[level-membership-for-basic-science-category]
Chapter 54 Mechanisms of Action of Antineoplastic Drugs
| Abbreviations | |
|---|---|
| Ara-C | Cytarabine |
| DNA | Deoxyribonucleic acid |
| FH4 | Tetrahydrofolate |
| 5-FU | 5-Fluorouracil |
| IV | Intravenous |
| MESNA | Sodium 2-mercaptoethane sulfonate |
| 6-MP | 6-Mercaptopurine |
| MTX | Methotrexate |
| RNA | Ribonucleic acid |
| 6-TG | 6-Thioguanine |
Therapeutic Overview
Antineoplastic agents are used to treat more than 100 types of neoplastic diseases, with the goal of destroying malignant cells. Additional drugs (see Chapter 55) are used to enhance host defense mechanisms to eradicate those tumor cells not killed by the antineoplastic drugs. In clinical practice nearly all neoplastic diseases are treated by using multiple drugs, although only individual drugs, which form the basis for multiple drug therapy, are discussed in this chapter; multiple-drug protocols are described in Chapter 53.
The effectiveness of antineoplastic drugs varies greatly with the following:
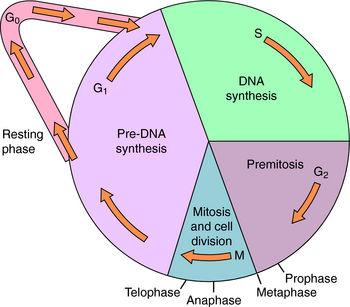
The end point used to evaluate effectiveness (e.g., tumor response, patient survival) is also important. Most antineoplastic agents, particularly chemotherapeutic agents, are more effective destroying cells that are progressing through the cell cycle (Fig. 54-1) than destroying cells that are resting in the G0 phase. The “growth fraction,” defined in Figure 54-1, is the fraction of cells progressing through the cycle. Besides tumor cells that may be proliferating, there are also some non-neoplastic cells undergoing division, particularly those of hair follicles, bone marrow, and intestinal epithelium. These rapidly dividing cells are especially sensitive to antineoplastic drugs and account for many of their undesirable side effects. It is believed that most, if not all, anticancer drugs kill cells primarily through a programmed, energy-dependent process called apoptosis, rather than through necrosis.
The number of cultured neoplastic cells that survive exposure to each drug typically shows a first-order relationship to the drug concentration (Fig. 54-2). This means that the same fraction of cells is killed with each drug dose and that a series of several doses does not kill 100% of them. This “log-cell kill” hypothesis is compatible with the clinical observation that a functional host immune system is needed for killing all neoplastic cells and curing a patient.
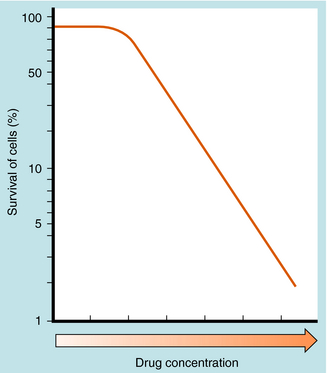
Endogenous cellular defenses, such as thiols or deoxyribonucleic acid (DNA) repair enzymes, however, can necessitate a threshold of drug concentration, or “shoulder,” in the survival curves of patients receiving these drugs (see Fig. 54-2).
| Therapeutic Overview |
|---|
| Goal |
Mechanisms of Action
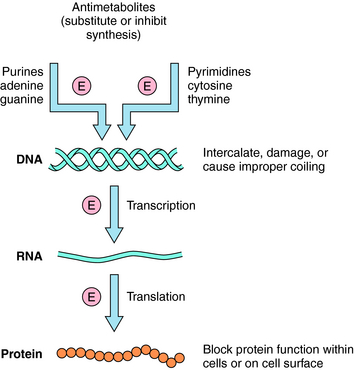
The basic mechanisms by which antineoplastic drugs kill tumor cells are summarized in Figure 54-3. Only compounds that show some selectivity for neoplastic cells are used clinically. In general, antimetabolites inhibit DNA synthesis, whereas alkylating agents, intercalators, and antibiotics damage or disrupt DNA, interfere with topoisomerase activity, or alter ribonucleic acid (RNA) structure. Steroids interfere with transcription, several plant alkaloids disrupt mitosis, agents such as asparaginase destroy essential amino acids needed for translation, and other drugs act through important growth factor signal transduction pathways. Many clinically used antineoplastic drugs must undergo either chemical or enzymatic modification to become actively cytotoxic.
