Chapter 4 Mechanisms of Re-entrant Arrhythmias
The most deadly cardiac arrhythmias result from re-entry, that is, electrical waves that rotate at a high frequency, in a self-sustaining manner. These waves give rise to electrical activity that propagates throughout the ventricles in complex ways. Spontaneous re-entry often occurs as a consequence of a wavebreak produced by the interaction of a propagating wavefront with a functional or anatomic obstacle.1 It is important to note, however, that the waves that break and initiate re-entry may be generated by electrical pacemaker discharges, triggered activity (i.e., early or delayed after-depolarizations), or another re-entry circuit. The objective of this chapter is to briefly examine the mechanisms of re-entrant arrhythmias. First, the chapter provides a brief historical perspective on the mechanisms of initiation and maintenance of reentry, including those mechanisms thought to underlie tachycardia and fibrillation. Emphasis is placed on the concepts derived from the theory of nonlinear wave of propagation in generic excitable media. This is followed by a summary of the work from the authors’ laboratory pertaining to functional re-entry in numerical simulations, engineered mouse models, and larger animals, as well as two-dimensional monolayers of neonatal rat ventricular myocytes. The aim of the chapter is to provide the interested reader with information to enhance didactic, clinical, or research endeavors.
What Is Re-entry?
In its simplest form, re-entry is the circulation of the cardiac impulse around an obstacle; this leads to repetitive excitation of the heart at a frequency that depends on the conduction velocity of the circulating impulse and the perimeter of the obstacle (Figure 4-1).2 According to the original description by George Mines, which was published in The Journal of Physiology in 1913, re-entry occurs around a fixed anatomic obstacle, and the physical disruption of the surrounding circuit will interrupt the activity. As illustrated in Figure 4-1, the initiation of the re-entrant activity depends on the occurrence of a unidirectional block in which activation takes place in only one direction within the circuit.
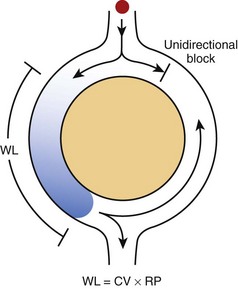
It is clear from Figure 4-1 that the rotation time around the circuit should be longer than the recovery period of all segments of the circuit. The extra time required for the impulse to successfully complete a rotation may result from a relatively large circuit, a relatively slow conduction velocity of the impulse, or the relatively short duration of the refractory period. Hence, the “wavelength,” which may be calculated roughly as the product of the refractory period and the conduction velocity, must be shorter than the perimeter of the circuit. An excitable region will separate the front of the impulse from its own refractory tail (i.e., excitable gap) and re-excitation will ensue.
The classic model of anatomically determined re-entry depicted in Figure 4-1 is directly applicable to specific cases of tachyarrhythmias. These include supraventricular tachycardias occurring within the AV node or those using accessory pathways, as well as bundle branch re-entrant tachycardia. However, other types of re-entrant arrhythmias require somewhat different explanations for their mechanisms. For example, the cellular basis of closely coupled ventricular extrasystoles initiated somewhere in the Purkinje fiber network can be explained by the so-called reflection mechanism described by Antzelevitch et al in 1980 (Figure 4-2, A).3 Recently, reflection to and from an accessory pathway was shown to be a potential mechanism for the initiation of atrial fibrillation in patients with manifest (pre-excited) Wolff-Parkinson-White (WPW) syndrome.4
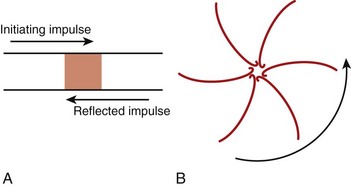
In the absence of a predetermined obstacle or circuit (Figure 4-2, B), however, many tachyarrhythmias that originate in the myocardium (atrial or ventricular) require mechanisms whereby re-entrant activation may occur as vortices of electrical excitation rotating around an area of myocardium. Accordingly, the impulse must circulate around a region of quiescence. In 1977, Allessie et al5 explained such functionally determined re-entry by proposing the so-called leading circle hypothesis, with its two variants of “anisotropic” re-entry described in 1988 by Dillon et al6 and “figure-of-8” re-entry proposed by El-Sherif7 in 1985. A somewhat different postulate for vortex-like re-entry, the “spiral wave re-entry” hypothesis, was put forth by Davidenko et al8 in 1992 and is derived from the theory of wave propagation in excitable media. Spiral wave re-entry attempts to provide a unifying explanation for the mechanisms of monomorphic and polymorphic ventricular tachycardias as well as the mechanism of fibrillation.
