[level-membership-for-critical-care-medicine-category]11
Mechanical Ventilation in Acute Respiratory Distress Syndrome
PHYSIOLOGIC BASIS OF MECHANICAL VENTILATION
Respiratory Mechanics, Chest Wall Elastance, and Lung Volume
MECHANICAL VENTILATION IN ACUTE RESPIRATORY DISTRESS SYNDROME: AVAILABLE EVIDENCE
INDIVIDUALIZING MECHANICAL VENTILATION IN PATIENTS WITH ACUTE RESPIRATORY DISTRESS SYNDROME
Since the first description of acute respiratory distress syndrome (ARDS) in 1967,1 mechanical ventilation has been the primary “buying time” treatment for acute lung injury (ALI). Mechanical ventilation is not a “gas exchanger,” but instead replaces, totally or partially, the force normally generated by the respiratory muscles to promote ventilation, providing muscle rest. The effects of mechanical ventilation on gas exchange are indirect and include (1) better clearance of carbon dioxide (CO2) by the power of the mechanical ventilator to expand the diseased and collapsed lung; (2) improvement in oxygenation by preventing alveolar hypoxia caused by hypoventilation; (3) improvement in oxygenation by increasing inspiratory oxygen fraction, which affects alveolar partial pressure of oxygen (PAO2); (4) improvement in oxygenation by opening lung regions otherwise collapsed (alveolar recruitment); and (5) improving oxygenation by maintaining positive end-expiratory pressure (PEEP), preventing the collapse of the lung regions previously recruited during the inspiratory phase.
History
Extensive reviews on the history of mechanical ventilation of ARDS can be found elsewhere.2,3 In the 1970s, mechanical ventilation was recommended and performed with low PEEP and high tidal volume: “. . . larger tidal volumes (10 to 15 per kilogram) are preferable, having been used in several thousand ventilated patients with no evidence of development of pulmonary damage.”4 Today, this advice seems inconceivable. At that time, the main concerns were the putative harm of high inspiratory oxygen fraction and the hemodynamic impairment. It was later recognized in experimental and clinical settings5–7 that high-volume/high-pressure mechanical ventilation could severely damage the lung parenchyma. Such lesions, primarily attributed to the excessive airway pressure, were collectively classified as barotrauma. In the same period, Suter and colleagues8 published a report that, for the first time, systematically described the interaction between PEEP, lung mechanics, gas exchange, and hemodynamics.
In the 1980s, based on the work of Dreyfuss,9,10 the focus progressively shifted from the potential harm of pressure to the harm of volume (overdistention), a concept that was popularized as volutrauma.11 In the mid-1980s, the application of computed tomography (CT)12,13 and the quantitative approach to CT analysis14 led to the concept of baby lung,15,16 which accounted for most of the previous observations on respiratory mechanics, gas exchange functionality, and potential harm of mechanical ventilation. The premise of this line of thought is that high pressure or excessive distention applied to a small fraction of the lung parenchyma (with a size similar to the lung of a baby) unavoidably leads to structural lesions of the lung regions open to ventilation. Total lung rest was achievable with the use of extracorporeal CO2 removal, targeted to prevent the damages of high-pressure/high-volume ventilation.17–19 The target of mechanical ventilation shifted toward lung protection, rather than normal gas exchange functionality. Hickling and associates20 proposed the “permissive hypercapnia” strategy for ARDS, providing “gentle treatment” of the portion of the lung that remains open to ventilation (the baby lung) through less aggressive mechanical ventilation, at the acceptable price of an abnormally higher arterial partial pressure of carbon dioxide (PaCO2).
A large amount of experimental and clinical data over the years has supported the approach of a lung protective strategy.21–25 Low tidal volume prevents the excessive global stress and strain of the baby lung, whereas higher PEEP prevents the regional excessive stress and strain by avoiding alveolar collapse and reopening during mechanical ventilation.26 These mechanical events are associated with an inflammatory reaction of the epithelial and endothelial lung cells (biotrauma/atelectrauma) as shown by the seminal study of Tremblay and colleagues27 (see also work of Dreyfuss and Saumon11 and Tremblay and Slutsky28). The literature to date supports the thought that the harm of mechanical ventilation is due to excessive global or regional stress and strain.29 This situation leads to two results—a physical rupture of the lung and a mechanically induced inflammation of the lung parenchyma, constituting VILI. The best “mechanical ventilation” would provide adequate gas exchange with the lowest amount of VILI.
Physiologic Basis of Mechanical Ventilation
Driving Force
To ventilate the respiratory system, force is required. The driving force for ventilation under normal circumstances is provided by the respiratory muscles. During spontaneous breathing, the thoracic cage expands. This causes a decrease in intrathoracic pressure (pleural pressure, Ppl) relative to the atmosphere (in normal conditions approximately 2 mm Hg of ΔPpl is sufficient to expand the thoracic cage by 0.5 L). Because the lungs are connected in series with the thoracic cage, their volume is expanded to a near equal extent (not considering the blood shift30). The force that distends the lung is the pressure difference between the alveoli and the pleural cavity (the transpulmonary pressure, PL). As the lung expands, the alveolar pressure becomes subatmospheric, and gas flow is generated (inspiration). When the respiratory muscles relax, the potential energy accumulated in the respiratory system (lung and chest wall) returns the chest wall and the lung parenchyma to the resting position (expiration). In spontaneously breathing subjects, the driving force (muscular pressure, ΔPmusc) is spent partly to expand the chest wall (ΔPpl), partly to expand the lung (ΔPL), and partly to overcome the resistances to the gas flow. In quasi-static conditions, in which the resistances to gas flow are negligible:
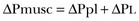
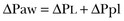
Transpulmonary Pressure
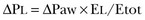
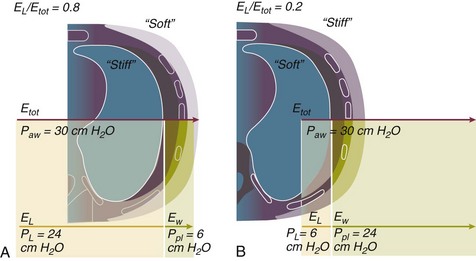
The transpulmonary pressure for a given driving pressure uniquely depends on the ratio of lung elastance to respiratory system elastance. In normal subjects EL/Etot is approximately 0.5, whereas in patients with ARDS it may range from 0.2 (e.g., in obese patients or in patients with high intra-abdominal pressure) to 0.8 (e.g., in patients with a very small baby lung and a normal chest wall elastance). This variability implies that for the same driving force applied and read on the ventilator display (e.g., 30 cm H2O), the resulting transpulmonary pressure may range from 6 to 24 cm H2O (Fig. 11.1).
Force-Bearing Structure of Lung Parenchyma
The transpulmonary pressure is applied to the force-bearing structure of lung parenchyma, the extracellular matrix, which constitutes the lung skeleton.31 The lung skeleton is a complex and metabolically active structure that includes a network of several components—elastin, collagen, and proteoglycans. All these molecules are involved in determining the mechanical characteristics of the respiratory system. The elastin may be considered as an elastic spring, whereas the unextensible collagen, which is folded at end expiration and completely unfolded at a lung volume equal to total lung capacity, acts as a stop-length fiber.32,33 The proteoglycans stabilize the collagen-elastin network, contributing to lung elasticity and alveolar stability at low and medium lung volumes.34
The matrix of elastin, collagen, and proteoglycans is arranged in two main fiber systems: (1) the axial system, which originates from the pulmonary hilum and runs deeply into the lung parenchyma down to the alveolar level, where it joins (2) the peripheral system, which originates from the visceral pleura and runs centripetally within the lung parenchyma.31 The lung skeleton may be considered as a continuous elastic structure that reaches its extension limits at total lung capacity, a lung volume equal to about threefold the lung resting volume. At this level of alveolar distention, the collagen is completely unfolded, and further expansion is prevented. The epithelial and endothelial cells do not directly bear the applied forces because they are anchored to the extracellular matrix by a series of structural proteins (integrins), which are connected to the cytoskeleton. During lung expansion, the epithelial and the endothelial cells modify their shape.
It is well documented that mechanically induced cellular deformation activates a series of mechanosensors with the production of several inflammatory mediators, such as cytokines (interleukin 6, tumor necrosis factor-α, and interferon-γ),27,35 metalloproteinases (enzymes involved in the remodeling of the matrix),36 leukotrienes,37 and interleukin 8,38–40 the most powerful attractor of neutrophils.41 Gross barotrauma (e.g., pneumothorax) is due to the stress at rupture of the lung skeleton, whereas intrapulmonary inflammation is primarily due to the excessive strain of the epithelial and endothelial cells.
Concept of Stress and Strain
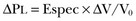
where ΔV is the volume variation applied to the lung (i.e., the tidal volume), and V0 is the lung resting volume (i.e., the functional residual capacity at atmospheric pressure [without any application of PEEP]). The lung-specific elastance is the transpulmonary pressure required to double the lung resting volume (i.e., the ΔPL when ΔV/V0 is equal to 1). In ARDS, lung-specific elastance is similar to normal,16,42 reinforcing the concept of the baby lung (lung is small and not stiff), and questions the use of normalizing the tidal volume to the ideal body weight. The same tidal volume per kilogram may result in completely different strain according to the size of the baby lung (the V0 of the previous equation). For example, a 70-kg man with ARDS may have, according to the severity of the lung injury, a residual baby lung equal to 60%, 40%, or 20% of his normal lung size. If the ventilator is set to deliver 10 mL/kg, the actual delivered tidal volume would generate an alveolar strain, which would result from the application, in normal lung, of a tidal volume equal to 17 mL/kg, 25 mL/kg, and 50 mL/kg, values associated with a significant lung injury in laboratory studies.11,29
Recently we attempted to quantify the relationship between stress-strain and VILI in healthy animals. We found that edema formation was a threshold phenomenon, induced by mechanical ventilation when the global strain reaches a critical value of about 2.43 This threshold roughly corresponds to the point where the stress-strain curve loses its linearity and starts an exponential growth, indicating that some lung regions reach their own total capacity and cannot expand any further. At this level of strain, in a period of 24 to 48 hours the mechanical ventilation is lethal and the increased lung weight (two to three times the baseline) is associated with a striking impairment of respiratory mechanics, gas exchange, hemodynamics, inflammation, distal organs damage, and 100% mortality rate. This lethal strain, and associated stress, however, is rarely applied in clinical practice. To explain VILI in a diseased lung, therefore, alternative phenomena must be taken into account as the lung dishomogeneity and the presence of stress rise, which will be discussed later.
Patient Characterization
Gas Exchange
Oxygen
PaO2, PaO2-to-fraction of inspired oxygen ratio (PaO2/FIO2), and Riley’s shunt fraction44 are the most commonly used variables to assess the severity of lung injury. In particular, the PaO2/FIO2 thresholds of 300 and 200 were used to define ALI (300) and ARDS (200) according to the American-European Consensus Conference on ARDS.45 Consequently, the PaO2/FIO2 ratio is perceived as a key variable by most intensive care unit (ICU) physicians: The lower the PaO2/FIO2 ratio, the greater the lung injury. This equivalence is highly questionable. First, in most large studies on ARDS, no association was found between hypoxemia and outcome.46 In the large study showing a better outcome with low tidal volume compared with higher tidal volume, the PaO2/FIO2 ratio was significantly lower in the low tidal volume group despite ending with better outcome.25 Finally, the PaO2/FIO2 ratio was not different in patients with early, intermediate, and late ARDS, suggesting that oxygenation was not dependent on the structural changes of lung parenchyma occurring with time.47 In a study48 in which the lung severity was assessed by CT scan (and defined as a fraction of the gasless tissue), we did not find significant changes of PaO2/FIO2 ratio over a wide range of nonaerated tissue (Fig. 11.2), and PaO2/FIO2 ratio was not associated with outcome.
