Chapter 3 Mechanisms of Action of Levetiracetam and Newer SV2A Ligands
Introduction
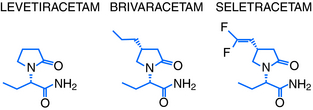
Levetiracetam (LEV; ucb L059; (S)-α-ethyl-2-oxo-pyrrolidine acetamide; Figure 3-1) is the (S)-enantiomer of the ethyl analog of the nootropic drug piracetam. Random screening in audiogenic susceptible mice initially showed that both systemic and central administration of LEV protect against seizure activity in these animals, whereas the R-enantiomer (ucb L060) and main metabolite (ucb L057) of LEV were inactive.1 This suggests that the parent compound mediates an antiepileptic effect by a central action. However, the therapeutic utility of this observation was challenged by other findings, which showed LEV to be inactive in the two conventional screening models in rodents for antiepileptic drugs (AEDs), the maximal electroshock and pentylenetetrazol seizure tests.2
Despite these conflicting results, UCB Pharma initiated a number of small, open-label studies in patients with treatment refractory seizures. The outcome suggested LEV to be an effective and well-tolerated add-on therapy devoid of clinically relevant pharmacokinetic interactions. This triggered a clinical development program resulting in approval of LEV as add-on treatment of adult patients with partial onset seizures by the FDA in 1999 and the EMEA in 2000.3 Both agencies later extended this approval to include children, and more recent approvals include adjunctive treatment of both myoclonic seizures in patients with juvenile myoclonic epilepsy and primary generalized tonic clonic seizures in patients with idiopathic generalized epilepsy and monotherapy treatment of patients with new onset epilepsy (by EMEA only).3 Taken together, these studies demonstrate LEV’s efficacy as a broad-spectrum add-on therapy as well as a monotherapy treatment for epilepsy.
More than 1 million patients had been treated with LEV by 2006, and by 2007 the drug had become the most prescribed new AED for the treatment of epilepsy.3 Together with its novel pharmacological profile and unique mechanisms of action, this triggered significant drug discovery activities at UCB Pharma targeting the identification of LEV analogs with improved mechanistic and antiepileptic properties. The first successful outcome of these efforts was the discovery of brivaracetam (BRV) and seletracetam (SEL) (Figure 3-1). These two clinical AED candidates currently undergo phase III and II studies, respectively, as add-on treatment of drug-refractory adult patients with partial onset seizures. This chapter will briefly describe the epilepsy pharmacology and review the mechanisms of action of levetiracetam, brivaracetam, and seletracetam.
Levetiracetam
EPILEPSY PHARMACOLOGY
The lack of activity of LEV in the maximal electroshock and pentylenetetrazol seizure test appears to reflect a general absence of anticonvulsant activity in rodents in acute seizure tests employing either maximal electroshocks or administration of CD97 doses of chemoconvulsants.2 This contrasts a significant ability of LEV to suppress seizures in animals with an acquired, chronic epilepsy, as revealed by LEV’s potent seizure protection in a number of different kindling models, or in various genetic animal models of epilepsy.4 These results reveal a selective, broad-spectrum action of LEV in animal models of epilepsy that mimics both partial and generalized seizures in man—a profile that it does not share with any other AED.
Several studies have shown LEV’s remarkable ability to counteract kindling acquisition, induced by either PTZ administration to mice1 or electrical stimulation of amygdala in rats.5 Two independent experiments in the latter model showed that LEV permanently abolishes the kindling-induced increase in afterdischarge duration, even after cessation of treatment.5,6 This persistent effect against kindling acquisition after prolonged treatment distinguishes LEV from other AEDs7 and suggests that it does not simply mask the expression of kindled seizures through seizure suppression, but potentially possesses antiepileptogenic properties.
Systemic administration of high doses of LEV to rodents only induces minor sedative and ataxic effects.2 Psychomimetic behaviors are absent2 and cognitive performance unaltered.8 Combined with the potent seizure suppression by LEV in animal models of epilepsy, this results in a very high separation between doses inducing seizure protection and CNS-related adverse effects.2
The findings summarized earlier indicate that the pharmacological properties of LEV in animal models of seizures and epilepsy are unique and distinguish it from all other AEDs.4 LEV is the only AED to reveal an absence of anticonvulsant activity in conventional screening tests. This contrasts broad-spectrum seizure suppression and kindling inhibition with a wide safety margin in animal models of acquired and genetic epilepsy. This novel preclinical profile has nourished the desire to determine LEV’s mechanism of action.