Circus Movement Re-entry
Functionally Determined Re-entry
In 1924, Garrey9 presented in an article in Physiological Reviews the first description of re-entrant excitation in the absence of anatomic obstacles in experimental studies on circus movement in the turtle heart. Garrey’s observations suggested that point stimulation of the atrium was sufficient to initiate a regular wave of rotation around the stimulus site. Subsequently, in 1946, Wiener and Rosenblueth10 developed the first mathematical model of circus movement re-entry, which supported waves of rotation around a sufficiently large barrier, but they could not demonstrate re-entry in the absence of an obstacle. This prompted Wiener and Rosenblueth to suggest that perhaps Garrey may have unwittingly produced a transient artificial obstacle near the stimulation site.
The “Leading Circle” Model
According to the leading-circle concept of Allessie and colleagues, in the absence of an anatomic obstacle, the dynamics of re-entry are determined by the smallest possible loop in which the impulse can continue to circulate. Under these conditions, the wavefront must propagate through relatively refractory tissue, in which case no “fully excitable gap” will be present and the wavelength will be very close to the length of the circuit. The leading circle idea paved the way for major advances in the understanding of functional re-entry. It served as a platform for developing a unifying hypothesis (spiral wave re-entry) that explains most of the major properties of functionally determined re-entry, which are commonly observed in normal cardiac muscle in experiments. This includes the phenomenon of re-entry “drift” described by Davidenko et al8 in 1992 and Pertsov et al11 in 1993. Re-entry drift results in beat-to-beat changes in the location of the rotation center (see Drifting Vortices and Ventricular Fibrillation).
Anisotropic Re-entry
In 1986, Spach and Dolber12 implicated microscopic structural complexities of the cardiac muscle in the mechanism of re-entrant activation in both atria and ventricles, particularly in relation to the orientation of myocardial fibers, the manner in which the fibers and fiber bundles are connected to each other, and the effective electrical resistivities that depend on fiber orientation. Because of these structural properties, propagation velocity in the cardiac muscle is three to five times faster in the longitudinal axis of the cells than along the transverse axis. Mapping studies performed by Peters and Wit13 in 1998, using multiple extracellular electrodes have shown that, in the setting of myocardial infarction, re-entry may occur in the survival epicardial rim of tissue. Under such conditions, the wave circulates around a functionally determined elongated region of block, the so-called line of conduction block (Figure 4-4, A). Based on the orientation of the line of block, it was thought that anisotropic propagation played a major role both in the initiation as well as in the maintenance of re-entry in ventricular tissue surviving a myocardial infarction (Figure 4-4, B). In addition, propagation velocity is exceedingly slow at the edges of the lines of block, which has also been attributed to anisotropic propagation.
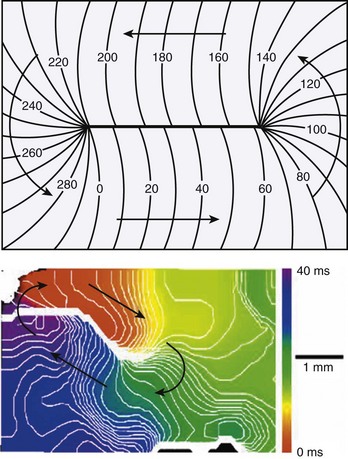
Figure-of-8 Re-entry
Figure-of-8 re-entry, described in 1987 by El-Sherif, Gough, and Restivo, has been recognized as an important pattern of re-entry in the late stages of myocardial infarction. In most cases, two counter-rotating waves coexist at a relatively short distance from each other (Figure 4-5). As described for the case of single re-entrant circuits, each wave of the figure-of-8 re-entry circulates around a thin line or arc of block. The region separating the lines of block is called the common pathway. A detailed description of the common pathway is of great practical importance, since there is evidence that it could be a strategic region for surgical or catheter ablation in this type of re-entry. In fact, unlike other forms of functionally determined re-entry, figure-of-8 re-entry may, indeed, be interrupted by physical disruption of the circuit. Several studies have attempted to describe the characteristics of propagation in the common pathway. However, the properties of the common pathway are still not clearly defined. It effectively behaves like an isthmus limited by two functionally determined barriers. In addition, there are two wavefronts that interact in the common pathway. As a result, propagation may be determined by a combination of factors other than those analyzed in most experimental studies such as anisotropy. The study of propagation across an isthmus and the influence of wavefront curvature may have significant implications in understanding the properties of the common pathway, as discussed in the next section.