Most data suggest that PaO2/FIO2 ratio is a weak indicator of the overall lung severity, with compensatory rearrangement of perfusion during ARDS limiting the deterioration of oxygenation. The same limits apply when PaO2/FIO2 ratio changes are used to assess lung recruitment. Because this maneuver is unavoidably associated with changes of perfusion (global or regional), the increase of PaO2/FIO2 ratio may be partly due to decrease of perfusion, as shown in the 1980s.49–51 Most data suggest that the use of oxygenation variables alone to assess lung severity is misleading.
Carbon Dioxide
Although less considered, the variables derived from CO2, as the total or alveolar deadspace, seem to be of greater value in assessing lung severity. It has been shown in ALI/ARDS patients that deadspace at entry is a strong predictor of outcome,52 and that PaCO2 for the same total ventilation steadily increases from early to intermediate and to late ARDS, reflecting the lung structural changes.47 The PCO2 response to prone position (in contrast to PO2 response) is a strong prognostic index of mortality.53 Most data suggest that CO2-related variables (deadspace), more than PaO2/FIO2 ratio, reflect the severity of lung injury (and associated mortality rate) at the time of presentation and the structural changes of lung parenchyma occurring with time (fibrosis, Pneumocystis, and possibly perfusion defects).
Respiratory Mechanics, Chest Wall Elastance, and Lung Volume
In the original description of ARDS,1 the low compliance (i.e., high elastance) of the respiratory system was a landmark of the syndrome. The respiratory system compliance is not considered in the current definition of ARDS, however.45 For years, the low compliance was attributed uniquely to the lung component (lung stiffness and lack of surfactant). Quantitative CT shows, however, that the respiratory system compliance primarily reflects the size of lung open to gases (the baby lung14,16), suggesting that the intrinsic functioning lung elasticity in ARDS is close to normal (the lung is “small” rather than “stiff”). More than gas exchange, the respiratory system compliance indirectly assesses the lung injury severity (the smaller baby lung, the greater lung injury),54 as confirmed in animal experiments.55 Another variable also must be taken into account—the elastance of the chest wall. It has been shown in a significant portion of ALI/ARDS patients that the chest wall elastance is greater than normal because of increased intra-abdominal pressure, obesity, or severe edema.56 In patients with extrapulmonary ARDS57 and in obese patients,58 the high elastance of the respiratory system may be due to the chest wall and to the lung derangement. Measurement of intra-abdominal pressure should be considered when selecting mechanical ventilator settings.
Severity of Lung Injury and Lung Recruitability
CT can be used to assess the severity of lung injury by measuring the fraction of nonaerated lung tissue at end expiration (end expiration pause at 5 cm H2O). This fraction includes the lung tissue that is “consolidated” (i.e., not openable at 45 cm H2O airway pressure) and the tissue that is collapsed but openable at 45 cm H2O. These values were chosen to produce a minimal risk during the maneuver and because this is the most frequently used in the literature for recruitment maneuver.59 In patients with elevated chest wall elastance, the resulting transpulmonary pressure could be insufficient, however, to overcome the opening pressure of some pulmonary units.
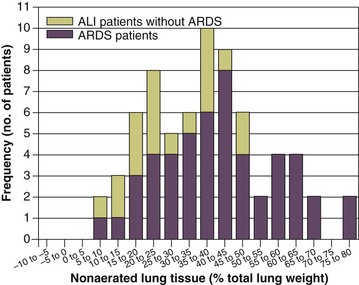
The total fractions of nonaerated lung tissue (consolidated plus recruitable) and the recruitable tissue alone (tissue that regains aeration at 45 cm H2O airway pressure) are strongly associated with mortality rate. The data of Figure 11.2 show the inadequacy of the term ALI/ARDS, as currently defined, to describe the lung injury.48 The data refer to a population of ALI/ARDS patients, classified according to the American-European Consensus Conference on ARDS,45 in which a CT-based quantitative analysis of the whole lung parenchyma at 5 cm H2O PEEP was performed. As shown in Figure 11.3, the fraction of the nonaerated lung tissue (consolidated or collapsed or both) may range from 5% to 70% of the entire lung parenchyma. Patients meeting ALI/ARDS criteria may have a baby lung size very close to that of normal subjects, or a baby lung that is just a small fraction of the expected normal lung. When the distending force is applied to the lungs by the mechanical ventilator, previously collapsed lung regions may open. Lung recruitability may be expressed as the amount of lung tissue regaining aeration when increasing the applied driving force from 5 to 45 cm H2O. As shown in Figure 11.3, in some patients, lung recruitability was almost negligible, whereas in others it was equal to 25% to 35% of the entire lung parenchyma. Lung recruitability was strongly associated with the fraction of nonaerated tissue, suggesting that the greater the inflammatory edema, the greater the lung collapse. It seems that the best way to assess the overall lung severity and the related lung recruitability, both strongly associated with mortality rate, is the CT scan analysis. Physiologic variables are poor indicators of the severity of the lung injury. This relationship is shown in Figure 11.4—for a large variation of lung injury, ranging from 20% to 60% of the lung parenchyma, the values of PaO2/FIO2 ratio, lung compliance, and PaCO2 greatly overlap.
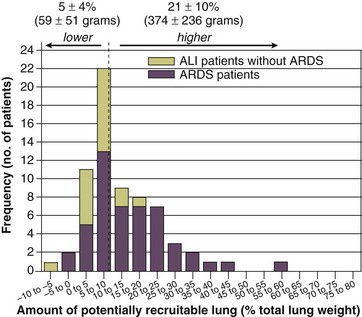
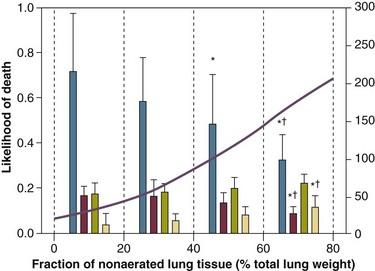
Figure 11.4 Mean ± standard deviation values of respiratory variables of a population of 68 patients with acute lung injury/acute respiratory distress syndrome. Blue columns represent PaO2/FIO2 ratio at positive end-expiratory pressure (PEEP) 5 cm H2O (mm Hg), red columns represent respiratory system compliance at PEEP 5 cm H2O (mL/cm H2O), green columns represent deadspace fraction (%), and gold columns represent alveolar deadspace fraction (%). *P < 0.05 versus patients with a fraction of nonaerated tissue recorded at 5 cm H2O PEEP less than 0.2. †P < 0.05 versus patients with a fraction of nonaerated tissue recorded at 5 cm H2O PEEP ranging from 0.2 to 0.4. The continuous purple line represents the likelihood of death predicted by the fraction of nonaerated lung tissue recorded at 5 cm H2O PEEP (P = 0.015). Weight is a poor surrogate of strain because ARDS patients with similar body weights may have completely different lung sizes and consequently different levels of strain at equal tidal volumes. This is confirmed by the plateau pressures measured in these different studies (see Fig. 11.5). As shown for the same tidal volume per ideal body weight, the plateau pressures are widely distributed with huge overlap between the studies. This reflects a wide distribution of the respiratory system compliance and consequently of the lung size.
Ards Classification
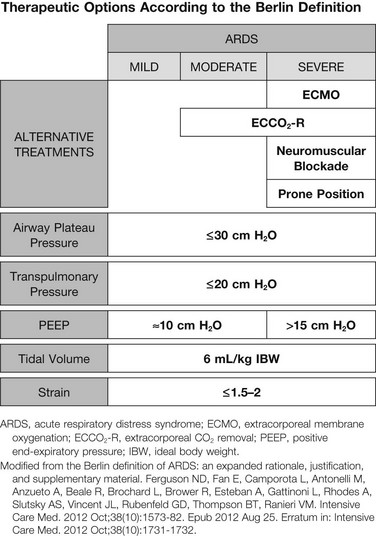
Although, in our opinion, the definition of ARDS should include a quantitative estimate of the lung edema, because this is not feasible in most of the ICU, alternative ways for defining ARDS have been sought. ARDS was first defined by Ashbaugh and coworkers in 1967.1 Several other definitions have been introduced afterward, up to the American-European Consensus Conference (AECC) in 1994.45 This conference defined ARDS as the acute onset of hypoxemia (PaO2/FIO2 ≤ 200) with bilateral infiltrates seen at x-ray study and no evidence of left atrial hypertension, and these parameters were largely used for the enrollment of patients in clinical trials. In 2011, a panel of experts, under the initiative of the European Society of Intensive Care Medicine endorsed from the American Thoracic Society and Society of Critical Care Medicine, convened to develop what has been called the “Berlin definition.”60 The final Berlin definition is reported in Table 11.1. Stages of mild, moderate, and severe ARDS were related to increased mortality rates and increased median duration of mechanical ventilation in survivors.
Table 11.1
| Defining Features | Criteria |
| Timing | Within 1 week of a known clinical insult or new/worsening respiratory symptoms |
| Chest imaging* | Bilateral opacities, not fully explained by effusions, lobar/lung collapse, or nodules |
| Origin of edema | Respiratory failure not fully explained by cardiac failure or fluid overload Need for objective assessment (e.g., echocardiography) to exclude hydrostatic edema if no risk factor is present |
| Oxygenation† | |
| Mild | 200 < PaO2/FIO2 ≤ 300 with PEEP or CPAP ≥ 5 cm H2O‡ |
| Moderate | 100 < PaO2/FIO2 ≤ 200 with PEEP ≥ 5 cm H2O |
| Severe | PaO2/FIO2 ≤ 100 with PEEP ≥ 5 cm H2O |
*Chest x-ray study or computed tomography scan.
†If altitude is higher than 1000 m, the following correction factor should be used: PaO2/FIO2 × (barometric pressure/760).
‡This may be delivered noninvasively in the “Mild” group.
From The ARDS Definition Task Force. Acute Respiratory Distress Syndrome—The Berlin definition. JAMA 2012;307(23):2526-2533.
Mechanical Ventilation in Acute Respiratory Distress Syndrome: Available Evidence
Setting Tidal Volume
The main results of several outcome studies specifically designed to compare different tidal volumes are summarized in Table 11.2. A survival benefit was found in the study comparing 6 mL/kg tidal volume versus 12 mL/kg, the highest tidal volume range tested,25 whereas no survival differences were found in the other studies, which compared intermediate values of tidal volumes.21,23,24 As previously discussed, the tidal volume per ideal body weight is a poor surrogate of strain because ARDS patients with similar body weight may have completely different lung sizes and consequently different strain at equal tidal volume. This is confirmed by the plateau pressures measured in these different studies (Fig. 11.5). As shown for the same tidal volume per ideal body weight, the plateau pressures are widely distributed with huge overlap between the studies. This reflects a wide distribution of the respiratory system compliance and consequently of the lung size.
