10 Management of Intramedullary Spinal Cord Tumors
Intramedullary spinal cord tumors (IMSCTs) are rare, a fact that is reflected by the paucity of large case series in the literature. Published accounts on the management of IMSCTs consist primarily of case reports and a handful of small case series. Current management strategies, therefore, are largely founded upon past experience, and expert opinion.1 The earliest expert opinion on the treatment of IMSCTs dates back to 1911 with a serendipitous observation in the operating room by Elsberg, who unintentionally made a myelotomy in the posterior spinal cord while opening the dura, resulting in the extrusion of tumor tissue. Realizing his error, he closed the wound without an attempt at tumor resection. One week later, the incision was reopened and a well-defined tissue plane was noted, which permitted total tumor resection. The patient, severely quadraparetic prior to surgery, was able to ambulate without assistance and use a typewriter eight months following the procedure.2 Based upon his experience, Elsberg advocated the following two stage method for resection of IMSCTs:
“After about a week the wound is reopened, and the tumor, which will in all probability be found outside the cord, can be removed by dividing the few adhesions which remain. When the tumor has been removed and all bleeding controlled, the dura, muscles, fascia, and skin are closed in the usual manner.”3
During the first half of the twentieth century, other surgeons did not share Elsberg’s early success. In 1969 Schneider asserted that when an intramedullary tumor is encountered that is not obviously cystic in a patient with little or no neurological deficit, “the dura is left open with no attempt made to perform a myelotomy or procure a biopsy.”4 Until recently IMSCTs were treated with biopsy or subtotal removal followed by irradiation—a therapy that is usually associated with early tumor recurrence and progressive neurological impairment.5 The evolution of diagnostic and surgical technologies now permits a more aggressive surgical role in the management of IMSCTs.
With MRI, IMSCTs are diagnosed more frequently, and in earlier stages of their disease progression.1 It has been shown that preoperative neurological function is the most important predictor of patient outcome following surgery for an IMSCT,6,7 and in this respect early detection with MRI is extremely helpful. Improved operative technologies such as neurophysiologic monitoring, the ultrasonic aspirator, and carbon dioxide laser have also facilitated the resection of IMSCTs.8 These recent surgical advances, in light of poor results in tumors treated solely with radiation and chemotherapy, have led many to advocate complete surgical resection, whenever possible, as the standard of care.1,5,8–10
Epidemiology and Presentation of Specific Intramedullary Spinal Cord Tumors
EPENDYMOMAS
Spinal ependymomas arise from the ependymal rests in the vestigial central canal, and, as a result, are centrally located within the spinal cord.11 Ependymomas are the most common IMSCT in adults, comprising 40% of a large series compiled by Fischer and Brotchi.12 In children, they are the second most common primary IMSCT (28%), second only to astrocytomas.13 However, there were no ependymomas in a series of IMSCTs in children under 3 years of age.14 There is an equal distribution among males and females. They occur throughout the spinal cord, but are most common in the cervical region.12,15 Myxopapillary ependymomas are a distinct subtype occurring in the conus medullaris and cauda equina, and have a slight male predominance.16 Genetic studies have suggested a possible link between neurofibromatosis type 2 and the development of spinal ependymomas.17,18 Families predisposed to the development of ependymal tumors have been shown to have a loss of heterozygosity on chromosome 22.19
These tumors are slow growing, with an average interval of 16 months between the onset of symptoms and diagnosis.15 Sixty-five percent of patients present with complaints of radiculopathy or regional neck pain accompanied by minimal motor or sensory deficit. Because these slowly growing tumors compress rather than invade adjacent neural tissue, they can take up a considerable volume within the spinal cord without causing significant motor deficit. Parasthesias and other sensory phenomena result from compression of the crossing spinothalamic fibers. Within the corticospinal tract, hand fibers are located medially and leg fibers are located laterally. A centrally located cervical IMSCT or associated cyst, therefore, may produce weakness and atrophy of the small hand muscles from anterior horn cell compression before lower extremity dysfunction becomes apparent. Cervical lesions rarely present with bowel or bladder dysfunction.15 Myxopapillary tumors arising from the conus, however, can compress sacral anterior horn cells and adjacent nerve roots in the cauda equina, resulting in bowel or bladder dysfunction in 20 to 25% of cases.16
ASTROCYTOMAS
Juvenile pilocytic astrocytomas (JPA) are a unique subclass of astrocytomas. Generally speaking, low-grade astrocytomas fall into two categories: World Health Organization (WHO) grades I and II. Pilocytic astrocytomas are WHO grade I tumors, while protoplasmic, gemistiocytic, fibrillary, and mixed astrocytomas are classified as WHO grade II. Separation of pilocytic astrocytomas into their own grade reflects the fact that they have a different prognosis and clinical course. The 10-year survival rate in patients with a pilocytic spinal cord astrocytoma is 81%, while the 10-year survival rate drops to 15% in patients with diffuse fibrillary astrocytomas.20
Astrocytomas are the most common pediatric IMSCT, representing 59% of the tumors in a compilation of 13 pediatric series.13 In adults, they are second to ependymomas in frequency, accounting for about 20% of tumors.12,21 Unlike intracranial astrocytomas, spinal cord astrocytomas are usually low-grade lesions in both children and adults. High-grade lesions (WHO grades III and IV) comprise only 10% to 15% of pediatric tumors and a modestly higher proportion in adults.22,23 There is a slight male predominance,12,20 and the cervical area is most frequently affected, followed closely by the thoracic region. These lesions span an average of six spinal levels, but total spinal cord involvement has been described.24 Genetic studies have shown a potential association between neurofibromatosis type I and the development of spinal astrocytomas.17,18,25
In contrast to ependymomas, astrocytomas are often infiltrative lesions that occupy an eccentric location within the spinal cord. Presenting symptoms typically consist of regional back or neck pain and sensory disturbances including dysesthesias and loss of sensation, unilateral or bilateral in nature, as well as motor deficit. In the pediatric population, pain remains the most common symptom, but gait deterioration, motor regression, torticollis, and kyphoscoliosis are common presenting findings.26 Symptoms resulting from low-grade lesions usually evolve over months to years.27,28 High-grade astrocytomas, however, present with a more rapid decline in motor function with progression to significant disability in only 3 to 5 months.22,28
GANGLIOGLIOMAS
Gangliogliomas are neoplasms containing both neoplastic neuronal and glial cells. They account for approximately 1.1% of all spinal neoplasms. Ten percent of intracranial gangliogliomas undergo malignant degeneration. Such malignant change is believed to be due to the glial component of the tumor. How this data regarding intracranial gangliogliomas translates to spinal cord gangliogliomas is unclear. They mainly occur in children and young adults, with both sexes affected in equal proportion.29–31 Symptoms include pain and weakness of the extremities, while examination findings may include myelopathy and kyphoscoliosis. Symptoms and imaging characteristics fail to distinguish these from other glial tumors.29–31
HEMANGIOBLASTOMAS
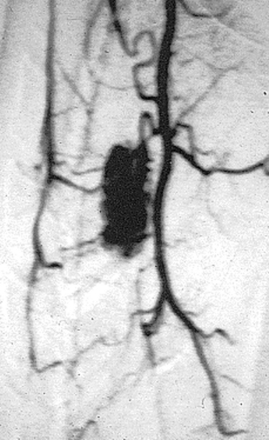
Hemangioblastomas consist of thin-walled blood vessels interspersed with large, pale stromal cells. They represent 3% to 11% of IMSCTs, with a slight male predominance.32 Up to one third of cases occur in association with von Hippel-Lindau (VHL) disease. VHL disease occurs in both an autosomal dominant and a sporadically inherited fashion. The autosomal dominant form results from a mutation of a tumor suppressor gene on chromosome 3p.33
Hemangioblastomas involving the spinal cord are occasional manifestations of VHL disease,34 and multiple lesions may be present, particularly in the posterior fossa. Symptom onset is typically in the fourth decade of life and the mean age at surgery is 40 years; childhood presentation is rare.35 The most frequent locations are thoracic (55%) and cervical (40%). Cyst formation occurs in 87% of cases.35
Hemangioblastomas differ from ependymomas and astrocytomas in that they generally are found on the dorsal or dorsolateral surface of the spinal cord. As a result, they often present with complaints of proprioceptive loss in addition to pain and sensory deficits.35
LYMPHOMAS
Intramedullary spinal cord lymphoma is an unusual entity. It is most commonly seen as part of a multifocal central nervous system lymphoma, or in patients immunosuppressed from AIDS or other causes.36 Pathologic studies have demonstrated that the vast majority of primary spinal cord lymphomas are of the non-Hodgkin B-cell variety.37,38 Reports of T-cell lymphomas involving the spinal cord are rare.39 Presentation can range from myelopathy to paresis,40,41 and can progress rapidly over a period of days to weeks.
