[level-membership-for-anesthesiology-category]
Mechanisms of hepatic drug metabolism and excretion
Hepatic clearance
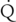 hepatic). Disease-induced or anesthetic-induced reductions in total hepatic blood flow are the principal causes for diminished hepatic clearance for a large number of drugs; the elimination of these drugs is termed flow-limited. Other factors affecting hepatic clearance include maximal hepatic metabolic activity, expressed as intrinsic clearance:
hepatic). Disease-induced or anesthetic-induced reductions in total hepatic blood flow are the principal causes for diminished hepatic clearance for a large number of drugs; the elimination of these drugs is termed flow-limited. Other factors affecting hepatic clearance include maximal hepatic metabolic activity, expressed as intrinsic clearance:< ?xml:namespace prefix = "mml" />
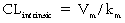
where Vm = maximal metabolic rate (mg/min) and km (Michaelis constant) = drug concentration producing the half-maximal metabolic rate (mg/L). In this case drug elimination is termed capacity-limited. In this situation, unlike the flow-limited condition, drug elimination may change as a function of free-drug concentration that is available for hepatic metabolization and may, thus, be affected by the amount of protein binding and disease-induced changes in protein binding. Whether the hepatic elimination of a drug is flow-limited or capacity-limited depends on the ratio of the free plasma concentration of the drug to km (flow-limited if < 0.5) and that of the CLintrinsic to total hepatic blood flow (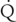 hepatic) of the drug, which determines the extraction ratio (ER) of the drug (ER = CLhepatic/
hepatic) of the drug, which determines the extraction ratio (ER) of the drug (ER = CLhepatic/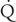 hepatic) according to the following formula (Figure 50-1):
hepatic) according to the following formula (Figure 50-1):
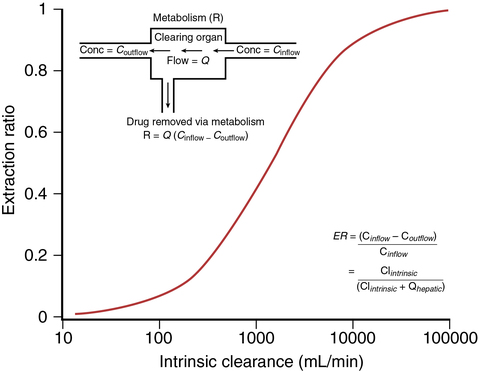
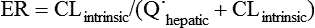
Depending on these ratios, different types of hepatic ERs have been described (Table 50-1).
Table 50-1
Flow-Limited Versus Capacity-Limited Elimination of Drugs by the Liver
| Type of Hepatic Elimination | Extraction Ratio (ER) | Rate of Hepatic Drug Metabolism |
| Flow-limited | High: At clinically relevant concentrations, most of the drug in the afferent hepatic blood is eliminated on first pass through the liver. | Rapid: Because drugs with a high ER are metabolized so rapidly, their hepatic clearances roughly equal their rates of transport to the liver (i.e., hepatic flow). |
| Capacity-limited | Low: Hepatic elimination of these drugs is determined by their plasma concentration. | Slow: When the capacity of the liver to eliminate a drug is less than the dosing rate, a steady state is unachievable; plasma levels of drug will continue to rise unless the dosing rate is decreased. Drug clearance has no real meaning in such settings. |
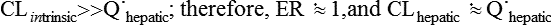
 hepatic (flow-limited). Drug elimination is diminished by conditions of decreased
hepatic (flow-limited). Drug elimination is diminished by conditions of decreased 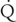 hepatic (arterial hypotension; increased splanchnic vascular resistance, including hepatic cirrhosis; hepatic venous congestion). If drug administration rate is not adjusted for changes in hepatic clearance, resulting drug concentrations increase in a reciprocal fashion. Examples of highly extracted drugs include propofol, ketamine, fentanyl, sufentanil, morphine, meperidine, lidocaine, bupivacaine, metoprolol, propranolol, labetalol, verapamil, and naloxone.
hepatic (arterial hypotension; increased splanchnic vascular resistance, including hepatic cirrhosis; hepatic venous congestion). If drug administration rate is not adjusted for changes in hepatic clearance, resulting drug concentrations increase in a reciprocal fashion. Examples of highly extracted drugs include propofol, ketamine, fentanyl, sufentanil, morphine, meperidine, lidocaine, bupivacaine, metoprolol, propranolol, labetalol, verapamil, and naloxone.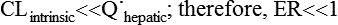
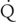 hepatic are of minimal significance. Hepatic enzyme activity may be affected (decreased or increased) by a variety of factors, including extremes of age, genetic factors (gene polymorphisms), environmental exposure (enzyme induction), and medication history (enzyme induction by phenobarbital, polycyclic hydrocarbons, rifampin, phenytoin, or chronic alcohol consumption; enzyme inhibition, by other substrates of the enzyme, or by drugs, such as cimetidine). Examples of poorly extracted drugs include thiopental, phenobarbital, hexobarbital, diazepam, lorazepam, phenytoin, valproic acid, ethanol, digitoxin, theophylline, acetaminophen, and warfarin.