Table 11.2
Different Tidal Volumes per Ideal Weight and Airway Plateau Pressures Investigated in Other Studies
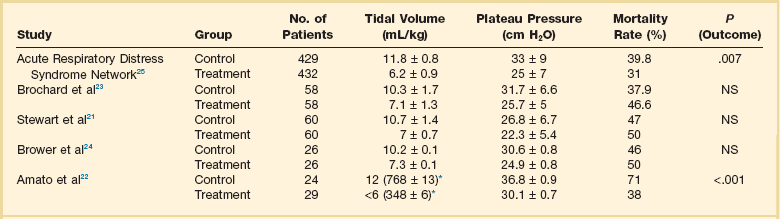
Studies cited in this table can be found in the complete list of references for the chapter, available online at www.expertconsult.com.
*Mean ± standard error values of tidal volume (mL/kg) were not provided in the report by Amato and colleagues. The value set by the physician and the mean ± standard error values of tidal volume in mL (over the first 36 hours) have been provided.
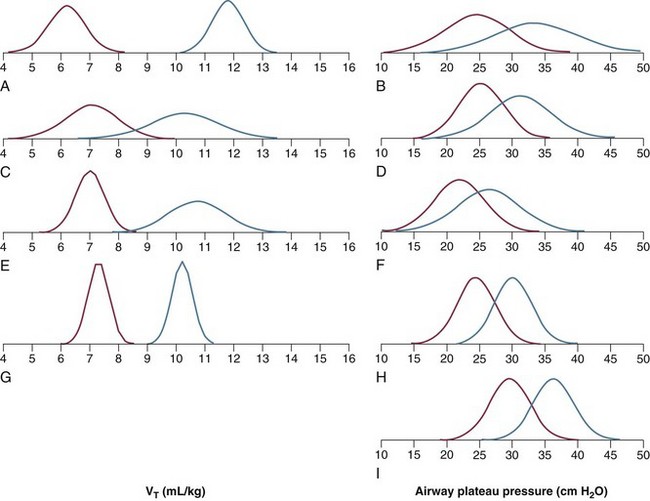
Figure 11.5 Distribution of tidal volume (VT) per ideal body weight (mL/kg) (left column) and of plateau pressure (right column) (red line = treated group; blue line = control group) in five randomized trials (computed from the reported mean ± standard deviation assuming a Gaussian distribution). Data taken from the following studies: A and B, Acute Respiratory Distress Syndrome Network;25 C and D, The Multicenter Trial Group on Tidal Volume Reduction in ARDS;23 E and F, Pressure- and Volume-Limited Ventilation Strategy Group;21 G and H, Brower and associates;24 I, Amato and associates.22 (With permission from Gattinoni L, Carlesso E, Cadringher P, et al: Physical and biological triggers of ventilator-induced lung injury and its prevention. Eur Respir J Suppl 2003;47:15s-25s.)
It has been suggested that a plateau pressure of 30 cm H2O represents the safe limit for plateau pressure.61 In a retrospective analysis, such a safe limit has been challenged, however.62 This is understandable if we consider that the airway plateau pressure is a poor surrogate of stress owing to the high variability of the chest wall compliance. The same plateau pressure, 30 cm H2O, may result in completely different stress values in different patients.
Setting Positive End-Expiratory Pressure
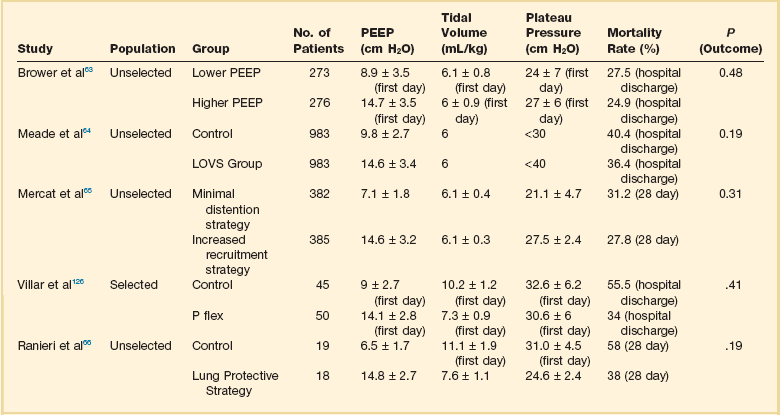
The available evidence on the effects of higher versus lower levels of PEEP in ALI/ARDS patients is summarized in Table 11.3. As shown, despite the fact that the experimental data provide striking evidence that PEEP may reduce the damages of mechanical ventilation, the largest studies comparing higher versus lower PEEP levels in unselected ALI/ARDS patients were unable to find any difference in outcome.63–66 It has been suggested that PEEP should be of benefit in patients with higher lung recruitability and useless or harmful in patients with low lung recruitability.67 Actually two meta-analyses seem to confirm this hypothesis. The meta-analysis by Phoenix and associates68 showed a trend toward improved survival in the high PEEP group with no evidence of increase in barotrauma. The other meta-analysis by Briel and colleagues69 on the largest three trials reported no treatment effect on hospital survival between higher and lower PEEP groups while a significant improved survival rate was found in patients in presence of ARDS defined as PaO2/FIO2 ratio equal or less than 200. In contrast, in patients with mild and moderate ARDS, higher PEEP seemed harmful. These results roughly account for what we know about the ARDS pathophysiology. In fact, the putative beneficial effect of PEEP on survival should be related to the prevention of excessive regional stress and strain by keeping open lung regions that would otherwise collapse.26,70 When we studied lung recruitability in an unselected ALI/ARDS population, we found that it varied from 0% to more than 50% of the whole lung.59 We found that severity of lung injury was associated with higher lung recruitability and more severe hypoxemia, greater deadspace, and lower compliance of the respiratory system. By arbitrarily dividing the study population into patients with higher or lower lung recruitability, we first observed that in the latter, the amount of recruitable lung was almost negligible, amounting to about 50 g of tissue weight. The response to the application of higher levels of PEEP was minimal and much lower than that observed in patients with higher lung recruitability.
Setting Respiratory Rate
Although extensive work has been devoted to understanding better how to set tidal volume and PEEP in ARDS, the potential importance of respiratory rate in the development of VILI has been scarcely investigated. Technology allows the use of respiratory rate from near zero breaths per minute using extracorporeal techniques to remove CO2 to 2000 to 3000 breaths per minute, by employing high-frequency oscillation. Each tidal volume delivered may be considered as a stress cycle. Increasing the number of stress cycles may increase the lung damage. In experimental ARDS models, it has been observed that decreasing the respiratory rate decreases edema formation in isolated rabbit lung,71 whereas an increase in respiratory rate, during spontaneous breathing, leads to an increase in edema formation.72 It is conceivable that the respiratory rate may play a role in the pathogenesis of VILI when associated with a harmful tidal volume or inadequate PEEP. A different issue is the use of high-frequency oscillatory ventilation (HFOV), a technique that uses very high respiratory frequencies in association with very low tidal volumes and very high mean airway pressure, minimizing inspiratory overdistention and end-expiratory lung collapse.73 Most patients treated with HFOV improved PaO2/FIO2 ratio and reduced oxygenation index over time. Clinical trials were not able to prove a survival benefit, however, a recent meta-analysis on six randomized controlled trials comparing HFOV to conventional ventilation suggested a survival benefit at 30 days.74 Two clinical trials are currently ongoing with the aim of comparing HFOV with lung protective mechanical ventilation (OSCAR trial in the United Kingdom [ISRCTN10416500] and OSCILLATE trial in Canada [ISRCTN87124254]). In the absence of definitive evidence HFOV remains investigational for routine management of ARDS and should be reserved for patients unresponsive to conventional ventilator strategies.
Setting Inspiratory-to-Expiratory Ratio
Mechanical ventilators provide a wide range of inspiratory-to-expiratory (I:E) ratios. Setting an adequate I:E ratio may be of great importance when ventilating patients with structural alterations of lung parenchyma, such as chronic obstructive pulmonary disease and emphysema, and during asthmatic exacerbation. In these conditions characterized by a time constant of the respiratory system greater than normal, a longer expiratory time must be provided to allow deflation of gas from the lungs before the next respiratory cycle, avoiding dynamic hyperinflation and intrinsic PEEP.75 During ALI/ARDS, no convincing evidence has been provided for the advantages of setting an I:E ratio different from 1:1.
In the past, inverse ratio ventilation76 was advocated as a tool to improve the effects of mechanical ventilation in ARDS.77–79 Studies primarily showed an improvement of arterial oxygenation. Increased mean airway pressure, intrinsic PEEP, and a decrease in cardiac output were problematic.80 Extreme forms of manipulating I:E ratio are not recommended; an I:E ratio in ALI/ARDS of 0.5 to 1.5 is appropriate.
Individualizing Mechanical Ventilation in Patients with Acute Respiratory Distress Syndrome
General principles underlying the application of mechanical ventilation in ALI/ARDS patients have been discussed. We now present the sequence of interventions that we believe are most appropriate for tailoring mechanical ventilation in an individual patient.81
Clinicians should be aware of the immediate possible consequences of intubation and initiation of mechanical ventilation. Sedation, either by itself or in association with muscle paralysis, produces a loss of respiratory muscle tone and a cranial shift of the diaphragm, promoting further lung collapse with immediate consequences in gas exchange. Mechanical ventilation produces an increase in intrathoracic pressure and decreased venous return. The intravascular volume status of the patient should be assessed before intubation, and hypovolemia should be corrected. The rate and the amount of fluid replacement should be decided for each individual patient. Inadequate fluid replacement may lead to a severe hemodynamic impairment immediately after beginning positive-pressure ventilation. An excessive fluid replacement in an inflamed lung with leaking capillaries may result in a dramatic increase of pulmonary edema with devastating consequences on gas exchange. Wiedemann and coworkers,82 in a study published in the New England Journal of Medicine, compared a conservative and a liberal strategy of fluid management in 1000 patients with ALI. Although the primary end point (60-day mortality) was not significantly different, patients treated with the conservative strategy had better lung function and shorter duration of mechanical ventilation and intensive care without increasing nonpulmonary organ failures. For these reasons, we prefer to tailor fluid replacement according to the results of a fluid challenge test. Echocardiography also may be helpful in assessing the volumetric status.83 After endotracheal intubation, the initial ventilatory setting employed is blind because the pathophysiologic characteristics of the patient have not yet been assessed. We usually set the ventilator (volume controlled) with a tidal volume equal to 6 to 8 mL/kg ideal body weight, FIO2 equal to 0.7, and a respiratory rate of 15 breaths per minute, with an I:E ratio of 1:1. As soon as hemodynamic stability is obtained, a PEEP trial (preceded by a recruitment maneuver) is performed by applying in a random sequence of 5 to 15 cm H2O PEEP with the patient under sedation and, sometimes, muscle paralysis. At each level of PEEP, we maintain tidal volume, respiratory rate, FIO2, and I:E ratio constant for about 20 minutes, after which we measure (1) arterial oxygenation, (2) respiratory system compliance, and (3) alveolar deadspace. Patients who, after the application of higher PEEP, improve at least two of these three parameters have a greater likelihood of having a higher lung recruitability. Alternatively, it is possible to target a specific saturation on the basis of pulse oximetry, if available, aiming at a hemoglobin oxygen saturation of 90%.