LIPOMAS
Intramedullary spinal lipomas, excluding those associated with dysraphism, comprise just 1% of all IMSCT. These tumors consist of ordinary adipose tissue, and are believed to arise from rests of ectopic tissue.42 Lipomas are often densely adherent to surrounding neural tissue, precluding complete resection.43
Most patients present in the second to fourth decade in life and there is no gender predilection.44 Clinical presentation is that of a slowly progressive myelopathy (58%), a syringomyelic syndrome (9.5%), or a Brown-Séquard syndrome (6.5%), with the remaining 26% having atypical features.42 Lipomas tend to have long indolent courses, followed by a rapid decline in neurological function.43,44 In females, neurological deterioration may follow pregnancy and delivery.45
CAVERNOUS ANGIOMAS
Cavernous angiomas, while not true neoplasms, can form mass lesions in the spinal cord parenchyma, and should be considered in the differential diagnosis of intramedullary spinal mass lesions. Commonly known as cavernomas, they represent 1% to 3% of IMSCTs. Cavernomas are angiographically occult vascular malformations consisting of a collection of enlarged vascular spaces surrounded by a rim of gliosis, without intervening neural tissue.46 Both sporadic and familial forms are recognized. The familial form is inherited in an autosomal dominant fashion and is associated with multiple angiomas.47,48 Molecular analysis has shown that a gene mutation in CCM1, encoding the KRIT1 protein, is largely responsible for the hereditary form of cavernous angiomas.49,50
Cavernomas can cause progressive myelopathy due to repeated hemorrhage, resulting in reactive gliosis.51,52 A large sudden hemorrhage, albeit uncommon, can lead to catastrophic neurological deterioration, and surgery is the only effective treatment.46 Asymptomatic patients do not benefit from surgical intervention. Once a patient becomes symptomatic, however, progressive neurological deterioration from repetitive hemorrhage is the rule and surgical intervention is advisable in most cases.52
METASTASES
In a large postmortem study, intramedullary spinal cord metastases were found in only 2% of 627 patients with systemic cancer.53 Other accounts estimate that metastases comprise 2% to 8% of all IMSCTs.54,55 The incidence of intra-cerebral metastases in cancer patients, in contrast, has been estimated at 25% to 35%.56 Because of the comparatively small volume of the spinal cord relative to the brain, metastases to the spinal cord are much less common.57 The most common sources of intramedullary spinal cord metastases are the lung and breast. The mechanism of metastatic spread to the spinal cord is thought to be hematogenous rather than direct invasion, since metastases to the spinal cord are not always associated with disease in the adjacent tissues.53,58,59 The diagnosis of spinal cord metastasis carries a grave prognosis, and 80% of patients die within three months. The presenting symptoms consist of pain and weakness. Rapid neurological deterioration is observed in almost half of all patients, progressing to cord hemisection or transection syndromes over days to weeks.59
Diagnostic Imaging
PLAIN X-RAYS
Plain x-rays have little place in the modern diagnosis of IMSCTs and are unremarkable in the majority of cases. However, an enlarged spinal canal with scalloping of the vertebral bodies, medial pedicle erosion, and thinning of the laminae may be seen.60 These findings are consistent with any long-standing intradural tumor that thins and remodels the surrounding bone and are not specific for IMSCTs.
Scoliosis is frequently seen in children with IMSCTs, often with the apex of the curvature to the left rather than the right. Dextroscoliosis, where the apex of the curvature is to the right, is more common in patients with idiopathic scoliosis. Although scoliosis is unusual in older adults, it may be the presenting symptom in young adults with asymptomatic onset of tumor growth during childhood. Plain x-rays are also valuable in assessing alignment as the presence of preoperative kyphosis or scoliosis may necessitate early fusion to prevent progressive deformity.61
SPINAL ANGIOGRAPHY
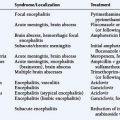
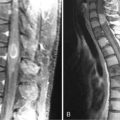
Spinal angiography may be considered when MRI suggests a hemangioblastoma (Figure 10-1). Although angiography will delineate the location of the vessels that supply and drain the hemangioblastoma, the vascular supply is generally evident at surgery and we have not found angiography to be important in the planning or execution of surgery. Cavernous angiomas are angiographically occult vascular lesions and, when suspected, angiography is not indicated.