Alkylation refers to the covalent attachment of alkyl groups to other molecules. Alkylating agents came to be used for cancer therapy as a result of observations of the effects of the mustard gases on cell growth. Although these compounds are too toxic for clinical use in cancer, the first effective antineoplastic agents, including mechlorethamine, were developed from related nitrogen mustard alkylating agents and are still used today.
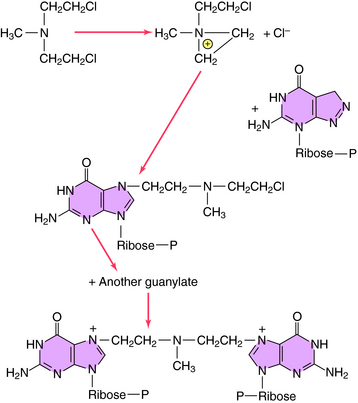
Alkylation takes place through chemical formation of a positively charged carbonium ion that reacts with an electron-rich site, particularly on DNA or RNA, to form modified nucleic acids. Most clinically used alkylating drugs have two active groups, which enable them to form covalent links between adjacent nucleic acid strands that are more difficult to repair than monofunctional adducts. These cross-links also prevent separation of the dual strands of DNA during cell cycling. For maximal kill, it is important to administer the maximally tolerated dose. The alkylation sequence for mechlorethamine reacting with the N-7 position of deoxyguanylate is shown in Figure 54-4. Although many other nucleophilic constituents, including RNA, proteins, and membrane components, become alkylated within cells, it is generally believed that the primary cytotoxic events occur through alkylation of DNA, especially by coupling to the N-7 position of the deoxyguanylates of either single- or double-stranded DNA.
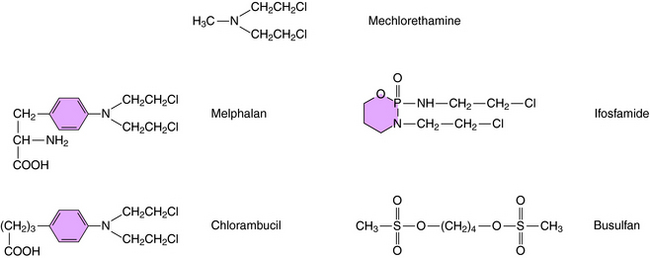
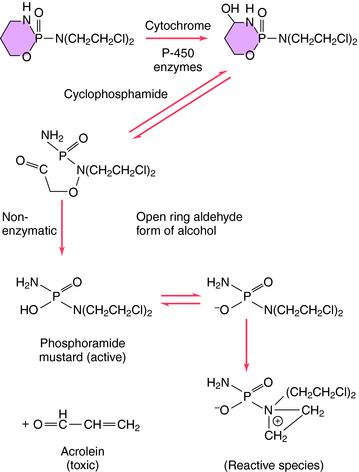
Structures of several clinically used alkylating agents are shown in Figure 54-5. Cyclophosphamide undergoes a combination of enzymatic and chemical activation to form the active phosphoramide mustard alkylating agent (Fig. 54-6). Exposure of cells to cyclophosphamide and other alkylating agents can also lead to carcinogenesis. For example, leukemia is a well-known long-term complication in patients with Hodgkin’s disease treated with a regimen including mechlorethamine.
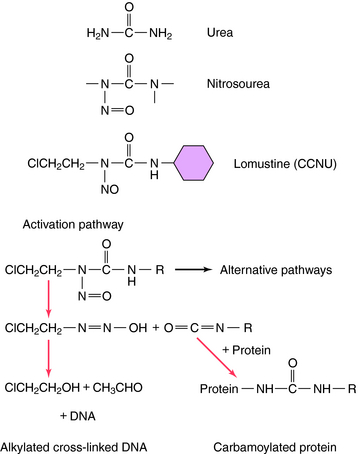
Another group of antineoplastic alkylating agents in clinical use is the nitrosoureas. The structures and primary mechanisms of activation for these compounds are shown in Figure 54-7. In addition to alkylation of DNA, the nitrosoureas also cause carbamoylation of proteins, which may play a role in cytotoxicity. The alkylation route, however, is the major source of cytotoxicity. Nitrosoureas are lipophilic and can cross the blood-brain barrier, so they are often used to treat brain tumors.