The improvement in arterial oxygenation alone, although commonly employed, may be misleading for the assessment of lung recruitment because it may be the result of a slight decrease in cardiac output or a modification of regional distribution of pulmonary blood flow or both. It has been shown that improvement in respiratory physiologic variables from a PEEP trial has low sensitivity (71%) and specificity (59%) in assessing lung recruitability;59 we prefer to obtain whole-lung CT scanning. In the CT scan facility, while maintaining the baseline ventilator setting, whole-lung CT scanning is performed at 5 cm H2O PEEP at end expiration and 45 cm H2O at end inspiration. The subsequent quantitative analysis of the CT scans allows us to obtain a precise assessment of lung recruitability. In patients with higher recruitability we apply PEEP greater than 15 cm H2O, up to 20 cm H2O and exceptionally greater than 20 but not exceeding 25 cm H2O, whereas in patients with lower lung recruitability we apply a PEEP level not greater than 10 cm H2O.
We attempt to keep the plateau pressure less than 30 cm H2O by modifying tidal volume and respiratory rate. As discussed earlier, however, tidal volume and plateau pressure are inadequate surrogates for alveolar stress and strain. We now measure, in each severe ARDS patient, the transpulmonary pressure by helium dilution technique, by employing an esophageal balloon (lung resting volume of 0 cm H2O or 5 cm H2O PEEP). With mechanical ventilation, a global pulmonary strain greater than about 1.5 (strain is dimensionless because it is the ratio of two volumes), or an end-inspiratory transpulmonary pressure greater than 20 cm H2O, may cause VILI, with irreversible respiratory failure to follow.11,29 These values of alveolar stress and strain are the ones at which the residual ventilatable lung reaches its near total lung capacity, with a full extension of the collagen fibers of the lung skeleton.
In some patients, we have observed that a tidal volume of 8 to 10 mL/kg ideal body weight determines a lung strain value less than 1 and a transpulmonary pressure value less than 14 cm H2O. In these patients, we employ a tidal volume greater than 6 mL/kg ideal body weight, avoiding excessive hypercapnia or need for sedation. In contrast, in the most severe patients, in which the baby lung is extremely small, even a ventilation with 6 mL/kg ideal body weight may result in values of alveolar stress and strain higher than those required to reach total lung capacity. Evidence from the literature and physiologic reasoning strongly indicate that for these patients a safe form of mechanical ventilation does not exist.84,85 We reserve to them the use of extracorporeal support. We acknowledge that measuring alveolar stress and strain may seem to be a physiologic curiosity or a research tool. Nonetheless, we have introduced them in our clinical practice as the most logical approach for VILI prevention.
Some authors advocate the use of other forms of mechanical ventilation in patients with ARDS.86,87 The rationale of pressure support ventilation in ARDS relies on the possibility of decreasing the need for sedation, while preserving the contribution of spontaneous breathing, and with possible advantages on gas and flow pulmonary distribution.88 During the full-blown phase of the illness, we prefer to keep the patient well sedated. This approach may help to reduce energy requirement and oxygen consumption and CO2 production.
Possible Adjuncts to Mechanical Ventilation
Prone Position
Prone positioning, first proposed in 197489 and first applied in ARDS patients in 1976,90 results in improved arterial oxygenation in most patients. After the introduction of CT scanning, showing lung consolidation located in the dependent lung regions and the aerated baby lung in the nondependent lung regions,12,16 we integrated prone positioning as standard practice in clinical treatment of ARDS patients to improve systemic oxygenation.91 The initial hypothesis was that better perfusion of the baby lung, located in the dependent lung regions after prone positioning, would provide advantages in gas exchange. The picture observed was quite different, however. We did observe an improvement in arterial oxygenation, but the mechanism was likely different because CT scans taken in the prone position showed a density redistribution toward the dependent lung areas.92
This observation led to our introduction of the “sponge model” as our pathophysiologic understanding of ARDS.93 Whatever the position of the patient, the increased weight of the nondependent lung tissue squeezes the gas out of the dependent regions of the lung.42 The mechanisms of improved gas exchange were different from that first hypothesized. It is not the aim of this chapter to discuss the possible physiologic mechanisms of prone positioning, which may be found elsewhere.94–98 Taken together, all of the studies, including small and large series of patients, consistently showed that in 70% of the patients systemic oxygenation improves in prone compared with supine positioning,98,99 without any change in the applied airway pressure. There is no doubt that in life-threatening severe hypoxemia a trial in the prone position is indicated.
A different issue is the effectiveness of the prone position in improving ARDS outcome. Is mechanical ventilation in ARDS less harmful in the prone compared with the supine position? Does mechanical ventilation induce less alveolar stress and strain in the prone position? There is a consistent physiologic rationale to believe that this is the case. In experimental settings100–103 and in normal subjects and patients affected by ARDS, CT scan shows a more homogeneous distribution of gas throughout the lung parenchyma in the prone compared with the supine position.98 This observation strongly suggests that the distribution of alveolar stress and strain is more homogeneous in the prone position. In experimental models of ARDS, there is evidence that prone positioning prevents or significantly delays the development of VILI.103,104 Two large randomized studies on prone positioning were unable to show a significant benefit on outcome;97,105 however, prone positioning was applied for only about a quarter of the day, and mechanical ventilation was not controlled. In a more recent trial,106 in which prone positioning was applied for 20 hours per day and mechanical ventilation was strictly controlled, a positive benefit was found for the patients treated with prone positioning. On these bases the Prone-Supine II study107 was organized to detect potential survival benefit of prone positioning avoiding the limitations of previous trials. Although, the study was not able to show a significant survival benefit in the general population, a favorable trend was detected in the subgroup of patients with severe ARDS. In a meta-analysis including 10 clinical trials on adults and children Sud and associates108 found that prone ventilation reduced mortality rate in severely hypoxemic patients (PaO2/FIO2 ≤ 100, p = 0.01) but not in patients with PaO2/FIO2 greater than 100 (p = 0.36). The authors’ suggestion was that prone position may provide benefits in severely hypoxemic patients, but it should not be routinely used in all patients affected by acute hypoxemic respiratory failure. In a pooled analysis107 of the four largest databases99,105–107 of trials on prone position, the absolute mortality rate reduction in severe ARDS patients treated in prone position was approximately 10% (log-rank = 0.03). On the contrary, in patients with moderate ARDS prolonged prone position may be useless or possibly harmful.
Extracorporeal Support
Extracorporeal support of ARDS was first applied in 1972.109 The first randomized trial ever performed in ALI/ARDS showed that patients treated with extracorporeal support or with conventional ventilation had similar mortality rates, equal to about 90%.110 In the 1980s, our center introduced extracorporeal CO2 removal in ARDS, aiming at lung rest with a suggestion of benefit.111,112 A randomized study performed in 1994 did not show any survival benefit with extracorporeal CO2 removal support.113 The results of this trial may have been significantly influenced by bleeding complications in patients undergoing extracorporeal CO2 removal. Despite the discouraging results, in Europe few centers continued to use veno-venous extracorporeal support as a last resource in selected series of patients.114 In the United States, Bartlett and colleagues115,116 continued to provide extracorporeal support associated with mechanical ventilation with less strict entry criteria and with encouraging results. The interest on extracorporeal membrane oxygenation (ECMO) renewed after the publication of CESAR trial in 2009,117 which showed clear benefits on outcome when severely hypoxemic patients were treated with extracorporeal support in an expert high case volume center when compared to nonspecialized hospitals. The rebirth of the technique, however, occurred with its use as a rescue therapy during H1N1 flu epidemics in Australia and New Zealand in severely hypoxemic patients untreatable with conventional methods.118 The approach was veno-venous with high blood flows. This report showed a survival rate higher than 70%, and an impressive number of centers in Europe, United States, South America, Canada, and Asia started to use ECMO in severely hypoxemic patients who do not receive benefits from maximal mechanical ventilation.119–124 Survival rate ranged between 56% and 79%.
The impressive diffusion of ECMO led to the great improvement of this technology, and there are increasing numbers of reports describing simple forms of extracorporeal support, primarily aiming at CO2 removal.125 The actual indications for ECMO depend on the patient’s need and the physician’s request. The choice of the technique may vary from low-flow bypass with CO2 removal to high-flow ECMO with total oxygenation support. If the aim is the treatment of life-threatening hypoxemia, the clear-cut indication is high-flow veno-venous ECMO. If the patient, however, presents with severe cardiac failure, veno-arterial ECMO must be used.
A simplified schema of the possible intervention is reported in Figure 11.6, which underlines the recent ARDS Berlin definition, which may be helpful in dictating the sequence of possible intervention when ARDS severity is increasing.
Key Points
• The targets of mechanical ventilation in ARDS have shifted from providing normal gas exchange to protecting the lung from VILI.
• Mechanical ventilation replaces the action of the respiratory muscles. The driving force of mechanical ventilation (airway pressure displayed on the ventilator) is spent partly to overcome the resistances to flow, partly to inflate the lung, and partly to inflate the chest wall.
• The distending force of the lung (the trigger of VILI) is the transpulmonary pressure (PL)—the difference between the airway pressure (Paw) and the pleural pressure (Ppl). For the same driving force, in normal conditions, the transpulmonary pressure equals the driving force multiplied by the ratio between the lung elastance and the respiratory system elastance (lung elastance + chest wall elastance): PL = Paw × EL/Etot. In normal subjects, this ratio is approximately 0.5; in ARDS patients, it may be 0.2 to 0.8. Consequently, the airway pressure alone may be misleading to assess the transpulmonary pressure imposed on the lung by mechanical ventilation (i.e., to assess the stress). We used airway pressure to indicate end-inspiratory pressure.
• The delivered tidal volume increases the lung volume and in the process produces strain on the lung. The ratio of change in lung volume over resting lung volume (ΔV/V0) is a rough approximation of the strain. Excessive strain causes shape changes of endothelial and epithelial cells inducing an inflammatory reaction. Because the resting volume (V0, the baby lung) may vary largely in ARDS, the same tidal volume per ideal body weight may induce largely different strain values.
• Stress and strain, the triggers of VILI, are linked by a constant, specific lung elastance (Espec), accounting for the following relationship: PL (stress) = Espec × ΔV/V0 (strain). The specific lung elastance is similar in normal subjects and in ARDS patients.
• Physiologic variables are generally poor indicators of the severity of lung injury, with the alveolar deadspace being the only variable associated with outcome. The severity of lung injury may be assessed by CT scan. The fraction of nonaerated lung tissue is strongly associated with mortality risk. The greater the lung injury, the smaller the baby lung, and the greater the stress-strain induced by mechanical ventilation.
• Among outcome studies testing different tidal volumes, only the study comparing the two extreme values tested (6 mL/kg versus 12 mL/kg) showed a significant benefit of lower tidal volume. Data from clinical studies and rationale from physiologic studies are strongly in favor of using the lowest tidal volume possible, likely associated with lower stress and strain, accepting hypercapnia as a side effect.