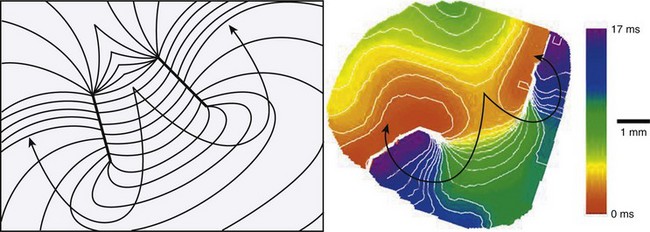
Spiral Wave Re-entry
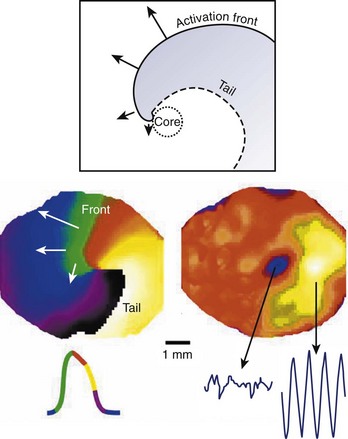
According to traditional concepts proposed by Allessie and colleagues, circus movement re-entry may be initiated in the heart because block is predetermined by the inhomogeneous functional characteristics of the tissue, whereas spiral waves could be formed in the heart even if cardiac muscle was completely homogeneous in its functional properties as shown by Davidenko et al8 in 1992, Pertsov et al11 in 1993, and Winfree14 in 1998. This is because the initiation of rotating activity may depend solely on transient local conditions (e.g., the conditions created by cross-field stimulation).1 Moreover, according to the traditional concept of re-entry, the circulation of the activity occurs around an anatomically or functionally predetermined circuit, and the rotating activity cannot drift. In other words, the circuit gives rise to and maintains the rotation. However, spiral waves occur due to initial curling of the wavefront; in fact, the curvature of the wavefront determines the size and shape of the region, called the core, around which activity rotates (Figure 4-6). Importantly, the core remains unexcited by the extremely curved activation front and it is readily excitable.1 This explains the mechanism underlying the drift of spirals.
Modes of Initiation of Spiral Wave Re-entry
In 1946, Weiner and Rosenblueth10 published a theoretical description of the mechanisms of initiation of flutter and fibrillation in cardiac muscle in the presence as well as in the absence of anatomic obstacles. They proposed that wave rotation around single or multiple obstacles was required for the initiation and maintenance of both types of arrhythmias, which they assumed to result from a single re-entrant mechanism.
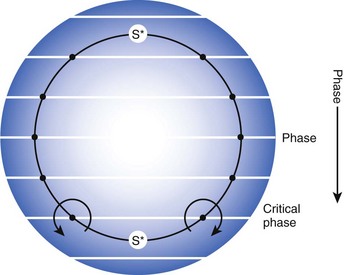
More than three decades later, another theory of initiation of vortices in two dimensions was suggested, and it has been supported by experiments in a number of different excitable media. It is based on Winfree’s “pinwheel experiment” protocol carried out in 1990.15 As shown in Figure 4-7, this protocol involves crossing a spatial gradient of momentary stimulus with a spatial gradient of phase (i.e., refractoriness, established by prior passage of an activation front through the medium). In accordance with this theory, when a stimulus of the right size (S*) is given at the proper time, mirror image vortices begin to rotate around crossings of critical contours of transverse gradients of phase and stimulus intensity. On the basis of this theory, a vulnerable domain was described. Its timing occurred just before complete recovery from previous excitation. Thus, with its limits of timing and stimulus intensity, the idea of vulnerable domain was similar to the empirical concept of the vulnerable period. In a 1988 study, Shibata et al16 demonstrated the application of Winfree’s theory to the induction of ventricular fibrillation (VF) in the heart. They concluded that the response to administered shocks during the vulnerable period is a complex one. However, in accordance with theory, during pacing of the ventricles, if a shock of the proper amplitude and delay is applied during the vulnerable period, two counter-rotating vortices can be formed. Thus, as predicted by theory, vortices can be formed even in the normal myocardium. Subsequently, in 1989, Frazier et al17 used an extracellular recording array with a modification of the pinwheel experiment, the so-called twin-pulse protocol, to demonstrate the mechanism of re-entry and fibrillation in the canine heart. They used the term critical point to refer to a phase singularity and provided strong support for what is referred to as the critical point hypothesis for the initiation of vortex-like re-entry and fibrillation. They also demonstrated that an upper limit of vulnerability for VF exists and that during the vulnerable period, if shock with a strength that is larger than a certain limit is applied, then VF will most likely not be induced.
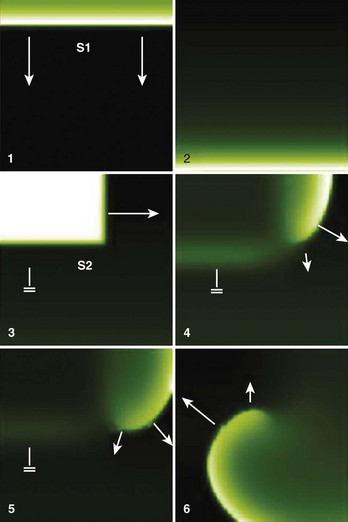
Another approach for initiating vortices is the cross-field stimulation protocol. This method is different from the pinwheel protocol in that it does not require a large stimulus. As shown in Figure 4-8, in cross-field stimulation, a conditioning stimulus (S1) is used to initiate a plane wave propagating in one direction. Subsequently, a second stimulus, S2, is applied perpendicular to S1 and timed in such a way as to allow interaction of the S2 wavefront with the recovering tail of the S1 wave. The S2 wavefront cannot invade the refractory tissue at the site of the interaction with the S1 wave tail; consequently, a wavebreak or phase singularity is formed at the end of the S2 wave, and rotation about this point occurs.