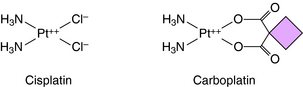
Temozolomide is the first new alkylating agent approved for treatment of malignant gliomas in decades. It has a structure similar to dacarbazine and is rapidly absorbed after oral administration. Unlike many other alkylating agents, temozolomide crosses the blood-brain barrier. It methylates guanine and adenine, resulting in misincorporation of thymidine (across from the methylated guanidine), which cannot be easily corrected by the mismatch repair system. Resistance occurs by up regulation of components of the DNA mismatch repair system. Other compounds that form covalent bonds with DNA are cisplatin and its analog carboplatin, whose structures are shown in Figure 54-8. Cisplatin is a square planar complex of platinum with two ammonia molecules and two chloride ions at the corners of the plane. The reaction sequence of the active species is complex and not completely understood. Replacement of a chloride with a hydroxyl must occur before the platinum-nitrogen bond can interact with DNA. Subsequently, the second chloride is aquated and reacts with DNA. The stereochemistry of the complex enables the cis, but not the trans isomer, to form two covalent platinum-nitrogen bonds, primarily at two adjacent deoxyguanylates of DNA. This intrastrand cross-link prevents DNA replication and is cytotoxic. Cisplatin also forms interstrand cross-links and protein-DNA cross-links. Similar DNA cross-links are formed with carboplatin. Oxaliplatin is an organoplatinum compound constructed to overcome resistance to cisplatin by binding the platinum atom to 1,2 diaminocyclohexane. Oxaliplatin undergoes conversion to reactive metabolites that covalently bind to either adjacent guanines, adjacent adenine-guanines, or guanines separated by an intervening nucleotide. This creates interstrand and intrastrand cross-links and inhibits DNA replication and transcription. Oxaliplatin is approved for treatment of metastatic colorectal carcinoma, has activity in lung cancer, and may have activity in breast and esophageal cancers and lymphoma.
The specific reaction sequences by which dacarbazine or procarbazine alkylate DNA are not well understood. Melphalan is a phenylalanine derivative and is actively transported into the cell by the carriers that transport leucine and glutamine. Melphalan is associated with induction of secondary leukemias. Chlorambucil is structurally similar to melphalan and is used primarily in treatment of chronic lymphocytic leukemia. Ifosfamide, like its analog cyclophosphamide, is activated by hepatic microsomes. Early studies with ifosfamide showed its use was associated with a significant incidence of hemorrhagic cystitis. Ifosfamide is now administered with a systemic thiol, sodium-2-mercaptoethane sulfonate (MESNA). MESNA becomes a free thiol after glomerular filtration and combines with the products responsible for causing the cystitis. Ifosfamide is active against several cancers, including small-cell lung cancer, sarcomas, lymphomas, testicular carcinoma, and gynecological cancers.
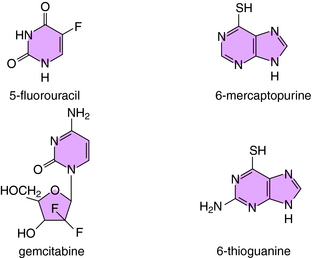
The antimetabolites are compounds that mimic the structures of normal metabolic constituents including folic acid, pyrimidines, or purines. The antimetabolites inhibit the enzymes necessary for folic acid regeneration or the pyrimidine or purine activation of DNA or RNA synthesis in neoplastic cells. Antimetabolites frequently kill cells in the S phase (see Fig. 54-1). Methotrexate (MTX), 5-fluorouracil (5-FU), cytarabine (Ara-C), 6-mercaptopurine (6-MP), gemcitabine, and 6-thioguanine (6-TG) are the primary antimetabolites used clinically.
Folic acid is essential for enzymatic reactions that transfer methyl and related groups during purine and pyrimidine synthesis. The antimetabolite MTX competitively inhibits the enzyme dihydrofolate reductase, which catalyzes the reduction of dihydrofolate to tetrahydrofolate (FH4; Fig. 54-9). As a consequence, FH4 regeneration is blocked, and the synthesis of purines and pyrimidines is prevented. MTX is specific for cells in the S phase of the cell cycle. Intracellular addition of several glutamates to MTX greatly enhances its inhibitory activity and also prevents cellular efflux. MTX is more toxic to tumor cells than normal cells, in part because of the greater polyglutamating enzyme activity in tumor cells. Thus a higher concentration of the more active MTX covalently linked to multiple glutamates is trapped within tumor cells, where it acts as an antimetabolite.