MAGNETIC RESONANCE IMAGING (MRI)
MR spectroscopy may allow for more definitive diagnosis in the future, although there are significant susceptibility artifacts because of the close proximity of tissues with different magnetic susceptibilities, such as spinal cord, CSF, bone, and muscle. Currently, this precludes evaluation by MR spectroscopy because magnetic field homogeneity is necessary for this technique.55
Ependymomas
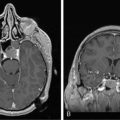
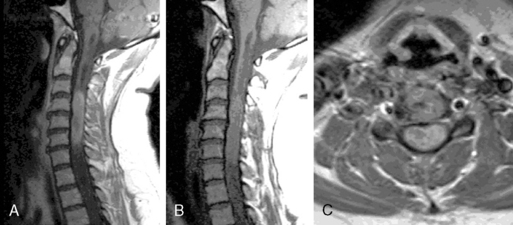
Ependymomas typically occupy the central regions of the spinal cord. They are characteristically isointense on T1-weighted images, hyperintense on T2-weighted images, and administration of gadolinium yields a strongly enhancing mass that is well defined from adjacent spinal cord. Cystic areas and regions of prior hemorrhage produce mixed signal intensity. Tumor-associated cysts are commonly seen at the rostral and caudal extremes of the tumor (Figure 10-2). Prior hemorrhage can produce a hypointense cap of hemosiderin on T2-weighted images, which is pathognomonic of ependymoma.62,63 The propensity of ependymomas to hemorrhage is attributed to their vascular connective tissue stroma.63
Astrocytomas
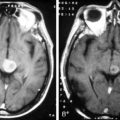
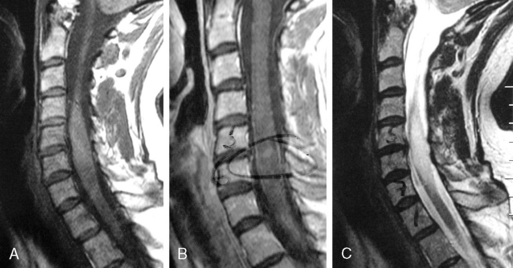
Astrocytomas may occupy the central regions of the spinal cord, or they may have an eccentric location. They are iso- to slightly hypointense on T1-weighted images, and hyperintense on T2-weighted images. Astrocytomas enhance to variable degrees after administration of gadolinium, typically to a lesser degree than ependymomas, and they are not as well defined from surrounding normal cord (Figure 10-3). Tumor-associated cysts are common, as is the case with ependymomas.
Hemangioblastoma
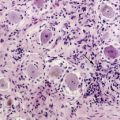
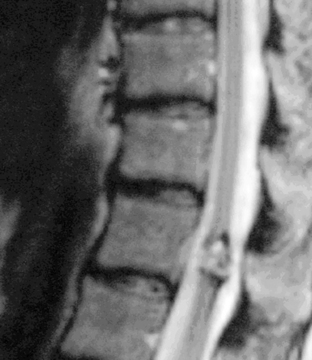
This appears as an intensely enhancing tumor nodule following the administration of gadolinium. Tumor-associated cysts are frequently larger than the tumor and do not enhance. These cysts may extend for multiple spinal levels, and contain protein-rich fluid, which is hyperintense on T2-weighted images. The lesions are usually located on the posterior or posterolateral surface of the spinal cord (Figure 10-4). Because patients with von Hippel-Lindau syndrome commonly have multiple lesions, the entire neuraxis should be imaged in the search for additional tumors.
Cavernous Angiomas
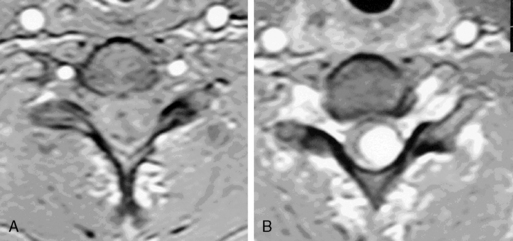
Cavernous angiomas contain hemorrhagic regions of differing ages. CT scan can also demonstrate calcific areas in the lesion. MRI reveals a T2 rim of low intensity surrounding a region of variegated T2 signal intensity (Figure 10-5). Multiple lesions may be present, particularly in familial cases.
Multiple Sclerosis
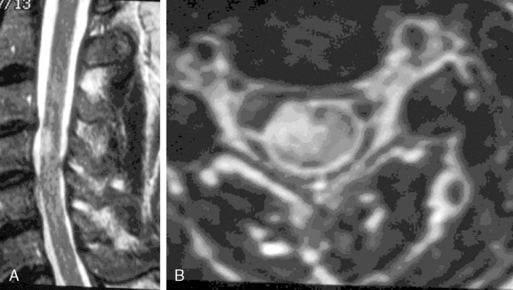
MS plaques are found in the white matter, are iso- to hypointense on T1-weighted images, and are hyperintense on T2-weighted images (Figure 10-6). During active demyelination, MS plaques may enhance upon administration of gadolinium. In cases of acute MS, a follow-up MRI in 4 to 6 weeks will show lessening mass effect, diminution of enhancement, and a decreased hyperintensity on T2-weighted images.
Differential Diagnosis
MULTIPLE SCLEROSIS
Multiple sclerosis (MS) affecting the spinal cord results in the following clinical findings: Lhermitte sign, limb weakness (usually asymmetric spastic paraparasis), and sensory dysfunction.64 The presence of oligoclonal bands in the CSF will help confirm the diagnosis of MS.65 Important clues differentiating MS from IMSCT can be obtained in a detailed history. IMSCTs often result in a gradual, steady deterioration in neurological function. MS, with the exception of the primary progressive variety (PPMS), typically follows a relapsing and remitting course.64 IMSCTs are usually accompanied by pain, whereas MS myelitis is usually painless.
SARCOIDOSIS
Sarcoidosis is a multisystemic granulomatous disease that affects the spinal cord in only 0.43% of patients.66 Spinal sarcoidosis can present with progressive mye-lopathy and sphincter dysfunction if the conus is involved.67–69 On MRI, spinal sarcoid often demonstrates regional enlargement of the spinal cord along with patchy, multifocal enhancing nodules in the spinal parenchyma.67,70 Most patients with sarcoid affecting the central nervous system have systemic disease, with the lungs and thoracic lymph nodes almost always affected. Bronchial biopsy or bronchialveolar lavage is useful to establish the diagnosis.70–72 Elevated angiotensin-converting enzyme levels in both the serum and the CSF can be helpful in confirming the diagnosis.70,73 Definitive diagnosis, however, is made only with a spinal cord biopsy. The mainstay of treatment is a prolonged course of corticosteroids, but other immunosuppressants such as cyclosporine and methotrexate are useful adjuncts in refractory cases.72,74,75
SYRINGOMYELIA
Syringomyelia, a cystic cavitation within the spinal cord that occurs following trauma or in association with Chiari malformations, may be confused with the cystic component of some intramedullary spinal cord tumors. A syrinx widens the spinal cord and clinically presents with slowly progressive deficits, classically affecting spinothalamic sensory modalities before dorsal columns. It can be difficult to distinguish from a slow growing IMSCT by history and neurological examination. After the administration of gadolinium, tumor tissue adjacent to the cyst will enhance whereas there will be no enhancement in syringomyelia. Tonsillar or brainstem herniation is seen with a cervical syrinx associated with a Chiari malformation but does not occur with IMSCTs.76
INFECTION
A variety of infectious agents can mimic intramedullary spinal cord tumors. Parasitic infections such as Angiostrongylus catonensis and Schistosoma haematobium can mimic spinal cord tumors both in clinical presentation and MRI imaging.77–79 Toxoplasmosis and tuberculosis of the spinal cord resembling an IMSCT, likewise, have been described.77,80
Surgical Management
SELECTION OF OPERATIVE CANDIDATES
The natural history of IMSCTs is that of progressive neurological deficit and early operative intervention is desirable, as postoperative functional outcome is closely correlated with the severity of the patient’s preoperative deficit.14,81 Ideal surgical candidates are ambulatory patients with minimal neurological deficit. Even patients with significant deficit may still derive benefits from surgery with preservation of sphincter function or retention of the ability to position in bed. Patients with complete loss of neurological function are not appropriate surgical candidates.