Spontaneous Formation of Rotors
A major contribution of wave propagation theory in excitable media to the understanding of the mechanisms of initiation re-entrant arrhythmias is the concept of wavebreak, which explains how the interaction of a wavefront with an obstacle can lead to wavefront fragmentation and rotor formation.14 The re-entrant wave can begin as a single vortex, as a pair of counter-rotating vortices, or as two pairs of counter-rotating vortices.
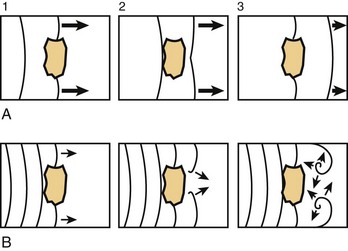
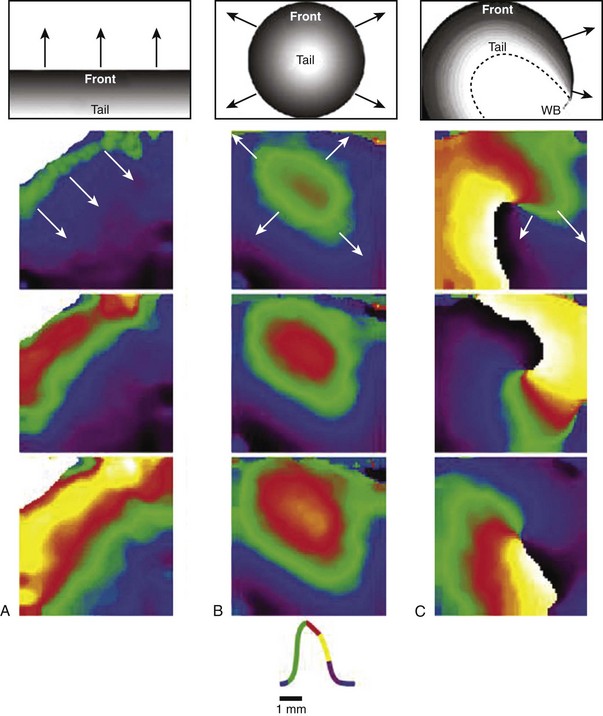
The concept of wavebreak is illustrated schematically in Figure 4-9, which shows the dynamics of the interaction of a wavefront with an anatomic obstacle in a two-dimensional sheet of cardiac tissue with two different excitability conditions. In A, when tissue excitability is normal, after circumnavigating the obstacle, the broken ends of the wave join together, the previous shape of the wavefront is regained, and the wave continues. However, in B, when excitability is low, the broken ends of the wave do not fuse. Instead, the broken ends rotate in the opposite direction. As illustrated by the diagrams in Figure 4-10, during “normal” propagation, which is initiated by a linear source (planar wave, A) or a point source (circular wave, B), the wavefront is always followed by a recovery band or wave tail. Under these conditions, the front and tail never meet, and the distance between them corresponds to the wavelength of the excitation. In contrast, as shown in C, the broken waves demonstrate a unique feature whereby the front and the tail meet at the wavebreak. In this situation, the wavefront curls, and its velocity decreases toward the wavebreak. In fact, at the wavebreak, the curvature is so pronounced that the wavefront fails to activate the tissue ahead. Consequently, the wavebreak effectively serves as a pivoting point, which forces the wavefront to acquire a spiral shape as it rotates around the core.1
Multitudes of obstacles, both anatomic and functional, are present in cardiac tissue. However, the excitation of the heart, which is triggered by signals that originate in the sinus node and subsequently propagate throughout the atria and the ventricles, occurs repeatedly in a rhythmic manner. This process occurs without the induction of arrhythmias because the normal sequence of activation through the His-Purkinje system prevents the formation of wavebreaks. Consequently, the presence of obstacles is not a sufficient condition for the establishment of re-entry. Using a voltage-sensitive dye in conjunction with a high-resolution video imaging system, Cabo et al demonstrated that certain critical conditions must be met in order for unexcitable obstacles to destabilize propagation and produce self-sustained vortices that result in uncontrolled high-frequency stimulation of the heart.18 They demonstrated that the critical condition is the excitability of the tissue such that when the tissue excitability is low, a broken wave will contract and vanish (i.e., conduction will be blocked). However, at an intermediate level of excitability, the broken wave detaches from the barrier and forms a vortex in a manner visually similar to the separation of the main stream from a body in a hydrodynamic system, where there is subsequent eddy formation during turbulence. Moreover, Cabo et al demonstrated that high-frequency stimulation, which decreases excitability, also resulted in the detachment of the broken wave and the generation of vortices in the presence of anatomic obstacles. This phenomenon has been termed vortex shedding. In summary, the dynamics of wavebreaks are determined by (1) the critical curvature of the wavefront (i.e., the curvature at which propagation fails), (2) the excitability of medium, and (3) the frequency of stimulation or wave succession.