• All studies comparing lower versus higher PEEP randomly applied to unselected ARDS populations failed to show benefits on survival. In contrast, benefits of higher PEEP were found in more severe selected ARDS patients.
• The lung recruitability may vary greatly within the ARDS population (5% to 50% of the lung parenchyma) and is correlated with the overall lung injury severity. Higher PEEP possibly should be reserved only for patients with higher recruitability.
• When treating patients with ARDS, individual characteristics (lung injury severity, gas exchange, lung mechanics, abdominal pressure) should be assessed to tailor the least harmful mechanical ventilation setting according to the available evidence and physiologic rationale, providing the lowest stress and strain possible.
• In the severe ARDS, in which mechanical ventilation is more harmful, associated therapy such as prone positioning and extracorporeal support must be sought.
References
1. Ashbaugh, DG, Bigelow, DB, Petty, TL, Levine, BE. Acute respiratory distress in adults. Lancet. 1967; 2(7511):319–323.
2. Bernard, GR. Acute respiratory distress syndrome: A historical perspective. Am J Respir Crit Care Med. 2005; 172(7):798–806.
3. Ware, LB, Matthay, MA. The acute respiratory distress syndrome. N Engl J Med. 2000; 342(18):1334–1349.
4. Pontoppidan, H, Geffin, B, Lowenstein, E. Acute respiratory failure in the adult, 3. N Engl J Med. 1972; 287(16):799–806.
5. Webb, HH, Tierney, DF. Experimental pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures. Protection by positive end-expiratory pressure. Am Rev Respir Dis. 1974; 110(5):556–565.
6. Kumar, A, Falke, KJ, Geffin, B, et al. Continuous positive-pressure ventilation in acute respiratory failure. N Engl J Med. 1970; 283(26):1430–1436.
7. Baeza, OR, Wagner, RB, Lowery, BD. Pulmonary hyperinflation. A form of barotrauma during mechanical ventilation. J Thorac Cardiovasc Surg. 1975; 70(5):790–805.
8. Suter, PM, Fairley, B, Isenberg, MD. Optimum end-expiratory airway pressure in patients with acute pulmonary failure. N Engl J Med. 1975; 292(6):284–289.
9. Dreyfuss, D, Basset, G, Soler, P, Saumon, G. Intermittent positive-pressure hyperventilation with high inflation pressures produces pulmonary microvascular injury in rats. Am Rev Respir Dis. 1985; 132(4):880–884.
10. Dreyfuss, D, Soler, P, Basset, G, Saumon, G. High inflation pressure pulmonary edema. Respective effects of high airway pressure, high tidal volume, and positive end-expiratory pressure. Am Rev Respir Dis. 1988; 137(5):1159–1164.
11. Dreyfuss, D, Saumon, G. Ventilator-induced lung injury: Lessons from experimental studies. Am J Respir Crit Care Med. 1998; 157(1):294–323.
12. Gattinoni, L, Mascheroni, D, Torresin, A, et al. Morphological response to positive end expiratory pressure in acute respiratory failure. Computerized tomography study. Intensive Care Med. 1986; 12(3):137–142.
13. Maunder, RJ, Shuman, WP, McHugh, JW, et al. Preservation of normal lung regions in the adult respiratory distress syndrome. Analysis by computed tomography. JAMA. 1986; 255(18):2463–2465.
14. Gattinoni, L, Pesenti, A, Avalli, L, et al. Pressure-volume curve of total respiratory system in acute respiratory failure. Computed tomographic scan study. Am Rev Respir Dis. 1987; 136(3):730–736.
15. Gattinoni, L, Pesenti, A. ARDS: The non-homogeneous lung; facts and hypothesis. Intensive Crit Care Digest. 1987; 6(1):1–4.
16. Gattinoni, L, Pesenti, A. The concept of “baby lung. ”. Intensive Care Med. 2005; 31(6):776–784.
17. Kolobow, T, Gattinoni, L, Tomlinson, TA, Pierce, JE. Control of breathing using an extracorporeal membrane lung. Anesthesiology. 1977; 46(2):138–141.
18. Kolobow, T, Gattinoni, L, Tomlinson, T, Pierce, JE. An alternative to breathing. J Thorac Cardiovasc Surg. 1978; 75(2):261–266.
19. Gattinoni, L, Kolobow, T, Tomlinson, T, et al. Control of intermittent positive pressure breathing (IPPB) by extracorporeal removal of carbon dioxide. Br J Anaesth. 1978; 50(8):753–758.
20. Hickling, KG, Henderson, SJ, Jackson, R. Low mortality associated with low volume pressure limited ventilation with permissive hypercapnia in severe adult respiratory distress syndrome. Intensive Care Med. 1990; 16(6):372–377.
21. Stewart, TE, Meade, MO, Cook, DJ, et al. Evaluation of a ventilation strategy to prevent barotrauma in patients at high risk for acute respiratory distress syndrome. Pressure- and Volume-Limited Ventilation Strategy Group. N Engl J Med. 1998; 338(6):355–361.
22. Amato, MB, Barbas, CS, Medeiros, DM, et al. Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med. 1998; 338(6):347–354.
23. Brochard, L, Roudot-Thoraval, F, Roupie, E, et al. Tidal volume reduction for prevention of ventilator-induced lung injury in acute respiratory distress syndrome. The Multicenter Trial Group on Tidal Volume Reduction in ARDS. Am J Respir Crit Care Med. 1998; 158(6):1831–1838.
24. Brower, RG, Shanholtz, CB, Fessler, HE, et al. Prospective, randomized, controlled clinical trial comparing traditional versus reduced tidal volume ventilation in acute respiratory distress syndrome patients. Crit Care Med. 1999; 27(8):1492–1498.
25. The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000; 342(18):1301–1308.
26. Lachmann, B. Open up the lung and keep the lung open. Intensive Care Med. 1992; 18(6):319–321.
27. Tremblay, L, Valenza, F, Ribeiro, SP, et al. Injurious ventilatory strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model. J Clin Invest. 1997; 99(5):944–952.
28. Tremblay, LN, Slutsky, AS. Ventilator-induced lung injury: From the bench to the bedside. Intensive Care Med. 2006; 32(1):24–33.
29. Gattinoni, L, Carlesso, E, Cadringher, P, et al. Physical and biological triggers of ventilator-induced lung injury and its prevention. Eur Respir J Suppl. 2003; 47:15s–25s.
30. Aliverti, A, Dellaca, R, Pelosi, P, et al. Optoelectronic plethysmography in intensive care patients. Am J Respir Crit Care Med. 2000; 161(5):1546–1552.
31. , Functional morphology of lung parenchyma American Physiological SocietyWeibel ER, ed. Handbook of Physiology a Critical, Comprehensive Presentation of Physiological Knowledge and Concepts. Waverly Press: Baltimore, MD, 1986:89–111.
32. Maksym, GN, Bates, JH. A distributed nonlinear model of lung tissue elasticity. J Appl Physiol. 1997; 82(1):32–41.
33. Maksym, GN, Fredberg, JJ, Bates, JH. Force heterogeneity in a two-dimensional network model of lung tissue elasticity. J Appl Physiol. 1998; 85(4):1223–1229.
34. Souza-Fernandes, AB, Pelosi, P, Rocco, PR. Bench-to-bedside review: The role of glycosaminoglycans in respiratory disease. Crit Care. 2006; 10(6):237.
35. Dos Santos, CC, Slutsky, AS. Invited review: Mechanisms of ventilator-induced lung injury: A perspective. J Appl Physiol. 2000; 89(4):1645–1655.
36. Haseneen, NA, Vaday, GG, Zucker, S, Foda, HD. Mechanical stretch induces MMP-2 release and activation in lung endothelium: Role of EMMPRIN. Am J Physiol Lung Cell Mol Physiol. 2003; 284(3):L541–L547.
37. Caironi, P, Ichinose, F, Liu, R, et al. 5-Lipoxygenase deficiency prevents respiratory failure during ventilator-induced lung injury. Am J Respir Crit Care Med. 2005; 172(3):334–343.
38. Pugin, J, Dunn, I, Jolliet, P, et al. Activation of human macrophages by mechanical ventilation in vitro. Am J Physiol. 1998; 275(6 Pt 1):L1040–L1050.
39. Yamamoto, H, Teramoto, H, Uetani, K, et al. Cyclic stretch upregulates interleukin-8 and transforming growth factor-beta-1 production through a protein kinase C-dependent pathway in alveolar epithelial cells. Respirology. 2002; 7(2):103–109.
40. Vlahakis, NE, Schroeder, MA, Limper, AH, Hubmayr, RD. Stretch induces cytokine release by alveolar epithelial cells in vitro. Am J Physiol. 1999; 277(1 Pt 1):L167–L173.
41. Belperio, JA, Keane, MP, Burdick, MD, et al. Critical role for CXCR2 and CXCR2 ligands during the pathogenesis of ventilator-induced lung injury. J Clin Invest. 2002; 110(11):1703–1716.
42. Gattinoni, L, D’Andrea, L, Pelosi, P, et al. Regional effects and mechanism of positive end-expiratory pressure in early adult respiratory distress syndrome. JAMA. 1993; 269(16):2122–2127.
43. Protti, A, Cressoni, M, Santini, A, et al. Lung stress and strain during mechanical ventilation: Any safe threshold? Am J Respir Crit Care Med. 2011; 183(10):1354.
44. Riley, RL, Cournand, A. “Ideal” alveolar air and the analysis of ventilation-perfusion relationships in the lungs. J Appl Physiol. 1949; 1:827–847.
45. Bernard, GR, Artigas, A, Brigham, KL, et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994; 149(3 Pt 1):818–824.
46. Luhr, OR, Karlsson, M, Thorsteinsson, A, et al. The impact of respiratory variables on mortality in non-ARDS and ARDS patients requiring mechanical ventilation. Intensive Care Med. 2000; 26(5):508–517.
47. Gattinoni, L, Bombino, M, Pelosi, P, et al. Lung structure and function in different stages of severe adult respiratory distress syndrome. JAMA. 1994; 271(22):1772–1779.
48. Gattinoni, L, Caironi, P, Cressoni, M, et al. Lung recruitment in patients with the acute respiratory distress syndrome. N Engl J Med. 2006; 354(17):1775–1786.
49. Dantzker, DR, Brook, CJ, Dehart, P, et al. Ventilation-perfusion distributions in the adult respiratory distress syndrome. Am Rev Respir Dis. 1979; 120(5):1039–1052.
50. Dantzker, DR, Lynch, JP, Weg, JG. Depression of cardiac output is a mechanism of shunt reduction in the therapy of acute respiratory failure. Chest. 1980; 77(5):636–642.
51. Matamis, D, Lemaire, F, Harf, A, et al. Redistribution of pulmonary blood flow induced by positive end-expiratory pressure and dopamine infusion in acute respiratory failure. Am Rev Respir Dis. 1984; 129(1):39–44.
52. Nuckton, TJ, Alonso, JA, Kallet, RH, et al. Pulmonary dead-space fraction as a risk factor for death in the acute respiratory distress syndrome. N Engl J Med. 2002; 346(17):1281–1286.
53. Gattinoni, L, Vagginelli, F, Carlesso, E, et al. Decrease in PaCO2 with prone position is predictive of improved outcome in acute respiratory distress syndrome. Crit Care Med. 2003; 31(12):2727–2733.
