50/200 mg three times daily is a reasonable goal before adding a dopamine agonist. If agonists are already being taken, and there is still an inadequate response, the dosage of levodopa should be increased gradually, switching to the 25/250 mg tablets as necessary. A dose of 25/250 mg four times daily may be required. A reasonably high dose before concluding that levodopa is ineffective is 2000 mg of levodopa per day.
A clinical trial was conducted to determine if entacapone should be given with levodopa when the latter is introduced. There was a widespread belief that continuous dopaminergic therapy, which is the method utilized to treat fluctuations and dyskinesias, would also prevent the development of these motor complications to levodopa. (For more detailed discussion on motor complications and continuous dopaminergic stimulation, see the section entitled “Treatment of advanced-stage Parkinson disease.”) Subjects requiring dopaminergic treatment were randomized to be on either levodopa with carbidopa (LC group) or with entacapone added to the tablet (LCE group). The opposite outcome than what was expected happened. The LCE group developed dyskinesias earlier, and more subjects in that group developed dyskinesias compared to the LC group (Stocchi et al., 2010). One possible explanation is that when levodopa dosage equivalents were calculated, the LCE group received higher dosages than the LC group. Based on the results of this study, we cannot recommend entacapone be administered at the beginning of levodopa therapy; it should be reserved for the treatment of fluctuations, as described in the section “Treatment of advanced-stage Parkinson disease.”
A patient’s response or lack of response to levodopa is a very important piece of information to help differentiate PD from parkinsonism-plus syndromes. If the response is nil or minor, it is most likely that the disorder is not PD (Marsden and Fahn, 1982). However, a beneficial response does not ensure that a diagnosis of PD is correct. All cases of presynaptic disorders (e.g., reserpine-induced, MPTP-induced, postencephalitic parkinsonism) will respond to levodopa. Also, patients in the early stages of multiple system atrophy and progressive supranuclear palsy may improve with levodopa; later in these diseases, when dopamine receptors are lost, the response is lost. Some patients with Shy–Drager syndrome and olivopontocerebellar atrophy may continue to have intact dopamine receptors in the striatum and continue to respond to levodopa. The only effective drugs in situations in which levodopa is not effective are the anticholinergics and amantadine, even though only mildly so.
It takes a mean of 9.3 ± 1.8 days to achieve maximum response when beginning treatment with Sinemet CR and 6.8 ± 3.0 days for the parkinsonism severity to return to baseline levels after stopping chronic Sinemet CR treatment (Barbato et al., 1997). This compares to a decay time of 6.2 ± 1.7 days following withdrawal of the dopamine agonist ropinirole (9–21 mg daily). These studies support the concept that the long-duration effect of levodopa and ropinirole might be due to some slowly evolving postsynaptic pharmacodynamic change in the CNS (Barbato et al., 1997).
It is important to avoid sudden withdrawal of levodopa, which is sometimes done for a surgical procedure (Stotz et al., 2004). A “neuroleptic malignant-like” syndrome can ensue, with fever, rigidity, and incoherence (Friedman et al., 1985; Hirschorn and Greenberg, 1988; Gordon and Frucht, 2001; Ueda et al., 2001). Tapering levodopa over 3 days appears to be safe (Parkinson Study Group, 2004b).
As PD worsens, the duration of effectiveness of a dose of levodopa becomes shorter (Contin et al., 1998a). The effective clinical half-life of a dose of levodopa declines in relation to both worsening of symptoms and duration of disease. While the pharmacokinetic half-life remains constant, around 90 minutes, the clinical half-life declines from a mean of 262 minutes in stage I patients, to 142 minutes in stage II, to 54 minutes in stage III. The rate of decline becomes smaller as the disease progresses. The mean annual reduction of the effective half-life slows down as the disease worsens, dropping by 37 minutes/year in stage I patients and by 6.5 minutes/year in stage III, and is about 17% per year of the disease. Contin and colleagues (1998a) determined this “effective” half-life by administering a standard oral fasting dose (100 mg) of levodopa (with a peripheral decarboxylase inhibitor). Motor response was measured by finger tapping speed and walking speed. The decline in effective half-life is equivalent to the loss of the long-duration response from levodopa to only a short-duration response as the disease worsens, and as first reported by Muenter and Tyce (1971).