Role of Wavebreaks in Ventricular Fibrillation
Under normal conditions of excitability and stimulation, the interaction of the wavefront with an obstacle does not produce a wavebreak. However, when the excitability is lowered, wavebreaks may be initiated and persist after the collision of the front with the appropriate anatomic or functional obstacles. As predicted by theory, the obstacle size must be equal to or greater than the width of the wavefront for perturbation of propagation to occur. At a propagation speed of 50 cm/s, the wavefront width (i.e., the spatial spread of the action potential upstroke) in normal cardiac muscle is approximately 1 mm.1 Consequently, obstacles of 1 mm or larger have the potential to generate wavebreaks in the propagating waves and producing vortex-like re-entry.
Clearly, numerous conditions can lead to the formation of wavebreaks. However, what is the relationship between wavebreaks and ventricular fibrillation (VF)? The authors hypothesize that the numerous fragmented wavefronts observed during VF form as the result of the interaction of waves emanating from a high-frequency source with the obstacles present in cardiac tissue.1 Because of their lateral instability, some waves may shrink and undergo decremental conduction, but other waves may continue unchanged until annihilated by other waves. Still others may undergo curling and form new rotors. The final result is the fragmentation of the mother waves away from the source into multiple short-lived daughter waves that produce a complex pattern of propagation during VF.
Mechanisms of Maintenance of Ventricular Fibrillation
Is Ventricular Fibrillation Random or Organized?
On the basis of his cinematographic studies in 1940, Wiggers19 concluded that VF could not be adequately described as an asynchronous contraction of myocardial fibers. Wiggers observed that the lack of coordination and asynchrony initially involves comparatively large sections of the myocardium, which progressively multiply and decrease in size as fibrillation continues; however, the study showed that even in the later stages of fibrillation, asynchronous contraction of adjacent fibers does not seem to occur. These observations are in agreement with the notion that VF arises from wandering wavefronts that are ever changing in direction and number. Furthermore, it is possible that the fragmentation of the wavefront into multiple independent wavelets may arise from the interaction of the wavefront with obstacles and with the refractory tails of other waves. As the front breaks, some waves may shrink and cease to exist (i.e., decremental propagation), others may propagate until terminated by the collision with other waves or boundaries, and still others may give rise to new vortices.1 The product of such phenomena may be the complex patterns of propagation that characterize VF. However, currently, ample evidence in the literature suggests that VF is not entirely a random phenomenon. A summary of the studies that have documented “organization” during VF follows.
In 1981, Ideker and colleagues20 documented that ventricular activation during the transition to VF arises near the border of the ischemic–reperfused region of the canine heart and is organized as it passes across the nonischemic tissue, but the body surface electrocardiogram (ECG) appears disorganized as judged by the variable spacing between successive, coexistent activation fronts. In 1992, Damle et al21 demonstrated that epicardial activation during VF in a canine model of healing infarction is not random. Moreover, they showed that during VF, spatial as well as temporal “linking” of activation occurs; in this phenomenon, the same path of conduction is traversed by several consecutive wavefronts in a relatively rhythmic manner.
