Chapter 23 Mechanisms of ER Stress in Retinal Disease
The endoplasmic reticulum
As its name suggests, the ER is a membrane-bound labyrinth of tubes and sacs, where virtually all plasma membrane and secreted proteins begin their progression to the cellular surface. The ER is the largest membrane compartment within eukaryotic cells, frequently comprising more than half of the total membrane composition. The ER has a number of major physiological functions.1 It plays a key role in lipid and protein biosynthesis; it functions as a Ca2+ storage organelle. Importantly, it is the site where most secretory and transmembrane proteins fold into their native conformation. When proteins enter the ER, they may undergo formation of disulfide bonds through the action of folding enzymes such as protein disulfide isomerase (PDI).2 Proteins that are processed through the ER frequently undergo glycosylation, a posttranslational modification involving the addition of asparagine-linked oligosaccharides. There are two known chaperone systems in the ER, the calnexin/calreticulin system and binding protein/glucose-regulated protein 78 (BiP/GRP78). The calnexin/calreticulin system is particularly relevant to folding of glycoproteins, although some calnexin/calreticulin substrates can also bind BiP/GRP78. Protein folding is essential for protein function; therefore, sophisticated mechanisms have evolved to ensure that proper folding occurs or that irreversibly misfolded proteins are eliminated. Quality control is an ER surveillance mechanism to permit only properly folded proteins to exit the ER en route to other organelles.
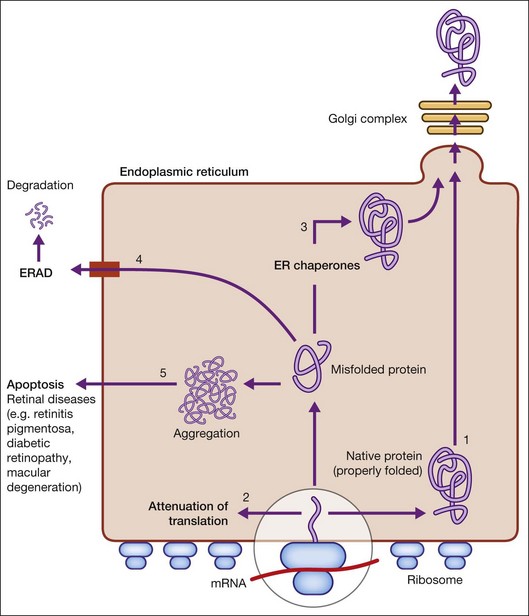
When there is perturbation of ER function (such as inhibition of glycosylation or disulfide bond formation, disruption of Ca2+ homeostasis, hypoxia, and infection), unfolded or misfolded proteins accumulate. This situation, termed ER stress, is defined as an imbalance between the cellular demand for ER function and ER capacity.2–4 When the influx of nascent unfolded polypeptides exceeds the folding and processing capacity of the ER, the normal physiological state of the ER is perturbed, activating signaling pathways, termed the ER stress response or the unfolded protein response (UPR), to return the ER to its normal physiological state.2–4 The possible fates for a protein in the ER are shown in Figure 23.1 and include: (1) proper folding and exit through the ER; or in the case of misfolded proteins: (2) translational attenuation whereby protein synthesis halts temporarily to prevent accumulation of unfolded proteins; (3) transcriptional induction of ER chaperone genes to increase protein folding capacity; (4) transcriptional induction of ER-associated degradation (ERAD) activity. If these strategies fail and protein aggregation is excessive, apoptosis (5) is induced to dispose of cells injured by ER stress, thereby ensuring survival of the organism. Recent studies implicate dysregulation of the UPR in neurodegenerative diseases, including retinal diseases.
ER stress and UPR signaling
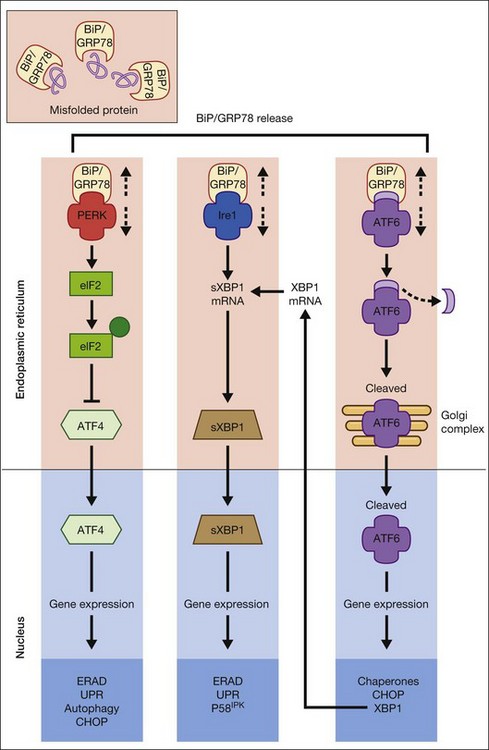
The UPR is a complex cellular signaling pathway that attempts to restore cellular homeostasis, but, in the face of prolonged ER stress, pathways may be activated that lead to cell death.5 The UPR involves three signaling pathways governed by three integral ER membrane proteins: PERK (protein kinase-like ER kinase, pancreatic ER eukaryotic translation initiation factor (eIF)-2a kinase), IRE-1 (inositol-requiring protein 1), and ATF6 (activating transcription factor 6). Research related to ER stress and retinal disease has focused on investigations of BiP/GRP78 and these downstream effector proteins.