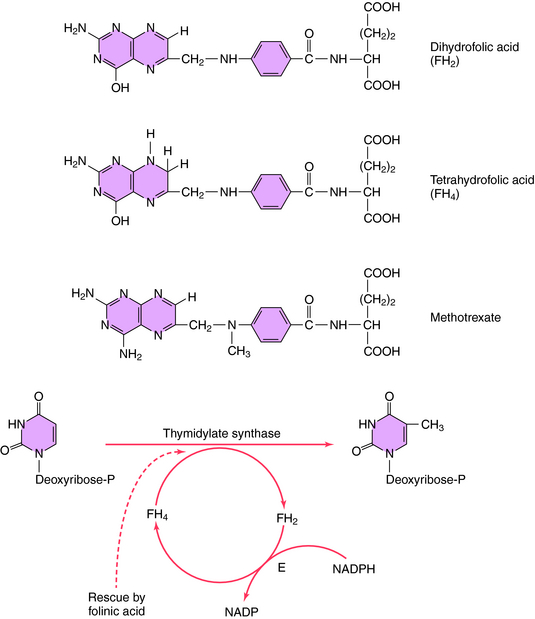
Another antimetabolite, 5-FU, acts primarily by inhibiting pyrimidine synthesis and thus DNA formation. Its structure is shown in Figure 54-10. 5-FU is metabolized to the 5-fluoro analog of deoxyuridylic acid, which inhibits thymidylate synthase by covalent coupling. Capecitabine, an oral fluoropyrimidine, is an inactive precursor of 5-FU. It is converted to 5-FU selectively in the liver and tumor tissues. Evidence suggests that thymidine phosphorylase, the enzyme responsible for the final step in conversion to active 5-FU, is overexpressed in neoplastic tissues.
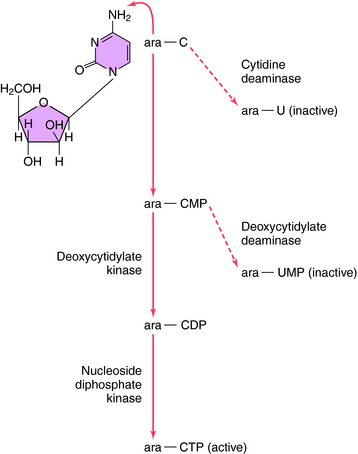
Ara-C also acts by inhibiting pyrimidine synthesis but through a more complex pathway (Fig. 54-11). The drug must undergo enzymatic conversion to the active cytosine triphosphate derivative, which is incorporated into DNA. At high doses, Ara-C also binds to and inhibits DNA polymerase competitively. Cytidine deaminase activity is high and deoxycytidylate kinase activity is low in some patients, resulting in considerable inactivation of the drug before conversion to its active form.
Gemcitabine is another pyrimidine antimetabolite (see Fig. 54-10). It is a prodrug that, once transported into cells, must be phosphorylated by deoxycytidine kinase to an active form that inhibits DNA synthesis. Cell death most likely occurs as a result of blockade of DNA strand elongation. Gemcitabine appears to have activity against adenocarcinoma of the pancreas.
The purine analogs 6-MP and 6-TG (see Fig. 54-10) also must undergo activation to form nucleotides, which then act as competitive inhibitors of several enzymes in purine synthesis pathways. The adenosine deaminase inhibitor pentostatin (2-deoxycoformycin) is highly active against hairy cell leukemia.
Several antibiotics of microbial origin are very effective in treatment of certain tumors. These antibiotics include doxorubicin, daunorubicin, bleomycin, actinomycin D, and mitomycin. The anthracycline structures of daunorubicin and doxorubicin are shown in Figure 54-12.
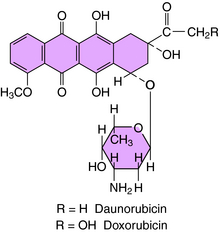
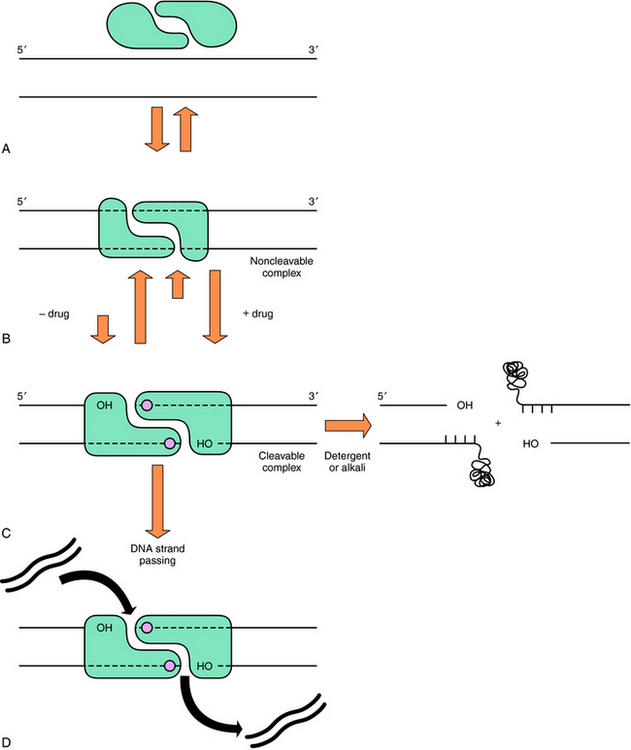
Bleomycin is a mixture of several basic glycopeptides, with one called A2 predominating. Among the common anticancer drugs, it has a unique mechanism of action in that it forms a tertiary complex with O2 and Fe++ to cause sequence-specific single- and double-stranded DNA cleavage. The double-stranded DNA cleavage that results is thought to be lethal. Doxorubicin and daunorubicin intercalate between the bases in double-stranded DNA, poison topoisomerase II, generate free radicals, and possibly disrupt the functioning of the cell membrane. It is generally believed that their poisoning of DNA topoisomerase II constitutes their major antitumor action (Fig. 54-13). DNA topoisomerase II is essential for DNA replication and catalyzes the uncoiling and breakage of both strands of double-stranded DNA to modify the number and the types of linkage twists. Doxorubicin and daunorubicin inhibit the enzyme by stabilizing a covalent complex of an enzyme-DNA intermediate, preventing the DNA breaks from rejoining and leading to cell death. Doxorubicin is the single most active agent against breast cancer; daunorubicin and idarubicin are frequently used to treat leukemias.