Garfinkel et al used nonlinear dynamics theory to study fibrillation in a computer model and three stationary forms of arrhythmias: (1) in human chronic atrial fibrillation, (2) in a stabilized form of canine VF, and (3) in fibrillation-like activity in thin sheets of canine and human ventricular tissues.22 They found that fibrillation arose through a quasi-periodic stage of period and amplitude modulation; thus, they concluded that fibrillation is a form of spatio-temporal chaos. Bayly et al explored several techniques to quantify spatial organization during VF.23 They used epicardial electrograms recorded from porcine hearts by using rectangular arrays of unipolar extracellular electrodes and concluded that VF is neither “low-dimensional chaos” nor “random” behavior but, rather, a high-dimensional response with a degree of spatial coherence.23
The development of an analytic technique by Gray et al, which markedly reduces the amount of data required to depict the complex patterns of fibrillation, has enabled investigators to study the detailed dynamics of wavelets and rotors, including their initiation, life span, and termination.24 Using a fluorescent potentiometric dye and video imaging, Gray et al recorded the dynamics of transmembrane potentials from a large region of the heart and determined that transmembrane signals at many sites exhibit a strong periodic component. With this analysis, the periodicity is seen as an attractor in two-dimensional phase space, and each site can be represented by its phase around the attractor. Using spatial phase maps at each instant in time, Gray et al revealed the “sources” of fibrillation in the form of topologic defects, or phase singularities (a term coined by Winfree in 1990), at several sites.24 Thus, they demonstrated that a substantial amount of spatial and temporal organization underlies cardiac fibrillation in the whole heart.24
The authors of this chapter, using isolated Langendorff-perfused rabbit heart, demonstrated organization during VF in the form of sequences of wave propagation that activated the ventricles in a spatially and temporally similar fashion.1 Furthermore, the frequency of the periodic activity was shown to correspond to the dominant peak in both the global bipolar electrogram and the optical pseudo-ECG, which suggests that the sources of the periodic activity are the dominant sources that maintain VF in this model. Moreover, quantification of wavelets revealed that during VF, wavebreaks underlie wavelet formation; however, the breakup of rotor waves was not a robust mechanism for the maintenance of VF. Overall, the results suggested that the organized activity of periodic sources is responsible for most of the frequency content of VF and is therefore important for the maintenance of this arrhythmia.1
Rotors and Ventricular Fibrillation
Rotors are thought to be the major organizing centers of re-entrant arrhythmias, so much investigation has focused on rotors as the underlying mechanism for VF in the heart. However, two schools of thought have emerged. On one hand, many recently proposed mechanisms for fibrillation have focused on the transience and instability of rotors. These mechanisms suggest that the breakup of rotors results in the “turbulent” nature of fibrillation. One such mechanism, the restitution hypothesis, suggests that fractionation of the rotor ensues when the oscillation of the action potential duration is of sufficiently large amplitude to block conduction along the wavefront.22,25 Another mechanism for breakup focuses on the fact that propagation within the three-dimensional myocardium is highly anisotropic because of the intramural rotation of fibers; this produces the twisting and instability of the organizing center (filament), which results in multiplication following repeated collisions with boundaries in the heart.26
Studies by the authors of this chapter have also focused on rotors as the primary engines of fibrillation. However, here the breakup of the rotor is not regarded as the underlying mechanism of VF. Rather, it is proposed that VF is a problem of self-organization of nonlinear electrical waves with both deterministic and stochastic components.1,24 This has led to the hypothesis that there is spatial as well as temporal organization during VF in the structurally normal heart, although there is a wide spectrum of behavior during fibrillation. On one end, it has been demonstrated that a single drifting rotor can give rise to a complex pattern of excitation that is reminiscent of VF.27 On the other end, it has been suggested that VF is the result of a high-frequency, stable source and that the complex patterns of activation are the result of the fragmentation of emanating electrical activity from that source (i.e., fibrillatory conduction).28,29 In the following sections, these two extremes are examined.
Drifting Vortices and Ventricular Fibrillation
Using optical mapping in the structurally normal isolated Langendorff-perfused rabbit heart, Gray et al studied the applicability of spiral wave theory to VF.24,27 In that study, they demonstrated the presence of a drifting rotor on the epicardial surface of the heart. Simultaneous recording of a volume-conducted ECG and fluorescence imaging showed that a single, rapidly moving rotor was associated with turbulent polymorphic electrical activity, which was indistinguishable from VF. It was assumed that rotors were the two-dimensional epicardial representation of a three-dimensional scroll wave. In addition, computer simulations incorporating a realistic three-dimensional heart geometry and appropriate model parameters demonstrated the ability to form a rapidly drifting rotor similar to that observed in the experiments.24,27 Frequency analysis of the irregular ECGs for both experiments and simulations showed spectra that were consistent with previously published data. Furthermore, Gray et al confirmed, through the Doppler relationship, that the width of the frequency spectrum can be related to the frequency of the rotation of the rotor, the speed of its motion, and the wave speed.30
Fibrillatory Conduction
Some forms of fibrillation depend on the uninterrupted periodic activity of discrete re-entrant circuits. The faster rotors act as dominant frequency sources that maintain the overall activity. The rapidly succeeding wavefronts emanating from these sources propagate throughout the ventricles and interact with tissue heterogeneities, both functional and anatomic, leading to fragmentation and wavelet formation.1
Zaitsev et al,28 using spectral analysis of optical epicardial and endocardial signals for sheep ventricular slabs, have provided additional evidence suggesting that fibrillatory conduction may be the underlying mechanism of VF. Zaitsev and colleagues presented data showing that the dominant frequencies of excitation do not change continuously on the ventricular surfaces of slabs. Rather, the frequencies are constant over regions termed domains; moreover, only a small number of discrete domains are found on ventricular surfaces. Zaitsev and colleagues28 also demonstrated that the dominant frequency of excitation in the adjacent domains is often related to the fastest dominant frequency domain in 1 : 2, 3 : 4, or 4 : 5 ratios, and this was suggested to be the result of intermittent, Wenckebach-like conduction block at the boundaries between domains.28 Thus, they concluded that in their model, VF could have resulted from a sustained high-frequency, three-dimensional intramural scroll wave, which created complex patterns of propagation as the result of fragmentation when waves emanating from a high-frequency scroll interacted with tissue heterogeneities.