MECHANISM OF ACTION
Electrophysiological Properties
A vast number of both in vitro and in vivo electrophysiological studies in rodents has consistently shown an absence of effect of LEV on normal neuronal responses and neuronal characteristics.9 This contrasts several other studies reporting that LEV exerts a preferential action against hypersynchronization of epileptiform activity.
It has been observed that LEV differs from other AEDs by its ability to reduce increases in the amplitude of evoked population spikes, reflecting hypersynchronization, in rat hippocampal slices expressing epileptiform activity due to perfusion with high K+/low Ca2+.10 Further exploration with simultaneous dual extra- and intracellular recordings in the same model showed that LEV was the only AED to decrease the number of population spikes per extracellular response, without altering the number of action potentials per intracellular burst.11 These results suggest that LEV may counteract the transition from interictal to ictal activity. This could explain both its absence of activity against acute seizures induced by immediate, ictal activity in normal animals as well as its selective seizure protection, and low induction of adverse effects in animals with acquired and genetic epilepsy.2
Conventional AED Mechanisms
The antiepileptic action of AEDs traditionally relates to a primary action on one or more of three main mechanisms. These consist of facilitation of GABAA/BZ receptors; inhibition of voltage-gated Na+ channels, and inhibition of low voltage-gated (T-type) Ca2+ channels. Numerous studies have failed to show a direct interaction of LEV with any of these three mechanisms.9 This confirms that LEV’s unique profile in epilepsy pharmacology must reflect a novel mechanism of action.
Other Mechanisms
It has repeatedly been observed that LEV possesses an ability to inhibit AMPA-gated currents in rat hippocampal and cortical neurons.12,13 This effect only becomes significant at concentrations above therapeutic relevance and can therefore not be expected to contribute to LEV’s antiepileptic action. However, other studies conducted at therapeutically relevant concentrations have discovered two novel cellular effects that probably contribute to LEV’s unique mechanism of action.
LEV was shown to differ from other AEDs by an ability to reverse the inhibition of Zn2+ and β-carbolines of both GABA-gated currents in rat hippocampal and dentate granule neurons and glycine-gated currents in spinal cord neurons (Table 3-1).14 This distinguishes LEV from other AEDs and associates it with a potentially novel mechanism, in particular when viewed in the context of the “sprouted mossy fiber/Zn2+-sensitive GABAA receptor” hypothesis.15 Indeed, it has been proposed that epileptogenesis involves alterations in both the subunit composition of the GABAA receptor as well as mossy fiber sprouting from dentate granule cells with Zn2+ containing terminals. This is believed to induce a vicious circle of disinhibition in hippocampus resulting in epileptic discharges—a condition that LEV may counteract.
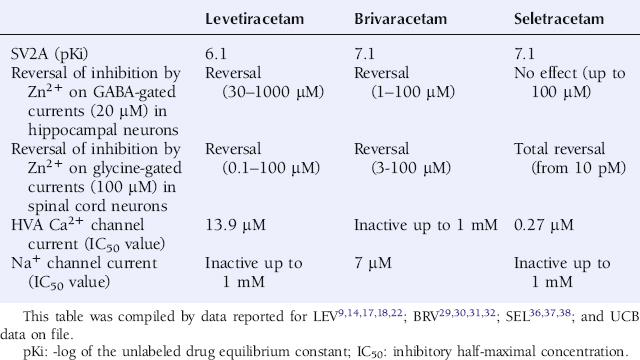
LEV was also been shown to reduce high-voltage gated Ca2+ currents.16,17 This effect appears to relate to a selective modulation of N-type Ca2+ channels.18 Furthermore, several studies have reported on LEV’s ability to reduce both ryanodine and IP3-receptor mediated Ca2+ release from the endoplasmic reticulum.19,20 Taken together, these results suggest a novel effect of LEV on Ca2+ transients by a dual action on both N-type Ca2+ channels, incorporated in the plasma membrane, as well as on intraneuronal Ca2+ stores.