Binding protein/glucose-regulated protein 78
BiP/GRP78 is a calcium-dependent resident ER protein that facilitates the transfer of proteins into the ER through the ER translocator.2,3,6 It is a heat shock protein with a molecular weight of 78 kDa. As a key ER stress regulatory protein, it binds to exposed hydrophobic amino acid sequences of polypeptides that would normally be buried in the interior of correctly folded proteins. Under nonstress conditions, BiP/GRP78 binds to the luminal domains PERK, IRE1, and ATF6, maintaining them in an inactive state (Fig. 23.2). When misfolded proteins accumulate, BiP/GRP78 preferentially binds to these misfolded proteins and dissociates from the UPR sensor proteins allowing their activation. BiP/GRP78 and other UPR gene targets such as GRP94 and calreticulin contain an ER stress response element (ERSE, CCAAT(N9)CCACG) that is required and sufficient to activate the UPR. The absolute requirement for BiP/GRP78 for survival is underscored by evidence that absence of BiP/GRP78 results in perimplantation embryonic lethality in mice.7 Upregulation of BiP gene expression is considered by some investigators as a marker for ER stress induction.
PERK
The signaling events mediated by PERK represent the most immediate response to ER stress in metazoan cells. PERK is an ER-associated transmembrane serine/threonine protein kinase. When unfolded proteins accumulate in the ER lumen, BiP/GRP78 dissociates from PERK, which then dimerizes and is subsequently autophosphorylated, triggering phosphorylation of eukaryotic translation initiation factor 2 on the alpha subunit (eIF2α). Phosphorylation of eIF2α attenuates mRNA translation, thereby preventing influx of newly synthesized polypeptides into the ER lumen of the stressed cell.6 Interestingly, although phosphorylation of eIF2α inhibits translation initiation in general, it is required for selective translation of several mRNAs such as ATF4. Activation of ATF4 can increase levels of chaperones, restore cellular redox homeostasis, and help the ER to fold proteins or degrade them. Lack of the gene encoding PERK is not lethal, but does result in increased hypersensitivity to ER stress.8 It has been reported that activation of PERK induces transcription of ~1/3 of UPR-dependent genes.9
IRE1
IRE1 is a 100-kDa bifunctional protein with kinase and endoribonuclease (RNase) activity. It was the first component in the UPR pathway to be identified10 and is evolutionarily the oldest branch of the UPR. Under nonstress conditions, the protein kinase is maintained in a monomeric form through its interactions with BiP/GRP78. IRE1 can bind members of the tumor necrosis factor (TNF) receptor family and can activate protein kinases that are implicated in immunity, inflammation, and apoptosis. Under conditions of ER stress, when unfolded proteins are accumulating, IRE1 is released from BiP/GRP78, dimerizes, and autophosphorylates to activate its RNase activity.2–4 In mammals, there are two forms of the protein, IRE1α and IRE1β. IRE1α is expressed in most cells and tissues (including retina), while IRE1β is primarily in intestinal epithelial cells. Upon activation of the UPR, IRE1 RNase activity initiates removal of a 26-nucleotide intron from X-box binding protein (XBP1) mRNA. Spliced XBP1 is a transcriptional activator that activates many UPR target genes through its interaction with ERSE and it can activate genes required for ER-associated degradation (ERAD). IRE1 may be a focal point for the BCL-2 family of proteins that regulates cell death. BCL-2-associated X protein (BAX) and BCL-2 antagonist/killer (BAK) interact physically with IRE1α, modulating the UPR. If either IRE1 or XBP1 is absent in mouse models, the result is embryonic lethality.
ATF6
ATF6α and ATF6β are bZIP family transcription factors. In the absence of ER stress, BiP/GRP78 binds to the luminal domain of ATF6, tethering it to the ER membrane. When unfolded proteins accumulate, BiP/GRP78 releases ATF6, which then translocates to the Golgi apparatus by vesicular transport.3,6 Unlike PERK and IRE1α, ATF6α and ATF6β do not undergo oligomerization, rather in the Golgi ATF6 is cleaved by proteases and the resultant cytoplasmic portion translocates to the nucleus, where it (like XBP1) binds to ERSE to activate transcription of ER chaperone genes such as BiP/GRP78, GRP94, and calreticulin. Thus, ATF6 activation can increase ER chaperone activity. If both ATF6α and ATF6β are deleted in mouse, the result is early embryonic lethality.