Samie et al29 presented new evidence in the isolated Langendorff-perfused guinea pig heart that strongly supports the hypothesis that fibrillatory conduction from a stable high-frequency re-entrant source is the underlying mechanism of VF. Samie et al29 obtained optical recordings of potentiometric dye fluorescence from the epicardial ventricular surface along with a volume-conducted “global” ECG. Spectral analysis of optical signals (pixel by pixel) was performed, and the dominant frequency (DF, peak with maximal power) from each pixel was used to generate a DF map. Pixel-by-pixel fast Fourier transformation (FFT) analysis revealed that DFs are distributed throughout the ventricles in clearly demarcated domains. The highest frequency domains are always found on the anterior wall of the left ventricle. Correlation of rotation frequency of rotors and the fastest DF domain strongly suggests that rotors are the underlying mechanism of the fastest frequencies. Further analysis of optical recordings has demonstrated that fragmentation of wavefronts emanating from high-frequency rotors occurs near the boundaries of the DF domains. The results demonstrate that in the isolated guinea pig heart, a high-frequency re-entrant source that remains stationary in the left ventricle is the mechanism that sustains VF.
Moreover, experiments and simulations have attributed the stabilization of the high-frequency rotors driving VF in the left ventricle of the guinea pig heart to the presence of a gradient in the inwardly rectifying potassium (K) current (IK1) whereby IK1 is larger in the left ventricle compared with that in the right ventricle.29 IK1 is a K current that (1) contributes to the resting membrane potential, (2) controls the approach of the membrane voltage to the range where sodium (Na) channels activate to give rise to the upstroke, and finally (3) modulates the final phase of repolarization. However, in the experiments of Samie et al, there no direct link was demonstrated at the molecular level between the stability of rotors in the left ventricle of the fibrillating guinea pig heart and IK1.29 As a result, Noujaim and colleagues31 used a murine model of cardiac-specific Kir2.1 upregulation, where IK1 density was consequently increased by about 12-fold to investigate rotor behavior in such a substrate. It was found that the increased IK1 serves to stabilize and accelerate rotors responsible for re-entrant VT and VF.31 The experiments and numerical simulations suggested that during re-entry, the larger IK1 accelerates conduction velocity, decreases the core size, and hyperpolarizes the resting membrane potential.31 The combination of these factors leads to the generation of very fast and stable rotors. As discussed in earlier sections, wavebreaks and fibrillatory conduction can be characteristics of VF. The authors of this chapter recently examined the role of the slow component of the delayed rectifier K current (IKs) in wavebreak formation and fibrillatory conduction.32 In single cells, IKs has been shown to contribute to repolarization and postrepolarization refractoriness, where because of its slow kinetics of activation and deactivation, IKs accumulates in a deactivated state in response to fast, repetitive stimuli.32 As a result, the pool of deactivated channels will serve to oppose a carefully timed depolarization stimulus, even after the myocyte has fully repolarized and the Na channels have recovered from inactivation; this leads to the failure of action potential generation, hence the notion of postrepolarization refractoriness. Using numerical simulations and monolayers of neonatal rat ventricular myocytes expressing IKs via viral transfer of KvLQT1-minK fusion protein (the respective α- and β-subunits of the channel responsible for carrying IKs), Munoz et al showed that IKs is an important player in the formation of wavebreaks and fibrillatory conduction during excitation patterns that closely resemble those recorded during ventricular fibrillation.32
1 Jalife J. Ventricular fibrillation: Mechanisms of initiation and maintenance. Annu Rev Physiol. 2000;62:25-50.
2 Mines GR. On dynamic equilibrium in the heart. J Physiol. 1913;46:349-383.
3 Antzelevitch C, Jalife J, Moe GK. Characteristics of reflection as a mechanism of reentrant arrhythmias and its relationship to parasystole. Circulation. 1980;61:182-191.
4 Schwieler JH, Zlochiver S, Pandit SV, et al. Reentry in an accessory atrioventricular pathway as a trigger for atrial fibrillation initiation in manifest Wolff-Parkinson-White syndrome: A matter of reflection? Heart Rhythm. 2008;5(9):1238-1247.
5 Allessie MA, Bonke FI, Schopman FJ. Circus movement in rabbit atrial muscle as a mechanism of tachycardia. III. The “leading circle” concept: A new model of circus movement in cardiac tissue without the involvement of an anatomical obstacle. Circ Res. 1977;41:9-18.
6 Dillon S, Allessie MA, Ursell PC, Wit AL. Influence of anisotropic tissue structure on reentrant circuits in the subepicardial border zone of subacute canine infarcts. Circ Res. 1988;63:182-206.
7 El-Sherif N. The figure 8 model of reentrant excitation in the canine post-infarction heart. In: Zipes DP, Jalife J, editors. Cardiac electrophysiology and arrhythmias. Orlando, FL: Grune & Stratton; 1985:363-378.