Although early surgical intervention is recommended, surgical candidates must maintain realistic expectations of surgical outcomes. Published postoperative results suggest that 10% to 40% of patients remain stable, 40% to 80% improve, and 10% to 20% worsen neurologically.82,83 It is reasonable, therefore, to defer surgery in a patient with very mild symptoms. Should serial examinations (approximately every 3 to 6 months) demonstrate decline in neurological function, both patient and physician might become more accepting of the surgical risks.
EVOKED POTENTIAL MONITORING
Most surgeons employ evoked potential monitoring in the hope that intraoperative data will guide the extent of the surgical resection and predict postoperative deficits. Somatosensory evoked potentials (SSEPs) are used to monitor the integrity of the dorsal columns and spinothalamic tracts. Significant intraoperative changes in SSEPs are demonstrated to be predictive of postoperative neurological deficits.84 Their usefulness in improving outcome, however, may be limited.85 Furthermore, preoperative neurological deficit may result in failure to obtain baseline readings. Variability in user skill and equipment error can be other causes for unsatisfactory physiologic data. Most important, however, neurological damage can result in the brief, 10 to 60 second delay between the time of spinal cord injury and evoked potential changes generated from computer averaging techniques. Such neurological injury may be irreversible, in contrast to evoked potential changes seen in the course of scoliosis surgery, which are usually reversible by repositioning of the instrumentation.
Motor evoked potentials (MEPs) are a newer technique used to assess the integrity of the corticospinal tracts during IMSCT surgery and provide “real time” intraoperative data. Utilizing scalp electrodes in combination with epidural electrodes, the presence of MEPs is believed to correlate better with surgical outcome than the preoperative neurological exam.86 One surgical group reports the use of a 50% decline in the amplitude of MEPs as a mark at which to interrupt dissection.87 The benefit of this technique was limited, however, by the fact that MEPs could not be measured in a large proportion of patients, many of whom had baseline neurological compromise and stood to benefit most from such monitoring.88 Kothbauer et al. believe that evoked potential monitoring is an essential adjunct to surgery.87 The use of evoked potential monitoring allows prediction of outcome after surgery.86 However, controlled case studies supporting the efficacy of evoked potential monitoring in preventing neurological deterioration and improving the outcome from surgery are lacking. In short, there is little downside to the use of evoked potentials and we routinely employ both MEP and SSEP monitoring, but we are not convinced by our own experience or data from the literature that monitoring results in improved outcome.
Postoperative Complications
INCREASED NEUROLOGICAL DEFICIT
Deterioration of motor function in the immediate postoperative period is reported in most series. These deficits generally are followed by recovery over a period of days to months. However, approximately 20% of patients experience a permanent increase in their deficits.6,15,28,82,87,89 Progressive deterioration in the postoperative period suggests spinal cord compression by hematoma or spinal cord swelling and compression by the dura in a patient with significant amounts of residual tumor who did not undergo duraplasty.
Patients with more severe motor deficits preoperatively are less likely to sustain recovery and are more likely to experience further deterioration than those with lesser degrees of impairment. Because most astrocytomas infiltrate neural tissue, resection of astrocytomas (with the exception of juvenile pilocytic astrocytomas) inevitably results in injury to functional neural tissue. For this reason, increased permanent postoperative deficit is more common with astrocytomas than with ependymomas. Innocenzi reports that at discharge from the hospital, the proportion of children with neurological deterioration from their preoperative status was greater in those with astrocytomas than those with ependymomas.90
Loss of proprioception can occur as a result of injury to the dorsal columns from the myelotomy, and is more likely with larger tumors and longer myelotomies.21 The spinothalamic tracts also may be injured during dissection at the lateral margins of a centrally placed tumor. Intraoperative somatosensory evoked potentials can provide an early warning of disturbance to either the dorsal columns or the spinothalamic tracts. Dysesthesia, hyperesthesias, and anesthesia are feared complications of surgery. Their presence may render a functional extremity useless and prevent a patient with minimal or no motor deficit from returning to a former occupation or resuming a normal social life. In general, sensory deficits resolve within 3 months after surgery, after which point any residual deficit is usually fixed. Motor deficits, however, are not fixed and can continue to gradually improve beyond the 3-month postoperative window.91
SPINAL DEFORMITY
In children, deformities of the thoracic and lumbar spine may represent the initial manifestation of an IMSCT months or years before the appearance of neurological signs and symptoms.92 It is unclear whether the exacerbation of these deformities following operation results from the effects of the tumor or of laminectomy. Whether present preoperatively or not, progressive postoperative kyphoscoliosis of the thoracic spine and swan-neck deformity of the cervical spine are seen with great frequency in children, but are rare in adults if not present preoperatively.13,93 The incidence varies with the spinal level involved; the cervical area is affected more frequently than the thoracic area and lumbar deformity is exceptional.94
Osteoplastic laminotomy is favored in children to retard or prevent the development of kyphoscoliosis.95 In the cervical spine, early fusion at the first sign of flexion deformity is indicated. Progression of kyphoscoliosis may be a sign of tumor recurrence, and this is not prevented by laminoplasty.26 Thus, appropriate follow-up to identify early development of spinal deformities is an essential part of the postoperative management. In the thoracic and lumbar spine, instrumentation and fusion are indicated when progressive deformity is recognized and the presence of recurrent tumor is ruled out.
Radiation Therapy
Radiation is reserved for cases of subtotal removal, recurrence, and otherwise inoperable infiltrating tumors. It is inappropriate for use in ependymomas which are almost always totally resectable. Radiation results in arachnoiditis as well as gliosis and fibrosis within the neoplasm, obscuring the cleavage plane between tumor and cord. Furthermore, the microvasculature of the spinal cord is obliterated by radiation, further increasing its sensitivity to surgical manipulation.83 Numerous surgeons have noted an association between poor postoperative outcome and preoperative radiation therapy.82,83 Radiation therefore increases the difficulty and risk of surgery substantially, and is reserved for use when surgery is not felt to be beneficial. The use of postoperative radiation therapy has not been validated in a prospective, controlled study, but many reports describe its beneficial effects upon recurrence and survival.20,97 There is disagreement regarding its utility in instances where gross total resection is accomplished.
The use of the Cyberknife to treat IMSCTs is experimental, and only two cases are reported in the literature. Ryu et al. describe the treatment of a hemangioblastoma and a cavernous angioma with total radiation doses approaching 25 Gy.98 Both of these intramedullary lesions, however, are extremely amenable to surgical resection, and we question the use of radiation in their treatment. Recent reports have only demonstrated that the Cyberknife is a feasible and safe treatment modality.98,99 Further study is needed to compare the efficacy of Cyberknife to other existing treatments. The Cyberknife will still result in radiation changes to the spinal cord, making surgery difficult.