ER-associated degradation
The ER employs ERAD to clear aggregated, misfolded, or unassembled proteins. Target proteins are selected through the ER quality control system, retrotranslocated to the cytosol, and degraded by the ubiquitin proteosome system. The four steps associated with ERAD are recognition, retrotranslocation, ubiquitination, and degradation.3 In the recognition step, glycosylated proteins are bound to ER degradation-enhancing α-mannosidase-like protein that can discriminate folded from unfolded proteins. Misfolded proteins destined for the retrotranslocation machinery associate with PDI and BiP/GRP78 to cleave disulfide bonds and to unfold the partially folded protein. Proteins are translocated to the cytoplasm where they undergo ubiquitination by the E1–E2–E3 ubiquitin system. The proteins are then deglycosylated and degraded by the proteosome.
Retinal diseases associated with ER stress
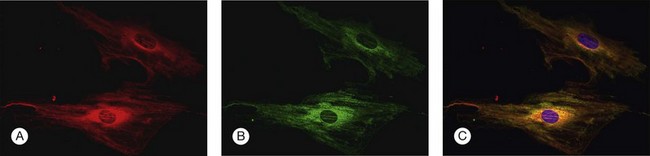
To investigate the role of ER stress genes and proteins in retina, a number of research tools (e.g., antibodies, molecular probes) are available commercially or have been developed by individual laboratories. Figure 23.3 shows immunocytochemical studies performed in freshly isolated mouse retinal Müller cells to detect two ER proteins. PDI is a known ER-resident protein and hence an excellent marker of this organelle (Fig. 23.3A). In this dual-label experiment, a second antibody was used to detect BiP/GRP78, the key ER stress-regulatory protein (Fig. 23.3B). The final panel shows the merged image of PDI and BiP/GRP78; there is considerable overlap in the expression of these two proteins (Fig. 23.3C). Immunodetection methods, gene expression analysis, and elegant genetic manipulation methods have formed the basis of investigations of the ER stress genes/proteins in various retinal diseases.
Retinitis pigmentosa and other photoreceptor dystrophies
RP is an inherited retinal dystrophy in which loss of photoreceptors leads to progressive vision loss. The prevalence of nonsyndromic RP is ~1/3500–4000. The most common form of RP is a rod–cone dystrophy, characterized initially by night blindness, followed by progressive loss in the peripheral visual field in daylight, eventually leading to blindness after several decades.11 To date mutations in over 40 genes have been implicated in RP.12 In some cases, the genes are specific to photoreceptor cells, including rhodopsin, rod cGMP phosphodiesterase, peripherin, and rod outer-segment membrane protein-1, while others are expressed in retinal and nonretinal cells.
Rhodopsin mutations
Among the genes associated with RP, mutations within the rhodopsin gene account for approximately 25% of cases of autosomal dominantly inherited RP (adRP). AdRP is a human protein folding disease that is frequently caused by a proline-to-histidine mutation at position 23 of rhodopsin (P23H rhodopsin) that leads to its retention in the ER.13,14 In vitro studies have shown that cells transfected with P23H rhodopsin increased expression of BiP mRNA to a level greater than in cells transfected with wild-type rhodopsin.13,14 Interestingly, activation of the IRE1α pathway was protective, while prolonged PERK activation was associated with cell death. Elegant studies by Gorbatyuk and colleagues14 provide immunohistochemical data showing that when HeLa cells are transfected with mutant rhodopsin (P23H), the protein was not able to traffic to the cell membrane and was localized in the cytoplasm – clear evidence of the retention in the ER of a misfolded protein.14 These cell culture observations have been extended to studies in an animal model of adRP (transgenic rat expressing P23H rhodopsin at high levels). During retinal development, levels of BiP mRNA were high, but diminished with age; however, levels of CHOP indicative of apoptosis increased significantly with age as the retinopathy advanced in the adRP rat model.13 Subsequent experiments in which BiP/GRP78 was overexpressed using adenoviral vector-mediated delivery in the rat adRP model resulted in improved a- and b-wave amplitudes of the scotopic electroretinogram (ERG) and a reduction in photoreceptor cell loss.14 The field of microribonucleic acid (micro-RNA or miRNA) expression profiling and bioinformatics has identified miR-708 in the homeostatic regulation of ER function in photoreceptor cells, specifically preventing excess rhodopsin from entering the ER. Investigators speculate that miR-708 may function analogously to UPR control proteins such as PERK and IRE1α.15
cGMP-PDE mutations
Mutations of the gene coding for the β subunit of the rod photoreceptor-specific cGMP phosphodiesterase 6 (PDE6-β) underlie cases of autosomal recessive RP (arRP) and account for ~1–2% of all cases of human RP.16 The rd1 mouse carries a nonsense mutation of this gene and has proven to be a very useful model for understanding the pathogenic mechanisms of this form of RP. The absence of phosphodiesterase activity leads to increased accumulation of cGMP in photoreceptors, which leads to increased influx of Na+ and Ca2+ through cGMP-gated cation channels. The uncontrolled influx of Ca2+ triggers apoptosis of photoreceptor cell nuclei so that between postnatal days 10 and 21, rod photoreceptor cells are lost. The number of rows of cells in the retinal outer nuclear layer decreases from ~10–12 to ~1–2, representing remaining cones. Interestingly, as the rod cells are lost there is an increase in expression of BiP/GRP78, phosphorylated eIF2α, phosphorylated PERK and caspase-12 over postnatal days 10–14, but levels decrease by postnatal day 21.17 These data clearly implicate ER stress in the pathogenesis of RP caused by mutations of PDE-β.