Synaptic Vesicle Protein 2A
LEV (10 µM) has not been found to displace radioligands specific for a variety of receptors, ion channel proteins, reuptake sites, and second messenger systems.21 This contrasts to the observation of a specific binding site for 3H-LEV in rat brain membranes, which has become known as the Levetiracetam Binding Site (LBS).21 LEV was found to bind saturably, reversibly, and stereospecifically to LBS. A strong correlation also existed between the affinity of a series of LEV analogs to the LBS and their seizure protection in epilepsy models.21 This suggests an important functional role for LBS in the antiepileptic mechanism of LEV and provided a strong rationale to identify its molecular nature. Approximately 10 years after the discovery of LBS, it was finally documented that synaptic vesicle protein 2A (SV2A) is the molecular correlate to LBS.22
SV2 is a 12-transmembrane protein incorporated in the membrane of synaptic vesicles, present in the presynaptic terminal. It exists in three isoforms, SV2A, SV2B, and SV2C, of which SV2A is the most widely distributed in the brain and also present on many neuroendocrine cells.23 The role of SV2 in the modulation of synaptic events remains obscure, but it is presumed to exert a modulatory effect on maturation and/or fusion of vesicles with the plasma membrane of the presynaptic terminal.23,24 Further evidence that the SV2A isoform has an impact on neurotransmission is derived from studies on animals lacking SV2A. These have shown that SV2A homozygous knockout (KO) mice express a lethal seizure phenotype,23 and that SV2A heterozygous KO mice reveal accelerated kindling acquisition.25 This supports the theory that SV2A has an important role in the control of vesicle exocytosis and may be involved in the pathophysiology of epilepsy.
LEV has been documented to bind to SV2A expressed in fibroblasts (Table 3-1) but reveals no significant binding to SV2B or SV2C.22 Binding of 3H-LEV to brain membranes and purified synaptic vesicles from SV2A KO mice was absent, further supporting that SV2A is the molecular correlate of LBS (Figure 3-2). A strong correlation for LEV analogs was confirmed between their affinity for SV2A and LBS as well as between their affinity for SV2A and seizure protection in animal models of epilepsy (Figure 3-3). No other AED tested, up to 100 µM, revealed any significant affinity for SV2A.
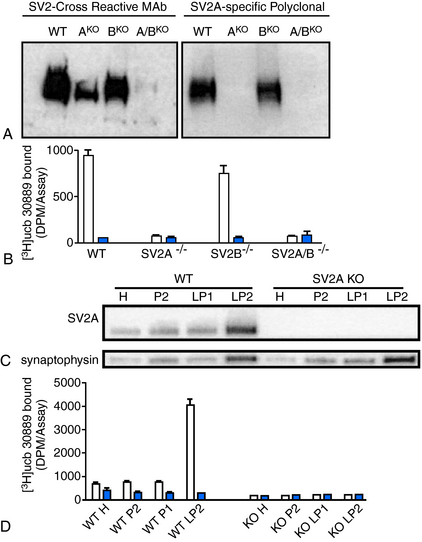
 , [3H]ucb 30889 alone;
, [3H]ucb 30889 alone;  , [3H]ucb 30889 plus 1 mM levetiracetam. Error bars are the SD of experiments performed with five WT brains and four KO brains, with three replicates within each experiment. C, Purification of synaptic vesicles enriched for the synaptic vesicle proteins and levetiracetam binding. Shown are blots of mouse brain homogenate (H), crude synaptosomes (P2), plasma and heavy membranes (LP1), and synaptic vesicles (LP2) (2 μg of each fraction) that were probed for the synaptic vesicle proteins SV2A (Upper) and synaptophysin (Lower). The synaptic vesicle fraction from WT animals displays enrichment of both synaptic vesicle proteins and LEV-binding proteins, whereas material from SV2A KOs shows enrichment of synaptophysin only. D, Binding to the different fractions using [3H]ucb 30889 shows significant binding only to the WT LP2 fraction, containing SV2A-rich synaptic vesicles. Shown are [3H]ucb 30889 alone (open bars) and [3H]ucb 30889 plus 1 mM levetiracetam (filled bars). Shown are representative examples of two experiments. Error bars are the SD of two replicates.