54. Gattinoni, L, Pesenti, A, Baglioni, S, et al. Inflammatory pulmonary edema and positive end-expiratory pressure: Correlations between imaging and physiologic studies. J Thorac Imaging. 1988; 3(3):59–64.
55. Henzler, D, Pelosi, P, Dembinski, R, et al. Respiratory compliance but not gas exchange correlates with changes in lung aeration after a recruitment maneuver: An experimental study in pigs with saline lavage lung injury. Crit Care. 2005; 9(5):R471–R482.
56. Malbrain, ML, Chiumello, D, Pelosi, P, et al. Prevalence of intra-abdominal hypertension in critically ill patients: A multicentre epidemiological study. Intensive Care Med. 2004; 30(5):822–829.
57. Gattinoni, L, Pelosi, P, Suter, PM, et al. Acute respiratory distress syndrome caused by pulmonary and extrapulmonary disease. Different syndromes? Am J Respir Crit Care Med. 1998; 158(1):3–11.
58. Pelosi, P, Croci, M, Ravagnan, I, et al. Total respiratory system, lung, and chest wall mechanics in sedated-paralyzed postoperative morbidly obese patients. Chest. 1996; 109(1):144–151.
59. Gattinoni, L, Caironi, P, Cressoni, M, et al. Lung recruitment in patients with the acute respiratory distress syndrome. N Engl J Med. 2006; 354(17):1775–1786.
60. The ARDS Definition Task Force: Acute respiratory distress syndrome—The Berlin definition. JAMA. 2012; 307(23):2526–2533.
61. Tobin, MJ. Culmination of an era in research on the acute respiratory distress syndrome. N Engl J Med. 2000; 342(18):1360–1361.
62. Hager, DN, Krishnan, JA, Hayden, DL, Brower, RG. Tidal volume reduction in patients with acute lung injury when plateau pressures are not high. Am J Respir Crit Care Med. 2005; 172(10):1241–1245.
63. Brower, RG, Lanken, PN, MacIntyre, N, et al. Higher versus lower positive end-expiratory pressures in patients with the acute respiratory distress syndrome. N Engl J Med. 2004; 351(4):327–336.
64. Meade, MO, Cook, DJ, Guyatt, GH, et al. Ventilation strategy using low tidal volumes, recruitment maneuvers, and high positive end-expiratory pressure for acute lung injury and acute respiratory distress syndrome: A randomized controlled trial. JAMA. 2008; 299(6):637–645.
65. Mercat, A, Richard, JC, Vielle, B, et al. Positive end-expiratory pressure setting in adults with acute lung injury and acute respiratory distress syndrome: A randomized controlled trial. JAMA. 2008; 299(6):646–655.
66. Ranieri, VM, Suter, PM, Tortorella, C, et al. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: A randomized controlled trial. JAMA. 1999; 282(1):54–61.
67. Gattinoni, L, Caironi, P. Refining ventilatory treatment for acute lung injury and acute respiratory distress syndrome. JAMA. 2008; 299(6):691–693.
68. Phoenix, SI, Paravastu, S, Columb, M, et al. Does a higher positive end expiratory pressure decrease mortality in acute respiratory distress syndrome? A systematic review and meta-analysis. Anesthesiology. 2009; 110(5):1098–1105.
69. Briel, M, Meade, M, Mercat, A, et al. Higher vs lower positive end-expiratory pressure in patients with acute lung injury and acute respiratory distress syndrome systematic review and meta-analysis. JAMA. 2010; 303(9):865–873.
70. Mead, J, Takishima, T, Leith, D. Stress distribution in lungs: A model of pulmonary elasticity. J Appl Physiol. 1970; 28(5):596–608.
71. Hotchkiss, JR, Jr., Blanch, L, Murias, G, et al. Effects of decreased respiratory frequency on ventilator-induced lung injury. Am J Respir Crit Care Med. 2000; 161(2 Pt 1):463–468.
72. Mascheroni, D, Kolobow, T, Fumagalli, R, et al. Acute respiratory failure following pharmacologically induced hyperventilation: An experimental animal study. Intensive Care Med. 1988; 15(1):8–14.
73. Hager, DN. High-frequency oscillatory ventilation in adults with acute respiratory distress syndrome. Curr Opin Anesthesiol. 2012; 25(1):17–23.
74. Sud, S, Sud, M, Friedrich, JO, et al. High frequency oscillation in patients with acute lung injury and acute respiratory distress syndrome (ARDS): Systematic review and meta-analysis. BMJ. 2010; 18:340.
75. Pepe, PE, Marini, JJ. Occult positive end-expiratory pressure in mechanically ventilated patients with airflow obstruction: The auto-PEEP effect. Am Rev Respir Dis. 1982; 126(1):166–170.
76. Lachmann, B, Haendly, B, Schulz, H, Jonson, B. Improved arterial oxygenation, CO2 elimination, compliance and decreased barotrauma following changes of volume generated PEEP ventilation with inspiratory/expiratory (I/E) ratio of 1/2 to pressure generated ventilation with I/E ratio of 4:1 in patients with severe adult respiratory distress syndrome (ARDS). Intensive Care Med. 1980; 6:64.
77. Lessard, MR, Guerot, E, Lorino, H, et al. Effects of pressure-controlled with different I:E ratios versus volume-controlled ventilation on respiratory mechanics, gas exchange, and hemodynamics in patients with adult respiratory distress syndrome. Anesthesiology. 1994; 80(5):983–991.
78. Mercat, A, Titiriga, M, Anguel, N, et al. Inverse ratio ventilation (I/E = 2/1) in acute respiratory distress syndrome: A six-hour controlled study. Am J Respir Crit Care Med. 1997; 155(5):1637–1642.
79. Zavala, E, Ferrer, M, Polese, G, et al. Effect of inverse I:E ratio ventilation on pulmonary gas exchange in acute respiratory distress syndrome. Anesthesiology. 1998; 88(1):35–42.
80. Mang, H, Kacmarek, RM, Ritz, R, et al. Cardiorespiratory effects of volume- and pressure-controlled ventilation at various I/E ratios in an acute lung injury model. Am J Respir Crit Care Med. 1995; 151(3 Pt 1):731–736.
81. Marini, JJ, Gattinoni, L. Ventilatory management of acute respiratory distress syndrome: A consensus of two. Crit Care Med. 2004; 32(1):250–255.
82. Wiedemann, HP, Wheeler, AP, Bernard, GR, et al. Comparison of two fluid-management strategies in acute lung injury. N Eng J Med. 2006; 354(24):2564–2575.
83. Vignon, P. Hemodynamic assessment of critically ill patients using echocardiography Doppler. Curr Opin Crit Care. 2005; 11(3):227–234.
84. Terragni, PP, Rosboch, G, Tealdi, A, et al. Tidal hyperinflation during low tidal volume ventilation in acute respiratory distress syndrome. Am J Respir Crit Care Med. 2007; 175(2):160–166.
85. Terragni, PP, Del Sorbo, L, Mascia, L, et al. Tidal volume lower than 6 ml/kg enhances lung protection role of extracorporeal carbon dioxide removal. Anesthesiology. 2009; 111(4):826–835.
86. Ferguson, ND, Cook, DJ, Guyatt, GH, et al. High-frequency oscillation in early acute respiratory distress syndrome. N Engl J Med. 2013; 10:795–805.
87. High-frequency oscillation for acute respiratory distress syndrome. N Engl J Med. 2013; 368:806–865.
88. Henzler, D, Pelosi, P, Bensberg, R, et al. Effects of partial ventilatory support modalities on respiratory function in severe hypoxemic lung injury. Crit Care Med. 2006; 34(6):1738–1745.
89. Bryan, AC. Conference on the scientific basis of respiratory therapy. Pulmonary physiotherapy in the pediatric age group. Comments of a devil’s advocate. Am Rev Respir Dis. 1974; 110(6 Pt 2):143–144.
90. Piehl, MA, Brown, RS. Use of extreme position changes in acute respiratory failure. Crit Care Med. 1976; 4(1):13–14.
91. Langer, M, Mascheroni, D, Marcolin, R, Gattinoni, L. The prone position in ARDS patients. A clinical study. Chest. 1988; 94(1):103–107.
92. Gattinoni, L, Pelosi, P, Vitale, G, et al. Body position changes redistribute lung computed-tomographic density in patients with acute respiratory failure. Anesthesiology. 1991; 74(1):15–23.
93. Bone, RC. The ARDS lung. New insights from computed tomography. JAMA. 1993; 269(16):2134–2135.
94. Lamm, WJ, Graham, MM, Albert, RK. Mechanism by which the prone position improves oxygenation in acute lung injury. Am J Respir Crit Care Med. 1994; 150(1):184–193.
95. Lee, DL, Chiang, HT, Lin, SL, et al. Prone-position ventilation induces sustained improvement in oxygenation in patients with acute respiratory distress syndrome who have a large shunt. Crit Care Med. 2002; 30(7):1446–1452.
96. Pelosi, P, Tubiolo, D, Mascheroni, D, et al. Effects of the prone position on respiratory mechanics and gas exchange during acute lung injury. Am J Respir Crit Care Med. 1998; 157(2):387–393.
97. Albert, RK. Prone position in ARDS: What do we know, and what do we need to know? Crit Care Med. 1999; 27(11):2574–2575.
98. Gattinoni, L, Valenza, F, Pelosi, P, Mascheroni, D. Prone positioning in acute respiratory failure. In: Tobin MJ, ed. Principles and Practice of Mechanical Ventilation. New York: McGraw-Hill; 2006:1081–1092.
99. Gattinoni, L, Tognoni, G, Pesenti, A, et al. Effect of prone positioning on the survival of patients with acute respiratory failure. N Engl J Med. 2001; 345(8):568–573.
100. Du, HL, Yamada, Y, Orii, R, et al. Beneficial effects of the prone position on the incidence of barotrauma in oleic acid-induced lung injury under continuous positive pressure ventilation. Acta Anaesthesiol Scand. 1997; 41(6):701–707.
101. Nishimura, M, Honda, O, Tomiyama, N, et al. Body position does not influence the location of ventilator-induced lung injury. Intensive Care Med. 2000; 26(11):1664–1669.
102. Johansson, MJ, Wiklund, A, Flatebo, T, et al. Positive end-expiratory pressure affects regional redistribution of ventilation differently in prone and supine sheep. Crit Care Med. 2004; 32(10):2039–2044.
103. Valenza, F, Guglielmi, M, Maffioletti, M, et al. Prone position delays the progression of ventilator-induced lung injury in rats: Does lung strain distribution play a role? Crit Care Med. 2005; 33(2):361–367.
104. Broccard, A, Shapiro, RS, Schmitz, LL, et al. Prone positioning attenuates and redistributes ventilator-induced lung injury in dogs. Crit Care Med. 2000; 28(2):295–303.
105. Guerin, C, Gaillard, S, Lemasson, S, et al. Effects of systematic prone positioning in hypoxemic acute respiratory failure: A randomized controlled trial. JAMA. 2004; 292(19):2379–2387.