LOW-GRADE ASTROCYTOMA
There is considerable controversy regarding the use of radiation therapy in cases of low-grade astrocytomas. Kopelson recommended radiation for all low-grade astrocytomas without regard to the extent of resection.100 Epstein et al., however, concluded that radiation should be reserved for cases where subtotal resection is performed,28 and in a subsequent study by this group, they concluded that less than 80% resection was associated with a significantly worse prognosis.28,101 While Guidetti et al. did not find any consistent benefit from radiation therapy, others contend that postoperative radiation therapy will reduce the relapse rate after partial resection of low-grade gliomas.102–105 Given the changes in the architecture of the spinal cord following radiation therapy, however, it should be reserved for instances when surgery is no longer a treatment option.
For astrocytomas in adults, total resection is usually difficult, because astrocytomas are infiltrative and poorly defined from the normal spinal cord. For low-grade astrocytomas we prefer to follow patients with MRI and consider radiation when imaging shows tumor growth. However, others have reported that subtotal removal followed by 45 Gy given in a local field will result in satisfactory motor function and survival in low-grade astrocytomas in adults.103
MALIGNANT ASTROCYTOMAS
The outcome of patients with high-grade intramedullary gliomas remains dismal in spite of the progress in neurosurgical and radiotherapy techniques, and it is not clear that radiation therapy beneficially affects survival or retards the onset of neurological impairment. Aggressive radiotherapy in doses that cause “radiation cordotomy” has been reported by Cohen et al. to result in an occasional survival at 4 years after the initial surgery.27,103,106 Radiation cordotomy may be one option for patients with high-grade astrocytomas who have already poor motor function at the level of the thoracic or conus medullaris, providing that the malignant nature of the tumor is histologically confirmed.
EPENDYMOMAS
Gross total resection is the most efficacious treatment in the management of ependymomas, and radiation therapy is unnecessary if complete removal has been accomplished. Patients who have had incomplete removal should be followed closely with frequent MRI and treated with reoperation rather than radiation for recurrence. In one large series, radiotherapy had no effect on disease progression or recurrence when patients with and without radiotherapy were compared.107 Although Isaacson et al. recommended up to a total dose of 50.40 Gy in 1.80 Gy fractions for residual benign ependymomas using local fields,108 we disagree with the use of radiation therapy for this benign, potentially curable tumor.
SIDE EFFECTS OF RADIATION
Radiation to IMSCTs is a potential hazard to the spinal cord, bone growth, fertility, and the gastrointestinal tract. The spinal cord has a tolerance dose reported as 45 to 50 Gy in conventional fractionation, considerably lower than the brain. Doses of more than 50 Gy have been proposed,106 but such treatment is not recommended because of the risk of radiation myelopathy. The tolerance of the spinal cord in children may be lower than in adults. Isaacson et al. recommended reducing radiation dose in children by 10%.22 Because of the harmful effect of radiation therapy on development in children, most authorities do not irradiate pediatric patients who are believed to have had a gross total resection of their tumors.
Chemotherapy
Chemotherapy has become a subject of interest in the pediatric population, since children are more sensitive than adults to the deleterious effects of radiation. Treatment protocols for intramedullary gliomas are based upon regimens currently used for intracranial neoplasms.109–111 Two small case series have demonstrated some promise. Lowis et al. report their experience in two pediatric patients with WHO grade II and III astrocytomas using carboplatin and vincristine. Both patients improved neurologically, and the disappearance of contrast-enhancing tumor was noted on follow-up MRI.112 Doireau et al. reported progression-free intervals ranging from 16 to 59 months in five of eight children with predominantly low-grade glial tumors treated using a six drug chemotherapy regimen including carboplatin, procarbazine, vincristine, cyclophosphamide, etoposide, and cisplatin.113 While chemotherapy has shown some promise, the reported numbers of patients treated are small, and there is lack of comparison with other treatment groups. A multicenter study will be necessary to define efficacy and the ideal chemotherapy drug regimen.
Outcome
Long-term postoperative neurological outcome correlates most closely with the preoperative functional status. Significant neurological improvement rarely occurs in the face of long-standing deficit, and even if there is some improvement in patients who are severely impaired, change in the clinical functional grade is exceptional.21
EPENDYMOMAS
The outcome for ependymomas is generally good. There is a clear relationship between the extent of resection and the rate of recurrence and survival.15,96 Gross total resection is attainable, and, when achieved, recurrence is rare. Hoshimaru reports that in 36 patients with spinal cord ependymomas at a mean postoperative follow-up of 56 months, 39% were neurologically improved, 47% were stable, and only 14% worse, using McCormick’s functional status scale.15,114
The histological grade of ependymomas does not appear to affect outcome.115 In cases of gross total removal, radiation therapy is withheld, as surgery for recurrence is feasible and less difficult to accomplish in a nonradiated field. If postoperative imaging reveals significant residual tumor that is resectable, reoperation should be undertaken. Tumors that have been subtotally resected are followed with serial MRI scans and treated with reoperation when there is evidence of tumor growth.
ASTROCYTOMAS, GRADE I AND II
Although low-grade astrocytomas are categorized as “benign,” recurrences occur and neurological outcomes are generally far less satisfactory than is the case with ependymomas. Tumor recurrence is associated with progressive neurological deficit, and eventual paraplegia or quadriplegia. Patients with tumor recurrence can encounter numerous health problems that affect the life expectancy of debilitated and immobile individuals such as septicemia, pulmonary emboli, and pneumonia.116 Five-year survival was 57% in a series of 21 patients, 18 of whom had a pathological grade of I or II.117 In another series, 4 of 11 patients with grade I or II astrocytomas died within the follow-up period and only four of these were not worse in functional grade as compared with their preoperative status.96
A correlation between extent of resection and tumor recurrence is controversial and poorly demonstrated in the literature.6,20,117,118 The infiltrating nature of these tumors complicates the assessment of extent of resection, subjecting the surgeon’s estimates to inaccuracy. Even when a surgeon believes that a gross total resection was accomplished and MRI fails to demonstrate residual tumor, tumor fragments most likely remain.119 Cristante notes that 16 of 22 low-grade astrocytomas radically or “quasiradically” resected were tumors that had a discrete tumoral plane, and most had a fibrillary histology with adjacent cystic areas.21
Histological subtype is also reported to have prognostic value, as those with pilocytic features enjoy better outcomes than those with the diffuse fibrillary subtype.20 This probably accounts, at least in part, for the more favorable prognosis for the pediatric age group seen by Sandler, as the pilocytic tumors represent a larger proportion of their tumors.117
ASTROCYTOMAS, GRADE III AND IV
The prognosis for high-grade astrocytomas (WHO grades III and IV), like their intracranial counterparts, is extremely poor, and all patients will eventually die as a consequence of progressive disease. Like patients with malignant intracranial astrocytomas, widespread leptomeningeal metastases and hydrocephalus are common, occurring in 58% of patients.27 The terminal event in many malignant astrocytomas is tumor growth to the cervicomedullary area with respiratory paralysis. Pulmonary embolus and pneumonia may prove fatal to patients bedridden by progressive disease.