Carbonic anhydrase mutations
Carbonic anhydrase IV (CA IV) is another gene that, when mutated, leads to human adRP that involves ER stress. This form of RP (RP17) is caused by an arginine-to-tryptophan (R14W) mutation in the signal sequence of carbonic anhydrase IV.18–20 This protein is a glycosylphosphatidylinositol-anchored membrane protein expressed in the choriocapillaris of the eye and in the renal epithelium. Interestingly, the mutation results in an exclusively ocular phenotype. In vitro studies have shown that in cells, R14W mutation, which is the RP17 form of adRP, leads to accumulation of CAIV as unfolded protein in the ER. This gene defect is associated with increased expression of BiP/GRP78, PERK, and CHOP, leading to cell death.19
LRAT mutations
An inherited retinal dystrophy that preferentially affects cones before rods is Leber congenital amaurosis (LCA), the most severe retinal dystrophy in early childhood.21 Mutations in RPE65 or lecithin-retinol acyltransferase (LRAT) disrupt 11-cis-retinal recycling, causing this devastating disease. LRAT catalyzes the esterification of all-trans retinol (vitamin A) to all-trans retinyl esters, which are the substrate for RPE65, to produce 11-cis retinol. In studies of a murine model of LCA (LRAT−/− mouse), large quantities of M and S opsins are mislocalized to the inner regions of cones, creating an extra burden on the cell.22 Interestingly, mislocalized M opsin is degraded, whereas S opsin is resistant to proteasome degradation, resulting in far more toxic aggregation of S opsin in the ventral and central retina than of M opsin in the dorsal retina. Furthermore, aggregation of S opsin leads to CHOP activation. The UPR in cones copes with mislocalized M opsin more effectively than mislocalized S opsin. Thus, M opsin is degraded by the ERAD pathway, which relieves ER stress. S opsin was resistant to ERAD, resulting in aggregation/accumulation, which induces apoptosis.
The apparent role of ER stress and UPR in some forms of photoreceptor disease has prompted suggestions that therapeutic approaches, which modulate these pathways, may prove beneficial.22–24
Diabetic retinopathy
Diabetic retinopathy is a major complication of diabetes mellitus, a complex metabolic disorder characterized by deficiency of or insensitivity to insulin. Diabetic retinopathy is a neurovascular disease.25,26 The microvascular characteristics, which are visible clinically, include pericyte dropout, microaneurysms, intraretinal hemorrhages, capillary nonperfusion, intraretinal microvascular abnormalities, and neovascularization. The neuronal component is characterized by death of ganglion cells, loss of cells in the inner nuclear layer,27,28 and functional changes detected, particularly using multifocal ERG.29 ER stress has been implicated in diabetic retinopathy and investigations have examined features of ER stress in retinal vascular cells and retinal neurons.30
Regarding the vasculature, retinal homeostasis is regulated in part by the blood–retinal barrier. The outer barrier is comprised of the tight junctions between retinal pigment epithelial (RPE) cells while the inner blood–retinal barrier is composed of tight junctions between vascular endothelial cells. Breakdown of this barrier is characteristic of diabetic retinopathy. TNF-α is a major proinflammatory cytokine induced by diabetes that plays a key role in endothelial cell injury in diabetic retinopathy. Investigators have shown that ER stress plays a pathogenic role in retinal inflammation and vascular leakage in diabetic retinopathy.31–33 Interestingly, preconditioning human retinal endothelial cells with very low levels of ER stress-inducing agents such as tunicamycin actually alleviate TNF-α-induced endothelial adhesion molecule expression, retinal leukostasis, and vascular leakage in vitro.34 The beneficial effects of ER stress preconditioning require that XBP1, a major regulator of the adaptive response to ER, must be activated if ER stress is to be protective of endothelial cell function.34 There are reports showing that P58IPK, a 58-kDa inhibitor of protein kinase, which has been shown to be important in ER stress, reduced the level of TNF-α in endothelial cells.35 Thus, the potential role of modulating ER stress may prove therapeutically useful for the endothelial cell alterations observed in diabetic retinopathy.