106. Mancebo, J, Fernandez, R, Blanch, L, et al. A multicenter trial of prolonged prone ventilation in severe acute respiratory distress syndrome. Am J Respir Crit Care Med. 2006; 173(11):1233–1239.
107. Taccone, P, Pesenti, A, Latini, R, et al. Prone positioning in patients with moderate and severe acute respiratory distress syndrome. A randomized controlled trial. JAMA. 2009; 302(18):1977–1984.
108. Sud, S, Friedrich, JO, Taccone, P, et al. Prone ventilation reduces mortality in patients with acute respiratory failure and severe hypoxemia: Systematic review and meta-analysis. Intensive Care Med. 2010; 36(4):585–599.
109. Hill, JD, O’Brien, TG, Murray, JJ, et al. Prolonged extracorporeal oxygenation for acute post-traumatic respiratory failure (shock-lung syndrome). Use of the Bramson membrane lung. N Engl J Med. 1972; 286(12):629–634.
110. Zapol, WM, Snider, MT, Hill, JD, et al. Extracorporeal membrane oxygenation in severe acute respiratory failure. A randomized prospective study. JAMA. 1979; 242(20):2193–2196.
111. Gattinoni, L, Agostoni, A, Pesenti, A, et al. Treatment of acute respiratory failure with low-frequency positive-pressure ventilation and extracorporeal removal of CO2. Lancet. 1980; 2(8189):292–294.
112. Gattinoni, L, Pesenti, A, Mascheroni, D, et al. Low-frequency positive-pressure ventilation with extracorporeal CO2 removal in severe acute respiratory failure. JAMA. 1986; 256(7):881–886.
113. Morris, AH, Wallace, CJ, Menlove, RL, et al. Randomized clinical trial of pressure-controlled inverse ratio ventilation and extracorporeal CO2 removal for adult respiratory distress syndrome. Am J Respir Crit Care Med. 1994; 149(2 Pt 1):295–305.
114. Lewandowski, K, Rossaint, R, Pappert, D, et al. High survival rate in 122 ARDS patients managed according to a clinical algorithm including extracorporeal membrane oxygenation. Intensive Care Med. 1997; 23(8):819–835.
115. Kolla, S, Awad, SS, Rich, PB, et al. Extracorporeal life support for 100 adult patients with severe respiratory failure. Ann Surg. 1997; 226(4):544–564.
116. Bartlett, RH, Roloff, DW, Custer, JR, et al. Extracorporeal life support: The University of Michigan experience. JAMA. 2000; 283(7):904–908.
117. Peek, GJ, Mugford, M, Tiruvoipati, R, et al. Efficacy and economic assessment of conventional ventilatory support versus extracorporeal membrane oxygenation for severe adult respiratory failure (CESAR): A multicentre randomised controlled trial. Lancet. 2009; 374(9698):1351–1363.
118. Davies, A, Jones, D, Bailey, M, et al. Extracorporeal membrane oxygenation for 2009 influenza A(H1N1) acute respiratory distress syndrome. JAMA. 2009; 302(17):1888–1895.
119. Roch, A, Lepaul-Ercole, R, Grisoli, D, et al. Extracorporeal membrane oxygenation for severe influenza A (H1N1) acute respiratory distress syndrome: A prospective observational comparative study. Intensive Care Med. 2010; 36(11):1899–1905.
120. Norfolk, SG, Hollingsworth, CL, Wolfe, CR, et al. Rescue therapy in adult and pediatric patients with pH1N1 influenza infection: A tertiary center intensive care unit experience from April to October 2009. Crit Care Med. 2010; 38(11):2103–2107.
121. Ugarte, S, Arancibia, F, Soto, R. Influenza A pandemics: Clinical and organizational aspects: The experience in Chile. Crit Care Med. 2010; 38(4 Suppl):e133–e137.
122. Freed, DH, Henzler, D, White, CW, et al. Extracorporeal lung support for patients who had severe respiratory failure secondary to influenza A (H1N1) 2009 infection in Canada. Can J Anaesth. 2010; 57(3):240–247.
123. Kao, TM, Wu, UI, Chen, YC. Rapid diagnostic tests and severity of illness in pandemic (H1N1) 2009, Taiwan. Emerg Infect Dis. 2010; 16(7):1181–1183.
124. Liong, T, Lee, KL, Poon, YS, et al. The first novel influenza A (H1N1) fatality despite antiviral treatment and extracorporeal membrane oxygenation in Hong Kong. Hong Kong Med J. 2009; 15(5):381–384.
125. Bein, T, Prasser, C, Philipp, A, et al. [Pumpless extracorporeal lung assist using arterio-venous shunt in severe ARDS. Experience with 30 cases]. Anaesthetist. 2004; 53(9):813–819.
126. Villar, J, Kacmarek, RM, Perez-Mendez, L, Aguirre-Jaime, A. A high positive end-expiratory pressure, low tidal volume ventilatory strategy improves outcome in persistent acute respiratory distress syndrome: A randomized, controlled trial. Crit Care Med. 2006; 34(5):1311–1318.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]11
Mechanical Ventilation in Acute Respiratory Distress Syndrome
PHYSIOLOGIC BASIS OF MECHANICAL VENTILATION
Respiratory Mechanics, Chest Wall Elastance, and Lung Volume
MECHANICAL VENTILATION IN ACUTE RESPIRATORY DISTRESS SYNDROME: AVAILABLE EVIDENCE
INDIVIDUALIZING MECHANICAL VENTILATION IN PATIENTS WITH ACUTE RESPIRATORY DISTRESS SYNDROME
Since the first description of acute respiratory distress syndrome (ARDS) in 1967,1 mechanical ventilation has been the primary “buying time” treatment for acute lung injury (ALI). Mechanical ventilation is not a “gas exchanger,” but instead replaces, totally or partially, the force normally generated by the respiratory muscles to promote ventilation, providing muscle rest. The effects of mechanical ventilation on gas exchange are indirect and include (1) better clearance of carbon dioxide (CO2) by the power of the mechanical ventilator to expand the diseased and collapsed lung; (2) improvement in oxygenation by preventing alveolar hypoxia caused by hypoventilation; (3) improvement in oxygenation by increasing inspiratory oxygen fraction, which affects alveolar partial pressure of oxygen (PAO2); (4) improvement in oxygenation by opening lung regions otherwise collapsed (alveolar recruitment); and (5) improving oxygenation by maintaining positive end-expiratory pressure (PEEP), preventing the collapse of the lung regions previously recruited during the inspiratory phase.
History
Extensive reviews on the history of mechanical ventilation of ARDS can be found elsewhere.2,3 In the 1970s, mechanical ventilation was recommended and performed with low PEEP and high tidal volume: “. . . larger tidal volumes (10 to 15 per kilogram) are preferable, having been used in several thousand ventilated patients with no evidence of development of pulmonary damage.”4 Today, this advice seems inconceivable. At that time, the main concerns were the putative harm of high inspiratory oxygen fraction and the hemodynamic impairment. It was later recognized in experimental and clinical settings5–7 that high-volume/high-pressure mechanical ventilation could severely damage the lung parenchyma. Such lesions, primarily attributed to the excessive airway pressure, were collectively classified as barotrauma. In the same period, Suter and colleagues8 published a report that, for the first time, systematically described the interaction between PEEP, lung mechanics, gas exchange, and hemodynamics.
In the 1980s, based on the work of Dreyfuss,9,10 the focus progressively shifted from the potential harm of pressure to the harm of volume (overdistention), a concept that was popularized as volutrauma.11 In the mid-1980s, the application of computed tomography (CT)12,13 and the quantitative approach to CT analysis14 led to the concept of baby lung,15,16 which accounted for most of the previous observations on respiratory mechanics, gas exchange functionality, and potential harm of mechanical ventilation. The premise of this line of thought is that high pressure or excessive distention applied to a small fraction of the lung parenchyma (with a size similar to the lung of a baby) unavoidably leads to structural lesions of the lung regions open to ventilation. Total lung rest was achievable with the use of extracorporeal CO2 removal, targeted to prevent the damages of high-pressure/high-volume ventilation.17–19 The target of mechanical ventilation shifted toward lung protection, rather than normal gas exchange functionality. Hickling and associates20 proposed the “permissive hypercapnia” strategy for ARDS, providing “gentle treatment” of the portion of the lung that remains open to ventilation (the baby lung) through less aggressive mechanical ventilation, at the acceptable price of an abnormally higher arterial partial pressure of carbon dioxide (PaCO2).
A large amount of experimental and clinical data over the years has supported the approach of a lung protective strategy.21–25 Low tidal volume prevents the excessive global stress and strain of the baby lung, whereas higher PEEP prevents the regional excessive stress and strain by avoiding alveolar collapse and reopening during mechanical ventilation.26 These mechanical events are associated with an inflammatory reaction of the epithelial and endothelial lung cells (biotrauma/atelectrauma) as shown by the seminal study of Tremblay and colleagues27 (see also work of Dreyfuss and Saumon11 and Tremblay and Slutsky28). The literature to date supports the thought that the harm of mechanical ventilation is due to excessive global or regional stress and strain.29 This situation leads to two results—a physical rupture of the lung and a mechanically induced inflammation of the lung parenchyma, constituting VILI. The best “mechanical ventilation” would provide adequate gas exchange with the lowest amount of VILI.
Physiologic Basis of Mechanical Ventilation
Driving Force
To ventilate the respiratory system, force is required. The driving force for ventilation under normal circumstances is provided by the respiratory muscles. During spontaneous breathing, the thoracic cage expands. This causes a decrease in intrathoracic pressure (pleural pressure, Ppl) relative to the atmosphere (in normal conditions approximately 2 mm Hg of ΔPpl is sufficient to expand the thoracic cage by 0.5 L). Because the lungs are connected in series with the thoracic cage, their volume is expanded to a near equal extent (not considering the blood shift30). The force that distends the lung is the pressure difference between the alveoli and the pleural cavity (the transpulmonary pressure, PL). As the lung expands, the alveolar pressure becomes subatmospheric, and gas flow is generated (inspiration). When the respiratory muscles relax, the potential energy accumulated in the respiratory system (lung and chest wall) returns the chest wall and the lung parenchyma to the resting position (expiration). In spontaneously breathing subjects, the driving force (muscular pressure, ΔPmusc) is spent partly to expand the chest wall (ΔPpl), partly to expand the lung (ΔPL), and partly to overcome the resistances to the gas flow. In quasi-static conditions, in which the resistances to gas flow are negligible:
Transpulmonary Pressure
The transpulmonary pressure for a given driving pressure uniquely depends on the ratio of lung elastance to respiratory system elastance. In normal subjects EL/Etot is approximately 0.5, whereas in patients with ARDS it may range from 0.2 (e.g., in obese patients or in patients with high intra-abdominal pressure) to 0.8 (e.g., in patients with a very small baby lung and a normal chest wall elastance). This variability implies that for the same driving force applied and read on the ventilator display (e.g., 30 cm H2O), the resulting transpulmonary pressure may range from 6 to 24 cm H2O (Fig. 11.1).