, [3H]ucb 30889 plus 1 mM levetiracetam. Error bars are the SD of experiments performed with five WT brains and four KO brains, with three replicates within each experiment. C, Purification of synaptic vesicles enriched for the synaptic vesicle proteins and levetiracetam binding. Shown are blots of mouse brain homogenate (H), crude synaptosomes (P2), plasma and heavy membranes (LP1), and synaptic vesicles (LP2) (2 μg of each fraction) that were probed for the synaptic vesicle proteins SV2A (Upper) and synaptophysin (Lower). The synaptic vesicle fraction from WT animals displays enrichment of both synaptic vesicle proteins and LEV-binding proteins, whereas material from SV2A KOs shows enrichment of synaptophysin only. D, Binding to the different fractions using [3H]ucb 30889 shows significant binding only to the WT LP2 fraction, containing SV2A-rich synaptic vesicles. Shown are [3H]ucb 30889 alone (open bars) and [3H]ucb 30889 plus 1 mM levetiracetam (filled bars). Shown are representative examples of two experiments. Error bars are the SD of two replicates.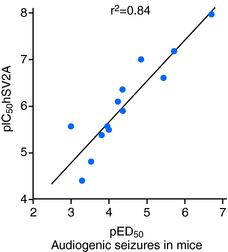
These results clearly prove that SV2A is the binding site of LEV and represents the novel and primary mechanism of action of this AED. However, LEV only possesses a moderate affinity for SV2A (Table 3-1). Together with the correlation between SV2A affinity and seizure protection, this provided a strong rationale to pursue a drug discovery program aimed at identifying high-affinity SV2A ligands with antiepileptic properties potentially superior to LEV.
In this quest, approximately 12,000 compounds were screened in vitro for SV2A binding affinity, 900 compounds were examined for seizure protection in audiogenic susceptible mice, and 30 compounds were characterized more widely in animal models of seizures and epilepsy. As a first outcome of these efforts, brivaracetam (BRV; ucb 34714; (2S)-2-[(4R)-2-oxo-4-propylpyrrolidin-1-yl] butanamide; Figure 3-1) and seletracetam (SEL; ucb 44212; (2S)-2-[(4S)-4-(2,2-difluoro-vinyl)-2-oxo-pyrrolidin-1-yl]-butyramide; Figure 3-1) were discovered.
Brivaracetam
EPILEPSY PHARMACOLOGY
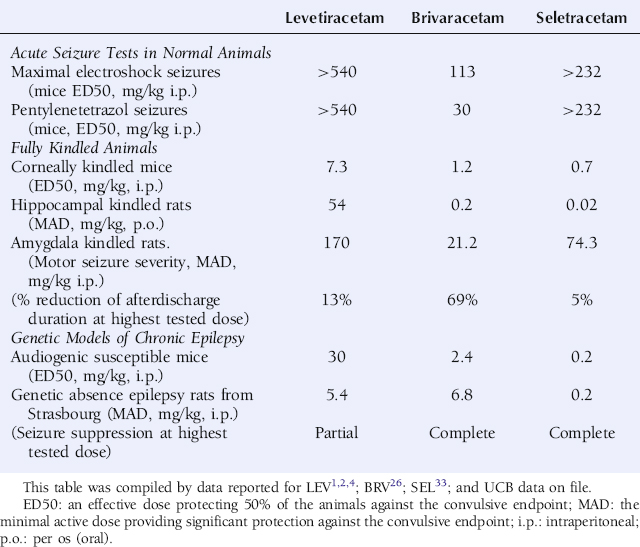
BRV differs from both LEV and SEL by inducing seizure protection, albeit at relatively high doses, against maximal electroshock and pentylenetetrazol seizures in mice (Table 3-2).26 In kindling models, BRV shows a more potent seizure protection from LEV and a more complete seizure suppression than both LEV and SEL (Table 3-2).26 The latter is reflected by a superior ability to reduce both motor seizure severity as well as afterdischarge duration in fully amygdala-kindled rats (Table 3-2). BRV is also associated with a more potent seizure protection than LEV in genetic animal models of epilepsy, like audiogenic susceptible mice, and reveal a more complete suppression of cortical spike-and-wave discharges than LEV in an experimental model of absence epilepsy, the Genetic Absence Epilepsy Rat from Strasbourg (GAERS) (Table 3-2).26
TABLE 3–2 Levetiracetam, Brivaracetam, and Seletracetam: Seizure Protection in Animal Models of Seizures and Epilepsy
Intravenous bolus administration of BRV has also been assessed for its ability to interfere with the process of self-sustaining status epilepticus (SSSE). An SSSE was induced in rats by perforant path stimulation. The cumulative duration of seizure activity was reduced dose-dependently to 11% and 8% of controls at 20 and 300 mg/kg, respectively.27 This compares favorably to the 35% and 15% obtained with 200 mg/kg of LEV and 10 mg/kg of diazepam, respectively.27