Vascular endothelial growth factor (VEGF) plays a key role in the development and progression of diabetic retinopathy.36 Investigators have shown that homocysteine, which induces ER stress, increased expression of VEGF along with BiP/GRP78.37 Incubating RPE cells with homocysteine in vitro caused transient phosphorylation of eIF2α and increased ATF4 protein level. In addition to its effects on vasculature, excess homocysteine induces death of ganglion cells and, in a mouse model of hyperhomocysteinemia, diabetes accelerates the loss of these retinal neurons.38
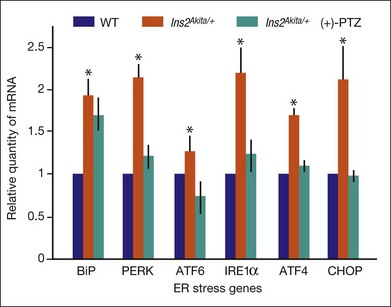
Retinal neurons die in diabetic retinopathy, as evidenced by decreased contrast sensitivity,29 decreased blue–yellow color sensitivity,39 and reduced electrical responses in full-field and multifocal ERG.40 ER stress has been implicated in the death of retinal neurons, particularly ganglion cells. Studies subjecting the retinal neuronal cell line RGC-5 to oxidative stress, as observed in diabetic retinopathy, demonstrated increased expression of the ER stress gene BiP/GRP78 as well as PERK, IRE1α, ATF6, and the apoptosis gene CHOP.41 Upregulation of several of these ER stress markers has been detected in neural retinas of the Ins2Akita/+ mouse.31,41 The Ins2Akita/+ mouse is an endogenous model of diabetic retinopathy characterized by marked apoptosis of retinal neurons, including ganglion cells and cells of the inner nuclear layer and vasculopathy.42,43 Interestingly, treatment of this mouse with a ligand for sigma receptor 1 (σR1), a molecular chaperone that binds BiP/GRP78, provided marked neuroprotection against neuronal cell death when it was administered over a period of several weeks.44 Examination of the ER stress genes in retinas of these mice (using quantitative reverse-transcriptase polymerase chain reaction) showed that BiP/GRP78 expression increased in diabetic mice compared with wild-type mice, as did PERK, ATF6, IRE1, ATF4, and CHOP (Fig. 23.4). The retinas of Ins2Akita/+ diabetic mice treated with the σR1 ligand (+)-pentazocine showed ER stress gene expression levels that were very similar to the age-matched wild-type mice (Fig. 23.4). The data suggest that targeting ER stress may hold promise in the treatment of diabetic retinopathy.
Macular degeneration
The macula is the cone photoreceptor-dense region of the retina; dystrophy of this area is collectively termed “macular degeneration.” Macular degeneration can develop at an early age through single-gene mutations or can present later in life as the much more common multifactorial age-related macular degeneration (AMD).12 Clinically, patients present with progressive loss of visual acuity, abnormal color vision, and central scotomas. ER stress has been implicated in the pathogenesis of both genetically inherited, early-onset macular dystrophies and multifactorial age-related macular degenerations.
Early-onset macular dystrophies
Mutations of at least two genes leading to juvenile macular dystrophy are associated with ER stress, ELOVL4 and EFEMP1. ELOVL4 encodes a 314-amino-acid ER-bound transmembrane protein associated with the long-chain fatty acid synthesis machinery. Retinal tissue has a unique fatty acid composition; the lipid environment is critical for normal retinal functions. While the precise role of ELOVL4 in photoreceptors is not known, mutations of ELOVL4 result in an autosomal dominant atrophic macular dystrophy resembling Stargardt macular degeneration; hence the disease is referred to as Stargardt-like macular dystrophy. Wild-type ELOVL4 is localized to the ER; however the mutant form of the protein accumulates in the Golgi apparatus.45 Investigators have transfected cells with the mutant forms of ELOVL4, known to cause Stargardt-like macular dystrophy, and observed an increase in the expression of BiP/GRP78 and the UPR apoptosis-associated gene, CHOP.46
EFEMP1 (epithelial growth factor (EGF)-containing fibulin-like extracellular matrix protein 1) encodes an extracellular matrix protein, fibulin-3. Fibulin-3 is a glycoprotein that typically undergoes proper folding in the ER, is transported to the Golgi, and then secreted. A missense point mutation (arginine-to-tryptophan (Arg345Trp)) results in an early-onset autosomal dominant maculopathy, known as Doyne honeycomb retinal dystrophy (also termed malattia leventinese). Mutant forms of the protein accumulate aberrantly in the ER of RPE, hindering proper secretion to the extracellular milieu.47,48 To determine the consequences of the Arg345Trp mutation on ER stress, investigators transfected the human ARPE-19 cell line with the mutated EFEMP1 and demonstrated an upregulation of BiP/GRP78; indeed, the level of BiP/GRP78 expression paralleled the intracellular levels of fibulin-3. Additional evidence that the UPR was activated by this mutation was increased IRE-1 endonuclease activity and XBP-2 mRNA processing.48
Age-related macular degeneration