Surgical intervention usually results in worsened or, at best, the same level of neurological function. Median survival after surgery for a grade IV tumor is 6 months, and length of survival does not correlate with the extent of resection.27,28 A series of pediatric patients (median age of 11) fared slightly better, with a median survival of 13 months.120
GANGLIOGLIOMAS
Gangliogliomas, like ependymomas, are amenable to surgical cure. Recurrences, however, occur in 25% to 33% of cases, at which point a second resection is pursued. Histological grading has been undertaken but does not significantly correlate with outcome. Adjuvant chemotherapy or radiation is not recommended.14,31
1. J. Brotchi. Intrinsic spinal cord tumor resection. Neurosurgery. 2002;50(5):1059-1063.
2. C.A. Elsberg, BE. The operability of intramedullary tumors of the spinal cord. A report of two operations with remarks upon the extrusion of the spinal cord. Am J Med Sci. 1911;142:636-647.
3. CA, E. Diagnosis and Treatment of Surgical Diseases of the Spinal Cord and Its Membranes. 1916.
4. R. Schneider. Intraspinal Tumors. In: R. Schneider, editor. Correlative Neurosurgery. Springfield, Ill: Charles C. Thomas; 1969:444-463.
5. P.R. Cooper, F. Epstein. Radical resection of intramedullary spinal cord tumors in adults. Recent experience in 29 patients. J Neurosurg. 1985;63(4):492-499.
6. M. Samii, J. Klekamp. Surgical results of 100 intramedullary tumors in relation to accompanying syringomyelia. Neurosurgery. 1994;35(5):865-873. discussion 873
7. U.K. Chang, et al. Surgical outcome and prognostic factors of spinal intramedullary ependymomas in adults. J Neurooncol. 2002;57(2):133-139.
8. G.I. Jallo, D. Freed, F. Epstein. Intramedullary spinal cord tumors in children. Childs Nerv Syst. 2003;19(9):641-649.
9. D.C. Bowers, B.E. Weprin. Intramedullary Spinal Cord Tumors. Curr Treat Options Neurol. 2003;5(3):207-212.
10. M.C. Chamberlain. Ependymomas. Curr Neurol Neurosci Rep. 2003;3(3):193-199.
11. F.G. Moser, et al. Ependymoma of the spinal nerve root: case report. Neurosurgery. 1992;31(5):962-964. discussion 964
12. G. Fischer, BJ, A. Chignier, et al. Clinical Material. In: BJ, G. Fischer. Intramedullary Spinal Cord Tumors. Stuttgart: Thieme; 1996:10-20.
13. R. Reimer, B.M. Onofrio. Astrocytomas of the spinal cord in children and adolescents. J Neurosurg. 1985;63(5):669-675.
14. S. Constantini, et al. Intramedullary spinal cord tumors in children under the age of 3 years [see comments. J Neurosurg. 1996;85(6):1036-1043.
15. P.C. McCormick, et al. Intramedullary ependymoma of the spinal cord. J Neurosurg. 1990;72(4):523-532.
16. J.S. Schweitzer, U. Batzdorf. Ependymoma of the cauda equina region: diagnosis, treatment, and outcome in 15 patients. Neurosurgery. 1992;30(2):202-207.
17. K.L. Roos, M. Muckway. Neurofibromatosis. Dermatol Clin. 1995;13(1):105-111.
18. M. Lee, et al. Intramedullary spinal cord tumors in neurofibromatosis. Neurosurgery. 1996;38(1):32-37.
19. T. Yokota, et al. A family with spinal anaplastic ependymoma: evidence of loss of chromosome 22q in tumor. J Hum Genet. 2003;48(11):598-602.
20. K.J. Minehan, et al. Spinal cord astrocytoma: pathological and treatment considerations. J Neurosurg. 1995;83(4):590-595.
21. L. Cristante, HH. Surgical management of intramedullary spinal cord tumors: functionaloutcome and sources of morbidity. Neurosurgery. 1994;35:69-76.
22. J.C. Allen, et al. Treatment of high-grade spinal cord astrocytoma of childhood with “8-in- 1” chemotherapy and radiotherapy: a pilot study of CCG-945. Children’s Cancer Group. J Neurosurg. 1998;88(2):215-220.
23. P.C. McCormick, SB. Intramedullary tumors in adults. Neurosurg Clin North Am. 1998;1:687-700.
24. F. Epstein, N. Epstein. Surgical management of holocord intramedullary spinal cord astrocytomas in children. J Neurosurg. 1981;54(6):829-832.
25. T. Yagi, et al. Intramedullary spinal cord tumour associated with neurofibromatosis type 1. Acta Neurochir (Wien). 1997;139(11):1055-1060.
26. S. Constantini, EF. Intraspinal Tumors in infants and Children. YJ, editor. Neurological Surgery, vol. 4. Philadelphia: WB Saunders. 1996:3123-3133.
27. A.R. Cohen, et al. Malignant astrocytomas of the spinal cord. J Neurosurg. 1989;70(1):50-54.
28. F.J. Epstein, J.P. Farmer, D. Freed. Adult intramedullary astrocytomas of the spinal cord. J Neurosurg. 1992;77(3):355-359.
29. C. Hamburger, A. Buttner, S. Weis. Ganglioglioma of the spinal cord: report of two rare cases and review of the literature. Neurosurgery. 1997;41(6):1410-1415. discussion 1415–6
30. D.C. Miller, F.F. Lang, F.J. Epstein. Central nervous system gangliogliomas. Part 1: Pathology. J Neurosurg. 1993;79(6):859-866.
31. F.F. Lang, et al. Central nervous system gangliogliomas. Part 2: Clinical outcome. J Neurosurg. 1993;79(6):867-873.
32. T. Murota, L. Symon. Surgical management of hemangioblastoma of the spinal cord: a report of 18 cases. Neurosurgery. 1989;25(5):699-707. discussion 708
33. J.S. Nelson. Inhereted Tumor Syndromes Involving The Nervous System. In: J.S. Nelson, et al, editors. Principles and Practice of Neuropathology. New York: Oxford University Press; 2003:448-458.
34. S. Wizigmann-Voos, K.H. Plate, Pathology, genetics and cell biology of hemangioblastomas. Histol Histopathol, 11. 1996;4:1049-1061.
35. J. Brotchi, FG. Treatment. In: B.J. Fischer, G. Intramedullary Spinal Cord Tumors. Stuttgart: Thieme; 1996:60-84.
36. I. Landan, J. Gilroy, D.E. Wolfe. Syringomyelia affecting the entire spinal cord secondary to primary spinal intramedullary central nervous system lymphoma. J Neurol Neurosurg Psychiatry. 1987;50(11):1533-1535.
37. N.W. Hautzer, A. Aiyesimoju, Y. Robitaille. “Primary” spinal intramedullary lymphomas: a review. Ann Neurol. 1983;14(1):62-66.