hepatic are of minimal significance. Hepatic enzyme activity may be affected (decreased or increased) by a variety of factors, including extremes of age, genetic factors (gene polymorphisms), environmental exposure (enzyme induction), and medication history (enzyme induction by phenobarbital, polycyclic hydrocarbons, rifampin, phenytoin, or chronic alcohol consumption; enzyme inhibition, by other substrates of the enzyme, or by drugs, such as cimetidine). Examples of poorly extracted drugs include thiopental, phenobarbital, hexobarbital, diazepam, lorazepam, phenytoin, valproic acid, ethanol, digitoxin, theophylline, acetaminophen, and warfarin.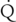 hepatic, hepatic enzyme activity, and free-drug concentration). Examples of these drugs are methohexital, midazolam, alfentanil, and vecuronium.
hepatic, hepatic enzyme activity, and free-drug concentration). Examples of these drugs are methohexital, midazolam, alfentanil, and vecuronium.Hepatic metabolic reactions
Phase 1 reactions
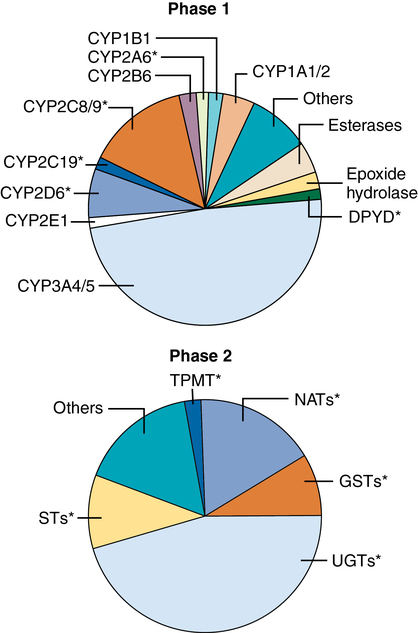
Phase 1 reactions are oxidative, reductive, or hydrolytic reactions performed by more than 50 microsomal cytochrome P-450 enzymes (belonging to 17 distinct families, Figure 50-2) that are responsible for more than 90% of all hepatic drug biotransformation reactions. These processes act by inserting or unmasking polar OH, NH2, or SH chemical groups through hydroxylation, N-dealkylation or O-dealkylation, deamination, desulfuration, N– or S-oxidation, epoxidation, or dehalogenation. The resulting more hydrophilic metabolites are passively returned to blood, flow through the liver, and may serve as substrate for subsequent nonmicrosomal (phase 2) conjugation reactions. Phase 1 reactions are quite variable, exhibiting greater than fourfold differences in maximal metabolic rate, even among healthy people (due to genotype and drug or environmental exposure causing enzyme induction), and are further affected by nutrition status and hepatic disease, including a risk of oxidative stress related to the preferential centrilobular location of phase 1 reactions in zone 3, the area most vulnerable to the development of tissue hypoxia.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Mechanisms of hepatic drug metabolism and excretion
Hepatic clearance
< ?xml:namespace prefix = "mml" />
where Vm = maximal metabolic rate (mg/min) and km (Michaelis constant) = drug concentration producing the half-maximal metabolic rate (mg/L). In this case drug elimination is termed capacity-limited. In this situation, unlike the flow-limited condition, drug elimination may change as a function of free-drug concentration that is available for hepatic metabolization and may, thus, be affected by the amount of protein binding and disease-induced changes in protein binding. Whether the hepatic elimination of a drug is flow-limited or capacity-limited depends on the ratio of the free plasma concentration of the drug to km (flow-limited if < 0.5) and that of the CLintrinsic to total hepatic blood flow (hepatic) of the drug, which determines the extraction ratio (ER) of the drug (ER = CLhepatic/hepatic) according to the following formula (Figure 50-1):
Depending on these ratios, different types of hepatic ERs have been described (Table 50-1).
Table 50-1
Flow-Limited Versus Capacity-Limited Elimination of Drugs by the Liver
| Type of Hepatic Elimination | Extraction Ratio (ER) | Rate of Hepatic Drug Metabolism |
| Flow-limited | High: At clinically relevant concentrations, most of the drug in the afferent hepatic blood is eliminated on first pass through the liver. | Rapid: Because drugs with a high ER are metabolized so rapidly, their hepatic clearances roughly equal their rates of transport to the liver (i.e., hepatic flow). |
| Capacity-limited | Low
Buy Membership for Anesthesiology Category to continue reading. Learn more here
[/not-level-membership-for-anesthesiology-category] |