AMD is the leading cause of visual impairment in elderly persons. The macula is the photoreceptor-dense retinal region which, when affected by this disease, results in central vision loss. The RPE cells sustain photoreceptor cells through myriad activities, including the transport of needed vitamins such as vitamin A and folate, removal of waste, and phagocytosis of shed outer-segment discs. RPE cells are vascularized via the choriocapillaris. A hallmark of AMD is the accumulation of lipofuscin and extracellular deposits known as drusen. The retina and RPE are exposed constantly to oxidative stress through intense light exposure, high metabolic activity, oxygen consumption, and the high concentration of polyunsaturated fatty acids, making them particularly susceptible to ER stress.49
Clinically, AMD is classified as either atrophic (dry) AMD or exudative (wet) AMD,50 based upon whether neovascularization has developed. A major trigger for exudative AMD is upregulation of VEGF expression and ER stress can trigger this upregulation.37,48,51 Also implicated in the development of AMD is smoking52,53; cigarette smoke extract contains benzopyrene, a potent inducer of ER stress via the PERK pathway.54 A number of studies performed using human ARPE-19 cells have implicated cigarette smoke in VEGF upregulation and in induction of ER stress.55–57
In vivo studies of AMD implicate ER stress in the disease pathogenesis. For example, Dr C. Chan (National Eye Institute, National Institutes of Health) developed a mouse model of AMD that has defects in two genes which are involved in immunologic processes.58 One gene encodes CX3CR1, the receptor for CX3CL1/fractalkine chemokine, which is expressed in RPE, Müller cells, and microglia. Two single-nucleotide polymorphisms of CX3CR1 coupled with a decreased number of CX3CR1 transcripts and protein in AMD maculas are associated with AMD.59 The second gene, Ccl2 (MCP-1, a CC chemokine), is thought to play a homeostatic, immunoregulatory role in AMD pathogenesis.60 Aged mice with deficient Ccl2 or Ccr2, the corresponding receptor, develop many cardinal features of AMD, including drusen formation, RPE accumulation of lipofuscin and complement factors, and choroidal neovascularization.61 To mimic closely the pathologic features of AMD, mice with mutations in these two genes have been crossed to generate Ccl2/Cx3cr1 mice.62 Fundoscopic examination of Ccl2−/−/Cx3cr1−/− mice as early as 4–6 weeks of age revealed drusen-like lesions that progressed to large, confluent areas of yellow deposits in the deep retina and subretinal space by 4–6 months of age and flattened atrophic areas by 6 months of age. Studies of the retinas of these mice suggest that the pathogenesis of AMD may be mediated by ER stress and protein misfolding. Indeed, there is decreased expression at the mRNA and protein level of ERp29, a molecular chaperone protein.58 Given that AMD is multifactorial and there is no single mutated protein to be targeted, Sauer and colleagues hypothesized that enhancing chaperone activity through the use of chemical and/or pharmacological chaperone compounds may prove beneficial to many individuals suffering from this devastating sight-threatening disease.49 A promising example of this concerns the chaperone protein, αB crystallin, a 20-kDa member of the small heat shock protein (HSP) family. HSPs prevent aggregation of folded proteins and facilitate intracellular protein trafficking. αB crystallin is secreted from RPE cells and is taken up by adjacent photoreceptor cells conferring neuroprotection.63 As increased aβ crystallin is a biomarker for AMD, it may be fruitful to exploit increased secretion toward neuroprotective effects.
1 Chen X, Karnovsky A, Sans MD, et al. Molecular characterization of the endoplasmic reticulum: insights from proteomic studies. Proteomics. 2010;10:4040–4052.
2 Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18:716–731.
3 Yoshida H. ER stress and diseases. FEBS J. 2007;274:630–658.
4 Ni M, Lee AS. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007;581:3641–3651.
5 Tsai YC, Weissman AM. The unfolded protein response, degradation from endoplasmic reticulum and cancer. Genes Cancer. 2010;1:764–778.
6 Kaufman RJ. Regulation of mRNA translation by protein folding the endoplasmic reticulum. Trends Biochem Sci. 2004;29:152–158.
7 Luo S, Mao C, Lee B, et al. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol Cell Biol. 2006;26:5688–5697.
8 Harding HP, Zhang Y, Bertolotti A, et al. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904.
9 Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274.
10 Cox JS, Walter P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell. 1996;87:391–404.
11 Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. 2006;1:40.
12 Berger W, Kloeckener-Gruissem B, Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. 2010;29:335–375.
13 Lin JH, Li H, Yasumura D, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949.
14 Gorbatyuk MS, Knox T, LaVail MM, et al. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc Natl Acad Sci U S A. 2010;107:5961–5966.
15 Behrman S, Acosta-Alvear D, Walter P. A CHOP-regulated microRNA controls rhodopsin expression. J Cell Biol. 2011;192:919–927.