Kindling acquisition, induced by corneal stimulation in mice, was counteracted to the same extent by pretreatment with LEV (1.7–54 mg/kg i.p.) and BRV at 10-fold lower doses (0.21–6.8 mg/kg i.p.).26 However, continuation of corneal stimulation following cessation of treatment showed a significantly more persistent inhibition of the kindling process by BRV than by LEV.
BRV is devoid of any psychomimetic reactions in amygdala-kindled rats and demonstrates a higher separation than LEV between doses inducing adverse effects and seizure protection in that model. BRV also possesses a high margin between these two parameters in corneally kindled mice, despite a separation inferior to LEV.26
Taken together, the profile of BRV in animal models of seizures and epilepsy suggests that it may represent a well-tolerated broad-spectrum agent for the symptomatic treatment of epilepsy in humans. BRV may provide a more potent and efficacious seizure protection than LEV and SEL against partial seizures and status epilepticus and possess a promising potential in animal models to also affect the course of the epileptic disease process.
MECHANISM OF ACTION
BRV has been reported to possess a more potent and efficacious action than LEV against epileptiform responses in rat hippocampal slices expressing epileptiform activity due to perfusion with high K+/low Ca2+.28 BRV (3.2 µM) was more potent and active against repetitive firing than LEV (32 µM) and also inhibited spontaneous field bursting, an epileptiform marker resistant to the action of LEV.
BRV (10 µM) was not observed to induce any significant displacement of radioligands specific for various receptors, ion channel proteins, reuptake sites, and second messenger systems. Instead, BRV possesses a high and selective affinity for SV2A (Table 3-1) and does not reveal any significant affinity for SV2B or SV2C.29
Patch clamp studies in cultured hippocampal neurons have shown that BRV is devoid of any direct interaction with excitatory and inhibitory neurotransmission, with the exception of a weak and minor inhibition of the NMDA receptor current.30 BRV is also devoid of effect on low- (T-type) and high-voltage-gated Ca2+ currents, the latter being different from the action of both LEV and SEL (Table 3-1).31 Another study has shown BRV to be without impact on voltage-gated K+ currents in cultured mice hippocampal neurons (UCB data on file). In contrast, BRV has a potent ability, like LEV, to reverse the inhibitory action of Zn2+ on both GABA- and glycine-gated current in hippocampal and spinal cord neurons, respectively (Table 3-1).30 Interestingly, BRV was also shown to produce a concentration-dependent inhibition of voltage-gated Na+ currents, recorded in rat cortical neurons in culture, with an IC50 value of 7 µM and a maximal inhibition of approximately 65% appearing from a concentration of 30 µM (Table 3-1).31
Seletracetam
EPILEPSY PHARMACOLOGY
SEL resembles LEV, but differs from BRV, by its absence of activity against maximal electroshock and pentylenetetrazol seizures in mice (Table 3-2).33 In kindling models, mimicking partial epilepsy in man, SEL shows a very potent protection against secondarily generalized seizure activity, superior to both LEV and BRV, but only reveals modest activity against the focal/partial seizure activity (Table 3-2). The latter is illustrated by its inability to reduce epileptiform afterdischarges in supramaximally stimulated amygdala-kindled rats (Table 3-2). In genetic animal models, mimicking primary generalized epilepsy in man, SEL also reveals a very potent suppression of seizure activity in audiogenic susceptible mice and of cortical spike-and-wave discharges in GAERS rats—an action superior to both LEV and BRV (Table 3-2).33 In the latter model, SEL is also superior to LEV in its ability to induce complete suppression of the spike-and-wave discharges.