38. A.C. McDonald, J.A. Nicoll, R. Rampling. Intramedullary non-Hodgkin’s lymphoma of the spinal cord: a case report and literature review. J Neurooncol. 1995;23(3):257-263.
39. D.K. Lee, et al. Multifocal primary CNS T cell lymphoma of the spinal cord. Clin Neuropathol. 2002;21(4):149-155.
40. A. Bekar, et al. A case of primary spinal intramedullary lymphoma. Surg Neurol. 2001;55(5):261-264.
41. P.A. Caruso, et al. Primary intramedullary lymphoma of the spinal cord mimicking cervical spondylotic myelopathy. AJR Am J Roentgenol. 1998;171(2):526-527.
42. A. Mrabet, et al. Cervicobulbar intramedullary lipoma. Apropos of a case with review of the literature. Neurochirurgie. 1992;38(5):309-314.
43. W. Jarmundowicz, J. Sakowski, E. Wilska. Own experience in surgical treatment of intramedullary spinal cord lipomas. Neurol Neurochir Pol. 1998;32(4):969-978.
44. M. Lee, et al. Intramedullary spinal cord lipomas. J Neurosurg. 1995;82(3):394-400.
45. F. Fujiwara, et al. Intradural spinal lipomas not associated with spinal dysraphism: a report of four cases. Neurosurgery. 1995;37(6):1212-1215.
46. L. Cristante, H.D. Hermann. Radical excision of intramedullary cavernous angiomas [see comments. Neurosurgery. 1998;43(3):424-430. discussion 430–1
47. J.M. Zabramski, et al. The natural history of familial cavernous malformations: results of an ongoing study. J Neurosurg. 1994;80(3):422-432.
48. D. Rigamonti, et al. Cerebral cavernous malformations. Incidence and familial occurrence. N Engl J Med. 1988;319(6):343-347.
49. S. Laberge-le Couteulx, et al. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet. 1999;23(2):189-193.
50. T. Sahoo, et al. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum Mol Genet. 1999;8(12):2325-2333.
51. H. Deutsch, JG, A. Faktorovich, F. Epstein. Spinal intramedullary cavernoma: clinical presentation and surgical outcome. J Neurosurg. 2000;93(Suppl. 1):65-70.
52. D. Zevgaridis, MR, C. Hamburger, H.J. Steiger, H.J. Reulen. Cavernous haemangiomas of the spinal cord. A review of 117 cases. Acta Neurochir (Wien). 1999;141:237-245.
53. D.A. Costigan, M.D. Winkelman. Intramedullary spinal cord metastasis. A clinicopathological study of 13 cases. J Neurosurg. 1985;62(2):227-233.
54. H. Chigasaki, PJ. A long term follow-up study of 128 cases of intramedullary spinal cord tumors. Neurol Med Chir(Tokyo). 1968;10:25-66.
55. R.N. Edelson, DM, J.B. Posner. Intramedullary spinal cord metastasis. Neurology. 1979;22:1222-1231.
56. P. Kehrli. [Epidemiology of brain metastases.]. Neurochirurgie. 1999;45(5):357-363.
57. P.C. McCormick, SB. Spinal Cord Tumors in Adults. In: Y, JR. Neurological Surgery. Philadelphia: WB Saunders; 1996:3102-3122.
58. K. Jellinger, et al. Intramedullary spinal cord metastases. J Neurol. 1979;220(1):31-41.
59. J.L. Grem, J. Burgess, D.L. Trump. Clinical features and natural history of intramedullary spinal cord metastasis. Cancer. 1985;56(9):2305-2314.
60. J.W. Thorpe, et al. Spinal MRI in patients with suspected multiple sclerosis and negative brain MRI. Brain. 1996;119(Pt 3):709-714.
61. L.I. Malis. Intramedullary spinal cord tumors. Clin Neurosurg. 1978;25:512-539.
62. G. Fischer, BJ. Intramedullary Spinal Cord Tumors. Stuttgart: Thieme, 1996.
63. M.J. Fine, et al. Spinal cord ependymomas: MR imaging features. Radiology. 1995;197(3):655-658.
64. J. Miller. Multiple Sclerosis. In: L. Rowland, editor. Merritt’s Neurology. Philadelphia: Lippincott Williams and Wilkins, 2000.
65. T.E. Feasby, et al. Spinal cord swelling in multiple sclerosis. Can J Neurol Sci. 1981;8(2):151-153.
66. A. Vighetto, et al. Intramedullary sarcoidosis of the cervical spinal cord. J Neurol Neurosurg Psychiatry. 1985;48(5):477-479.
67. R.M. Pascuzzi, et al. Sarcoid myelopathy. J Neuroimaging. 1996;6(1):61-62.
68. T. Morimoto, et al. Spinal cord sarcoidosis without abnormal shadows on chest radiography or chest CT diagnosed by transbronchial lung biopsy. Nihon Kokyuki Gakkai Zasshi, 39. 2001;11:871-876.
69. K. Prelog, S. Blome, C. Dennis. Neurosarcoidosis of the conus medullaris and cauda equina. Australas Radiol. 2003;47(3):295-297.
70. J. Peltier, et al. Sarcoidosis revealed by a spinal cord lesion. Rev Neurol (Paris). 2004;160(4 Pt 1):452-455.
71. V. Pierre-Kahn, et al. Intramedullary spinal cord sarcoidosis. Case report and review of the literature. Neurochirurgie. 2001;47(4):439-441.
72. F.C. Vinas, S. Rengachary. Diagnosis and management of neurosarcoidosis. J Clin Neurosci. 2001;8(6):505-513.
73. A.J. Tahmoush, et al. CSF-ACE activity in probable CNS neurosarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2002;19(3):191-197.
74. J.P. Zajicek. Neurosarcoidosis. Curr Opin Neurol. 2000;13(3):323-325.
75. G.I. Jallo, et al. Intraspinal sarcoidosis: diagnosis and management. Surg Neurol. 1997;48(5):514-520. discussion 521
76. P. Schubeus, et al. Spinal cord cavities: differential-diagnostic criteria in magnetic resonance imaging. Eur J Radiol. 1991;12(3):219-225.
77. A.A. Cohen-Gadol, et al. Spinal cord biopsy: a review of 38 cases. Neurosurgery. 2003;52(4):806-815. discussion 815–6
78. S. Petjom, et al. Angiostrongylus cantonensis infection mimicking a spinal cord tumor. Ann Neurol. 2002;52(1):99-101.
79. G. Samandouras, A. King, A.J. Kellerman. Schistosoma haematobium presenting as an intrinsic conus tumour. Br J Neurosurg. 2002;16(3):296-300.
80. M. Mehren, et al. Toxoplasmic myelitis mimicking intramedullary spinal cord tumor. Neurology. 1988;38(10):1648-1650.
81. T.H. Schwartz, MP. Intramedullary ependymomas: clinical presenttion, surgical treatment strategies and prognosis. J Neurooncol. 2000;47:211-218.