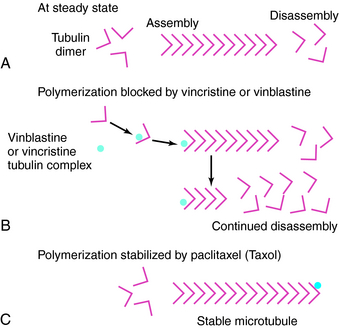
The primary plant alkaloids, vincristine and vinblastine, bind avidly to tubulin, block microtubule polymerization, and disrupt mitotic spindle formation during mitosis at the M phase of the cell cycle (see Figs. 54-1 and 54-14). Cell death results from an inability to segregate chromosomes properly. Paclitaxel, which acts as a mitotic inhibitor, binds specifically and reversibly to tubulin, but unlike other antitubule drugs, it stabilizes microtubules in the polymerized form. Paclitaxel is active against solid tumors, including ovarian carcinomas. Etoposide is a semisynthetic derivative of podophyllotoxin that is prepared from the mandrake plant (mayapple). Etoposide and teniposide, a close analog, also inhibit topoisomerase II. Etoposide has significant activity against small-cell cancer of the lungs and testicular carcinoma and is used in most first-line regimens for these diseases. Teniposide is active against acute leukemias in children.
In resistant subjects the reduced effectiveness can often be attributed to a decreased intracellular concentration of drug, repair of drug-induced damage, or a modification of drug targets. Increased expression of proteins that block the energy-dependent process of apoptosis, including oncogenes such as BCL2, can also cause resistance to many agents. Several mechanisms account for these differences, as indicated in Box 54-1.
BOX 54–1 Possible Mechanisms for the Development of Resistance to Antineoplastic Agents
One mechanism of resistance is decreased drug uptake by cells, especially to drugs such as MTX, which requires carrier proteins for transmembrane transport. Actinomycin D resistance also results from decreased uptake. A second mechanism is lack of drug activation. Cyclophosphamide requires metabolic activation, and in the absence of this pathway, tumor cells can be resistant. A third mechanism is the enhanced conversion of the active agent to an inactive metabolite. For example, increased activity of aldehyde dehydrogenase leads to enhanced metabolism of cyclophosphamide and drug resistance.
Pharmacokinetics
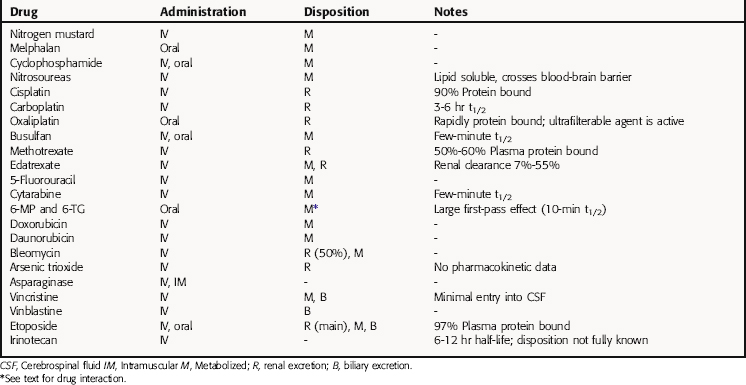
Numerous measurements have been performed to determine the plasma concentration decay curves for antitumor drugs. A standard compartment model is used to explain the plasma concentration versus time decay curves. It is a sum of one to three exponentials and is useful in providing guidance in planning dosing schemes that maximize drug tumor contact but minimize drug tissue contact. For antitumor agents that disappear rapidly from the plasma, continuous IV infusion rather than bolus injection often is needed to obtain a high enough concentration to achieve a therapeutic effect. The modes of administration and disposition of several antineoplastic agents are listed in Table 54-1.