Force-Bearing Structure of Lung Parenchyma
The transpulmonary pressure is applied to the force-bearing structure of lung parenchyma, the extracellular matrix, which constitutes the lung skeleton.31 The lung skeleton is a complex and metabolically active structure that includes a network of several components—elastin, collagen, and proteoglycans. All these molecules are involved in determining the mechanical characteristics of the respiratory system. The elastin may be considered as an elastic spring, whereas the unextensible collagen, which is folded at end expiration and completely unfolded at a lung volume equal to total lung capacity, acts as a stop-length fiber.32,33 The proteoglycans stabilize the collagen-elastin network, contributing to lung elasticity and alveolar stability at low and medium lung volumes.34
The matrix of elastin, collagen, and proteoglycans is arranged in two main fiber systems: (1) the axial system, which originates from the pulmonary hilum and runs deeply into the lung parenchyma down to the alveolar level, where it joins (2) the peripheral system, which originates from the visceral pleura and runs centripetally within the lung parenchyma.31 The lung skeleton may be considered as a continuous elastic structure that reaches its extension limits at total lung capacity, a lung volume equal to about threefold the lung resting volume. At this level of alveolar distention, the collagen is completely unfolded, and further expansion is prevented. The epithelial and endothelial cells do not directly bear the applied forces because they are anchored to the extracellular matrix by a series of structural proteins (integrins), which are connected to the cytoskeleton. During lung expansion, the epithelial and the endothelial cells modify their shape.
It is well documented that mechanically induced cellular deformation activates a series of mechanosensors with the production of several inflammatory mediators, such as cytokines (interleukin 6, tumor necrosis factor-α, and interferon-γ),27,35 metalloproteinases (enzymes involved in the remodeling of the matrix),36 leukotrienes,37 and interleukin 8,38–40 the most powerful attractor of neutrophils.41 Gross barotrauma (e.g., pneumothorax) is due to the stress at rupture of the lung skeleton, whereas intrapulmonary inflammation is primarily due to the excessive strain of the epithelial and endothelial cells.
Concept of Stress and Strain
where ΔV is the volume variation applied to the lung (i.e., the tidal volume), and V0 is the lung resting volume (i.e., the functional residual capacity at atmospheric pressure [without any application of PEEP]). The lung-specific elastance is the transpulmonary pressure required to double the lung resting volume (i.e., the ΔPL when ΔV/V0 is equal to 1). In ARDS, lung-specific elastance is similar to normal,16,42 reinforcing the concept of the baby lung (lung is small and not stiff), and questions the use of normalizing the tidal volume to the ideal body weight. The same tidal volume per kilogram may result in completely different strain according to the size of the baby lung (the V0 of the previous equation). For example, a 70-kg man with ARDS may have, according to the severity of the lung injury, a residual baby lung equal to 60%, 40%, or 20% of his normal lung size. If the ventilator is set to deliver 10 mL/kg, the actual delivered tidal volume would generate an alveolar strain, which would result from the application, in normal lung, of a tidal volume equal to 17 mL/kg, 25 mL/kg, and 50 mL/kg, values associated with a significant lung injury in laboratory studies.11,29
Recently we attempted to quantify the relationship between stress-strain and VILI in healthy animals. We found that edema formation was a threshold phenomenon, induced by mechanical ventilation when the global strain reaches a critical value of about 2.43 This threshold roughly corresponds to the point where the stress-strain curve loses its linearity and starts an exponential growth, indicating that some lung regions reach their own total capacity and cannot expand any further. At this level of strain, in a period of 24 to 48 hours the mechanical ventilation is lethal and the increased lung weight (two to three times the baseline) is associated with a striking impairment of respiratory mechanics, gas exchange, hemodynamics, inflammation, distal organs damage, and 100% mortality rate. This lethal strain, and associated stress, however, is rarely applied in clinical practice. To explain VILI in a diseased lung, therefore, alternative phenomena must be taken into account as the lung dishomogeneity and the presence of stress rise, which will be discussed later.
Patient Characterization
Gas Exchange
Oxygen
PaO2, PaO2-to-fraction of inspired oxygen ratio (PaO2/FIO2), and Riley’s shunt fraction44 are the most commonly used variables to assess the severity of lung injury. In particular, the PaO2/FIO2 thresholds of 300 and 200 were used to define ALI (300) and ARDS (200) according to the American-European Consensus Conference on ARDS.45 Consequently, the PaO2/FIO2 ratio is perceived as a key variable by most intensive care unit (ICU) physicians: The lower the PaO2/FIO2 ratio, the greater the lung injury. This equivalence is highly questionable. First, in most large studies on ARDS, no association was found between hypoxemia and outcome.46 In the large study showing a better outcome with low tidal volume compared with higher tidal volume, the PaO2/FIO2 ratio was significantly lower in the low tidal volume group despite ending with better outcome.25 Finally, the PaO2/FIO2 ratio was not different in patients with early, intermediate, and late ARDS, suggesting that oxygenation was not dependent on the structural changes of lung parenchyma occurring with time.47 In a study48 in which the lung severity was assessed by CT scan (and defined as a fraction of the gasless tissue), we did not find significant changes of PaO2/FIO2 ratio over a wide range of nonaerated tissue (Fig. 11.2), and PaO2/FIO2 ratio was not associated with outcome.
Most data suggest that PaO2/FIO2 ratio is a weak indicator of the overall lung severity, with compensatory rearrangement of perfusion during ARDS limiting the deterioration of oxygenation. The same limits apply when PaO2/FIO2 ratio changes are used to assess lung recruitment. Because this maneuver is unavoidably associated with changes of perfusion (global or regional), the increase of PaO2/FIO2 ratio may be partly due to decrease of perfusion, as shown in the 1980s.49–51 Most data suggest that the use of oxygenation variables alone to assess lung severity is misleading.
Carbon Dioxide
Although less considered, the variables derived from CO2, as the total or alveolar deadspace, seem to be of greater value in assessing lung severity. It has been shown in ALI/ARDS patients that deadspace at entry is a strong predictor of outcome,52 and that PaCO2 for the same total ventilation steadily increases from early to intermediate and to late ARDS, reflecting the lung structural changes.47 The PCO2 response to prone position (in contrast to PO2 response) is a strong prognostic index of mortality.53 Most data suggest that CO2-related variables (deadspace), more than PaO2/FIO2 ratio, reflect the severity of lung injury (and associated mortality rate) at the time of presentation and the structural changes of lung parenchyma occurring with time (fibrosis, Pneumocystis, and possibly perfusion defects).
Respiratory Mechanics, Chest Wall Elastance, and Lung Volume
In the original description of ARDS,1 the low compliance (i.e., high elastance) of the respiratory system was a landmark of the syndrome. The respiratory system compliance is not considered in the current definition of ARDS, however.45 For years, the low compliance was attributed uniquely to the lung component (lung stiffness and lack of surfactant). Quantitative CT shows, however, that the respiratory system compliance primarily reflects the size of lung open to gases (the baby lung14,16), suggesting that the intrinsic functioning lung elasticity in ARDS is close to normal (the lung is “small” rather than “stiff”). More than gas exchange, the respiratory system compliance indirectly assesses the lung injury severity (the smaller baby lung, the greater lung injury),54 as confirmed in animal experiments.55 Another variable also must be taken into account—the elastance of the chest wall. It has been shown in a significant portion of ALI/ARDS patients that the chest wall elastance is greater than normal because of increased intra-abdominal pressure, obesity, or severe edema.56 In patients with extrapulmonary ARDS57 and in obese patients,58 the high elastance of the respiratory system may be due to the chest wall and to the lung derangement. Measurement of intra-abdominal pressure should be considered when selecting mechanical ventilator settings.
Severity of Lung Injury and Lung Recruitability
CT can be used to assess the severity of lung injury by measuring the fraction of nonaerated lung tissue at end expiration (end expiration pause at 5 cm H2O). This fraction includes the lung tissue that is “consolidated” (i.e., not openable at 45 cm H2O airway pressure) and the tissue that is collapsed but openable at 45 cm H2O. These values were chosen to produce a minimal risk during the maneuver and because this is the most frequently used in the literature for recruitment maneuver.59 In patients with elevated chest wall elastance, the resulting transpulmonary pressure could be insufficient, however, to overcome the opening pressure of some pulmonary units.
The total fractions of nonaerated lung tissue (consolidated plus recruitable) and the recruitable tissue alone (tissue that regains aeration at 45 cm H2O airway pressure) are strongly associated with mortality rate. The data of Figure 11.2 show the inadequacy of the term ALI/ARDS, as currently defined, to describe the lung injury.48 The data refer to a population of ALI/ARDS patients, classified according to the American-European Consensus Conference on ARDS,45 in which a CT-based quantitative analysis of the whole lung parenchyma at 5 cm H2O PEEP was performed. As shown in Figure 11.3, the fraction of the nonaerated lung tissue (consolidated or collapsed or both) may range from 5% to 70% of the entire lung parenchyma. Patients meeting ALI/ARDS criteria may have a baby lung size very close to that of normal subjects, or a baby lung that is just a small fraction of the expected normal lung. When the distending force is applied to the lungs by the mechanical ventilator, previously collapsed lung regions may open. Lung recruitability may be expressed as the amount of lung tissue regaining aeration when increasing the applied driving force from 5 to 45 cm H2O. As shown in Figure 11.3, in some patients, lung recruitability was almost negligible, whereas in others it was equal to 25% to 35% of the entire lung parenchyma. Lung recruitability was strongly associated with the fraction of nonaerated tissue, suggesting that the greater the inflammatory edema, the greater the lung collapse. It seems that the best way to assess the overall lung severity and the related lung recruitability, both strongly associated with mortality rate, is the CT scan analysis. Physiologic variables are poor indicators of the severity of the lung injury. This relationship is shown in Figure 11.4—for a large variation of lung injury, ranging from 20% to 60% of the lung parenchyma, the values of PaO2/FIO2 ratio, lung compliance, and PaCO2 greatly overlap.
Figure 11.4 Mean ± standard deviation values of respiratory variables of a population of 68 patients with acute lung injury/acute respiratory distress syndrome. Blue columns represent PaO2/FIO2 ratio at positive end-expiratory pressure (PEEP) 5 cm H2O (mm Hg), red columns represent respiratory system compliance at PEEP 5 cm H2O (mL/cm H2O), green columns represent deadspace fraction (%), and gold columns represent alveolar deadspace fraction (%). *P < 0.05 versus patients with a fraction of nonaerated tissue recorded at 5 cm H2O PEEP less than 0.2. †P < 0.05 versus patients with a fraction of nonaerated tissue recorded at 5 cm H2O PEEP ranging from 0.2 to 0.4. The continuous purple line represents the likelihood of death predicted by the fraction of nonaerated lung tissue recorded at 5 cm H2O PEEP (P = 0.015). Weight is a poor surrogate of strain because ARDS patients with similar body weights may have completely different lung sizes and consequently different levels of strain at equal tidal volumes. This is confirmed by the plateau pressures measured in these different studies (see Fig. 11.5). As shown for the same tidal volume per ideal body weight, the plateau pressures are widely distributed with huge overlap between the studies. This reflects a wide distribution of the respiratory system compliance and consequently of the lung size.
Ards Classification
Although, in our opinion, the definition of ARDS should include a quantitative estimate of the lung edema, because this is not feasible in most of the ICU, alternative ways for defining ARDS have been sought. ARDS was first defined by Ashbaugh and coworkers in 1967.1
[/not-level-membership-for-critical-care-medicine-category]