82. J. Brotchi, et al. A survey of 65 tumors within the spinal cord: surgical results and the importance of preoperative magnetic resonance imaging. Neurosurgery. 1991;29(5):651-66;. discussion 656–7
83. Q.W. Xu, et al. Aggressive surgery for intramedullary tumor of cervical spinal cord. Surg Neurol. 1996;46(4):322-328.
84. L.A. KearseJr, et al. Loss of somatosensory evoked potentials during intramedullary spinal cord surgery predicts postoperative neurologic deficits in motor function [corrected]. J Clin Anesth, 5. 1993;5:392-398, published erratum appears in, J Clin Anesth. 1993 Nov-Dec;5(6):529.
85. D.C. Adams, et al. Monitoring of intraoperative motor-evoked potentials under conditions of controlled neuromuscular blockade [see comments. Anesth Analg. 1993;77(5):913-918.
86. T. Murota, SL. Surgical management of hemangioblastoma of the spinal cord: a report of 18 cases. Neurosurgery. 1989;25:699-708.
87. K. Kothbauer, V. Deletis, F.J. Epstein. Intraoperative spinal cord monitoring for intramedullary surgery: an essential adjunct [see comments. Pediatr Neurosurg. 1997;26(5):247-254.
88. JS. Comment. Neurosurgery. 1997;41:1336.
89. H.D. Herrmann, M. Neuss, D. Winkler. Intramedullary spinal cord tumors resected with CO2 laser microsurgical technique: recent experience in fifteen patients. Neurosurgery. 1988;22(3):518-522.
90. G. Innocenzi, et al. Intramedullary astrocytomas and ependymomas in the pediatric age group: a retrospective study. Childs Nerv Syst. 1996;12(12):776-780.
91. G. Fischer, BJ. Functional Results. In: B.J. Fischer, G. Intramedullary Spinal Cord Tumors. Stuttgart: Thieme; 1996:85-90.
92. J.K. Houten, H.L. Weiner. Pediatric intramedullary spinal cord tumors: special considerations. J Neurooncol. 2000;47(3):225-230.
93. A.L. DeSousa, et al. Intraspinal tumors in children. A review of 81 cases. J Neurosurg. 1979;51(4):437-445.
94. S. Yasuoka, H.A. Peterson, C.S. MacCarty. Incidence of spinal column deformity after multilevel laminectomy in children and adults. J Neurosurg. 1982;57(4):441-445.
95. R. Abbott, FN, J.H. Wisoff, F.J. Epstein. Osteoplastic laminotomy in children. Pediatr Neurosurg. 1992;18:153-156.
96. P.R. Cooper. Outcome after operative treatment of intramedullary spinal cord tumors in adults: intermediate and long-term results in 51 patients. Neurosurgery. 1989;25(6):855-859.
97. C. O’Sullivan, et al. Spinal cord tumors in children: long-term results of combined surgical and radiation treatment. J Neurosurg. 1994;81(4):507-512.
98. S.I. Ryu, et al. Image-guided hypo-fractionated stereotactic radiosurgery to spinal lesions. Neurosurgery. 2001;49(4):838-846.
99. P.C. Gerszten, et al. CyberKnife frameless stereotactic radiosurgery for spinal lesions: clinical experience in 125 cases. Neurosurgery. 2004;55(1):89-98. discussion 98–9
100. G. Kopelson, et al. Management of intramedullary spinal cord tumors. Radiology. 1980;135(2):473-479.
101. S. Constantini, MD, J.C. Allen, L.B. Rorke, D. Freed, F.J. Epstein. Radical excision of intramedullary spinal cord tumors: surgical morbidity and long-term follow-up evaluation in 164 children and young adults. J Neurosurg. 2000;93(Suppl. 2):183-193.
102. R. Jyothirmayi, MJ, M.K. Nair, B. Rajan. Conservative surgery and radiotherapy in the treatment of spinal cord astrocytoma. J Neurooncol. 1997;33:205-222.
103. H. Shirato, KT, K. Hida, I. Koyanagi, Y. Iwasaki, K. Miyasaka, et al. The role of radiotherapy in the management of spinal cord glioma. Int J Rad Oncol Biol Phys. 1995;33:323-328.
104. S.J. Whitaker, et al. Postoperative radiotherapy in the management of spinal cord ependymoma. J Neurosurg. 1991;74(5):720-728.
105. B. Guidetti, S. Mercuri, R. Vagnozzi. Long-term results of the surgical treatment of 129 intramedullary spinal gliomas. J Neurosurg. 1981;54(3):323-330.
106. M.P. McLaughlin, BJ, R.B. Marcua, B.L. Maria, P.J. Mickle, A. Kedar. Outcome after radiotherapy of primary spinal cord glial tumors. Rad Oncol Invest. 1998;6:276-280.
107. S. Sgouros, MC, A. Jackowski. Spinal ependymomas-the value of postoperative radiotherapy for residual disease contro. Br J Neurosurg. 1996;10:559-566.
108. S.R. Isaacson. Radiation therapy and the management of intramedullary spinal cord tumors. J Neurooncol. 2000;47(3):231-238.
109. J.L. Finlay, et al. Randomized phase III trial in childhood high-grade astrocytoma comparing vincristine, lomustine, and prednisone with the eight-drugs-in-1-day regimen. Childrens Cancer Group. J Clin Oncol. 1995;13(1):112-123.
110. C. Balmaceda. Chemotherapy for intramedullary spinal cord tumors. J Neurooncol. 2000;47(3):293-307.
111. R.J. Packer, et al. Carboplatin and vincristine for recurrent and newly diagnosed low-grade gliomas of childhood. J Clin Oncol. 1993;11(5):850-856.
112. S.P. Lowis, et al. Chemotherapy for spinal cord astrocytoma: can natural history be modified? Childs Nerv Syst. 1998;14(7):317-321.
113. V. Doireau, et al. Chemotherapy for unresectable and recurrent intramedullary glial tumours in children. Brain Tumours Subcommittee of the French Society of Paediatric Oncology (SFOP). Br J Cancer. 1999;81(5):835-840.
114. M. Hoshimaru, et al. Results of microsurgical treatment for intramedullary spinal cord ependymomas: analysis of 36 cases. Neurosurgery. 1999;44(2):264-269.
115. CEd Rawlings, et al. Ependymomas: a clinicopathologic study. Surg Neurol. 1988;29(4):271-281.
116. M.J. DeVivo, et al. Cause of death for patients with spinal cord injuries. Arch Intern Med. 1989;149(8):1761-1766.
117. H.M. Sandler, et al. Spinal cord astrocytomas: results of therapy [see comments. Neurosurgery. 1992;30(4):490-493.
118. H.H. Hardison, et al. Outcome of children with primary intramedullary spinal cord tumors. Childs Nerv Syst. 1987;3(2):89-92.
119. F. Epstein. Surgical management of intramedullary spinal cord tumors: functional outcome and sources of morbidity (Comments). Neurosurgery. 1994;35:69-76.
120. T.E. Merchant, et al. High-grade pediatric spinal cord tumors [In Process Citation. Pediatr Neurosurg. 1999;30(1):1-5.