6-Mercaptopurine undergoes enzyme-catalyzed metabolism, with xanthine oxidase as the principal enzyme. Allopurinol, a drug used in the treatment of gout, also is metabolized by the same enzyme, and a drug interaction occurs if the two compounds are given concurrently. Allopurinol also lengthens the half-life of cyclophosphamide and increases myelotoxicity, possibly resulting from the decreased renal elimination of cyclophosphamide metabolites. MTX and weak organic acids such as nonsteroidal antiinflammatory agents compete for plasma binding and for renal tubular excretion, and significant increases in MTX concentrations have been noted in patients receiving these drugs.
Relationship of Mechanisms of Action to Clinical Response
Most antineoplastic drugs are used in multiple-agent protocols in which the cytolytic effects of the different agents interact in a complex manner. The clinical use of combination chemotherapy is discussed in Chapter 53.
Pharmacovigilance: Side Effects, Clinical Problems, and Toxicity
Typical undesirable side effects of many antitumor drugs are listed in Table 54-2. Many of these side effects reflect actions of the drug on rapidly proliferating normal cells. Nausea and vomiting are quite common and can be attributable to effects on both cycling and noncycling cells. Strategies for ameliorating these effects are available (see Chapter 53).
TABLE 54–2 Typical Undesirable Side Effects of Antineoplastic Drugs in Humans*
| Tissue | Undesirable Effects |
|---|---|
| Bone marrow | Leukopenia and resulting infections |
| Immunosuppression | |
| Thrombocytopenia | |
| Anemia | |
| Gastrointestinal tract | Oral or intestinal ulceration |
| Diarrhea | |
| Hair follicles | Alopecia |
| Gonads | Menstrual irregularities, including premature menarche; impaired spermatogenesis |
| Wounds | Impaired healing |
| Fetus | Teratogenesis (especially during first trimester) |
* Many of these effects are caused by drug action on nontumor cells that usually are growing (i.e., cycling).
Nephrotoxicity, peripheral neuropathy, and ototoxicity remain the major side effects of cisplatin, although the severity of the renal toxicity can be reduced through hydration of the patient and administration of mannitol (see Chapter 21). Renal damage arises from the toxic effects of cisplatin on renal tubules, resulting in decreased glomerular filtration rates and increased reabsorption. Nephrotoxicity does not develop until a week or two after treatment is begun and may be worsened if nephrotoxic agents are coadministered—for example, if an aminoglycoside is coadministered for treatment of an infection. Cisplatin-induced neuropathy occurs mainly in large sensory fibers and results in numbness and tingling, followed by loss of a sense of joint position and a disabling sensory ataxia. The toxicity is reversed upon discontinuation of the drug, but it may take a year or longer for it to resolve. Nephrotoxicity is more common in patients who receive a bolus injection of cisplatin. Fractionating the dose over several days has been observed to reduce the intensity of this toxicity. Cisplatin neuropathy is a more recently observed cumulative dose-limiting side effect. Carboplatin causes less neurotoxicity and nephrotoxicity but more pronounced myelosuppression than cisplatin.
Cyclophosphamide, which is metabolized to an active compound and other reactive metabolites (see Fig. 54-6), occasionally causes hemorrhagic cystitis. The risk of this can be largely eliminated through vigorous hydration of patients during treatment. As little as a single IV dose of cyclophosphamide can produce cystitis. MESNA is used as a protectant in patients receiving very high doses of IV cyclophosphamide. The cystitis appears to be caused by acrolein, which is produced as a toxic byproduct of the metabolism of cyclophosphamide. This side effect is not age- or sex-related and leads to a 9 to 45 times greater risk of bladder cancer. The risk of bladder cancer is not as great in those given cyclophosphamide orally as opposed to IV.
Nearly all antineoplastic drugs have side effects that patients consider very objectionable.
New Horizons
There have been enormous advances in our understanding of the molecular and cellular biology of cancer. As a result, a large number of new targets have become available, including targets associated with oncogenic kinases and phosphatases, growth factors, and growth factor receptors involved in signal transduction (see Chapter 55). Bioreductive alkylating agents and radiation sensitizers have become subjects of considerable interest as a result of the observation that malignant cells within the center of the tumor are hypoxic, and hypoxia is a cause of drug resistance. Compounds designed to affect angiogenesis and hormone receptors and agents targeted toward redox systems are also emerging as exciting new therapeutic approaches to cancer. Many currently available
antineoplastic agents require IV administration, and considerable effort has been directed toward the development of orally active compounds that would permit patients to be treated outside of a hospital setting. Cancer vaccines and gene therapy continue to be actively investigated. The next 5 years may provide important clues concerning which of the aforementioned approaches are likely to yield effective new anticancer drugs.