SEL is devoid of any psychomimetic reactions in amygdala-kindled rats and reveals an exceptionally high CNS tolerability.33 Previous studies with LEV revealed that it possesses a very high separation between doses inducing significant CNS-related adverse effects, measured by impairment of performance in the rotarod test, and seizure protection when compared with reference AEDs. The ratio between these two parameters for LEV in corneally kindled mice and GAERS rats was 148 and 235, respectively.2 For relevant reference AEDs, they were between 2-21 and 2-5,2 respectively. The similar numbers for SEL are 1048 and 3075, respectively.33
Taken together, the data generated with SEL in animal models of seizures and epilepsy suggest that it may possess an outstanding tolerability and a broad spectrum potential for the symptomatic treatment of epilepsy in humans. Compared to both LEV and BRV, SEL appears to provide a particular benefit by a very potent suppression of both primary- and secondary-generalized seizures.
MECHANISM OF ACTION
SEL (3.2 µM) has been reported to induce a more potent suppression than LEV (32 µM) of epileptiform responses in rat hippocampal slices expressing epileptiform activity due to perfusion with high K+/low Ca2+.34 In that model, LEV has been reported to differ from other AEDs by its significant ability to reduce increases in the amplitude of population spikes. Interestingly, SEL has been observed to possess a more potent and complete effect than both LEV and BRV on this measure of hypersynchronization.35
SEL (10 µM) was not observed to induce any significant displacement of radioligands specific for various receptors, ion channel proteins, reuptake sites, and second messenger systems. Instead, SEL possesses a high and selective affinity for SV2A (Table 3-1) and does not reveal any significant affinity for SV2B and SV2C (UCB data on file).
Patch clamp studies in cultured hippocampal neurons have shown that SEL is devoid of any direct interaction with excitatory and inhibitory neurotransmission.36 SEL is also without effect on voltage-gated K+ currents in cultured mice hippocampal neurons (UCB data on file) and differs from BRV by an absence of effect on voltage-gated Na+ currents in cultured rat cortical neurons (Table 3-1).37 Like LEV and BRV, SEL is also devoid of effect on low-voltage-gated (T-type) Ca2+ currents (UCB data on file) but is distinct from BRV by a more potent ability than LEV to inhibit high-voltage-gated Ca2+ currents (Table 3-1).38
The ability of LEV and BRV to oppose the inhibitory action of Zn2+ on both GABA- and glycine-gated currents potentially contributes to their mechanism of action. Interestingly, SEL reveals a different profile by a very potent and selective effect only against the inhibition by Zn2+ on glycine-gated currents (Table 3-1).36
Conclusion
Brivaracetam and seletracetam represent the first successful outcome of efforts focused on generating optimized, high-affinity SV2A ligands. They are both superior to LEV with respect to their affinity for SV2A and their ability to provide seizure protection in vivo. But they differ significantly, in particular by their effect on other relevant antiepileptic mechanisms, a finding further supported by their distinct profile in in vitro and in vivo models of epilepsy. Taken together, this highlights the promise that both may represent an important addition to the existing armamentarium of AEDs available for the treatment of epilepsy.
1. Gower A, Noyer M, Verloes R, Gobert J, Wülfert E. ucb L059, a novel anti-convulsant drug: pharmacological profile in animals. Eur J Pharmacol. 1992;222:193-203.
2. Klitgaard H, Matagne A, Gobert J, Wülfert E. Evidence for a unique profile of levetiracetam in rodent models of seizures and epilepsy. Eur J Pharmacol. 1998;353:191-206.
3. Klitgaard H, Verdru P. Levetiracetam: the first SV2A ligand for the treatment of epilepsy. Expert Opin on Drug Discov. 2007;2:1537-1545.
4. Klitgaard H. Levetiracetam: the preclinical profile of a new class of antiepileptic drugs? Epilepsia. 2001;42(suppl 4):13-18.