16 Bowes C, Li T, Danciger M, et al. Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature. 1990;347:677–680.
17 Yang LP, Wu LM, Guo XJ, et al. Activation of endoplasmic reticulum stress in degenerating photoreceptors of the rd1 mouse. Invest Ophthalmol Vis Sci. 2007;48:5191–5198.
18 Rebello G, Ramesar R, Vorster A, et al. Apoptosis inducing signal sequence mutation in carbonic anhydrase IV identified in patients with the RP17 form of retinitis pigmentosa. Proc Natl Acad Sci U S A. 2004;101:6617–6622.
19 Stams T, Nair SK, Okuyama Pandor A, et al. Cell-specific differences in the processing of the R14W CAIV mutant associated with retinitis pigmentosa 17. J Cell Biochem. 2010;111:735–741.
20 Datta R, Waheed A, Bonapace G, et al. Pathogenesis of retinitis pigmentosa associated with apoptosis-inducing mutations in carbonic anhydrase IV. Proc Natl Acad Sci U S A. 2009;106:3437–3442.
21 den Hollander AI, Roepman R, Koenekoop RK, et al. Leber congenital amaurosis: Genes, proteins and disease mechanisms. Prog Retin Eye Res. 2008;27:391–419.
22 Zhang T, Zhang N, Baehr W, et al. Cone opsin determines the time course of cone photoreceptor degeneration in Leber congenital amaurosis. Proc Natl Acad Sci U S A. 2011;108:8879–8884.
23 Mendes CS, Levet C, Chatelain G, et al. ER stress protects from retinal degeneration. EMBO J. 2009;28:1296–1307.
24 Farrar GJ, Palfi A, O’Reilly M. Gene therapeutic approaches for dominant retinopathies. 3. Curr Gene Ther. 2010;10:381–388.
25 Gardner TW, Abcouwer SF, Barber AJ, et al. An integrated approach to diabetic retinopathy research. Arch Ophthalmol. 2011;129:230–235.
26 Barber AJ, Gardner TW, Abcouwer SF. The significance of vascular and neural apoptosis to the pathology of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2011;52:1156–1163.
27 Barber AJ, Lieth E, Khin SA, et al. Neural apoptosis in the retina during experimental and human diabetes: early onset and effect of insulin. J Clin Invest. 1998;102:783–791.
28 Abu-El-Asrar AM, Dralands L, Missotten L, et al. Expression of apoptosis markers in the retinas of human subjects with diabetes. Invest Ophthalmol Vis Sci. 2004;45:2760–2766.
29 Ng JS, Bearse MA, Jr., Schneck ME, et al. Local diabetic retinopathy prediction by multifocal ERG delays over 3 years. Invest Ophthalmol Vis Sci. 2008;49:1622–1628.
30 Oshitari T, Hata N, Yamamoto S. Endoplasmic reticulum stress and diabetic retinopathy. Vasc Health Risk Manag. 2008;4:115–122.
31 Li J, Wang JJ, Yu Q, et al. Endoplasmic reticulum stress is implicated in retinal inflammation and diabetic retinopathy. FEBS Lett. 2009;583:1521–1527.
32 Yang H, Liu R, Cui Z, et al. Functional characterization of 58-kilodalton inhibitor of protein kinase in protecting against diabetic retinopathy via the endoplasmic reticulum stress pathway. Mol Vis. 2011;17:78–84.
33 Ikesugi K, Mulhern ML, Madson CJ, et al. Induction of endoplasmic reticulum stress in retinal pericytes by glucose deprivation. Curr Eye Res. 2006;31:947–953.
34 Li J, Wang JJ, Zhang SX. Preconditioning with endoplasmic reticulum stress mitigates retinal endothelial inflammation via activation of X-box binding protein 1. J Biol Chem. 2011;286:4912–4921.
35 Yang H, Liu R, Cui Z, et al. Functional characterization of 58-kilodalton inhibitor of protein kinase in protecting against diabetic retinopathy via the endoplasmic reticulum stress pathway. Mol Vis. 2011;17:78–84.
36 Praidou A, Androudi S, Brazitikos P, et al. Angiogenic growth factors and their inhibitors in diabetic retinopathy. Curr Diabetes Rev. 2010;6:304–312.
37 Roybal CN, Yang S, Sun CW, et al. Homocysteine increases the expression of vascular endothelial growth factor by a mechanism involving endoplasmic reticulum stress and transcription factor ATF4. J Biol Chem. 2004;279:14844–14852.
38 Ganapathy PS, Roon P, Moister TK, et al. Diabetes accelerates retinal neuronal cell death in a mouse model of endogenous hyperhomocysteinemia. Ophthalmol Eye Dis. 2009;1:3–11.
39 Daley ML, Watzke RC, Riddle MC. Early loss of blue-sensitive color vision in patients with type I diabetes. Diabetes Care. 1987;10:777–781.
40 Fortune B, Schneck ME, Adams AJ. Multifocal electroretinogram delays reveal local retinal dysfunction in early diabetic retinopathy. Invest Ophthalmol Vis Sci. 1999;40:2638–2651.