Approved Oncology Drugs. U.S. Food and Drug Administration Center for Drug Evaluation and Research. http://www.fda.gov/cder/cancer/approved.htm.
O’Connor 2007 O’Connor R. The pharmacology of cancer resistance. Anticancer Research. 2007;27:1267-1272.
Swanton C. Cell-cycle targeted therapies. Lancet Oncol. 2004;5:27-36.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 54 Mechanisms of Action of Antineoplastic Drugs
| Abbreviations | |
|---|---|
| Ara-C | Cytarabine |
| DNA | Deoxyribonucleic acid |
| FH4 | Tetrahydrofolate |
| 5-FU | 5-Fluorouracil |
| IV | Intravenous |
| MESNA | Sodium 2-mercaptoethane sulfonate |
| 6-MP | 6-Mercaptopurine |
| MTX | Methotrexate |
| RNA | Ribonucleic acid |
| 6-TG | 6-Thioguanine |
Therapeutic Overview
Antineoplastic agents are used to treat more than 100 types of neoplastic diseases, with the goal of destroying malignant cells. Additional drugs (see Chapter 55) are used to enhance host defense mechanisms to eradicate those tumor cells not killed by the antineoplastic drugs. In clinical practice nearly all neoplastic diseases are treated by using multiple drugs, although only individual drugs, which form the basis for multiple drug therapy, are discussed in this chapter; multiple-drug protocols are described in Chapter 53.
The effectiveness of antineoplastic drugs varies greatly with the following:
The end point used to evaluate effectiveness (e.g., tumor response, patient survival) is also important. Most antineoplastic agents, particularly chemotherapeutic agents, are more effective destroying cells that are progressing through the cell cycle (Fig. 54-1) than destroying cells that are resting in the G0 phase. The “growth fraction,” defined in Figure 54-1, is the fraction of cells progressing through the cycle. Besides tumor cells that may be proliferating, there are also some non-neoplastic cells undergoing division, particularly those of hair follicles, bone marrow, and intestinal epithelium. These rapidly dividing cells are especially sensitive to antineoplastic drugs and account for many of their undesirable side effects. It is believed that most, if not all, anticancer drugs kill cells primarily through a programmed, energy-dependent process called apoptosis, rather than through necrosis.
The number of cultured neoplastic cells that survive exposure to each drug typically shows a first-order relationship to the drug concentration (Fig. 54-2). This means that the same fraction of cells is killed with each drug dose and that a series of several doses does not kill 100% of them. This “log-cell kill” hypothesis is compatible with the clinical observation that a functional host immune system is needed for killing all neoplastic cells and curing a patient.
Endogenous cellular defenses, such as thiols or deoxyribonucleic acid (DNA) repair enzymes, however, can necessitate a threshold of drug concentration, or “shoulder,” in the survival curves of patients receiving these drugs (see Fig. 54-2).
| Therapeutic Overview |
|---|
| Goal |
Mechanisms of Action
The basic mechanisms by which antineoplastic drugs kill tumor cells are summarized in Figure 54-3. Only compounds that show some selectivity for neoplastic cells are used clinically. In general, antimetabolites inhibit DNA synthesis, whereas alkylating agents, intercalators, and antibiotics damage or disrupt DNA, interfere with topoisomerase activity, or alter ribonucleic acid (RNA) structure. Steroids interfere with transcription, several plant alkaloids disrupt mitosis, agents such as asparaginase destroy essential amino acids needed for translation, and other drugs act through important growth factor signal transduction pathways. Many clinically used antineoplastic drugs must undergo either chemical or enzymatic modification to become actively cytotoxic.
Alkylation refers to the covalent attachment of alkyl groups to other molecules. Alkylating agents came to be used for cancer therapy as a result of observations of the effects of the mustard gases on cell growth. Although these compounds are too toxic for clinical use in cancer, the first effective antineoplastic agents, including mechlorethamine, were developed from related nitrogen mustard alkylating agents and are still used today.
Alkylation takes place through chemical formation of a positively charged carbonium ion that reacts with an electron-rich site, particularly on DNA or RNA, to form modified nucleic acids. Most clinically used alkylating drugs have two active groups, which enable them to form covalent links between adjacent nucleic acid strands that are more difficult to repair than monofunctional adducts. These cross-links also prevent separation of the dual strands of DNA during cell cycling. For maximal kill, it is important to administer the maximally tolerated dose. The alkylation sequence for mechlorethamine reacting with the N-7 position of deoxyguanylate is shown in Figure 54-4. Although many other nucleophilic constituents, including RNA, proteins, and membrane components, become alkylated within cells, it is generally believed that the primary cytotoxic events occur through alkylation of DNA, especially by coupling to the N-7 position of the deoxyguanylates of either single- or double-stranded DNA.