5. Löscher W, Hönack D, Rundfeldt C. Antiepileptogenic effects of the novel anticonvulsant levetiracetam (ucb L059) in the kindling model of temporal lobe epilepsy. J Pharmacol Exp Ther. 1998;284:474-479.
6. Stratton S, Large C, Cox B, Davies G, Hagan R. Effects of lamotrigine and levetiracetam on seizure development in a rat amygdala kindling model. Epilepsy Res. 2003;53:95-106.
7. Silver JM, Shin C, McNamara JO. Antiepileptogenic effects of conventional anticonvulsants in the kindling model of epilepsy. Ann Neurol. 1991;29:356-363.
8. Lamberty Y, Margineanu D, Klitgaard H. Absence of negative impact of levetiracetam on cognitive function and memory in normal and amygdala-kindled rats. Epilepsy Behav. 2000:333-342.
9. Margineanu DG, Klitgaard H. Levetiracetam mechanisms of action. In: Levy RH, Mattson RH, Meldrum BS, Perucca E, editors. Antiepileptic Drugs. 5th. Philadelphia: Lippincott Williams & Wilkins; 2002;:419-427.
10. Margineanu DG, Klitgaard H. Inhibition of neuronal hypersynchrony in vitro differentiates levetiracetam from classical antiepileptic drugs. Pharmacol Res. 2000;42(4):281-285.
11. Niespodziany I, Klitgaard H, Margineanu DG. Desynchronizing effect of levetiracetam on epileptiform responses in rat hippocampal slices. Neuroreport. 2003;14(9):1273-1276.
12. Hans G, Nguyen L, Rocher V, Belachew S, Moonen, Matagne A, Klitgaard H. Levetiracetam: no relevant effect on ionotropic excitatory glutamate receptors. Epilepsia. 2000;41(Suppl 7):35.
13. Carunchio I, Pieri M, Ciotti MT, Albo F, Zona C. Modulation of AMPA receptors in cultured cortical neurons induced by the antiepileptic drug levetiracetam. Epilepsia. 2007;48(4):654-662.
14. Rigo JM, Hans G, Nguyen L, et al. The anti-epileptic drug levetiracetam reverses the inhibition by negative allosteric modulators of neuronal GABA- and glycine-gated currents. Br J Pharmacol. 2002;136(5):659-672.
15. Coulter DA. Mossy fiber zinc and temporal lobe epilepsy: pathological association with altered “epileptic” gamma-aminobutyric acid A receptors in dentate granule cells. Epilepsia. 2000;41(Suppl 6):S96-S99.
16. Niespodziany I, Klitgaard H, Margineanu DG. Levetiracetam inhibits the high-voltage-activated Ca(2+) current in pyramidal neurons of rat hippocampal slices. Neurosci Lett. 2001;306(1-2):5-8.
17. Pisani A, Bonsi P, Martella G, et al. Intracellular calcium increase in epileptiform activity: modulation by levetiracetam and lamotrigine. Epilepsia. 2004;45(7):719-728.
18. Lukyanetz EA, Shkryl VM, Kostyuk PG. Selective blockade of N-type calcium channels by levetiracetam. Epilepsia. 2002;43(1):9-18.
19. Ängehagen M, Margineanu DG, Ben-Menachem E, Ronnback L, Hansson E, Klitgaard H. Levetiracetam reduces caffeine-induced Ca2+ transients and epileptiform potentials in hippocampal neurons. Neuroreport. 2003;14(3):471-475.
20. Cataldi M, Lariccia B, Secondo A, Di Renzo G, Annunziato L. The antiepileptic drug levetiracetam decreases the inositol 1,4,5-trisphosphate-dependent [Ca2+] increase induced by ATP and bradykinin in PC12 cells. J Pharmacol Exp Ther. 2005;313(2):720-730.
21. Noyer M, Gillard M, Matagne A, Henichart JP, Wülfert E. The novel antiepileptic drug levetiracetam (ucb L059) appears to act via a specific binding site in CNS membranes. Eur J Pharmacol. 1995;286(2):137-146.
22. Lynch BA, Lambeng N, Nocka K, et al. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci U S A. 2004;101(26):9861-9866.