8 Davidenko JM, Pertsov AV, Salomonsz R, et al. Stationary and drifting spiral waves of excitation in isolated cardiac muscle. Nature. 1992;355:349-351.
9 Garrey WE. Auricular fibrillation. Physiol Rev. 1924;4:215-250.
10 Weiner N, Rosenblueth A. The mathematical formulation of the problem of conduction of impulses in a network of connected excitable elements, specifically in cardiac muscle. Arch Inst Cardiol Mex. 1946;16:205-265.
11 Pertsov AM, Davidenko JM, Salomonsz R, et al. Spiral waves of excitation underlie reentrant activity in isolated cardiac muscle. Circ Res. 1993;72:631-650.
12 Spach MS, Dolber PC. Relating extracellular potentials and their derivatives to anisotropic propagation at a microscopic level in human cardiac muscle. Evidence for electrical uncoupling of side-to-side fiber connections with increasing age. Circ Res. 1986;58:356-371.
13 Peters NS, Wit AL. Myocardial architecture and ventricular arrhythmogenesis. Circulation. 1998;97:1746-1754.
14 Winfree AT. Evolving perspectives during 12 years of electrical turbulence. Chaos. 1998;8(1):1-19.
15 Winfree AT. Vortex action potentials in normal ventricular muscle. Ann N Y Acad Sci. 1990;591:190-207.
16 Shibata N, Chen PS, Dixon EG, et al. Influence of shock strength and timing on induction of ventricular arrhythmias in dogs. Am J Physiol. 1988;255:H891-H901.
17 Frazier DW, Wharton JM, Wolf PD, et al. Mapping the electrical initiation of ventricular fibrillation. J Electrocardiol. 1989;22(Suppl):198-199.
18 Cabo C, Pertsov AM, Davidenko JM, et al. Vortex shedding as a precursor of turbulent electrical activity in cardiac muscle. Biophys J. 1996;70(3):1105-1111.
19 Wiggers CJ. The mechanism and nature of ventricular fibrillation. Am Heart J. 1940;20:399-412.
20 Ideker RE, Klein GJ, Harrison L, Smith W, Kassell J, Reimer K, Wallace A, Gallagher J. The transition to ventricular fibrillation induced by reperfusion after acute ischemia in the dog: A period of organized epicardial activation. Circulation. 1981;63:1371-1379.
21 Damle RS, Kanaan NM, Robinson NS, et al. Spatial and temporal linking of epicardial activation directions during ventricular fibrillation in dogs. Evidence for underlying organization. Circulation. 1992;86:1547-1558.
22 Garfinkel A, Chen PS, Walter DO, et al. Quasiperiodicity and chaos in cardiac fibrillation. J Clin Invest. 1997;99(2):305-314.
23 Bayly PV, KenKnight BH, Rogers JM, et al. Spatial organization, predictability, and determinism in ventricular fibrillation. Chaos. 1998;8(1):103-115.
24 Gray RA, Pertsov AM, Jalife J. Spatial and temporal organization during cardiac fibrillation. Nature. 1998;392(6671):75-78.
25 Weiss JN, Garfinkel A, Karagueuzian HS, et al. Chaos and the transition to ventricular fibrillation—a new approach to antiarrhythmic drug evaluation. Circulation. 1999;99(21):2819-2826.
26 Fenton F, Karma A. Vortex dynamics in three-dimensional continuous myocardium with fiber rotation: Filament instability and fibrillation. Chaos. 1998;8(1):20-47.
27 Gray RA, Jalife J, Panfilov AV, et al. Mechanisms of cardiac fibrillation. Science. 1995;270(5239):1222-1223. author reply 1224–1225, 1995
28 Zaitsev AV, Berenfeld O, Mironov SF, et al. Distribution of excitation frequencies on the epicardial and endocardial surfaces of fibrillating ventricular wall of the sheep heart. Circ Res. 2000;86(4):408-417.
29 Samie FH, Berenfeld O, Anumonwo J, et al. Rectification of the background potassium current: A determinant of rotor dynamics in ventricular fibrillation. Circ Res. 2001;89(12):1216-1223.
30 Gray RA, Jalife J, Panfilov A, et al. Nonstationary vortexlike reentrant activity as a mechanism of polymorphic ventricular tachycardia in the isolated rabbit heart. Circulation. 1995;91(9):2454-2469.
31 Noujaim SF, Pandit SV, Berenfeld O, et al. Up-regulation of the inward rectifier K+ current (IK1) in the mouse heart accelerates and stabilizes rotors. J Physiol. 2007;578(Pt 1):315-326.
32 Munoz V, Grzeda KR, Desplantez T, et al. Adenoviral expression of IKs contributes to wavebreak and fibrillatory conduction in neonatal rat ventricular cardiomyocyte monolayers. Circ Res. 2007;101(5):475-483.