Structures of several clinically used alkylating agents are shown in Figure 54-5. Cyclophosphamide undergoes a combination of enzymatic and chemical activation to form the active phosphoramide mustard alkylating agent (Fig. 54-6). Exposure of cells to cyclophosphamide and other alkylating agents can also lead to carcinogenesis. For example, leukemia is a well-known long-term complication in patients with Hodgkin’s disease treated with a regimen including mechlorethamine.
Another group of antineoplastic alkylating agents in clinical use is the nitrosoureas. The structures and primary mechanisms of activation for these compounds are shown in Figure 54-7. In addition to alkylation of DNA, the nitrosoureas also cause carbamoylation of proteins, which may play a role in cytotoxicity. The alkylation route, however, is the major source of cytotoxicity. Nitrosoureas are lipophilic and can cross the blood-brain barrier, so they are often used to treat brain tumors.
Temozolomide is the first new alkylating agent approved for treatment of malignant gliomas in decades. It has a structure similar to dacarbazine and is rapidly absorbed after oral administration. Unlike many other alkylating agents, temozolomide crosses the blood-brain barrier. It methylates guanine and adenine, resulting in misincorporation of thymidine (across from the methylated guanidine), which cannot be easily corrected by the mismatch repair system. Resistance occurs by up regulation of components of the DNA mismatch repair system. Other compounds that form covalent bonds with DNA are cisplatin and its analog carboplatin, whose structures are shown in Figure 54-8. Cisplatin is a square planar complex of platinum with two ammonia molecules and two chloride ions at the corners of the plane. The reaction sequence of the active species is complex and not completely understood. Replacement of a chloride with a hydroxyl must occur before the platinum-nitrogen bond can interact with DNA. Subsequently, the second chloride is aquated and reacts with DNA. The stereochemistry of the complex enables the cis, but not the trans isomer, to form two covalent platinum-nitrogen bonds, primarily at two adjacent deoxyguanylates of DNA. This intrastrand cross-link prevents DNA replication and is cytotoxic. Cisplatin also forms interstrand cross-links and protein-DNA cross-links. Similar DNA cross-links are formed with carboplatin. Oxaliplatin is an organoplatinum compound constructed to overcome resistance to cisplatin by binding the platinum atom to 1,2 diaminocyclohexane. Oxaliplatin undergoes conversion to reactive metabolites that covalently bind to either adjacent guanines, adjacent adenine-guanines, or guanines separated by an intervening nucleotide. This creates interstrand and intrastrand cross-links and inhibits DNA replication and transcription. Oxaliplatin is approved for treatment of metastatic colorectal carcinoma, has activity in lung cancer, and may have activity in breast and esophageal cancers and lymphoma.
The specific reaction sequences by which dacarbazine or procarbazine alkylate DNA are not well understood. Melphalan is a phenylalanine derivative and is actively transported into the cell by the carriers that transport leucine and glutamine. Melphalan is associated with induction of secondary leukemias. Chlorambucil is structurally similar to melphalan and is used primarily in treatment of chronic lymphocytic leukemia. Ifosfamide, like its analog cyclophosphamide, is activated by hepatic microsomes. Early studies with ifosfamide showed its use was associated with a significant incidence of hemorrhagic cystitis. Ifosfamide is now administered with a systemic thiol, sodium-2-mercaptoethane sulfonate (MESNA). MESNA becomes a free thiol after glomerular filtration and combines with the products responsible for causing the cystitis. Ifosfamide is active against several cancers, including small-cell lung cancer, sarcomas, lymphomas, testicular carcinoma, and gynecological cancers.
The antimetabolites are compounds that mimic the structures of normal metabolic constituents including folic acid, pyrimidines, or purines. The antimetabolites inhibit the enzymes necessary for folic acid regeneration or the pyrimidine or purine activation of DNA or RNA synthesis in neoplastic cells. Antimetabolites frequently kill cells in the S phase (see Fig. 54-1). Methotrexate (MTX), 5-fluorouracil (5-FU), cytarabine (Ara-C), 6-mercaptopurine (6-MP), gemcitabine, and 6-thioguanine (6-TG) are the primary antimetabolites used clinically.
Folic acid is essential for enzymatic reactions that transfer methyl and related groups during purine and pyrimidine synthesis. The antimetabolite MTX competitively inhibits the enzyme dihydrofolate reductase, which catalyzes the reduction of dihydrofolate to tetrahydrofolate (FH4; Fig. 54-9
[/not-level-membership-for-basic-science-category]