23. Crowder KM, Gunther JM, Jones TA, et al. Abnormal neurotransmission in mice lacking synaptic vesicle protein 2A (SV2A). Proc Natl Acad Sci U S A. 1999;96(26):15268-15273.
24. Janz R, Goda Y, Geppert M, Missler M, Sudhof TC. SV2A and SV2B function as redundant Ca2+ regulators in neurotransmitter release. Neuron. 1999;24(4):1003-1016.
25. Leclercq K, Dassesse D, Lambeng N, Klitgaard H, Matagne A. Unaltered seizure susceptibility contrasts accelerated kindling acquisition in heterozygous SV2A KO mice. Program No. 82.7 2006, Abstract viewer/itinerary planner. Washington DC, Society for Neurosciences 2006. On-line.
26. Matagne A, Kenda B, Michel PH, Klitgaard H. ucb 34714, a new pyrrolidone derivative: comparison with levetiracetam in animal models of chronic epilepsy in vivo. Epilepsia. 2003;44(suppl 9):261.
27. Wasterlain C, Suchomelova L, Matagne A, Klitgaard H, Mazarati A, Shinmei S, Baldwin R. Brivaracetam is a potent anticonvulsant in experimental status epilepticus. Epilepsia. 2005;46(suppl 8):219.
28. Margineanu D, Kenda B, Michel Ph, Matagne A, Klitgaard H. ucb 34714, a new pyrrolidone derivative: comparison with levetiracetam in hippocampal slice epilepsy models in vitro. Epilepsia. 2003;44(suppl 9):261.
29. Gillard M, Fuks B, Lambeng N, Chatelain P, Matagne A. Binding characteristics of brivaracetam, a novel antiepileptic drug candidate. Program No.3.186. 2007 Abstract Viewer. Philadelphia, PA: American Epilepsy Society.
30. Rigo JM, Nguyen L, Hans G, et al. ucb 34714: effect on inhibitory and excitatory neurotransmission. Epilepsia. 2004;45(suppl 3):56.
31. Kostyuk PG, Lukyanetz EA, Klitgaard H, Margineanu DG. ucb 34714, a new pyrrolidone derivative, without impact on high- and low-voltage activated calcium currents in rat isolated neurons. Epilepsia. 2004;45(suppl 7):141-142.
32. Zona C, Pieri M, Klitgaard H, Margineanu DG. ucb 34714, a new pyrrolidone derivative, inhibits Na+ currents in rat cortical neurons in culture. Epilepsia. 2004;45(suppl 7):146.
33. Matagne A, Margineanu DG, Michel Ph, Kenda B, Klitgaard H. Seletracetam (ucb 44212), a new pyrrolidone derivative, reveals potent activity in in vitro and in vivo models of epilepsy. J Neurol Sciences. 2005;238(suppl 1):S133.
34. Margineanu DG, Michel Ph, Kenda B, Matagne A, Klitgaard H. Seletracetam (ucb 44212), a new pyrrolidone derivative, inhibits epileptiform responses in hippocampal slices in vitro. Epilepsia. 2005;46(suppl 6):121.
35. Margineanu D, Klitgaard H. The novel SV2A ligands brivaracetam and seletracetam manifest different effects against the epileptiform markers of field potentials in a “high K+-low Ca2+” rat hippocampal slice model. Epilepsia. 2006;47(suppl 3):74-75.
36. Rigo JM, Nguyen L, Hans G, et al. Seletracetam (ucb 44212): effect on inhibitory and excitatory neurotransmission. Epilepsia. 2005;46(suppl 8):110.
37. Zona C, Niespodziany I, Pieri M, Klitgaard H, Margineanu DG. Seletracetam (ucb 44212), a new pyrrolidone derivative, lacks effect on Na+ currents in rat brain neurons in vitro. Epilepsia. 2005;46(suppl 8):116.
38. Pisani A, Bonsi P, Martella G, Cuomo D, Klitgaard H, Margineanu DG. Seletracetam (ucb 44212), a new pyrrolidone derivative, inhibits high-voltage-activated Ca2+ currents and intracellular [Ca2+] increase in rat cortical neurons in vitro. Epilepsia. 2005;46(suppl 8):119.