41 Ha Y, Dun Y, Thangaraju M, et al. Sigma receptor 1 modulates endoplasmic reticulum stress in retinal neurons. Invest Ophthalmol Vis Sci. 2011;52:527–540.
42 Barber AJ, Antonetti DA, Kern TS, et al. The Ins2Akita mouse as a model of early retinal complications in diabetes. Invest Ophthalmol Vis Sci. 2005;46:2210–2218.
43 Gastinger MJ, Kunselman AR, Conboy EE, et al. Dendrite remodeling and other abnormalities in the retinal ganglion cells of Ins2Akita diabetic mice. Invest Ophthalmol Vis Sci. 2008;49:2635–2642.
44 Smith SB, Duplantier JN, Dun Y, et al. In vivo protection against retinal neurodegeneration by the sigma receptor 1 ligand (+)-pentazocine. Invest Ophthalmol Vis Sci. 2008;49:4154–4161.
45 Ambasudhan R, Wang X, Jablonski MM, et al. Atrophic macular degeneration mutations in ELOVL4 result in the intracellular misrouting of the protein. Genomics. 2004;83:615–625.
46 Karan G, Yang Z, Howes K, et al. Loss of ER retention and sequestration of the wild-type ELOVL4 by Stargardt disease dominant negative mutants. Mol Vis. 2005;11:657–664.
47 Marmorstein LY, Munier FL, Arsenijevic Y, et al. Aberrant accumulation of EFEMP1 underlies drusen formation in malattia leventinese and age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:13067–13072.
48 Roybal CN, Marmorstein LY, Vander Jagt DL, et al. Aberrant accumulation of fibulin-3 in the endoplasmic reticulum leads to activation of the unfolded protein response and VEGF expression. Invest Ophthalmol Vis Sci. 2005;46:3973–3979.
49 Sauer T, Patel M, Chan CC, et al. Unfolding the therapeutic potential of chemical chaperones for age-related macular degeneration. Expert Rev Ophthalmol. 2008;3:29–42.
50 Jager RD, Mieler WF, Miller JW. Age-related macular degeneration. N Engl J Med. 2008;358:2606–2617.
51 Salminen A, Kauppinen A, Hyttinen JM, et al. Endoplasmic reticulum stress in age-related macular degeneration: trigger for neovascularization. Mol Med. 2010;16:535–542.
52 Klein R, Peto T, Bird A, et al. The epidemiology of age-related macular degeneration. Am J Ophthalmol. 2004;137:486–495.
53 Kabasawa S, Mori K, Horie-Inoue K, et al. Associations of cigarette smoking but not serum fatty acids with age-related macular degeneration in a Japanese population. Ophthalmology. 2011;118:1082–1088.
54 Hengstermann A, Müller T. Endoplasmic reticulum stress induced by aqueous extracts of cigarette smoke in 3T3 cells activates the unfolded protein-response-dependent PERK pathway of cell survival. Free Rad Biol Med. 2008;44:1097–1107.
55 Bertram KM, Baglole CJ, Phipps RP, et al. Molecular regulation of cigarette smoke induced oxidative stress in human retinal pigment epithelial cells: implications for age-related macular degeneration. Am J Physiol Cell Physiol. 2009;297:C1200–C1210.
56 Pons M, Marin-Castaño ME. Cigarette smoke-related hydroquinone dysregulates MCP-1, VEGF and PEDF expression in retinal pigment epithelium in vitro and in vivo. PLoS ONE. 2011;6:e16722.
57 Pons M, Cousins SW, Csaky KG, et al. Cigarette smoke-related hydroquinone induces filamentous actin reorganization and heat shock protein 27 phosphorylation through p38 and extracellular signal-regulated kinase 1/2 in retinal pigment epithelium: implications for age-related macular degeneration. Am J Pathol. 2010;177:1198–1213.
58 Tuo J, Bojanowski CM, Zhou M, et al. Murine ccl2/cx3cr1 deficiency results in retinal lesions mimicking human age-related macular degeneration. Invest Ophthalmol Vis Sci. 2007;48:3827–3836.
59 Tuo J, Smith B, Bojanowski CM, et al. The involvement of sequence variation and expression of CX3CR1 in the pathogenesis of age-related macular degeneration. FASEB J. 2004;18:1297–1299.
60 Forrester JV. Macrophages eyed in macular degeneration. Nat Med. 2003;9:1350–1351.
61 Ambati J, Anand A, Fernandez S, et al. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat Med. 2003;9:1390–1397.
62 Chan CC, Ross RJ, Shen D, et al. Ccl2/Cx3cr1-deficient mice: an animal model for age-related macular degeneration. Ophthalmic Res. 2008;40:124–128.
63 Sreekumar PG, Kannan R, Kitamura M, et al. αB crystallin is apically secreted within exosomes by polarized human retinal pigment epithelium and provides neuroprotection to adjacent cells. PLoS ONE. 2010;5:e12578.